Note: Descriptions are shown in the official language in which they were submitted.
p3() .5~ 3 0 ~3
P~-~AR~ CEUTICALS
n.e present invention rel;ltes to novel compounds which ~rc ot po[~n[i~l u~ as
5 antiviral agents, [o prucesses lor Lheir prepara~ion and to their use ~s ph~rm~ccuticals.
EP-A-319228 and EP-A-353955 (Beecham Group p.l.c.) disclose a oroup of purine
derivatives containing a 9-[2-(phosphoncmethoxy)alkoxy] substituent. which are
described as having antiviral activity.
- EP-A-3S3955 (Ceslcoslovenska akademie ved) discloses a group of
9-(phosphonomethoxyalkyl)adenines, which are described as havin~ antiviral activity.
'Nucleotide An~looues as Antiviral Agents' ACS Symposium Series 401, Editor J.C.
15 Martin, published by the American Chemical Society, Washington DC (1989) Chapters
4 and 5 discloses, a number of (phosphonometho~yalkyl) derivatives of purines and
pyrimidines and their antiviral activity.
Particular compounds of interest are adenine or guanine having a 9-substituent as
2~ follows:
(HO)2POCH20cH2cH2o- Ex.2, EP-A-319~
(HO)2POCH20CH2CH(CH20H)O- Ex.16, EP-A-206~59
(H0)2POCH20CH2CH2- PMEA/PMEG
- (H0)2POCH20CH(CH20H)c~2- HPMPA/HPMPG
It nas now been discovered that certain derivatives of these compounds are prodru~,s
therefor, having improved gastrointestinal absorption properties.
`3 '`' ~c~
O 93/19075 2 ~ ' 2 3 ~ 2 - rC~ /GI;93/005~'
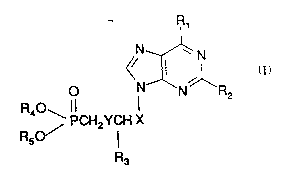
Accordingly, ~he present invennon provides a compound of fomlula (I), or a
pharmaceutically accep~able salt thereof:
o ~XN~'1R
PCH2YCH X
R5
R3
S (I)
wherein
X is -CH2O, -CH2 or -CH(CH20R6)0 where R6 is hydrogen or acyl;
YisOo~S;
10 Rl is hydroxy or amino;
R2 is amino or hydrogen;
R3 is hydrogen or, when X is CH2O and Y is O, R3 may be CH20R7 where R7 is
hydrogen or acyl; and
R4 and Rs are both phenyl, ~bromophenyl, 4-methylphenyl, 4-methoxyphenyl, 2-
lS acctoxyphenyl or 2^me~ylphenyl. :
When Rl is hydroxy and R2 is amino9 the compound of formula (~) is a guaninedcrivative;
20 When Rl is amino and R2 is hydrogen, the compound of foImula (I) is an adenine
deriva~ve;
Whcn Rl is hydroxy and R2 is hydrogen, the compound of formula (1) is a
h~xanthine denvahve; and
25 - ~
Whcn Rl and R2 aIe both arnino groups, thc compound of formula (1) is a
2,6 diaminopmine desivative~
Often, d~e compound of formula (I) is a guanine or adenine derivative.
R4 and Rs arc prcfesably both phenyl.
~13~3~3
93/1907' PCI /GB9.~/()O~
- 3 -
Suitable examples of R6/R7 when acyl include ca~>oxylic acyl, such as Cl 7 aLkanoyl
and benzoyl optionally substituted in ~e phenyl nng by one, two or three groups or
atoms selected from halogen, such as fluoro, chloro, bromo, and Cl 4 alkyl or Cl 4
5 aL~coxy whereln the alkyl moiety is selected from methyl, ethyl, n- and iso-propyl, n-,
sec-. is~ and tert-butyl. Prefe~ed acyl groups include acetyl, propionyl, butyryl,
heptanoyl and hexanoyl.
There are groups of compounds of interest wherein:
i) X is ~CH2O and R3 is hydrogen.
ii) X is -CHzO and R3 is CH20R7 as defined.
iii) X is -CH2(CH20R6)0 as defined and R3 is hydrogen.
iv) X is -CH2 and R3 is hydrogen.
v) X is -CH2 and R3 is C~H20R7 as defined.
Y is preferably O.
Examples of pharmaceutically acceptable salts of the compound of formula (I) are acid
~ddition salts formed with a phannaceu~cally acceptable acid such as hydrochloric
25 ~d, or~ophosphoric acid and sulphuric acid. Pha~naceu~acally acceptable s~ts also
include ~ose fo~med with organic bases, preferably with amines, such as
ethanol~es or diamines; and alkali metals, such as sodium and po~ssium.
It will be ap~ated that some o~ the compounds of formula (1), especially those
30 whercin R3 is o~er dlan hydrogen, have an asymmetric centre, and therefore are
capablc of cxis~ng in more than one stereoisomeric f~nn. The inven~on extends toeach o~ thcsc f~ms individually and to mixtures thereof, including racemates. The
isome~s may be scparatcd convcntionally by chromatographic methods or using a
rcsohnng agent. Altematively, dle individual isomers may be prepared by asymmc~ric
35 sSmthcsis using chiral intelmediates.
Thc compounds of fc~rmula (I) including their alka.li metal salts may form solvates such
as hydrates and thesc are included wherever a compound of folmula (I) or a salt thsrcof
wo 93l1907~ pcrtGB9~ o~
2 ~ 3 æ 3 9 4
is herein refelTed tO.
It will be appreciated that, when Rl is hydroxy in formula (I~ the compound exists in
the predominant tautomeric form of structure (LA):
S O
< X'',l
0 N N R2
R4CL--PCH YCH
- R50
R3 :
(l[A) .. ,
The invention also provides a process for the preparation of a compound of formula (I),
10 or a pharmaceutically acceptable salt thereof, which pr~cess comprises reacting a
compound of formula ~
<
o N N R2
Cl~ll
, PCH2YCH X
~1 1
R3
with R40H and RsOH and Rl, R2, and R3 are as defined in formula (I), and thereafter
optionally fo~ning a pharmaceutically acceptable salt thereof.
. . .
The reaction takes place in a suitable inert solvent such as dichloromethane at
20 tcmperatures with cooling 0 to 3C, under an inert atmosphere.
As appropriate or necessary, Rl/R2/R3 may be pr~tected. Suitable examples of
protecting groups and their removal, are as described in EP-A-242482. A par~cularly
suitable protecting group for R3 when other than hydrogen is the t-butyldiphenylsilyl
~.
)93/19075 - - ~ 10 .3 PCT/GB93/00560
group removable by conventional methods.
The compounds of formula (~) may be generated from the co~esponding compounds
of formula (I~, but wherein R4/Rs is replaccd by an ethyl group, by trea~nent with
5 bromotrimethylsilane in dichloromethane followed by chlorina~on with PCls in
dichl~methane: caIbon tetrachlo~ide.
It will bc appreciated th~t the aboYe convcrsions may takc place in any desired or
neccssary order, having rcgard t~ thc final desired compound of formula (1).
Thc R4~ = edlyl compounds of the formula (1) are prepared as described in
A-313289 and the aforcmentioned publications, the subject matter of which are
inc~npora~ herein by rcfcrcncc.
15 Whcn R6~R7 is hydroxy, appropriate selective protection may be required, eg using
acetate.
.
Pharmaceutically acceptable salts may be prepared in conventional manner, for
cxample, in d~e case of acid addition salts, by reaction with the appropriate ~rganic o~
20 inorganic acid.
lt will be appreciated that the inv~ntion provides a process for the preparation of a
compound of fonnula (I) wherein R6J~7 is hydrogen which process comprises the
deprotection of a correspondin~ compound of f~ula (I) wherein Rg/Rg is protected25 hy~xy. Methods for deprotec~on, are conventiona~ for the protecting group
conccrne~
The compounds of ~e invcnnon are of potendal use in the ~eatment of infections
- caused by viruses, in pa~cular DNA viruses and ~ oviruses. Examples of DNA
30 viruses include he~pesviruses such as herpes simplex types 1 and 2, varicdla-zoster
virus, Eps~ Ba~ virus and ~rtomegalo~us. Examples of retr~viruses include
len~iviruses such as visna ~nrus and human imIslunodeficiency virus (strains 1 and 2).
The compounds may also be inhibitors of tumorogenic viruses and~or of potential use
35 in thc =ent of ncoplastic diseases, i.e. cancer.
WO 93/19075 PCl /ÇR93/0056'
`~ 3~3~3 -6-
Compounds of the invention may be fo~nulate~ for use in a pharrnaceutical
composition. Accordingly, in a further aspect of the invention, there is provided a
pharmaceutical composition which comprises a compound of formula (I) or
pharmaceuticaDy acceptable salt thereof together with a pharmaceutically acceptable
5 ca~Ticr or excipicn~
A composidon which may be administered by the oral route to humans may be
Impoonded in thc form of a syrup, tablct o~ capsule. ~Vhen thc composition is in the
foIm of a tablct, any pharmaccutical calTicr suitablc fo~ fo~mulating such solid10 c~ositions may be uscd, for examplc magncsium stearate, starch, lactosc, glucose,
r,icc, flour and chall~ Ibe composidon may also be in the form of an ingcstible
capsulc, for cxample of gclatin, to contaîn thc compound, or in thc fo~m of a syrup, a
soludon ora suspcnsion. Suitablc liquid pharmaccutical camers include ethyl alcohol,
glyc~inc, saline and watcr to which flavouring o~ colouring agents may be added to
15 form syrups. Thc compounds may also be prcscnted with a sterile liquid ca~ier for
injcction.
lhc composition may also be fo~mulated for topical apph;cation to the skin or cyes.
20 For topical application to thc sl~n, the composition may be in the form of a cream,
lotion o~ ointmen~ These fo~mulations may be conventional foqmulations well known
in thc art, f~3r cxample, as dcscribed in standard books of pharmaccutics and cosmctics,
such as Ha~y's Cosmeticology published by Lconard Hill Books and the British
Pharmacopaeia
Thc composition for application to the eyes may be a conventional eye-drop
composition well h~own in the art, or an ointmenl composition.
Preferably, the composition of this invention is in unit dosage fo~m or in some other
form ~at may be administcr~ in a single dose. A suitable dosage unit might contain
30 fr~m 50 mg to 1 g of active ingredient, for example 100 to 500 mg.
- Such doses may be ad~nisterd 1 to 4 times a day or more usually 2 or 3 dmes a day.
The cffcc~ve dose of compound will in general be in the range of from 1.0 to 20 mg/kg
of body wdght pcr day or more usually 2.0 to 10 mg/lcg per day.
No unacceptablc to~icological effcctsi are indicated at the above dcscribcd dosagc
lc~rcls.
393/1907s ~ 3 ~ 3 r~/GB9~/00560
The invention also provides a method of treating viral infections/diseases in a human or
non-human animal, which comprises administering to the animal an effective,
non-toxic amount of a compound of formula (I) or a pharmaceutically acceptable salt
thereof.
S
Ihc invcntion also provides a compound of formula (I) or a pharmaceutically
acccptablc salt dlereof for use as an active the~apcutic substance, in particular for the
t~cnt of viral infections/discases`.
The compounds of the invcntion are also believed to exhibit a synergistic antiviral - ~
cffcct in conjuncdon with intcrfcrons; and combination products comprising these two ~ --
- componcnts for sequcntial or concomitant administration, by the same or differcnt
routcs, arc thcrcfo~c ~ithin the ambit of the present invention
15 Thc following cxamplcs illustrate the invention.
.:
,-'~'.
wo 93/19û75 ~, pcr/GB93/ou56
8-
Examples
The following compoun~ls of ~ormula (I) are prepared.
Example R1 R2 R~ R~, X Y
NH2 H H C~ 2
II NH2 H H ~Br-C~ I20 0
m NH2 H H 4-CH~,-C~ H20 0
IV N~I2 H H ~CH?,~C~H4 CH~O O
V NH2 H H 2,-CH~C02-C~H4 CH20 0
VI NH2 H H 2~ C~
In cxamples I-VI, the follo~g general synthehc pr~cedure was used:
~NH2 i)~MSBr,CH2CI2 1 2
~N iD~ ~~ cc~ <N_lî N
~ J~ d iii)ROH.Et3N,CH2cl2~ NJ~ ~J
O N N l-met~yl~nidazole 1l I N
(EtO)21lVO l (R)2PV l
R = C6H5 -~
II R = 4-Br-C6H4
m ~ = 4 cH3-C6H4
IV R = 4-CH30 C6H4
V R = 2- CH3CQ C6H4
VI R = 2-CH3-C6H4
.-.
) 93/1907~ 3 PCI /GB9?./00~i6()
,
~xamples I-Vl
To a solution of the diethyl ester ( 1 .45Ir~nol) in dry dichloromethane (5ml)
bromotnmethylsilane (14.5mmol) was added. The reaction mixture was stiITed at RTS for 2h and the solvents were evaporated. The residue was coevaporated with drytoluene (2x5ml) and the resulting glass was dissolvcd in dry dichloromethane: carbon
tetrachloridc ~.olution (3:1, lOml). Phospho¢us pentachloride (3.045mmol) was then
addcd and the rcaction mixture was stirrcd at RT for 3h. The solvcnts were evaporated. ~-
the rcsiduc was coevaporated with dry toluene (lxlOml). The resulting phosphonyldichloridatc dcri~rativc was suspendcd in dry dichloromcthane (lOml), the appropriate `~
aloohol (3.045mmol) was added and thc solution was coolcd to 0-3C undcr argon.Thc reaction mixture was thcn treated with triethylamine (3.625mmol) followed by 1-
methylimidazole (5.8mmol), stirred at 0 -3C for lOmin and then at RT for l.Sh. The
plecipitate was filtered, washed with dichloromcthane and the solvents were ~ ~ -
cvaporatcd. Thc residue was coevaporated with tolucne (2x20ml), dissolved in
chlorofoIm (801rd), washed succcssivcly with aqueous sodium hydrogen car'oonate
(2x20ml) and water (2x20ml). The product was purified by column chromatography
on silica gd dudng with chlorofonn-ethanol to give the compound examples I-VI in12-40% yicld.
Example I, R=C6Hs, 36% yield
~H t(CD3)2SO~ 3.97 (2H, m, C~20CH2P)? 4.35 (2H, d, J--7.15Hz, CH2P), 4.55 (2H, ~-
m, NOCH2), 7.21-7.44 (12H, m, Ar and NH2. D20 exchangeable), 8.15 (lH, s, H-2),
8.31 (lH, s, H-8).
Found: C, 50.84; H, 4.20; N, 14.59%. C2bH20osp x 0.35 CHC13 requires C, 50.59,
H, 4.24; N, 14.49%.
Example II, R=4-Br-C6H4, 31% yield
~H [(CD3)2S01 3.96 (2H, m~ CH2OCH2P), 4.38 (2H, d, J=7.42Hz, CH2P), 4.54 (2H,
m, NOCH2), 7.2()-7.63 (8H, m, Ar), 7.39 ~2H, br.s, D20 cxchangeable, NH2), 8.15
(lH, s, H-2), 8.31 (lH, s, H-8).
Found: C, 38.50; H, 2.88; N, 10.84%. C2~ gNsOsPBr2 x 0.3 CHCl3 requires C,
38.4, H, 2.90, N, 10.87%.
Elcamplc m, ~CH3-C~14, 12% yield
WO 93/1907-~ PCI /~ 9~/~)0~
H ((CD3)2SO] 3.31 (6H, s, CH3), 3.94 (2H, m, Ç~2ocH2p)7 4.38 (2H, d, J=7.15Hz,
CH2P), 4.54 (2H, m, NOCH2), 7.08-7.21 (~H, m, Ar), 7.38 (2H, br.s, D20
exchangeable, NH2), 8.15 (lH, s, H-2), 8.31 (lH, s, H-8).
Found: M+H (C.I.) 470.1594; C22H24NsOsP requires M~H 470.1594.
Example IV, R~CH3~C6H4, 26% yield
~H [(CD3)2SO] 3.73 (6H, s, OCH3), 3.95 (2H, m, ~120C~I2P). 4.27 (2H, d,
J=7.15Hz, CH2P), 4.55 (~ m, NOCH2), 6.68-7.32 (8H, m, Ar), 7.38 (2H, b~.s, D20
exchangeable, NH2), 8.15 (lH, s, 2-H), 8.33 (lH, s, 8-H).
- Found: C, 50.37; H, 5.50, N, 13.92%, M+H (C.I.) 502.1492; C22H24NsO7P x H20
- s~quires C, 50.87; H, 5.05; N, 13.48%, M+H 502.1492. ~:
Exampl~ V, R=2-CH3(~O2-C6H4, 14% S ield
~H [(CD3)2S03 2.20 (6H, s, CH3C02), 3.96 (2H, m, ~120CH2P~t 4.36 (2H, d,
J=6.87H~, CH2P), 4.55 (2H, m. NOCH2), 7.27-7.84 (lOH, m, Ar and NH2, D~O
exchangeable), 8.15 (lH, s, H-23, 8.30 (lH, s, H-8).
Found: C, 50.50; H, 4.31; N, 12.21%. C24H24NsOgP x 0.1 CHCl3 requires
C,50-84;H, 4.26; N, 12.30%.
Example VI, R=2-CH3-C6H4, 40% yaeld
~H 1(CD3)2S0~ 3-32 (6H, s, CH3), 3.98 ~2H, m, ~20CH2P), 4.38 (2H, d, J--7.25Hz,
CH2P), 4.54 (2H, m, NO~H2), 7.09-7.25 (8H, m, Ar), 7.33 (2H, br.s, D2C)
exchangeable,NH2), 8.15 (lH, s, H-2), 8.31 (lH, s, H-8).
- Found: C, 53.14; H, 5.80; N, 13.87%. C22H24NsOsP x l.S H20 requ~res C, 53.23;
H, ~.48; N, 14.10%.
213~30~
~ 93/1907~ r~ /Gss~
- 1 l -
9-[2-lBis(phenoxy)phosphorylmethoxy lethoxy~adenine
(alternative preparation method)
A mixture of 9-[1phosphonomethoxy)ethoxy]adenine (1.0 g, 3.46 mmol) and thionyl
chlo~ide (50 ml) was heated undcr reflux for 2 h. The solution was cooled to room
temperature and the solvent was removed under reduc~d pressure (wi~h exclusion of
moisn~re) to give an oily residue. The residue was coevapolated with dry
dichlc~romethane and then it was redissolved in dry dichloromethane ~15 ml). Phenol
(Q.72 g, 7.61 mmol) was added to the solution and the rcsulting mixture was cooled to
0C under argon. Tnethylamine (1.06 ml, 7.61 mmol) was added dropwise (over 2
min) followed by l-medlylimidazole (l.1 ml, 13.84 mmol). The reactants were sti~ed
at 0C fc~r 15 min and dlen at room temperature for 1 h. The precipitate was filte~ed
off, washed with cold dichloromethane (lO ml). The combined filsrate and washingw~e diluted with dichloromedlane (100 ml), washed with san~rated ~queous NaHCO3
(30 ml), water (30 ml) and dried (MgSO4). The solYent was removed under reduced
pressure, the ~duct was p~ified by column chromatogra~hy, elu~ng with 6% edlanolin chloroform, to gi~re thé title compound as a colourless gum; yield 0.52 g, 34%.
lH NMR ~ (DMSO-d6)
3.97 (2H,m,CH2CH2O), 4.35 (2H,d,J=7.42,CH2P), 4.55 (2H,m,CH2CH2O), 7.22-7.44
(12H,m,aromatic protons and NH2,D20 exchangeable), 8.15 and 8.31 (lH,s and
lH,s~-2 and H-8). Pound: (CI),M+,441.1202;C2~H2oNsOs P r~quires M+, 441.1201
- Anal. Calcd for C2~H20Nsos PØ2 CHC13: C,52. 15; H, 4.38; N, 15.05. Found: C,
25 52.37; H, 4.42; N, 14.83.
wO 93/1907~ pcr/GB9~/oo~
c~,~ 3'~ 12 -
9-[2-Bis(phenoxy)phosphorylmethoxylethoxy~adenine hydrochloride
9-[2-Bis(phenoxy)phospho~ylrnethoxy]ethoxy~adenine (l.S g, 3.4 mmol) was dissolved
in dry dichloromethane (10 ml) and a saturated (0C) solution of hydrogen chloride in
S dichloromethane (30 ml) was addcd. Aft:er being stirred at rt>om temperature for 5
min, ~e solution was conccntrated (with exclusion of moisture) and d~e residue was
coevaporated vnth dry dichloromethane (3 ~c 50ml) to give the diphenyl ester
hydr~chlo~idc as a colourless solid; yicld 1.6 g (99%). A sample of the compound (S0
mg) was evaporated ~vith dry tolucnc (20 ml) and dried in ~acuo to give a crystalline
soL 139-143C.
H NMR ~ ~DMSO d6)
' ..:,.
4.01 (2H,m,CH2CH20), 4.32 (2~d,J=7.42,CH2P), 4.62 (2H,m,CH2CH20), 7.1~7.44
(lOH,m,a~ma~ic protons), 8.49 and 8.69 (lH,s and lH,s, H-2 and H-8), 8.7-9.25
(2H,v.b.d,NFI2,DzO oxchangeable). Found: (CI), MH+, 442.1280; ~2tjH20Nsos P
equircs MH+, 442.1280. Anal. Calcd for (: 2bH2bNsOs P.H2O. 1.2 HCl: C, 47.74; H,4.64; N, 13.92; Cl, 8.45. Found: C, 47.42; H, 4.54; N, 13.90; Cl, 8.~2.
2 1 3~303
93/l907:~ PCl /C~E~93/00~i60
- 13-
Biological Evaluation Procedures
Compounds were administered by oral gavage to female Balb/C mice weighing 20g, as
single 0.1ml doses of 0~mmoVkg. These solu~ons were made by dissolving the
5 compounds in DMP and diluting with 1% carboxymethyl cellulose and 1% Tween 80,to a final DMF concentration of 5%. Pood was withheld from the mice for 18 hoursprior to thc start of thc e~cpe~iment. Blood was collectcd by cardiac puncturc using
hcpa~inised syringcs 15, 60 and 180 mins aftcr dosing. Equal volumes (0.2ml) ~om 3
rnice werc pooled at each timc point and 0.6ml of ice-cold ethanol was added.
10 Following chilling at -20C and centrigugation, 0.5ml of supernatant was dried undcr
- ~eduoed p~ssure. Thc samplc was then rcconstituted with 0.5ml of 0.4M NH4OAc
(pH 6.0) and analysed by HPLC.
Thc results arc as shown in thc table below
9-12-(Phosphonometho~cy)etho~y]adenine concn. (~lM) in blood at time (min) after dosing
: Compound of Example No. 15 60
II 7
m 4 ~
IV 9 1 : .
V 7 2
- V~l ~ 10 5