Note: Descriptions are shown in the official language in which they were submitted.
W093/26019 ~ 1~ 71~ d ~ PCT/~'S93/05595
1 PREPARATION OF CONTROLLED SIZE INORGANIC PARTICLES
2 ~OR USE IN SEPARATIONS, AS MAGNETIC MOLECULAR SWITC~ES,
3 AND AS INORGANIC LIPOSOMES FOR MEDICAL APPLICATIONS
4 --
Field of the Invention
6 This invention relates to a method for producing
7 inorganic oxides of substantial}y uniform particle size
8 distribution, coating said particles with various
9 ~unctional moieties, and clustering said moieties together
via controllably degradable chemica~, complex, or ionic
11 bonds. More particularly,~this invention relates to a
12 method of producing magnetic inorganic oxide particles of
13 substantially uniform size, or organic coated particle
1~ beads, linkins the particle or particle bead toqether to
form a large aggregate cluster with different chemical,
16 physical, or magnetic properties than the unit particle or
17 bead, and con$rollably and predictably revising the
18 cluster bac~ to unit bead or particle si~e and -~ice versa.
19 The substantially uniform size inorganic oxides also
allow for the preparation of novel inorganic core liposome
21 compositions for in vivo and in ~itro m~dical
22 applications.
~- 23 Backqround of the Invention
2~ Separations of all types ~are routinely done by the
exploitation of physical and chemical differences in the
26 various species to be separated. Size exclusion, boiling
27 point, and chemical affinity are techniques that have been
28 used for separations of particles, chemical species, and
29 biological moieties for hundreds of years. More recently,
the use of magnetism has been used as a tool for
31 separation of ~arious species material from one another.
- 32 By the early 1960's, the first stable magnetic fluid
33 colloid had been described. Later research led to the
34 development of a separations de~ice based on magnetic
3S density gradients in magnetic fluid columns. By 1979,
36 magnetic particles coated with appropriate functional
~ .
W0~3/26019 2 1 3 7 1 ~ 5 PCT/US93/OS~.
--2--
;
1 chemical groups for affinity chromatographv separations
2 were reported. The first commercial application of
3 magnetic separations was described by Chagnon et al in
4 U.S. Patent No. 4,628,037. The Chagnon p~tent describes
S the use of amine terminated silane coupled magnetic
6 particles for immunodiagnostic applications. The
7 materials described in the Chagnon et al patent are now
8 used commercially in medical diagnostic kits.
9 Magnetic separations have not been exclusively applied
to in ~itro apDlications. The use of magnetic separations
11 for in vivo applications is becoming increasingly more
12 acceoted and important as a therapeutic and diagnostic
13 tool. By the early 1980's, published reports described
14 the magnetic targeting and isolation of chemotherapeutic
drugs into rat-tail sarcoma. Widder (U.S. Patent Nos.
16 4,84g,210; 4,2~7,406; and 4,230,685) describe the use of
17 magnetic albumin spheres for ultrasound contrast media and
18 magnetic drug targeting. Schroeder (U.S. Patent ~o.
19 4,501,726) reports a method of preparing magnetic starch
beads for use in MRI imaging for the separation of TltT2
21 relaxation signals.
~2 In all of this previous work, the ~se of magnetic
23 separations has been done on magnetic particles or varying
24 particle size distribution. Thè magnetic particle is
coated with an organic compound, and used either as a
26 signal (e.g., M~I), targeting agent (e.q. in drug
27 delivery) or for separation in a magnetic field (e.g. in
28 vitro separations). However, an advantage in enhanced
29 separations, for example, could be achieved if the
magnetic particle could alte_ its s1ze, shape or magnetic
31 properties while in use in a controlled fashion.
32 Various methods have been reported for preparing
33 inorganic or inorganic oxide particles of some degree of
34 particle size control:
35- U.S. Patent 5,071,076 describes a method for producing
,
~ 36 magnetic microparticles from metallocenes. The method
W O 93/26019 PC~r/US93/05595
1 involves combining an aqueous slurry of the ~etallocene
2 and an aqueous slurry of a metal hydroxide ~d milling the
3 slurries together.
4 U.S. Patent 4,987,012 describes a process for
S preparing spherical particles of hydroxide having a
6 particle diameter from 0.1 to lOµm by adding a
7 corres~onding metal alkoxide to a dispersion of a water-
8 alcohol system having dispersed therein a metal oxide or
9 hydroxide as a seed, under alkaline conditions and
allowing a decomposition product from said metal alkoxide
11 to attach onto said seed to effect par~icle growth of the
12 seed. The improvement reported comprises maintaining said
13 dispersion at a substantially constant pH within the range
14 between 10 and 13 during the addition of the metal
alkoxide to said dispersion and the subseouent particle
16 growth of the seed, thereby to prepare mono-dispersed
17 particles substantially free from particle aggregation
18 having a sharp particle size distribution of a standard
19 deviation of not greater than 0.5.
; ~ 20 U.S. Patent 4,985,273 describes a method of producing
21 fine inorganic particles. The method comprises ~he steps
22 of reacting an inorganic fine particle on the entire
23 surface thereof with a silane type surface active agent
24 containing a straight hydrocarbon chain and a functional
group to form a monomolecular film on the entire surface
26 of said inorganic fine particle, thereafter making the
27 inorganic fine particles covered with the monomolecular
28 film in a predetermined density on a substrate, and
29 thereafter subjecting the monomolecular film to physical
or chemical treatment to allow the functional groups to be
31 chemically bonded to each other.
32 U.S. Patent 4,945,049, reports on a method for
33 preparing magnet~c powder comprising homogeneous and fine
34 particles using an alkali-producing enzyme. Particles
ha~ing a particle size ranging from 50 to 500 nm's were
36 reported.
, ` .
r~J ,~, ~i ~ 1 ' ;J
WO93/26019 PCT/US93/0C~S
--4--
1 U.S. Patent 4,702,775 describes the control of
2 particle size in the preparation of magnetite pigments.
3 The mean particle size was brought to a value within the
4 range of 0.06 to 0.5 µm by means of a residence stage
S between the precipitation stage and the oxidation stage.
6 Various other disclosures describe the preparation of
7 microporous membranes, primarily for a filtration purpose,
8 which~}imit the passage of selected size molecules within
9 à particular li~uid medium. For example, U.S. Patent
4,943,374 concerns the use of a microporous membrane
11 constructed of a polyether sulfone and hydrophilization
12 agent having a pore size which is within the range of 0.1
13 and 1.2 microns for the filtration of beer. U.S. Patent
14 4,954,381 describes the preparation of porous substrates
having well defined~morphology. U.S. Patent 4,g64,992
~ 16 describes a membrane filter having predetermined :~
; ~ 17 controlled porosit~y and to the method for making such a -~
18~membrane~filter~. U.S.~Patent 5,057,226 describes a method
l9~ ~of~;removing~a~constltuent of a biological fluid including
20~ a~blood~component,~ said method including f lowing the
21 biological fluid past one side of a first semipermeable
22 membrane, fl wing solution containing a first
23 precipitation agent past a second side of the membrane so
24 ~ as~to cause transfer of the precipitation agent through
25~ the membrane to;the biological fluid so as to improve`
26~ precipitàt;ion characteristics ~of the fluid; and
27 precipitating the constituent~ . ~
~ 28 ~What~emerges~from~the above, therefore, is the lack of
`~ 29 a convenient method to control~inorganic oxide particle
size, such that particle size control can then be further
31 utilized to manufacture novel aggregate particle clusters
32 with unique ch-mic~al or physical-chemical properties.
33 ~ Accordingly, it 1s an object of this invention to
34 prov~de a method~;for producing inorganic oxides of
substantially uniform particle size, coating said
36 ;particles w1th various functional moieties, and clustering
~., .
W093/26019 ~ 7~ 4~ PCT/US93/OSS95
1 said moieties together ~ia controllably degradable
2 chemical, complex or ionic bonds.
3 It is also an object of this invention to provide a
4 method of producing magnetic particle or organic coa~ed
~artlcle beads, linking said particle or particle beads
6 together to form a large aggregate cluster with different
7 chemical, physical, or magnetic properties than the unit
8 particle or bead from which it is derived, and
9 controllabiy and predictably revising the cluster back to
unit bead or particle, and vice versa.
ll It is also a further object of this invention to
12 provide a method of producing unit magnetic crystals of
13 small, substantiaily uniform part~cle slze for use in
14 preparing magnetic-molecular switches and apply such to
lS several in vitro and in vivo medical ahd biological
16 applications.
17 Nomenclature
18 The term "magnetic crystal" is defined as a particle
19 lOA to lO,OOO A in diameter comprised of iron oxide, iron
metal, cobalt metal, nickel metal, magnetic ferrites,
2l magnetic alloys, or~mixed lattice magnetic metals or metal
22 oxides. The term "magnetic bead" is defined as a magnetic
23 crystal or population of crystals coated by an organic
24 moie * or polymer or inorganic moiety or polymer to form a
25- bead of lOA to 500,000 A in diameter. The term "magneto-
26 molecu}ar switch" is defined as a cluster of magnetic
27 crystals or beads formed by the attachment of organic
28 moieties to the surface of the crystal or beads that link
29 the beads or crystals together via controllably degradable
chemical, complex, or ionic bonds.
31 As used herein the term:
32 I'Polyalkylether'' refers to polyethyleneglycol and
33 relate~ homopoIymers, such as polymethylethyleneglycol,
34 polyhydroxypropyleneglycol, polypropyleneglycol, poly-
3S methylpropyleneglycol, and polyhydroxypropyleneoxide, and
36 to heteropolymers of small alkoxy monomers, such as
, .
.: ~
,~ .
, ~:
~, .
WO93/26019 ~J 1 3
PCr/US93/
--6--
1 polyethylene/polypropyleneglycol, such polymers having a
~ molecular weight of at least about 120 daltons, and up to
3 about 20,000 daltons.
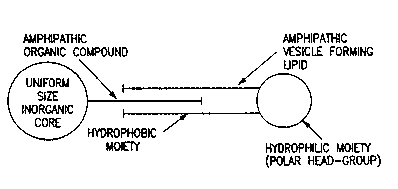
4 ~Amphipathic organic compound~ refers to any organic
compound containing both a hydrophobic and hydrophilic
6 moiety.
7 ~Amphipathic vesicle forming lipid" refers to any
8 lipid having a hydroohobic unit and hydrophilic unit, the
9 hydrophobic group typically including two acyl hydrocarbon
chains, the hydrophilic group containing a reactive
11 chemical group such as amine, acid, ester, aldehyde, or
12 alcohol group by which the lipid can be derivatized, e~g.
13 to a polyalkylether.
14 Summary of the Invention
This invention provides a method for preparing novel
16 precipitated inorganic oxide crystal particles of
17 substantially uniform particle size distribution. The
lB method comprises contacting aqueous solutions of an
19 inorganlc salt and an inorganic base across a porous
membrane wherein the membrane contains a plurality of
21 pores which allows for precipitation of substantially
22 mono-dispersed inorganic oxide particles on one side of
23 the membrane and precipitation of a salt of the
24~ corresponding base on a second side of the membrane.
WXen~the inorganic oxide crystal particles produced
26 accordinq to this method is an iron oxide particle of
27 reduced particle size (e~g. Fe3O4), which are non-
28 magnetic, they can be aggregated into one embodiment of
29 the magneto-molecular switch which comprises attachment of
organic moieties to the surface of the crystals that link
31 the crystal together to from controllably degradable
32 chemical, complex or ionic bonds. It has also been found
33 that aggregate cIusters of crystals can be prepared by air
34 or inert gas drying of the crystal particles along with
several different solution encapsulation techniques.
36 In a further embodiment of the magneto-molecular
`'
W093/~6019 PCT/US93/05595
l switch, the individual crystal particles or population of
2 crvstals so prcduced are coated by polymer encapsulation,
3 adsorbtion of monomer followed by crosslinking, or by
4 a~plying organo-metallic polymer coatings which are
covalently bonded or adsorbed on~o said particles, to form
6 a non-reversibly coated bead of lOA to 500,000 A in
7 diameter. Accor~ingly, the beads themselves can be
8 aggregated into controllably de~radable bead clusters by
9 the organic moieties that may ~e present on the beads, or
bv further attachment of organic moieties to the bead
ll surface, which in either case allow the beads to link
12 'ogethe~ to form controllably degradable chemical,
13 complex, or ionic bonds.
14 The present invention relates in one aspect to a
coated magnetically responsive particle comprising a
16 maanetic core particle comprising a magnetically
17 responsive metai, metal alloy or metal oxide and an
18 organo-metallic polymer coating covalently bonded to said
l9 particle wherein the bonding does not depend on the
presence of hydroxy functionality on the surface of said
21 particle, and wherein the organo-metallic polyme_ coating
22 is capable of bindin~ at least one type of bioaffinity
23 adsorbent. In addition to covalent bondin~, the organo-
24 metallic polymer can be adsorbed. The coated magnetically
r~sponsive particles have utility for either the
26 s_?aration or directed movement of biological molecules
27 from a surrounding medium.
28 The organo-metallic polymer is formed from an organo-
29 metallic monomer, which is applied to the metal particle,
and thermally cross-linked in situ to form an adsorbed or
3l a covalently bound polymer coating. Organo-titanium
32 polymers are preferred, however, organo-metallic polymers
33 formed from coordinate complexes of other transition
34 metals, such as zirconium (Zr), hafnium (Hf), vanadium
(V), tantalum (Ta) and niobium (Nb) or post-transition
36 metals, such as tin (Sn) and antimony (Sb), can be used.
WO93/26019 PCT/US93/0'
--8--
l A wide variety of bioaffinity adsorbents can be covalently
2 bonded to the organo-metallic polymer coating through
3 selected coupling chemistries.
4 More particularly, the invention relates to methods
for the preparation of magnetically responsi~e pzrticles
6 comprising a metal, metal alloy or metal oxide core and an
7 organo-metallic coating having an aliphatic moiety and an
8 organic functionality to which a variety of organic and/or
9 biological molecules can be coup~ed. The particles,
coupled or uncoupled, can be dispersed in a~ueous media
11 forming a colloidal dispersion which is stable, that is,
12 the particles resist rapid gravitational settling. The
13 particles can be reclaimed from the media by applying a
14 magnetic field.
Preferably, the particles are superparamagnetic; that
16 is, they exhibit no reminent magnetization after remo~al
}7 of a magnetic field which allows the particles to be
18 redispersed without magnetic aggregate formation.
l9 The organo-metallic coated magnetically responsive
particles of the invention may be coupled through the
21 organic functionality to biological or organic molecules
22 with affinity for, or the ability to adsorb, or which
23 interact with, certain other biological or organic
24 molecules. Particles so coupled may be used in a ~ariety
of in vitro or in vivo ~systems involving separations steps
26 or the directed movement of coupled molecules to
27 particular sites, including immunological assays, other
28 biological assays, biochemical or enzymatic reactions,
29 affinity chromatographic purification, cell sorting and
diagnostic and therapeutic uses.
31 In connection with the above, and in a ~urther aspect
32 of the present invention, a method of measuring analytes
33 in a sample is disclosed comprising the steps of: (a)
34 contacting a sample containing an unknown concentration of
the analyte with a known amount of a labeled analyte in
36 the presence of magnetic particles comprising: (l) a
WO93/26019 ~l ~ 7 ~ ~ a PCT/US93/0559
l magnetic core particle comprising a magnetically
2 r~sponsive metal, metal alloy or metal oxide; and (2) an
3 organo-metallic pol~mer coating covalently bonded to~said
4 particle wherein the bonding does not depend on the
presence of hydroxy functionality on the surface of said
6 particles, and wherein said organo-metallic coating has a
7 bioaffinity adsorbent covaLentIy ~oupled thereto, said
8 bioaffinity adsorbent is capable of binding to or
9 interacting with both the unlabeled and the labeled
analyte; (b) maintaining the mixture in step (a) under
ll conditions sufficient for said binding or interaction to
12 oc~ur; (c) magnetically separating the magnetic particles;
13 and (d) measuring the amount of label associated with the
14 magnetic particles and determining the concentration of
analyte in solution.
16 The present organo-metallic coated magnetic particles
17 ~provide superior composition, size, surface area, coupling
18~ ~ersatility, settling properties, and m~gnetLc behavior
l~ for use in biological separations. The magnetic particles
of this invention are suitable for many of the assays,
21 enzyme immobilization, cell sorting and affinity
22 chromatography procedures reported in the literature and,
23 in fact overcome many of the problems associated with
24 particle settling and reuse experienced in the past with
such procedures.
26 It has now been found that the inorganic oxides of
27 substantialiy uniform particle size can be used to prepare
28 a liposome composition comprising a substantially uniform
29 size inorganic core coated with an amphipathic organic
compound and further coated with a second amphipathic
31 vesicle forming lipid. The inorganic core is again
32 prepared by contacting aqueous solutions of an inorganic
33 salt a~d an inorganic base across a porous membrane
34 wherein the membrane contains a plurality of pores which
allows for precipitation of substantially monodispersed
36 size inorganic oxide particles on one side o~ the membrane
WO93/26019 ~ t ~ 7 ~ ? PCT/US93/05 `
--10--
l and precipitation of a salt of the corresponding base on a
2 second side of the membrane. Inorganic cores are also
3 prepared bv the reaction of metallocènes with aqueous
4 metal hydroxide slurries followed by milli~g to uniform
particle size. The class of inorganic cores include
6 Fe304, Fe203, A1203, Ti02, ZnO, FeO, and Fe.
7 The amphipathic vesicle forming lipid is preferably a
8 lipid having two hydrocarbon chains, including acyl
9 chains, and a polar head group. Included in this class
are the phos~holipids, such a pnosphatidylcholine (PC),
11 phosphatidic acid (PA), phosphatidylinositol (Pl),
12 sphingomyelin (SM), and the glycolipids, such as
13 cerebroside and gangliosides.
14 The amphipathic vesicle forming lipid can also be a
novel synthetic phenyl lipid compound having the
16 structural formula:
17 ~
18 R2 ~ (F4)n~H3
21
22 R~
23 wherein two of Rl, R2 and R3 represent a saturated or
24 unsaturate~d straight-chain or branched chain alkyl or acyl
group, the other béing hydrogen, therein pro~iding at
26 least two hydrocarbon chains attached to the phenyl
27 moiety, wherein the two hydrocarbon chains are typically
28 between about 14-22 carbon atoms in length, and have
29 varying degrees of unsaturation. R4 represents the
repeating unit of either a poly(alkylene oxide) polymer,
31 preferably ethylene, propylene and mixtures thereof, or
32 the repeating unit of poly(vinyl alcohol). The number of
33 alkylene oxide or vinyl alcohol groups in the polymer,
34 designated as n, may vary from 0 to about 200 or more.
In a further aspect, the invention includes an
36 inorganic core liposome composition for administering
.
~ WO93/2601~ P~T/US93/05~9~ ~'
-11- ,
;:
1 drugs via the bloodstream, comprising a substantially
2 uniform size inorganic core coated with an amphipathic
3 organic comDound and further coated with l-Z0 mole p~ercent
4 of an amphipathic vesicle-forming lipid derivatized with a
S hydrophilic polymer, and containing the compound in
6 liposome-entrapped form.
7 It has now aIso been found that liposome compositions
8 can be prepared to comprise a wave absorbing magnetic core ~-
9 coated with an amphipathic organic compound and further
coated with a second amphipathic vesicle forming lipid.
11 Tn a preferred em~odiment, the wave absorbing magnetic
12 core particles comprise ferrite or mixed ferrite
13 materials, prefe~ra~ly`of a uniform, controllable size and
14 narrow size distribution, wherein the primary component, I
the oxide, is of the formula M2(+3)M(~+2)04, wherein M(+3)
16 is Al, Cr or Fe, and M(+2) is Fe, Ni, Co~ Zn, Zr, Sr, Ca,
; 17 Ba, Mg,~Ga, Gd, Mn or Cd. In a further aspect, the oxides
18 can be advantageously mixed with LiO, MaO and KO, or with
19 Fe2O3 and Fe3O4. ~ i
The oreparation of substantially uni'orm size oxides,
21 1 to SQ,000 nm ir. diameter, is achieved by conversion of
22 hydrous; oxide gels, in a multi-step process, wherein
23 alkali is added to indivri~ual M(+3) and M(+2) aqueous
24 solutions, which separately precipitate the corresponding
metal hydroxide. The two precipitates are then coarsely
26 mixed to provide micron size amphorous gel particles, or
27 the gels can be finally mixed by ball milling, for
~28 example, to a particle size of about 100 A in diameter.
29 Tnese particles are then heated to effect dehydration, in
the presence of oxygen or air, wherein the dehydration
31 temperature, time of dehydration, and concentration of
32 oxygen or air operate to control the particle size of the -
33 oxide crystals therein produced.
; 34 In a further aspect, the invention includes a process
for the treatment of cancer cells by application of
36 external electromagnetic energy capable of the generation
:, ' ,
WO93/26019 ~ ~ri ~ a ~ pCT/US93/05
-12-
1 of heat in int_acellular particles to induce selective
2 thermal death of cancer cells comprisinq intravenously
3 injecting into the patient a wave absorbing magnetic core
4 particle coated with an amphipathic organic compound and
S further coated with a second amphipathic vesicle forming
6 lipid, absorbing said coated wave absorbing magnetic core
7 ?article intracellulary into the cancer cells, subjecting
8 the patient to an alternating electromagnetic field to
- 9 inducti~ely heat the magnetic core particle and thereby
10 the cancer cells, and continuing the inductive heating of
11 said magnetic core particle to attain an increase in
12 _ntracellular temperature to selecti~ely kill the cancer
13 cells.
14 Brief Description of the Figures
15 Fig. 1 is a drawing of a precipitation chamber used in r
16 accordance with the present invention.
17 ~ Fig. 2 illustrates the general liposome com~osition
18 comprising a substantially uniform size inorganic core
19 coated with an amphipathic organic compound and further
20 coatedlwith an amphipathic vesicle forming lipid.
21 Fig. 3 is a reaction scheme for preparing a phenyl
2Z lipid derivatized with polyethyleneglycol.
23 Fig. 4 illustrates the general liposome aomposition
24 comprising a wave~absorbing magnetic core particle coated
25 with an amphipathic organic compound and further coated
26 ~with an amphipathic vesi;cle forming lipid.
27 Detailed DescriDtion of The Invention
:
28 The magnetically responsive particles of this
29 invention-overcome problems associated with the size,
30 surface area, gravitational settling rate and magnetic
31 character of previously developed magnetic particles.
32 Gravitational settling times in excess of about 24 hours
33 can be a~hieved with the present magnetic particles. The
34 gravitational settling time is defined to be the time for
35 the turbidity of a dispersion of particles to fall by
36 fifty percent in the absence of a magnetic field gradient.
' .
WO 93/26019 ~ 1 ~ 7 ~ ~ 5 PCT/US93/~S~95
-13-
The present magnetlc parelcles comprise a core of a
magnetically responslve metal, metal alloy or metal~
o~ide, coaeed with organo-metallic polymer, wh$ch is
capable of binding reactl~e groups or agents, for
05 e~ample, che~ically react~ve gro~ps, biologically
react~ve gro~ps or b~oafinlty agent~. The organo-
metallic polymer is adsorbed onto or covalenely bound to
the magnetlc part~cle. The term n~agnetical~ly responsive
particle~ or "magnetic partlcle" is defined as any
part~cle dispersible or suspendibLe ln aq~eous med~a
without s~gniflcant grav~tacLonal settling, and separable
from suspension by application of a ~agnetic field.
The term "magnetic core" ~s defined as a crystal or
group (or cluseer) of crystals of a cransltion metal,
alloy or magnetlc metal oxide having ferrosplnel
structure and comprising tri~alen~ and ti~alent catlons
of the same oF dlfferent transition metals or magnetLc
metal crystal group. Metals, alloys and oxides which are
- useful as magnet~c core materlal in the present ln~ention
- 20 include the metals, alloys and oxidos b3sed on m~cals
which appear ln th- Periodic Table ln Gro~ps 4a and b, 5a
and b, 6a and 7a. TheQe lnclude, for exampLe, dl~alent
trans~tlon metals, sucb as lron, magnesium, manganese,
cobalt, nickel, zlnc and copper, alloys of ebese metals
such as iron alloys or oxites (e.g., ~ron magnesiu~
oxide, iron mangane-Qe oxide, iron cobalt oxide, iron
nickel oxide, iron zinc oxide and iron copper o~ide),
cobalt ferrite, samnrium cobalt, barlum ferrite, and
aluminum-nic~el-cobalt ant metal oxides including
magnetite (Fe304), h-maelte ~Fe203) and chromium d~oxide
~CrO2). By way of illustraelon, magnetlc core may be
~'
.
WO93/26019 ~ ~ 7 ~ PCT/USg3/05r
-14-
comprised of a cluster of superparamagnetic crystals o~
iron oxide, or a cluster of superparamagnetic or
ferromagnetic crystals of irons or oxide, or may consist
of a single superparamagnetic or ferromagntic crystal of
05 an iron oxide or metal alloy.
It has now been found that the Fe3O4 affords a crystal
lattice which contains primarily triva1ent iron (Fe+3) at
or near the surface of the crystal. These "surface
trivalent" elements of the lattice contain imperfections
which make them available for direct covalent attachment
of the orqanometallic compounds of the formula Ti(OR)4
according to the following general equation:
.
~: 15
,
~ ~ :
. , ;
. ~ ,
., ~ , .
,~-
:
.
.:~
~ 4 ~
W O 93/26019 PC~r/US93/05595 !;'''
O O (> O ' ''"
Fe~F ~ e Fe-F--F~ ~
'I 'I 1 o I I
F~-Fe~Fe ~T~O ~ ~ F~e-Fe
I l I o o o
1o Fo-Fe~Fe F~F~Fe -~
O O O O '~
Fe~F~F- ¦ SURF.4fE Fe~e-Fe
~ ~ ¦ ~PERFECTIONS I 1
O O SURFACECOA~ING
t -r~r~
. O~d~ion l I
l r ' ~.
O O o O
25 ~ ~
Fe-Fe~Fe Fe-Fe~Fo
I l HydrdY~
I f ~ 1 1
Fe-Fe-Fe Fe~e~Fe
/
Fe-Fo~Fe Fe~Fe-Fe
¦ HYDROXIDE FUNCTIONALITY
' ON OH . .
-
WO93/26019 PCT/US93/0 q
-16-
It should be noted that the imperfections of th~
surface trivalent iron are somewhat short-lived, and if
organo--metallic coatin~ is delayed, oxidation and
hydrolysis can oc~ur causing the development of surface
05 hydroxyls which preclùde direct covalent attachment of the
orqano-metallic moiety. For example, freshly ~ade Fe3O4
will spontaneously react; Fe3O4 material after 24 hours
reacts but requires about l hour of dwell time; after 48
hours the coupling reaction takes place very slowly and is
~enerally incomplete.
organo-metallic compounds are preferably of the
formula Ti(OR)4 wherein R is an alkyl group and the
dissociation to the reactive component follows the
following general reaction criterion: -
'.
~,o--r. --a-, FAS~ o SLOW ~ ~_T; --O
Accordingly, Rl, R2, R3 and R4 are sèlected so that rapid
dissociation of~the first radical (Rl) is fast, and
~ dissociation of subsequent radicals ~R2-~4) is slow. It
:~ 25 has been~found that when the radicals Rl-R4 are
collectively alkyl type, the dissociation is linear with
respect to the length of the chain (the shorter the chain,
~he faster the dissociation). Therefore it is possible to
shift the reactivity of such organo-metallic compounds by
simply replacing shorter alkyl substituents with longer
alkyl substitution. It has also been found that when R is
an aryl moiety, dissociation is relatively 910w. Other
~:: " ' ' ' '
'~ ~ ' ' .
~- WO93/26019 PCTJUS93/05~95
-17-
moieties (e.g. esters, ketones) have been found to provide
intermediate dissociation constants.
The present particles are preferably between about
0.003 and about 1.5 microns in diameter, and have a
05 surface area of from about 50 to 150 meters/gm, which
provides a high capacity for coupling of a bioaffinity
adsorbent, chemical or biochemical reactive group.
Magnetic particles of this size range overcome the rapid
settling problems of larger particles, but obviate the
10 need for large magnets to generate the magne_ic fields and
magnetic field gradients re~uired to separate smaller
particles. For exampl-, magnets used to effect
separations of the magnetic particles of this invention
need only generate magnetic fields between about 100 and
15 about 1000 Oersteds. Such fields can be obtained with
permanent magnets which are smaller than the container
which holds the dispersion of magnetic particles and,
thus, are suitable for benchtop use.
Particles with superparamagnetic behavior are
20 preferred since superparamagnetic particles do not
exhibit the magnetic aggregation associated with ferro-
magnetic particles and permit redispersion and reuse.
The term "superparamagnetism" is de~ined as that magnetic
behavior exhibited by iron, cobalt, nickel or other metal
25 alloys or metal oxides having a crystal size of less than
about 300A, which behavior is characterized by
~ 3
W093/26019 PCT/US93/0~; ~
-18- !.
respons{venesq eo a ~a~nettC fleld withou~ re~inane
~agnetlzation.
Ferromagnetic p~rticle~ may be u~eful in certain
applicat~ons af the in~entlon. The term "ferromagnetlsm"
05 i5 defined as that ~agnetic behavlor exhlb~ted by iron,
iron alloys or iron oxides with 8 cry~tal size gre~cer
than about 500A, whlch behavior ~s characterlzed by
responqiveness to a magnetic field with a re~inane
magnetizaeion of greater than abo~t 10 ~auss upon removal
of the magnetic field.
The partlcles or cry-qtaLs are t~en coated ~ith an
organo-metallic mono~er materlal capable of atsorpeive or
covalently bondlng to the ~agnetlc part~cles. Organo-
~etalllc mono~er~ u~eful for the present coated particles
are organic coort~nate complexes of selected transltion
andtor post Cransitton metalg which are capable of
form~ng a stable coordination co mpound, and organic
llgands, which can be adsorbed onto or covalentiy bound
to the magneeic part~cle and, crosslinked ln situ on the
part~cle s~rface, thereby for2~ng the organo-~etall~c
polymer coat~ng. The organo-~etallic monomer must be
able to be functlonalized or deri~atized in a ~anner ehac
allows the polymer formed thcrefro~ eo for~ covalene
bonds with bioaffinity or che~cal affinley adsorbents.
For ehis purpose, the organo-aetallic poly~er ls post-
functionalized or terivitized with an aliphaeic ~spacer
arm" which is t~rminated with an organic funct~onal group
captble of coupling wlth bioaffinicy adsorbents, She
n spacer armn i3 an aliphatic hytrocarbon having rom
about 2 to about 60 atoms, ~ . g., carbon, nierogen and/or
oxygen ato~s, The purpose of the spacer ar~ is to
: '' ' '' ' ` ~
WO93/26019 ~ ~ PCT/US93/0559
-19-
provide a non-reactlve llnker (or sp~cer~ between ehe~
organic group whlch reacts with the che~ical group,
biochemical group or bioafElnlty adsorbent and the
polymer chaLn, and to i~pare an appropriate de~ree of
05 hydroph~lic/hydrophobic balance to the surfac~ of the
coaced part~cle. The organLc ~roup ~g gonerally a
reactlve group ~uch a~ sn a~ine (NH2), carboxyl group
~COOH), cyanaee ~CN), pho-qphaee -(Po3a), qulfate (SO3H),
thiol (SH), hydroxyl (OH) group, ~inyl (C-C), nitrate
(NO2), aldehyde, epoxide, succlnamide or anhydride group
coupled to an aliphatic or aro~atlc moiety.
Partlcularly u~eful organo-metallic compounds are
coordlnaee co~plexes formed from selected transition
metals (e.g., Tl, Zr, Hf, V, Zn, Cd, ~n, Te, Re, Ta, Nb)
and/or post-transition metals (e.g., Sn, Sb, Al, Ga, In,
Ge). Organo-ti~aniuo compounts aro parelcularIy
preferred~ Organo-tltanium co~pount~ which are useful
lnclut~ng, or exa~ple, t~taniu~-tetra-isopropoxide,
amino-hexyl-titanlum-trl-isopropoxide, amino-propyl-
eitanium-tri isopropoxide and carboxyl-hexyl titan$um-
tri-isopropoxide. In ono ombodlmone of the presene
~ invention, a~ino-noxyl-tltanium-erl-l~soproxlde is coaeed
~~ onto the magnetic part~cle of cholce, and thermally
crosslInked to for~ sn organo-tltaniu~ polymer coating
having an aliphatlc spacer arm (the he~yl moieey) and
organic functlonal group (the a~ine group).
The coated particle is post-functionalizod, if
necossary, In a manner that allowq the organo-~-talllc
polymor to form co~al-nt bonds with bioaffinlty or
chemlcal afflnley adsorbenes. In ono e~bodioene of the
present meehod, an org~no~eltan~u~ poly~er, ~uch as
" ~ ' i
, ' . . . ;
, ~'
:,~
~ L~ J
W093t26019 PCT/US93/0~'` ;
-20-
ticanium-tecra-lsopropoxld~ which lacks the spacer a~m
and organic functional group, iq coated on~o the magnetic -
par~icle of choice and partly crosslinked at about 40-C
for a period of t$me sufficient to allow the
oS organotitanium polymer to become ad~orbed on~o ehe
pareicle surface. The or~anocitanium coated magnetLc
particle ls then acti~ated by react~on with an agent such
as l-hydroxy-6-amino he~ane, to form the amino-hexyl-
eieanium-tri-isopropoxide~ The coating is then
crosslinked at elevated te~peratures eo form an
organotîtanium poly~er coating having an aliphatic spacer
arm and an organic functionality (i.e., the amine group).
Ihe functionalLzed particle can then be reacted or
coupled, with the bioaffinlty adsorbent of choice. ~ ~
The magnetic core particleR are prepared according '`
to the following general proceture: metal salts are
- precipitated in a base to form fine ~agnetic metal oxide
crystals. Ihe crystals are retispcrsed, then washed in
water and in an electrolyte. Magnetic separation can be
used to collect the crystal~ becween washes if ehe
crystals are superparamagnetic.
In one e~bodl~ent of ehe presene ln~entlon, super-
pa~amagneeic iron oxide parcicles ar~ made by precipita-
tion of dLvalene (Fe2 ) snd trlvalent (Fe3~) iron salts,
2S for example, ferrous ammoniu~ sulfate, Fe2(NH2)(S04) and
ferric sulfate, Fe2(S04)3, in aqueous base. The ratio of
Fe2 and Fe3 and counterion can be varied without
substantial changes in the final produce by increasing
the amount of Fe2 whil~ msLntaLning a constant mo}ar
a~ount of iron. Counte~ion5 ~ncluding nitrate, sulfate,
~: ~ chloride o~ hydroxide ~ro useful ln th~ methot. A
; ~ .
~ ' ' .
; ~VO 93/26019 ~ ~ 3 7 1 4 ~ PC~r/US93/OS595
-21-
Fe2 /Fe3 ratio of about 2:1 to abouc 4:1 is useful in
the present invention; a racio o f abouc 2 :1 Fe2 :Fe3 is
particularly useful. An Fe /Fe ratio o f 1:1 produces
~agneeic pareicles of sllghtly inferlor quality to those
05 resulting fro~ the higher Fe2 /Fe3 ratios, the particle
slze is more hoterogeneous than that resultLng from
Fe /Fe of 2:1 or 4:1.
In this embodimont, aqueous solutlons of the iron
salts are mixed in a base, such as smmonium, sodium or
potassium hydroxide, which results in the formation of a
crystallLne precipitate of superparamagnetic iron oxide.
The precipltate is washed repeatedly wsth water by
magnetically separating and redispersing it until a
neutral pH is reachod. The precipitate is then washed
lS w~th abouc five oq~ual portions of a water misciblo
solvent, such as cetone, methanol or ethanol that has !;
been dried over molecular sieves to remove all of the
watcr~.
. ,
The repoated use of magnetic fields to separate the
iron oxide from suspension turing tho w-shing seops is
facilieated by the superparamagnetic properties of the
cryseals. Regartless of how many times the particles are
; sub~ected to msgnetlc fields, thoy nov-r become
m-gnetically ag~lome~ratet and consoquently, can be
2S redisp~ersed by ~ild agitation. Ferrooagnetic particles
cannot be prepared by this washing proc~dure as they tend
to magnotically `aggrega~to after exposure to magneeic
fielts ant cannot be ho~ogeneously redispersed.
Other divalent transltion meeal salts such as
ma~nesium, manganese, cobalt, nickel, zinc and copper
salts may bo substltutet for iron salts ~ tho
, "~
~ ~ :
.~,, , . ,
,~
,~ -
,: : :
~lc3~Ll~
WOg3/26019 PCT/US93/0
precipieation or milling procedure to yield magneeic
metals or ~etal oxLdes. For example, the substitution of
divalent cobalt chloride (CoC12) for FeCl~ in the abo~e
proceture produced ferromsgnetic mecal oxide pare~cles.
os Ferromagnetic mecal oxide particles such as those
produced with CoC12 can be washed in the absence of
magnetic fields by employing convenclonal techniques of
centrifugation or filtration between washings to avoid
magnetlzing the particles. As long as ehe resulting
ferromagneeic metal oxides are of sufficieneLy small
diameeer to remain dispersed ~n aqueous media, they can
also be coated with tho organo-metallic poly~er and
coupled to bioaffinity adsorbonts for use in systems
requiring 8 single magnetic separation, e.g., certain
radioimmunoassays. Ferromagneti3m limits particlo
usefulness in tho~e applications requiring redispersion -
or reuse.
~ In another embodiment of the present inveneion, the
-~ magnetic core partlcles can be made by precipieaeing
meeal powders and roducing the pareicle size by millin~
the resulting precipitate, for example, in a ball m~ll.
In th~is procoss, ths metal powder is precipitated from an
aqueous solution of, for example, F- 2 or ~e~3 sale wieh
sodium borohydrid-. For oxample, an aqueou~ solueion of
;~ 2S ferrous chlorido (FoC12) is mi~ed wieh sodium borohydride
(NaBH~) co for~ a ftne iron precipitate. The resulting
properties of the motal powder Are unaffecte~d by the
va}ance of tho counter ion or lron meeal salt elected.
Complete precip~taeion occurs spontaneously upon
`~ 30 borohydride attition. The magnetic meeal powder is then
- - ~ collected by flltraeion ant wa-hed with about flve equal
.,:
: :
, ~
~'
~ .
WO93/26019 PCT/US93/0~595
-23-
volumes of water to remove all soluble salts, then washed
with five equal volumes of dried acetone to remove all
residual water. The particle is added as an aqueous
slurry in a concentration of about 1-25% to a commercial
05 ball mill filled half way with 1~4" stainless steel balls
and milled for 3-30 days. At the completion of the
milling period, a superparamagnetic metal slurry is formed
and coated and functionalized as the superparamagnetic
particles described in the previous section.~ i
In another embodiment of the present invention, the
magnetic core particles are made by reacting a
metallocene, e.g., particulate ferrocene
(dicyclopentadenyliron, Cl0Hl0Fe) with iron (II)
hydroxide. In this embodiment, an aqueous ferrocene (or
l5 other metallocene) slurry is prepared, and an aqueous
slurry of iror. (II) hydroxide is prepared separately. The
ferrocene slurry is prepared, for example, by milling a
mixture of ferrocene and water in a ball mi}l. The iron
II) hydroxide slurry can be prepared, for exampie, by-
20 precipitating an a~ueous solution of ferrous~sulfate withammonium hydroxide to form ferrous hydroxide. The two
slurries are then combined and milled, for example,
forming fine magnetite particles. Other metallocene
compounds (e.g. nickelocene, cobaltocene) can be mixed
25 with the ferrocene to produce ~arious magnetic ferrite
particles. This process is described in detail in U.S.
Patent No. 5,071,076, the teachings of which are hereby
incorporated by reference.
;
'~ ' , ' ' ' - .
WO93/26019 ~ 3~ PCT/USg3~0
--24--
In one embodi~ent of the present ~nveneion the
coa~ing around the magnet~c core particle is amino-pro-
pyl-titanium-tri-isopropoxide `The polymerization is
performed by redispersing the ~agnetic part$cle in an
acetone solution, adding the organo-titanium monom~er,
then crossLinking with heat Th;e terms "coupled magne-
tically responsi~e part~cle~ or ~coupled magnetic
par.icle" refer to any magnetic pareicle to which one or
; more types of bioaffinity adsorbents are coupled by
lQ covalent bonts, which co~alent b~onds may be amide, ester,
ether sulfonamide, tisulfite, az~o or other suitable
organic linkages depending on the functionalities a~ail-
able for bonding on both the coating of the magnetic
particle and the bioaffin~ty~adsorbents
15~ ~ Prcferred m-gnetic~ally responsi~e particles of the
pres~ent~invention~ha~e metal ox~de~ cores composet of
cluseers of superparamagnetic crystals affording
efficiene separation of the particles in low magnetic
fields ~OO-lOQO Oerst-ads) while maineaining super-
20 ~paramagneeic propertios Aggregaéion of particles iscontro~lled turing~particle synthosis to produce particles
whic~h~ar~e~pref~er~ably small enou~h to avoid s bstantial
gra~itational settling o~er t~nes suf~icien~ to permit
tispersions of~the p`a~rt~icles to be used in an intented
25 biological~assay or o~her application The ad~antage of
- havLng~s~uperpar~a~magne;tic cores in magneeically responsi~e
particles is thae such particles can be repeatodly
exposed to magnotic fiolds Sup-rparamagneeic particles
do noe xhibit rem~nent m~gnetization ~nd ha~e no
co-rcive strongth, and, th~refore, do not nagnotically
~ aggrogatc, thu , th- particles can be redisperset ant
'''', ' ~ ~ :~ ,
.
.
"., :'' ::
,
~,:
;" ~
:5~ :-
j ~093/26019 2 1 3 7 1 -Q ~ PCT/US93/0559S
-25-
i
reused. Even after coating, preferred pareicles of the
inven~ion ha~ing cores made up of clusters o~f crystals
exhibit a remarkably high surface area per unie weighe
and a ~enerally corresponding high co~pling capaciey,
oS which indicaees that such particles ha~e an open or
porous struceure.
The bioaffinity adsorbents can be covalently bonted
. to the organo-~etallic coated magnetic particles of this
in~ention by con~entional coupling chemistries. Several
coupling reactions can be perfor~ed. For e~ample:
(a) If the l~gand to be coupled contains an amino
group, ie can be couplcd directly to the acti~ated
organo-metallic polymer. If a tifferene funceionality is
- desired, it can be introduced, for e~ample, by adding a
~- 15 spacer arm containing the functionality by sequential
~ reactlon~of the organo-metallic poly~er (e.g., titanium-
-~ tetra~-isopropox$te) with any o~ega-functional higher
moleou1ar weight alcohol. The anino group on ehe ligand
can then be coupled to the free funct$onal group on tho
spacer arm; or
(b) If the ligand contains an altehyde group
instead of an a~ino group, $e can be coupled d$rectly to
the free;am~no group o an amino alkane (that is, an
aIkaQ~e spacer ar~ haYing an a~ino functionality) on the
coated;~agneeic particle.
The ~tern ~bLoaffinity adsorbene~ is definet as any
biological or o~her organ$c molecule capable of spec$fic
or nonspecific binding or interaction with another
biological ~olecule, wh~ch bind~ng or inter-ce$on may be
referred to as ~lig8nd/ligate~ binding or interaction and
is exemplified by, but not limited to, antibody/antigen,
~, ' . . .
, .
,
,
':~
WO 93/26019 ~ ~ 3 714 5 PCT/US93/0
antibody/hapeen, enzym~/substrate, carrie~ procein/sub-
strate, lectin/carbohydrate, receptor/hormone, receptor/
effector or repressor/inducer bintings or interactions.
The coupled organo-meeallic coated magnetic
05 particles of the present inveneion can be used in immuno-
assays or other binding assays for the measuremene of
analytes in solution. Thc eerm ~i~munoassay" is defined
as any meehod for measuring the concenoration or amount
of an analyte in a solution based on the immunological
binding or interaction of a polyclonal or monoclonal
antibody and an antigen, which method (a) requires a
separation of bound from unbound analyte; (b) employs a
radioisotopic, fluoromctric, enzymatic, chcmiluminescent
or other label as the means for measuring the bound
and/or unbaund analytc; and (c) may be described as
"competiti~e~ if thc amount of bound measurable label is
generaLly inversely proportional to the amount of analyte
originaLly in solution or ~non-competiti~e~ if the amount
of bound measurable label is generally directly propor-
tional to the amount of analyee originally ~n the solu-
eion. Label may be in the aneigen, ehe antibody, or in
double aneiboty methods, the second ant~body. Immuno-
assays are ex-mplified by, but are not limieed to,
radioim~unoassays (~IA), immunoradiometric assays tIR~A).
fLuoro$mmunoassays (FIA), enzyme immunoassays (EIA), and
sandwich method i~munoassays. The analyte or the
bioaffini.y adsorbene can include, for e~ample, anti-
boties, antigensj haptens, enzymes, apoenzymes, enzymatic
subserates, enzymatic inhibitors, cofactors, nucl~ic
acids, binding proeeins, c~rrier proteins, compounds
bound by binding proteins, compounds bound by carrier
,
, . . .
,,
.~
'd
~, '
;WO93/26019 ~1 3 7 ~ Q ~ PCT/US93/05595 ` ~
,.~.
-27-
pr~eeins, lectins, monosacchasides, polysaccharides,
hormones, receptors, repressors and inducers.
Such assays are preferably carried out by mixing a
sample containing an unknown conccneration o analyte ~-
05 with a known amount o labeled analy~ in the presence of
magnetic particles coupled to a bioaffinity adsorben~
capable of binding to, or interacting with, both
unlabeled and labeled analyte, all`owing the binding or
interaction to oacur, magneticalLy separating the
10 particles, measuring the amount of label associated with `~
.he magnecic particles and comparing the amount of label
to a standard c~rve to detcrmine the conceneration of
analyte in the sample.
The term "binding assay~ or non-immune assay" is -
definet as any ~ethod for m-asuring th- conccntration or
amount o an analyte $n solution based on the specific or
nonspecifLc binding or interaceion~ other than antibody/
antigen binding or interaction, or a bioaffinity ad-
sorbent and another biological or or~anic molecule, which
20 ~ethod (a) requires a separat~on of bount fro~ unbound `;
analyte; (b) employs a radioisotopic, fluoro~etric, ~:~
enzymaeic, chemiluminescent or other label as as the
means for ~easuring the bound and/or unbound analyte; and
(c) may be described as "competitive~ if the amoun~ of
bound measurable label is generally in~erscly propor-
tional to the amount of analyte originally in solution or
"non-competLti~e" if the a~ount of bound measurable label
is gcnerally originally in solution.
The magnetic or~ano-metallLc-coated particleç of
30 this invention arc useful in lmmobilLzet enZymo systems,
particularly where en2ymc recycLing is desired~ The term
:`
.
,.
~ . .
W093/26019 ~ PCT/US93/05
-28-
, ~
. . ~ .
"immobilized enzyme sysee~" is defined as any enzymati-
cally catalyzed biochemical con~ersion or synthesis or ,
degradation wherein the enzyme ~olecule or acsi~e site
thereof is noe ~reely soluble bu~ is adsorpei~ely or
05 co~alenely bound to a solid phase support, ~hich support
- is suspended in or contactet 7ith the surrounding medium
and which ~ay be reclainet or separated fro~ said meehod.
In this embodiment, enzymatic reaceions are carried out
by dispersing enzyme-coupled magnetic particles in a .
l~ reaction mi~ture cvntaining one or more substrates, under
condieions sufficient for the reaction between ehe enzyme
and substraee to occur, magnetically separating the
enzyme-magnctic particle from the reaceion mixture `
coneain~ng products ant unreacted substrates and, if ,~
desired, redispersing the particles in fresh substrates
thereby reusing the cnzyme.
Affinity chromatography separations and cell sorting
can be pcrformed using the ~agnetic particles of this -~
inveneion. Tho term ~affinity ch~onatography~ is defined
as a meehod or separatlng, isolating, and/or purifying a
seleceet molecule from its surrounding medium on ehe
basis of ~ts binding or interaction wi~h a bioaffinity ~.`
adsorbent adsorptively or covalenely bound to a solid
phase support, ~hich support is sus-pended in or contaceed i,^
2~ with the surrountins medium and which may be reclai~ed or
:, ~,;;.
separated fro~i said nedium by dispersing bioaf:inity
adsorbent coupled ~agnetic particles in solutions or `
suspensions containing moleculeis or cells to be isolated
. . .
and/or p~rified, allowing the bioaffiniey adsorbent and ""
30 the desired molccules or cells to ~nteract, ma~netically ,~
separatlng the particles from the solutions or suspension
:` .
. .
: ' , ~j` '.'
.
, WO93/26019 2 ~ 3 7 1~ ~ PCT/US93/05~9~ ~
_~9_
.,
and recovering the isolated moLecules or cells fro~ ehe
magneeic particles. -~
I~ is further coneemplated thae the organo-metallic
coa~ed magnecic particles of this in~ention can be used
05 in ~n vivo syste~s for ehe diagnostic localization of ~;;
cells or tissues recognized by the particular bioaffinity.``
atsorbent coupled to the particle and also for magneti-
cally direc~ed delivery of therapeutic agents coupled eo
ehe particles eo pathologica} sites.
Hagneeic separation tines of iess than aboue ten
minutes can bc achievcd with magnetic particles of the
invention by contacting a vessel containing a dispersion
of the particles with a pole face of a permanent magnet ~-
no larger in volune than the volume of the vessel.
}5 Magnecic sepàration time is tefinet to be ~he eime for ,''!
the turb$dity of thQ tispers$on to iall by 95 percene.
Further~ore, thc use of functionalized organo-
metallic polymers as the coaeing surrounding the mceal
oxidc core of the magn~tic particles describcd herein ~-.
~ 20 make pos~sible th~ coupling of a wide varieey of molecules
;~ under an equally wide variety of coupling coniitions
compared to other ~agnetic particle coaeings known in the
art with more li~ited coupling funceionalities.
The invention $s further illustraeed by ~he follow-
ing Examples-
.:
EXA~PLES `
Examole 1: ~5~p3~5~ f SuoerParama~neeic_Maenetiee '~
Part~clos ~`
200 gra~s (1.58 nol~5) of ferrous chloride (VUR t`'`
Sciçntific) and 3i5 gra~s ~2.0 moles) o ferric chloride Iii
,. .. .. . . . .
: '~
'~:: ,`:
: , . ..
.
WO 93/26019 2137~ ~ PCT/US93/0~ ;
. ..
-30-
: .
were dissolved in 3 liters of water 2000 grams of
am~onium hydroxide (~WR Sciencific) concentrate were
added at a race of SO ml/minute ~nder constant agitation
turing which ti~e the eemperaeure of the solution was
0~ kept becJcen 25 and 40 C After the addition of ehe
ammonium hydroxide was complete the ~agnetic particle
(Fe304) a~ueous slurry was allowed to coo} to room
te~perature
~Ex~moLe 2 Preoaration of Am~no-Hex~l-Titanium Tri-
Iso~roooxide
_______ _
0 1 moles of tieanium-tri~isOprOpOxide (Tyzor ~PT
Dupont Uilmington DE) and 0 1 moles of 6-amino-1-
hexanol were addet to a 50 ml beaker and seirred at room
temperature for 1 minute to form 0 1 ~ole of a~ino-hexyl-
15~ titanium-tri-isopropoxide The reaction mixeure was
heated to 70 C for 10 minut-s to evap~rate the isopropyl
alcohol formed during the reaction
- The material was coolcd to room eemperature and used
as a monomer in making the tetra~al~nt titanium organo-
me~aLlic coaelng in Examp}e #3
E am~l _3 Pre~aEatLon of Am~ne Function-l Or~ano
ti~anate Coatet Masnetic Particle
According to the proccdure set ou~ in Example 1 4
moles FeC13 and L mo~les~of FcC12 were dissol~ed in 4 L of
2S distilled waeer and precipitated wieh 16 moles of
;~ ammonium hytroxide~ The precipitate;was w-shed S times
wieh water and 3 tLmes with ac-eone N,N-dimethyl
ior~amide (DHF) was added to th- precipitate in the
following ratio lO ml of DHF per gram of Fe304 The
~ 30 mixture was loaded ineo a Eiger Mill and milled
's`"'.''
. , .
'.~''
~',' '
-~
',~
"~
W O 93/26019 2 1 3 7 1 ~ ~ PC~r/US93/05595
continuously for LO minutes. The mixture was then
eransferred to a beaker and heaeed with stirring for 30
~inutes ae lOO-C. The amine functional organo-titanaee
prepared in Example 2 was im~ediately added after
05 preparaeion with constant stirring to the mixture in a
ratio of 1 g dry Fe3O4 per 3 g of amine funceional
organo-titanate.
Th~s mixeure was then heat~d with stirring for 20
, minutes at 65-C and then passed through the Eiger Mill
¦ 10 for two passes. The resulting material was washed fi~e
s with water, the coaeed particles were collected
I with an external magnetic ficld of 2000 gauss and the
I aqueous waste was decanted.
-~ Exam~le ~: Pre~arat$on of An Alternatins Functional-Non
-~ ~ 15 Funct'onal Or~ano-Tlea_-te Monomer
The proceture tescribet in Example 2 was followed
except that the organo-citanaee was reacted with a
co~i~ture of amino-functional hexanol and hexanol to
produce a mono~er ha~ing reduced amine funceionality.
Hexanol and 6-amino-1-hexanol in a molar ratio of 6:1
were mixed in a 50 mL beaker for one minute. Tyzor TPT
was adted to the alcohol mixture ~n the ratio of 1 mole
~- ~ of alcohol per ~ole of Tyz or TPT. The reaction mixture
~ was stirred or one minute, heatod to 70-C for lO 3inutes~ ~ 2S to evaporate the isopsopyl alcohol produced oy the reac-
tion and cooled ~o room temperature. Ihe resulting
compound was an organotitanate, 6-auino-hexyl-t~eanium-
tsi-isopropox$de ha~ing alternaeing non-functiona~ he~yl
groups, ehat ~s, hexyl chains lac~ing the amino group.
Ihe weighe ratios o 6-am~no-l-hexanol:Tyzor TPT:hexanol
-:
,'~, ' .
.~
~ WO93/26019 PCT/US93/05~
~.
-32-
~ ~.
were 1:26:9.6. This compound was used as a monomer to
make an organo-~ieanium coati~g as described in Example
5.
'~a~le 5: Pre~aration of Amine Functional_0r ano
05 titanate Maenetic Particles
The procedure describet in Examp}e 3 was followed
except that the amine-functional or~ano-titanate was the
material preparet.in Example 5. The ~ixt~re of magnetic
particLes and organo-titanaee monomer was heated to 95-C
for one hour with constant stirring and milled in an
Ei~er ~ill for 4 ~inutes. The mixture was washet nine
times wieh water. Atipic acid was atted in the ratio of
0.S moles o adipic acid per mole of total particles.
One mole of carbodiimide (CDI) was addet, and the ~ixture
- 15 was mixed for 30 ~inute- on a ball mill. 1,6 he~ane-
diamine was attet in the ratio of 0.5 moles of 1,6
he~ane-diamine per mole of total pareicles. One mole of
CDI was atded and the ~ixture was mixet for 30 minuees.
The resulting material was washed five times with water,
the part~cles were colleceed using an external magnetic
field of 2000 gauss and éhe aqueous waste was decan~ed.
Examole 6: Pre~aration of Subdomain MaEnetite ~articles
bv Reactlon of Partlculate Ferrocene and
__ _ _ . __
Iron(II) HYdroxide
A 100 g of a slurry containing 20~ ferrocene (by
~ weight) (ticyclopenèadenyliron; Strem Chemical Co.,
Newburyport, MA) in water was propared by mixing the
ferroceno with tho waeer, Th- slurry was added to a
commercial ball mill, Tho mill was filled halfway wi~h
~" stainless stecl balls and the slurry was milled for a
~ ' , .
~::
,
,
'
~-
WO93/26019 ~ 1 ~ 7 1 ~ 5 PCT/US93/05595
-33-
period of 2 hours. .~
A second ferrous hydroxite slurry (iron (II) ~`
hydroxide) was made according to the following procedure. ~ `
An aqueous solution containing 20g of ferro~s sulfate
05 (VWR Scien~ific) ~as precipita~ed using 50g o~ ammoniu~
hydroxide concentrate to form gelatinous ferrous
hydroxide. The gel was fileered and the filtrate washed
wieh 5 to lOOg ~olu~es of water. The washed gel was then
made into a 10% aqueous slurry and milled as pre~iously
lO described for 5 hours. .~
Ihe ferroc-ne and hytroxide slurries were mixed, and ~-
the mixture was ~illed for one day to for~ fine Fe30~ -
particles. The particles were abouc lO0 A in diameter -;
and were responsi~e to a magnetic field. These particles
15 can be coated as describet ~n Examples 2-5 above. ;:`-
. , ~
Exam~le 7: Preoaraclon of Subdomain Nic~el-Ferrlte
Par~ c l e 5
Subdo~ain nickel-ferr~te particles were prep~red
according to the procedure set out in Example 6, excepe
20 that a mix~ure of 50g a 20~ nickelocene slurry `~
(dicyclopentadenyLnlckol; Strem Chemical Co., ;~;
Newburyport, ~A) and 50~ of a 20~ f~rrocene slurry were
uset in lieu of the lOOg of ehe ferroccne slurry in
Examplo 6. Nagnetically responsi~e nickel-ferrite :-`
particl-s having a part~cle size of about lO0 A were
produced by ehis ~ethod.
Exam~le 8: PreParation Subdomain Cobalt-Ferrite
Particles `
Subdo~ain cobalt-forr~eo psrticles wero prepsred
according to the procedure set out in Example 6, except
that a mixture of 50g of a 20~ (by we~) cobaleo~e~e
~''"`.''~
.~
WO93/~6019 ~ 1 37~ PCT/Us93/0~
; -34-
;':.'
:,,, '-
.~ '''
::,
slurry ~dicyclopentatenylcobalt; Strem Che~ical Co., ~-~
Newburyport, ~A ) and 50g of the ferrocene slurry were
used in lieu of 100g of the errocene slurry in Example!,~`- .`
6. Magnetically responsi~e cobalt-ferrite pare~cles
05 having a particle size of about 100 A were produced by -
this method. ~'
- Exam~le 9: Pre~araion of Subdomain Metal Partic}es_by
Sodium Borohrdride-Reducion and_SiZ_
Reduction b~ ~illin~
200 gm (1.58 moles) of errous chloride was
dissolved in 1 lieer of wacer. 500 g~ of dry sotium i;-
borohydride were added to the solution to form a fine l`
iron po~der prec~pitate. Ihe precipitate was washed with `
water and collected by filtration. The f~ltered powder
was res~spended in watcr and re-filtered. The washing
procedure was done 4 additional times. On ehe final
suspension, the sl~rry was ad~usced to a eoncentraee of
20~ and milled as descrlbed in Example 6 for a period of
75 days to produce particles wlth a ~ean diameeer of less
than 50 A.
. .~ ~, .
,, ~,
....,~
.
"
. `,
W093/26019 ~ PCT/US93/0~95
-35-
1 DescriDtion of the Sub lOOA Ferrite Particle
;
2 Sub lOOA ferrites have been prepared by the co-
3 precipitation of metal(+Z) and metal(+3) salts in aqueous
4 solutions with aqueous base across a porous or dialysis
5 membrane. The metal salt solutions are put into a `-
6 dialysis bag and the bag is sealed. The bag containing ;
7 the metal salt solution is then immersed in an aqueous
8 solution of base (i.e. ammonium hydroxide) over a period
9 of several minutes to several days, depending on the
concentration of the various reactants, and a precipitate
~1 of metal oxide forms inside of the dialysis bag. The size
12 of the particles thus prepared is controlled by:
13 concentration of the metal salt solution; concentration o~
14 the base solution; pore size of the membrane; temperature !';`'~
of the various solutions; ionic strengths (or ionization
16 constant) of solutions; and the contact times of each
17 solution across the dialysis membrane. - ~`
18 It has further been discovered that metal oxide
19 particles of various controlled size can also be formed by ,~
20 contacting an aqueous solution of metal salts with a ,~
21 dialysis bag filled with aqueous base. In this case, the
22 desired metal oxide product will form outside of the ,~`
23 dialysis bag.
24 In a preferred embodiment, the inorganic ~ase and the
25 inorganic salt solutions are maintained in large volume ,~
26 chambers separated by a porous membrane. Accordingly, '~
27 large amounts of inorganic oxide of controlled particle
28 size can be produced. As can be seen from Figure 1, a
29 large volume chamber (10) contains a partition (12), a ~-`
semi-permeable membrane (14), an opening (16), a support
31 (18) for mounting of the membrane, and portals (20) for
32 draining. The metal salt solution is placed on the ',~-
33 membrane side of the chamber, such that the metal oxide '~
34 particles precipitate on that side of the large volume
35 chamber. `- -~
W093/26019 ~13 7 ~ d ~ PCT/US93/05l ~
-36- ~
;,
1 It has also been discovered that the size of the
2 cationic moiety on the base side of the membrane controls
3 the size of the precipitated inorganic oxide particle so ~ -
4 produced near the surface of the membrane within the -
inorganic salt solution. Apparently, the speed of
6 dissociation of the inorganic base is believed to be
7 controlled by the size of the cationic moiety; the larger
8 the cationic moiety the slower the dissociation to -
9 cationic and anionic component. When the dissociation is ~ `
10 relatively slow, a relatively low concentration of anionic `
11 moiety is present, providing a relatively low ` ``
12 concentration of anion diffusing across the porous
13 membrane and into the inorganic salt solution.
14 Accordingly, the cationic component (of the inorganic
salt) exists in large excess, tnereby surrounding the
16 slowly diffusing anion, resulting in precipitation of many
17 smali-sized inorganic oxide particles.
18 By contrast, if the cationic moiety of the inorganic
19 base is relatively small, the speed of dissociation is
relatively fast, providing a relatively large
21 concentration of anionic moiety diffusing across the
22 porous membrane and into the inorqanic salt solution. At
23 the surface of the membrane within the inorganic salt l:;~
24 solution the cationic component (of the inorganic salt)
once again exists in large excess. Accordingly, while the
26 cationic component surrounds those anionic moieties which -`~
27 have diffused across the membrane, the elevated ``
28 concentration of diffusing anionic moiety rapidly finds
29 its way to the cationic surface of such a growing
particle, so that a further layer of ionic bonding can
31 result, thereby producing larger overall particle size
32 prior to precipitation from solution.
33 It has been found, for example, that ~OH in contact
34 with an aqueous solution of FeC12/FeC13 affords iron oxide
3S particles (Fe3O4) that are smaller in size as compared to
36 iron oxide particles produced when LiOH is employed as the
4 a .~ ~ ~
W093/26019 PCT/US93/05595
~; .
-37- ~
1 inorganic base. This would comport with the above insofar ;~`
2 as the R+ ion is known to be relatively larqer than the
3 Li+ ion.
4 With respect to the foregoing, NH40H, KOH, LiOH, NaOH
5 and other hydroxides formed by elements in group Ia of the ;~
6 periodic table serve as suitable inorganic base~compounds. 'i
7 Inorganic salt solutions based on mixtures of the type ~`
~8 M(+3)VJ~(+2)y include those whereLn Y is selected from the ~;
9 group~consisting of Cl, Br, I, S04, No3 and PQ4. M can be
selected from the group consisting~of Fe, Co, N~i, Zn, Mn,
11 Mg, Ca, Ba, Sr, Cd, ~g, Al, B, Sc, Ga, V and In. The
12 preferred inorganic salts are those which are readily
13 productive in an aqueous medium of an anion and a cation
14 which can combine with the aforementioned diffusing
15 hydroxide anion to form an inorganic oxide. ~^;
16 Accordingly, inorganic oxide particles of the~formula
17~ M304 are prepared wherein M is selected from the group
18 consisting of Fe, Co, Ni, Zn, Mn, Hg, Ca, Ba, $r, Cd, Hg,
19 Al, B, Sc,~Gaj V and In and mixtures thereof. It will
also be appreciated that for a given~M304 particlé, the
21 metal (M~ may often be a combination of different
22 ;oxidation states of the same metal component. For
23~ examp1e, and in the preferred embodiments,~Fe304 particles
24 are prepared and represent a mixed Fe(+2)Fe(+3) oxide of
the formul~a tFe~+2)~tFe(+3)J204.
26 With~respect to the foregoing, reference is made to
27 the following~
28 I.~ The~Effe¢t of~Alternative Base Counter Ions
29 The~effect of alternative base counter ions on crystal ~`~
properties such as~size, distribution, magnetics, etc. was
31 established as follows: Three experiments were conducted.
32; All experimental conditions were identical except for the
33 type of base. Experiment A utilizes NaOH, B with LiOH and
34 ~ C with ~OH. For each~experiment: 1. Wash a Spectra/Por~
35~ 5 dialysis membrane (cellulose ester based membrane
36 a~ailable from Spectrum Medical Industries, Inc.) and
.
, :~
'. ~.
r;~ , a
WO93/26019 PCT/US93/0
.'.,~
-38-
1 secure over the opening in the dialysis chamber; 2. Fill
2 both sides of the tank with 20 liters of distilled H2O (at `
3 room temperature); 3. Dissol~e 12.5g FeC12~4H2O in 2
4 liters of distilled H2O. Add 20 g FeC13 and stir until `-
5 dissolved; 4. Decant all iron solution into the membrane ~'~
6 side of chamber; 5. For A dissolve 55g NaOH in 2 liters ~ -
7 of distilled H2O. For B dissolve 55g LiOH in 2 liters of
8 distilled H2O. For C dissolve 60.6g KOH in 2 liters of
9 distilled H2O. Decant base solution into opposite side of
dialysis chamber. After 70 hours contact time, remove the
11 crystal precipitate solution for evaluation. The results
12 are listed below in Table 1.
13 Table 1
14 Base Crystal Cluster Magnetics Iron Conc. % Total
Sam~le Ion ~Size(A) Size~A) (Gaus~s) (mg/ml) Solids
16 A Na 60-80 300 340 7.0 1
17 B Li 120-140 170-250 275 6.44
18 C K 40 500 187 6.10 1
}9 II The Effect of Base Concentration
...
The effect of base concentration on crystal properties
21 such as size, distribution, magnetic response, etc. was `;~
22 established as follows: Two experiments were conducted.
23 All experimental conditions were identical except for base
24 concentration- Experiment A was conducted at 0-5~O NaOH.
Experiment B was conducted at 0.25~ NaOH. For each
26 experiment: 1. Wash a Spectra/Por~ 5 dialysis membrane and
27 secure over the opening in the dialysis chamber; 2. Fill `~
28 both sides of the tank with 20 liters of H2O (at room
29 temperature); 3. Dissolve 12.5g FeC12~4H2O in 2 liters of ~-
distilled H2O. Add 20g FeC13 and stir until dissolved; 4.
31 Decant all iron solution into the membrane side of chamber;
32 5. For concentration A: Dissolve 120g NaOH in 2 liters
33 distilled H2O. Decant into opposite side of tank; For
34 concentration B: Dissolve 55g NaOH in 2 liters distilled
H2O. Decant into opposite side of tank; 6. A~ter 70-80
36 hours contact time remove iron solution and precipitated
37 crystals for evaluation. The results are listed below in
..::
`,','~
~,.
.
W093/26019 ~ PCT/US93/0559
-39-
..'''''' .
1 Table 2.
2 Table 2 ,;,;i,
4 Crystal Cluster Magnetics % Total ^''
5 Samole Size(A) Size(A) Gauss Iron Conc. Solids ''~'
6 A 70-80 500 360 7.Omg/ml 1.0
7 s 60-80 300 340 0.39mg/ml 0.085 '`,',,
8 It has also been found that the size of the particles ,'~-
9 may be effected by the following additional variable: the !''.~`"',
10 temperature of the solutions; whether the particles formed ,~,
11 are removed (including magnetic removal, if the particles '~
12 are of the appropriate size) from the immediate surface of
13 the membrane; the pore size of the membrane; and whether
14 or not the solutions are stirred. With respect to the
pore size, membranes of different molecular-weight cut-
16 offs (MWCO) have been examined. The MWCO represents a
17 limit on the size of the molecule allowed to pass through
18 the pore. M~CO's between 1000 and 500,000 have been ,~
19 investigated. The smaller the MWCO, the smaller the ;"
20 inorganic oxide produced. '`
21 Descri~tion of Magnetic Clusters l '
22 Iron oxide, for example, has been prepared using this ',''
23 technique in sizes of 80A, 50A and 20A, all with a narrow l',
24 (+/-10%) particle size distribution. A product that '~
25 agrees with x-ray diffraction patterns for Fe304 has been '`~
26 prepared in 100, 80, 50 and 20A crystal sizes. The supra ~''
27 50A particles of Fe304 have domain magnetization, when "'
28 measured by a Vibrating Sample Magnetometer (VSM), of 5660
29 gauss. This result is in agreement with the literature. -''
30 The sub 50A Fe304 crystals' surprisingly have a very low ,~
31 magnetization. In fact, crystals of 20A Fe304 have domain
32 magnetization of less than 100 gauss. This low
33 magnetization observed in sub 50A Fe304 crystals is li~ely
34 the result of having insufficient mass for spin coupling ,',
35 and the absence of domain,wall formation. ',
'36 Surpr'isingly, when non-magnetic sub 50A crystals of ~
37 Fe304 are clustered together to form aggregates of 250A or '~,
W093~26019 2 t 3 7 ~ ~ 5 PCT/US93/0~ ,
-40-
.
~;.
1 greater, the aggregate particles are strongly magnetic. zl-
2 Ag~regate particles of 500A or greater in diameter, when
3 measured by VSM, have domain magnetizations in excess of ;~
4 4000 gauss.
It has been further discovered that if the aggregates
6 of magnetic crystals are returned to non-aggregated unit
7 sub 50A crystal size, the effect if reversed, that is, the
8 magnetization is returned to nominally 0.
9 The e~act size at which the onset of superparamagnetic
behavior occurs in the unit crystal, is a function of the
11 crystal structure, shape, and composition.
12 Several different cubic ferrites have been prepared
13 with several different crystal sizes each. The onset of
14 superparamagnetic behavior occurs at various size unit
crystals depending on the exact composition. Table 3 is
16 an estimate of the size where supermagnetic behavior
17 begins for several different crystal compositions. -~-
18 TABLE 3 ~;
19 MINIMUM SIZE FOR -
CRYSTAL COMPOSITION SUPERPARAMAGNETIZATION
21 Fe34 50A
22 Fe2.sZno.5O4 80A
23 Fe2Zno4 120A
24 Fe2.sMno.so4 100A
Fe2MnO4 50A
26 Fe2Sro.2sAlo.so420A ~
27 The substantially uniform size Fe3O4 affords a crystal ~;
28 lattice which contains primarily trivalent iron (Fe+3) at
29 or near the surface of the crystal. It has~been found
that these "surface trivalent" elements of the lattice
31 contain imperfèctions which make thèm available for direct
32 covalent attachment of the organo-metallic compounds of
33 the formula Ti(OR)4 according to the following general
34 equation:
WO 93/26019 ~ 1. 3 ~ i 4 ~ PCl`/US93/05~95 ~`'"''
--4 1-- . ~
.. .
'`
Z o 1 0 I F~:e F~ ' ''
Fe ~ ~ F~F~
6 1 1 1 1 `~
r. ~ SURFACI~ F
IMPERFECTIONS I o ¦ SUR~ACE S
12 ~ / J COATING
13
14
15 It should be noted that the imperfections of the surface C'~`
16 trivaler.t iron is somewhat short-lived, and if organo- ~-
17 metallic coating is delayed, oxidation can occur causing -
18 the development of surface hydroxyls, which can hydrolyze, 1-
19 to provide an FeO coating, precluding direct covalent
20 ~attachment of the organo-metallic moiety. For example, ~`;
21 freshly made ~e3O4 will spontaneously react; Fe3O
22 material after 24 hours reacts but requires about 1 hour .
23 of dwell time; after 48 hours the coupling reaction takes
24 place very slowly and is generally incomplete.
Organo-metallic compounds are preferably of the
26 formula Ti(OR)4 wherein R is an alkyl group and the -
27 dissociation to the reactive component follows the
28 following general reaction criterion:
29 `~
o~ o~ o~ :;
32 f--~ F~ST ~ o, SLOW ~ o~
33 o~ o~ o ~
34 ` -i;`
35 Accordingly, Rl, R2, R3 and R4 are selected so that rapid .
36 dissociation of the first radical (Rl) is fast, and
W093/26019 ~ 1 3 7 1 ~ PCT/US93/0~ 'a~
-42-
1 dissociation of subsequent radicals ~R2-R4) is slow. It
2 has been found that when the radicals Rl-R4 are
3 collectively alkyl type, the dissociation is linear~with
4 respect to the length of the chain (the shorter the chain,
the faster the dissociation). Therefore, it is possible
6 to shift the reacti~ity of such organo-metallic compounds
- 7 by simply replacing shorter alkyl substituents with longer
8 alkyl substitution. It has also been found that when R is
9 an aryl moiety, dissociation is relatively slow. Other
10 moieties (e.g. esters, ketones) have been found to provide `
11 intermediate dissociation constants. ~`
: ,
12 Description of Chemical Bond Magneto Clusters
13 Aggregate clusters of sub 50A non-magnetic ferrites
14 were prepared by several techniques including air drying
of the particles to form agglomerates, argon drying at
16 room temperature, several different solution encapsulation -~
17 techniques and by covalent coupling of surface modified
18 crystals. All of the techniques employed provided
19 particle clusters of at least 250A diameter and mostly of
2~ 500A or greater. In all cases, surprisingly, the particle
21 clusters of non-magnetic ferrite crystal were magnetic. -`
22 Organo-metallic coating with monomer material capable
23 of adsorptive or covalent binding to iron oxide particles
24 (of less controlled particle size) is reported in U.S.
patent application no. 556,169, filed August 10, 1990.
26 According to the instant invention, such coatings can now
27 advantageously be applied to inorganic oxide çrystal ~-~
28 particles of substantially uniform particle size
29 distribution. For example, substantially uniform sub 50A
Fe304 was treated with titanium tetra-isopropoxide and
31 subsequently terminated with a C-6 carboxylic acid and a
32 second population was terminated with a C-6 amîne. When `~
33 mixed together and measured for magnetic response, no
34 magnetic moment was observed. However, upon addition of
35 methyl diisocyanate, the amine and carboxyl terminus ~``
36 groups spontaneously caused clustered aggregates of ~
; WO93/26019 ~ PCT/US93/05595 ~
.. .
-43-
~;
.-. .
1 magnetic particles to form and a magnetic moment
2 proportional to the concentration of methyl diisocyanate
3 added was observed until saturations occurred when ~ll of
the amine and/or carboxyl reagent was exhausted. -
Description of the Magnetic Molecular Switch
6 Another application for the magnetic cluster is the `
7 so-called magneto-molecular switches. Sub 50A non-
8 magnetic Fe3O4 particles are treated by mixing them in a
9 non-a~ueous solvent, such as dimethyl formamide and with
titanium tri-isopropoxy-3,4-dihyroxy phenoxide.
11 The particles prepared in this fashion, are titanium
12 oxide coated with o-dihyroxy benzene termination and are
13 non-magnetic in an applied field. Upon addition of a ~
14 solution of a transition metal, sodium molybdate and ~"
15 tungsten, for example, a 2:1 coordination complex forms ~`~
16 between 1 metal clustered and 2 o-hydroxy benzene atoms
17 causing the particles to become clustered and giving rise
18 to a magnetic signal that is proportional to the
19 concentration of metal ion coupling formed.
Surprisingly, a slight change in pH causes the complex
21 to decompose and the resulting magnetization return to 0.
22 A return to the pH favorable for the formation of the ~`
23 complex results in a renewed magnetization of equi~alent "~
24 field strength to that achieved after initial addition of
metalate ion. This so called magneto-molecular switch is
26 useful for, but not limited to: magnetic tracers for in
27 vitro analysis, magnetic tracers for in t~ivo~diagnostics,
28 magnetic processing by metals (especially for group ~I
29 transition metals), analysis of metals, filtering aids, -`
magneto chromatography, and cell sorting.
31 Descri~tion of the Applications
32 The inorganic oxide crystal particles of substantially
33 uniform particle size distribution may be coupled to ~`
-34 biological or organic molecules with affinity for or the
35 ability to adsorb or which interact with certain other -~
36 biological or organic molecules. Particles so coupled may
':
WO93/26019 ~ 1 3 7 1 4 ~ PCT/US93/05
-44-
I`r~
1 be used in a variety of in vitro or in vivo systems -~
2 involving seDaration steps or the directed movement of
3 coupled molecules to particular sites, including, but not
4 limited to, immunological assays, other biological assays, ~;
biochemical or enzymatic reactions, affinity
6 chromatographic purifications, cell sorting and diagnostic
7 and therapeutic uses.
8 Magnetic In vitro Tracers
9 Controlled size inorganic oxide particles of this ~
10 invention can be covalently bonded by conventional -`
11 coupling chemistries to bioaffinity adsorbents including,
12 but not limited to, antibodies (ligands, e.g., anti-
13 thyroxine, anti-triiodothyronine, anti-thyroid stimulating
14 hormone, anti-thyroid binding globulin, anti-l ~-
thyroglobulin, anti-digoxin, anti-cortisol, anti-insulin,
16 anti-theophylline, anti-vitamin B-12, anti-folate, anti- `
17 ferritir$ anti-human chorionic gonadotropin, anti-follicle ~`
18 stimulating hormone, anti-progesterone, anti-testosterone, ~`
19 anti-estriol, anti-estradiol, anti-prolactin, anti-human ~;
placental lactogen, anti-gastrin and anti-human growth
21 hormone antibodies), antigens (ligates, e.g. hormones,
22 peptides, pharmacological agents, vitamins, cofactors,
23 hematolgical substances, virus antigens, nucleic acids and
24 nucleotides) and specific bonding proteins, which coupled
particles can be used in immunassays or other binding
26 assays for the measurement of analytes in solution. In
27 broad aspect, when such controlled size inorganic oxide ~
28 particles are non-magnetic, and bound to a given species ~-
29 having specific affinity for a corresponding biochemical
moiety, the màgnetic response becomes directly
31 proportional to the concentration of the biochemical
32 moiety causing the complexation.
33 For example, crystals are prepared that are, as
34 explained earlier, below the critical size for the
development of superparamagnetic behavior. The non~
36 magnetic crystals are then coated with an organo-metallic
...
VO93/26019 PCT/US93/05~95
s
-45-
1 coating, for example, amino-hexyl-titanium-tri-
2 isopropoxide, and thermally crosslinked to form an organo-
3 titanium polymer coating having an organic sp~cer arm (the -
4 hexyl moiety) and organic functional group (i.e., the
5 amino-group). Anti-T-4 (thyroid hormone) with carboxylic ``
6 acid terminal functionality is then coupled to the non-
7 magnetic crystal in the presence of CDI (carbodimide
8 catalyst) thereby forming an amide linkage between Anti-T-
9 4 and the coated particle. Upon the addition of T-4
10 hormone, clusters are formed, and magnetic properties are -~
11 detected.
12 In a further embodiment, an antibody, such as IgG, is -
13 coupled to the non-magnetic crystals, followed by addition
14 of antitithiophillene. Upon addition of thiophillene,
magnetic clusters are formed.
16 In ~ivo Tracers
17 A surface modification is put on the surface of non- ,~
18 magnetic Fe3O4. The modified reagent is injected into a -
19 patient and a complex is formed at a specific site in the l~`
body. The patient is imaged by MRI, or other suitable
21 magnetic detection techniques.
22 Magnetic Metal ProcessinglMetal Anal~sis
23 Non-magnetic Fe304 is coupled to chelating agents and
24 put into contact with the process stream. The complex '~
25 forms and gives rise to a magnetic moment on the cluster ~
26 thus formed. The cluster and metal of choice are `
27 collected with a magnet. The pH is changed to strip the
28 metal and the product is collected. For example, the non- ~,
29 magnetic crystals are prepared as described above, with an
organo-titanium polymer coating having an organic spacer
31 arm and a terminal amino functionality. The particles are
32 then reacted, by and through the amino functionality, with `
33 2,3-dihydroxy-S-benzoic acid (upon addition of CDI1 to
34 form an amide coupled product with 2,3-dihydroxy-benzene
termination. When such dihydroxy functionality is brought
36 into contact with metals such as Tu, or Mo, under
......
~. .
.
W093t26019 ~1 ~ 7
PCr/US93/oS~
-4~-
...~
1 controlled pH (6-8) a complex forms and gives rise to the , -,
2 magnetic moment, In a similar manner, 2,3-dithio-
3 5-benzoic acid can be employed, providing terminal dithio ,--~
4 functionality, for more selective chelating with, e.g.,
5 Mo. ,
6 EXAMPLES
7 Exam~le 1 ,''
8 PREPARATION OF 25A DIALYZED IRON OXIDE CRYSTAL
9 A stock of solutlon of iron salt is prepared by first
dissolving 2.5g FeC12.H2O (Aldrich) in 37.5g of tap water
11 at 65C, then adding 4g FeC13 (Aldrich) to the solution
12 and mixing until dissolved. The solution is dark orange
13 in color. From this stock solution a ~ilute solution is ''
14 prepared for dialysis by adding 3g of the stock iron
~15 solution to 297g of warm (50C) water. 50g of this 1%
16 ~solution lS- sealed in cellulose dialysis tubing (Sigma
17 MW12000) that~has been prepared in the following manner: ~`
18 A 12 inch strip of tubing is soaked in warm water for 30 `
19 minutes, rinsed thoroughly in warm water and stored in
20 ~cool water until the addition of iron solution. '~;'
21;~ The~dialysis tubing containing 50 g of the 1% iron ,"',
22 ~solution is scaled and then placed in a 2% ammonium
23 hydroxide solution~
24 6g~NH40H (Ashland Chemical 28-30%) in 294g cool water ''
25 The~container holding the NH40H solution,and dialysis ';`
~26 sack of iron~solution is covered tightly and allowed to ~
27 dialyze at room temperature until equilibrium is reached -'
28 (4-6 hours). An orange precipitate of iron oxide forms '`
29 inside the dialysis sack, white precipitate of~,ammonium
30 chloride forms outside the sack. The precipitate is ';
31 decanted from the tubing and washed by centrifuging,
32 decanting the supeFnatant~ and adding water. This step is ~;,
33; repeated three times. ~,`"
' ' `,`,'
.
' '
WO93/~6019 ~ PCr/US93/0559
-47-
1Exam~le 2
2PREPARATION OF 50A DIALY2ED IRON OXIDE CRYSTAL
3A stock of solution of iron salt is prepared by~first `~
4dissolving 2.5g FeC12.4H2O (Aldrich) in 37.5g of tap water `-~
5 at 65C, then adding 4g FeC13 (Aldrich) to the solution '~
6 and mixing until dissolved. The solution is dark orange
7 in color. From this stock solution a dilute solution is ``~
8 prepared for dialysis by adding 6g of the stock iron
9 solution to 295g of warm (50C) water. 50g of this 2%
solution is sealed in cellulose dialysis tubins (Sigma
11 MW12000~ that has been prepared in the following manner:
12 A 12 inch strip of tubing is soaked in warm water for 30
13 minu*es, rinsed thoroughly in warm water and stored in
14 cool water until the addition of iron solution.
T~e dialysis tubing containing 50g of the 2% iron
16 solution is sealed and then placed in a 4% ammonium
17 hydroxi~e solution~
18 ~12g NH40~ (Ashland Chemical 28-30%) in 288g cool water
19 ~ The~container holding the NH40H solution and dialysis
sack of iron solution is covered tightly and allowed to
21 dialyze at room temperature until equilibrium is reached 'li?''.
22 (4-6 hours). A dark orange precipitate of iron oxide
23 forms~inside the dialysis sack, white precipitate of
24- ammonium~-~chloride forms outside the sack. The precipitate
25 is decanted from the tubing and washed by centrifuging, !r;~
26 decanting the supernatant, and adding water. This step is ` `~`~
27 repeated three times. `:
28 ~ ~Exam~le 3 ~'
29 PREPARATION OF 75A DIALYZED IRON OXIDE CRYSTAL ^;`
A stock solution of iron salt is prepared by first ~`
31 dissolving 2.5g FeC12.4H40 (Aldr ch) in 37.5g of tap water ,
32 at 65C, then adding 4g FeC13 (Aldrich) to the solution
33 and mixing until dissolved. The solution is dark orange ~``
34 in color~ From this stock solution a dilute solution is
35 prepared for dialysis by adding 9g of the stock iron ~"
'~
WO 93/26019 ~ 1 3 7 1 A~ ~ PCI'/US93/o'!~ :
-48-
1 solution to 291g of warm (50C) water. 50g ~f this 3%
2 solution is sealed in cellulose dialysis tubing (Sigma
3 MW12000) that has been prepared in the following ma~nner:
4 A 12 inch strip of tubing is soaked in warm water for 30 ,
5 minutes, rinsed thoroughly in warm water and stored in -`
6 cool water until the addition of iron solution.
7 The dialysis tubing containing 50g of the 3% iron
8 solution is sealed and then placed in a 4% ammonium
9 hydroxide solution:
12g NH40H (Ashland Chemical 28-30%) in 288g cool water
11 The container holding the NH40H solution and dialysis
12 sack of iron solution is covered tightly and allowed to
13 dialyze at room temperature until equilibrium is reached L'`"
14 (4-6 hours). A brown precipitate of iron oxide forms
inside the dialysis sack, while precipitate of ammonium
16 chloride forms outside the sack. The precipitate is
17 decanted from the tubing and washed by centrifuging,
18 decanting the supernatant, and adding water. This step is ~`
19 repeated three times. ;
Example 4
21 SYNTHESIS OF TITANIUM COATED 100A MAGNETIC PARTICLES
22 Titanium coated magnetite, Fe3O4, is prepared using
23 the following method:
24 Iron salts, FeC12.4H2O and FeC13 (41g) are each
dissolved in 1000 cc of water. The solutions are combined
26 into a 2 liter beaker and 70 ml of ammonium hydroxide is
27 added while mixing. The beaker containing the resulting ~
28 precipitate, 28 gm of Fe3O4, is then placed onto a ``
29 permanent magnet to magnetically separate the magnetic ~-
particle from the salt by-products. After resting on the
31 magnet for 5 minutes, the clear salt solution is decanted.
32 The precipitate is then resuspended in a total of 1500 cc ;~
33 of water and placed on a permanent magnet for 5 minutes
34 before decanting. The above washing process is repeated
three additional times. After the final decanting, the
~093/26019 ~~ ~ 71 4 5 PCTtUS93/05S95
.
-49-
''`, .;~
1 magnetite is suspended in 1500 cc of dry acetone and
2 magnetically separated as above. The particles are
3 acetone washed a total of 3 times. After the final
4 decanting, the particles are suspended in 500 cc of N,N
5 dimethyl formamide. ~-~
6 The solution, 250 cc, is poured into a horizontal bead ~-
7 motor mill and milled for 10 minutes to ensure efficient
8 dispersion. Titanium isopropoxide, 35 gm, dissolved in 50 ~ ~
9 cc of N,N dimethyl formamide is slowly pipetted into the ~-
10 funnel of the operating motormill and milled for 15 ~;
11 minutes. `~
12 The d$spersion is removed from the mill, magnetically
13 separated, decanted and water washed 5 times with 1000 cc
14 of distilled water. ``-~
Example 5 -
16 SYNTHESIS OF TITANIUM COATED 20A NON MAGNETIC PARTICLES
17 This example illustrates the preparation of
18 organometallic, titanium isopropoxide, coated non-magnetic ~-~
19 20A ferrites. A dispersion of non-magnetic 20A particles
is water washed five times and anhydrous methanol washed
21 three times by centrifugation. A total of 5.0 g of
22 particle is suspended in 250 ml of N,N dimethyl formamide ~`
23 and milled in a bead motormill for 15 minutes. 12.0g `~
24 titanium isopropoxide dispersed in 30.0 g N,N- dimethyl
25 formamide is slowly pipetted into the operating mill and `
26 milled for another 15 minutes. The product is then
27 removed to form the mill and water washed five times by -
28 centrifugation and resuspended in distilled water. ~-
29 Example 6 `',`~!'"'
30 SYNTHESIS OF AMINE TERMINATED MAGNETIC PARTICLES ~-
.
31 Magnetite coated with an organometallic, Ti, a`nd
32 terminated with a C-6 amine is prepared using the `;
33 following method.
34 The precipitation, washing and coating wit~
organometa~lic, titanium isopropoxide, is conducted in the
36 exact manner as described above. After the washed
W093/26~19 ~l~c~ 7 ~ ~ 5 PCT/US93/Os~
-50~
1 magnetite particle, N,N- dimethyl formamide and titanium
2 isoproxide have milled for 15 minutes, 15 gm of 6-amino
3 l-hexanol dissolved in 30 cc of N,N dimethyl formam~de is -~
4 pipetted into the operating mill. After milling for 15
5 minutes, the dispersion is heated for 20 minutes at 100C f.``-~
6 with occasional mixing. The dispersion is then allowed to -~
7 cool, magnetically separated and washed five times with ~`
8 1,000cc of distilled water. I
g Example 7
SYNTHESIS OF CARBOXYL TERMINATED MAGNETIC PARTICLES
, . . ., ~
11 Magnetite coated with an organometallic, Ti, and
12 terminated with a C6 carboxyl group is prepared as -
13 follows:
14 1~.2 g of 4-hydroxy butyric acid sGdium salt dispersed
15 in 30 cc of N,N-dimethyl formamide is slowly pipetted to -
16 the 250 cc of washed organometallic coated magnetic --
17 particles as described above in Exam~ple 4. After milling `~
18 for 15 minutes, the dispersion is heated for 20 minutes !`:S
~19 at 100C with mixing. The solution, at room temperature,
20 is~ magnetically separated and washed five times with 1,000 `~
21 cc of distilled water.
22 ExamPle 8
23 SYNT~ESIS OF DIHYDROXY AROMATIC TERMINATED MAGNETIC PARTICLES
1 .
24 This example iliustrates the preparation of dihydroxy~
25 aromatic terminated magnetic particle. 5 g of magnetite -~
26 coated with titanium isopropoxide and 6-amino-1-hexanol, `~
27 prepared as above, is dispersed in sodium metabisulfite
28 and distilled water solution, 300 cc. The sodium
29 metabisulfite solution has been pretreated with nitrogen
gas`to prevènt oxidation of the particles. 78 g of gallic
31 acid, and 1.0 g of carboddimide is combined with the
32 amine-terminated magnetic particle with mixing. After
33 incubating for one hour, the product is magnetically
34 separated and water washed.
~ .
~15 ~14~) ..
WO ~3/26019 PCI/US93/0559~
~.
-51- '~
Exam~le 9
2MAG~ETIC T~ACE~S FOR IMMUNO ASSAY I
320A non-magnetic ferrite particles were washed 4 times
4 with water, 3 times with acetone and 3 times with
anhydrous methanol by collecting the particles after
6 centrifugation and resuspendins the particles by vigorous
7 agitation.
8 Tyzor (titanium tetra-isopropoxide), dissolved in j~
9 anhydrous methanol was added to 0.53 g dry of particles at
10 25 g Ti/9.6 g dry particles. Steel balls were added and ~;
11 the particles were milled in a ball mill for one hour.
12 The particles were then amine terminated by adding
13 6-amino-1-hexanol dissolved in anhydrous methanol to the -
14 Tyzor coated particles. For every 9.6 g dry partlcles,
15 .088 mol amine was used. This was added to the Tyzor ,
16 coated particles and milled on the ball mill far 3 1/2
17 hours. The magnetics were tested on a vibrating sample
18 magnetometer. The particles were found to be non-
19 magnetic.
The sample was divided into 4 equal dry parts of 0.13
21 g each. 1,6 diisocyanato-hexane was added to particles in `
22 four concentrations: 0, .5, 4, 8 lm 1,6 diiso./.5 g dry.
23 The particles were milled overnight in the ball mill ~j`
24 without using steel balls. ~
. . ,
The magnetics were tested again on the VSM. It was
26 determined that the increase in 1,6 diisocyananatohexane
27 resulted in a proportional increase in magnetivity.
28 ExamPle 10
29 ENCAPSULATION BY A POLYMER `-~
20A non-magnetic ferrite particles were washed 4 times
31 with water, 5 times with acetone, (collecting with a
32 centrifuge between washes). The acetone slurry is then
33 washed 5 times with hexane. A solvent borne solution of `
34 the polymer (e.g., polystyrene, polyurethane, poly(vinyl
chloride)) from about 0.1%-10% by weight in an amount
36 equal to about 1:10 to 10:1 partLcle:polymer ratio is then
. .
O 93/26019 .~ 1 37~ ~5 PC~r/US93/05.
-52-
l added. Mixing continues for about l0 minutes in a high
2 shear mixer to allow the,crystals to coat uniformly with '~'
3 polymer. Water is then added in a volume equal to about ~-
4 l0-l00 times the amount of solvent to flocculate the ,'~
5 polymer. The beads are then collected. In the case of '~,',
6 polyurethane, it has been found the THF is the solvent of '
7 choice. '"
8 Example ll
9 ADDITION OF MONOMER FOLLOWED BY CROSSLINKING ~;
l0 A particle slurry is prepared as in Example l0. Oleic ~'
ll acid is then added to the hexane slurry of particles and
12 mixed in a high shear mixer for about 20 minutes. A ,~
13 volume of acetone is then added, equal to approximately 5 ,
14 times the amount of hexane to the oleic acid coated
particle dispersion, in order to flocculate. The
16 resulting residue is collected and mixed in water in a ''
~7 high; shear mixer for about l hour to produce oleic acid
18 coated crystal beads. The bead slurry is then exposed to
19 3-beam generator (Energy sources, Woburn, MA), from 1-20 '~`
meg Rad for about 0.25-0.5 sec., to crosslink through the '
2l unsaturated group.
22 ' Exam~le 12 1`
23 ~ PREPARATION OF SUB 10 NM, PARTICLES IN A TWO-SIDED
24 'DIALYSIS~TANK
25 2 nm diameter uniform magnetic crystals were prepared ,
26 by controlled contact of a base solution and iron salt
27 solution across a semipermeable membrane, resulting in an ,'-
28 iron oxide crystal precipitate of defined size within a ':
29 narrow size distribution range. A Spectra/Por~ 5 dialysis `'
membrane (flat sheet) was affixed in a manner as to
3l separate two equal sized chambers of a two sided Dialysis
32 reaction tank. Both sides of the tank were filled with 20
33 liters of distilled H2O at 20C. 12.5g FeCl2 4H20 and 20g
34 FeCl3 were added to one chamber of the tank and stirred, `
until dissolved. 60.6g NaOH were dissolved in 2 liters of
36 H20 and added to the solution into the opposite chamber in ,
......
..' '`
'~093/26019 ~. 3 7 1 4 ~ PCT/US93/05~95
-53-
`; .,
l the tank. Both sides were. agitated by a mechanical paddle
2 stirrer for 15 min. After 70-80 hours of contact time,
3 the iron solution and precipitated crystals were removed
4 from the tank and the magnetic crystals were collected by
5 centrifugation and measures by TEM to be 2 nm average .. ~-
,~
6 diameter. ~.`
7 Uniform size inorganic core particles can be prepared
8 by the preferred method reported in U.S. Patent
9 Application Serial No. 894,260, filed June 8, 1992, the
teachings of which are incorporated by reference. As
11 described therein, aqueous solutions of an inorganic salt
12 and an inorganic base are contacted across a porous
13 membrane wherein the membrane contains a plurality of
14 pores which allows for precipitation of substantially
monodispersed inorganic oxide particles on one side of the
16 memb:rane and precipitation of a salt of the corresponding
17 base~ on a second side of the membrane. Particle size
18 diameter can range between 5-1000 Angstroms, and in a -
19 preferred embodiment, 5-100 Angstroms, with a particle `
20 size distribution of +/- 10%. The inorganic salts are of .-
21 the formula MY, wherein M is selected from the group ~
22 consisting of Fe, Co, Ni, Zn, Mn, Mg, Ca, Ba, Sr, Cd, Hg, ```
23 Al, B, Sc, Ga, V, In, and mixtures thereof, with Y being -.
24 selected from the group consisting of C1, Br, I, SO4, NO3, -;~
PO4 and mixtures thereof. The inorganic base is selected
26 from the group consisting of NH40H, KOH, LiOH, NaOH, CsOH, ~.
27 RbOH and mixtures thereof. Accordingly, and in a ~.
28 preferred embodiment, Fe3O4 is prepared (a mixed `~
29 Fe(+2)Fe(+3) oxide of the formula [Fe(+2)]~Fe(+3)]2O4) l -
with a uniform sub 100 Angstroms diameter serving as the
31 inorganic core of the liposomes described herein. ~-
, ...
32 Inorganic core particles can also be prepared
33 according to the following general procedure: metal
34 salts, or organometallocenes are precipitated in base at
35 high temperature and pressure to form fine magnetic metal ~`
36 oxide crystals. The crystals are redispersed, then washed
',''. -`
'';
~::
WO93/26019 2 1 3 7 1 ~ ~ PcT/us93/oc~
-54-
',:
1 in water and an electrolyte. Magnetic-s~paration can be
2 used to collect the crystal between wa~s. The cr~stals
3 are then milled to a more controlled particle size, for
4 example, in a ball mill, under conditions sufficient to
5 form 50 Angstroms or lower particle size. See, U.S. ~
6 Patent No. 5,071,076, and U.S. Patent Application Serial -`
7 No. 806,478, filed December 31, 1991, the teachings of
8 which are incorporated by reference.
9 III. Am~hipathic Orqanic Com~ounds ~`
., ~
The amphipathic organic compounds which can be
11 used in forming the inorganic core liposome of the -~
12 invention may be selected from a variety of organic
13 compounds which contain both a hydrophobic and hydrophilic
14 moiety. According to one important aspect of the
invention, it has been discovered that the hydrophilic
16 moiety is adsorbed or coordinated onto the surface of the
17 inorganic oxide, whereas the hydrophobic moiety of the
18 molecule extends outwardly to associate with the
19 amphipathic vesicle forming lipid compounds. Preferred
amphipathic organic compounds include fatty acids selected
21 from the group consisting of oleic, stearic, linoleic,
22 lionlenic, palmitic, nyristic and arachidonic acid.
23 IV Am hi~athic Vesicle Formin~ LiEid Components
~ .
24 The lipid components used in forming the ~-~
25 inorganic core liposomes of the invention may be selected --
26 from a variety of vesicle forming lipids, typically
27 including phospholipids, such as phosphatidylcholine (PC),
28 phosphatidic (PA), phosphatidylinositol (Pl),
29 sphinogomyelin (SM), and the glycolipids, such as
30 cerebroside and gangliosides. The selection of lipids is '
31 guided by consideration of (a) drug release rate is serum,
32 (b) drug-entrapment effi~iency, (c) liposome toxicity, and
33 (d) biodistribution and targeting properties. A variety
3~ of lipids having selected chain compositions are
commercially available or may be obtained by standard
.
:'`,'
,
WO93/~6019 ~ 3 7 1~ PCT/US93/05~9
-5~-
!, ~....'
1 lipid isolation procedures. See, e~g. U.S. Patent No.
2 4,994,213. .
3 The lipids may be either fluidic lipids, e.g.
4 phospholipids whose acyl chains are relatively '"~"'J'~"
unsaturated, or more rigidifying membrane liplds, such as
6 highly saturated phospholipids. Accordingly, the vesicle ^`
7 forming lipids may also be selected to achieve a selected ;`~
8 degree of fluidity or rigidity to control the stability of
9 the liposome in serum and the rate of release of entrapped
drug from the liposome in the bloodstream. See, e.g. U.S.
11 Pat. No. 5,013,556.
12 In a preferred embodiment, the vesicle forming lipid
13 include those phospholipids in which the polar-head-group
14 region is modified by the covalent attachment of `~
15 polyaIkylene ether polymers of various molecular weights. r-.'``
16 The attachment of the relatively hydrophilic polyalkylene
17 ether polymer, particularly polyethylene oxide, alters the
18 hydrophilic to hydrophobic balance within the phospholipid
19 in order to give unique solubility to the phospholipid
compound in an aqueous environment. See, e.g. U.S. Pat.
21 No. 4,426,330. The polyalkyl ether lipid is preferably ~-
22 employed in the inorganic core liposome composition in an ~-
23 amount between about 1-20 mole percent, on the basis of ~ `
24 moles of derivatized lipid as a percentage of total moles
25 of vesicle-forming lipids. The polyalkylether moiety of -`
26 the lipid preferably has a molecular weight between about ~
27 120-20,000 daltons, and more preferably between about '`~!`'~`
28 1000-5000 daltons. ~
29 In yet another embodiment of the present invention, a `
new series of phenyl lipid compounds are described which
`- ```
~....
, ~ . ^ ~ .
~, .
~, .
W093/260l9 ~ 1 3714~ PCT/US93/05~.
-56
1 have the following structural formula:
3 ~ `
~ R~ C~3
8 :
9 Rt ~ ~
'.`
11 wherein two of Rl, R2 and R3 represent a saturated or
12 unsaturated straight-chain or branched;chain hydrocarbon
13 group, the other being hydrogen, therein providing at
14 least two hydrocarbon chains attached to the phenyl ...
15 moiety~, wherein the two hydrocarbon chains are typically `'~
16 between abou`t 14-22 carbon atoms in length, and have .
17 varying degrees of unsaturation. R4 represents the .
18 repeating unit of either a poly(alkylene oxide) polymer, ~.;.
19 preferably ethylene, propylene and mixtures thereof, or -:
20 :the repeating unit of poly(vinyl alcohol~. The number of `
21 alkylene oxide or vinyl alcohol groups in the polymer, `i`~
22 designated as n, may vary from 0 to about 200 or more.
23-
24 V. Preparinq the Inorqanic Core Li~osome Composition ~
25 One p~referred method for producing the uniform ~.
26 size inorganic core liposome composition begins with first
27 coating the magne:tic particles described above in Section `.
. .
. ..
WO93/26019 ~ 71 ~ ~ PCT/US93/05595 -~
`~
i _57~
1 II with an amphipathic organic compound which contains
2 both a hvdrophillic and hydrophobic moiety. For exa~mple, ..
3 fatty acids, such as oleic acid, linoleic acid or
4 linolenic acid, dispersed in an organic solvent, are
S directly added to the particles at a ratio of dry `
6 Fe304:acid equal to 2:1 weight percent. After
7 mechanically milling the mixture for 1 to 1.5 hours on a
8 ball mill with 4 mm glass media, the acid coated particles ;
9 collapse around the media allowing for easy removal of
~...
water without the loss of the particles. The coated
11 particles are then dispersed in an organic solvent by `
12 addition of 700 ml of hexane, toluene or chloroform and
13 mechanically milling with glass media overnight (15 hrs). `
14 Absorbing a phospholipid onto the fatty acid coated
15 particles was accomplished by addition of a synthetic l~
16 polyethylene~glycol terminated phosphatidyl ethanolamine ~;
17 to the above dispersion and mechanically mixing for 3
18 hours. The ratio of fatty acid:pure lipid is about 1
: :.
l9 weight percent.
20 To transfer the particles from an organic phase to an i
21 aqueous phase, 7 mls of the dispersion was placed into a
22 14 ml glass vial with 3 ml of distilled water. The vial
23 was placed in warm, 35C sonicating wàter bath with N2
24 bubbling through it to evaporate the solvent. Once the
solvent has evaporated, the aqueous dispersion was then
26 suspended in a total of lO mls of autoclaved water,
27 sonicated for one hour, and centrifuged for 5 minutes. i
28 The supernatant was removed and brought to 20 mg ~`
29 particle/ml solution with autoclaved water.
30 VI. UtilitY ';~
31 From the above, it can be appreciated that the
32 present invention offers a number of advantages over prior
33 art liposome-methods. The preparation of uniform size
34 inorganic core particles by dialysis and precipitation i;~
35 across a semi-permeable membrane is unique in its ability --
36 to allow for the production of uniform size liposomes
. .
W0~3/26019 ~ 1 3 7 1 4 ~ PCT/US93Jo'~
t; ~ .
',
-58- ;
.. ..
1 without the requirement for extrusion or other additional
2 liposome sizing techniques. The ability to selecti~ely
3 vary the average size of liposomes, according to lipid
4 composition and/or ionic strength, is another useful
feature of the invention. While the present invention
6 provides inorganic core liposomes with a size~range of
7 about 5-5000 nm, one selected size range, between about ~-
8 100-300 nm, is particularly useful for a variety of
9 parenteral uses, as discussed. ~`L
One general class of drugs include water-soluble
11 liposome permeable compounds which are characterized by a
12 tendency to partition preferentially into the~aqueous
13 compartments of the liposome suspension, and to
14 e~uilibrate, over time, between the inner liposomal spaces
and outer bulk phase of the suspension. Representative
16 drugs in this class include terbutaline, albuterol,
~17 stropine methyl nitrate, cromolyn sodium, propracalol, ~-
18~ funoisolide, ibuprofin, geniamycin, tobermycin,
19 pentamidine, penicillin, theophylline, bleomycin,
20 etopoxide, captoprel, n-acetyl cystein, verapamil, .
21 vitamins, and radio-opaque and particle-emitter agents,
22 such as chelated metaIs. 3ecause of the tendency of these
23 agents to equilibrate with the~aqueous composition of the ;~`~
- 24 medium; it is preferred to store the liposome composition
25 in lyophilized form, with rehydration shortly before `~
26 administration. ~`
27 A second general class of drugs are those which are
28 water-soluble, but liposome-impermeable. For the most `~
29 part, these are peptide or protein molecules, such as
30 peptide hormones, enzymes, enzyme inhibitors, ~`
3I apolipoproteins, and higher molecular weight carbohydrates `~`
32 characterized by long-term stability of encapsulation.
33 Representative compounds in this class include calcitonin,
34 atriopeptin, -1 antitrypsin (protease inhibitor),
interferon, oxytocin, vasopressin, insulin, interleukin-2,
36 superoxide dismutase, tissue plasminogen activator (TPA), ,
!:,
- WO93/26019 ~ 1~ 7 1 ~ 5 PCT~S93/0~95 . ~
-59-
'~
1 plasma factor 8, epidermal growth factor, tumor necrosis ~`~
2 factor, lung surfactant protein, interferon, lipocortin,
3 ~ -interferon, macrophage colony stimulating factor, and `~
4 erythroprotein.
5 A third class of drugs are lipophilic molecules. The `~
6 drugs in this class are defined by an oil/water partition
.~
7 coefficient, as measured in a standard oil/water mixture
8 such as octanol/water, of greater than 1 and preferably `~
9 greater than about 5. Representative drugs include
prostaglandins, amphotericin B, progesterone, isosorbide
11 dinitrate, testosterone, nitrogIycerin, estradiol,
12 doxorubicin, epirubicin, beclomethasone and esters,
13 vitamin E, cortisone, dexamethasone and esters, and
14 betamethasone valerete. -
15 In another application, the inorganic core liposome 1'`5
16 composition is designed for targeting a specific target
17 tissue or organ. For example, this feature allows for i~`
18 targeting a tumor tissue, for drug treatment by
19 intravenous administration to a tumor-bearing subject.
As another example, the inorganic core liposomes may
21 be~prepared with surface-bound ligand molecules, such as
22 antibodies, which are effective to bind specifically and
23 with high affinity to ligand-binding molecules`such as
24 antigens, which are localized specifically on target
25 cells. ~;~
26 ~ A variety of methods for coupling ligands to the
27 surface of liposomes are known, including the
28 incorporation of ligand-derivatized lipid components into
29 liposomes or coupling of ligands to activated liposome
surface components.
31 The targeted inorganic core liposomes may be prepared
32 to include cancer chemotherapeutic agents, such as those
33 listed above. In one preferred embodiment, the liposomes
34 are prepared to include PEG-PE and PG, to a final
.,
,
. ;
WO93/26019 ~1 3 7 ~ 4 5 PCT/US93/05
-60-
l concentration of charged lipids up to 40 mole percent, ~`~
2 doxorubicin, and remainder neutral phospholipids or~
3 neutral phospholipids and cholesterol. `~
4 In an inorganic core liposome composition which is
5 useful for radio-imaging of solid tumor reglons, the -
6 liposomes are prepared with encapsulated radio-opaque or
7 particle-emission metal, typically in a chelated form -
8 which substantialIy prevents a permeation through the
9 liposome bilayer.
l0 In still another application, the liposome composition -
ll is designed to enhance uptake of circulating cells or -
12 other blood-borne particles, such as bacteria, virus
13 infected blood cells and the like. Here the long-life `-;
14 liposomes are prepared to include surface-bound ligand
15 mole;cules, as above, which bind specifically and with high ~`
16 af~finity to the selected blood-borne cells. Once bound to
17 the blood-borne particles, the liposomes can enhance
18 uptake by the RES.
l9 Polyalkylether moieties on the liposomes may be i`~
der~vatized by the associated amphipathic lipid by an
21 ester, peptide, or disulfide bond which can be cleaved,
22 ~after liposome binding,~to the target cells, to further !,'~'';`
23 enhance RES particle clearance.
24 Studies performed in support of the present invention ;
25 indicate that the inorganic core liposome composition of ii
26 the invention provides an enhancement in blood circulation ~`
27~ lifetime which is equal, and in some cases superior, to
28 the most effective RES-evading rigid-lipid liposomes which ~`
29 have been reported heretofore, including liposomes
containing GMI and membrane-rigidifying lipids.
31 The blood circulation lifetimes achieved in the
32 present invention should be substantially greater than ``
33 with fluid-core liposomes.
34 The following examples illustrate methods of
preparation of inorganîc core liposomes with enhanced
36 cirFulation times, and for accessing circulation times in
.. .
.:.,
~093/26019 2 1 3 7 i ~ PCT/US93/~559
-61-
1 vivo and invitro. The examples are intended to illustrate --
2 specific inorganic-core liposome compositions and methods ~`~
3 of the invention, but are in no way intended to limit the -~
4 scope thereof.
DESCRIPTION OF THE EMBODIMENTS
6 EXAMPLE 1
7 Preparation of Magnetic Particles by Co-precipitation
8 of Fe+2/Fe+3 with Excess Base ``
9 Magnetic particles of 100 Angstroms in diameter are
prepared using the following method. Iron salts, FeC12~,
11 3H2O, (25g), and FeC13 (41g) are each dissolved in 1000 cc `
12 of water. The solutions are combined into a 2 liter -`~
13 beaker and 70ml of ammonium hydroxide is added while
14 mixing. The resultlng black magnetic precipitate yields
28gm of magnetite, Fe304.
16 EXAMPLE 2
17 Preparation of sub 10 nm particles
18 ~ 2 nm diameter unlform magnetic crystals were prepared
l9~ by controlled contact of a base solution and iron salt
solution across a semipermeable~membrane, resulting in an
21 iron oxide crystal precipitate of defined size within a
22 narrow size distribution range.
23 A Spectra/Por S dialysis membrane (flat sheet) was
24 affixed in a manner as to separate two equal sized
chambers of a two sided Dialys`is reaction tank. Both
26 sides of the tank were filled with 20 liters of distilied
27 H20 at 20C. 12.5 g FeC12~ 4H20 and 20g FeC13 were added
28 to one chamber of the tank and stirred until dissolved.
29 60.6g NaOH were dissolved in 2 liters of H20 and added to ;
30 the solution into the opposite chamber in the tank. Both `
31 sides were agitated by a mechanical paddle stirrer for 15
32 min. After 70-80 hours of contact time, the iron solution
33 and precipitated crystals were removed from the tank and
34 the magnetic crystals were collected by centrifugation and
measures by TEM to be 2nm average diameter.
;~ ~
'
WO93/26019 ~`~ PCT/US93/05~
,~
-62-
1 EXAMPLE 3
2 Preparation of Oleic Acid Coated Magnetite
3 Magnetic particles, Fe304, coated with oleic acid are
4 prepared using magnetite as precipitated in Example 1.
The magnetite is water washed by successive additions of
6 distilled water to a slurry concentrate of magnetite. The
7 beaker containing the magnetite slurry is place onto a
8 permanent magnet to magnetically separate the magnetic `
9 particle from the salt by-products between each successi~e -
addition of water. After resting the slurry on the magnet
11 for 5 minutes, the aqueous salt solution is decanted. The ;`
12 precipitate is then resuspended with agitation in a total l~
...
13 of 1500 cc of water and placed on a permanent magnet for 5
14 minutes before decanting. The above washing process is ~-
repeated three additional times with water. After the
16 final water wash is decanted, the particles are acetone -
17 washed and hexane washed a total of 5 times each in the
18 above manner. ~ ;~
19 Oleic acid is added to the magnetic hexane slurry in a ;`
20 ratio of oleic acid:dry particle equal to 2:1 weight ~,
21 percent. The mixture is adjusted to 15% total solids with
22 hexane and mechanically milled overnight in a glass jar ;`~
23 half filled with 3mm stainless steel media.
24 EXAMPLE 4 ?'-'"'',
25 Preparation of Oleic Acid Coated Dialyzed ;`
26 Magnetic Particles "~
27 Dialyzed particles coated with oleic acid are prepared `
28 using particles as prepared in Example 2. 0.1 grams of
29 particles are~washed with three 200 ml; volumes of `
30 distilled water and acetone by suspending approximately `
31 O.lgm dry particle in 200 ml of acetone and centrifuging
32 for 45 minutes to collect particles between each washing. ~
33 Oleic acid was added to the acetone slurry in a ratio - `-
34 of oleic acid:dry particle equal to 2:1 weight percent and
mechanically milled overnight in a glass jar half filled
36 with 3mm glass media. ~
,:
,
~- W093/26019 2 ~ 3 7 1 4 ~ PCT/US93/05~95 ~
-63- ;
~ ., .~.
~i~,....
1 EXAMPLE S
2Preparation of Magnetite Core Liposomes using
3Phosphatidyl Choline
410 gms Oleic acid coated magnetite as prepared in
Example 3 was dispersed in 100 cc hexane. The phosphate
6 lipid is absorbed onto the particle by dissolving
7 phosphatidyl choline (Sigma, P-364~4, L-2, lechithin, 45%
8 PC) into -hexane with heating to create a 15% solution.
9 The PC/hexane solution is combined with the
mzgnetic/hexane solut~ion at a ratio of pure phosphatidyl
11 choline:oleic acid equal to 1:2 weight percent.
12 The solution was mixed in a glass jar (without mediaj
13 on a jar roller for two hours. After mixing, the lipid
14 was absorbed onto the particle by adding three times as
much acetone than hexane and collecting the~lipid coated
16 parèicles over a magnet.~ After the~coated magnetic
17 particles~were separated from the solvents,~the solvents
18 were decanted, distilled water was àdded to produce a 2.0%
19 TS slurry. The slurry is~heated in a beaker on a hot
20~ plate to 100C for 10 min. From 0.5 to 50 grams of triton
21 x-I14 (Union Carbide) was added~to disperse the lipidized
22 magnetic particles in an aqueous system. A ratio of
23 triton x114:1ipid particle equal to 1:6 weight percent was
24 the optimum level for the dispersion. The dispersion was
mixed on a laboratory vortex mixer for 2 minutes and
26 placed~in an ultrasonic bath (Branson 1200, YWR) for two ;`
2~7~ hours. The final dispersion is adjusted to 0.2% TS
28 (2mg/ml). Particles were;measured on a Nycomp laser
29 part`icle size analyzer and were found to be approximately
200 nm in diameter.
31 ~ EXAMPLE 6
32 Preparation of Phenyl Lipid
33 A. Synthesis of a m-isophthalic acid based phenyl
34 lipid. ~
35 The starting material for this synthesis if ;
36 5-Aminoisophthalic acid. The 5-aminoisophthalic acid is `~`~
37 not soluble in dioxane alone. It is soluble in a mixture
` ,:' `''
~ .
:' :
W093/26019 7 1 3 7 1 4 5 PCT/US93/05~--
-64-
1 of dioxane and triethylene glycol. 5-aminoisophthalic
2 acid (145 mg) was dissolved in 5 ml. of dioxane and~2 ml.
3 of triethylene glycol, and the pH was adjusted to 10 with
4 NaOH. Methoxypolyoxyethylene imidazoly carbonyl, average
5 mol. wt. 5,000 from Sigma (2.0g) was dissolved in 2ml of ,~
6 H20, l.Oml of lN Na2C03, and 2.0 ml of triethylene
7 glycol. This solution was added to the 5-aminoisophtlalic
8 acid solution and stirred for 36 hours at room ,
9 temperature. The reaction mixture was then dialyzed `i
10 overnight against 2 liters of H20. The dialyzed reaction ~;
11 mixture was mixed with lOOml of pyridine and the liquids
12 removed via rotary evaporation. The resulting yellow oil
13 was placed in the refrigerator. After several days a
14 white precipitate formed. The precipita.e contains both
15 coupled and uncoupled PEG.
16 Oleyl alcohol can be coupled to the above isophthalic
17 acid derivative using thionyl chloride. The thionyl
18 chloride can be used to activate the oleyl alcohol for
19 ester formation with the carboxyl groups of the "
20 isophthalate. See. Fig. 2.
21 B. Synthesis of ortho phenyl lipids
22 The ortho analog of the phenyl lips can be ¦`
23 synthesized starting with either 3,4 dihydroybenzaldehyde
24 or 3,4 dihydraxybenzoic acid. The aldehyde group can be
25 coupled to an amino group by formi~g the Schiff's base and
26 then reducing it with NaBH4. Olegic acid could then be `
27 coupled to the hydroxyl groups using thionyl chloride to `
28 pro~ide: `
29 i-
C~(OC~C~)"-N~C~ o-cotc~l2)~cH~cl~ctl2)~c~l3
32 O-CO-~CI`~ C~C~C~
33 ` `
34 . 3 ,4 dihydroxybenzolic acia could be coupled through `
35 its carboxyl group to amino-terminated PEG using
. .
i'~
,
~093/26019 PCT/US93/0559~ j
,'~`';'.
-65-
1 dicyclohexyl carbodiimide. Oleic acid could then be
2 coupled as above. ~ `~
3 Since both amino and carboxyl PEG derivatives as well
4 as both oleic acid and oleylamine are available, the PEG
and oleic acid groups can be easily interchanged in the
6 above compounds.
7 VII. Preparation of Wave Absorbinq Magnetic Core
8 Particles
10 The wave absorbing magnetic core particles suitable in ;~
11 the present invention are those particles which, upon
12 application of an electromagnetic field, create inductive
13 heat local to the particle. In a preferred embodiment,
14 the wave absorbing magnetic co-- particles comprise
ferrite or mixed ferrite mater~ ls, preferably of a
16 uniform, controllable size, and narrow size distribution, `
17 wherein the primary component, the oxide, is of the
18 formula M2(+3)M(+2)O4, wherein M(+3) is Al, Cr or Fe, and
19 M(+2) is Fe, Ni, Co, Zn, Ze, Ca, Ba, Mg, Ga, Gd, Mn or Cd. !`
20 In a further aspect, the oxides can be advantageously -
21 mixed with LiO, NaO and RO, or with ~ or ~ Fe2O3 and ~;-
~22 Fe3O4- ,-
23 The preparation of substantially uniform si2e oxides,
24 1 to 50,000 nm in diameter, is achieved by conversion of
hydrous oxide gels, in a multi-step process, wherein
26 alkali is added to individual M(+3) and M(12) aqueous
27 solutions, which separately precipitate the corresponding ~`~
28 metal hydroxide. The two precipitates are then coarsely
29 mixed to provide micron size amorphorus gel particles, ~
30 which can be milled to form hydrous oxide gel part -les ~-
31 about 100 A in diameter. These particles are then Aeated
32 to effect dehydration, in the presence of oxygen or air,
~33 wherein the dehydration temperature, time of dehydration,
3~ and concentration of oxygen or air operate to control the
particle size of the oxide crystals therein produced.
:,.
.--. . .
WO93/26019 ~ 3 7 1 ~ ~ PCT/US93/oS~
;, `:.~
i - .,
-66-
, ., .,, ~
1 For example, in connection with the above, a u
2 dehydration temperature of 100C, at a ti~e of abou~ 6 i3'
3 hours, in the presence, of oxygen, provides oxides :,
4 particles of about 70A diameter. Alternatively, a i`',
S dehydration temperature of about 65C, at a time of about `,,,
6 24-36 hours, in the presence of oxygen, affords oxide '~
7 particle sizes of about 1000-2000A. Accordingly, by ',~
8 recognizing that short dwell times and high temperature
9 promote small oxide particle formation, and that long
dwell times and low temperature promote large particle
11 formation, oxide particles from SOA to several microns in
12~ diameter have been produced.
13 Heretofore, the use of ferrite materials as a
14 protective medium for electromagnetic radiation reflecting '-
15 surfaces was well known. In the present invention, ~,
16 however, it has been found that very small ferrospinal
17 particles provide a high degree of absorbtion of
18 electromagnetic waves. It has also been found that the
1, complex permeability of certain ferromagnetic metallic ,~
20 oxides varies with frequency in such a way as to provide '~
21 high absorption of electromagnetic magnetic radiation over ','-
22~ wide frequency ranges without using large amounts of ':-
23 absorber material. Upon exp'osure to electromagnetic ,','
24 waves, these ferrites generate significant infra-red
25 radiation over short distances local to the ferrite ','`,
26 particle's surface. -`"
27 Tn general, those ferrites suitable for use in the -'
28 present invention are cubic crystalline materials
29 characterized-by a spinal structure containing Fe2O3 and
30 at least one other oxide, usually of a bivalent metal, '~
31 e.g. lithium oxide, cadmium oxide, nickel oxide, .ron
32 oxide and zinc oxide. `
33 The ferrite materials of this invention can also be -
34 prepared by a thermal process, in which they are mixed ' '
together then ground together mixed and fired at about
36 1200C in a tube furnace for four hours or made by
.
,
W 0 93/26019 ~ . 4 ~ PC~r/US93/OS~95
-67 i~r
. . j .,
;;.~,~.
1 oxidation of ferrite powders from metal hydroxide gels.
2 The imaginery permeability must be high enough to pr~oduce
3 a large loss. For high frequencies, it has been found
4 that nickel can replace lithium and for narrow bands zinc
can replace cadnium.
6 One preferred mixed ferrite having the composition
7 0,45 LiO, 0,5 Fe203 + 0.30 CdFe204 1 0.25 Fe304 yielded
8 the following results: ~
9 -,,
10 Frequency Range (mHz) % Absorbance Surface Temp `
11 .~i
12 1800-2500 98 230 1"
13 -
14 As noted above, ferrites of interest to this invention
15 can also be prepared by conversion of hydrous oxide gels ~-~
16 in a multi-s~ep process. In one particular preferred
17 example, alkali is added to a ferrous sulphate solution at
18 a temperature between 15 and 40C, in a stoichiometric `~
19 amount adapted to precipitate ferrous hydroxide, from the ~-
Fe++ ion. At the conclusion of said precipitation, air is
21 blown into the slurry, thus oxidizing ferrous hydroxide to
22 goethite, FeO(OH).
23 During a second step, alkali is added to the slurry
24 obtained in the first step. The remaining Fe++ is
25 precipitated in the form of ferrous hydroxide, and tne -
26 slurry is heated to a temperature between 70C and 100C
27 thus causing the formation of ferrite which is then -~
28 separated from the solution.
29 Accordingly, the present inventioz ~rovides a process
suitable for treating ferrous sulphat~ olutions in order
31 to obtain ferrite exhibiting an equiaxial morphology with
32 a narrow particle size distribution.
33 VIII. AmPhiPathic Organic Compounds
34 The amphipathic organic compounds which can be used in -
35 formin,g a liposome composition comprising the wa~e ;~
.
.
a
W093/26019 PCT/US93/05~
.,
-68-
,,
1 absorbing magnetic core particle may be selected from a
2 variety of organic compounds which contain both a
3 hydrophobic and hydrophilic moiety. According to one
4 important aspect of the invention, it has been discovered
5 that the hydrophilic moiety is adsorbed or coordinated -
6 onto the surface of the wave adsorbing magnetic core ~,;J"`~
7 particle, whereas the hydrophobic moiety of the molecule
8 extends outwardly to associate with amphipathic vesicle
9 forming lipid compounds. ~
10 When the wave absorbing magnetic core particle is `--
11 freshly made Fe3O4, it has been found, as reported in U.S. ;~
12 Patent Application Serial No. 894,260, filed June 8, l99Z,
13 that surface trivalent elements of the core particle ~`
14 contain imperfections which makes them available for -
15 direct covalent attachment with organometallic compounds -
16 of the formula Ti(OR)4, wherein R is an alkyl group. --
17 Accordingly, the wave absorbing magnetic core particle can -`~
18 be coated with an organometallic coating covalently bonded ~`
19 to said particle wherein the bonding does not depend upon
20 hydroxy functionality on the surface of said particle. ;
21 Such coated particles can then be associated with an
22 amphipathic vesicle forming lipid.
23 Preferred amphipathic organic compounds include fatty ;
24 acids selected from the group consisting of oleic, -~
25 stearic, linoleic, lir.olonic, palmitic, myristic and -i
26 arachidonic acid. -`-
27 IX. Amphipathic Vesicle Forminq Lipid
28 The lipid components used in forming the wave
29 absorbing magnetic core particle liposomes of the `~
invention may be selected from a variety of vesicle
31 forming lipids, typically including phospholipids, such as
32 phosphatidylcholine ~PC), phosphatidic (PA),
33 phosphatidylinositol (Pl), sphinogomyelin (SM), and the
i4 glycolipids, such as cerebroside and gangIiosides. The
35 selection of lipids is guided by consideration of liposome -
36 toxicity and biodistribution and targeting properties. A
:"'
~.``'
WO93~6019 2 i :~ 7 ~ 4 ~ PCT/US93/0559~ ~
; -69-
~
1 variety of lipids having selected chain compositions are
2 commercially available or may be obtained by standard ,~`?~
3 lipid isolation procedures. See, e.g. U.S. Patent No. ?
4 4,994,213. ;
5 The lipids may be either fluidic lipids, e.g. ~,
6 phospholipids whose acyl chains are relatively ``~~
7 unsaturated, or more rigidifying membrane lipids, such as ~
8 highly saturated phospholipids. Accordingly, the vesicle ;~^-
9 forming lipids may also be selected to achieve a selected
10 degree of fluidity or rigidity to control the stability of ~-
11 the liposome in serum. See, e.g. U.S. Pat. No. 5,013,556.
12 In a preferred embodiment, the vesicle forming lipid `~
13 include those phospholipids in which the polar-head-group `-
14 region is modified by the covalent attachment of
15 polyalkylene ether polymers of various molecular weights.
16 The attachment of the relatively hydrophilic polyalkylene
17 ether polymer, particularly polyethylene oxide, alters the `;~
18 hydrophilic to hydrophobic balance within the phospholipid ~
19 in order to give unique solubility to the phospholipid ~`
20 compound in an aqueous environment. See, e.g. U.S. Pat.
21 No. 4,426,330. The polyalkyl ether lipid is preferably
22 employed in the wave absorbing magnetic core particle
23 liposome composition in an amount between about 1-20 mole
24 percent, on the basis of moles of derivatized lipid as a
25 percentage of total moles of vesicle-forming lipids. The `
26 polyalkylether moiety of the lipid preferably has a !,-`
27 molecular weight between about 120-20,000 daltons, and -`
28 more preferably between about 1000-5000 daltons.
2~ In yet another em~odiment of the present invention,
30 phenyl lipid compounds (as reported in U.S. Application
31 Serial No. 958,646) can be employed as amphipathic vesicle
32 forming lipid components. These phenyl lipids have the
~ ; 4J
WO93/26~19 PCT/US93/05
-70-
i ,:
~.
1 structural formula: -~
2 ~-
3 R3
6 Fi2 ~ (F~4)n~CH3 :~
7 \ /
8 >=/ `.
9 / .",
1o R1 "~j~
11 wherein two of Rl, R2 and R3 represent a saturated or
12 unsaturated straight-chain or branched chain hydrocarbon ~-
13 group, the other being hydrogen, thereir providing at
14 least two hydrocarbon chains attached to the phenyl
moiety, wherein the two hydrocarbon chains are typically
16 between about 14-22 carbon atoms in length, and have `
17 varying degrees of unsaturation. R4 represents the
18 repeating unit of either a poly(alkylene oxide) polymier,
1~ preferably ethylene, propylene and mixtures thereof, or ~
20 the repeating unit of poly(~inyl alcohol), or a -
21 polycarbohydrate. The number of alkylene oxide or vinyl
22 alcohol groups in the polymer, designated as n, may vary ~;
23 from 0 to about 200 or more.
24
X. Pre~arlng the Wave Absorbin~ Magnetic Core
`26 Particle LiPosome ComPosition i~
27 One preferred method for producing the wave absorbing ~-`
28 magnetic core liposome composition begins with first ~;
29 coating the magnetic particles described above in Section
II with an amphipathic organic compound which contains
31 both a hydrophillic and hydrophobic moiety. For example,
32 fatty acids, such as oleic acid, linoleic acid or
33 linolenic acid, dispersed in an organic solvent, are
34 directly added to the particles at a ratio of dry
ferrite:acid equal to 2:I weight percent. After
36 mechanically milling the mixture for 1 to 1.5 hours on a
wo 93/26019 ? 1~ ~ 7 1 ~ 5 PCT/US93/OS~95
-71-
1 ball mill with 4 mm glass media, the acid coated particles
2 collapse around the media allowing for easy removal Df
3 water without the loss of the particles. The coated -~
4 particles are then dispersed in an organic solvent by
5 addition of 700 ml of hexane, toluene or chloroform and -
6 mechanically milling with glass media overnight (15 hrs).
7 Absorbing a phospholipid onto the fat vT acid coated
8 particles was accomplished by addition o- ~nthetic
9 polyethylene glycol terminated phosphati ~hanolamine
10 to the above dispersion and mechanically ~g for 3 ~`
11 hours. The ratio of fatty acid:pure lip ab~
12 weight percent. -~
13 To transfer the particles from an org E ,e t-
14 aqueous phase, 7 mls of the dispersion was aced `~
14 ml glass vial with 3 ml of distilled wa~er. T _i
16 was placed in warm, 35C sonicating water bath with
17 bubbling through it to evaporate the solvent. Once the
18 solvent has evaporated, the aqueous dispersion was then
19 suspended in a total of 10 mls of autoclaved water,
sonicated for one hour, and centrifuged for 5 minutes.
21 The supernatant was removed and brought to 20 mg
22 particle/ml solution with autoclaved water. ,~-
23 `;``
24 XI. ~ y ~`~
.
The targeted wave absorbing magnetic core liposome may
26 be prepared to include ferrites useful as cancer
27 chemotherapeutic agents. In one method of synthesis, the
28 magnetic core liposomes are prepared to include PEG-PE and
29 PG on the liposome backbone to aid in targeting to
30 specific areas and to avoid RES uptake. ;~
31 Magnetic liposome compositions are also useful for ,-
32 radio-imaging or MRI imaging of solid tumor regions prior
33 to EM wave exposure and cell destruction. The magnetic
34 liposomes are prepared with encapsulated radio-opaque or
particle-emission metal oxides or ferrites which
WO93/26019 ~ 1 3 7 1 q 5 PCT/US93~05 ~
` .
-72~
1 substantially prevents permeation through the magnetic
2 liposome bilayer.
3 In sti~l another application, the magnetic liposome
4 , composition is designed to enhance uptake of circulating
cells or other blood-borne particles, such as bacteria,
6 virus-infected blood cells and the like. Here the long- ~
7 life masnetic liposomes are ?repared to include surface- ~-
8 bound ligand molecules, as above, which bind specifically
9 and with high affinity to the selected blood-borne cells.
Once bound to the blood-borne particles, the magnetic
11 liposomes can be exposed to EM fields for specific cell or
1~ ~irus destruction.
13 Other objects and advantages of this invention will
14 become apparent upon consideration of the following ~`
15 working examples. ;`~
16 `
17 EXAMPLE 1 - !`
18 Pre~aration of Absor~inq Ferrite by Thermal Processes ~-
19 A mixture consisting of nic~el oxide (NiO), zinc oxide
(ZnO), ferric oxide (Fe2O3) was mixed in a muller for 1
21 hour. The resulting powder was then screened through a 20
22 mesh screen. The powder was then treated in an oven at
23 350 degrees C. for 48 hours. The powder was then sintered
24 at 1260 degrees C. in contact with air for 24 hours, and
then cooled to room temperature o~er a period of 24 hours.
26 Powders of different compositions were manufactured by -~
27 varying the ratio of nickel oxide and zinc oxide in --
28 accordance with the relationship NiOxZnO-Fe2O4 where x is
29 varied between 0.3 and 1Ø Frequency range absorbances
are specified for some of the compositions in the
31 following table.
'~
"
.,
~V0 93~26019 2 1 3 7 ~ 4 5 PC~r/US93/0559~ ~;
_73- ;
':
1 Table 1
3 COMPOSITION (X) FREQUENCY RANGE (mHz) LABSOR8ANCE ;~
NiOZnOFe2O4 .3 55 - 105 89
7 NiOZnOFe304 .6 145 - 1040 66 ~`~
9 NiOZnOFe2O4 .9 530 - 2750 105 ;~
12 EXAMPLE 2
13 Pre~aration of Ferrite ~v Hvdroxide Gel Process ;
: :.
14 .148 moles of FeC13 was dissolved in 50ml distilled ;
water then precipitated with 150ml of lM NaOH. .037 moles
16 of FeC12:4H2O was dissolved in 50 ml distilled water then ~
17 precipitated with 25ml of lM NaOH. .0185Moles CaC12 was ~ -
18 dissolved in 50ml distilled water and precipitated with `--~
19 25ml of lM NaOH. .0185 moles ZnC12 was dissolved in 50ml
20 distilled water and precipitated with 25ml of lM NaOH. ,--
21 All four precipitated solutions were added together in a -
22 large beaker and mixed vigorously for foùr min. in an `
23 industrial blender. The resulting gel was heated at 90
24 degrees C for 6 hours. 2 was bubbled through the
solution for the entire 6 hours.
26
27 EXAMPLE 3 ;`~-
28 Pre~aration of Ferrite bv H~droxide Gel Process
29 .148 moles of FeC13 was dissol~ed in 50 ml distilled ;
water then precipitated with 150ml of lM NaOH. .037 moles -~
31 of FeC12:4H2O was dissolved in 50ml distilled water then ~-~
32 precipitated with 25ml of lM NaOH. .037 Moles MnC12 was
33 dissolved in 50ml distilled water and precipitated with ,f-
34 25ml of lM NaOH. All three precipitated solutions were !: ~ `
added together in a large beaker and mixed vigorously in a
36 blender for four min. The resulting gel was heated at 90 ,
37 degrees C for 6 hours. 2 was bubbled through the `~
38 solution for the entire 6 hours.
39
WO93/26019 ~ 1~ 7 1 ~ ~ PCT/US93/05~
-74- -
, '.:
1 EXAMPLE 4
2 PreDaration of Ferrite bY Hvdroxide Gel Process ;~
~ .~
3 .148 moles of FeC13 was dissolved in 50ml distilled
4 water then preci~itated with 100ml of 0.lM LiOH. .037 -
moles of FeC12:4H2O was dissolved in 50ml distilled water
6 then precipitated with 25ml 0.lMLiOH. Both precipitated `~
7 solutions were added together in a large beaker and mixed
8 vigorously for four min. The resulting gel was heated at i
9 90 degrees C for 6 hours. 2 was bubbled through the l`;
solution for the entire 6 hours.
11
12 EXAMP~E S ;
13 Pre~aration of Ferrites from Hvdroxide Gels - l-
14 A reactor provided with a heat exchange coil~ and a ``
radial stirrer, was fed with 3600 ml of ferrous~sulphate
16 solution having a concentration of 40 glliter.
17 Subseguently, 290 ml of ammonia solution (200 glliter of
18 NH3) were added thereto, whiie stirring at 100 rpm. Such
I9 stirring was carried on throughout the first step. Air
was blown into the reactor at a flow rate of 100 cc/hr.
2~1 ~and the temperature was kept at about 30 Deg. C by cooling ~
22 the heat exchange coi} with water. The first step of the ~`:``?
23 reaction was concluded when the pH value decreased to 3.5
24 and the platinum electrode, with respect to the calomel ;`~
electrode, indicated +llOmV. This occurred about 7 hours
26~ after the beginning of the flowing in of air.
,~ ~
27 The analysis of the slurry was as follows: :~
28 Fe++ = ll.lglliter; Fe - 37.1 g/liter. ~-
29 160 ml of a ferrous sulphate solution (63.5 g/liter of `~`
Fe++) were admixed with the slurry. After this `
31 adjustment, the analysis of the slurry was as follows:
32 Fe+l = 13.1 g/liter; Fe = 38.5 g/liter, the FeII/FeIII
33 ratio being 0.52.
34 The rëactor was fed with 155 ml of an ammonia solution ``
(195 g/liter of NH3) with stirring at 110 rpm. This
36 stirring was continual throughout the second step. The
' ':'
~'~
,~
W093/26019 2 1 ~ 7 1 4 ~ PCT/US93/05~95 ;
1 temperature was brought to 90 degrees C. by conveying
2 steam into the heat exchange coil, and the temperature was
3 kept constant by means of a thermostat. During the
4 reaction the pH value decreased from 8 to about 6.5. The
second step of the reaction was terminated when the redox
6 potential rose from -700 to about -450 mV. This occurred
7 about 3 hours from the beginning of the heating. At the `,`
8 end, the ferrous iron present as Fe(0~)2 was 0.34 g//liter
9 of FeII. The slurry was acidified to a pH value = 4 to
10 remove ferrous hydroxide. The magnetic particles, once ~
11 filtered, washed and dried, exhibited the following l~-
12 characteristics~
13 Morphology Cubic
14 Average Diameter
dlO 0.182
16 Numerical variancy
17 Coefficient 22.0%
18 Mg content 0.04%
19 S content 0.61% -
Specific surface 6.52 m2/g
...
21 magnetization 5680 G/domain
22
23 EXAMPLE 6
24 PreDaration of Oleic Acid Coated Ma~netic Particles
Wave absorbing magnetic particles, coated with olèic acid
26 were prepared using the ferrites prepared in Examples 1-5. `~
27 The ferrite powder is dispersed in a beaker with `
28 approximately 1500 cc distilled water, adjusted to a
29 concentration of approximately 10 wt k and stirred with a
paddle stirrer for about 5 minutes. The beaker containing
31 the ferrite slurry is then placed onto a permanent magnet, t
- 32 separating the wave absorbing magnetic particle from the
33 aqueous salt waste solution. After resting the slurry on
34 the magnet for 5 minutes, the aqueous salt solution is
decanted. The precipitate is then resuspended by
36 agitation in an additional 1500 cc of fresh distilled -~
W093/26019 ~ 4~ PCT/U593/O5l'-
;-76- ;~
l water. After the final water wash is decanted, the
2 particles are suspended in acetone and the above washing
3 procedure is repeated 5 times. The particles are then
4 washed with hexane a total of five times each in the above
manner.
6 Oleic acid is added to the magnetic particle/hexane
7 slurry in a ratio of 2:1 oleic acid:dry particle. The !~`!;,~
8 mixture is adjusted to 15h total solids with hexane and
9 milled overnight on a mechanical jar roller in a glass jar
half filled with 3mm stainless steel balls.
11 The samples were labeled l-5 to correspond to the
12 ferrites prepared in Examples i-s .
13
14 EXAMPLE 7
PreDaration of Phenvl Li~id --~
16~ A. Synthesis of a m-isophthalic acid based phenyl
17 lipia.
18 The starting material for this synthesis if
19 5-Aminoisophthalic acid. The S-aminoisophthalic acid is
20 not soluble in dioxane alone. It is soluble in a mixture ,~i
21 of dioxane and triethylene glycoI. 5-aminoisophthalic
22 acid (145 mg) was dissolved in 5 ml. of dioxane and 2 ml.
23 of~triethylene glycol, and the pH was adjusted to lO with ~`
24~ NaOH. Methoxypolyoxyethylene imidazoly carbonyl, average ;~
25 mol. wt. 5,000 from Sigma (2.0g) was dissolved in 2ml of
26 H20, l-Oml of lN Na2C03, and 2.0 ml of triethylene glycol. r~ r3
27 This solution was added to the 5-aminoisophthalic acid
28 solution and stirred for 36 hours at room temperature.
29 The reaction mixture was then dialyzed overnight against 2 `~
30 liters of H20. The dialyzed reaction mixture was m~ixed -
31 with lOOml of pyridine and the liquids removed via rotary `
32 evaporation. The resulting yellow oil was placed in the
33 refrigerator. After several days a white precipitate ~
34 formed. The precipitate contains both coupled and -~,
35 uncoupled PEG. ` `~
36 Oleyl alcohol can be coupled to the above isophthalic
- `~
Wo93/26019 2137145 PCT/US93/~559~ ~
-77- ~
.,..- ,:
1 acid derivative using thionyl chloride. The thionyl ~`
2 chloride can be used to activate the oleyl alcohol for
3 ester formation with the carboxyl groups of the ~ -
4 isophthalate. See. Fig. 2.
B. Synthesis of ortho phenyl lipids
6 The ortho analog of the phenyl lipids can be
7 synthesized starting with either 3,4 dihydroybenzaldehyde ;`-
8 or 3,4 dihydroxybenzoic acid. The aldehyde group can be `
9 coupled to an amino group by forming the Schiff's base and
then reducing it with NaBH4. Oleic acid could then be
11 coupled to the hydroxyl groups using thionyl chloride to
12 provide:
13 -~
4 C~r(OC~2C~n-NH-C~2 ~ o-Co-(c~2)rc~=c~-~cH2)
16 0-C0-(CH~rCH=CH-(CH~rCH,
17 ;~
18 3,4 dihydroxybenzolic acid could be coupled through
19 its carboxyl group to amino-terminated PEG using -
dicyclohexyl carbodiimide. Oleic acid could then be
21 coupled as above. ~
22 Since both amino and carboxyl PEG derivatives as well 1 --
23 as both oleic acid and oleylamine are available, the PEG
24 and oleic acid groups can be easily interchanged in the
above compounds.
26
27 EXAMPLE 8
28Preparation of Magnetic Li~osomes
29Usinq Phosphatidyl Choline
3010 grams of each of the oleic acid coated ferrite as
31 prepared in Example 6 were dispersed in 100 cc hèxane.
32 The phospholipid was absorbed onto the particle by
33 dissolving phosphatidyl choline (Sigma, P-3644, L-2
34 lecithin, 45% PC) into hexane with heating ~ ~ate a 1-~ - '
35 solution. The PC/hexane solution was comb th t~- `
36 magnetic particles/hexane solution at a ra: pur~
~ ~ ~t ~ ~ 4 ~
W093/26019 PCT/US93/0~^'` ;
-78-
.
1 phosphatidyl choline:oleic acid equal to 1:2 weight
2 percent. , ~
3 The solution was mixed in a glass jar ~without media)
4 on a jar roller for two hours. 50 cc of distilled water ,~
were added to the jar and mixing was continued for an
6 additional 1 hour. The jar and its contents were then `~
7 transferred to an ultrasonic bath and treated by
8 ultrasound for an additional 30-60 minutes.
9 The slurry was transferred to a 200 cc beaker and
heated on a hot plate to 100 dcg C for 10 min. From .05
11 to 50 grams of triton x-114 (Union Carbide) was added to
12 disperse the lipidized ferrite in water. A ratio of
13 triton X114:lipid particle equal to 1:6 weight percent was
14 the~optimum le~el for the dispersion.~ The dispersion was
mixed on a laboratory vortex mixer for 2 minutes and
16 placed in an ultrasonic bath (Branson 1200, VWR) for two ;:~
17 hours. The final dispersion was adjusted to 0.2% TS
18 (2mg/ml). Particles were~measured on a Nycomp laser
19 particle size analyzer and were found to be approximately
200 nm in diameter.
21
22 EXAMPLE 9
.
23 PreDaration of Ma~netic Li~osomes usinq Phenyl LiDid
24 Samples were prepared using particles from Examples
25 1-5 exactly as described in Example 8 except that phenyl i"`
26 lipids prepared in Example ~7 was used in place of PC..
27 Samples were labeled for later i.d. 6-10 to correspond ~
28 with the particles as prepared in Examples 1-5. Samples s
29 were measured for~particle size on a nycomp particle
analyzer and found to be approximately 200 nm in diameter.
31 EXAMPLE 10
32 Pre~aration of MDCK Cell Cultures
33 Upon the arri~al, ampules of CCL34, MDCX cells (NBL-2
34 canine ki~ney) from ATCC, are quickly thawed. Using a
35 sterile Pasteur pipette the contents of the ampule are `
36 transferred to a flask containing at least 10 volumes of
- WO93t26019 ~ 7~ 45 PCT/US93/05~9~
` ~ '
-79-
1 culture medium (Eagles ME~) previously adjusted to pH 7.4.
2 The cells are incubated for 24 hours~ the media is
3 withdrawn, discarded and replaced. Cells are incubated at
4 36.5 degrees C. in a CO2 incubator for approximately 7
days. Another medium change may be necessary if indicated
6 by a drop in pH or high cell concentration. ~;
7 Cells are transferred during log phase, once ;~
8 confluence has been reached. The procedure is as follows:
9 The media is wlthdrawn and discarded. A PBSA (Sml/25cm 2)
prewash is added to the flask opposite the cell monolayer.
11 To avoid disruption the cells are rinsed and the solution
12 discarded. Next, 3 ml/25 cm 2 trypsin is added to the
13 flask (opposite of cells). The flask is turned to expose
14 the cells to the trypsin for 15-30 seconds, then the
15 trypsin is discarded making sure the monolayer is not ~ "-
16 detached. The cells are incubated until the monolayer
17 will slide down the flask surface when tipped.
18 (Approximately 5-15 min.) MEM medium is used to disperse
19 the cells by repeated pipetting. Cells are diluted to
10-100 cells/ml and seeded in transwells as follows:
21 Costar 6 well transwell-COL(3418) with pore size of 3.0
22 micron and well and l.Sml of c~lture (media and cells) are
23 added to the inside of the transwell. The wells are
24 covered and incubated until the monolayer i~ established ~
25 on the membrane. The cell cultures thus p-~ ared were ~-
26 used for all further experiments. ~-
27 EXAMPLE 11 ~
28 Ferrites were prepared as described in Examples 1-5, ;~;`
29 coated with oleic acid as in Example #6 and treated with a
second layer of pheny~l lipid as described in Example #7.
31 A culture of MDCK cells were prepared as described in ~
32 Example #10. The lipid coated ferrites and uncoated ` t` ;-
33 ~bare) ferrite controls were put in contact with the MDCK ~`
34 cells grown above a colony of rat crain cancer cells
~'
W093/26019 ~ PCT/US93/OS,~
'~",:
-80
1 (neuroblastoma) as detailed in the figure below~
MDCK Cell Film
S \~\~ ~'
8 Brain Càncer Cells
~ ~;^'`v,
The sample was allowed to incubate at room temperatu~e
11 for a period of 1 hour, then exposed to a freauency o~
12 20000 mHz for 3 minutes. None of the bare ferrite were
13 permeable to the endothelial cell tMDCK) membrane and had
14 no effect on the cancer cell colony.
Ferrites as prepared in Example 1, 2, 3 and 4 rapidly
16 heated upon exposure to the EM wave and all the brain
17 cells in the culture were killed.
18 Ferrites as prepared in Sample #5 were able to cross "-~
19 the endothelial cell barrier, however, because they are
20 all iron, do not absorb EM waves and had no effect on the --~
21 neuroblastoma cells. `
~,
~, . .
'''`'
,~ .
.:,
~`~
,. '''' ,' ., ~.