Note: Descriptions are shown in the official language in which they were submitted.
w o 93/17019 21~0563 PCI/US93/00880
2,6-Methano-2H-quinol~z~n der~vat~ve as 5-HT3-receptor antagon~st
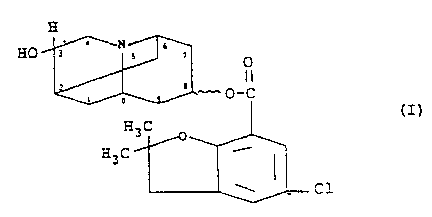
This invention relates to 5-chloro-2,3-dihydro-2,2-
dimethylbenzofuran-7-carboxylic acid-octahydro-3-hydroxy-
2,6-methano-2H-quinolizin-8-yl ester, a novel 5-HT3-receptor
antagonist, its method of preparation, and to its end-use
application in the treatment of conditions responsive to
5-HT3 receptor antagonism such as radio- and chemo-therapeu-
tically-induced nausea and vomiting, ~ the treatment of
10 pain associated with migraine, in the treatment of cognitive
disorders such as Alzheimer's Disease, the treatment of
hallucinatory endogenous psychoses of the type manifested in
patients suffering from schizophrenia and mania, the
treatment of irritable or inflammatory bowel syndrome, or to
1~ combat drug abuse.
~ ore specifically this invention relates to compounds of
the formula
HO--3~ -~ ~
~O-C
H ~
~ Cl
WO93/17019 PCT/US93/00880
2 1305 63 - 2 -
its tautomers, stereo- and geometrical isomers, and mixtures
thereof, and to the pharmaceutically acceptable salts
~hereof.
5 As used herein, the wavy line bonding the oxygen atom of the
ester moiety to the 8-position of the octahydro-2,6-methano-
2H-quinolizin moiety ~hereinafter sometimes referred to as
the methano bridged quinolizinyl moiety) indicates that the
bonding may be in the endo (trans) or the exo (cis)
10 configuration. The preferred configuration is endo.
Preparation of such geometric isomers may be effected by the
processes and techniques of U.S. Patent 4,906,755 which is
incorporated herein by reference. A chirality exists at the
3-position of the methano-bridged quinolizinyl moiety
15 presenting d- or l-isomers and racemate mixtures thereof.
When desired resolution of said racemates may be effected by
standard procedures and techniques well known in the art.
The (+) enantiomer is preferred.
The pharmaceutically acceptable acid addition salts
referred to above can be non-toxic salts with suitable acids
such as those with inorganic acids, for example,
hydrochloric, hydrobromic, nitric, sulfuric or phosphoric
acids; or with organic acids such as organic carboxylic
25 acids, for example, acetic, propionic, glycolic, maleic,
hydroxymaleic, malic, tartaric, citric, salicylic, 2-acetyl-
oxybenzoic, nicotinic or isonicotinic; or organic sulfonic
acids, for example, methanesulfonic, ethanesulfonic, 2-
hydroxyethanesulfonic, 4-toluenesulfonic or 2-naphthalene-
30 sulfonic.
The preparation of the compounds of the presentinvention may be illustrated by the following example.
2130~63
WO93/17019 PCT/US93/00880
- 3
EXAMPLE
5-CHLORO-2,3-DIHYDRO-2,2-DIMETHYLBENZOFURAN-7-CARBOXYLIC
ACID ~RANS-OCTAXYDRO-3-HYDROXY-2,6-METHANO-2H-QUINOLIZIN-8-
5 YL ESTER
STEP A: Using triphosgene
5-Chloro-2,3-dihydro-2,2-dimethylbenzofuran-7-carboxylic
acid trans-octahydro-3-oxo-2,6-methano-2H-quinolizin-8-Yl
l0 ester
A solution of triethylamine (1.79 g), 4-dimethylamino-
pyridine (l.09 g) and 5-chloro-2,3-dihydro-2,2-dimethyl-
benzofuran-7-carboxylic acid (3.8 g) in dichloromethane
15 (75 ml) was slowly added to a stirred solution of tri-
phosgene (5.28 g) in dichloromethane (l00 ml) at 0~C,
nitrogen being continuously bubbled through the mixture.
After the addition, the mixture was stirred at room
temperature overnight under nitrogen, filtered and
20 evaporated.
A suspension of the residue in anhydrous toluene was
refluxed with a stirred suspension of hexahydro-8-hydroxy-
2,6-methano-2H-quinolizin-3(4H)-one (3.25 g) overnight. The
25 cooled solution was filtered, the toluene washed with
aqueous potassium carbonate, dried over magnesium sulphate
and evaporated to give a residue (4.8 g) containing the
title compound. It was then purified by partitioning between
ethyl acetate (200 ml) and lN methanesulphonic acid (40 ml).
30 Basification of the separated methanesulphonic acid solution
with a saturated aqueous solution of potassium carbonate
gave a crystalline solid which was further purified by
crystallization, e.g., from aqueous methanol or by silica
gel ch~omatography to give a pure sample of the title
35 compound
W093/17019 PCT/US93/OOX80
2~30s63
Step A' (Alternate to Step A): Using trichloromethyl
chloroformate
5 5-Chloro-2,3-dihydro-2,2-dimethylbenzofuran-7-carboxylic
acid trans-octahydro-3-oxo-2,6-methano-2H-quinolizin-8-yl
ester
A solution of trichloromethyl chloroformate (1.76 g,
10 1.08 ml) in dichloromethane (10 ml) was added to a stirred
solution of 5-chloro-2,3-dihydro-2,2-dimethylbenzofuran-7-
carboxylic acid (2 g) and triethylamine (1.4 ml) in
dichloromethane (30 ml) at 0~C. The mixture was stirred
overnight at room temperature, washed with an ice cold
15 saturated aqueous solution of ammonium chloride, dried over
magnesium sulphate and evaporated to give an oil (2.2 g).
A stirred mixture of the oil ((2.2 9), hexahydro-8-hydroxy-
2,6-methano-2H-quinolizin-3(4H)-one (1.6 g) and toluene
(30 ml) was refluxed overnight. The cooled mixture was
20 washed with a saturated aqueous solution of potassium
carbonate and then four times with water to remove any
unreacted hexahydroquinolizine one. Evaporation of the dried
toluene solution (MgSO4) gave a solid residue which was
recrystallized from ethyl acetate/hexane to give the title
25 compound (0.35 g).
STEP B:
5-Chloro-2,3-dihydro-2,2-dimethylbenzofuran-7-carboxylic
acid trans-octahydro-3-hydroxy-2,6-methano-2H-quinolizin-8-
yl ester
Sodium borohydride (1.52 g) was slowly added to astirred solution of 5-chloro-2,3-dihydro-2,2-dimethylbenzo-
furan-7-carboxylic acid trans-octahydro-3-oxo-2,6-methano-2H-
35 quinolizin-8-yl ester (3.9 g) in ethanol (75 ml) at room
2130563
WO93t17019 PCT/US93/00880
temperature and the mixture stirred at room temperature
overnight. The ethanol was evaporated, the residue dissolved
in a mixture of water (20 ml) and 2N hydrochloric acid
(20 ml), and the acidic solution almost immediately basified
5 by the addition of an excess of a saturated solution of
aqueous potassium carbonate. Extraction with a mixture of
tetrahydrofuran-ethyl acetate (l.l) and evaporation of the
dried extract gave the title compound. m.p.: 192-193~C.
Conversion to the methane sulphonate salt was effected
by the addition of one equivalent of an alcoholic solution
of methanesulphonic acid and evaporation of the solvent.
The racemic mixture can be separated into its separate
15 enantiomers by standard techniques.
WO93/17019 PCT/US93/00880
~3~S~3 - 6 -
The compounds of the present invention block the M
receptors for 5-hydroxytryptamine (5HT) on afferent sensory
neurons otherwise known as 5HT3-receptors. The activity of
the compounds of this invention against the 5HT3-receptor
5 can be assessed by determining their PA2 values in the
isolated rabbit heart as described by J. R. Fozard et al.,
Eur. J. Pharmacol. 59, 195-210 (1979). The in vivo
5HT3-receptor antagonist activity can be assessed by
measurement of the effect of the compound on the Von Bezold-
10 Jarisch reflex induced by 5HT injected intravenously intothe rat (see Paintal A. S., Physiol. Rev. 53, 159-227
(1973); J. R. Fozard, Naunyn-Schmiedeberg's Arch. Pharmacol.
326, 36-44 (1984).
Using the foregoing standard procedures, as well as
other standard procedures recognized to illustrate
5HT3-receptor antagonist activity for the above-stated end-
use applications, as well as by comparison with other
5~T3-receptor antagonists known to be useful for such
20 purposes, the compounds of this invention may be utilized in
a variety of treatments.
Generally, the compounds of the present invention are
useful in treating conditions responsive to 5-HT3 receptor
25 antagonism. Examples of conditions responsive to 5-HT3
receptor antagonism are well known to those skilled in the
art. Some examples of these conditions are treating anxiety,
psychosis, glaucoma and for stimulating gastric motility
(U.S. Patent No. 5,011,846), treatment of panic disorders
30 and/or agoraphobia or obsessive compulsive disorders (Patent
No. EP 422,154); treatment of autism or other disorder
originating in childhood in which there is mental
retardation (Patent No. EP 450,757); treatment of cognitive
disorders such as Alzheimer's Disease (U.S. Patent
35 Application Serial No. 806,987); production of orexiogenic
- 7 ~ 2 1 30 5 63
effect (U.S. Patent Application Serial No. 742,951);
serotonin-induced nasal disorders, rhinitis or impaired
approach-oriented behavior in stressful situations or for
increasing vigilance (Patent No. GB 2,193,633); relief or
prevention of withdrawal syndrome resulting from addiction
to and/or for the suppression of dependence on a drug or
substance (Patent No. EP 279,114 and Gs 2,206,788); treat-
ment of cough and/or bronchoconstriction (Patent No. EP
340,270); treatment of nausea, bradycardia, and/or hypo-
tension associated with myocardial instability (Patent No.WO91/09593); treatment of urinary incontinence (Patent No.
EP 467,365); and combination therapy with ACE inhibitors
(Patent No. EP 477,625 and EP 477,624), with 3-[2-(dimeth-
ylamino)ethyl]-N-methyl-lH-indole-5-methanesulphonamide
(Patent No. EP 433,043); and histamine H2-receptor antago-
nist (Patent No. EP 275,699).
Preferred uses for the compounds of the present inven-
tion include the treatment of nauseas and vomiting, particu-
larly radio- and chemo-therapeutically induced in those
patients being treated for cancer, in the treatment of pain
associated with migraine and neuralgia, in the treatment of
cognitive dysfunctions such as memory and learning disorders
as well as dysfunctions in selective attention, in the
treatment of hallucinatory endogenous psychoses of the type
manifested in patients suffering from schizophrenia and
mania, for the treatment of irritable or inflammatory bowel
syndrome, as well as to be useful in combating drug abuse.
Doses administered to patients are within the range of
about 0.01 to about 10 mg per kilogram of body weight, with
0.01 to 1 mg per kilogram of body weight being preferred for
~2
WO93/17019 PCT/US93/00880
2130~ 63 - 8 -
parenteral administration and 0.25 to l mg per kilogram of
body weight upon enteral administration. Of course, the
dosage required for the treatment of the foregoing disease
states will depend upon such factors as the severity and
5 stage of the particular disease, the age and condition of
the patients as such other normal factors taken into
consideration by the attending diagnostician.
The term "patient" means warm-blooded animals such as
lO rats, mice, dogs, cats, guinea pigs, primates and humans.
The term "treat" means to prevent or alleviate the patient's
disease or condition.
The compounds of Formula (I) can be administered in
15 various manners to achieve the desired effect. For oral
administration, the compounds can be formulated into solid
or liquid preparations such as capsules, pills, tablets,
lozenges, melts, powders, suspensions, or emulsions. Solid
unit dosage forms can be capsules of the ordinary gelatin
20 type containing, for example, surfactants, lubricants and
inert fillers such as lactose, sucrose, and cornstarch or
they can be sustained release preparations. In another
embodiment, the compounds of Formula (I) can be tableted
with conventional tablet bases such as lactose, sucrose, and
25 cornstarch in combination with binders, such as acacia,
cornstarch, or gelatin, disintegrating agents such as potato
starch or algenic acid, and a lubricant such as stearic acid
or magnesium stearate. Liquid preparations are prepared by
dissolving the active ingredient in an aqueous or non-
30 aqueous pharmaceutically acceptable solvent which may alsocontain suspending agents, sweetening agents, flavoring
agents, and preservative agents as are known in the art.
For parenteral administration, the compounds may be
35 dissolved in a physiologically acceptable pharmaceutical
- 9 - 2 1 305 6 3
carrier and administered as either a solution or a suspen-
sion. Illustrative of suitable pharmaceutical carriers are
water, saline, dextrose solutions, fructose solutions, eth-
anol, or oils of animal, vegetative, or synthetic origin.
The pharmaceutical carrier may also contain preservatives,
buffers, etc. as are known in the art.
The compounds of this invention can also be adminis-
tered topically. This can be accomplished by simply prepar-
ing a solution of the compound to be administered, prefer-
ably using a solvent known to promote transdermal absorption
such as ethanol or dimethyl sulfoxide (DMSO) with or without
other excipients. Preferably topical administration will be
accomplished using a patch either of the reservoir and por-
ous membrane type or of a solid matrix variety.
Some suitable transdermal devices are described in U.S.Patent Nos. 3,742,951, 3,797,494, 3,996,934, and 4,031,894.
These devices generally contain a backing member which de-
fines one of its face surfaces, an active agent permeableadhesive layer defining the other face surface and at least
one reservoir containing the active agent interposed between
the face surfaces. Alternatively, the active agent may be
contained in a plurality of microcapsules distributed
throughout the permeable adhesive layer. In either case,
the active agent is delivered continuously from the reser-
voir or microcapsules through a membrane into the active
agent permeable adhesive, which is in contact with the skin
or mucosa of the recipient. If the active agent is absorbed
through the skin, a controlled and predetermined flow of the
active agent is administered to the recipient. In the case
of microcapsules, the encapsulating agent may also function
as the membrane.
r~
D
- lO 2 1 3 0563
In another device for transdermally administering the
compounds in accordance with the present invention, the
pharmaceutically active compound is contained in a matrix
from which it is delivered in the desired gradual, constant
and controlled rate. The matrix is permeable to the release
of the compound through diffusion or microporous flow. The
release is rate controlling. Such a system, which requires
no membrane is described in U.S. Patent No. 3,921,636. At
least two types of release are possible in these systems.
Release by diffusion occurs when the matrix is non-porous.
The pharmaceutically effective compound dissolves in and
diffuses through the matrix itself. Release by microporous
flow occurs when the pharmaceutically effective compound is
transported through a liquid phase in the pores of the
matrix.
Specific formulations of the present invention are
prepared in a manner well known per se in the pharmaceutical
art and usually comprise one or more active compounds of the
invention in admixture or otherwise in association with a
pharmaceutically acceptable carrier or diluent therefor.
The active ingredient will usually be mixed with a carrier,
or diluted by a diluent, or enclosed or encapsulated in a
capsule, sachet, cachet, paper or other container. A car-
rier or diluent may be solid, semisolid or liquid materialwhich serves as a vehicle, excipient or medium for the ac-
tive ingredient. Suitable carriers or diluents are well
known per se. See Remington's Pharmaceutical Sciences, Mack
Publishing Company, Easton, Pennsylvania, for a description
of the preparation of such formulations.
1~