Note: Descriptions are shown in the official language in which they were submitted.
63Y PCI/US93/02803
~ocesses and Interrnediates foq the Pre~a~ation of 2-Substituted E~çnzaldehvdes
Field of thc Invendon
This invention rclates to novel intennediates and p¢ocesses foq preparing use~ulinteln~ediates in dle syn~esis of ph~maceutically active agcnts.
Back~und
2-Substi~ted benzaldehydes aIe useful in~nediates fo~ preparing phannaceuticallyactive compounds. For ex~mple, certain compounds which are leuko~iene antagonis~ and
5 useful in the treatrnent of as~ma may be prepared fi~m 2-subs~tuted benzaldehyd~s of dle
general foq~nula ~
o
R2 ~JI
A Rx
(Ia)
20 whe~ein:
Rx is ~L)a-(cH2)b-~I)~M;
a is 0 or 1;
b is 3 to 14;
WO 93/19033 PCr/US93/028r-
6 ~ 9
- 2 -
c is 0 or 1;
L and T are independently sulfur, oxygen, or CH2; and
M is Cl 1alkyl, ethynyl, trifluoromethyl, isopropenyl, furanyl, Lhienyl, cyclohexyl
or phenyl optionally mono substituted with Br, Cl, CF3, Cl~alkyl, Cl4aLtcoxy,
methylthio, or trifluoromethylthio;
R2 and A arc independently selccted from H, CF3, Cl~alkyl, Cl~alkoxy, F, Cl,
Br, I, OH, NO2 or NH2;
or Rl and A are H and R2 is (L)a-(cH2)b-(T)c-M wherein a, b, c, L, T, and M arc
as dcfined abovc.
0 Such compounds are discloscd, for instance in U.S. Patent 4,820,719, U.S. Patent
4,874,792 and EP-A 0 296 732, the disclosures of which are incorporated herein by
rcference. Accordingly, two general methods for preparing the 2-substituted
benzaldehydes are repo~ed therein: 1) palladium catalyzed addition of a substituted 1- -
aLkynyl compound to a 2-halo benzaldehyde effects a coupling to pro~ide a 2-(1-
aLtcynyl~benzaldehyde direcdy, and 2) a 2-methoxy-benzoic acid may be converted to
2-(2-methoxy-phenyl~4,4-dimethyl-oxazoline and treated with an aL~cyl or aralkyl Grignard
reagent to prepare the corresponding 2-(2-all~yl phenyl)-4,4dimethyl-oxazoline or 2-(2-
aralkyl phenyl) 4,4dimethyl-oxazoline (subsequent treatment of the 2-substitutedoxazoline with methyl iodide, reducdon with sodium barohydride and subsequent acid
hydrolysis produces dle co~responding 2-substituted benzaldehyde). The lattcr method is
based upon methods disclosed by Meyers et al., ~. Org. Chem., 43, 1372 (1978). Similar
methods forpIe~aring 2-substituted benzaldehydes are disclosed by Perchonock e~al., J.
Med. Chem., 28, 1145 (1985). In general, thesc mcthods cmploy reagents which
functionally displace substituents upon the aryl ring.
2S Methods for adlding an o~ho substituent to an a~yl ring by rendenng the aryl nng
nucleophilic are also known. Org. Reactions, 26, 43-61 (19 79) discloses that certain
functional groups which contain nitr~gen hcteroatoms and aIe attached t~ phenyl rings can
stabi}ize a phenyl ring toward lithiation, preferably in the ortho position. The ~thiated sitc
may then be trea~ed with a suitable clectroq~hilic reagent to effect substltution. Functional
groups which are re~orted dlerein to ~e particularly effective for this purpose arc mon~ o~
di-aLkyl amides, amines, N,N~ialk~ . hyd~zones, irnidazolines and oxazolines. De Silva
el al., Tetrahedron Lett., 5107 (197 ~1, report an o~ho~ iation of a benzamide using sec-
butyllithium and a diisopropyl amine, and Trécourt et al., J. Org. Chem., 53, 1367 (1988),
repart o~lithiation of 2-mcthoxy-p~idine with methyllithium and a catalytic amount o~
3S diiso~ylamine. Arylcarbimhcs, lu)wcvcr, arc repor~d to have limited synthctic utili~
duc to thcir tendency t~ suffcr ~n rcaction at the azomethine linkage and alpha-dcprotona~on. Sce Org. Reactions, 26, 57-58 (1979). Zeigler et al., J. Org. Chem., 41,
VO 93/19033 ~ 1 3 ~ 6 ~ 9 PCT/US93/02803
1564 (1976) report that arylcarbimines may be induced to undergo ortho-lithiation if an
adjacent ether substituent is present.
In addition, it has been reported that methyl groups can be lithiated if located in the
o~ho posiion of benzamides, 2-phenyl imidazolines and 2-phenyl oxazolines. Thus,Watanabe e~ al., J. Org. Chem., 49, 742 (1984) report chain extension via an ortho-
toluamide in the synthesis of isocoumarins; Gschwend, et al., J. Org. Chem., 40, 2008
(1975), rcpo~t benzylic chain extension vh lithiadon of 2-(o-tolyl)oxazolines; and
Houlihan, U.S. Patent 4,100,165, reports condensation of a dilithiated
2-(o-tolyl)imidazoline with csters and acyl halidcs. ~;
Currcnt mcthods for the synthesis of the 2-substituted benzaldehydcs of this
invention employ expcnsivc reagents o~ multiple process steps which makc thcrn
unattractivc for commcrcial prcparadon of 2-subsdtuted benzaldehydes. Thcrc is thcrcforc
a nced for an cfficicnt altemative method for the preparadon of 2-substituted
bcnzaldehydcs.
Summanr of thc Invcntion
It is an objcct of this invcntion to providc a ncw and efficient proccss fo~ thcp~eparation of compounds of foqmula ab):
o
~R
A
(Ib)
wherein: -
Rl is CH~CH2-(Ll)p-(CH2)q~(L2)~ CH2-(T)S-Z;
Ll andL2 are independently CH2C~I2, C~I=CH or C5C;
qisOto8;
p, r and s arc independently O or l;
T is 0, S, CH2, CH=CH, C-C; and
Z is Cl4alkyl, cthynyl, trifluoromethyl, isopropenyL furanyl, thienyl, cyclohexyl
orphenyl opdonally mono subsdtuted with CF3, C~ cyl, Cl 4alkoxy, methylthio, or
rifluo~methylthio"~nd
R2 and A are independently H, CF3, C~ cyl, F, Cl, Br or I.
Onc feature of this invention is a proocss foq- plep~ing a compound of thc fonnula: -
WO 93/19033 '~ fi 3 9 PCI/US93/02X
R
~R
A
(rb)
whcrein A, Rl, R2, Ll, L2, q, p, r, s, T and Z are as defined above for formula (Ib),
which comprises reacdng a compound of the formula
5 ~
N-R3
A~XJCH3
(m
whercin:
R2 and A are as dcfined above for foqmula ab);
R3 is Cl~ alkyl, C3~ cycloaLlcyl, (CH2)~phcnyl or N(R')~;
R' is Cl~ alkyl, C3~ cycloalkyl or (CH2)~phcnyl; and
tisOorl;
with a basc and a co~npound of the fonnula
X-CH2~ )r(CH2)q-(L2)rCH2~(T)s~Z
~V)
whcrcin:
Ll, L2, p, q, r, s, T and Z are as defined ab~ve for forrnula ab), and X is a
displaceable gTOUp;
and hcadng dle product thercof wi~ acid.
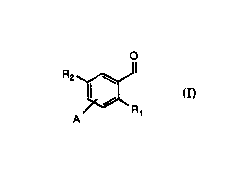
Another feature of this invention is a novcl intennediate accoq~ing to fo~mula (II):
I R3
A~JRl
(~
25 whcrein:
Rl, R2 and A a~c as dcfined for fonnula (Ib);
R3 is Cl~ alkyl, C3~ cycloall~l, (GH2)~phenyl or N(R')2;
R' is Cl~ alLyl, C3~ cycloalkyl or (CH2)tphenyl; and
t is O OT 1,
~vo 93/19033 ~ 1 3 ~ 6 3 9 Pcr~US93/02XO3
- 5 - -
Another feature of this invention is a process for the preparation of the novel
inter nediate of formula (II), which comprises reacting a compound of the formula (III):
N-R3 :~
A~CH3
S (III)
wherein A, R2 and R3 are as defined for formula (II);
with a base and a compound of the formula (IV):
X-CH2-~1)p-(~2)q~~2)r~2~~s~Z
(IV)
wherein, Ll, L2, p, q, r, s, T and Z are as defined above for for nula (Ib); and X is a displaceable group.
Yet anodlcr feature of this invention is an improved process for preparing a
compound of the formula a~. which comprises adding a catalytic amount of an organic
aminc to thc ~ac~on mixture pQior to addi~on of thc basc. -~
Still anothcr featurc of this invention is an improved process for prepanng a
compound of thc fonnula aI), which comprises adding a sodium or potassium aL~coxide to
~c ~action mDcture.
Ano~er fea~e of this invention is an improved process for prepanng a compound
of thc foImula (II), which comprises conduc~ng the ~action wi~in a specifled temperature
range.
De~ailed D~scr~tion of the Inven~on
The present invention discloses useful intennediates and a process for the
~eparation of compounds of formula (Ib):
~R,
(Ib) '~
whcran:
Rl is CH2GH2~ )p-(CH2)~(L2)rcH2~(T)~Z;
Ll and L2 àrc independently (~H2CH2, CH=CH or C-C;
WO 93/ 1 9033 PCl`/US93/028'
fi 3 9 - 6 -
qis0to8;
p, r and s are independently O or l;
T is 0, S, CH2, CH=CH, C-C; and
Z is C14aL~cyl, ethynyl, trifluoromethyl, isopropenyl, furanyl, thienyl, cyclohexyl
or phenyl optionally mono substituted with CF3, Cl4aLtcyl, C14alkoxy, methylthio, or
trifluoqomethylthio; and
R2 and A are independently H, CF3, Cl 1aL~yl, F, Cl, Br or I,
which comprises reacting a compound of the forrnula:
N-R3
R2 ~JI .
~CH3
A
(III) :
wherein.
R2 and A are as defined above;
R3 is Cl~ alkyl, C3~ cycloalkyl, (cH2)lphenyl or N(R )2;
R' is C~ l, C3~ cycloaLtcyl o~ (CH2)~phenyl; and
tisOor l;
with a base and a compound of the formula
X-cH2~ H2)q-(L2~rcH2-(T)sz
(IV)
whe~ein:
Ll, L2, p, q, r, s, T and Z are as defined above; and
X is à displaceable group;
and treating the product thereof ~vith acid.
Accordingly, this invention discloses novel inteImediates according to fonnula (II):
N-R3
R2 ~JI
~R
A
(Il)
wherein:
Rl is CH2CH2-(LI)p-(CH2)q-~L2)~CH2-(T)~Z;
Ll and L2 arc in~cndently CH2CH2. CH=CH or C-C;
qisOto8;
6 3 ~ PCI/US93/02803
p, r and s are independently O or 1;
T is 0, S, CH2, CH=CH, C-C; and
Z is Cl~aLkyl, ethynyl, trifluoromethyl, isopropenyl, furanyl, thienyl, cyclohexyl
or phenyl optionally mono subs~ituted with CF3, C14allcyl, Cl~aLkoxy, methylthio, or
trifluoromethylthio;
R2 and A are independently H, CF3, Cl 1aLkyl, F, Cl, Br or l;
R3 is Cl~ aLlcyl, C3~ cycloaL~yl, (CH2)tphenyl or N(R')2;
R' is Cl~ alkyl, C3~ cycloaLkyl or (CH2)tphenyl; and
tisOorl.
Suitably Z is phenyl and Ll and L2 are CH2CH2.
Suitably R3 is t-butyl.
Suitably p, r and s are 1.
Suitably q is 0-2.
Suitably T is CH2 or C~C.
A prefemed compound is N-[2-(8-phenyloctyl)phenyl)-methylene~-1,1-
dimethylethanamine.
The novd intermediatcs of formula aI) are prcpa2ed by a process which compsiscs
reacting a compound of fnula (II~:
N-R3
R2 ~ JI
YC~CH3
2û A
(III)
wherein:
R2 and A are independently H, CF3, C14aL~cyl, F, Cl, Br ~r I;
R3 is C~ yl, C3~ cycloalkyl, (cH2)d~henyl or N(R )2;
R' is Cl~ al~yl, C3~ cycloaL~cyl or (CH2)~phenyl; and
t is O o~
wid~ a base and a compound of the fonnula (IV):
X-C H2~ p-(CH2)q-~2)r~H2-~s-Z
whe~in:
L~ C itldepcndcDdy CH2CH2~ CH=CH o~r C5C;
qi~Oto8;
p, r ai~d s are independcntly O or 1;
T is 0, S, CH2, GH=CH or C~C; and
WO 93/19033 PCI/US93/02P `
2 1i~ , 3 9 - 8 -
Z is Cl 1aL~cyl, ethynyl, trifluoromethyl, isopropenyl, furanyl, thienyl, cyclohexyl
or phenyl optionally mono substituted with C~:3, C14alkyl, C14aL~coxy, methylthio or
trifluoromethylthio; and
X is a displaceable group.
ln a preferred embodiment, this invention discloses a process for preparing a
compound of fv..,.ula (Ib) which comprises reacting a compound of formula (III) with a
base and a compound of formula (IV) and treating the reaction mixture with acid. Thus, in
the preferrcd embodiment, the overall conversion is accomplished in a single reaction
vessel without isolation of the intermediate produc~ This process utilizes readily availablc
0 materials and proceeds in cfficient yield in a minimum number of process st~ps.
Compounds of formula (III) are hydrazones and imines, or Schiff bases, and are
generally prepa~d by any means cornmon to the art for prep~ng such compounds. One
mcthod for preparing the imines comprises rcacting a compound of formula (V):
o
R2~
~CH
A 3
(V)-
with an amine or a hydrazine of the for nula, R3-NH2. Such reactions are nonnally
conductod by admi~ing the reactants in a non-aqueous solvent and optionally heatin~ the
two ~ants. Dehydra~ng agents may be used to drivc the reaction towards product if
neccssary. Cornmon dehydrating agents are, for instance, molecular sieves or magnesium
sulfate. Alternatively, dehydration may be effected by aæotroping dle wate~ produced by
thc ~eaction ~om an app~opriate solvcnt, such as benzene or toluene. The group R3 is C
aL1cyl, C3~cycloal~yl, benzyl, phenyl or N(R')2. Cyclohexylamine, t-butyl amine, a~iline
and N,N dimcthyl hydra~ne are s utable reagents. t-Butyl amine is prefer~ed.
The electrophilc, givcn by formula aV), is prepared by conventional methods, such
as thosc disclosed in U.S. Patent 4,820,719, U.S. Patent 4,874,792, EPA 0 296 732 and
Perchonock et al., J. Med. Chem., 28, 1145 (1985) which are incorpoqated herein by
referen~e. The X moiety of the electrophile represents a displaceable group, which may be
any grolYp capable of being displaced by ~e carbon nuskophile prepared from the
compound of formula (II~. A large llwnber of displaceable groups a~e suitable, such as
allcyl and aryl sulfonates, aL~yl and aralkyl acctates, benzoates and halogens.
Rcp~e~cntati~c of thc class are Cl, Br, I, R4SO3 and R4CO2, whcrein R4 is Cl4aLIcyl,
ully subsdtutcd by 1-5 fluorine atoms, or phenyl, optionally substituted by onc ~
two halogen, C14alkyl. C14alkoxy or nitro groups. Representative displaceable groups
arc tolucncsulfonate, bromobenzenesulfonate, nitrobenzene-sulfonate, methanesulfonate,
vo 93/19033 2 1 ~ ~ 6 3 9 Pcr/usg3/n2Xo3
trifluoromethanesulfonate, acetate. chloroacetate, trifluoroacetate, benzoate,
bromobenzoate, chlorobenzoate, nitrobenzoate, chloro, bromo and iodo. Chloro andbromo are prefer~ Chloro is especially prefer~
In general, the X g~up of the compounds of fonnula (IV), if not present in the
5 precursor. is prepaled from the corresponding alcohol by reaction with an appropriate acyl
halide, anhyWe, sulfonyl halide or appropriate halogenating agent. Typical of such
reagents are tolucnesulfonyl chloride, bromobenzenesulfonyl chloride,
nitrobenzenesulfonyl chlaride, mcthanesulfonyl chlonde, acetyl chloride, chloroacetyl
chloride, trifluo~oacetic anhydride, bcnzoyl chloride, bromobenzoyl chlonde,
10 chlarobenzoyl chloridc, nitrobenzoyl chloqide, oxallyl chloride or bromide, hydrochloric
acid, hydrobromic acid, hydroiodic acid, phosphorous tribromide, phosphorous
trichlondc, phosphorous oxychlo~ide and carbon tetrabromide with triphcnyl phosphinc.
Compounds of the formula HO~CH2~(LI)p~(CH2)q~(L2)rCH2~('I )~Z, wherein T is CH2,Ll or L2 arc CH2CH2, and Z is Cl4alkyl or phenyl are generally availablc commercially.
15 Compounds wherein T is 0, S or CSC, may be prepared by reacting thc compound H-T-Z
with a compound of thc structurc X-CH2-(LI)p-(cH2)q-(L2)rcH2-x whcrcin X, Ll, L2,
T, p, q and r a e as dcfincd abovc, in thc prcscncc of an appropriatc basc. Compounds
wherein T is CH~H may bc prcpa~ed by scmi-hydrogcnation of compounds whcrcin T is
C~C, such as with Lindlars catalyst or S% paLladium on barium sulfate and hydrogcn.
20 Hydrogcnadon with a palladium catalyst, such as 5% palladium on carbon, yiclds thc
compound whcrein T is (~H2. Whcn Ll or L2 are C=C or CH=CH thc ~csulting productmay b? roduccd at a subsequent time to yield a product wherein Ll or L2 are CH~ or
CH2CH2. Forcxamplc, l-bromo-7-phcnylhcptanc is prcpared frtnn 1,5~ibromopcntanc
and phcnylacetylenc in ~e prescnce of n-butyl lithium, followed by reduction with
2s hydrogen over a palladium catalyst. In an altcrnate example, l-bromo o~ l~hloro 7-
phenylheptane may be prepared via a coppcr mediated coupling of benzyl magnesiumhalide with 1,6 dibromohexane or l-bromo~chlo~ohexane.
Alkyla~on of ~e carbimine of fo~mula (III) is ini~iated by ~ng a compound of
foQmula (m) wi~ a stTong base to deprotonate the o~tho methyl group. Since the
30 metallated intesmediate is teactive with water, the activa~on reaction is suitably ca~ied ou~
in an inert, dry atmosphere, such as nitrogen or argon, although dry air is sufficienL
The activation reaction is ca~ried out in an aprotic solvent. Suitable solvents for this
reaction ale common aliphatic or aromatic hydmcar~on solvents which are umeactive to
strong bases. Rcprescntativc of such solvents are dicthyl ether, tetrahydrofuran, dioxane,
35 dimcthoxyc~e, toluene, benzenc, pentane, hcxanc and petrokum cthers, d mixnlres
thor~of. Dic~yl c~er, dioxanc and tctrahydrofuran arc prefellcd. Tctrahyd~uran is
espccially p~efe~ed. '
WO 93~19033 PCI/US93/02~ .
~3~)39 - lo-
A base of sufficient strength to deprotonate the ortho methyl group is required
Any base capable of effecdng such deprotonadon without causing apprcciable side
rcacdons is suitable. Typical of such bascs arc an alkali metal alkyl, an alkali mctal aminc
(e.g., a salt of an organic or inorganic aminc), or an alluli metal a~yl. Represcntative of
such bases are n-butyl lithium, sec-butyl lithium, methyl lithium, phenyl lithium, lithium
dusopropylamide, lithium tetramethylpiperidide, lithium diethylamide or lithium amidc, or
the corrcsponding sodium or potassium salt of any of these spccics. Alkyl lithium rcagcnts
are espocially suitablc. ~Butyl lithium, lithium tetramcthylpipcrididc and lithium
diisopropylamidc are prefc~ It is also within the contemplation of this invcndon that the
lo metal of the base initially used may bc exchanged f anothcr metal, for instance anothcr
alkali mctal, coppcr, magnesium or zinc. It is oftcn hclpful to use a slight molar exccss of
basc, such as 1% to 25%, to cnsurc complete mctallation. About onc molar equivalcnt is
nonnally satisfacto~y. It will bc apparent to one skillcd in thc art that cestain of thcsc bascs,
such as allcali mctal alkyl or aryl, may bc incompatiblc with a halogen substituent h the
carbimhe, and tbat othcr bases, such as lithium diisopropylamidc would be more suitable.
The ~action of a co npound of f~nula (Ill) with the basc is ca~ied out by
admixing the two ~ea~ants. lhe leaction sho~d be camed out at a te np~We suf~ ;cnt to
causc tbc basc to dqn~n~ thc o~o me~yl group, yet not so high as to cause adverse
sidc ~ ons Thus, the optimum tcmpe~ature will be dependent upon the base uscd and
the iminc ~ If thc ba~e is a lithium dialkyl amide, typ~ally the ~ion is carried out
be~cn about -2lPC and 60C; suitably, the leaction is carned out bctwecn about ^10C
and40DC
It has becn found that surprisingly improved yields are obtained when thc reaction
is run using an organolithium base at between about 15C to about 35C Typically, when
2s st ong bases arc reacted with compoundr. which possess a moiety which is susceptible to
nucleophilic attack, such as a earbimine function, thc reactions are conduc~d attcmpc~es o~about 0C and lowcr. These low tempcraturcs are bclieved to prcvcnt
undesirablc sidc ~ons, sueh as nuclcophilie attaclc upon d~c labilc carbiminc
funetionality by thc base itsclf or by thc anion crcatcd by thc aetion of thc basc.
Uncxpeetodly, with celtain bases, such as n-butyllithium optionally with a catalytic amount
of dii~ncpyl aminc or dicyclohexylamine, side rcactions a~c mininuzed and yields ale
~eased by adding the base to thc carbimine at about 15C to about 35C. Conducting the
~on bctween about 20C to about 30C is espeeially suitablc. Tempe~res abovc 55C
gena~ly Iemlt L an cnh~ncement of ~ablc side rcaetions.
3S Thc cbc~ c, X~ L2~2-(~I~Z, is typically added u pon
oomple~on of the mctallaion Ieaction. Although thc clec~ophilc may bc addcd ncat, it is
VO 93/19033 PCr/US93/02803
213~639
11
conveniently added in a solvent such as that which has been used to form the metallated
interrnediate. The reacion is then allowed to sir for about 15 min to about 24 h.
If the irnine is to be isolated, the reacion solution is diluted with an appropriate
solvent. washed with water and concentrated in vacuo to an oil. If a purifled product is
S desired, the product is purified by distillaion, or, if appropriate, by crystallization.
Conversion of a compound of fonnula (lI) to a benzaldehyde is accomplished by
stirring the imine with any acid of sufficient strength to cause hydrolysis of the C=N bond.
~thin the context of this invention mineral acids, organic acids and the like are considered
to be sufficiently strong acids. For example, methanesulfonic acid, toluenesulfonic acid,
trifluoroacetic acid, benzoic acid, acetic acid, hydrofluoric acid, hydrochloric acid,
hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid and phosphoric acid, are all
suitable. Mineral acids are preferred Hydrochloric acid is especially preferr~
In the preferred process, the reaction mixture containing the product (Il) is
hydrolyzed direcdy by the addition of acid to the reacdon mixture. Generally, the reacdon
mixture is added to a cooled soludon of the acid and thereafter allowed to warm to room
tempcrature. The reaction mixture may be monito~ed for formation of the desired
benzaldehyde, such as by analy~cal chromatography, but typically the ~eaction is s~red
~n about 1 h to about 24 h. The product is then isolated by conventional techniques,
such as extracdve workup.
An improved process for preparing a compound of the formula (II), comprises
adding a catalydc amount of an organic amine to the Ieaction mixture prior to addition of the
base, panicularly when an all~l lithium reagent is used as the base. Higher yields are
obtained when a catalytic amount of an oqganic amine is used than when the amine is absent
o¢ when a full molar equivalent of the amine is employe~ Suitably the oTganic amine is a
secondary amine. Rep~escntatiw amines are diethylamine, diiso~opylarnine,
dicyclohexylamine, piperidine, 2,~dimethylpiperidine, and 2 7,6,~te~amethylpipcridinc.
DiisopropyL~nine, dicyclohcxylaminc, and 2,2,6,~tctramethylpipcridine a~ especially
suitable. The cat~lytic amount may be from about 0.01 to about 0.3 molar equivalcnts of
~rganic amine relative to thc carbimine. About 0.01 to about Q15 molar equivalents is
suitaUe. About 0.01 to 0.1 molar equivalents is typical, depending on the amine use~
For instance, about 0.01 to about Q05 equivalents aIe useful for diisopropylamine and
2,2,6,~tetrarnethylpipe~idine.
Sill another feature of this invenion is an imp~ved process for preparing a
compound of the fonnula (II), which cainprises preparing a sodium or potassium salt of
~c csbim~ of formula (m~ and reacting the product with a compound of the fonnulaaV). Far instancc, thc 2-mcthyl-phenyl car~iminc of formula aII) may bc treated with a
basc such as n-butyllithium or lithium diiso~ropylamide, to form the lidlium salt, and
Wo93/19033 i ~ 3'~,639 - 12- Pcr/uss3/o~r
further treated with a sodium or potassium base or salt to form the desired salt by a metal
exchange reaction. Sodium or potassium aL~coxide, or sodium or potassium trifiuoroacetate
are representative baseslsalts. Reaction of thc carbimine salt with a compound of formula
(IV), such as 7-phenylheptylchloride, effects aLlcylation at lower temperatures, and with
fewcr side reactions, than obtained with the comparable lithium salt of the carbimine. Use
of the potassium salt is especialiy suitable.
~.
The examplcs which follow illustrate how to make and use the compounds and
processes which consdtutc this inventdon.
~xam~lcs
The nomcnclatu~ and abbreviadons common to the chcmical art are used in the
examples. Unless othcrwise noted, reagents were obtained from commcrcial suppliers and
wcrc uscd without further purification. Tetrahydrofuran, if used as a reacdon solvent, was
dricd ovcr 4A molecular sievcs if neccssary. All other solvents were obtained f~om
commc~ial supplicrs as Rcagcnt gTade and were used without furtherpurification. All
non-aqueous ~actions were perf~ned undcr an atmosphe~c of dry nitro~en. Mclting
points werc ta~en on a Thomæ-Hoover capillary melting point apparatus and are
unanr~ l~uid cluomatography was conducted on a Whatman P,artisil~Z9 5 ODS 3
RAC IL Gas chromatographic analysis was pe~formed on a I)B-1 30 m X QS3 mm
capillary column. IR spec~a were ~ W on a Pa~in-Elmcr Model 283 infr~ed
spec~ophotomcter. ~;T-IR spectTa werc obtained on a Nicolet 6000 FT in~ared
spectromcter. Combustion analyses wer~ run on a Per~n-Elmer 240 C elemental analyzcr.
Unless othenvise indicated all lH-N~ (proton magnetic resonance) spec~a wcre obtained
at 400 MHz, using a Brul~er Instruments WM 400 spectromet~ in deuterochlo~oform
soluion. ~3C-NMR spcctra werc obtained at 100 MHz. Chemical shifss ~c repo~tcd in
ppm (~) downfcld ~om tetramethylsilanc. Annotations to lH-NMR are as follows: s,singlet, d, doublet, t, triplet, br, b~oad; m, multiplet; J, coupling constant in Hertz.
Exam~lc 1
aration of 1-b~omo-7-phenvlhe~tane.
To a sti~ed solution of 1500 mL (0.15 mol) of ().lM Li2CuCl~, and
1,6-dibromohacanc (456.8 g, 1.87 mol, 1.25 eq) in tc~ahydro~uran, at -5 to 0C, was
added a solu~on of bcnzyl magncsium chloridc (750 mL, 2 M in tc~ahydrDfuran, 1.5 mol)
over a 90 min pcriod. Thc reaction mixturc was stiITed at 0C fo~ 90 min, ~cn quenched
9033 ~ fi 3 9 Pcr/uss3/o28o3
- 13-
carefully with 2.0 L of saturated aqueous ammonium chloride. The internal reaction
temperature was kept below 20C during the quench. The mixture was stirred for 1 h at
room temperaturc and the layers separated. The organic layer was washed with 20%aqueous sodium chloride (4 X 500 mL). The organic layer was dried (magnesium sulfate),
S filtered, and conccn~rated in vacuo at 45-50C to an amber oil. Purification by fractional
vacuum distillation through a 12-inch vacuumjacketed Vigreux column gave the desi
product as a colorlcss oil (198.2 g,52%). An analytical samplc was prepared by
rcdistillation: bp 123-124C (1.5 mm Hg); FI-IR (neat film) 3100 3000,30~2800,
2000 1700, 1604, 1496, 748, 699, 644 cm~l; IH NMR (CDCl3, 400 MHz) ~ 7.29-7.16
10 (m, S H), 3.40 (t, 2 H), 2.6û (t, 2 H), 1.88-1.81 (m, 2 H), 1.63- 1.60 (m, 2 H), 1.43-
1.32 (m, 6 H); 13C NMR (CDC13, 100 MHz) ~ 142.7, 128.4, 128.2, 125.6, 35.9, 34.0,
32.8, 31A, 29.1, 28.6, 28.1. Anal. Calcd for C13HIgBr: C, 61.19; H, 7.50, Br, 31.31.
Found: C, 61.25; H, 7.59; Br, 31.47.
. Exam~lc 2
epara~ion of 7-chlor~l-phenylheptane
a) I-bromo-6-chlor~hexanc.
~0 A mixturc of 1,6-hcxanediol (30 kg, 254 mol),4896 hydrobromic acid (51.0 kg,
302 mol) and toluenc was heatcd to reflux. Water (34.5 kg) was removed under azeotrcpic
condidons. When disdllation ccased the mixturc was coolcd to 20C and extracted with a
soludon of concent~ted hydrochloric acid (69.9 kg) and watcr (60 L). Thc phascs wcrc
sepa~ated and thc organic phase dried by rchcating and removing water by azeotropic
25 disdllation. The mixture was cooled to 65C and dimethylfonnamide (1.11 kg) was addcd
Thionyl chloQide (31.41 kg, 264 mol) was added ove~ 45 min while maintaining thetempcraIure between 65-68C. The mLxture was heated to 109C over 1.25 h and cooled to
20C. It was then washed successively with 20% sodium hydroxide solution (100 L) and
water (2 x 150 L, 1 x 100 L). Toluene (400 L) was removed under vacuum to yield the
30 bromochlo~ohcxane as a toluene solution (85.5 kg,55% w/w by assay, 93% yield).
b) 7-chloro-1-pheny~ne
A solution of lithium tetrachlorocuprate [1~ 33 L, lithium chlo~ide (0.87 kg, 19.3
mol), cup~ic chloQide (1.4 kg, 10.4 mol)] was added to a solution of benzylmagnesium
35 chla¢ide (160 L of 1.86 M, 298 mol) in tetrahydrofuran at 15C, and the mixturc stined for
30 min. Bromochlo~hcxanc in tolucnc (85.5 kg of solution,55% w/w by assay,47.11cg,
236 mol) was added ovcr 3 h whilc maintaining thc tempcraturc bctwecn 15-20C Stirring
was con~nucd for a funher 1.25 h. 10% Ammonium chloridc solution (263 L) was added
ovcr 1 h, maintaining thc t~mpcraturc bclow 30C. Thc phæs wcrc scparatcd and thc
wo 93/l9033 Pcr/uss3/o2xr
2~ 639
- 14-
organic phase further washed with ammonium chloride solution (170 L) and 20% sodium
chloride solution (3 x 197 L). The organic solution was concentrated under vacuum to
leave an oil (56.8 kg, 77% pure by HPLC assay, 88% colTected yield), which was distilled
(b.p. 129-132C, 2 mbar) to yield the title compound (70%, 99% pure by GC assay).
s
Example 3
c~aradon of N-r(2-methyl~henvl)methylenel-1.1-dimethvl~an~mine.
A sti~Ted s~lution of o-tolualdehyde (25 g, 0.21 mol), and t-butylamine (27.75 g,
10 0.38 mol) in toluene (250 mL) was refluxed under standard Dean-Starlc condidons for 20
h. Thc soludon was evaporated to an oil which was vacuu n disdlled (bp 70-73C, 0.6 mm
Hg) to afford 33.9 g (93%) of product: IR (neat) 2980, 1645, 1605, 1460, 1375, 1210,
960, 910 cm~l; lH NMR (400 MHz, CDC13) ~ 8.S6 (s, 1 H), 7.86-7.83 (m, 1 H), 7.25-
7.11 (m, 3 H), 2.46 (s, 3 H), 1.30 (s, 9 H); 13C NMR (CDC13, 100 MHz) ~ 153.7,
15137.1~ I35.1, 130.5, 129.6, 127.1, 126.4, 57.5, 29.8, 19.2; GC RT 7.6 min (DB-l, 30
m X QS3 mm, p~ogram: 100C fo~ S min,10~260C a~ 15C/min, hold at 260C for 12
m~).
Exam~lc 4
Pn~paration of 2-mcthvl~cnzaldehvdc dimcthvl hvd~zone
A sti~ed solution of o-tolualdehyde (25.0 g, 0.21 mol), and l,l-dimethyl
hydra~ne (25.2 g, 0.42 mol) was rcfluxed in toluene (200 mL) for 24 h. Thc solution was
concentrated in vacuo and thc residual oil was vacuum distilled (51-60C,0.2 mm Hg) to
25 affo~d the tided product (31.98 g, 94%): IR (neat) 2950, 2850. 1580, 1550, 1455, 1025.
745 cm-l; lH NMR (CDC13, 400 MHz) ~ 7.8-7.6 (m, lH), 7.~7.3 (m, lH), 7.1-6.9 (m,3H), 2.9 (s, ~), 2.4 (s, 3H).
Example 5
~ration of N-r(2-(8-phe~loctvl)phenyl~methylenel-l .l~imethvlcthanamin~
To a sti~d solution ~f diisopropylamine (29.14 g,Q289 mol) in tctrahyd~ofuran
(450 mL) cooled to -5C was added n-butyllithium (2.5 M,114.3 mL, 0.286 mol) at a late
which maiDtained the solution tempcrature below 10C. After the addition was c~mplcte,
35 thc solution was stirrd 15 min with cooling. To this solution was added N-[(2-
mcthylphcnyl~mcthylcnc]-l,l dimethylcthanamine (50.0 g, 0.286 mol) in tct~ahydrofuran
~65.0 mL) at such a ratc as to keep thc reaction tempcrature below 5C. The ~on was
~/O 93/19033 ~ 1 3 ~ fi 3 ~ PCl/US93/02X03
stirred for lS min with cooling then l-bromo-7-phenylheptane (72.9 g, 0.286 mol) in
tetrahydrofuran (75 mL) was quickly added. The reaction mixture was stir~d for 1 h with
cooling then allowed to warm to room temperature and stin~d for an additional 14 h. The
reacion mixture was assayed by gas chromatography for product imine (RT 19.8 min.,
s DB-1, 30 m X 0.53 mm, program, 100C for S min, l0~260C at 15C/min, hold at
260C for 12 min.). The product was isolated by diludon of thc reaction mixture with
watcr and mcthylenc chloridc, quickly washing the organic mixturc with water, and ;
concentrating the solution to an oil. The oil was purified by distillation.
0 Example 6
Pre~a~ation of ~-L~2-1~-phenyloctvl~henyl~methvlene~-1.1-dimcthylcthanamine
A stirred solution of 2-(8-phenyloctyl)bcnzaldehydc (10 g, 0.034 mol), and t-
butylamine (4.96 g, 0.068 mol) in tolucne (100 mL) was rcfluxcd under standard I)can-
Staric conditions for 16 h. The solution was cvaporatcd to an oil which was vacumn
distilled (bp 260C, O.lS mm Hg) to afford the dtled product (11.1 g,94%): GC RT 19.8
min (DB-1, 30 m X 0.53 mm, program, 100C for S min, 10~260C at 15C/min, hold at
260C for 12 min.); lH NMR (CDCI3, 400 MHz) ~ 8.58 (s, lH), 7.86 (d, J=7.S Hz, lH),
7.29-7.13 (m, 8H), 2.79 (t, J=7.5 Hz, 2H), 2.58 (t, J=7.5 Hz, 2H), l.S9-l.Sl (m, 12H),
1.30 (s, 9H).
Exa n~le 7
e~aration of 2-(8-~henvloctYl)benzaldehYde
25 Via hydrolysis of N-~(2-(8-phenyloctyl)phenyl)me~ylene]-1,1 dimc~ylethanamine
To a solunon of N-[(2-(8-phenyloctyl)phenyl)methylene]-1,1 dimcthyle~anaminc
(0.51 g, 0.0146 mol) in tetrahydrofuran (S mL) was added 10% aqucous hydrochlonc acid
(5 mL) and the mLl~ture stir~ed for 15 h at room temperature. Methylene chloqide (10 mL)
30 and water (10 mL) were added and the layers se~ated. The aqueous layer was extra~ted
with rnc~ylenc chlo~ide (1 x lS mL) and the combined organics were dried (magnesium
sulfate), filte~d and concentrated in Yacuo to an oil (0.405 g,97.4% pure by H~C assay,
92% wnecled yield): IR (neat) 2920, 2880,1695, 1600, 1455 cm~l; lH N~ (CDC13,
400 MHz) ~ 10.25 (s, 1 H), 7.80 (dd, 1 H, 1 = 1.2 and 7.7 Hz), 7AS (m, 1 H), 7.33-
35 7.13(m,7H),2.98(t,2H,1=77Hz),2.58(t,2H,1=7.7Hz). 1.58(m,4H), 1.30(m, 8 H).
wo 93/1~033 PCr/US93/028~'
~2i~fi39
-- 16 --
Example 8
e~aration of 2-(8-~henvloctvl)benzaldehvde.
Using one mole equivalent of a nitrogenous base and l-bromo-7-phenylheptane
To a stirred solution of dusopropylamine (29.14 g,0.289 mol) in tetrahydrofuran
(450 mL) cooled to -5C was added n-butyllithium (2.5 M, 114.3 mL, 0.286 mol) at a rate
which maintained the solution temperature below 10C. Afoer the addition was complete,
the solution was stir~d 15 min with cooling. To this solution was addcd N-[(2-
~o methylphenyl~methylene]-l,l~imethyle~anamine (50.0 g, 0.286 mol) in tetTahydrofuran
(65.0 mL) at such a rate æ to keep the rcacdon temperature below 5C. The ~on wæsti~ed fo~ 15 min with eooling then 1-blomo-7-phenylheptane (72.9 g, Q286 mol) in
tetrahydrofuran ~75 mL) was quiekly addul The reaction mixh~e was sti~ed for 1 h with
cooling then allowed to warm to rwm temperature and stiITed for an additional 14 h. The
reaedon mixture was ~uenched with aqueous 10% hydrochloqic acid soludon, and wasstined for 1 h at 0C, then at ambient temperature for 14 h. The ~eaction mixture was
pourcd int~ methylene chlaride (700 mL) and sti~d fo~ S min. The organic layer was
removed, and the aqueous layer ext~ac~d with methylene chloride (2 x 700 mL). The
combined organic layers were wæhed with 10% hydrochloric acid (2 X S00 mL) and
satmaocd brine (1 X 350 mL), then concentrated in vacuo to a golden oil. The crude
product was passed through a Pope Still (100C, 0.2 mm Hg) and the residue treated with
hexane (400 mL) with stimng for S min. The solution was allowed to setde, and dccantul
Thc hexane ~eatment was ~ed an addi~ional two tDnes, and the combined hexane
wæhes were then fil~ed through a Celite~) plug and concentrated to a ligh~ yellow oil
(72.5 g,92.4% pure by HPLC æsay,82~ conected yield). P~r analytical purposes, a
small sample was fi~her purified by Kugelr~hr distillation (250C, Q1 mm Hg): IR (neat)
2910, 1695, 1600, 1450, 1210, 1190 cm~l;lH NMR (CDC13, 400 MHz) o 10.25 (s, 1
H), 7.80 (dd, 1 H, J = 1.2 and 7.7 Hz), 7.45 (m, 1 H), 7.33-7~13 (m, 7 H), 2.98 (t, 2 H,
1 = 7.7 Hz), 2.S8 (t, 2 H, J = 7~7 Hz), 1.58 (m, 4 H), 1.30 (m, 8 H); 13C NMR (CDC13,
100 MHz) ~ 192.2, 145.7, 142.8, 133.7, 133.6, 131.3, 130.9, 128.3, 128.2, 126.3,125.5, 35.9, 32.4, 32.4, 31.4, 29.5, 29.4, 29.3, 29.2; HPLC RT 5.8 min (Wha~nan
Par~sil~D S ODS 3 RAC II, 4.6 mm ID. x 10 cm, 2 mLJmin,7:3 CH3CN:H20, W
detecaon at 211 nm3.
6 3 ~ Pcr/US93/02803
Exarnple 9
e~aration of 2-(8-~henvloctvl)benzaldehyde
Using two mole equivalents of imine and nitrogenous base and one mole equivalent of 1-
s chlor~7-phenylheptane
A solution of lithium diisopropylamide in THF (15.4 g, 0.024 mol) was added to
TH~ (30 mL) and cooled ~o -10C under a nitrogen atmosphere. A solution of N-[(2-
methylphcnyl)-mcthylene]-l,l-dimethylcthanamine (4.23 g, 0.024 mol) in THF (5 ml)
o was added and thc mixturc was stirred at -10C for 20 min. Phenylheptylchloridc (2.77 g,
0.012 mol) in T~ (5 ml) was added and the mixture was heated to 58C. GC analysis
showed no phenylhcptylchloridc remaining after 3 h. The mixtu~e was cooled to 0C and
dilute HCl (50 mL) was added such that the temperature was kept bclow 25C. The
solution was reheated to 58C where it was maintained for 1~ h. After cooling to 20C,
methylenc chloridc (100 ml) was added and the phascs were separatcd. Tbe aqucousphase was furthcr extracted with methylene chloride (50 ml) and the combined o~ganic
phases washcd with watcr (100 ml). Aftcr d~ying ovcr magnesium sulphate, filtering and
evaporation of thc solvcnt the product was obtained as an oil wdghing (6.96 g, 28.6%
pure by HPLC assay, 57% corrected yield).
Using the above condidons, but sdning the reaction mixture at ambient temp~re
for 20 h instead of ~cfluxing for 3 h, a coT~ectcd yield of 59% was obtained.
Using the above condihons, but cmploying one molar cquivalent of the carbimine
and amine base relative to the phenylheptylchloride, a corrected yield of 42% was obtained
2s Example 10
e~ara~ion of 2-!8-~henvloc~yl)benzaldeh~de
Exchanging potassium fo¢ lithium as basic coun~ion/using different imines
a) N-[(2-methylphenyl)methylene]-1,1-dimethylethanarnine (5.00 g, 29 mmol) was
added to a solution of lidlium diisopropylamide [28~5 mmol; ~repa~d from
diisopropylamine (4.0 mL, 2.89 g, 29 mmol) and n-butyl lithium (2.5 M, 11.43 mL, 28.5
mmol)] in THF (50 mL) at -10C. After sti~ring at this tempe~ e for 75 min, a solutioD
of potassium t-butoxide (1.49 M, 19.2 mL, 28.5 mmol) in THF was ad~ After a
furthcr 15 min, 1-chlo~7-phenylhcptanc (3.77 g, 17.9 mmo1) was added. Thc ~eaction
was allowed to warm to room temp~rature and sti~Ted for 16 h. Hydrochloqic acid (6M, S
mL) was added and the mixturc was refllL~ced for gO min. The aqueous layer was separated
WO 93/190~3'~, 6 3 ~ Pcr/lJlss3/o2gr
- 18 -
and extracted with hexane (2 x 200 mL). T~le combined organic fractions were dried over
sodium sulphate, filtered and the solvents removed by evaporation under ~duced pressure
to give an oil (7.44 g). Assay of this material showed it to cont~un 65% w/w
phenyloctylbenzaldehyde (4.84 g, 16.5 mmol, 92%).
s
b) Using the procedure of (a), except substituting N-[(2-methylphenyl)methylene]-
isopropylamine and N-[(2-methylphenyl)methylene]-n-butylamine, gave the folllowing
results: `
R3 ratio
imine substituent imine:PHC Yield(%)
i) t-Bu 1.6:1 92
ii) i-Pr 2.0:1 31
iii) n-Bu 2.0:1 ~10
Example 1 1
Pr~a~s~nof 2-(8-phenvloctvl)benzaldehyde
Usc of a catalytic amount of a nitrog~n base/companson of different electrophiles
a) phenylheptylbromide/phenylhep~yliodide
i.) To a solution of N-[(2-methylphenyl)methylenel-1,1-dimethyle~anasnine
(5.0 g, 0.03 mol) and N,N,N',N'-tetramethylethylene diamine ~3.31 g, 0.03 mol) in
tetrahydrofuran (40 mL), n-butyl lithium (2.5 M, 11.4 mL, 0.03 mol) at 0C was slowly
20 added. The solution was stirred fo~ an additional 30 min followed by the quick addition of
1-bromo-7-phenylheptane (7.28 g, 0.03 mol) in tetrahydrofuran (10 mL). The reaction
mixture was allowed to warm to room temperature and stirnng was continued fo~ 15 h.
The ~action mixture was quenched with 10% aqueous hyd~ochloqic acid (50 mL) and
stirred fo~ 30 min. The layers were separated, methylene chloTide (50 mL) wæ added to
25 the c~rganic layeT and dle organics were washed wi~ sat~ated b~ine solu~ion (50 mL). The
oqganics we~ then dried (magnesium sul~ate), and concentrated to an oil to yield 2-(8-
phenyloc~l)benzaldehyde (3.8 g, 45%): IR (neat film) 2920, 2880, 1695, 1600, 1455 cm~
I; lH NMR (CDC13, 400 MHz) ~ 10.25 (s, 1 H), 7.80 (dd, 1 H, J = 1.2 and 7.7 Hz),7.45 (m, 1 H), 7.33-7.13 (m, 7 H), 2.98 (t, 2 H, J = 7.7 Hz), 2.58 (t, 2 H, J = 7.7 Hz),
1.58 (m, 4 H), 1.30 (m, 8 H).
~VO 93/19033 2 1 3 ~ 6 3 9 PCl/US93/02803
19
ii) Using the procedure of (a)(i), except substituting l-iod~7-phenylheptane
for l-bromo-7-phenylheptane, 2-(8-phenyloctyl)benzaldehyde was prepared in 34% yield.
b) 1-bromo-7-phenylhcptane/1-chloro 7-phenylheptane
s i) A solution of N-[(2-methylphenyl)methylene]-l.l~imethylethanamine (2.8 g,
0.016 mol) and 2,2,6,6-tctramethylpiperidinc (0.23 g, 0.0016 mol) in tetrahydrofuran (10
mL) was coolcd to -5C. To this was added n-BuLi (1.6 M, 10 mL, 0.016 mol) ovcr 40
min maintaining the temperature at -5C. A solution of l-br~m~7-phcnylhcptanc (3.4 g,
0.0133 mol) in tetrahydrofuran (5 mL) was addcd quickly at -5C. The tcmperatureo quiclcly rose to 40C and after cooling to ambient tcmperature the mixture was stirrcd for 1
h. T~hc mixture was qwnched by the addidon of dilutc hydrochlonc acid and stirred at
ambicnt ~empcraturc for 16 h. Thc product was isolated in the usual manner (5.0 g, 75%
pure, 96% correctcd yield).
ii) Using the procedure of (b)(i), cxcept substituting l-chloro-7-phenylheptane for
1-bromo-7-phcnylheptane, phenyloctyl bcnzaldehyde was plepared in 879to corn~cted yield.
Examplc 12
cparation of 2-(~henvloctyl~benzaldchvdc
Effect of changing the temperaturc at which anion is formed for various nit~ogen bases.
a) A stirrcd solution of N-~(2-methylphenyl)methylene]-1,1 dimethylethanaminc (11.2
g, 0.064 mol) and 2,2,6,6-tetra-methylpiperidine (0.9 g, 0.0064 mol) in tetrahydrofuran
(40 mL) was cooled to -5C. To this was added n-BuLi (1.6 M, 40 mL, 0.064 mol) over
60 min fsom a syringe pump such that the tPmperature was maintained below ODC. The
mixture was sti~red for 30 min and 1-chloro-7-phenylheptane (11.23 g, Q053 mol) in
~etrahydrofuran (20 mL) quickly added The reaction mixture was heated at 5~55C f
2 h The reaction mixture was cooled to 40C and quenched by the slow addition of dilute
hydrochl~,ric acid (100 mL of acid diluted with 300 mL of water). Hydrolysis wascompleted by heating the mixture at 5~60C for 2.S h. The mixture was cooled to ambient
temperature and the organic phase separated. l~e aqueous phase was extracted with
hexane (100 mL) and the combined organic extracts washed with water (100 mL). The
ex~cts werc dned ovcr magncsium sulphate and after filtcring and washing the filtcr calce
wi~ hcxanc ~c organic solution was conccntratcd under vacuum to givc 2-(8-
phcnyloctyl)bcnzaldchydc as an oil (14.5 g, 69.3% purc by HPLC assay, 87% corrected
yicld),
wo 93/19033 Pcr/uss3/o28r-
639
- 20-
b) To a s~red solution of N-~(2-me~ylphenyl)methylene]-1,1-dimethylethanamine
(21.0 g, 0.12 mol) in tetrahydrofuran (75 mL), n-BuLi (1.54 M, 78 mL, 0.12 mol) was
added over 1 h such that the temperature was maintained between 20-30C with cooling.
The mixture was stirred for 30 min and l-chloro 7-phenylheptane (21.05 g, 0.1 mol) in
tetrahydrofuran (40 mL) was added quickly. The mixture was heated at 50~C for 3 h and
quenched by the slow addition of dilute hydrochloric acid. Hydrolysis was completed by
heating the mixture at 50-60DC for 2.5 h. The mixture was cooled to ambient temperature
and the organic phase separatcd. The aqueous phasc was extracted with hexane, and thc
0 combincd or~ic cxtracts were washed with water. The cxtracts were dlud over
magnesium sulphatc and, after filtering and washing the filter cake with hcxane, the organic
solution was concentratcd under vacuum to givc 2-(~phenyloctyl)benzaldehyde æ an oil
(34.56 g, 65.3% pure by HPLC assay, 77% corrected yicld).
c) Using thc same proccdure æ in (a) or (b), exccpt valying the nitrogcn base and the
temperature at which thc anion wæ formed, thc following results we~e obtaincd
Anion soludon impurity profilc
Aminc mp.(~2 vield (%~ (% PHE~ ~%
i) (i-Pr)2NH -5 55 7.2 17.3
ii) (i-Pr)2N~I 25 94 1.8 0
iii) DCAf -5 48 8.6 14.9
iv) DCAt 25 83 1.7 0. 1
v) TMPtt -5 87 0.6 0.4
vi) T~tt 25 89 ().7 0.7
~) -- O 45 ND ND
~i) -- 25 77 ND ND
DCAt = dicyclohexylamine P~ = phenylhcptene
TMPtt = tetramcthylpiperidine PHC~ = phenylhcptylchl~ide
d) lJsing thc p~cedure of (a) oq (b), except substituting l-br~m~7-phenylheptane,
dle following rcsults wcre obtained
WO 93/19033 ~ 1 3 2 6 3 9 PCr/lJS93/02X03
- Anion solution impurity profile
nine temp.(C) vield(%~ (% PHE~ (% PHB~
i) (i-Pr)2NH O 89 ND ND
ii) TMP 25 96 ND ND
ii) SSJl 195/1 19
PHB~ = phenylheptylbromide
S Many variations of thcse examples will be apparent to one skilled in the art and this
invendon is not limited to these examples, but includes all variations encompassed by the
claims which follow.