Note: Descriptions are shown in the official language in which they were submitted.
The present invention relates to a novel pyrazole
derivative and its salts, to a method of preparing it and
to the pharmaceutical compositions in which it is present.
Numerous pyrazole derivatives have been described
in the literature; more particularly, EP-A- 268554 and DE
A-3910248 claim pyrazoles possessing herbicidal properties,
EP-A-430186 and JP-A-3 031840 claim compounds useful for
photography, and EP-A-418845 claims pyrazoles possessing
antiinflammatory, analgesic and antithrombotic activity.
It has now been found that a N-piperidino-pyrazole-
3-carboxamide has a very good affinity for the cannabinoid
receptor and is useful in the therapeutic areas in which
cannabis is known to be involved.
O9-Tetrahydrocannabinol, or 09-THC, is the main
active constituent extracted from Cannabis sativa (Tuner,
1985; In Marihuana $~, Ed. Harvey, DY, IRL Press, Oxford).
The effects of cannabinoids are due to an
interaction with specific high-affinity receptors present
in the central nervous system (Devane et al., Molecular
Pharmacology, 1988, 34, 605-613) and peripheral nervous
system (Nye et al., The Journal of Pharmacology and
Experimental Therapeutics, 1985, ~, 784-791; Kaminski et
al., 1992, Molecular Pharmacology, ~, 736-742; Munro et
al., Nature, 1993, ~, 61-65).
Characterization of this receptor has been made
possible by the development of specific synthetic ligands
such as the agonists WIN 55212-2 (J. Pharmacol. Exp. Ther.,
1993, ~ø, 1352-1363) or CP 55,940 (J. Pharmacol. Exp.
Ther., 1988, ~, 1046-1051).
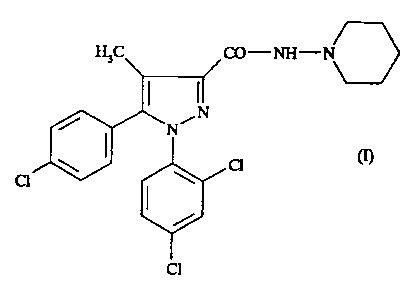
According to one of its features, the present
invention relates to the N-piperidino-5-(4-chlorophenyl)-1-
(2,4-dichlorophenyl)-4-methylpyrazole-3-carboxamide of the
formula .
2
._
H,C CO-NH-N
C1
CI
CI
to its pharmaceutically acceptable salts and to their
solvates.
The pharmaceutically acceptable salts of the
compound of formula (I) include the acid addition salts
such as the hydrochloride, hydrobromide, sulfate,
hydrogensulfate, dihydrogenphosphate, methanesulfonate,
methylsulfate, oxalate, maleate, fumarate, naphthalene-2-
sulfonate, glyconate, gluconate, citrate, isethionate and
paratoluene-sulfonate.
According to another of its features, the present
invention relates to a method of preparing the above
compound (I), its salts and their solvates, which comprises
treating a functional derivative of 5-(4-chlorophenyl)-1-
(2,4-dichlorophenyl)-4-methylpyrazole-3-carboxylic acid of
the formula .
H3C' ~COOH
CI
CI
can
CI
- 3 213~~393
with 1-aminopiperidine in an organic solvent in the
presence of a base, and optinally converting the resulting
compound into one of its salts or one of their solvates.
The acid chloride, the anhydride, the mixed
S anhydride, a straight or branched Cl-C4 alkyl ester, an
activated ester such as p-nitrophenyl ester, or the free
acid appropriately activated, for example by N,N
dicyclohexylcarbodiimide or benzotriazol-N-oxotris
(dimethylamino)phosphonium hexafluorophosphate (BOP) can be
used as the functional derivative of the acid (II).
Thus, in the method of the invention, 5-(4-
chlorophenyl)-1-(2,4-dichlorophenyl)-4-methylpyrazole-3-
carboxylic acid chloride, obtained by reaction of thionyl
chloride with the acid of formula (II), can be reacted with
1-aminopiperidine in a solvent such as dichloromethane in
an inert atmosphere, at a temperature between 0'C and room
temperature, in the presence of a base such as
triethylamine.
Alternately, the mixed anhydride of the acid of
formula (II) can be prepared by reaction of
ethylchloroformate with the said acid in the presence of a
base such as triethylamine, and then reacted with 1
aminopiperidine in a solvent such as dichloromethane in an
inert atmosphere, at room temperature, in the presence of a
base such as triethylamine.
The compound of formula (I) obtained in this way is
isolated, in the form of the free base or a salt or a
solvate, by the conventional techniques.
The compound of formula (I) can be isolated in the
form of one of its salts, for example the hydrochloride or
the oxalate; in this case, the free base can be prepared by
neutralizing said salt with a mineral or organic base such
as sodium or ammonium hydroxide or triethylamine, or with
an alkali metal carbonate or bicarbonate such as sodium or
potassium carbonate or bicarbonate, and converted into
--
another salt such as the methanesulfonate, fumarate or
naphtalene-2-sulfonate.
When the compound (I) is obtained in the form of
the free base, a salt is formed by treatment with the
chosen acid in an organic solvent. Treatment of the free
base, for example dissolved in an ether such as
diethylether, or in acetone, with a solution of the acid in
the same solvent gives the corresponding salt, which is
isolated by the conventional techniques.
The acid of formula (II) used as starting compound
in the method of the present invention can be prepared
according to conventionnal methods. Some of these methods
are illustrated therafter in the preparations.
Preparations 1 and 2 are similar. They are carried
out according to the reaction scheme below (route 1) .
Route 1
O 1) LiHMDS O-Li+
/ C1 O
Et 2) (COzEt~ ~ / NH-~z
CH3 COZEt Cl
O Cl \
3
'OEt Cl
/ N~N AcOH
O
w ~ CI Cl ~ / N-~ CI
CH3 COiEt
C1
1/ Toluene
KOH/Me~ 1~ C
2/ NaOH
(In
LiHMDS . lithium salt of hexamethyldisilazane
PTSA : paratoluenesulfonic acid.
The first step is carried out according to J.
Heterocyclic Chem., 1989, 26, 1389.
5
- ~~~6~9~
Preparation 3 is carried out according to the
scheme below (route 2) .
Route 2
TMSC1
O NaI
Cl ~3~ O- Sl (CH~3
\ ~ ~ Cl \ /
H
COzEt 1) ZnClz
COCI CHZCl2
Cl EtzO
Cl
Toluene
2) H20
\ / Cl
O O O
C1 \ ~ N-~ C1 HCl C C OEt
U _
~C~ ~C~
CH3 COzEt EtOH Cl \ ~ ~ II
Toluene ~3 O
Toluene
PTSA
110'C
OEt 1) MeOH
KOH
1 2) HCl
C1
O
~3
N.N
w I ~ C
TMSC1 . chlorotrimethylsilane
PTSA . paratoluenesulfonic acid
6
'~
The first step is carried out according to the
method described by E.S. SCHWEIZER in J. Org. Chem., 1987,
~, 1324-1332. The second step is carried out according to
the method described by R.E. TIRPAK et a1. in J. Org.
S Chem., 1982, ~, 5099-5102.
The other reagent used in the method of the present
invention, 1-aminopiperidine, is commercially available.
The compound of formula (I) exhibits a very good
affinity in vitro for the central cannabinoid receptors
under the experimental conditions described by Devane et
al., Molecular Pharmacology, 1988, ~ø, 605-613.
More particularly, the compound of the present
invention, as such or in the form of one of its
pharmaceutically acceptable salts, is a potent and
selective antagonist of the central cannabinoid receptors
and has a Ki of about 2nM. It is from 500 to 1000 times
more active on the central receptor as on the peripheral
receptor; it is also active upon oral administration and
penetrates the blood-brain barrier.
The good penetration of the compound of the present
invention in the central nervous system as well as its
antagonist character are confirmed by the results obtained
with the model of the antagonism of hypothermia induced by
an agonist of cannabinoid receptors. Especially, the
compound of the present invention antagonizes the
hypothermia induced by WIN 55212-2 in mice with an ED50 of
0.3 mg/kg i.p. and 0.4 mg/kg per os. In this test (Pertwee
R.G., 1985 . 263-277 ; in Marihuana $~, Ed. Harvey, D.Y.,
IRL Press, Oxford), the above compound exerted its action
for 8 to 10 hours after oral administration of a dose of
3 mg/kg.
In addition, the compound (I) alone, upon
subcutaneous administration, improves the memory capacities
of rats in the test of the central memory ( A P~rio et a1.
in Psychopharmacology, 1989, ~, 262-268).
~1~~~~~
Thanks to its remarkable properties, especially its
high affinity, its selectivity towards the central receptor
as well as its capacity to penetrate the blood-brain
barrier, the compound (I), as such or optionally in the
S form of a pharmaceutically acceptable salt or a solvate,
can be used as the active principle of drugs intended for
the treatment of the diseases of the central nervous system
in mammals.
The toxicity of compound (I) is compatible with its
use as a psychotropic drug, especially for the treatment of
thymic disorders, anxiety disorders, mood disorders,
vomiting, memory disorders, cognitive disorders,
neuropathies, migraine, stress, psychosomatic-induced
diseases, epilepsy, dyskinesia or Parkinson's disease.
The compound (I) according to the invention can
also be used as a drug for the treatment of appetite
disorders, especially as an anorexic, for the treatment of
schizophrenia, delirious disorders, psychotic disorders in
general as well as for the treatment of disorders
associated with the use of psychotic substances.
Furthermore, the compound (I) according to the invention
can be used in cancer chemotherapy.
The use of the compound according to the invention
as a drug for the treatment of appetite disorders, anxiety
disorders, mood disorders, schizophrenia, psychotic
disorders, memory disorders, cognitive disorders and
dyskinesia, as well as its use in cancer chemotherapy,
constitute a further feature of the present invention.
The compound according to the invention is
generally administered in dosage units.
Said dosage units are preferably formulated in
pharmaceutical compositions in which the active principle
is mixed with a pharmaceutical excipient.
Thus, according to another of its features, the
present invention relates to pharmaceutical compositions in
which a compound of formula (I) or one of its
8
pharmaceutically acceptable salts or one of their solvates
is present as the active principle.
The compound of formula (I) above and its
pharmaceutically acceptable salts can be used in daily
S doses of 0.01 to 100 mg per kilogram of body weight of the
mammal to be treated, preferably in daily doses of 0.1 to
50 mg/kg. In humans, the dose can preferably vary from 0.5
to 4000 mg per day, more particularly from 2.5 to 1000 mg,
depending on the age of the subject to be treated or the
type of treatment: prophylactic or curative.
In the pharmaceutical compositions of the present
invention for oral, sublingual, subcutaneous,
intramuscular, intravenous, transdermal, local or rectal
administration, the active principle can be administered to
animals and humans in unit forms of administration, mixed
with conventional pharmaceutical carriers. The appropriate
unit forms of administration include forms for oral
administration, such as tablets, gelatin capsules, powders,
granules and solutions or suspensions to be taken orally,
forms for sublingual and buccal administration, aerosols,
implants, forms for subcutaneous, intramuscular,
intravenous, intranasal or intraocular administration and
forms for rectal administration.
In the pharmaceutical compositions of the present
invention, the active principle is generally formulated as
dosage units containing from 0.5 to 1000 mg, preferably
from 1 to 500 mg, more preferably from 2 to 200 mg of said
active principle per dosage unit for daily administrations.
When preparing a solid composition in the form of
tablets, a wetting agent such as sodium laurylsulfate can
be added to the active principle optionally micronized,
which is then mixed with a pharmaceutical vehicle such as
silica, starch, lactose, magnesium stearate, talc or the
like. The tablets can be coated with sucrose, with various
polymers or other appropriate substances or else they can
be treated so as to have a prolonged or delayed activity
9
and so as to release a predetermined amount of active
principle continuously.
A preparation in the form of gelatin capsules is
obtained by mixing the active principle with a diluent such
S as a glycol or a glycerol ester and pouring the mixture
obtained into soft or hard gelatin capsules.
A preparation in the form of a syrup or elixir can
contain the active principle together with a sweetener,
which is preferably calorie-free, methylparaben and
propylparaben as antiseptics, as well as a flavoring and an
appropriate color.
The water-dispersible powders or granules can
contain the active principle mixed with dispersants,
wetting agents or suspending agents such as polyvinyl-
pyrrolidone, and also with sweeteners or taste correctors.
Rectal administration is effected using
suppositories prepared with binders which melt at the
rectal temperature, for example cacao butter or
polyethylene glycols.
Parenteral administration is effected using aqueous
suspensions, isotonic saline solutions or sterile and
injectable solutions which contain pharmacologically
compatible dispersants and/or solubilizers, for example
propylene glycol or polyethylene glycol.
Thus a cosolvent, for example an alcohol such as
ethanol or a glycol such as polyethylene glycol or
propylene glycol, and a hydrophilic surfactant such as
Tween~ 80, can be used to prepare an aqueous solution
injectable by intravenous route. The active principle can
be solubilized by a triglyceride or a glycerol ester to
prepare an oily solution injectable by intramuscular route.
Transdermal administration is effected using
multilaminated patches or reservoirs into which the active
principle is in the form of an alcoholic solution.
10
The active principle can also be formulated as
microcapsules or microspheres, optionally with one or more
carriers or additives.
The active principle can also be presented in the
form of a complex with a cyclodextrin, for example a-, B
or y-cyclodextrin, 2-hydroxypropyl-B-cyclodextrin or
methyl-B-cyclodextrin.
Among the prolonged-release forms which are useful
in the case of chronic treatments, implants can be used.
These can be prepared in the form of an oily suspension or
in the form of a suspension of microspheres in an isotonic
medium.
The following Examples illustrate the invention
without however implying a limitation.
The melting or decomposition points of the
products, M.p., were measured in a capillary tube
with a
Tottoli apparatus ; in some cases, differential scanning
calorimetry (DSC) was used to measure the melting
temperature.
The following abbreviations are used in the
preparations and in the examples .
THF . tetrahydrofuran
Ether . diethyl ether
Iso ether . diisopropyl ether
EtOH , ethanol
AcOEt . ethyl acetate
MeOH . methanol
DCM . dichloromethane
KOH . potassium hydroxide
AcOH . acetic acid
HC1 . hydrochloric acid
NaCl . sodium chloride
RT . room temperature
DSC . differential scanning calorimetry
M.p. . melting point
11 ~~3~~u~~
The following abbreviations are used in the
interpretation of the NMR spectra .
s . singlet
d . doublet
t . triplet
q . quadruplet
m . unresolved signals or multiplet
PREPARATION 1
A) Lithium salt of ethyl 4-(4-chlorophenyl)-3-methyl-4-
oxydo-2-oxobuten-3-oate
125 ml of a 1M solution of the lithium salt of
hexamethyldisilazane in THF are added under a nitrogen
atmosphere to 500 ml of ether. The mixture is cooled to
78' C and a solution of 21 g of 4-chloropropiophenone in
100 ml of ether is added dropwise. After stirring for 45
minutes, 19.2 ml of ethyl oxalate are added rapidly and the
mixture is stirred for 16 hours while allowing the
temperature to rise to RT. The precipitate formed is
filtered off, washed with ether and dried under vacuum to
give 12.6 g of the expected product.
B) Ethyl 5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-
methylpyrazole-3-carboxylate
9.8 g of 2,4-dichlorophenylhydrazine are added to a
solution of 12.6 g of the lithium salt obtained above in
70 ml of EtOH and the mixture is stirred for 16 hours at
RT. The precipate formed is filtered off, washed with EtOH
and then ether and dried under vacuum to give 12.6 g of
hydrazone. This is dissolved in 100 ml of AcOH, the mixture
is refluxed for 24 hours and then poured into 500 ml of
iced water. The mixture is extracted with AcOEt, washed
with water and then a saturated solution of NaCl. After
drying over magnesium sulfate and evaporation under vacuum,
the crude product is crystallized from iso ether to give
9.6 g of the expected product. M.p. - 124' C.
12 ~~ 36~~~
C) 5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-
pyrazole-3-carboxylic acid
A solution of 3.3 g of KOH in 70 ml of water is added to a
solution of 9.6 g of the ester obtained above in 70 ml of
S MeOH. The mixture is refluxed for 3 hours, poured into 200
ml of iced water and the reaction mixture is acidified to
pH = 1 upon addition of a 10 % solution of HCl. The
precipitate formed is filtered off, washed with water and
dried under vacuum to give 8.8 g of the expected acid.
M.p. = 211' C.
Preparation 1 is improved using the operating
conditions described in preparation 2 below .
PREPARATION 2
A) Lithium salt of ethyl 4-(4-chlorophenyl)-3-methyl-4-
oxydo-2-oxobuten-3-oate
2008 g of the lithium salt of hexamethyldisilazane are
dissolved, in a reactor, under a nitrogen atmosphere, in
10.1 1 of methylcyclohexane. A solution of 1686 g of 4
chloropropiophenone in 4 1 of methylcyclohexane is then
added slowly at 20 ~ 5' C. After stirring for 4 and a half
hours, 1607 g of ethyl oxalate are added over 35 minutes at
20 t 5' C. The mixture is stirred for 17 hours at the same
temperature. The solid formed is filtered off, washed with
methylcyclohexane and dried under vacuum to give 1931 g of
the expected product.
B) 5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-
pyrazole-3-carboxylic acid
1/ 1921 g of the lithium salt obtained above are dissolved,
in a reactor, under a nitrogen atmosphere, in 11 1 of EtOH.
1493 g of 2,4-dichlorophenylhydrazine hydrochloride are
then immediatly added at 20 * 5' C. The mixture is stirred
for 1 hour and 2.88 1 of deionized water are then added,
and stirring is continued for 17 hours at 20 t 5' C. The
precipitate formed is filtered off, washed with 80 % (v/v)
13
~~~6~~~
ethanol and dried under vacuum to give 2280 g of the
expected hydrazone. M.p. - 140' C.
2/ 2267 g of hydrazone are dissolved, in a reactor, under a
nitrogen atmosphere, in 11.3 1 of toluene. 201.6 g of
paratoluenesulfonic acid are then added and the mixture is
refluxed for 3 hours. The mixture is cooled to 20 t 5' C
and paratoluenesulfonic acid is removed by extraction with
deionized water. 120.75 g of benzyltriethylammonium
chloride and then a solution of 636 g of NaOH in 1180 ml of
deionized water are added to the toluene solution. The
mixture is heated for 4 hours at 68 t 3' C with vigorous
stirring, sodium hydroxide is then neutralized and the
reaction mixture is acidified with 1500 ml of HCl (d =
1.19). The mixture is cooled to 20 t 5' C, the precipitate
formed is filtered off, washed with toluene and then
deionized water, and dried under vacuum to give 1585 g of
the expected product. M.p. - 210' C.
PREPARATION 3
A) 1-(4-chlorophenyl)-1-trimethylsilyloxypropene
13.47 g of chlorotrimethylsilane are slowly added to
12.55 g of triethylamine, under a nitrogen atmosphere, at
20 * 3' C. 16.86 g of 4-chloropropiophenone (endothermic
mixture) and then a solution of 18.58 g of sodium iodide in
125 ml of acetonitrile are further added while maintaining
the temperature at 18 ~ 2' C. The mixture is then heated
for 3 hours at 40 ~ 5' C, the acetonitrile is removed under
reduced pressure and 150 ml of toluene are added to the
solid residue. 50 ml of solvent are distilled under reduced
pressure to drive the residual acetonitrile off. The
inorganic materials are extracted with 100 ml of iced
water, the organic phase is washed with 100 ml of iced
water and dried over magnesium sulfate. The toluene is
removed under reduced pressure and complete removal of the
14
~1~.3~~~~
solvents is performed for 15 hours at 60' C under a
pressure of 1 mbar to give 2~.7 g of an oil.
NMR run at 200 MHz (CDC13)
0.13 ppm . s . 9H
1.7 ppm . d . 3H
5.28 ppm . q . 1H
7.21-7.39 ppm . m . 4H.
B) Ethyl 3-(4-chlorobenzoyl)-3-methylpyruvate
g of anhydrous zinc chloride are suspended in 50 ml of
10 toluene under a nitrogen atmosphere. Residual water is
azeotropically driven off over 1 hour under atmospheric
pressure. 20 ml of toluene and then 11.5 ml of ethyl ether
are added to the mixture, cooled to 20 t 3' C. A solution
of 17 g of ethyl chloroglyoxylate diluted in 20 ml of
dichloromethane is then slowly added to the mixture cooled
to 0 t 2' C. 14.5 g of the product obtained in the previous
step are added over 1 and a half hours at the same
temperature. The temperature is then allowed to rise to RT
and the mixture is heated for 4 hours at 45' C. The organic
phase is washed with a solution of sodium hydrogen-
carbonate and then water, and dried over magnesium sulfate.
The solvents are removed under reduced pressure to give
17.6 g of an oil.
NMR run at 200 MHz (CDC13)
1.25 ppm . t . 3H
1.35 ppm . d . 3H
4.20 ppm . q . 2H
4.93 ppm . q . 1H
7.45-7.90 ppm . m . 4H.
C) Ethyl 5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-
methylpyrazole-3-carboxylate
13.3 g of 2,4-dichlorophenylhydrazine hydrochloride are
added to 17.6 g of the compound obtained in the previous
step and the mixture is stirred for 18 hours at 20 t 3' C.
Without isolating the hydrazone, 0.56 g of
paratoluenesulfonic acid are then added and the ternary
15
l c3
azeotrope (water, ethanol, toluene) is distilled. Toluene
reflux is continued for 1 hour and the reaction mixture is
then cooled to 20 ~ 3' C. The insoluble material is
filtered off and the toluene solution is then washed twice
with 100 ml of water. The solvents are removed under
reduced pressure to give a crude oil which is used as such
in the next step.
D) 5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-
pyrazole-3-carboxylic acid
8.1 g of KOH in pellets are added to a solution of the oil
obtained in the previous step in 100 ml of MeOH. The
mixture is left for 1 hour at 25 ~ 3' C and the solvents
are then removed under reduced pressure. The residue is
taken up with 200 ml of water and 40 ml of toluene, the
mixture is heated at 60 ~ 3' C, decanted, and the aqueous
phase is extracted three times, at this temperature, with
40 ml of toluene. Hydrochloric acid is then added to the
aqueous phase until pH = 1.5. The white crystals formed are
filtered off, washed with water and then iso ether and
dried under vaccum to give 9.9 g of the expected product.
M.p. - 210' C.
EXAMPLE 1
N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-
methylpyrazole-3-carboxamide
A) 5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-
pyrazole-3-carboxylic acid chloride
5 ml of thionyl chloride are added to a suspension of 8.8 g
of the acid obtained in step C of preparation 1 in 90 ml of
toluene, the mixture is refluxed for 3 hours and then
evaporated to dryness under vaccum. The residue is taken up
in 90 ml of toluene and the solvent is evaporated again
under vacuum to give 8.0 g of the expected acid chloride
which is used as such in the following step.
16
~.~~~9
B) N-piperidino-5-(4-chlorophenyl)-1(2,4-dichlorophenyl)-4-
methylpyrazole-3-carboxamide,
A solution of 8.0 g of the acid chloride obtained above in
80 ml of DCM is added dropwise to a solution of 2.8 ml of
1-aminopiperidine and 3.6 ml of triethylamine in 100 ml of
DCM, cooled to 0' C. The mixture is stirred for 3 hours
while allowing the temperature to rise to RT and then
poured into 200 ml of iced water. The mixture is extracted
with DCM, washed with water and then a saturated solution
of NaCl, dried over magnesium sulfate and evaporated under
vaccum. The residue is purified by chromatography on silica
gel using AcOEt/toluene (10/90; v/v) as the eluent.
Crystallisation in iso ether gives 5.9 g of the expected
product. M.p. - 148' C.
The compound of example 1 can also be prepared using
operating conditions which are industrially more
accessible.
A suspension of 1568.6 g of the acid obtained in step B of
preparation 2 in 14.1 1 of methylcyclohexane is heated,
under a nitrogen atmosphere, to 83 t 3' C, and a solution
of 554.5 g of thionyl chloride in 1.57 1 of
methylcyclohexane is added thereto. The mixture is stirred
for 3 hours at 83 ~ 3' C and the temperature is then
increased over 2 hours up to the reflux temperature of
methylcyclohexane while removing the excess thionyl
chloride by distillation. The mixture is cooled to 10
3' C and a solution of 452.9 g of 1-aminopiperidine and
457.5 g of triethylamine in 3.14 1 of THF is then slowly
added. The mixture is stirred for 17 hours while allowing
the temperature to rise to 20 ~ 5' C, and the organic phase
is successively washed, at 20 ~ 5' C, with deionized water
and a 4% aqueous solution of acetic acid. The organic phase
is then washed, at 70 ~ 3' C, with a 1.5 % solution of NaOH
and then deionized water, and the THF and water are driven
off by azeotropic distillation under atmospheric pressure.
The mixture is allowed to cool to 20 * 5' C. The expected
17
~'~3~~~~
product crystallises, the precipate formed is filtered off,
washed with methylcyclohexane and dried under vaccum to
give 1627 g of the title compound.
DSC . endothermic peak centered at 155.5' C.
EXAMPLE 2
N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-
methylpyrazole-3-carboxamide (solvate with ethanol)
10 g of the compound obtained in example 1 are suspended in
60 ml of absolute ethanol and the mixture is refluxed until
complete dissolution of the compound. The mixture is
allowed to cool to 20 ~ 3' C and stirring is continued for
2 hours. The white crystals formed are filtered off, washed
with ethanol and dried under vacuum to give 9.6 g of the
expected product.
DSC . endothermic peak centered at 102.7' C.
% calculated C . 56.5 H . 5.29 N . 10.98
% found C . 56.43 H . 5.41 N . 11.05.
EXAMPLE 3
N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-
methylpyrazole-3-carboxamide hydrochloride
A saturated solution of gaseous HC1 in ether is added
dropwise to a solution of 5.9 g of the compound obtained in
example 1 in 50 ml of ether until pH - 1. The precipitate
formed is filtered off, washed with ether and dried under
vacuum to give 6.0 g of the expected hydrochloride. M.p.
224' C (decomposition).
EXAMPLE 4
N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-
methylpyrazole-3-carboxamide hydrochloride (solvate with
ethanol)
18
~'~~
40 g of the compound obtained in example 3 are suspended in
400 ml of absolute ethanol.,The mixture is heated to the
boiling point until complete dissolution of the compound
and then stirred for 20 hours while progressively cooling
S it to 20 t 3' C. The white crystals formed are filtered
off, washed with ethanol and dried under vacuum to give
29.6 g of the expected product.
DSC : broad endothermic peak (175-230' C)
Thermogravimetry . weight loss . about 8.2 % starting at
100' C .
% calculated C . 53.04 H . 5.16 N . 10.31
% found C . 52.68 H . 5.23 N . 10.34.
EXAMPLE 5
N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-
methylpyrazole-3-carboxamide methanesulfonate (hemisolvate
with acetone)
3.84 g of methanesulfonic acid are added at 20 t 3' C to a
solution of 18.55 g of the compound obtained in example 1
in 185 ml of acetone and the mixture is stirred for 20
hours at the same temperature. The white crystals formed
are filtered off, washed with acetone and dried under
vacuum to give 21.65 g of the expected product.
DSC . melting, recrystallisation at about 175' C then
melting at 191.5' C.
Thermogravimetry . weight less . about 5.2 % starting at
90' C.
% calculated C . 49.90 H . 4.75 N . 9.50
% found C . 49.70 H . 4.76 N . 9.44.
EXAMPLE 6
N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-
methylpyrazole-3-carboxamide hemifumarate
19
~~~6~~J
A solution of 0.038 g of fumaric acid in 6 ml of acetone is
added dropwise to a solution of 0.30 g of the compound
obtained in example 1 in 3 ml of acetone. The white
crystals formed upon cooling to 0' C are filtered off,
washed with acetone and dried under vacuum to give 0.23 g
of the expected salt. M.p. - 168 'C.
EXAMPLE 7
N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-
methylpyrazole-3-carboxamide hydrogensulfate
0.018 ml of concentrated sulfuric acid are added to a
solution of 0.30 g of the compound obtained in example 1 in
3 ml of acetone. The white crystals formed are filtered
off, washed with acetone and then ether, and dried under
vacuum to give 0.31 g of the expected salt. M.p. = 240' C.
EXAMPLE 8
N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-
methylpyrazole-3-carboxamide paratoluenesulfonate
7.61 g of paratoluenesulfonic acid are added at 20 t 3' C
to a solution of 18.55 g of the compound obtained in
example 1 in 185 ml of acetone and the mixture is stirred
for 20 hours at the same temperature. The white crystals
formed are filtered off, washed with acetone and dried
under vacuum to give 24.25 g of the expected product.
DSC . endothermic peak centered at 236.8' C
% calculated C . 54.76 H . 4.60 N . 8.72
$ found C . 54.11 H . 4.71 N . 8.69.
EXAMPLE 9
N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-
methylpyrazole-3-carboxamide dihydrogenphosphate
2° ~~~b~~~
4.61 g of 85 % phosphoric acid are added at 20 t 3' C to a
solution of 18.55 g of the compound obtained in example 1
in 185 ml of methylethylketone. Water is removed by
distillation under atmospheric pressure of the azeotrope
S methylethylketone/water. The mixture is progressively
cooled to 20 t 3' C while stirring for 20 hours. The white
crystals formed are filtered off, washed with
methylethylketone and dried under vacuum to give 21 g of
the expected product.
DSC . endothermic peak centered at 185.5' C
% calculated C . 47.04 H . 4.31 N . 9.97
% found C . 46.96 H . 4.62 N . 9.98.