Note: Descriptions are shown in the official language in which they were submitted.
.._ WO 93/25574 ~ ~ 3 _~g 3 2 PGT/US93/03625
-1-
INHIBITORS OF ANGIOTENSIN I CHYMASE(S)
INCLUDING HUMAN HEART CHYMASE
Background of the Invention
This invention relates to novel polypeptides. These compounds are useful for
treatment of hypertension, congestive heart failure, cardiac hypertrophy
including left
ventricular hypertrophy, peripheral vascular complications, diabetic
complications
including renal disease, and other degenerative disorders mediated by
angiotensin II.
Certain chymases, a subfamily of serine proteases, including human heart
chymase, human mast cell chymase, and human skin chymase (which if not all the
same, are very closely related enzymes), have been found to cleave the
naturally
occurring decapeptide known as angiotensin 1 to the octapeptide known as
angiotensin
II, without substantially degrading angiotensin II. These enzymes are termed
herein
angiotensin I chymases.
Angiotensin II is known to be a potent pressor substance, i.e. a substance
that
is capable of inducing a significant increase in blood pressure and is
believed to act
by causing the constriction of blood vessels and the release of the sodium-
retaining
hormone aldosterone from the adrenal gland. Angiotensin II is also produced
from the
decapeptide angiotensin I by the action of angiotensin converting enzyme
(ACE). The
ACE pathway is the target of a number of therapeutically useful
antihypertensive agents.
Therefore, angiotensin I chymases, including human heart chymase, provide an
ACE-independent pathway for formation of angiotensin II, and are thus
implicated as
a causative factor in certain forms of hypertension and congestive heart
failure.
Angiotensin II is also implicated as a causative agent in other diseases or
risk factors
associated with hypertension, such as cardiac hypertrophy (enlargement of the
heart),
myocardial infarction, vascular hypertrophy (proliferation of vascular smooth
muscle
cells), diabetic and nondiabetic renal disease (caused in part by hypertension
in the
kidney), and restenosis or vascular damage in blood vessels of patients
suffering from
aetheroma treated with angioplasty techniques or thrombolytics. It is a
current goal of
antihypertensive therapy to treat or prevent such damage to organs or vessels
in
addition to lowering blood pressure. Angiotensin I chymases, including human
heart
chymase, are thereby implicated as causative factors in the above-named
degenerative
disorders in addition to hypertension and congestive heart failure.
WO 93/25574 2 ~ 3,"~ . ,~,~ ; PCT/US93/0362~
_2-
One means of alleviating or preventing the above mentioned adverse effects and
diseases produced by angiotensin II is the administration of a substance
capable of
inhibiting the angiotensin I cleaving action of chymase, including human heart
chymase.
Summary of the Invention
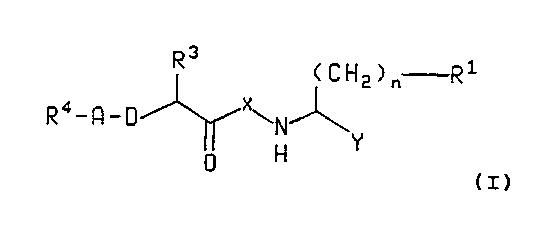
The present invention relates to compounds of the formula
3
R (CH2)n R1
Ra_R_D xwN~
i H Y
0
wherein n is one or hnro;
R' is phenyl, naphthyl, (C3-C,)cycloalkyl, unsaturated heterocycle, or benzo-
fused unsaturated heterocycle; wherein said unsaturated heterocycle is
selected from
pyrrolyl, pyrrolinyl, furyl, dihydrofuryl, thienyl, dihydrothienyl, oxazolyl,
oxazolidinyl,
isoxazolyl, isoxazolinyl, imidazolyl, imidazolinyl, thiazolyl, thiazolidinyl,
isothiazolyl,
isothiazolinyl, pyrazolyl, pyrazolinyl, triazolyl, tetrazolyl, 1,3,5-
oxadiazolyl, 1,2,4-
oxadiazolyl, 1,3,5-thiadiazolyl, 1,2,4-thiadiazolyl, pyridyl, pyranyl,
pyrazinyl, pyrimidinyl,
pyridazinyl, 1,2,4-triazinyl, 1,2,3-triazinyl, 1,3,5-triazinyl, 1,2,5-
thiadiazinyl, 1,2,5-
oxathiazinyl, and 1,2,6-oxathiazinyl; wherein said benzofused-unsaturated
heterocycle
is selected from benzoxazolyl, benzothiazolyl, benzimidazolyl, thianaphthenyl,
isothianaphthenyl, benzofuranyl, isobenzofuranyl, chromenyl, isoindolyl,
indolyl,
indazolyl, isoquinolyl, quinolyl, phthalazinyl, quinoxalinyl, quinazolinyl,
cinnolinyl and
benzoxazinyl; wherein each of said phenyl, naphthyl, unsaturated heterocycle
and
benzofused unsaturated heterocycle may optionally be substituted with from one
to
three substituents, said substituents being independently selected from bromo,
chloro,
fluoro, (C,-C5)alkyl, (C,-C5)alkoxy, (C,-C5)alkylthio, (C,-C5)alkylamino, (C,-
C,)alkylsulfonyl, (C,-C5)dialkylamino, hydroxy, amino, vitro, cyano,
trifluoromethyl,
O O O 0
II II II II
-CO(C,-C5)alkyl, -CNHz, -CNH(C,-C5)alkyl, -CN-di(C,-C5)alkyl, and formyl; and
wherein
said (C3-C~)cycloalkyl may optionally be substituted with from one to three
substituents,
WO 93/25574 ~ ~ ~ ~ PCT/US93/03625
-3-
said substituents being independently selected from bromo, chloro, fluoro, (C,-
C5)alkyl,
(C,-C5)alkylthio, (C,-C5)alkoxy, hydroxy, trifluoromethyl and oxo (O=);
R' is (C,-C5)alkyl, (C3 C~)cycloalkyl, (C3-Ca)cycloalkyl(C,-C5)alkyl, (C,
C6)alkoxy(C,-Cz)alkyl, (C,-C5)alkylthio(C,-C2)alkyl, phenyl, unsaturated
heterocycle,
phenyl(C,-Cz)alkyl, or unsaturated heterocycle(C,-Cz)alkyl; wherein said
unsaturated
heterocycle is as defined for R'; wherein said unsaturated heterocycle(C,-
Cz)alkyl is an
unsaturated heterocycle moiety as defined in R', wherein any one of the carbon
atoms
of said unsaturated heterocycle moiety is substituted with (C,-Cz)alkyl;
wherein said (C,-
C5)alkyl, (C3-Ca)cycloalkyl(C,-C5)alkyl and (C3-Ce)cycloalkyl may optionally
be
substituted with one or more fluorine atoms; wherein each of said phenyl,
unsaturated
heterocycle, phenyl(C,-CZ)alkyl, and unsaturated heterocycle(C,-CZ)alkyl may
optionally
be substituted on the ring atoms with from one to three substituents, said
substituents
being independently selected from the functionalities set forth in the
definition of R' for
the substituents on said phenyl;
R4 is selected from the functionalities listed in groups (a) - (d) below:
a) piperazino, piperidino, pyrrolidino, 3-azabicyclo[3.1.0)hex-3-yl and
azetidino,
wherein any of the carbon atoms of said piperazino may optionally be
substituted with
one or two substituents, said substituents being independently selected from
(C,-
C5)alkyl, (C,-C5)alkoxy(C,-C3)alkyl, hydroxy(C,-C3)alkyl, (C,-C5)alkylthio(C,-
C3)alkyl,
amino(C,-C3)alkyl, (C,-C5)alkylamino(C,-C3)alkyl, and (C,-C5)dialkylamino(C,-
C3)alkyl;
wherein the nitrogen in the 4 position of said piperazino may optionally be
substituted
with (C,-C5)alkyl, (C,-C5)alkoxy(Cz-C4)alkyl, hydroxy(Ci C4)alkyl, amino(C2
C4)alkyl, (C,-
C5)alkylamino(Ci C4)alkyl, (C,-C5)dialkylamino(CZ-C4)alkyl, and 2,2,2-
trifluoroethyl;
wherein any of the carbon atoms of said piperidino, pyrrolidino, 3-
azabicyclo[3.1.0]hex-
3-yl and azetidino may optionally be substituted with one or two substituents,
said
substituents being independently selected from chloro, bromo, fluoro, hydroxy,
(C,-
C5)alkyl, amino(C,-C,)alkyl, (C,-C5)alkylamino(C,-C3)alkyl, (C,-
C5)dialkylamino(C,-
C3)alkyl, (C,-C5)alkoxy(C,-C3)alkyl, (C,-C5)alkoxy, (C,-C5)alkoxy(C,-
C3)alkoxy, amino,
(C,-C5)alkylamino, (C,-C5)dialkylamino, (C,-C5)alkylthio, oxo (O=),
unsaturated
heterocycle, azetidino, pyrrolidino, piperidino, morpholino, 4-oxopiperdino, 4-
hydroxypiperidino and piperazino, wherein the nitrogen in the 4 position of
said
piperazino may optionally be substituted with (C,-C5)alkyl, (C,-C5)alkoxy(CZ
C4)alkyl,
hydroxy(C~-C4)alkyl, amino(Cz-C4)alkyl, (C,-C5)alkylamino(CZ-C4)alkyl, (C,-
WO 93/25574 ~ ~ ~ PCT/US93/03625
a
;.
C5)dialkylamino(Cz-C4)alkyl, or2,2,2trifluoroethyl; wherein said unsaturated
heterocycle
is as defined in R' ; wherein said unsaturated heterocycle may optionally be
substituted
with from one to three substituents independently selected from the
functionalities set
forth in the definition of R' for the substituents on said unsaturated
heterocycle;
b) 4-morpholino, 4-thiomorpholino, 1-oxothiomorpholino, or 1,1-
dioxothiomorpholino; wherein any of the carbon atoms of said 4-morpholino, 4-
thiomorpholino, 1-oxothiomorpholino, and 1,1 dioxothiomorpholino may
optionally be
substituted with one ortwo substituents, said substituents being independently
selected
from (C,-C5)alkyl, (C,-C5)alkoxy(C,-C3)alkyl, hydroxy(C,-C3)alkyl, (C,-
C5)alkylthio(C,-
C3)alkyl, amino(C,-C3)alkyl, (C,-C5)alkylamino(C,-C3)alkyl and (C,-
C5)dialkylamino(C,-
C3)alkyl;
c) (C,-C,)alkyl or (C3-C,)cycloalkyl; wherein said (C; C,)cycloalkyl may
optionally be substituted with from one to three substituents, said
substituents being
independently selected from halo, hydroxy, (C,-C5)alkoxy, (C,-C5)alkoxy(C,-
C3)alkyl,
hydroxy(C,-C3)alkyl, (C,-C5)alkylthio(C,-C3)alkyl, amino(C,-C3)alkyl, (C,-
C5)alkylamino(C,-C3)alkyl,(C,-CS)dialkylamino(C,-C3)alkyl(C,-C5jalkoxy(C,-
C3)alkyloxy,
amino, (C,-C5)alkylamino, (C,-C5)dialkylamino, (C,-C5)alkyfthio, azetidino,
pyrrolidino,
piperidino, piperazino, 4-(C,-C5)alkylpiperazino, morpholino, thiomorpholino,
oxothiomorphilino, dioxothiomorpholino, 4-oxopiperidino, 4-hydroxypiperidino,
and
unsaturated heterccycle, wherein said unsaturated heterocycle is defined as in
R' ;
wherein said unsaturated heterocycle may optionally be substituted with from
one to
three substituents independently selected from the functionalities set forth
in the
definition of R' for the substituents on said unsaturated heterocycle; wherein
said (C,-
C,)alkyl may optionally be substituted with one to three substituents, said
substituents
being independently selected from halo, hydroxy, (C,-C5)alkoxy, (C,-
C5)alkoxy(C,-
C3)alkyloxy, amino, (C,-C5)alkylamino, (C,-C5)dialkylamino, (C,-C5)alkylthio,
azetidino,
pyrrolidino, piperidino, piperazino, 4-(N)-(C,-C5)alkylpiperazino, morpholino,
thiomorpholino, oxothiomorpholino, dioxothiomorpholino, 4-oxopiperidino, 4-
hydroxypiperidino, and unsaturated heterocycle; wherein said unsaturated
heterocycle
is defined as in R'; wherein said unsaturated heterocycle may optionally be
substituted
with from one to three substituents independently selected from the
functionalities set
forth in the definition of R' for the substituents on said unsaturated
heterocycle;
d) (R5E)-, wherein E is oxygen, -NH or -N(C,-C5)alkyl, and wherein R5 is (C,-
.. WO 93/25574 ~ ~ ~ ~ '3 ~ PCT/US93/03625
-5-
O
C5)alkyl, 2,2,2 trifluoroethyl, R'(Cz-C4)alkyl, R'C(CZ-C4)alkyl,
O
R'C-N-(CZ-C4)alkyl, unsaturated heterocycle(Cz-C4)alkyl, amino(CZ C4)alkyl,
(C,-C5)alkyl
(C,-Cs)alkylamino(CZ-C4)alkyl, (C,-C5)dialkylamino(CZ-C4)alkyl, (C,-
C5)dialkylamino(Cz-
C,)alkyl, (C,-C5)alkoxy(Cz-C4)alkyl or hydroxy(CZ-C4)alkyl; wherein said
unsaturated
heterocycle(CZ-C4)alkyl is an unsaturated heterocycle moiety as defined in R'
, wherein
one of the ring atoms of said unsaturated heterocycle moiety of said
unsaturated
heterocycle(CZ C4)alkyl so defined is substituted with (CZ-C4)alkyl; wherein
said
unsaturated heterocycle(CZ-C4)alkyl may optionally be substituted on any of
the ring
atoms with from one to three substituents independently selected from the
functionalities set forth in the definition of R' for the substituents on said
unsaturated
heterocycle;
R' is azetidino, pyrrolidino, piperidino, piperazino, 4-(N)-(C,-
C5)alkylpiperazino,
thiomorpholino, oxothiomorpholino, dioxothiomorpholino or morpholino;
A is carbonyl or sulfonyl;
D is NH, N(C,-C5)alkyl, CHI, oxygen, CH(OH), or CH-O-(C,-C5)alkyl;
X is proline, 2-piperidinecarboxylic acid or 2-azetidinecarboxylic acid,
wherein
said proline, 2-piperidinecarboxylic acid and 2-azetidinecarboxylic acid may
optionally
be substituted with one or two substituents, said substituents being
independently
selected from bromine, chlorine, fluorine, (C,-C5)alkyl, (C,-C3)alkoxy, oxo,
and hydroxy;
O
Y is BFZ, B(OM)z, -C-Z or -C(OH)ZZ, wherein M is hydrogen, or (C,-C5)alkyl,
wherein the two M substituents may optionally form, together with the boron
atom and
the two oxygen atoms to which they are attached, a saturated heterocyclic ring
containing the boron atom, 2 oxygen atoms and 2 or 3 carbon atoms, and wherein
any
of the carbon atoms of said heterocyclic ring may optionally be substituted
with one or
two (C,-C5)alkyl groups;
WO 93/25574 ~ 1 ~ ~ 8 PCT/US93/03625
-g-
O O O
Z is selected from CFZR", CFZC-N-R'Z, -C-N-R'Z, -C-O-R'Z or a
i i
R,s R,a
heterocycle selected from 2-oxazolyl, 2-thiazolyl, 2-imidazolyl, 2-thienyl, 2-
furyl, 2-
pyrrolyl, 5-tetrazolyl, 2-benzothiazolyl, 2-benzoxazolyl, 2-benzimidazolyl, 2-
benzofuryl,
2-benzothienyl and 2-indolyl; wherein said heterocycle may optionally be
substituted
with one to three substituents, said substituents being independently selected
from (C,-
C,)alkoxy, bromo,
O O
chloro, fluoro, (C,-C3)alkyl, hydroxy, amino, vitro, cyano, -CO(C,-CS)alkyl, -
CNHZ,
formyl, (C,-C5)alkylthio, (C,-C5)alkylamino, -CF3, (C,-C4)alkyl-S02 ,
trifluoromethyl, and
(C,-C5)dialkylamino;
R" is hydrogen, fluorine, (C,-C5)alkyl, (C,-Ca)perfluoroalkyt, amino (C,-
C5)alkyl,
(C,-C5)alkylamino(C,-~5)alkyl, di(C,-C5)alkylamino(C,-C5)alkyl, (C,-
C5)alkoxy(C,-C5)-
alkyl or hydroxy(C,-C5)alkyl;
R'Z and R'3 are independently selected from hydrogen, (C,-C5)alkyl, (C3-
C5)alkenyl, and R'(Cz-C4)alkyl, wherein R' is defined as above;
with the proviso that (a) no carbon sipha to a ring nitrogen in the
substituent R'
may be directly bonded to a halogen, oxygen or nitrogen substituent, (b) when
X is
substituted proline, 2-piperidinecarboxylic acid or 2-azetidinecarboxylic
acid, then no
fluorine, oxo, (C,-C3)alkoxy or hydroxy substituent is present on either of
the ring
carbon atoms adjacent to the nitrogen atom of said proline, 2-
piperidinecarboxyfic acid
or 2-azetidinecarboxylic acid, and (c) the compound of formula I can not be
defined as
a compound wherein n is one, R' is phenyl, R3 is phenyl(C,-CZ)alkyl, R4 is
(R5E)-
wherein E is oxygen and R5 is (C,-C5)alkyl, A is carbonyl, D is NH, X is
proline and Y
is B(OM)Z.
Preferred compounds of formula I are those wherein:
n is 1;
R' is phenyl or (C3-C,)cycloalkyl, wherein said phenyl or (C; C,)cycloalkyl
may
optionally be substituted as defined in R' of formula I above;
WO 93/25574 1 '~ ~ PCT/US93/036Z5
-7-
R3 is (C,-C5)alkyl, hydroxy(C,-C5)alkyl, (C,-C5)alkoxy(C,-CZ)alkyl, (C,-
C5)alkylthio(C,-CZ)alkyl, phenylmethyl, 4-imidazolYlmethyl or 4-
thiazolYlmethyl; wherein
any of the carbon atoms of said (C,-C5)alkyl may optionally be substituted
with one or
more fluorine atoms; and wherein from one to three carbon atoms of the phenyl
moiety
of said phenylmethyl may optionally be substituted with any of the
functionalities set
forth in the definition of R' of formula I for the substituents on said
phenyl;
R4 is selected from the functionalities listed in groups (a)-(d) below:
a) piperidino, pyrrolidino, 3-azabicyclo[3.1.0]hex-3-yl and azetidino; wherein
the nitrogen in the 4-position of said piperazino may optionally be
substituted with any
of the functionalities set forth in the definition of R4(a) of formula I for
the substituents
on the nitrogen in the 4-position of piperazino; wherein any of the ring
carbon atoms
of said piperazino, piperidino, pyrrolidino, 3-azabicyclo[3.1.0]hex-3-yl and
azetidino may
optionally be substituted with one or two substituents, said substituents
being
independently selected from (C,-C5)alkyl, amino (C,-C3)alkyl, (C,-
C5)alkylamino(C,-
C3)alkyl,(C,-C5)dialkylamino(C,-C3)alkyl,hydroxy,oxo(O=),(C,-Cfi)alkoxy(C,-
C3)alkoxy,
amino, (C,-C5)alkylamino, (C,-C5)dialkylamino, azetidino, pyrrolidino,
piperidino,
piperazino, 4-N-(C,-C5)alkylpiperazino, morpholino, and unsaturated
heterocycle;
wherein said unsaturated heterocycle is as defined in R'; wherein said
unsaturated
heterocycle may optionally be substituted with from one to three substituents
independently selected from the functionalities set for in the definition of
R' of formula
I for the substituents on said unsaturated heterocycle;
b) morpholino optionally substituted with one or two substituents, said
substituents being independently selected from (C,-C5)alkyl, amino(C,-
C3)alkyl, (C,-
C5)alkylamino(C,-C3)alkyl, and (C,-C5)dialkylamino(C,-C3)alkyl;
c) (C,-C,)alkyl and (C,-C,)cycloalkyl; wherein said (C,-C,)alkyl may
optionally be substituted with from one to three substituents, said
substituents being
independently selected from amino, (C,-C5)alkylamino, (C,-C5)dialkylamino,
azetidino,
pyrrolidino, piperidino, piperazino, MN-(C,-C5)alkylpiperazino and morpholino;
wherein
said (C; C,)cycloalkyl may optionally be substituted with one to three
substituents, said
substituents being independently selected from amino, (C,-C5)alkylamino, (C,-
C5)dialkylamino, amino(C,-C3)alkyl, (C,-C5)alkylamino(C,-C3)alkyl, (C,-
C5)dialkylamino(C,-C3)alkyl, azetidino, pyrrolidino, piperidino, piperazino, 4-
N-(C,-
C5)alkylpiperazino, morpholino and unsaturated heterocycle, wherein said
unsaturated
213~83~
WO 93/25574 PCT/US93/03625
_g_
heterocycle is as defined above in R' of formula I; wherein said unsaturated
heterocycle
may optionally be substituted with from one to three substituents
independently
selected from the functionalities set forth in the definition of R' of formula
I for the
substituents on said unsaturated heterocycle;
d) (R5E)-, wherein E is oxygen or -N(C,-C5)alkyl, and wherein R5 is (C,
C5)alkyl, 2-(pyridyl)ethyl, di(C,-C5)alkylaminoethyl, di(C,-
C5)alkylaminopropyl, 2
O O
(I
(R'C)ethyl or 2-[R'CN(C,-C5)alkylJethyl;
R' is azetidino, pyrrolidino, piperidino, piperazino, 4-N-(C,-
C5)alkylpiperazino,
thiomorpholino, oxothiomorpholino, dioxothiomorpholino or morpholino;
A is carbonyl or sulfonyl;
D is NH, CH2 or oxygen;
X is proline;
O
Y is -C-Z or -C(OH)ZZ;
O O
II II
Z is CF3, COZR'Z, -C-N-R'Z, -CF2CN-R'2,
I I,
R, s R, 3
or a heterocycle selected from 2-oxazolyl, 2-benzoxazolyl, 2-thiazolyl, 2-
benzothiazoly!,
2-furyl, 2-benzofuryl, 2-thienyl and 2-benzothienyl; wherein said 2-oxazolyl,
2
benzoxazolyl, 2-thiazolyl, 2-benzothiazolyl, 2-furyl, 2-benzofuryl, 2-thienyl
and 2
benzothienyl may optionally be substituted with from one to three substituents
independently selected from (C,-C3)alkoxy, bromo,
O O
~~ II
chloro, fluoro, (C,-C3)alkyl, hydroxy, amino, vitro, cyano, -CO(C,-C5)alkyl, -
CNHZ,
formyl, (C,-C5)alkylthio, (C,-C5)alkylamino, -CF3, (C,-C4)alkyl-SOz-,
trifluoromethyl, and
(C,-C5)dialkylamino;
R'' and R'3 are independently selected from hydrogen, (C,-C5)alkyl, (C3-
C5)alkenyl, and R'(C2-C4)alkyl, wherein R' is defined as above;
with the proviso that (a) no carbon alpha to a ring nitrogen in the
substituent R'
may be directly bonded to a halogen, oxygen or nitrogen substituent, (b) when
X is
21~'~83~
WO 93/25574 PCT/US93/03625
_g_
substituted proline, then no fluorine, oxo, (C,-C3)alkoxy or hydroxy
substituent is
present on either of the ring carbon atoms adjacent to the nitrogen atom of
said
proline, and (c) when E is oxygen R5 is (C,-C5)alkyl; and (d) when E is N(C,-
C5)alkyl,
RS is selected
O
from 2-(pyridyl)ethyl, di(C,-C5)alkylaminoethyl, di(C,-C5)alkylaminopropyl, 2-
(R'C)ethyl
O
and 2-[R'CN(C,-C5)alkyl]ethyl.
More preferred compounds of formula I are the foregoing preferred compounds
wherein R' is cyclohexyl or phenyl; R3 is (C,-C5)alkyl, phenylmethyl, 4-
imidazolylmethyl,
or 4-thiazolylmethyl;
R' is piperazino, 4-N-(C,-C5)alkylpiperazino, morpholino, piperidino, 3-
azabicyclo[3.1.0]hex-3-yl, 2-(C,-C5)dialkylamino(C,-C3)alkylmorpholino, or (C3-
C,)cycloalkyl; wherein said piperidino and 3-azabicyclo[3.1.0]hex-3-yl may
optionally
be substituted with one or two substituents, said substituents being
independently
selected from (C,-C5)alkyl, amino(C,-C,)alkyl, (C,-C5)alkylamino(C,-C3)alkyl,
(C,-
C5)dialkylamino(C,-C3)alkyl, hydroxy, oxo, (C,-C5)alkoxy, (C,-C5)alkoxy(C,-
C3)alkyloxy,
amino, (C,-C5)alkylamino, (C,-C5)dialkylamino, azetidino, pyrrolidino,
piperidino,
piperazino, morpholino, 4-N-(C,-C5)alkylpiperazino and unsaturated
heterocycle;
wherein said unsaturated heterocycle is as defined in R' of formula I; wherein
said
unsaturated heterocycle may optionally be substituted with one to three
substituents
independently selected from the functionalities set forth in the definition of
R' of formula
I for the substituents on said unsaturated heterocycle; wherein said (C3-
C,)cycloalkyl
may optionally be substituted with from one to three substituents, said
substituents
being independently selected from hydroxy, oxo (=O), (C,-C5)alkoxy, amino, (C,-
C5)alkylamino, (C,-C5)dialkylamino, amino(C,-C,)alkyl, (C,-C5)alkylamino(C,-
C,)alkyl,
(C,-C5)dialkylamino(C,-C3)alkyl, azetidino, pyrrolidino, piperidino,
piperazino, 4-N-(C,-
C5)piperazino, morpholino, and unsaturated heterocycle; wherein said
unsaturated
heterocycle is as defined in R' of formula I; wherein said unsaturated
heterocycle may
optionally be substituted with one to three substituents independently
selected from the
functionalities set forth in the definition of R' of formula I for the
substituents on said
unsaturated heterocycle;
WO 93/25574 ~ ~ ~ ~ ~ PCT/US93/03625
-10-
A is carbonyl or sulfotiyl;
D is NH, CHz or oxygen;
X is proline;
O
~I
Y is -C-Z or -C(OH)2Z;
O O
Z is CF3, COzR'~, -C-N-R'2, -CF2CN-R'2,
R,s R,3
or a heterocycle selected from 2-oxazolyl, 2-benzoxazolyl, 2-thiazolyl, 2-
benzothiazolyl,
2-furyl, 2-benzofuryl, 2-thienyl and 2-benzothienyl; wherein said 2-oxazolyl,
2
benzoxazolyl, 2-thiazolyl, 2-benzothiazolyl, 2-furyl, 2-benzofuryl, 2-thienyl
and 2
benzothienyl may optionally be substituted with from one to three substituents
independently selected from (C,-C3)alkoxy, bromo,
O O
2o II II
chloro, fluoro, (C,-C3)alkyl, hydroxy, amino, vitro, cyano, -CO(C,-C5)alkyl, -
CNH2,
formyl, (C,-C5)alkylthio, (C,-C5)alkylamino, -CF3, (C,-C,)alkyl-S02-,
trifluoromethyl, and
(C,-C5)dialkylamino;
R'2 and R'3 are independently selected from hydrogen, (C,-C5)alkyl, (C3-
C5)alkenyl, and R'(Cz-C4)alkyl, wherein R' is defined as above;
with the proviso that (a) no carbon alpha to a ring nitrogen in the
substituent R'
may be directly bonded to a halogen, oxygen or nitrogen substituent,and (b)
when X
is substituted proline, then no fluorine, oxo, (C,-C3)alkoxy or hydroxy
substituent is
present on either of the ring carbon atoms adjacent to the nitrogen atom of
said
proline.
Specific preferred compounds of Formula I are:
N-[(1,1-dimethylethoxy)carbonyl]-L-alanyl-N-[2,3-dioxo-3-methoxy-1-
(phenylmethyl)propyl]-L-prolinamide;
N-[(1,1-dimethyethoxy)carbonyl]-L-valyl-N-[2,3-dioxo-3-methoxy-1-
(phenylmethyl)propyl]-L-prolinamide;
N-[4-[N-methylamino]piperidine-1 -carbonyl]-L-valyl-N-[3,3,3-
trifluoro-2-oxo-1 (S)-(phenylmethyl)propyl]-L-prolinamide hydrochloride;
2~~~s~z .
. . WO 93/25574 PCT/US93/03625
_11 _
N-[4-[N-methylamino]piperidine-1-carbonyl]-L-valyl-N-[3,3,3-trifluoro-2-oxo-
1 (S)-(phenylmethyl)propyl]-L-prolinamide hydrochloride;
N-[4-[N-methylamino] piperidine-1-carbonyl]-L-valyl-N-[2,3-dioxo-3-1-
methylethoxy-1 (S)-(phenylmethyl)propyl]-L prolinamide hydrochloride;
N-[4-[N-methyl amino]piperidine-1-carbonyl]-L-valyl-N-[2,3-dioxo-3-1-
methylethoxy)-1 (S)-(phenylmethyl)propyl]-L-prolinamide hydrochloride;
N-(4-oxopiperidine-1-carbonyl)-L-phenylalanyl-N-[2,3-dioxo-3-((1-
methyl)ethoxy)-
1-(phenylmethyl)propyl]-L-prolinamide; and
N-(4-oxopiperidine-1-carbonyl)-L-phenylalanyl-N-[2,3-dioxo-3-((1-methyl)
ethoxy)-1 (S)-(phenylmethyl)propyl]-L-prolinamide.
The .present invention also includes compounds of formula I wherein
a) R" is [1a, 5a, 6a]-6-(dimethylamino)-3-azabicyclo[3.1.0]hex-3-yl, A is
carbonyl, D is NH, R3 is isopropyl, X is proline, Y is C(=O)Z wherein Z is
CF3, n is 1
and R' is phenyl;
b) R4 is 4-methylpiperazin-1-yl, A is carbonyl, D is NH, R3 is isopropyl, X is
proline, Y is C(=O)Z wherein Z is CF3, n is 1 and R' is phenyl;
c) R4 is 4-methyl-1-piperazinyl, A is sulfonyl, D is CH2, R' is isopropyl, X
is
proline, Y is C(=O)Z wherein Z is CF3, n is 1 and R' is phenyl;
d) R4 is 4-(dimethylamino)piperidin-1-yl, A is CO, D is NH, R3 is isopropyl,
X is proline, Y is C(=O)Z wherein Z is CF3, n is 1 and R' is phenyl;
e) R° is 4-(methylamino)piperidin-1-yl, A is carbonyl, D is NH, R' is 2-
butyl,
X is proline, Y is C(=O)Z wherein Z is CF3, n is 1 and R' is phenyl;
f) R4 is 4-(methylamino)piperidin-1-yl, A is carbonyl, D is NH, X is proline,
0
Y is g/ , n is 1, R3 is isopropyl and R' is phenyl; or
0
g) R4 is 4-(methylamino)piperidin-1-yl, A is carbonyl, D is NH, R3 is
isopropyl, X is proline, Y is -BFZ, n is 1 and R' is phenyl;
The present invention also includes a method of treating or preventing a
disease
selected from hypertension, cardiac and left ventricular hypertrophy, coronary
artery
disease (including myocardial infarction, vascular hypertrophy, and vascular
damage
WO 93/25574 2 ~ 3 yg PCT/US93/03625
_12_
such as restenosis following angioplasty or aetheroma), diabetic renal
disease, and
non-diabetic renal disease comprising administering to a mammal, preferably a
human,
in need of such treatment a chymase inhibiting effective amount of a chymase
inhibiting
compound , or pharmaceutically acceptable salt thereof.
The present invention also includes a pharmaceutical composition for treating
or preventing a disease selected from hypertension, cardiac and left
ventricular
hypertrophy, coronary artery disease (including myocardial infarction,
vascular
hypertrophy, and vascular damage such as restenosis following angioplasty or
aetheroma), diabetic renal disease, and non-diabetic renal disease comprising
a
chymase inhibiting effective amount of a chymase inhibiting compound, or a
pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable
carrier.
The present invention also includes a method for treating or preventing a
disease selected from hypertension, cardiac and left ventricular hypertrophy,
coronary
artery disease (including myocardial infarction, vascular hypertrophy, and
vascular
damage such as restenosis following angioplasty or aetheroma), diabetic renal
disease,
and non-diabetic renal disease which comprises administering to a mammal,
preferably
a human, in need of such treatment an amount of a compound of the formula I or
a
pharmaceutically acceptable salt thereof, that is effective in treating or
preventing such
disease.
The present invention also includes a pharmaceutical composition for treating
or preventing a disease selected from hypertension, cardiac and left
ventricular
hypertrophy, coronary artery disease (including myocardial infarction,
vascular
hypertrophy, and vascular damage such as restenosis following angioplasty or
aetheroma), diabetic renal disease, and non-diabetic renal disease comprising
an
amount of a compound of the formula I that is effective in treating or
preventing such
disease, or a pharmaceutically acceptable salt thereof, and a pharmaceutically
acceptable carrier.
The present invention also includes a method for treating or preventing a
disease selected from hypertension, cardiac and left ventricular hypertrophy,
coronary
artery disease (including myocardial infarction, vascular hypertrophy, and
vascular
damage such as restenosis following angioplasty or aetheroma), diabetic renal
disease,
and non-diabetic renal disease which comprises administering to a mammal,
preferably
i 2137832
-13-
a human, in need of such treatment a chymase inhibiting amount of a compound
of the
formula I or a pharmaceutically acceptable salt thereaf.
The present invention also includes a pharmaceutical composition for treating
or preventing a disease selected from hypertension, cardiac and left
ventricular
hypertrophy, coronary artery disease (including myocardial infarction,
vascular
hypertrophy, and vascular damage such as restenosis following angioplasty or
aetheroma), diabetic renal disease, and non-diabetic renal disease comprising
a
chymase inhibiting amount of a compound of the formula I, or a
pharmaceutically
acceptable salt thereof, and a pharmaceutically acceptable carrier.
Preferred compositions comprise the foregoing preferred compounds.
The pharmaceutically acceptable salts of the present invention are those which
are non-toxic at the dosages administered. Since compounds of the invention
may
contain acidic or basic groups, acid or base addition salts are possible.
Pharmaceutically acceptable acid addition salts include, for example, the
hydrochloride,
hydrobromide, hydroiodide, sulfate, bisulfate, phosphate, acid phosphate,
acetate,
lactate, maleate, mesylate, fumarate, citrate, acid citrate, tartrate,
bitartrate, succinate,
gluconate and saccharate salts. Pharmaceutically acceptable base addition
salts are
for example sodium, potassium, calcium and magnesium salts.
All the natural amino acids contained in the structures of the compounds of
the
present invention are of the L configuration, the naturally occurring
configuration, unless
otherwise noted.
Unless otherwise indicated, the term 'alkyl', as used herein, may be straight
or
branched. The terms'di(C,-C5)alkylamino or dl(C,-C~)alkylamino', as used
herein, refer
to two alkyl groups independently selected from (C,-CS)alkyl or (C,-C,)alkyl.
Claimed in this application are (a) N-[4-(methyl-
amino)piperidine-1-carbonyl]-L-phenylalanyl-N-[3,3,3-
trifluoro-2-oxo-1(R)-(phenylmethyl)propyl]-L-prolinaminde,
(b) N-[4-(methylamino)piperidine-1-carbonyl]-L-valyl-N-
[3,3,3-trifluoro-2-oxo-1(8)-(phenylmethyl)propyl]-L-prolin-
amide, and
(c) N-[C1,1-dimethylethoxy)carbonyl]-L-alnyl-N-[2,3-
dioxo-3-methoxy-1-(phenylmethyl)propyl]-L-prolinamide,
and their pharmaceutically acceptable salts.
~w2) 3783
-l3a-
Detailed Description of the Tnvention
The compounds of the formula I may be prepared as
described in the following reaction schemes and discussion.
Unless otherwise indicated Rl, R3, R4, A, D, X, Y, Z and n in
the reaction schemes and discussion that follow are defined as
above.
WO 93/25574 213'7 8 3 2
PCT/US93/03625 -~~
-14-
Scheme 1
(CH2)n-R1
Z VIII
NH2
OH
( CH2 ) n-R1
R a-X- N H Z V I I
OH
3 (CH2)n-R1
R
R4 ~R~p X NH Z V I
0 OH
R3 (CH2>"-R1
~A~ X N H~ I
Y
0
30
WO 93/25574 ~ ~ ~ PCT/US93/03625
-15-
Scheme 2
R3
R4-R-D" 00 H X I
2
10
R3
Ra_A_D X V
0
VI
WO 93/25574 213'7 ~ 3 ~
PCT/US93/03625
-16-
Scheme 3
R1(CH2)n-CH2N02 + Z-C-OCH2CH3
I
OH
IV III
1
R1(CH2)n
Z
02N OH
R1(CH2)n
H2N Z
OH
VIII
..w. WO 93/25574 ~ ~ ~ PCT/US93/03625
_17_
Scheme 4
(CHp)n- R1
a
R NH CO~H X
(CH2)"- R1
RaNH ~ Xa
CO~R
(CH2)n- R1
RaNH CH0 X I I
(CH2)n- R1
RaNH ~~ CN ~ 1 1 I
OH
(CH~)n- R1
NHS ~ z V 1 I I
0 H
WO 93/25574
PCT/US93/03625
_ 18_
Scheme 5
( CH2 )"-R1
XII
RaNH~CHO
(CH2)"-R1
1o Z XIV
RaNH
HO
( CH2 ) n-R1
NHS Z VIII
HO
WO 93/25574
PCT/US93/03625
_19_
Scheme 6
R3
H-D~ C02Rb XV
,0
R3
a ~ CO~Rb XV I
~5 R -R-D
20 X I
WO 93/25574 PCT/US93/03625
21378~~
Scheme 7
R \t.v' R
3
b ~ b
G-R-D C02R --~~ R4-R-D C02R
XVII XVI
WO 93/25574 PCT/US93/03625
-21-
O
Scheme I refers to the synthesis of compounds of formula I wherein Y is -C-Z.
These compounds are prepared from compounds of formula VIII. The basic sub-
unit
of the prefer-ed chemical synthesis is the coupling or acylation of the
unprotected
amino group of the amine residue of a compound of formula VIII with an amino
acid
(e.g., proline) having an activated (for acylation purposes) carboxylic
function and a
suitable protecting group bonded to its own alpha-nitrogen to form a peptide
bond
between the two amino acid residues, followed by the removal of said
protecting group.
Such a coupling reaction is generally conducted at a temperature of about -30
to about 80°C, preferably about 0 to about 25°C. Examples of
suitable coupling
reagents which activate the carboxylic functionality of the amino acid are
dicyclohexylcarbodiimide/hydroxybenzotriazole (HBT), N-3-dimethylaminopropyl-
N'-
ethylcarbodiimide/HBT, 2-ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline (EEDQ),
carbonyl diimidazole (CDI)/HBT, and diethylphosphorylcyanide.
The coupling is conducted in an inert solvent, preferably an aprotic solvent.
Suitable solvents include acetonitrile, dichloromethane, chloroform, and
dimethytformamide. The preferred solvent is dichloromethane.
For a discussion of other conditions used for coupling peptides see Houben-
Weyl, Vol. XV, part II, E. Wunsch, Ed., George Theime Verlag, 1974, Stuttgart,
and
those described in M. Bodanszky. Principles of Peptide Synthesis, Springer-
Verlag,
Berlin (1984) and The Peptides. Analysis. Synthesis and BiologY (ed. E. Gross
and J.
Meienhofer), Vols 1-5. (Academic Press, New York) 1979-1983.
The coupling product of formula VII, wherein R° is any of the suitaoie
protecting
groups commonly used for amino group protection in peptide synthesis (Examples
of
such groups are carbobenzyloxy and t-butoxycarbonyl groups), is deprotected
using
conventional methods to provide a compound of formula VII wherein R° is
hydrogen.
For example:
(a) If the protecting group, of the compound of the formula VII is
carbobenzyloxy, the latter may be removed by hydrogenation with a noble metal
catalyst such as palladium or palladium hydroxide on carbon in the presence of
hydrogen. The hydrogenation is generally conducted at a temperature of about 0
to
about 100°C, preferably about 20 to about 50°C.
WO 93/25574 213 "~ PCT/US93/036Z5
-22-
(b) If the protecting group, R°, is t-butoxycarbonyl group, such group
may
be removed by acidolysis. Acidolysis may.,be conducted with HCI in dioxane or
with
neat tr'rfluoracetic acid at a temperature ~of about -30 to about 70°C,
preferably about -
to about 35°C.
5 (c) Ifthe protecting group, R°, is 9-fluorenylmethylenoxycarbonyl,
such group
may be removed by treatment with an amine base, preferably piperidine. This
reaction
may be run in piperidine as solvent at 10°C to about 100°C,
preferably at 25°C.
The compounds of formula VII (R° is hydrogen) are converted to
compounds of
formula VI by coupling, as described above, with an intermediate of the
formula R4-A-D-
CH(R')COZH (formula XI).
The compounds of formula VI are oxidized to compounds of formula I by
methods commonly known to those skilled in the art. Examples of oxidation
reactions
are the Swern oxidation and variants thereof, chromium based oxidations
(preferably
pyridinium dichromate), Pfitzner-Moffatt and modified variants thereof, and
the Dess-
Martin periodinane oxidation. A preferred method, to form the compounds of
formula
I, wherein Y is not COCF,, uses a Swem oxidation involving treatment of the
alcohol
of formula VI with oxalyl chloride, dimethyl sulfoxide and triethylamine in
methylene
chloride. In some cases, up to 10-20 equivalents of oxidizing agent is
preferred.
For all Z and especially when Z is -CF3, the more preferred oxidant is the
Dess-
Martin periodane. Four equivalents of 1,~,1,-triacetoxy-2,1-benzoxiodol-
3(3H)one is
added to a dry solution of the peptide in a non polar solvent such as
dichloromethane
and the mixture is stirred for a period of one to twenty-four hours. This
procedure is
described below as General Procedure C.
It will be understood that certain compounds of formula I contain tertiary
amine
functionality in the R4, R3, or R, moieties which, if present in a compound of
formula VI
could interfere with or be chemically reactive in the above-cited oxidation
step which
forms a compound of formula I. In such cases the Swern oxidation or
modifications
thereof, or Pfitzner-Moffatt oxidation, or modifications thereof are the
preferred methods
of oxidation, except for compounds wherein Z is CF3.
It will also be understood that some compounds of formula I contain primary or
secondary amine functional groups in the R4, R3 and R, moieties, and that
during the
synthesis of such compounds of formula I that this functionality is protected
where it
is present, in the R4, R3 or R, moieties of the intermediates of the formula
II, IV, V, VI,
WO 93/25574 213 ~ 8 3 ~, PCT/US93/03625
-23-
VIII, XI, XV or XVI. Suitable protecting groups for this purpose are those
suitable
protecting groups commonly used for amino group protection in peptide
synthesis
(such as N-tert-butoxycarbonyl, N-carbo-benzyloxy, and 9-fluorenylmethylenoxy-
carbonyl) which are also not chemically reactive under the coupling,
protection and
deprotection, or oxidation conditions described or referred to for the
synthesis of
compounds herein (for example, the oxidative conditions employed to convert
the thus-
protected intermediate of formula VI to the thus protected intermediate of
formula I).
The thus-protected intermediate of formula I is deprotected to the compound of
formula
I wherein the protecting group has been replaced by hydrogen. Methods are
commonly known to those skilled in the art and are described as above.
Examples 19
and 20 contained below illustrate the synthesis of such a secondary amine-
containing
compound of formula I wherein the intermediates of formula V and VI are
employed
having a methylamino functionality protected with the N-tart-butoxycarbonyl
protecting
group which is removed by treatment with HCI-dioxane to give a compound of
formula
I.
All compounds of formula I containing a tertiary amino functionality, in R4,
R3,
or R, including such compounds wherein Y is COF3, can also be prepared by
reductive
amination of a compound of formula I having secondary amine functionality at
the
corresponding position of R4, R3, or R" with an appropriate carbonyl compound.
A
suitable procedure for this reductive alkylation is the addition of the
appropriate
carbonyl compound (1 to 100 equivalents) to a mixture of the compound of
formula I
having a secondary amine functionality, sodium cyanoborohydride (1.4-2
equivalents),
and powdered 3 angstrom (~) molecular selves in absolute methanol at 0-
50°C,
preferably at 20-25°C. It will be clear to one skilled in the art that
amino, alkyiamino,
and dialkylamino substituted compounds of the formulae VI, V and C-terminally
protected compounds of formulae V, XVI and XI including the properly protected
variants thereof as described herein may be synthesized from the corresponding
oxo-
substituted compounds, by reductive amination of the latter with a salt of
ammonia
(e.g., NH4CI), an alkylamine, or a dialkylamine according to methods familiar
to those
skilled in the art, such as is illustrated for the conversion of an oxo-
substituted
compound of formula V to the corresponding methylamino-substituted compound of
formula V by reductive amination with methylamine in Example 19a, and
conversion of
the latter to a suitably protected R° amine-containing compound of
formula V in
WO 93/25574 PCT/US93/03625
213'83 ~
-24-
Example 19b. Thus, this reductive amination/protection method can be used not
only
for the synthesis of C-terminally protect~ii compounds of formula V, but also
for the
synthesis of amine-containing , compounds of formula VI, C-terminally
unprotected
compounds of formulae V, XI, and XVI and the properly N-protected variants
thereof
wherein the primary or secondary amine functionality is protected with one of
the
groups commonly employed for amine protection. Conditions for the introduction
and
removal of such groups are summarized by Greene in "Protecting Groups in
Organic
Synthesis', Wiley, NY, 1981.
Alternatively, compounds of formula VI may be synthesized as shown in Scheme
2, wherein a compound of formula XI is first coupled by standard peptide
coupling
methods referred to above to a C-terminally protected a-amino acid X such as,
for
example, proline benzyl ester to give a C-terminally protected compound of
formula V.
Suitable protecting groups are those commonly used for carboxyl group
protection in peptide synthesis. Examples of such groups are benzyl ester and
t-butyl
ester groups. The C-terminally protected compound of the formula V is
deprotected
using conventional methods to provide the C-terminally unprotected compound of
formula V. For example:
(a) If the carboxyl group of the compound of the formula V is protected as
a benzyl ester, the latter may be removed by hydrogenation with a noble metal
catalyst
such as palladium on carbon in the presence of hydrogen. The hydrogenation is
generally conducted at a temperature of about 0 to about 100°C,
preferably about 20
to about 50 ° C.
(b) If the carboxyl is protected as a t-butyl ester such group may be
deprotected by acidolysis. Acidolysis may be conducted with HCI in dioxane or
with
neat trifluoracetic acid at a temperature of about -30 to about 70°C,
preferably about -
5 to about 35 ° C.
(c) If the carboxyl protecting group is an alkyl ester, the group may be
removed by basic hydrolysis. Basic hydrolysis may be conducted with a suitable
base
(e.g., sodium hydroxide) at a temperature of about -30 to about 120°C,
preferably
about 0 to 80° C. The solvents used for removal of the protecting group
should be inert
solvents.
Suitable and preferred solvents are as described for the deprotection in
Scheme
1. The C-terminally unprotected compounds of the formula V so formed are then
_2~ i 2'! 37832
coupled with a compound of the formula VIII, from Scheme I by conventional
peptide
coupling reactions as described above (e.g., Procedure A described below) to
yield
compounds of the formula VI.
Scheme 3 refers to the synthesis of compounds of formula VIII wherein Z is
CF,.
Compounds of formula VIII can be prepared from compounds of formula II.
Compounds of formula II are prepared by Henry condensation (McBee, E.T., et
al. J. Amer. Chem. Soc. 78:4053 (1956) of an appropriate nitroalkane of
formula
R'(CHz)~CHzNOz (prepared by standard methods if not otherwise available) with
trifluoroacetaldehyde ethyl hemiacetal of the formula CF,CH(OH)OCHZCH, to
provide
a nitroalcohol of formula II which is obtained as a mixture of two racemic
diastereomers
[(2(RS), 3(RS) and [2(RS), 3(SR)]) (For example, see Example No. 18b).
Caution!
Similar compounds are reported to explode if distilled (US 5,055,450).
Reduction of the
vitro group in a compound of formula II with an appropriate reducing agent
affords a
compound of formula VIII as a mixture of two racemic diastereomers
([2(RS),3(RS)]
and [2(RS),3(SR)]). (For example, see Example 18c). This amine or its salt is
used
directly for further synthesis.
Attematively, compounds of formula VIII wherein Z is CF3 can also be prepared
by the method of Kolb et. al. (Vebings Ann. Chem. 1990, 1-6) as adapted by
Peet et.
al. (J. Med. Chem. 1990, 33, 394-407) which involves a) dehydration of an N-
aroyl (e.g.
N-benzoyl) amino acid derivative (Aryl)-CONH-CHCO~H with acetic anhydride, to
I
(CHz)~-R'
form an intermediate oxazolone, b) trifluoroacetylation of the resulting
oxazolone with
trifluoroacetic anhydride, c) dicarboxylation of this trifluoroacetyloxazolone
with oxalic
acid, d) reduction of the resulting trifluoromethyl ketone-(Aryl) CONH-
CH(CH2)~COCF,
(CHz),; R'
to the corresponding aroylaminotrifluoromethyl carbinol, and e) hydrolysis of
this
product to a compound of formula VIIII wherein Z is CF,. The instant step e)
above is
illustrated in Example 14a.
Compounds of formula VIII wherein Z is difluoromethyl,(C,-Ca)perfluoroalkyl
CFz(C,-CS)alkenyl and CFz(C,-C5)alkyl are also prepared by this method. The
oxazolone described in the instant step b) above is acylated with
difluoroacetic an
anhydride of the formula [(C,-C5)alkenyICFzCO]z0 as described by Kolb (ibid.,
above),
WO 93/25574 . PCT/US93/036Z5
21~~g~.2
-26-
an anhydride of the formula [(C,-C5)aIkyICF2~0]ZO or an anhydride of the
formula [(C,-
Ce)perfluoroaIkyICO]~O. These intermediates are converted to compounds of
formula
VIII by the above-cited steps c) and'd) as described by Kolb, and hydrolyzed
according
to the method of example 14A.
Compounds of formula VIII wherein Z is COz(C,-C6)alkyl are prepared as shown
in Scheme 4. Alpha-amino acids of formula X or N-protected analogs thereof if
not
commercially available may be prepared by the Strecker synthesis from an
aldehyde
of the formula R' (CHz)~CHO or by any of many literature methods, familiar to
one skilled
in the art, and esterified and protected with a suitable N-protecting group
(as described
above, eg. BOC or CBZ) to give a compound of formula Xa (R' is preferably
methyl or
ethyl). Many such compounds of formula Xa are also commercially available.
Aldehydes of formula XII are readily prepared from protected a-amino esters of
formula Xa by reduction with diisobutylaluminum hydride (DIBAH) or from
analogous
N-methoxymethylamides by reduction with lithium aluminum hydride (LAH).
Aldehydes of formula XII may be converted to cyanohydrins of formula XIII by
treating the aldehyde with a salt of cyanide, preferably potassium or sodium
cyanide,
in an aqueous solution with a cosolvent such as, tetrahydrofuran, ethyl
acetate, or
dioxane.
The cyanohydrin of formula XIII so formed can be converted by alcoholysis to
O
a compound of formula VIII wherein Z is -C-0(C,-Ce) alkyl. Alcoholysis of a
cyanohydrin of formula XIII is typically carried out by treating the
cyanohydrin with an
alcohol of the formula alkyi(C,-C8)OH with of a proton source, preferably
hydrogen
chloride gas. The protecting group is then removed (if still present) by one
of the
methods described above to give a compound of formula VIII. An illustration of
the
sequence described above wherein Boc-phenylalanine methyl ester is converted
to a
cyanohydrin of formula XIII is found in U.S. Patent 4,668,769.
An example of the conversion of a cyanohydrin of formula XIII to the
corresponding methyl ester of formula VIII with removal of protecting group
wherein Re
is BOC is Example 1 a. Other examples wherein this cyanohydrin is converted to
a
variety of lower alkyl esters of formula VIII are found in U.S. Patent
4,814,342.
WO 93/25574 ~ ' PCT/US93/03625
-27-
Compounds of formula VIII wherein Z is CONR'Z
R, 3
are prepared from compounds of formula VIII wherein Z is C02(C,-C5)alkyl by a)
protection of the nitrogen group with a protecting group R°, b)
hydrolysis of the ester
function with aqueous base as described above, c) coupling of the N-protected
hydroxy
acid to an amine of the formula HNR'2R'3 or its acid addition salt using
standard
peptide coupling methods described above (e. g. dicyclohexyl-
carbodiimide/HBT). The
protecting group R° is then removed by the methods of scheme 1 to yield
the amide
of formula VIII wherein Z is -CONR'ZR'3. Examples of synthesis of amides of
formula
VIII by this sequence are reported in U.S. Patent 4,814,342.
Alternatively, amides of formula VIII wherein Z is -CON-R' 2 may be
R, s
prepared by heating the corresponding esters of formula VIII wherein Z is
COZ(C,-
C5)alkyl with an excess of an amine of the formula R'ZR'3NH in a solvent such
as a
lower alcohol at 25-100°C, preferably in a closed system such as a
stainless steel
bomb. Suitable solvents are methanol, ethanol or isopropyl alcohol.
Compounds of formula VIII wherein Z is CFZCONR'ZR" are prepared as outlined
in Scheme 5. An aldehyde of formula XII is allowed to react with
ethylbromodifluoro-
acetate in the presence of zinc according to the procedures of Hallinan and
Fried
(Tetrahedron Lett. 1984, 25, 2301 ) or Thairivongs et al. (J. Med. Chem. 1986,
29, 2080-
7) or with ethyl bromodifluoroacetate, zinc, and titanium tetrachloride
(Hoover, U.S.
Patent 4,855,303) to yield a compound of formula XIV wherein Z is
CFZC02(CZHS). The
compound of formula XIV is then amidated by mixing with an amine of the
formula
R'~R"NH in a suitable polar, preferably erotic, solvent such as ethanol or
methanol to
give a compound of formula XIV wherein Z is CFzCONR'ZR". Removal of the amine
protecting group by the methods described in Scheme 1 yields the compound of
formula VIII wherein Z is CFZCONR' ZR".
Alternatively, the ester of formula XIV may be hydrolyzed to the corresponding
acid wherein Z is CF~COZH, and the latter amidated by coupling to an amine of
formula
R,zR,3NH by a standard peptide coupling method to give an amide of formula XIV
(Z=
CFZCONR,zR,3), which is then deprotected to yield a compound of formula VIII.
Examples of the synthesis of such compounds by these methods can be found in
WO 93/25574 ~ ~ ~ ~ '~ PCT/US93/03625
-28-
Example 12a, U.S. Patent 4,855,303, J. M~:d. Chem. 1992, 35, 2-14, and J. Med.
Chem.
1986, 29, 2080-7. By these methods, compounds of formula VIII wherein R" n,
and Z
= CFZCONR,ZR,3 as described~h~erein may be prepared.
Compounds of formula VIII wherein Z is a 2-substituted heterocycle or a benzo-
fused 2-substituted heterocycle such as 2-thiazolyl or 2-benzothiazolyl as
enumerated
above are also prepared as described in Scheme 5. Thus, an aldehyde of formula
XII
is allowed to react with a 2-metallo heterocycle to form a compound of formula
XIV
wherein Z is a 2-substituted heterocyclic moiety. Suitable solvents are inert
solvents,
preferably ether or tetrahydrofuran. The 2-metallo heterocycle is preferably a
2-lithio
heterocycle and is obtained by treatment of the parent heterocycle with a
suitable
organolithium reagent, such as n-butyllithium, methyllithium, sec-butyllithium
or tert-
butyllithium. 2-Metallo heterocycles may be alternatively prepared by
transmetallation
of a c-bromo or 2-iodo heterocycle with an organolithium reagent. Conditions
for the
formation and use of 2-metallo-heterocyclic reagents are dependent on the
particular
heterocycle. Methods for the formation of 2-metallo heterocycles are familiar
to those
skilled in the art. In general, the metallo heterocycle is formed and allowed
to react
with the aldehyde within 30 minutes at -78°C. Conditions for the
formation and
reaction of various 2-substituted heterocycles are described in Organic
Reactions
(Volume 26). Specific applications of this reaction t~ the synthesis of
compounds of
formula XIV whereir ~ Z is 2-thiazolyl or 2-benzothiazoyl are Examples 3a and
13c herein.
Compounds of formula XIV so formed are then converted to compounds of formula
VII I
by removing the protecting group by the methods described in Scheme 1, above.
Parent heterocycles having a free NH such as imidazole, tetrazole, or indole
are
N-protected with an appropriate protecting group, such as 1-ethoxyethyl or
trimethylsilylethyl prior to metallation of the heterocycle. Conditions for
introduction and
removal of these and other protecting groups suitable for this purpose are
found in
T. Greene 'Protecting Groups in Organic Synthesis", Wiley, 1981, N.Y.
Alternatively, compounds of formula VIII wherein Z is substituted or
unsubstituted 2-benzoxazolyl or substituted or unsubstituted 2-oxazolyl may be
prepared from a cyanohydrin of formula XIII and ar appropriate ortho-
aminophenol or
aminoethanol derivative, respectively, by the method of Edwards et. al. (J.
Am. Chem.
Soc. 1992, 114, 1854-1863).
.. WO 93/25574 ~ y ~ 2 PCT/US93/03625
_29_
The synthesis of compounds of formula I wherein Y is B(OM)2 is accomplished
by coupling a compound of formula V with an aminoboronic acid ester of the
formula
NHZCHB(OMZ). Compounds of the formula I wherein Y is B(OH)Z are synthesized
(CHZ)~ R'
from compounds of formula I wherein Y is B(OM)Z (e.g. M+M equal to -C(CH3)-
C(CH3)z-)
by reaction of the latter with diethanolamine to yield a diethanolamine ester
of formula
I wherein Y is B(OM)Z having M+M equal to -CHZCHzNHCHzCH~-, followed by
hydrolysis of the diethanolamine ester in the presence of the acid form of an
ion
exchange resin. An example of the synthesis of an aminoboronic ester of
formula XIV
(wherein R' (CHZ)~ is phenylmethyl) and methodology described above for the
synthesis
of the corresponding compound of formula I is described by Kettner et. al. (J.
Biol.
Chem. 1984, 259, 15106-15114).
Kinder and Katzellenbogen (J. Med. Chem. 1985, 28, 1917-1920) report that
acylamino boronic acids of the formula RCONHCH(R)B(OH)z are converted to the
difluoroboranes of the formula RCONHCH(R)BF~ upon brief exposure to excess
hydrogen fluoride (HF) in water and extraction with an organic solvent (such
as ethyl
acetate). Compounds of formula I wherein Y is BFZ may be made by the same
methods from the analogous compounds wherein Y is B(OH)Z.
Compounds of formula XI wherein A is C0; D is NH, O, or N(C,-C5)alkyl, and
R° is an N-linked moiety are prepared according to scheme 6. A carboxyl
protected a-
amino or hydroxy ester of the formula XV, is activated by treatment with a
phosgene
equivalent such as carbonyldiimidazole and condensed with an amine of the
formula
R4-H (or a protected analog thereof, if R' contains additional primary or
secondary
amine functionality) which is generally commercially available or can be
readily
prepared by literature methods familiar to one skilled in the art. The coupled
product
is then carboxyl-deprotected as described above in Scheme 2 to give a compound
of
formula XI. An example of this sequence are Examples 10a and b. Alternatively
a
compound of formula XV may be directly activated with phosgene or
trichloromethyl
chloroformate _to yield an isocyanate (Lombardino and Gerber, J. Med. Chem.
1964, 7,
97-101 ), N-carbamoylchloride, or chloroformate derivative wherein D is NH, N-
alkyl, or
O, respectively, and condensed with amine or protected amine of the formula
R4H to
yield a compound of the formula XVI.
WO 93/25574 ~ ~ ~~ ~ PCT/US93/03625
-30-
Many compounds of formula XI wherein R°, A, D, and R3 are as
described
instantly above are known in the literature and have been prepared by one of
these
methods. Compounds of formula XI wherein R', A, and D are as described above
and
R' contains a protected amine functionality suitable for the synthesis of a
compound
of formula I possessing a basic nitrogen atom in the R4 moiety by the strategy
outlined
above are also known to those skilled in the art. For example, U.S. Patent
4,814,342
describes the synthesis of intermediates of formula XI wherein R° is
morpholino, 4-
oxopiperdino, piperazino, 4-formylpiperazino, 4-methylpipera2ino,
thiomorpholino or
methylamino; A-D is CONH and R' is phenylmethyl. Additionally, European Patent
438,233 describes the synthesis of compounds of formula XI wherein R4 is
piperidino,
pyrrolidino, or azetidino optionally substituted with amino, alkylamino,
dialkylamino,
alkylaminodialkyl, or dialkylaminoalkyl, wherein the amine functionality is
suitably
protected if necessary, and wherein A is >C=O and D is oxygen or NH. Compounds
of formula XI so formed can be converted to compounds of formula I by the
methods
of Schemes 1 and 2. . This methodology as described in EP-438,233 is suitable
for
the synthesis cf other compounds of formula XI and XVI, by those skilled in
the art,
wherein R' is as described herein and D is oxygen or NH, according to Scheme
6,
when the corresponding starting material of formula XV and amine or
corresponding
amine salt of formula R°-H (or appropriately protected derivative
thereof) is employed.
European Patent 476,515 describes the preparation of a compound of formula
XVI wherein R° is 2-(2-pyridyl)ethyl(methyl)amino, A is carbonyl, D is
NH, R3 is
phenylmethyl and Rb is phenylmethyl by the methods of Scheme 6.
Compounds of formula XVI wherein R° is a nitrogen-linked
substituent or a
protected variant thereof, A is carbonyl or sulfonyl, and D is NH, and R' and
Rb are as
described herein may also be prepared according to Scheme 6. An (suitably
protected)
amine R°-H is first activated by its conversion to the isocyanate or
sulfamoyl chloride
(in the case of primary amines), or to the carbamoyl chloride or sulfamoyl
chloride (in
the case of secondary amines), and this derivative is allowed to react with a
compound
of formula XV wherein D is NH or N(C,-C5 alkyl). The reaction is conducted in
an inert
solvent such as dichloromethane, chloroform, dimethylformamide, or
tetrahydrofuran
in the presence of a tertiary amine base, preferably triethylamine or N,N-
diisopropylamine. Rosenberg et al. (EP 456,185) describes the synthesis of the
compound of formula XVI wherein R' is 4-methyl-1-piperazino, A is sulfonyl, D
is NH,
WO 93/25574 ~ ~ ~ PCT/US93/03625
-31-
and R3 and R4 are phenylmethyl by reaction of 1-methylpiperazin-4-yl) sulfonyl
chloride
(prepared by the method of Matier, J. Med. Chem., 1972, 15, 538) with
phenylalanine
benzyl ester p toluenesulfonic acid salt. Compounds of formula XI may also be
prepared by reaction of unprotected a-amino acids of formula NH2-CH(R')COZH
with
sulfamoyl chlorides. Wegler et al. (Ann. Chem., 1959, 624, 24-29) describe the
synthesis of the compound of formula XI wherein R' is morpholino, A is
sulfonyl, D is
NH, and R3 is phenylmethyl by this synthetic strategy. These methodologies are
adaptable by one skilled in the art to the synthesis of other compounds of
formula XVI
and XI wherein R' is as described herein or a protected variant thereof, A is
sulfonyl or
carbonyl, D is NH or N(C,-C5 alkyl), R° is an N-linked substituent or
protected variant
thereof as described herein.
Compounds of formula XI and XVI wherein R°-A is alkylcarbonyl,
alkoxycarbonyl
(C; C,)-cycloalkylcarbonyl, alkylsulfonyl, (C3-C,)-cycloalkylsulfonyl, and
substituted or
appropriately protected substituted variants of these five groupings and D is
NH, N-(C,-
C5)alkyl or oxygen may be prepared by the method of scheme 6 by acylation or
sulfonylation of a compound of the formula XVI with the appropriate activated
carboxylic
acid or sutfonyl chloride derivative. Acylations of this type are accomplished
via the
acid chloride R4COCI or by one of the peptide coupling methods described
above.
Sulfonylation is accomplished in an inert solvent, preferably dichloromethane,
in the
presence of a tertiary amine base, preferably triethylamine or
diisopropylethylamine.
Compounds of formula XI and XVI wherein R°A is substituted or
unsubstituted
alkoxycarbonyl may be prepared by acylation of a compound of the formula XVI
with
the appropriate chloroformate or carbonate derivatives.
Compounds of formula XVI and XI wherein A is CO and D is CH2 are
synthesized according to Scheme 7, by coupling of compounds of formula XVII
wherein
G is OH, A is CO, D is CHz and Rb is a suitable carboxyl protecting group to
an amine
or appropriately protected amine R°-H. Compounds of formula XVII
wherein G is OH,
A is CO, D is CHZ and Rb is a suitable carboxyl protecting group are prepared
by
literature methods. Plattner et al. (J. Med. Chem. 1988, 31, 2277-2288)
describe the
synthesis (Scheme V therein) of the compound of formula XVII wherein G is OH,
A is
CO, D is CHz, R3 is phenylmethyl, and Rb is phenylmethyl in the enantiomeric
form
preferred for the R3-bearing carbon in compounds of formula I herein. Other
compounds of formula XVII wherein G is OH, D is CH2, and R3 and R4 are as
described
WO 93/25574 ~ ~ 3'~ g ',~ PCT/US93/03625
-32-
herein may be prepared by this method by:one skilled in the art, starting with
a suitably
carboxyl-activated derivative of the appropriate acid R3CH2COzH and the amine
R°-H,
wherein R3 and R4 are as descrjbed herein or suitably protected variants
thereof. For
example, Hoover et. al. (EP-438,233) describes the synthesis by this method of
compounds of formula XI wherein A is CO, D is CHZ, R' is phenylmethyl and R4
is 4-
oxopiperidino, 2-morpholinoethyl(methyl)amino, 4-dimethylaminopiperidino, 4-
pyrrolidinopiperidino, 4-piperidinopiperidino, 4-
dimethylaminomethylpiperidino, 4-
piperidinomethylpiperidino, and 4-N-t-Boc(methyl)aminomethylpiperidino, and
compounds of formula XI wherein A is CO, D is CHZ, R° is 4-
oxopiperidino, and R3 is
2-thienylmethyl, 4-iodophenylmethyl, and 3-thienylmethyl. Additionally,
European Patent
416,393 describes the synthesis of compound of formula XVI wherein RQ is 4-
trifluoroethylpiperazino, 4-methylpiperazino, and 2-(2-
pyridyl)ethyl(methyl)amino by this
method. Compounds of formula XI wherein A is CO and D is CHz are also prepared
by other methods, such as a) Stobbe condensation of a carbonyl compound with a
succinic acid diester to give a 2-dihydrosuccinate 1-monoester, b) coupling of
the free
carboxyl to an amine R4-H, and c) reduction (e.g., catalytic hydrogenation) of
the olefin
to a compound of formula XI. Steps a and b of this method are described by
Plattner
et al. (J. Med. Chem. 1988, 31, 2277-2288) for the synthesis of an olefin
which upon
hydrogenation would give a compound XI wherein R° is morpholino, A is
CO, D is CH2,
and R' is phenylmethyl. lizuka et. al. (J. Med. Chem. 1990, 33, 2707-27 ~ 4)
report a
closely related method whereby, by varying the carbonyl compound giving rise
to R3
and the amine or protected amine R°-H, compounds of formula XI wherein
A is CO, D
is CH2, R3 is not an aryl group or tertiary radical, and R4 is an amine-linked
substituent
as described herein may be prepared.
European Patent 416,393 describes methods for the synthesis of compounds
of formula XI and XVI wherein R' is unsaturated
heterocyclicethyl(methyl)amino, A is
carbonyl, D is CHZ, and R3 is phenylmethyl, by N-alkylation of a five membered
unsaturated heterocycle such as imidazole and pyrazole. One skilled in the art
can use
this method to prepare other such compounds of formulae XVI and XI by varying
the
R'-substituted succinate monoester, and by choosing the appropriate cu-hydroxy-
(C2-
C4)alkyl(C,-C5)alkylamine, and by choosing the appropriate heterocycle.
Compounds of formula XVI wherein R4 is a nitrogen-linked substituent as
described above or a protected variant thereof, A is suifonyl and D is CHZ may
also be
-.,. WO 93/25574 ~ ~ ~ ~ ~ PCT/US93/03625
-33-
prepared according to Scheme 7, by coupling an amine R"-H or protected variant
thereof with a sulfonyl chloride of the formula XVII wherein G-A is CI-SOZ-.
Certain such
compounds of formula XVII and XVI are known in the literature. Rosenberg et
al. (EP
456,185) described the preparation of the compound of formula XVI, wherein G
is CI,
A is S02, D is CHz, R3 is phenylmethyl and Rb is methyl or phenylmethyl, and
their
reaction with selected amines of formula R4-H including morpholine, N-
benzylpiperazine
(as a protected form of piperazine) and 1-methylpiperazine. This methodology
is
adaptable by one skilled in the art to the synthesis of other compounds of
formula XVI
wherein R3 is as described herein or a protected variant thereof, A is
sulfonyl, D is CHZ
and R° is an N-linked substituent or protected variant thereof as
described herein.
Compounds of formula XVI wherein R° is substituted or unsubstituted
alkyl or
cycloalkyl, A is sutfonyl, D is CHZ, and R' and Rb are as defined herein may
be
prepared starting from 2-(R3)-substituted acrylic acid esters by the method
described
by Buhlmayer et al. (J. Med. Chem. 1988, 31, 1839-1846) wherein a compound of
formula XVI wherein R4 is t-butyl, A is sulfonyl, D is CHZ, R3 is
phenylmethyl, and Rb is
ethyl is synthesized by a) conjugate addition of ROSH (tent-butyl mercaptan)
to benzyl
2-benzylacrylate, and b) oxidation of the sulfide to the sulfone. The
requisite 2-(R3)-
substituted acrylic acid esters are prepared by the method of Stetter et al.
(Synthesis
1979, 29) from 2-(R3) substituted-malonic diesters or by other methods cited
therein.
2-(R') substituted-malonic diesters are commercially available or prepared by
literature
methods.
Compounds of formula XI wherein R° is substituted or unsubstituted
alkyl or
cycloalkyl, A is carbonyl, D is CHz, and R3 is as defined herein may be
prepared by
alkylation of a 2-(R3) substituted-malonic diester with a bromoketona of
formula
R°COCHzBr; followed by hydrolysis and decarboxylation of this product.
Buhlmayer et
al. (J. Med. Chem. 1988, 31, 1839-1846) report a method for synthesis of a
compound
of formula XVI wherein R° is t-butyl, A is carbonyl, D is CHz, and R'
is phenylmethyl is
synthesized by a) alkylation of diethyl benzylmalonate with 1-bromo-3,3-
dimethyl-2-
propanone b) sodium hydroxide hydrolysis, and c) NCI-induced decarboxylation
of the
Intermediate 2-benzylmalonic acid. Bromoketones of formula R4COCHzBr may be
prepared, as will be known to one skilled in the art, by many methods such as
a)
reaction of an activated acid R"COON (such as the acid chloride or mixed
anhydride)
217832
with diazomethane to give the diazomethyl ketone which is b) treated with
anhydrous
hydrogen bromide.
European Patent 476,515 describes a method for the preparation of compounds
of the formulae XVI and XI wherein R' is 2-(R'CON(CH3))ethyl(methyl)amino, A
is
carbonyl, D is NH or O, and R' is phenylmethyl, wherein R' is thiomorpholino,
piperidino, or dialkylamino, by acylation of an appropriate
methylaminoethyl(methyl)aminocarbonyl-phenylalanine or phenyllactate
derivative with
R'COCI. This method may be used by one skilled in the art for the synthesis of
other
compounds of the formula XVI and XI wherein the precursor containing R', the
monoprotected (C,-Cs)alkylamino(Cz-C,)alkyl(C,-C5)alkylamine, and the R'COCI
reagent are appropriately chosen.
Unless indicated otherwise, the pressures of the foregoing reactions are not
critical. Generally, the reaction pressures will be about 0.5 to about 2
atmospheres,
preferably ambient pressure i.e., generally at about one atmosphere).
The activity of the active compounds of the present invention as inhib'ttors
of the
angiotensin i-cleaving activity of angiotensin I chymase(s) may be determined
by
studying their ability to inhibit the angiotensin I-cleaving activity of an
angiotensin I
chymase isolated and semipurified from the heart of the marmoset. Thus, left
ventricles
were removed from necropsied marmoset monkeys. Tissues were frozen in liquid
nitrogen and stored at -70°C. The tissue was thawed and homogenized in
10 volumes
(w/v) of 20 mM Tris-HCI, pH 7.4 with a polytron set at 8. The homogenate was
centrifuged at 40,000 Xg for 30 min. The pellet was washed twice by
homogenization
and centrifugation. The final pellet was suspended in 10 volumes of 20 mM Tris-
HCI,
pH 7.4 with 196 Tritori X-100 and 10 mM KCI using a polytron. The homogenate
was
incubated at 4°C for 1 hr and centrifuged at 40,000 Xg for 30 min. The
pellet was
homogenized in 20 mM Tris-HCI, pH 8.0 with 196 Tritori X-100 and 0.5 M KCI,
incubated
and centrifuged. The resulting pellet was suspended in 20 mM Tris, HCI, pH 8.0
with
196 TritonX-100 and 2 M KCI, incubated and centrifuged. The supernatant was
the
source of chymase and was frozen in liquid nitrogen and stored at -
70°C. Protein
concentration was determined (Bradford, Anal. Biochem. 1976, 72, 248-254).
Inhibition
of angiotensin I chymases can be determined by an angiotensin-radioreceptor
assay.
In this assay, angiotensin I is incubated with the chymase in 20 mM Tris-HCI,
pH 8.0
with 0.2596 TritonX-100 and 0.5 M KCI in a final volume at 1001. Samples were
* Trade-mark
._ WO 93/25574 ~ ~ '~ ~ ~ PCT/US93/03625
-35-
incubated at 37°C and a 4°C control was included. The reaction
was terminated by
the addition of 100NI of 2 mM phenylmethylsulfonyl chloride (PMSF) in 50 mM
Tris-HCI,
pH 7.2 with 5 mM MgCl2 and 0.2596 bovine serum albumin and placing on ice. The
concentration of angiotensin II formed was measured by displacement of
radiolabeled
angiotensin II from preformed rat liver microsomes saturated with radiolabeled
angiotensin II. Rat liver microsomes are a known source of angiotensin II
receptors.
These microsomes were isolated and purified from the livers of sacrificed rats
which
were removed and homogenized in 10 volumes of 10 mM Tris-HCI, pH 7.4 with 200
mM
sucrose and mM ethylenediaminetetraacetic acid (EDTA) using 10 strokes of a
teflon
pestle in a glass tube. The homogenate was centrifuged at 3000 Xg for 10 min.
The
resulting supernatant was centrifuged at 12,000 Xg for 13 minutes. This
supernatant
was separated and centrifuged at 104,000 Xg for 1 hour. The resulting pellet
was
suspended in 50 mM Tris-HCI, pH 7.2 with 5 mM MgCl2. The microsomes were
assayed for protein (Bradford, ibid) and frozen at -20°C until use.
Radiolabeled '~51
Sarlle angiotensin II (0.125 nM) was incubated with the rat microsomes (30~g,
100 ~I)
in 50 mM Tris-HCI, pH 7.2 with 5 mM MgCl2, 1 mM PMSF and 0.2596 8SA for 40 min
at ambient temperature at a final volume of 200 NI. The reaction was
terminated by
filtration of the suspension through GF/B filters pretreated with
0.2°.6 PEI and dried. The
angiotensin II levels were determined from an angiotensin II standard curve.
The ICSo
of chymase inhibition was defined as the concentration of the inhibitor that
inhibited
5096 of the enzyme activity and was determined by increasing concentration of
inhibitor.
The colorimetric assay is a less time intense alternative method for measuring
the inhibitory activity of the compounds of this invention against the
angiotensin I
cleaving action of chymases. In this assay, the experimental sample is
prepared by
mixing inhibitor (90 NI, in 10°ro methanol) with enzyme, (90 NI, in 20
mM Tris, pH 8.0, 2
M KCI, 196 Triton X-100 (47 Ng/well)) and is pre-incubated at 37°C for
20 minutes. A
control sample of the enzyme, (90 NI, in 20 mM Tris, pH 8.0, 2 M KCI, 1
°~ Triton X-100
(47 Ng/well)) is separately prepared. To each of these samples is added a
solution of
a peptidyl para nitroanilide substrate (N-succinoyl-Phe-Val-Pro-Phe-p-
Nitroanilide) (180
wl volume of 400 NM) in 30 mM Tris, pH 8.0 (200 NM final concentration). The
final
buffer concentration is 20 mM Tris, pH 8.0 with 0.5 M KCI and 0.25°~
Triton X-100.
Cleavage of the para nitroanilide moiety by the chymase produces a color
change. As
the reaction of experimental and control samples are incubated at 37°C
for 3 hours,
WO 93/25574 21 ~'~ ~ ~ ~ PCT/US93/03625
-36-
the color change is continuously recorded:. by the increase in absorbance at
410
manometers (NM). The rate reaction is.expressed as mOD/minutes. The IC50 of
the
chymase inhibitors was defined as the concentration of the inhibitor that
inhibited 50°~
of the enzyme activity and was determined by increasing concentration of
inhibitor.
The following examples illustrate the invention but are not to be construed as
limiting the same. All melting points are uncorrected. In the Examples, 'boc'
refers to
t-butoxycarbonyl and 'diboc" to di-t-butoxy-carbonyl.
The active compounds of the present invention can be administered as
antihypertensive agents, agents for the treatment of congestive heart failure,
cardiac
and vascular hypertrophy including left ventricular hypertrophy and diabetic
and non
diabetic renal disease by either the oral or parental routes of
administration, with the
former being preferred for reasons of patient convenience and comfort. In
general,
these compounds are normally administered orally in dosages ranging from about
0.1
to about 50 mg per kg of body weight per day, preferably about 0.1 to about 20
mg per
kg of body weight per day, and about 0.05 mg to about 10 mg per kg of body
weight
per day, preferably about 0.05 to about 2 mg per kg of body weight per day,
when
given parenterally; variations will necessarily occur depending upon the
condition of the
subject being treated and the particular compound being administered.
Typically,
treatment is commenced at a low daily dosage and increased by the physician
only if
necessary. It is to be noted that these compounds may be administered in
combination with pharmaceutically acceptable carriers by either of the routes
previously
indicated, and that such administration can be carried out in both single and
multiple
dosages.
The active compounds of the present invention can be orally administered in a
wide variety of different dosage forms, i.e., they may be formulated with
various
pharmaceutically acceptable inert carrier in the form of tablets, capsules,
lozenges,
troches, hard candies, powders, sprays, aqueous suspensions, elixirs, syrups
and the
like. Such carriers include solid diluents or fillers, sterile aqueous media
and various
non-toxic organic solvents, etc. Moreover, such oral pharmaceutical
formulations can
be suitably sweetened and/or flavored by means of various agents of the type
commonly employed for such purposes. In general, the active compounds of the
present invention are present in such oral dosage forms at concentration
levels ranging
2137832
from about 0.596 to about 9096 by weight of the total composition, in amounts
which
are sufficient to provide the desired unit dosages.
For purposes of oral administration, tablets containing various excipients
such
as sodium citrate, calcium carbonate and calcium phosphate may be employed
along
with various disintegrants such as starch (preferably potato or tapioca
starch), alginic
acid and certain complex silicates, together with binding agents such as
polyvinylpyrrolido~e, sucrose, gelatin and acacia. Additionally, lubricating
agents such
as magnesium stearate, sodium lauryl sulfate and talc and compositions of a
similar
type may also be employed. Lactose or milk sugar as well as high molecular
weight
polyethylene glycols may be employed as fillers in soft and hard-filled
gelatin capsules.
When aqueous suspensions and/or elixirs are desired for oral administration,
the
essential active ingredient therein may be combined with various sweetening or
flavoring agents, coloring matter or dyes and, if so desired, emulsifying
agents and/or
solvents such as water, ethanol, propylene glycol, glycerin or combinations
thereof.
One or more other active compounds may be added to the formulations
described above to provide formulations for combination therapy. Such
compounds
include antihypertensives such as diuretics, beta-adrenergic blocking agents,
central
nervous system-acting agents, adrenergic neuron blocking agents, vasodilators,
renin
inhibitors, angiotensin II antagonists, and angiotensin I converting enzyme
inhibitors.
A preferred antihypertensive agent for administration together with a compound
of the
present invention is a diuretic.
EXAMPLES
Amico~ silica 30 NM, 60 ~ pore size, was used for column chromatography.
Melting points were taken on a Buchi~510 apparatus and are uncorrected.
Proton and carbon NMR spectra were recorded on a VarianXL-300, Bruke~ AM-300,
or
Bruke~ AM-500 at 25°C. Chemical shifts are expressed in parts per
million downfield
from trimethylsilane. Liquid secondary ion mass spectra (LSIMS) were obtained
on a
Kratos Concept~l S high resolution spectrometer using cesium ion bombardment
on
sample dissolved in a 1:5 mixture of dithioerythritol and dithivthreitol in
methanol. For
initial sample dissolution chloroform, methanol, or ethanol were employed.
Reported
data are sums of 3-20 scans calibrated against cesium iodide. FAB-MS spectra
were
obtained on a Kratos~MS-80RFA spectrometer operating in the FAB mode on sample
dissolved in a thioglycerol matrix. This layer chromatography (TLC) analyses
were
* Trade-mark
213832
~a~
performed using E. Merck 4Geselgel 60 F254 silica plates visualized (after
elution with
the indicated solvent(s)) by staining with 1596 ethanolic phosphomolybdic acid
and
heating on a hot plate. HPLC was pertormed with 214 nm detection on a (system
A)
150 mm Waters Novapak~Cl B column eluted at 0.8 ml/min, or (System B) 250 mm
Rainiri Microsorb~Cl8 column eluted at 1.0 ml/min by a two-pump/mixer system
supplying the indicated mixture (v:v) of acetonitrile and aqueous pH 2.1
(H3P0,) 0.1 M
KH~PO, respectively. The terms 'concentrated' and 'coevaporated' refer to
removal of
solvent at water aspirator pressure on a rotary evaporator with a bath
temperature of
less than 40°C. Organic solutions were dried over magnesium sulfate
unless specked
otherwise.
General Procedure A-jPeptide Couplin4 Usinq DEC1
A solution of the primary amine (0.2-0.5 M, 1.0 equivalent) in dichloromethane
(or a primary amine hydrochloride and 1.0-1.3 equivalents of triethylamine) is
treated
sequentially with the carboxylic acid coupling partner (1.0-1.2 equivalents),
hydroxybenzotriazole hydrate (HBT) (1.5-1.8 equivalents), and 1-(3-
dimethylaminoaropyl)-3-ethylcarbodiimide hydrochloride (DEC) (1.0-1.2
equivalents,
stoichiometrically equivalent to the quantity of carboxylic acid) and the
mixture is stirred
overnight in an ice bath. The ice bath is allowed to warm, thus the reaction
mixture is
typically held at 0-20°C for 4-6 hours and 20-25°C for the
remaining period. The
mixture is diluted with ethyl acetate or other solvent as specified, and the
resulting
mixture was washed twice with 1 N NaOH, twice with 1 N HCI, once with brine,
then
dried over magnesium sulfate (MgSO,), and concentrated to give the crude
product
which is purified as specified. The carboxylic acid component can be used as
the
dicyclohexylamine salt in coupling to the primary amine or hydrochloride of
the latter;
in this case no triethylamine is employed.
General Procedure B. (HCI-Dioxane Cleavage of a t-Boc-Protected Amine)
A cold (0-10°C) solution of 4N HCI-dioxane is added by syringe to the
solid t-
Boc amine (typically about 10 mL per gram amine) and the resulting solution is
stirred
at 25°C for 0.25-2 hours. The time required for complete disappearance
of the starting
material to a more polar product as judged by TLC. The resulting solution or
suspension is then concentrated, and the residue coevaporated several times
with
added ether, and then dried in vacuo. If specified, the solid hydrochloride is
washed
further with solvent.
* Trade-mark
WO 93/25574 PCT/US93/03625
21.3'~~~~
-39-
General Procedure C. Periodinane Oxidation of a peptidyl a-Hydroxyester,
Difluorostatine. or Trifluoromethyl Carbinol to the Corresponding Ketone.
The procedure of Burkhart, et al. (Tetrahedron Lett. 1988, 29, 3433-3436) for
the
oxidation of a-hydroxyesters by the Dess-Martin periodinane (J. Org. Chem.
1983, 48,
4155) was employed with several slight modifications (use of 4 equiv
periodinane
reagent and longer reaction times, and variation in extraction solvent). Thus,
a solution
of the peptidyl a-hydroxy ester (e.g., 1 mmol) in dry dichloromethane (about 5
mL) was
treated with the above referenced periodinane (4 equiv), and the reaction
mixture was
stirred overnight (about 16 hours) at 25 ° C. If the reaction was not
complete (TLC),
additional periodinane was added as specified. When complete reaction was
verified
the mixture was diluted with the specified extraction solvent and water (20-
100 mL
each/mmol substrate), NazSZ03~5H~0 (1.3-3 g/mmol substrate) and NaHC03 (2.5-3
g/mmol substrate) were added, and the resulting solution stirred 1 to 2 hours
or until
both layers clarified. The separated organic layer was washed with aqueous
NaHC03,
brine, and combined with one extract (same solvent) of the separated aqueous
layers.
The combined organic layers were dried (MgSO,), concentrated, and the residue
chromatographed on silica eluted with the specified solvent mixture.
WO 93/25574 PCT/US93/036~5
2137832
-40-
Example 1
N-((1 1-dimethylethoxy)carbonyll-L-phenylalanyl-N-(2.3-dioxo-3-methoxy-1-
jphenylmethyl)propyll-L-prolinamide
A. Methyl-3(S) 2(R)-3-amino-2-hydroxy-4-phenylbutanoate
3(S),3(R)-N-((1,1-dimethylethoxy)carbonyl]-3-amino-2-hydroxy-4-
phenylbutyronitrile (U.S. Patent 4,668,769, 9.13 g, 33.0 mmol) was added in
several
portions at 0°C to a stirred solution of anhydrous hydrogen chloride
(60 g) in absolute
methanol (250 mL). The resulting solution was warmed to 25°C for 5
minutes and the
flask was sealed with a plastic stopper and placed behind a safety shield
(Caution!).
After 63 hours at 25°C, slight pressure was relieved by piercing the
stopper with a
syringe needle, and the mixture was concentrated and dried in vacuo. The
resulting
solid (9.94 g) was suspended in saturated aqueous NaHCO, (about 250 mL) and
the
mixture extracted with chloroform (10 times 50 mL). The extracts were dried
(MgS04)
and concentrated. The residue (6.22 g) was recrystallized from 1:2 ethyl
acetate-
hexanes (120 mL, by dissolving in hot ethyl acetate and adding hexanes at
reflux). The
solid was collected at 0°C, washed with chilled 1:2 ethyl acetate-
hexanes, and dried
in vacuo at 56°C (5.31 g, 7796): m.p. 106-107°C. 'H NMR (CDCI3)
d 1.5 (br, 3H), 2.73
(dd, 1 H, J = 8.3, 13.3 Hz), 2.92 (dd, 1 H, J = 6.6, 13.3 Hz), 3.37 (m, 1 H),
3.79 (s, 3H),
4.08 (d, 1H), 7.2-7.35 (m, 5H). Anal. Calcd for (C"H,5N03) C, 63.14; H, 7.23;
N, 6.69.
Found: C, 63.16; H, 7.01; N, 6.61.
B. N'-((1 1-dimeth)rlethoxy)carbonyll-N°-f2(R)-hydroxy-3-methoxy-3-oxo-
1 (S)-
(phenylmethyl)propyll-L-prolinamide
The following illustrates a specific application of General Procedure A. A
solution of the product of Example 1 (3.81 g, 18.2 mmol) in dichloromethane
(125 mL)
was treated sequentially at 0°C with N-t-butoxycarbonyl-L-proline (4.30
g, 20.0 mmol),
1-hydroxybenzotriazole hydrate (HBT, 4.20 g, 27.3 mmol), and 1-(3-
dimethylaminopropyl) ethylcarbodiimide hydrochloride (DEC, 3.83 g, 20.0 mmol).
The
mixture was stirred 40 hours during which time the temperature rose to
25°C.
Dichloromethane (200 mL) was added and the resulting solution washed with 2N
NaOH
(2 .timas 70 mL), 1 N HCI (70 mL), and then dried (MgS04) and concentrated.
The
residue (6.0 g) was recrystallized from 1:1 chloroform-hexanes giving 5.75 g
(78%) of
a colorless solid, m.p. 167-168.5°C. 'H NMR (CDCI3) 6 1.25 (m, ca. 1
H), 1.48 (s, 9H),
1.6-2.0 (m, ca. 3H), 2.13 (br, ca. 0.5 H), 2.86 (dd, 1 H, J = 8.3, 13.7 Hz),
2.96 (m, 1 H),
WO 93/25574 PCT/US93/03625
-41-
3.18 (m, exchanges with DZO), 3.3-3.4 (m, 2H), 3.76 (s, 3H), 4.11 (d, 1 H, J =
5.1 Hz,
collapses to s with D20), 4.18 (br, 1 H), 4.58 (br, 1 H), 6.23 (br, ca. 0.5
H), 7.02 (br, ca.
0.5H), 7.15-7.35 (m, 5-6H). LSIMS m/e (rel. intensity) 407 (M++H, 70), 351
(20), 307
(100). Anal. Calcd for (CZ, H3°N208~0.5H20) C, 60.70; H, 7.52; N, 6.74.
Found: C,
60.92; H, 7.12; N, 6.43.
C. N-f2(R)-h~droxy-3-methoxy-3-oxo-1 (S)-(phenylmethyl)propvll-L-
prolinamide Hydrochloride
The following preparation illustrates a specific application of Procedure B. A
5°C solution of anhydrous hydrogen chloride in p-dioxane (30 mL of 4M)
was added
in one portion to the product of the preceding example (3.24 g, 7.97 mmol) and
the
resulting solution was warmed to 25°C. After 30 minutes, the mixture
was
concentrated, and the residue dried in vacuo and triturated with ether (3 x 6
mL). The
resulting colorless solid was dried in vacuo at 56°C for 1.5 hours
(2.82 g, 103%). 'H
NMR (DZO) 1.8-2.1 (m, 3H), 2.38 (m, 1 H), 2.85 (dd, 1 H, J = 9.9, 13.8 Hz),
3.02~(dd, 1 H,
J = 5.8, 13.9 Hz), 3.31 (m, 2H), 3.68 (s, 3H), 4.15 (dd, 1 H, J = 5.8, 8.6
Hz), 4.41 (d,
1 H, J = 2.0 Hz), 4.53 (ddd, 1 H, J = 2.0, 5.8, 9.9 Hz), 7.2-7.4 (m, 5H).
LSIMS 307
(M''+H, 10096).
D. N-f(1 1-dimethylethoxy)carbonyll-L-phenylalanyl-N-f2(R)-hydroxv-3-
methoxy-3-oxo-1 (Sl-(phenlrlmethylpropyll-L-prolinamide
According to general procedure A, the product of Example 1 C (250 mg) was
coupled to N-(1,1-dimethylethoxy)carbonyl-L-phenylalanine (1.1 equiv) giving
350 mg
of crude product which was chromatographed on silica eluted with ethyl acetate-
hexanes giving the title substance (306 mg, 76°~6). Anal. Calcd for
(Csol"IsaN30~~0.5Hz0): C, 64.04; H, 7.17; N, 7.47. Found: C, 64.09; H, 7 .14;
N, 7.22.
E. N-f(11-dimethylethoxy)carbonyl]-L-phenylalanyl-N-f2.3-dioxo-3-methoxy-1-
~phenylmethyl)propyll-L-arolinamide
The following preparation illustrates an application of General Procedure C
except that less periodinane was initially added. Dess-Martin periodinane (265
mg, 0.61
mmol) was added to a solution of the product of the preceding example (168 mg,
0.30
mmol) in dichloromethane (10 mL) and the mixture was stirred 16 hours at
25°C.
Additional periodinane (265 mg, 0.61 mmol) was added and the mixture was
stirred at
25°C another 72 hours. The mixture was diluted with ethyl acetate (30
mL) and a
solution of NaHCO, (0.70 g) and NaZS203~5HZ0 (2.2 g) was added, and the
mixture
WO 93/25574 PCT/US93/03625
2~3'~8~'~
-42-
,,
was stirred until both layers became clear. The organic layer was separated
and
washed with saturated aqueous NaHCO, (10 mL) and brine (10 mL). The combined
aqueous layers were extracted once with ethyl acetate. The combined organic
layers
were dried (MgSO,), and concentrated to yield 144 mg of a colorless foam which
was
chromatographed on 5 g silica packed in 0.5:100 ethanol-dichloromethane and
eluted
with 50 mL portions of 1:100, 2:100, and 4:100 ethanol-dichloromethane to
yield the title
substance (70 mg, 4296) as a colorless powder, (TLC Rf 0.55, ethyl acetate-
silica).
LSIMS 552 (3096, MH+), 496 (1096), 452 (1000.
Example 2
N-f(1.1-dimethylethoxy)carbonyll-L-histidyl-N-f2,3-dioxo-3-methoxy-1-
~phenylmethyl)propyll-L-prolinamide
A. N°N'-Bisf(1.1-dimethylethoxyycarbonyll-L-histidyl-L-
prolinephenylmeth~
ester
According to General Procedure A, L-proline phenylmethyl ester hydrochloride
(8.0 g) was coupled to N°N'-bis-[(1,1-dimethylethoxy)carbonyl]-L-
histidine (11.8 g) and
the crude product (11.7 g, oil) was chromatographed on 200 g silica eluted
with 2:1
ethyl acetate-hexanes followed by ethyl acetate. 3.1 g of the less polar
product was
isolated and identified by NMR as Boc-Pro-OBn; the more polar substance, a
colorless
foam (6.89 g, 39°6) was identified as the title compound:
LSIMS 543 (7596, MH+), 487 (30°~), 443 (25%), 387
(40°r°), 343 (75°~). Anal.
Calcd. for (CZBH38N4O,~O.SHZO): C, 60.96; H, 7.12; N, 10.16. Found: C, 61.02;
H, 6.86;
N, 10.08.
B. N°N'-Bisf(1.1-dimethylethoxy,carbonyll-L-histidyl-L-proline
A solution of the product of the preceding example (1.0 g, 1.84 mmol) in ethyl
acetate was shaken with 2096 Pd(OH)2/C under 50 p.s.i. hydrogen pressure for 3
hours.
The mixture was filtered through Supercel (trademark) and the cake washed with
ethyl
acetate. The filtrate was concentrated giving 0.87 g of a colorless foam, (TLC
R, 0.5 in
18/2/1 HCCh/Ethanol/HOAc).
C. N°N'-Bisf(1.1-dimethylethoxylcarbonyll-L-histidyl-N-f2(R)-
hydroxy-3-
methoxy-3-oxo-1 yS)-(phenylmethyl)prop IY 1-L-prolinamide
2-Ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline(EEDQ,247 mg, I.OOmmol)was
added to a solution of the product of the preceding Example (450 mg, 1.00
mmol) and
the product of Example 1 a (210 mg) in dichloromethane (5 mL) and the
resulting
21~'~8.~2
WO 93/25574 PCT/US93/03625
-43-
solution was allowed to stir at 25°C for 48 hours. The reaction mixture
was diluted with
100 mL ethyl acetate and the resulting solution was washed with 1096 aqueous
citric
acid (2 x 40 mL), saturated aqueous NaHC03 (2 x 40 mL), dried (MgS04) and
concentrated to yield the crude product as a colorless foam (602 mg,
94°~6), (TLC R,
0.19 in ethyl acetate). LSIMS 644 (MH+, 20°~), 544 (10096).
D. N°N'-Bisf(11-dimethylethoxy)carbonyll-Lfiistidyl-N-f2,3-dioxo-3-
methoxy-
~S)-(phenylmethyl~propyll-L-prolinamide
The compound was prepared in a manner according to the procedure described
as General Procedure C, except that only 2 equiv of periodinane was used, and
that
chloroform, rather than ethyl acetate, was used as the extraction solvent. The
product
of the preceding example (642 mg) was converted to the title substance (530
mg, 83%)
which was not further purified. (TLC R, 0.22, ethyl acetate).
E. N°-f(1 1-dimethylethoxy~carbonyll-L-histidyl-N-f2.3-dioxo-3-methoxy-
1 (S)-
~phenylmethyl)propyll-L-prolinamide
The product of the preceding example (151 mg, 0.236 mmol) was dissolved in
acetic acid (1 mL) and water (1 mL) for 18 hours at 25°C. Concentration
and drying
gave 139 mg of solid which was dissolved in a mixture of methanol (2 mL) and
0.5 mL
saturated aqueous NaHC03. Silica (3 g) was added and the solvent was removed
in
vacuo. The solid was added to the top of a 10 g silica column which was eluted
with
100 mL portions of 196 and 296 ethanol-dichloromethane, and 50 mL portions of
4%,
896 and 2096 ethanol-dichloromethane to yield the title substance (82 mg,
64%), (TLC
R, 0.55, 9:1 dichloromethane-ethanol containing aq. NH40H).
LSIMS 696 (MH++dithiothreitol, 100°~), 542 (MH+, 5096).
Example 3
N (morpholino-1-carbonyl)-L-phenylalanyl-N-f2-(2-thiazolyl)-2-oxo-1-
~phenylmethyl)ethyllL-prolinamide
A. 1 (R S) 2(S)-1-(2-thiazolyl)-2-fN-f(1 1-dimethylethoxy)carbonyllaminol-3-
phenyl-1-propanol
N-Butyllithium in hexane (7.1 mL, 17.7 mmol) was added dropwise to a -
78°C
solution of thiazole (2.1 g, 17.7 mmol) in tetrahydrofuran (20 mL) and the
resulting
mixture was stirred at -78°C for 30 minutes. A solution of 2-[N-(1,1
dimethylethoxy)carbonylJaminoJ-3-phenyl-1-propanal was added and the mixture
was
stirred for 45 minutes at -78°C. The mixture was warmed to 0°C
and treated with
WO 93/25574 PCT/US93/03625
213'~$3~
saturated aqueous NH4CI. Ethyl acetate was added and the resulting solution
was
washed twice with water, brine, dried (MgS04) and concentrated giving 2.2 g of
crude
product which was purified on silica eluted with ethyl acetate-hexanes giving
the title
product (580 mg, 2296). LSIMS 335 (MH+, 10096), 279 (50°6).
B. ~R.S). 2(S)-1-(2-thiazolyl~2-amino-3-phenyl-1-a~ropanol hydrochloride
According to Procedure B, the product of the preceding Example (546 mg) was
converted to the title hydrochlorides (500 mg), TLC Rf 0.75 and 0. 72 (18/2/1
CHZCIZ-
EtOH-NH40H). LSIMS 235 (10096, MH+)
C. N(morhholino-1-carbon I~phenylalanyl-Nf2-(2-thiazolyl)-1(R,S)-hydroxy-
1-(S)-phenylmethyl)ethyll-L;prolinamide
The product of the preceding Example (200 mg) was coupled to the product of
Example 13B (free acid, 304 mg) using General Procedure A, giving 265 mg of
crude
product which was purified on silica eluting with a gradient (1-16°~)
of ethanol in
dichloromethane. Yield, 194 mg, 43~, TLC Rf 0.73 (9:1 dichloromethane-
ethanol).
LSIMS 592 (MH+, 4096), 309 (45°~).
D. N-~morpholino 1-carbonyl)~-L~henvlalanyl-N-f2-(2-thiazoly,~-2-oxo-1-
(phenylmethyl)ethyll-L-prolinamide
According to General Procedure C, the product of the preceding Example (194
mg) was treated with the periodinane, and the crude product (133 mg), isolated
by
ethyl acetate extraction, was purified on silica eluted with ethyl acetate
giving the title
product (76 mg, 4296), TLC Rf 0.85 in 18/2/1 CHZCIz-EtOH-NHQOH. LSIMS 590
(MH+,
9096), 330 (7096).
Example 4
N-f (1 .1-dimethylethox~carbonyll-L-leucyl-N-f2,3-dioxo-3-methoxy-1-
~phenylmethylZpropyll-L-prolinamide
A. N-f (1,1-dimethylethoxy)carbonyll-L-leucyl-N-f2(R)-hydroxy-3-methoxy-3-
oxo-1 (Sy-(phenylmethyl)propyll-L-prolinamide
The compound was prepared in a manner according to the procedure described
as General Procedure A. The product of Example 1 C (250 mg) was coupled to Boc-
L-
leucine to yield 346 mg of crude product which was purified on silica eluting
with ethyl
acetate-hexanes and ethyl acetate to yield the title substance (303 mg,
80°~).
LSIMS 520 (MH+, 95°~), 420 (100°~).
Anal. (CZ~H4, N;O~~0.5 H20) C, H, N, were within 0.496 of the calculated
value.
WO 93/25574 ~ ~ '~ ~' 3'~ PCT/US93/03625
-45-
B. N-f~(1.1-dimethylethoxyycarbonyll-L-leucyl-N-(2.3-dioxo-3-methoxy-1-
jphenylmethyllpropyll-L-prolinamide
The compound was prepared in a manner according to the procedure described
as General Procedure C, with the following exception: 200 mg of the product of
the
preceding Example was treated with a total of 6 equiv of the periodinane which
was
added in 2 equiv portions over 7 days, until the reaction was complete by
HPLC. Ethyl
acetate was employed in the workup and the crude product (164 mg) was
chromatographed on silica eluted with a gradient of 0.5-496 ethanol in
dichloromethane
to yield the title substance (148 mg).
(TLC R, 0.52, ethyl acetate).
LSIMS 672 (2096, MH++matrix), 564 (35°0, MH++CzH50H), 518 (50%,
MH+),
462 (3596), 418 (10096).
Anal. (CZ,H39N30,~0.6 H20) C, H, N were within 0.4% of the calculated value.
Example 5
N-f(1.1-dimethylethoxy)carbonyl]~4-thiazolyl)-L-alanyl-N-f2.3-dioxo-3-methoxy-
1-
jphenylmeth)rl)propyll-L-prolinamide
A. N-f(1.1-dimethvlethoxy)carbonyll~-(4-thiazolyl)-L-alanyl-N-f2(R)-hydroxy-3-
methoxy-3-oxo-1-(phenYlmethyl)prop ly 1-L-prolinamide
The compound was prepared in a manner according to the procedure described
as General Procedure A. The product of Example 1 C (266 mg)was coupled to Boc-
3-
(4-thiazolyl)-L-alanine) (200 mg) and the crude product (370 mg) purified by
chromatography on silica eluted with ethyl acetate-hexanes, ethyl acetate, and
ethanol-
ethyl acetate to yield the title substance (195 mg, 46°~), (TLC R,
0.25, ethyl acetate).
LSIMS 561 (MH+, 100°ro).
B. N-f(1 1-dimethylethoxy)carbonyl)-3-(4-thiazolyl)-L-alanyl-N-f2.3-dioxo-3-
methoxy-1-(phenylmethyl)propyll-L-prolinamide
The compound was prepared in a manner according to the procedure described
as General Procedure C. The product of the preceding Example (190 mg) was
oxidized
with the periodinane (ethyl acetate used for extraction) to yield 183 mg of
crude product
which was purled on silica eluted with ethyl acetate-hexanes followed by ethyl
acetate,
to yield 136 mg (72°6) of the title substance (TLC R, 0.35 in ethyl
acetate).
' H NMR (CDCI3) d 1.43 (s, 9H), 1.8 (m, 3H), 2.12 (m, 1 H), 2.9 (m, 1 H), 3.00
(dd,
1 H, J = 9.6, 14.1 Hz), 3.12 (m, 2H), 3.35 (dd, 1 H, J = 14.2 Hz), 3.40 (m, 1
H), 3.86 (s,
WO 93/25574 PCT/US93/03625
-46-
3H), 4.58 (m, 1 H), 4.60 (dd, 1 H), 5.11 (m, 2H), 5.48 (m, 1 H), 7.08 (s, 1
H), 7.19 (d, 2H),
7.2-7.35 (m, 3-4H), 8.62 (s, 1 H).
LSIMS 713 (MH++C4H,oO2S2, 40°~), 559 (MH+, 90°~6), 127
(100°~).
Anal. (CZ~H34N40,S~9/8 Hz0) C, H, N were with 0.4°0 of the calculated
value.
Example 6
N-f (1 1-dimeth~ethoxy~carbonyll-L-alanyl-N-f 2.3-dioxo-3-methoxy-1-
~phenylmethyl)propyll-L-prolinamide
A. N-f(11-dimethylethoxy)carbonyl]i-L-alanyl-N-f2(R1-hydroxy-3-methoxy-3-
oxo-1-(phenylmethylZpropyll-L a~rolinamide
The compound was prepared in a manner according to the procedure described
as General Procedure A. The product of Example 1 C (250 mg) was coupled to Boc-
L-
alanine (141 mg) to yield 280 mg of crude product which was purified on silica
eluted
with ethyl acetate-hexanes to yield the title product (191 mg, 56°~),
(TLC R, 0.29 in ethyl
acetate) .
Anal. Calcd for Cz4H35N3~7~O.25 HZO: C, 59.79, H, 7.42; N, 8.71. Found: C,
59.82; H, 7.66; N, 8.46.
B. N-f(1 1-dimethylethoxv)carbonyll-L-alanyl-N-f2.3-dioxo-3-methoxy-1-
(phenylmethyl)prop~rll-L-prolinamide
The compound was prepared in a manner according to the procedure described
as General Procedure C. The product of the preceding Example (130 mg) was
treated
with periodinane (4.6 equiv) and the crude product, isolated by chloroform
extraction,
was purified on silica eluted with ethyl acetate-hexanes followed by ethyl
acetate, to
yield 86 mg (6796) of the title substance (TLC Rf 0.34 in ethyl acetate).
'H NMR (CDCI3 d 1.13 (d, 3H, J = 6.9 Hz), 1.43 (s, 9H), 1.85 (m, 1 H), 1.95
(m,
2H), 2.32 (m, 1 H), 2.97 (dd, 1 H, J = 8.0, 14.2 Hz), 3.23 (dd, 1 H, J = 5.6,
14.2 Hz) 3.38
(m, 1 H), 3.58 (m, 1 H), 3.85 (s, 9H), 4.38 (dq, 1 H), 4.57 (dd, 1 H), 5.27
(m, 2H), 7.14 (dd,
1 H), 7.2-7.4 (m, 3-4H). A set of resonances presumed due to a minor rotamer
(ca.
1096) were also observed: a 3.8 (s), 4.96 (m), 4.22 (m).
LSIMS 476 (MH+, 5096), 420 (50%), 376 (10096).
Anal. Calcd for CZ4H33N3O,: C, 60.62; H, 6.99; N, 8.84. Found: C, 60.55; H,
7.08; N, 8.84.
WO 93/25574 PCT/US93/03625
21~'~~3~
-47-
Example 7
N-f(1 .1-dimethylethoxy, carbonyll-L-prolyl-N-f2.3-dioxo-3-methoxy-1-
~phenylmethyl~,propyrll-L-prolinamide
A. N-j(1.1-dimethylethoxyycarbonyll-L-prolyl-N-f2(R)-hydroxy-3-methoxy-3-
oxo-1-(phenylmethy~propyll-L-prolinamide
The compound was prepared in a manner according to the procedure described
as General Procedure A. The product of Example 1 C (250 mg) was coupled to Boc-
L-
proline (173 mg) to yield 291 mg of crude product which was purled on silica
eluted
with ethyl acetate-hexanes followed by ethyl acetate to yield 245 mg (67~) of
the title
product.
Anal.. Calcd for CzeH3,N,0,~0.5 HZO: C, 60.91; H, 7.27; N, 8.20. Found: C,
61.03; H, 7.63; N, 8.04.
B. N-f(1.1-dimethylethox~)carbonyll-L-prolyl-N-f2.3-dioxo-3-methoxy-1-
~phenyrlmethyl)prop~rll-L-prolinamide
The compound was prepared in a manner according to the procedure described
as General Procedure C. The product of the preceding Example (204 mg) was
treated
with periodinane and the crude product, isolated by chloroform extraction, was
purified
on silica eluted with ethyl acetate-hexanes, to yield 172 mg (85~) of the
title substance,
(TLC R, 0.24 in ethyl acetate).
LSIMS 502 (MH+, 2296), 402 (10096).
Anal. Calcd for CZeH35N3O7~1.5 HzO; C, 59.07; H, 7.24; N, 7.95. Found: C,
58.85; H, 6.85; N, 7.70.
Example 8
N-f(1 1-dimethyethoxy)carbonyll-L-valyl-N-f2,3-dioxo-3-methoxy-1-
~phenylmethyl propyll-L-erolinamide
A. N-f(11-dimethyethoxy)carbonyll-L-valyl-N-f2(R)-hydroxy-3-methoxy-3-oxo-
1-(phenvlmethyl)propy_Ij-L-prolinamide
The compound was prepared in a manner according to the procedure described
as General Procedure A. The product of Example 1 C (250 mg) was coupled to Boc-
L
valine (174 mg) to yield 276 mg of crude product which was purified on silica
eluted
with ethyl acetate-hexanes to yield 189 mg (5196) of the title product.
LSIMS 506 (M++H, 4596), 450 (10~), 406 (100°6), 307 (70°~).
Anal. Calcd for
C~aH39N30,: C, 61.76; H, 7.77; N, 8.31. Found: C, 62.23; H, 7.43; N, 8.25.
WO 93/25574 PCT/US93/03625
2137~3~ ,
-4$-
B. N-f(1,1-dimethylethoxy carbonyll-L-valyl-N-f2(R!-hydroxyl-methoxy-3-oxo-
1-(phenylmethyl),propyll-L-prolinamide
According to General Procedure C, the product of the preceding example (140
mg) was oxidized and the crude product (137 mg), isolated by chloroform
extraction;
was purified on silica eluted with ethyl acetate-hexanes giving 107 mg (77~)
of the title
product, TLC Rf O.n 1:1 ethyl acetate-hexanes.
'H NMR (CDCI3) 0.82 (d, 3H, J = 6.7 Hz), 0.83 (d, 3H, J = 6.7 Hz), 1.42 (s,
9H),
1.83 (m, 2H), 1.94 (m, 2H), 2.29 (m, 1 H), 3.02 (dd, 1 H, J = 7.2, 14.0 Hz),
3.18 (dd, 1 H,
J = 6.1, 14.0 Hz), 3.52 (m, 1 H), 3.68 (m, 1 H), 3.82 (s, 3H), 4.21 (dd, 1 H,
J = 6.3, 9.2
Hz), 4.56 (dd, 1 H, J = ca. 8, ca. 3 Hz), 5.14 (d, 1 H, J = 9.5 Hz), 5.30 (dt,
1 H, J = 6.3
6.9 hz), 7.14 (m, 2H), 7.2-7.4 (m, ca. 3-4H). A set of resonances presumed due
to a
minor rotamer (ca. 1096) were also observed: d 0.99 (d, J = 6.7 Hz), 1.00 (d,
J = 6.7
Hz), 3.34 (s), 3.91 (m).
LSIMS 504 (M++H, 30°6), 448 (30°~6), 404 (100°~6),
305 (7596).
Anal. Calcd for CzeH3,N3O,~O.S HZO: C, 61.45; H, 7.34; N, 8.27. Found: C,
61.56; H, 7.61; N, 8.16.
Example 9
N-(mor~holino-1-carbonyl)-L-phenylalanyl-N-f2.3-dioxo-3-methoxy-1-
(phenylmethyl~propylLprolinamide
A. N-(morpholino-1-carbonyl)-L-phenylalanyl-N-f2(R)-hydroxy-3-method-3-
oxo-1-(phenylmethy~propyll-L-prolinamide
The compound was prepared in a manner according to the procedure described
as Procedure A. The product of Example 1 C (500 mg) was coupled to N-
(morpholino-
1-carbonyl)-L-phenylalanine (USP 4,814,342, 446 mg) and the crude product (828
mg)
was purified by chromatography on silica and eluted with 1 °~-
32°~ ethanol in
dichloromethane to yield the title substance (186 mg, 22°0), (TLC R,
0.41 in 5°~ ethanol-
dichloromethane).
LSIMS 567 (MH+, 10096), 442 (60°~), 307 (70%).
B. N-(morpholino-1-carbonyl)-L-phenylalanyl-N-f2.3-dioxo-3-methoxy-1-
y~henylmeth I~prop~l]-L-prolinamide
The compound was prepared in a manner according to the procedure described
as General Procedure C. The product of the preceding Example (168 mg) was
treated
with periodinane and the crude product (146 mg) was isolated by extraction
using ethyl
WO 93/25574 ~ ~ 3'~ ~ PCT/US93/036~5
-49-
acetate and was purled on silica eluted with 1-3296 ethanol in dichloromethane
to yield
the title substance (107 mg, 6396).
Anal. Calcd for C3°H,eN4O,~0.5 HzO: C, 62.81; H, 6.50; N, 9.77.
Found: C,
62.96; H, 6.45; N, 9.88.
Example 10
N-(4-oxopiperidine-1-carbonyl-L-phenylalanyl-N-(2.3-dioxo-3-methoxy-1-
jphenvlmethyl)propyll-L-prolinamide
A. N-(4-oxopiperidine-1-carbonyy-L-phenylalanine benzyl ester
L-Phenylalanine benzyl ester p-toluenesulfonate (420 g, 0.982 mol) was added
to a stirred mixture of 1 N NaOH (1.5 L) and dichloromethane (0.5 L) at
25° C. After the
solid dissolved the organic layer was separated, dried over MgS04, and added
over 1
hour to a stirred 0-5°C slurry of imidazole (135 g, 1.96 mol, 2.0
equiv) and
carbonyldiimidazole (175 g, 1.08 mol) in dichloromethane (1.6 L). The
resulting clear
solution was warmed rapidly to 25°C and stirred 1 hour at this
temperature. 4-
Piperidone hydrate hydrochloride (200 g, 1.28 mol, 1.3 equiv) and
triethylamine (178
mL, 1.28 mol, 1.3 equiv) were added sequentially, each in one portion, and the
mixture
was stirred overnight at 25°C. The mixture was washed with 1 N HCI (3 x
800 mL), and
the resulting organic layer (partially emulsified) was diluted with
dichloromethane (2 L)
and divided into two equal portions. Each half was washed with 2N HCI (0.6 L).
The
combined aqueous layers were washed with dichloromethane (1.6 L), and the
organic
layers were combined, washed with brine, dried (MgS04), and concentrated to a
viscous yellow oil which was coevaporated twice with added ether to give an
off-white
solid (352 g, 9496). This material was dissolved in hot 2:1 (v:v) ethyl
acetate-hexanes
(1.2 L) and the resulting near-solution was filtered through a cotton plug.
Crystallization
rapidly ensued as the mixture was allowed to stand undisturbed for 1 hour.
After the
mixture was chilled in an ice bath the mass was filtered, washed with cold 2:1
ethyl
acetate-hexanes and hexanes and dried to yield 260 g (70°6) of a
colorless crystalline
solid, homogeneous by TLC (Rf 0.27 in 2:1 ethyl acetate-hexanes) and HPLC
(2.56 min
in 70:30 MeCN-pH 2.1 phosphate): m.p. 104-107°C; NMR (CDCI3, ppm) 2.39
(m, 4H),
3.11 (dd, A of ABX, 1 H, J = 5.7, 13.8 Hz), 3.16 (B of ABX, 1 H, J = 5.7; 13.8
Hz), 3.61
(m, 4H), 4.85 (-dt, 1 H, J = 7.6, 5.8 Hz), 4.97 (d, 1 H, J = 7.6 Hz), 5.11 (A
of AB, 1 H,
WO 93/25574 PCT/US93/03625
213783'
-50-
J = 12.1 Hz), 5.21 (B of AB, 1 H, J = 12.1 Hz), 7.00 (dd, 2H, J = 2.5, 5.9
Hz), 7.18-7.37
(m, 8H); IR (CHCI3) 3435, 2990, 1731, 1651, 1496, 1401, 1176, 982 cm-' . Anal.
(C22H24N204) Ci H~ N.
B. N-(4-oxopiperidine-1-carbonyl)-L-phenylalanine
The product of the preceding example (115 g, 0.605 mol) was added to 12 g
1096 Pd/C in methanol (900 mL) and acetic acid (100 mL), and the resulting
mixture
was shaken under 30 p.s.i. hydrogen for 35 minutes and then filtered through
washed
Celite which was washed well with methanol. The filtrates and washings were
combined and concentrated in vacuo leaving a viscous yellow oil which was
coevaporated twice with added toluene and twice with added ether. The residue
was
dissolved in chloroform (500 mL) and the resulting solution was washed with
water (4
x 250 mL), brine, dried over MgS04, and stirred with 7 g of decolorizing
carbon (Darco,
trademark) G-60 at 25°C for 20 minutes. The mixture was filtered
through
diatomaceous earth (Supercel, trademark) and the filtrates concentrated in
vacuo and
dried at 0.2 mm and 25°C for 16 hours leaving a colorless foam which
was pulverized
to an off-white powder (72.4 g, 8396). An impurity (696 by RP-HPLC) was
present which
was identified by NMR as the corresponding dimethyl ketal. This material (63.7
g)was
dissolved in tetrahydrofuran (190 mL) and aqueous 1 N HCI (19 mL) at
25°C. After 2.5
hours at 25°C and 16 hours at 0°C the mixture was concentrated
to near dryness, the
residue dissolved in ethyl acetate (700 mL) and the resulting solution was
washed twice
with 1 N HCI. The aqueous extracts were combined, extracted twice with ethyl
acetate,
and the combined organic layers dried (MgS04) and concentrated. The colorless
foam
was pulverized and dried in vacuo (60.1 g, 7896 projected overall yield). 'H
NMR (300
mHz, CDC13) d 2.33 (m, 4H), 3.02 (dd, 2H, J = 7.2, 13.8 Hz), 3.21 (dd, 2H, J =
4.9,
13.8 Hz), 3.55 (m, 4H), 4.69 (dt, 1 H, J = 7.0, 12.5 Hz), 5.40 (d, 1 H, J =7.1
Hz), 7.12-
7.27 (m, 5H), 8.88 (br, 1 H); '3C NMR (75.4 mHz, CDCI3) a 37.5, 40.4, 42.7,
54.8, 127.2,
128.6, 129.3, 136.3, 157.2, 175.0, 207.6; HRMS (CI, isobutane) m/e 291.1358
(MH+,
calcd for C,SH,BNz04: 291.1346).
C. N-(4-oxopiperidine-1-carbonyl)-L-phenylalan -girl N-f2(R)-hydroxy-3-methox
r~-
3-oxo-1 (S)-(phenylmethyl)propyll-L-prolinamide
The compound was prepared in a manner according to the procedure described
as General Procedure A. The product of the preceding Example (281 mg) was
coupled
with the product of Example 1 C (300 mg) to yield crude product (427 mg) which
was
WO 93/25574 ~ ~ ~ ~ ~ ~ PCT/US93/03625
-51-
chromatographed on silica eluted with ethyl acetate and a 1-496 ethanol in
ethyl acetate
gradient to yield the title compound (254 mg), (TLC R, 0.5 in 18/2/1 CHCI3-
ethanol-
NH4OH).
LSIMS 579 (MH+, 100°6), 307 (7096).
Anal. Calcd for C3,H38N40,~HzO: C, 62.40; H, 6.76; N, 9.39. Found: C, 62.28;
H, 6.44; N, 9.23.
D. N-(4-oxopiperidine-1-carbonyl)-L-phenylalanyl-N-f2.3-dioxo-3-methoxy-1-
(phenylmethyl~propyll-L-prolinamide
The compound was prepared in a manner according to the procedure described
as General Procedure C. The product of the preceding Example (246 mg) was
treated
with periodinane. The product (215 mg, 87°6), was isolated by
extraction using ethyl
acetate.
LSIMS 731 (MH++C4H,o02S2, 896), 577 (MH'', 1596), 309 (100°6).
Anal. Calcd for C3,H3aN40,~1.25 HZO: C, 62.14; H, 6.48; N, 9.35. Found: C,
62.05,; H, 6.09; N, 8.95.
Example 11
N'- f4f4-(methvlamino)piperidinyll-1.4-dioxo-2(R)-(phenylmethyl)butyll-
N°-f2,3-
dioxo-3-methoxy-1-~(phenylmethyl~eropyll-L-prolinamide hydrochloride
A. N'-f4-f4-fN-f(1.1-dimethylethoxy)carbonyllmethylaminolpiperidinyll-1,4
dioxo-2(RL(phenylmethyl)butyll-N°-f2(R)-hydroxy-3-methoxy-3-oxo-1
(phen~methyl)prop~rll-L-prolinamide
The compound was prepared in a manner according to the procedure described
as General Procedure A. The product of Example 1C (85 mg) and 4-[4-[N-[(1,1-
dimethylethoxy)carbonyl]methylamino]piperidinyl]-2{R)-(phenylmethyl)-1,4-
butanedioic
acid (100 mg) were coupled to give 160 mg of crude product which was purified
on
silica eluted with ethyl acetate-hexanes followed by ethyl acetate, to yield
110 mg (64°~)
of product, (TLC R, 0.85 in 18/2/1 HCCI3-ethanol-NH,OH). LSIMS 693 (MH+,
100%),
387 (9096).
B. N'-f4-f4-fN-f(11-dimethylethoxy)carbonyllmethylaminolpiperidinyll-1,4-
dioxo-2(R)~-(phenylmethyl)butyll-N°-f2,3-dioxo-3-methoxy-1-
(phenylmethyl)propyll-L-
prolinamide
The compound was prepared in a manner according to the procedure described
as General Procedure C. The product of the preceding Example (90 mg) was
treated
WO 93/25574 PCT/US93/03625
213'~83~
-52-
with the periodinane, and the crude product (78 mg) isolated by ethyl acetate
extraction
was purified on silica, eluted with ethyl acetate followed by 2°6 and
496 ethanol in ethyl
acetate to yield the title product (45 mg, 50%), (TLC R, 0.22 in ethyl
acetate). LSIMS
691 (MH+, 4096), 387 (10096).
C. N'-f4-f4-(methylamino)piperidinyll-1.4-dioxo-2(RL(phenylmethyl)butyl]-
N°-
j2,3-dioxo-3-methoxy-1-(phenylmethyl)propyll-L-prolinamide hydrochloride
The compound was prepared in a manner according to the procedure described
as General Procedure B. The product of the preceding Example (37 mg) was
treated
with HCI-dioxane for 1 hour at 25°C to give 30 mg of the title
substance.
LSIMS 623 (MH++CH30H, 48°~), 591 (MH+), 287 (1000.
Example 12
N-f2,2-Difluoro-4-(N-fN-f(1,1-dimethylethoxy)carbonyll-L-leucyl-L-
prolyllaminol l ,3-
dioxo-5-phenylpent-1-yll methylamine
A. N-f2,2-Difluoro-4-fN-f(1,1-dimethylethoxy)carbonyllamino-3(R)-hydroxy-1-
oxo-5-phenylpent-1-yll methvlamine
A large excess of anhydrous methylamine was introduced into a solution of
ethyl
2,2-difluoro-4-[N-[(1,1-dimethylethoxy)carbonyl]amino]-3(R)-hydroxy-5-
phenylpentanoate
(J. Med. Chem. 1986, 29, 2080-2087, 200 mg) in ethanol (2 mL) at 0°C.
After 2 hours
at 25°C the mixture was concentrated and dried to yiaid 182 mg
(94°~) of a colorless
solid, (TLC R, 0.59 in ethyl acetate).
LSIMS 359 (MH+, 60~°), 303 (70%), 259 (100°~).
B. N-f2.2-Difluoro~l-amino(R)-hydroxy-1-oxo-5-phenylpent-1-yllmethylamine
~drochloride
The product of the preceding example (173 mg) was converted by General
Procedure B to the title substance (138 mg, 100%), (TLC R, 0.5 in 18/2/1
CHzCI~-
ethanol-NH40H).
LSIMS 259 (MH'", 10096).
C. N-f2,2-Difluoro-4-ffN ~1,1-dimethylethoxy)carbonyll-L-prolyllaminol-3(R)-
~droxy-1-oxo-5-phenylpent-1-yll methylamine
The compound was prepared in a manner according to the procedure described
as General Procedure A. The product of the preceding Example (123 mg) and Boc-
L-
proline (86 mg) were coupled to yield 175 mg of crude product which was
suspended
WO 93/25574 2 ~ ~ ~ g ~ 2 PCT/US93/03625
-53-
in dichloromethane and filtered. The filtered solid weighed 80 mg and was
homogeneous by TLC (R, 0.41 in 1:1 ethyl acetate-hexane).
LSIMS 456 (MH*, 9096), 356 (100°~).
D. N-f22-Difluoro-4.-ffL-prolylaminol-3(R)-hydroxy-1-oxo-5-phenylpent-1-yll
methylamine hydrochloride
The compound was prepared in a manner according to the procedure described
as Procedure B. The product of the preceding example (60 mg) was treated with
HCI-
dioxane to yield 66 mg of the title substance, (TLC R, 0.46 in 18:2:1 CHZCIZ
ethanol-
NH40H). ' H NMR showed that the sample contained p-dioxane.
LSIMS 356 (MH+, 10096).
E. N-f2 2-Difluoro-4-fN-fN-f(1.1-dimethvlethoxylcarbonvll-L-leucvl-L-
prolyllaminol-3 R)-hydroxY 1-oxo-5-phenvlpent-1-yll methvlamine
The compound was prepared in a manner according to the procedure described
as General Procedure A. The product of the preceding example (60 mg) was
coupled
to Boc-L leucine (38 mg) and the crude product (84 mg) was purified on silica
eluted
with ethyl acetate-hexane to yield the title substance (49 mg, 58°~),
TLC (R, 0.32 ethyl
acetate).
LSIMS 569 (MH+, 100°~), 469 (85°~), 356 (50°6).
F. N-f2 2-Difluoro-4-fN-fN-f~~1 1-dimethylethoxv)carbonvll-L-leucvl-L-
prolyllaminol-1 3-dioxo-5-phenYlpent-1-yll methylamine
The compound was prepared in a manner according to the procedure described
as General Procedure C. The product of the preceding Example (46 mg) was
treated
with the periodinane, and the crude product (54 mg), isolated by ethyl acetate
extraction, was purified on silica eluted with ethyl acetate-hexanes, to yield
the title
product (28 mg, 62~). LSIMS 585 (MH++HzO, 20°~), 567 (MH+,
60°0), 467 (1000, 354
(3096).
Example 13
N-(morpholino-1-carbonyl)-L-phenylalanyl-N-f2-(2-benzothiazolyl)-2-oxo-1-
jphenylmethyllethyll-L-prolinamide
A. N~mort~holino-1-carbonyl-L-phenylalanyl-L-proline benzvl ester
N-(morpholino-1-carbonyl)-L-phenylalanine (12.6 g) and L-proline benzyl ester
hydrochloride (10.0 g) were coupled according to General Procedure A and the
crude
WO 93/25574 PCT/US93/03625
2 ~3'~8'~~
product purified on silica gel eluted with ethyl acetate-hexanes followed by
ethyl acetate
to yield 10.0 g (5396) of the title product, (TLC R, 0.26 in ethyl acetate).
LSIMS 466 (MH+, 2896), 309 (5096);' 119 (10096).
B. N-(morpholino-1-carbonyl)-L-phenylalanyl-L-proline
and
~momholino-1-carbonyl)-L-phenylalanyl-L-proline dicyclohexylamine salt
A solution of the product of the preceding Example (9.9 g, 21.0 mmol) in
methanol (90 mL) and acetic acid (10 mL) was shaken with 1096 palladium-on-
carbon
(1.1 g) at 25°C under 45 p.s.i. hydrogen pressure for 30 minutes. The
mixture was
filtered through Supercel (trademark), the cake washed with 10:1 methanol-
acetic acid,
and the filtrate concentrated. The resulting oil was dissolved in chloroform
(200 mL)
and the resulting solution washed with water (3 x 40 mL), brine, dried (MgS04)
and
concentrated to yield 6.44 g (8296) of the free acid, (TLC R, 0.52 in 18/2/1
HCCI3-
ethanol-acetic acid). A portion (0.5 g) of this material was dissolved in
dichloromethane
and 0.26 mL (1.0 equiv) dicyclohexylamine was added. The mixture was
concentrated
and the residue dissolved with heating in ethyl acetate (5 mL). Hexane (15 mL)
was
added while the mixture was kept at reflux. Cooling (0°C) produced a
solid which was
recrystallized in like fashion from ether (2 mL) and hexanes (20 mL) to yield
430 mg of
a granular powder.
Anal. Calcd for C,9Hz5N305~C,zHZ3N~H20: C, 64.78; H, 8.77; N, 9.75. Found:
C, 64.86; H, 8.67; N, 9.71.
c. l~R,s~. 2cs~-1-(2-benzothiazolyl)-2-jN-U1.1-
dimethylethoxy)carbonyllaminol-3-phenyl-1-propanol
N-Butyllithium in hexane (1.88 mL, 4.7 mmol) was added dropwise to a -
78°C
solution of benzothiazole (0.62 g, 4.6 mmol) in tetrahydrofuran (5 mL) and the
resulting
mixture was stirred at -78 ° C for 30 minutes. A solution of 2-[N-[(1,1-
dimethylethoxy)carbonyl]amino)-3-phenyl-1-propanal was added and the mixture
was
stirred for 30 minutes at -78°C. The mixture was warmed at 0°C
and treated with
saturated aqueous NH4CI (6 mL). Ethyl acetate (100 mL) was added and the
resulting
solution was washed twice with water, brine, dried (MgS04) and concentrated to
yield
1.17 g of crude product which was purified on silica eluted with ethyl acetate-
hexanes
to yield the title compound (220 mg, 12%).
LSIMS 385 (MH+, 20%), 309 (100%).
~1.3'~~3~
.. WO 93/25574 PCT/US93/03625
-55-
D. 1(R S) 2(S)-1-(2-benzothiazoly,,-2-amino-3-phenyl-1-propanol
~drochloride
The compound was prepared in a manner according to the procedure described
as Procedure B. The product of the preceding Example (215 mg) was converted to
the
title hydrochlorides (189 mg), (TLC R, 0.75 and 0.72), 18/2/1 CHZCIZ-ethanol-
NHQOH).
EI-MS 284 (M+).
E. N-(momholino-1-carbonyl)-L-phenylalanyl-N-f2-(2-benzothiazolvl)-1 (R,S)-
hydroxy-1-(S)-phenylmethyl ethyll-L-prolinamide
The product of Example 13B (DCHA salt) (395 mg) was coupled to the product
of the preceding Example (189 mg) using General Procedure A, to yield 285 mg
of
crude product which was purified on silica eluting with a gradient (0.5-496)
of ethanol
in dichloromethane. Yield, 191 mg, 5096, (TLC R, 0.74, 9:1 dichloromethane-
ethanol).
RP-HPLC (50/50 System A) showed a 1.2:1 mixture of isomers.
LSIMS 642 (MH~, 3096), 309 (10096).
F. N Imomholino-1-carbon)~-L-c~henylalaryl-N-f2-(2-benzothiazolyl)-2-oxo-1-
~henylmethyl)ethvll-L-prolinamide
The compound was prepared in a manner according to the procedure described
as General Procedure C. The product of the preceding Example (191 mg) was
treated
with the periodinane, and the crude product (100 mg), isolated by ethyl
acetate
extraction, was purified on silica eluted with ethyl acetate to yield the
title product (30
mg, 1696), (TLC R, 0.56 in 18/2/1 CHZCIZ-ethanol-NHQOH).
LSIMS 640 (MH+, 90°6), 380 (4096), 309 (4096).
Example 14
N (morpholino-1-carbonyl)-L-phenylalanyl-N-f3 3.3-trifluoro-2-oxo-1 (S)-
jphenylmethyl)propyll-L-prolinamide ,
A. N f3 3 3-Trifluoro-2-hydroxv-1-(phenylmethyl)propyllbenzamide
Sodium borohydride (1.43 g, 37.7 mmol) was added to a solution of N-[3,3,3-
trifluoro-2-oxo-1-(phenylmethyl)propyl]benzamide (racemate, J. Med. Chem.
1990, 33,
394-407, compound 4c therein) in absolute ethanol (100 mL) at 25°C.
After 4 hours
the mixture was poured into a mixture of ice, excess 6N HCI, and chloroform,
and the
resulting mixture extracted repeatedly with chloroform until no more solid
remained.
The combined organic layers were dried and concentrated to yield the title
compound
(9.51 g, 7896).
WO 93/25574 PCT/US93/03625
2137$2
a.
-56-
LSIMS 324 (MH'", 10096).
B. 3-Amino-4-phenyl-1,1,1-trifluoro-2-butanol Hydrochloride
The product of the preceding Example (9.5 g, 29.6 mmol) was dissolved in a
mixture of 12N HCI (400 mL), H20 (200 mL) and ethanol (200 mL) and the
resulting
solution was heated at reflux for 24 hours. The mixture was concentrated and
the
residue, a colorless solid, was dissolved in HzO. The resulting solution was
extracted
with ether (5 x 100 mL) and the aqueous layer concentrated in vacuo. The
resulting
light yellow solid was recrystallized from ethyl acetate-hexanes to yield the
title
substance (3.9 g, 52~), (TLC R, 0.70 in 18/2/1 CHZCIz-ethanol-NH40H). Further
material precipitated and was filtered from the mother liquors (3.1 g).
C. N-(momholino-1-carbonyl-L-phenylalanyl-N-f2i(R.S)-hydroxy-3.3.3-trifluoro-
llS~-(phenylmethyl)propyll-L-prolinamide
and
N-(momholino-1-carbonyl)-L-phenylalanyl-N-f2(R.S)-hydroxy-3,3,3-trifluoro-
~R)-(phenylmethyl~propyll-L-prolinamide
The product of Example 13B (free acid, 404 mg) and the product of the
preceding Example (250 mg) were coupled according to General Procedure A, to
yield
500 mg of crude product which was purified on silica eluting with ethyl
acetate-hexanes.
Three fractions were thus obtained (distinguished by T~C): less polar material
(135 mg,
2496, TLC R, 0.21 in ethyl acetate), more polar material (164 mg, 29%, TLC R,
0.16 it
ethyl acetate), and a mixture (119 mg, 2196) of the above. Less polar band:
LSIMS 577
(100°6, MH+), 317 (50°~), 261 (50°~), 233 (40%). More
polar band 577 (MH+, 100%),
317 (80%), 261 (80°~), 233 (65°6).
D. N-(morpholino-1-carbon~rl)-L-phenylalanyl-N-f3.3,3-trifluoro-2-oxo-1(S)-
~phenylmethyl)propyll-L-prolinamide
The less polar product of the preceding Example ((125 mg) was converted by
Procedure C (ethyl acetate extraction) to 117 mg of crude material which was
purified
on silica gel eluted with ethyl acetate-hexanes followed by ethyl acetate to
yield 72 mg
(57°6) of the title substance, (TLC R, 0.40 in ethyl acetate). By HPLC
(50/50 System B),
less than C.596 of the isomeric product of Example ? 5 was present in this
sample. No
isomerization of the title substance to the product of Example 15 occurred in
24 hours
at 25°C in 1:1 acetonitrile-pH 7 phosphate buffer (HPLC, System B). In
contrast, a
WO 93/25574 ~ ~ 3'~ g ~ PCT/US93/03625
-57-
sample stored in 1:1 acetonitrile-pH 9 borate buffer for 18 hours isomerized
to a 1.2:
1 mixture of the title substance and that of Example 15, respectively.
LSIMS 593 (MH++H20, 50%), 575 (MH+, 40%), 261 (90%), 233 (10096).
Example 15
N-(morpholino-1-carbonyl)-L-phenylalanyl-N-(3.3.3-trifluoro-2-oxo-1 (R)-
(phenylmethyl)prop~rll-L-prolinamide
The more polar product of the preceding Example (155 mg) was converted by
Procedure C (ethyl acetate extraction) to 119 mg of crude material which was
purified
on silica gel eluted with ethyl acetate-hexanes followed by ethyl acetate to
yield 55 mg
(3596) of the title substance, (TLC R, 0.20 in ethyl acetate). By HPLC (50/50
System B),
about 596 of the product of Example 14D was present in this sample (retention
times
4.96 (product of the instant Example) and 5.46 minutes (product of Example
14d). This
material stored in 1:1 acetonitrile-pH 9 borate buffer for 18 hours isomerized
to a 1:1.2
mixture of the title substance and that of Example 14D, respectively.
LSIMS 593 (MH''+H20, 60%), 575 (MH+, 20%), 333 (40%), 261 (70%), 233
(4096).
Example 16
N-( 4-oxopiperidine-1-carbonyl)-L-phenylalanyl-N-(3.3.3-trifluoro-2-oxo-1 (S)-
iphenvlmethyl~propyll-L-prolinamide
A. N-(4-oxopiperidine-1-carbonyl)-L-phenvlalanyl-L-proline benzyl ester
The product of Example 10B (30.5 g) was coupled to proline benzyl ester using
General Procedure A and the crude product (45.3 g) triturated three times with
hexanes
to yield 43.4 g of an orange solid. A portion (38.1 g) of this material was
dissolved with
heating in 260 mL ethyl acetate and 100 mL hexanes was added. The resulting
solution
on standing 15 hours at 25°C deposited light beige crystals which were
collected by
filtration and washed with hexanes to yield 5.44 g of the title substance,
(TLC R, 0.16
in ethyl acetate). Anal. Calcd for CZ~H3, N3O5: C, 67.91; H, 6.54; N, 8.80.
Found: C.
67.35; H, 6.37; N, 8.67.
B. N-(4-oxopiperidine-1-carbonyl)-L-phenylalanvl-L-aroline
The product of the preceding Example (3.0 g, 6.28 mmol) was shaken with 300
mg of 2096 Pd(OH)2/C in 30 mL methanol and 3 mL acetic acid for 1 hour at
25°C and
50 p.s.i. hydrogen pressure. The resulting mixture was filtered through
Supercel
(trademark) and the cake washed well with methanol. The filtrate was
concentrated and
PCT/US93/03625
-58-
the residue dissolved in ethyl acetate.(50 mL). The resulting solution was
washed with
water (3 x 25 mL) and brine (25 mL). Tetrahydrofuran (25 mL) and 1 N HCI (25
mL)
were added, and the resulting mixture stirred at 25°C for 15 minutes.
The aqueous
layer was separated and the organic layer dried and concentrated. The dried
residue
weighed 510 mg (2196) and was homogeneous by TLC (R, 0.3 in 18/2/1 CHCI3-
ethanol-
acetic acid).
LSIMS 388 (MH+, 8596), 273 (100%), 245 (55°~). The aqueous layers
were
saturated with NaCI and extracted twice with chloroform to yield an additional
1.80 g
(7496) of product.
C. N-(4-oxopiperidine-1-carbonylLphenylalanyl-N-12(R,S1-hydroxy-3,3.3-
trifluoro-1 (S)-(phenylmethyl)propyll-L-prolinamide
and
N-(4-oxopiperidine-1-carbony,-L-phenylalanyl-N-12(R,S)-hydroxy-3.3,3-
trifluoro-1 (R)-(phenylmethyl)propyll-L-prolinamide
The product of the preceding Example (500 mg) and the product of Example
14B (300 mg) ~r:ere coupled according to General Procedure A, to yield 659 mg
of
crude product which was purified on silica eluting with ethyl acetate-hexanes,
ethyl
acetate, and 196 ethanol in ethyl acetate. Three fractions were thus obtained
(distinguished by TLC): less polar material (184 mg, 27°~; TLC R, 0.58
in 18/2/1
CHZCIZ ethanol-NH40H), more polar material (228 mg, 33%; TLC R, 0.44 in 18/2/1
CHZCIZ ethanol-NH40H), and a mixture (121 mg, 17~) of the above.
D. N-(4-oxopiperidine-1-carbonyll-L-phenylalanyl-N-(3,3.3-trifluoro-2-oxo-1
(S)-
(phenylmethyl)propyll-L-prolinamide
The less polar product of the preceding example (163 mg) was oxidized
according to General Procedure C, and the crude product (116 mg), isolated by
chloroform extraction, was purified on silica eluted with ethyl acetate-
hexanes, ethyl
acetate, and 196 ethanol in ethyl acetate to yield the title substance (98 mg,
84°~), (TLC
R, 0.16 in ethyl acetate). By RP-HPLC (40/60 System B) this product contained
3°~ of
the less-retained product of Example 17.
LSIMS 605 (20°~6, MH''+HZO), 587 (40%, MH+), 333 (80%), 315 (70%),
273
(7096), 245 (10096).
Anal. Calcd for C3°H33N405F3~1.5 HZO: C, 58.71; H, 5.91; N, 9.13.
Found: C,
59.04; H, 5.82; N, 8.96.
WO 93/25574 ~ ~ ~ ~ ~ PCT/US93/03625
-59-
Example 17
~4-oxopiperidine-1-carbony~~-L-phenylalanyl-N-13.3.3-trifluoro-2-oxo-1 (Rl-
~ahenylmethyl)aropyl-L-prolinamide
The more polar product of Example 16D was (153 mg) was oxidized according
to General Procedure C, and the crude product (90 mg), isolated by chloroform
extraction, was purified on silica eluted with ethyl acetate-hexanes, ethyl
acetate, and
196 ethanol in ethyl acetate to yield the title substance (35 mg,
23°~). RP-HPLC
indicated this product contained 796 of the more retained product of Example
16D.
LSIMS 633 (MH'+CzH50H, 4096), 605 (3096, MH'+H20), 587 (MH+,
40°~6), 361
(6096), 333 (4096), 315 (6096), 273 (70°r6), 245 (70~).
Examele 18
N-(,morpholino-1-carbonyl)-L-phenylalanyl-N-f 3.3, 3-trifluoro-2-oxo-1 !S)-
jcyclohex~methyl)pro~yll-L-prolinamide
A. 2-Nitro-1-cyclohexylethane
2-cyclohexylethyl bromide (50 g, 0.26 mol) was added to a mixture of sodium
nitrite (31 g, 0.45 mol) in dimethylformamide (500 mL) at 5 ° C and the
mixture was
stirred for 10 hours at 5°C and for 8 hours at 20°C. The
resulting solution was poured
into ice-water (1.5 L) and the mixture extracted with petroleum ether (3 times
150 mL).
The organic layers were combined and washed with water, dried, and
concentrated.
The residue was distilled through an 8 inch fractionating column to yield a
lower-boiling
material and 18.3 g of colorless liquid, by 70-80°C at 2 Torr. The
latter was redistilled
in the same apparatus with further separation of a low boiling fraction to
yield 16.4 g
of colorless liquid, by 45-55°C at 0.5 Torr, containing less than
2°0 of the lower boiling
impurity which was identified as the corresponding nitrite ester. Anal. Calcd
for
CeH,5NOZ: C, 61.12; H, 9.62; N, 8.91. Found: C, 60.57; H, 9.47; N, 9.37.
B. 3-Nitro-4-c~clohexyl-1.1.1-trifluoro-2-butanol
CAUTION! A caution has been raised that the distillation of substances of this
type can lead to explosion (USP 5055450).
A mixture of the product of the previous Example (15.2 g, 96.7 mmol) and
anhydrous potassium carbonate (200 mg, 1.45 mmol) was treated at 25 ° C
with
trifluoroacetaldehyde hydrate (16.8 g, 145 mmol and the mixture was stirred at
50°C
for 4 hours and at 60°C for 8 hours. The mixture was chromatographed on
100 g silica
eluted with 1:10 ether-hexanes to yield 24 g of impure product which was
WO 93/25574 PCT/US93/03625
213'~8~~
-60-
chromatographed on 300 g silica eluted with 1:15 and 1:10 ether-hexanes to
yield 20.2
g (8296) of the title product as a light: yellow oil, (TLC R, 0.50 and 0.42 in
1:2 ether-
hexanes).
C. 3-Amino-4-cyclohexYl-1.1,1-trifluoro-2-butanol
A solution of the product of the preceding Example (20.1 g, 0.078 mol) in
ethanol
was shaken with water-wet Raney nickel (5 g) (Aldrich Chemical Co.) under 45
p.s.i.
hydrogen pressure at 25°C for 16 hours. The mixture was filtered
through Supercel,
and the cake washed well with methanol. The filtrates were concentrated to
yield a
greenish waxy solid which was washed on the filter with hexanes and dried;
11.02 g
(6296) of the title substance was thus obtained as a colorless solid, (TLC R,
0.1 (major
substance) and 0.3 (minor substance) in ethyl acetate).
D. N-(morpholino-1-carbonyl)-L-phenylalanyl-N-i2-hydroxy-3.3.3-trifluoro-1-
(cyclohexylmethyl)propyll-L-prolinamide
The product of Example 13B (363 mg) was coupled to the product of the
preceding Example (239 mg) by General Procedure A, and the crude product (514
mg)
was purified on silica and sequentially eluted with ethyl acetate-hexanes,
ethyl acetate,
196 and 296 ethanol in ethyl acetate to yield 446 mg (79°.6) of the
title substance as a
mixture of (presumably four) isomers, (TLC R, 0.61 and 0.59 in 18/2/1 CH2CI2-
ethanol-
NH40H).
LSIMS 583 (MH+, 60°'0), 323 (50%), 307 (100%).
E. N-(morpholino-1-carbonyl-L-phenylalanyl-N-13.3,3-trifluoro-2-oxo-1 (S)-
~cyclohexylmethyl)propyll-L-prolinamide
The product of the preceding Example (419 mg) was oxidized by General
Procedure C to give 372 mg of crude product, isolated by dichloromethane
extraction,
which was purified on silica and sequentially eluted with 1 %, 2°~ and
4°~ ethanol in 1:1
ethyl acetate-dichloromethane. Two fractions were obtained, the less polar
title
substance (39 mg 9°~6; TLC Rf 0.21 in 1:1:0.1 ethyl acetate-CHZCIz-
ethanol), and the
product of the following Example.
LSIMS 613 (MH++CH30H, 10%), 599 (MH++HZO, 10%), 581 (MH+, 40%), 261
(100°~), 233 (6096).
WO 93/25574 ~ ~ ~ ~ ~ c°'! ~ PCT/US93/03625
-61-
F. N-~mornholino-1-carbonyl)-L-phenylalanyl-N-f3.3,3-trifluoro-2-oxo-1 (R.S)-
Lyclohexylmethyl)propyll-L-prolinamide
The second product to elute from the chromatography of the product of the
preceding example was a mixture of the above substance and the more polar 1
(R)
isomer (294 mg, 7096; TLC R, 0.15 in 1:1:0.1 ethyl acetate-CH2CIz-ethanol).
The two
substances contained in this product were not distinguishable by RP-HPLC in
System
B (60/40).
LSIMS 627 (MH++CZH~OH, 599 (MH++HZO), 581 (MH+ 33°~), 261
(100°~), 233
(6096) .
Anal. Calcd for CZ9H3sNoOsFs'2.3 H20: C, 55.99; H, 7.06; N, 9.01. Found: C,
56.17; H, 6.67; N, 8.54.
Example 19
N-(4-(methylamino)piperidine-1-carbonyl)-L-phenylalanyl-N-f 3.3,3-trifluoro-2-
oxo-
~S)-(phenylmethvl)prop~rll-L-prolinamide
A. N-(4-(methylamino)piperidine-1-carbony,-L-phenylalanyl-L-proline benzyl
ester
Sodium cyanoborohydride (441 mg, 1.2 equiv) was added to a mixture of the
product of Example 16A (2.79 g, 5.84 mmol), methylamine hydrochloride (1.972
g, 29.2
mmol), anhydrous sodium acetate (4.79 g, 58.4 mmol), and 3~ molecular selves
(600
mesh, 2.8 g) in methanol (30 mL) at 0°C and the mixture was stirred at
25°C for 16
hours. The mixture was filtered through Supercel (trademark) and the filtrate
concentrated. The residue was dissolved in ethyl acetate (125 mL) and the
resulting
solution washed with 1 N NaOH (3 times 20 mL), dried and concentrated. The
residue
was chromatographed on 100 g silica packed in 1 °~6 ethanol/1 % NH40H-
dichloromethane and eluted with this solvent, followed by 2~, 5°~,
10°~, 20°~, and 40%
ethanol-dichloromethane (500 mL portions, all containing 196 NH40H), to yield
the title
substance (2.45 g, 8596), (TLC Rt 0.35 in 1:4 ethanol-dichloromethane
containing 1 °~
NH40H).
LSIMS 493 (MH', 10090, 288 (8096).
B. N-f4-fN-f(11-dimethylethoxy)carbonyllmethylaminolpiperidine-
_1-carbonyll-L-phenylalanyl-L-proline benzvl ester
Di-t-butyldicarbonate ('~.17 mL, 5.1 mmol) was added to a solution of the
product of the preceding Example (2.29 g, 4.64 mmol) in tetrahydrofuran (15
mL) at
WO 93/25574 PCT/US93/03625
21~'~8:~~
-62-
25°C and the resulting mixture was stirred at 25°C for 15 hours.
The mixture was
concentrated, the residue dissolved in ethyl acetate (100 mL) and the
resulting solution
was sequentially washed with 1 N HCI (20 mL), brine, dried, and concentrated
to yield
2.8 g of the title product, (TLC Rf 0.3 in ethyl acetate).
LSIMS 593 (MH+, 7096), 388 (10096).
C. N-f4-fN-f(1 1-dimethylethoxy)carbonyllmethylaminolpiperidine-1-
carbonyll-L-phenylalanyl-L-proline
A solution of the product of the preceding Example (2.50 g, 4.23 mmol) in
methanol (40 mL) was shaken with 300 mg 20~ Pd(OH)2/C at 50 p.s.i. hydrogen
pressure at 25°C for 15 hours. The mixture was filtered through
Supercel (trademark)
and the filtrate concentrated and dried to yield 2.36 g of the title
substance, (TLC R,
0.59 in 18/2/1 CHCI3 ethanol-HOAc).
LSIMS 503 (MH+, 50°~6), 388 (10096).
D. N-f4-fN-f(1 1-dimethylethoxy)carbonyllmethylaminolpiperidine-1-
carbonyll-L-ghenylalanyl-N-f2(R S~hydroxy-3 3 3-tr'rfluoro-1 (S)-
(cyclohexylmethyl)propyll-
L-prolinamide
and
N-f 4-f N-f ( 1 1-dimethylethoxy)carbonyll methylaminol piperidine-1-
carbonyll-L-phenylalanyl-N-f2(R S)-hydroxy-3 3 3-trifluoro-1 (R)-
(cyclohexylmethyl)propyll-
L-prolinamide
The product of the preceding Example (649 mg) was coupled to the product of
Example 14B (300 mg) by General Procedure A and the crude product (765 mg) was
purified on silica and sequentially eluted with 2.5°~, 5°~, 1096
and 20°~ ethanol in 1:1
dichloromethane-ethyl acetate to yield the less polar, first-titled product
(78 mg, 9%),
(TLC R, 0.19 in 0.1:1:1 ethanol-dichloromethane-ethyl acetate).
LSIMS 704 (35°6, MH+), 388 (100°~).
Also obtained was the more polar, second-titled product (194 mg,
23°~), (TLC
R, 0.09 in the same TLC system), LSIMS 704 (35°~), 388 (100°~).
A third fraction
containing 208 mg (2596) of a mixture of the two products was also obtained.
WO 93/25574 2 1 ~ ~ ~ PCT/US93/03625
-63-
E. N !4-!N-!(1 1-dimethylethoxy~carbonyllmethylaminolpiperidine-1-
carbonyll L phenylalanyl-N-!3 3 3-trifluoro-2-oxo-1 (S)-(phenylmethyl)propvll-
L-
prolinamide
The first titled, less polar product of the preceding Example (65 mg) was
oxidized by General Procedure C to yield crude product (6796), which was
isolated by
chloroform extraction. The crude product was purified on silica and eluted
with ethyl
acetate-dlchloromethane to yield 52 mg of the title substance, (TLC R, 0.24 in
0.1:1:1
ethanol-dichloromethane-ethyl acetate).
LSIMS 748 (MH++CZH50H, 596), 720 (MHt+HZO, 1096), 702 (MH', 18°x),
388
(10096).
The title product was also obtained alternatively by oxidation of the third
fraction
referred to in Example 19D (mixture of first and second-titled products of
Example 19D)
(188 mg) according to General Procedure C, to yield crude product (191 mg)
which
was isolated by chloroform extraction and purified on silica, and sequentially
eluted with
196, 296 and 496 ethanol in 1:1 dichloromethane-ethyl acetate. The less polar
substance
(91 mg) was identical by TLC and NMR to that obtained immediately above and
different from the product of Example 20A. A mixture of this substance and a
more
polar product was also obtained (99 mg).
F. N (methylaminolpiperidine-1-carbonyl~L-phenvlalanyl-N-13.3.3-trifluoro-2-
oxo 1 (S) (phenylmethy~propyll-L-prolinamide hydrochloride
The product of the preceding Example (78 mg) was dissolved in trifluoroacetic
acid (1 mL) at 25°C for 30 minutes. The mixture was concentrated, the
residue
dissolved in ethanol (1 mL) and the resulting solution treated with 0.20 mL 1
N HCI. The
solution was concentrated and the crude product ground to a fine powder,
triturated
with ether and dried (61 mg, 8796). The product of Example 20B could not be
detected
in this material by RP-HPLC (30/70), System B.
LSIMS 634 (MH++CH30H), 602 (MH+, 309 (33°~). A sample which was
heated
at 55°C for 18 hours showed about 20~ conversion to the product of
Example 208.
WO 93/25574 PCT/US93/03625
213783
Example 20
N (4-(methylamino)piperidine-1-carbonyl)-L-phenylalanyl-N-f3,3,3-trifluoro-2-
oxo-
1 (R)-(phenylmethvl)propyll-L-prolinamide
A. N-f4-fN-f(1 1-dimethylethoxyycarbonyllmethylaminolpiperidine-1
carbonyll L phenylalanyl-N-f3 3 3-trifluoro-2-oxo-1 (R)-(phenylmethyl)propyll-
L
~rolinamide
The second-titled, more polar product of Example 19E (174 mg) was oxidized
by General Procedure C to yield crude product (168 mg), isolated by chloroform
extraction, which was purified on silica eluted with 1 °~, 2%, and 496
ethanol-
dichloromethane to yield the title substance (124 mg, 71 °~6), (TLC R,
0.11 in 0.1:1:1
ethanol-dichloromethane-ethyl acetate).
LSIMS 748 (MH++C2H50H, 1096), 702 (MH+, 20%), 388 (100°6).
B. N-(4-(methylamino)piperidine-1-carbonY!Lphenylalanyl-N-f3.3,3-trifluoro-
2-oxo-1 (R)-(phenylmethy,,propyll-L-prolinamide
The product of the preceding Example (54 mg) was treated sequentially with
trifluoroacetic acid and 1 N HCI as described for the preparation of the
product of
Example 19F. The crude product was redissolved in ethanol, filtered,
concentrated,
and the residue triturated with ether and dried at 25°C to yield 35 mg
(72°~) of a
colorless powder. The product of Example 19F was distinguishable from and not
present in this product by RP-HPLC.
LSIMS 648 (MH++CZH50H, 5~), 634 (MH'+CH30H, 20%), 620 (MH++HzO,
10°~), 602 (MH~, 15°~), 288 (100%). Anal. Calcd for
C3,H39N504CIF,~3Hz0: C, 53.79;
H, 6.55; N, 10.12. Found: C, 53.82; H, 6.25; N, 9.97.
Example 21
N f4-fN-methylaminolpiperidine-1-carbonyll-L-valvl-N-f3.3,3-
trifluoro 2 oxo 1 (S)-(phenylmethyl)propyll-L-prolinamide hydrochloride
A. N-(4-[N-[(1,1-dimethylethoxy)carbonyl]methylamino)piperidine-1-carbonyl)
-L-valine benzyl ester
A solution of L-valine benzyl ester p-toluenesulfonate (7.13 g, 18.8 mmol) and
triethylamine (2.90 mL, 20.6 mmol) in dichloromethane (40 mL) was added to a
stirred
0-5°C slurry of carbonyldiimidazole (3.34 g, 20.6 mmol) and imidazole
(2.56 g, 37.6
mmol) in dichloromethane (95 mL) and the resulting mixture was stirred at
25°C for 30
min. A solution of 4-[N-[(1,1- dimethylethoxy)carbonyl]methylamino)piperidine
(EP
... WO 93/25574 ~ 1 ~ ~ g ~ 2 PCT/US93/03625
-65-
457,686, Example 11 B therein, 4.00 g, 18.8 mmol) in dichloromethane (20 mL)
was
added and the mixture stin-ed at 25°C for 7 days. The resulting
solution was washed
with 2N HCI (2 x 100 mL), 1 N NaOH, dried, and concentrated. The residue was
chromatographed on silica eluted with ethyl acetate-hexanes giving the title
substance
(5.54 g, 6696). ' H NMR (CDCI3) a 0.84 (d, 3H, J= 6.9 Hz), 0.91 (d, 3H, J =
6.9 Hz),
1.4.4 and 1.45 (s, 1:9 ratio, 9H total), 1.5-1.7 (m, ca. 4H), 2.14 (m, 1 H),
2.68 and 2.71
{s, ca. 9:1 ratio, 3H total), 2.85 (m, 2H), 4.04 (br and m, 3H total), 4.48
(dd, 1 H, J = 4.7,
8.4 Hz), 4.94 (d, 1 H, J = 8.3 Hz), 5.10 (d, A of AB, 1 H, J = 12.2 Hz), 5.20
(d, B of AB,
1 H, J = 12.2 Hz), 7.34 (m, 5H). Anal. Calcd for C2,H3,N305~0.25 H20: C,
63.76; H,
8.36; N, 9.29. Found: C, 63.98; H, 8.11; N, 9.07.
B. N y4 fN ((1 1-dimethylethoxy)carbonyllmethylamino)piperidine-1-carbonyl)
-L-valine
The product of the preceding example (5.14 g, 11.5 mmol) was added to 1.5 g
of Pd(OH)2/C (2096 Pd content, Aldrich) in methanol (65 mL) and the resulting
mixture
was shaken under 30 p.s.i. hydrogen pressure for 30 min. The mixture was
filtered
through methanol-washed ~Iter aid and the filtrates were concentrated giving
the title
product as a colorless foam (3.96 g, 9796). ' H NMR (CDCI3) a 0.94 (d, 3H, J =
6.8 Hz),
0.98 (d, 3H, J = 6.8 Hz), 1.45 (s, 9H), 1.5-1.75 (m, 4H), 2.19 (m, 1 H), 2.69
(s, 3H,
NCH3), 2.85 (m, 2H, CONCHZ), 4.2- 4.0 and 4.04 (br and m, 3H total, CONCH and
CONCHz), 4.25 (dd, 1 H, NCHCO), 5.07 (d, 1 H, J = 8.1 Hz, NH). Anal. Calcd for
C"H3,N,05: C, 57.12; H, 8.74; N, 11.76. Found, C, 55.56; H, 8.17; N, 10.92.
C. N (4 fN f(1 1 dimethylethoxy)carbonyllmethylamino)piperidine-1-carbonyl)
-L-valyl-L-proline benzyl ester
The compound was prepared in a manner according to the procedure described
as General Procedure A. The product of the preceding F~cample (2.26 g, 6.32
mmol)
was coupled to proline benzyl ester hydrochloride (1.53 g, 6.33 mmol) to yield
crude
product (2.96 g, 86°6) which was used without further purification. 'H
NMR (CDC13) a
0.90 (d, 3H, J = 6.7 Hz), 0.98 (d, 3H, J = 6.7 Hz), 1.44 (s, 9H), 1.5-1.7 (m,
ca. 4H),
1.9-2.1 (m, ca. 4H), 2.20 (m, 1 H), 2.68 (s, 3H), 2.80 (m, 2H), 3.65 (m, 1 H),
3.88 (m, 1 H),
4.0-4.2 (m, 3H), 4.45 (dd, 1 H, J = 6.7, 8.7 Hz), 4.54 (dd, 1 H, J = 5.2, 8.8
Hz), 5.12 (d,
A of AB, 1 H, J = 12.3 Hz), 5.17 (d, B of AB, 1 H, J = 12.3 Hz), 7.32 (m, 5H).
Anal.
Calcd for Cz9H,,4N408~0.5H20: C, 62.90; H, 8.19; N, 10.11. Found: C, 62.88; H,
8.16;
N, 9.99.
WO 93/25574 PCT/US93/03625
2~.~'~~~~
-66-
D. N-(4-fN-f(11-dimethylethoxy)carbonyl]~methytamino)piperidine-1-carbonyl)
-L-valyl-L-proline
According to the procedurefor the preparation of the compound of Example 21 b
the product of the preceding Example (2.83 g, 5.35 mmol) gave 2.31 g (95
~°) of the
title substance as a colorless foam. 'H NMR (CDCI3) a 0.93 (d, 3H, J = 6.6
Hz), 0.98
(d, 3H, J = 6.6 Hz), 1.44 (s, 9H), 1.5-1.7 (m, 4H), 2.05 (m, 3H), 2.20 (m,
2H), 2.68 (s,
3H), 2.80 (m, 2H), 3.62 (m, 1 H), 3.92 (m, 1 H), 4.05 in 4.0-4.2 (m over br,
3H total), 4.41
(m, 1 H), 4.53 (m, 1 H), 4.9-5.3 (br, 1 H), 5.50 (d, 1 H, J = 8.6 Hz). LSIMS
455 (MH+,
3090), 340 (100°~), 284 (30%), 256 (20°~), 185 (38°~).
Anal. Calcd for CzZH38N406~H20:
C, 55.91; H, 8.53; N, 11.86. Found: C, 55.74; H, 8.46; N, 11.57.
E. N-f4-fN-f(1 1-dimethylethoxy carbonyl, meth)~laminolpiperidine-1-carbonyll
-L valyl-N-f2(R S)-h~droxy-3 3 3=trifluoro-1 (R.S)-(cyclohexylmethyl)propyll-L-
prolinamide
The compound was prepared in a manner according to the procedure described
as General Procedure A. The product of the preceding Example (463 mg, 1.02
mmol)
was coupled to the product of Example 14B to give 673 mg (103°~) of the
title
substance as a mixture of diastereomers which was used without further
purification.
'H NMR (CDCI3, partial) a 0.9-1.0 (doublets, 6H, (CH3)ZCH), 1.45 (s, 9H,
(CH,)3C), 2.7
and 2.72 (singlets, 1:1 ratio, 3H total, NCH3), 7.1-7.4 (m, 5H, aromatic). '9F
NMR
showed four doublets (J = ca. 7 Hz) in 3:1:2:5 ratio at -74.54, -75.02, -
75.18, and -75.5
ppm, respectively. Four additional doublets were observed (8% of the total
integration)
between -76.4 and -77.5 ppm. LSIMS 656 (MH+, 20~), 340 (100%), 317 (20%), 284
(2096), 256 (18~), 185 (3596).
F. N-f4-fN-f(11-dimethylethoxy)carbonyl]methylaminolpiperidine-1-carbonyll
-L-valyl-N-f3 3 3-trifluoro-2-oxo-1 (S)-(phenylmethyl)propyll-L-prolinamide
The product of the preceding Example (560 mg, 0.87 mmol) was oxidized by
General Procedure C to give 514 mg of crude product, isolated by chloroform
extraction, which was purified on silica eluted with 1:1 dichloromethane-ethyl
acetate,
followed by 1 °~6, 296, 4°~, and 10 96 ethanol in 1:1
dichloromethane- ethyl acetate.
Three fractions were obtained, the less polar title product (151 mg), the more
polar
product of the following example, and a mixture of the two substances (127
mg). The
less polar title product contained less than 1 ~ of the more polar product of
the
following example by HPLC. 'H NMR (CDCI3, 300 mHz) a 0.79 (d, 0.4H, J = 6.8
Hz),
0.83(d,0.4H,J=6.8Hz),0.94(d,2.6H,J=6.7Hz),0.99(d,2.6H,J=6.6Hz), 1.46
WO 93/25574 ~ ~ ~ ~ PCT/US93/03625
-67-
(s, 9H), 1.55-2.0 (m, ca. 7H), 2.16 (m, 1 H), 2.68, 2.69, and 2.72 (s, ca.
2:2.5:9 ratio, ca.
3H total), 2.75-3.1 (m, ca. 3H), 3.35 (dd, 1 H), 3.88 (m, 1 H), 3.95-4.15 (m,
ca.3H), 4.37
(d, 1 H, J = 7.6 Hz), 4.55 (dd, ca. 0.2H), 4.64 (m, 1 H), 4.89 (d, 1 H, J =
7.8 Hz), 5.04 (m,
ca 0.5H), 7.1-7.35 (m, 5H), 7.4 (d, 1H). 'aF NMR (282.4 mHz, CDC13) d -76.0, -
76.5;
-82.4, -82.5 (0.5: 2:5.7:1.3 intensity ratio, respectively). The ratio of the
combined
integrals of the -82.4 and -82.5 resonances (presumably hydrate) to the -76.5
and -76.0
resonances (presumably ketone) was 6.33. It is presumed that nonequivalent
rotational
isomers were observed. LSIMS 700 (MH++CZH50H, 8~), 672 (MH'+HzO, 8~), 654
(MH+, 10°~), 340 (100°~), 284(20°6), 256 (18°~),
185 (38°6). Anal. Calcd for
1O C3zH48N5OeF3 ~ 1.25 H20: C, 56.83; H, 7.23; N, 10.36. Found: C, 56.68; H,
7.16; N,
10.14.
G. N f4-fN-f(1 1-dimethylethoxy)carbon~rllmethylaminolpiperidine-1-carbonyll
_L valyl N f3 3 3-trifluoro-2-oxo-1 (R)-(phenylmethyl)propyll-L-prolinamide
A second substance eluted from the chromatography of the product of the
preceding example (103 mg). HPLC showed it to contain about 10°~ of the
product of
the preceding example. 'H NMR (CDCI3, 300 mHz) d 0.8-1.0 (overlapping d, 6H
total),
1.45 and 1.46 (s, 9H total), 1.5-2.0 (m, ca 7 H), 2.3 (m, ca. 0.5H), 2.68 and
2.69 (s, 3H
total), 2.75- 2.9 (m, ca. 2H), 3.11 (m, ca. 0.5H), 3.21 (dt, 1 H), 3.38 (m,
ca. 0.5H), 3.5 (m,
1 H), 3.77-4.25 (m, ca. 4.5H), 4.25-4.5 (m, 1.5H), 4.55 (d, 0.5H), 5.0-5.2 (m,
1 H), 6.05
(d, 0.2H), 6.75 (d, 0.3H), 7:1-7.35 (m, 5H), 7.75 (d, 0.2H). '9F NMR (CDCI3,
282.4 mHz)
a -75.9 (s, 0.45 F), -76.1 (s, 0.12 F), -76.5 (0.03 F), -76.6 (s, 0.72 F), -
77.0 (s, 0.12F),
-81.5 (s, 0.03F), -81.7 (s, 0.87 F), -82.3 (s, 0.21 F), -82.7 (s, 0.45 F). The
-76.5 and -82.3
resonances (0.25 F, ca. 1096 of total) are attributed to the 1 (S)
diastereomer (product
of Example 21f) which was present by HPLC at approximately the 10°~
level. LSIMS
700 (MH''*C2H50H, 1096), 654 (MH+, 10%), 340 (100°~), 284 (20%), 256
(20%), 185
(3696). Anal. Calcd for C,2H48N5OeF3 ~ 1.25 HzO: C, 56.83; H, 7.23; N, 10.36.
Found:
C, 56.81; H, 7.18; N, 10.20.
H. N f4-fN methylaminolpiperidine-1-carbonyll-L-valyl-N-f3.3.3-trifluoro-2-oxo-
1 (S) (c~henylmethyl)propyll-L-prolinamide hydrochloride
The product of Example 21f (122 mg, 0.186 mmol) was dissolved at
0°C in
trifluoroacetic acid (4 mL) and the resulting solution was stirred at this
temperature for
15 min. The mixture was evaporated and the residue dried at 0.05 mm and
dissolved
in several mL isopropyl alcohol. The resulting solution was treated with 1 N
HCI (0.2
WO 93/25574 PCT/US93/03625
~i3'~~~~
-68-
mL) and concentrated. The residue was dried~at 0.05 Torr for 30 min and the
residual
oil triturated to a colorless solid with ether. Drying gave the title
substance (105 mg,
9696) as a colorless solid. HPLC (retention time 5.93 min, 30/70 System B)
showed
none of the isomeric substance of Example ' H NMR (D20, 300 mHz) a 0.84 (d,
3H, J
= 6.8 Hz), 0.87 (d, 3H, J = 6.8 Hz), 1.44 (m, 2H), 1.64 (m, 1 H), 1.7-1.95 (m,
3H), 2.00
(m, 1 H), 2.08 (m, 2H), 2.66 (s, 3H), 2.73 (dd, 1 H, J = ca. 9, 11 Hz), 2.88
(dt, 2H), 3.25
(m, 2H), 3.56 (m, 1 H), 3.75 (m, 1 H), 4.05 (m, 2H), 4.14 (d, 1 H, J = 8.2
Hz), 4.30 (dd,
1 H, J = 4.8, 8.5 Hz), 4.38 (dd, 1 H, J = ca. 3, 8.4 Hz), 7.1-7.25 (m, 5H).
Resonances
attributable to the ketone form (present at 1096 by'9F NMR) were not
distinguished in
this or another sample where 33°~ of the ketone was present. '9F NMR
(D20, 282.4
mHz) d -76.1 (s, 0.3 F), -82.3 (s, 2.7F). This spectrum was taken on the
identical
sample used to obtain the'H spectrum above. In a separate sample, the ratio of
these
resonances was 1:3, respectively. Two additional singlets at -82.5 comprising
1396 of
the total integration were also observed in the latter sample, and in equal or
lesser
amount in the former sample. LSIMS 554 (100, M++H), 440 (45), 315 (50), 240
(50).
Anal. Calcd for Cz,H38N5O,F, ~ HCI ~ 3.5 H20: C, 49.65; H, 7.10; N, 10.72.
Found: C,
49.96; H, 6.57; N, 10.44.
Example 22
N-f4-fN-methylaminolpiperidine-1-carbonyll-L-valyl-N-(3,3,3-trifluoro-2-oxo-1
(R)-
~phenylmethyl)propyll-L-prolinamide hydrochloride
By the procedure of the preceding example (21 h), the product of Example 21 g
(99 mg, 0.137 mmol) was deprotected with trifluoroacetic acid and converted to
the title
hydrochloride, which was triturated with ether, and thus obtained as a
colorless solid
(70 mg, 8696). HPLC (retention time 6.77 min, 30/70 system B) showed
10°~ of the
isomeric product of Example 21 h (5.9 min retention time). ' H NMR (D20, 300
mHz) a
0.8-1.1 (overlapping d, 6H total), 1.5 (m, 2H), 1.61 (m, 1 H), 1.75 (m, 1 H),
1.93 (m, ca.
3H), 2.12 (m, ca. 2H), 2.73 (s, 3H) overlapping 2.7 (m, 1 H), 2.92 (m, 2H),
3.30 (m, ca
2H), 3.46 (m,1 H), 3.78 (m, 1 H), 4.06 (m, 2H), 4.18 (d, 1 H, J = 8.4 Hz),
4.22 (t, 1 H, J =
ca. 8 Hz), 4.53 (dd, 1 H), 7.2-7.4 (m, 5H). '9F NMR (D20, 282.4 mHz) d -76.1
(s, 0.75F),
-82.1 (s, 2.25F). An additional resonance was observed at -82.3, comprising
10°~ of
the total integration, which is attributed to the hydrate resonance of the L
isomer,
product of Example 21 h, known to be a contaminant in this sample at the
10°~6 level by
HPLC. LSIMS 554 (70, M++H), 440 (45), 300 (100), 240 (70). Anal. Calcd for
.. WO 93/25574 2 ~: ~ ~ ~ PCT/US93/03625
-69-
Cz,H38N fiO4F3 ~ HCI ~ 3.5 HZO: C, 49.65; H, 7.10; N, 10.72. Found: C, 49.58;
H, 6.77;
N, 10.43.
Examele 23
N-f4-f N-methylaminol piperidine-1-carbonyll-L-valyl-N-f2,3-dioxo-3-1-
methvlethoxy-1 (S)-(phenylmethyl)propyll-L-prolinamide hydrochloride
A. N-f4-fN-f(11-dimeth~lethoxy)carbon~rllmethylaminolpiperidine-1-carbonyll
-L-val~l-N-f2(R)-hYdroxy-3-1-methylethoxy-3-oxo-1 (S)-(phenylmethyl)propyll-L-
prolinamide
The compound was prepared in a manner according to the procedure described
as General Procedure A. The product of Example 21 d (766 mg, 1.68 mmol) was
coupled to isopropyl 3(S), 2(R)-3-amino-2-hydroxy-4- phenylbutanoate (USP
4,814,342,
400 mg, 1.68 mmol) to give 1.08 g (9796) of crude product which was purified
on silica
gel eluted with 50~, 6796, 7596, and 10096 ethyl acetate in hexanes giving the
title
substance (669 mg, 6096). 'H NMR (CDCI3, partial) showed two rotamers in
approximately 1:1 ratio, d 0.94-1.01 (3-4 d, 6H total, J = ca. 7 Hz , valyl
CH3's),
1.18-1.27 (3-4 d, 6H total, J = ca. 7 Hz, OCH(CH3)z), 1.45 (s, 9H, Boc), 2.69
(2 s, 3H
total, NCH3), 4.91 and 4.99 (septets, 1 H total, J = ca. 7 Hz, OCH(CH3)z),
5.14 (d, 1 H,
J = ca. 9Hz, partially exchanges in 8h with added Dz0), 6.98 (d, 1 H, J = 9.6
Hz, little
exchange in 8h with added DZO), 7.44 (d, 1 H, J = ca. 10 Hz, little exchange
in 8h with
added DZO), 7.2-7.4 (m, 5H). LSIMS 674 (MH+, 3096), 340 (85°~), 335
(100%), 284
(15~), 256 (22°~), 185 (5096). Anal. Calcd for C35H55N508~ C. 62.39; H,
8.23; N, 10.39.
Found: C, 62.16; H, 8.21; N, 10.08.
B. N-f4-fN-f(1 1-dimethylethoxy)carbonyllmethylaminolpiperidine-1
carbonyll-L-valyl-N-f2 3-dioxo-3-1-methylethoxy-1 (S)-(phenylmethyl)propyll-L-
prolinamide
The compound was prepared in a manner according to the procedure described
as General Procedure C. The product of the preceding Example (186 mg, 0.28
mmol)
was treated with the periodinane, and the crude product, isolated by
chloroform
extraction, purified on silica eluted with 2:1 ethyl acetate-hexanes, ethyl
acetate, and 1
ethanol in ethyl acetate, giving the title substance (153 mg, 82~). ' H NMR
(CDCI3, 300
mHz) a 0.83 (d, ca. 2.2H, J= 7 Hz), 0.84 (d, ca. 2.2H, J = 6.5 Hz), 0.9 (m, ca
2H), 0..93
(d, ca. 0.7 Hz, J = ca. 7 Hz), 0.98 (d, ca. 0.7 Hz, J = ca. 7 Hz), 1.28 (d,
ca. 4.5 H, J
= 6.3 Hz), 1.29 (d, ca. 1.5 H, J = 5.7 Hz), 1.44 (s, 9H), 1.45-1.7 (m, ca 4H),
1.80 (m,
1 H), 1.88 (m, 2H), 2.2 (m, ca 1 H), 2.68 (s, 3H), 2.78 (m, 2H), 3.01 (dd, 1
H, J = 6.5, 13.8
Hz), 3.16 (dd, 1 H, J = 6.4, 14.0 Hz), 3.45 (m, 1 H), 3.72 (m, 1 H), 4.0 (m,
ca. 3H), 4.16
WO 93/25574 PCT/US93/03625
21~'~~~2
-70-
(dd, 1 H), 4.50 (m, 1 H), 5.02 (m, ca 2H), 5.24 (q, 1 H), 7.05-7.3 (m, 5H).
LSIMS 690
(MH++H20, 596), 672 (MH+, 20%), 340 (100%), 333 (3096), 284 (20%), 256 (20%),
185
(4096). Anal. Calcd for C35Hs3Ns0e ~ HzO: C, 60.94; H, 8.04; N, 10.15. Found:
C,
60.96; H, 7.82; N, 10.29.
C. N f4-fN-methylaminolpiperidine-1-carbonyll-L-valyl-N-f2.3-dioxo-3-(1-
methylethoxy) 1 (S)-(phenylmethyl)pro~eyll-L-prolinamide hydrochloride
By the procedure of example 21 h, the product of the preceding Example (120
mg) was deprotected with trifluoroacetic acid and converted to the title
hydrochloride,
which was triturated with ether, and thus obtained as a colorless solid (92
mg, 84°~).
'H NMR (DZO, 300 mHz, partial) d 0.91 (d, 3H, J = 6.7 Hz), 0.96 (d, 3H, J =
6.7 Hz),
1.00 (d, ca. 1 H, J = 7.1 Hz), 1.27 (d, 3H, J = 6.5 Hz), 1.32 (d, 3H, J = 5.9
Hz), 1.54
(m, 2H), 1.67 (m, 1 H), 1.8-2.1 (m, ca 3H), 2.15 (m, 2H), 2.7 (dd, 1 H, J =
11, 14 Hz),
2.74 (s, 3H), 2.95 (m, 2H), 3.18 (dd, 1 H, J = 12, 14 Hz), 3.46 (m, 1 H), 3.64
(m, 1 H),
3.78 (m, 1 H), 4.1 (m, 2H), 4.22 (d, 1 H, 7.9 Hz), 4.35 (dd, 1 H, J = 4.5, 8.5
Hz), 4.48 (dd,
1 H, J = 3, 11 Hz), 4.98 (septet, 1 H, J = ca. 6 Hz), 7.2-7.4 (m, 5H). LSIMS
572 (70,
M++H), 458 (45), 354 (70), 315 (65), 250 (100). Anal. Calcd for C3oH45N5~e ~
HCI
4Hz0: C, 52.97; H, 8.00; N, 10.30. Found: C, 53.02; H, 7.33; N, 10.25.
Example 24
N (4-oxopiperidine-1-carbonyl)-L-phenylalanyl-N-f2 3-dioxo-3-((1-
methyl)ethoxy)-
1-(phenylmethyllpropyll-L-prolinamide
A. N (4-oxopiperidine-1-carbonyl)-L-phen)rlalanvl-N-f2(R)-hydroxy-3-((1-
methyl)ethoxy) 3 oxo 1 (S)-(phenylmethyl)propy,-L-prolinamide
The compound was prepared in a manner according to the procedure described
as General Procedure A. The product of Example 16b (250 mg, 0.645 mmol) was
coupled with isopropyl 3(S),2(R)-3-amino-2-hydroxy-4- phenylbutanoate (EP 4
814 342,
168 mg, 0.710 mmol) to yield crude product (316 mg) which was chromatographed
on
silica packed in 2:1 ethyl acetate-hexanes and eluted with ethyl acetate
followed by 3%,
1096, and 20°~ ethanol in ethyl acetate, giving the title substance
'(242 mg, 62%).
LSIMS 607 (40, M++H), 335 (100), 281 (70), 221 (75), 207(80). Anal. Calcd for
C33H42N4O7 ~ 3/4 H20: C, 63.91; H, 7.07; N, 9.03. Found: C, 63.95; H, 7.23; N,
8.85.
WO 93/25574
PCT/US93/03625
-71 _
B. N-(4-oxopj~eridine-1-carbonyl)-L-phenylalanyl-N-f2.3-dioxo-3-((1-methyl)
ethoxy)-1 (S)-(phenylmethyl)propyll-L-prolinamide
The compound was prepared in a manner according to the procedure described
as General Procedure C. The product of the preceding Example (172 mg, 0.283
mmol)
was treated with the periodinane, and the crude product, isolated by
chloroform
extraction, was purfied on silica eluted with 296, 496, and 10°~
ethanol in
dichloromethane giving the title substance as a colorless foam (110 mg,
64°io). LSIMS
605 (24, M+*H), 333(50), 207(55), 147 (100).
Example 25
N-(4-oxopiperidine-1-carbonyl)-L-phen~alanyl-N-f3-amino-2,3-dioxo-1 (S)-
then I~i methyl)propyll-L-prolinamide
A. N-f4-oxopiperidine-1-carbonyl i-L-phenylalanyl-N-f2(R)-hydroxy-3-lamino)-
3-oxo-1 (S)-(phenylmethy~propvll-L-prolinamide
The compound was prepared in a manner according to the procedure described
as General Procedure A. The product of Example 16b (250 mg, 0.645 mmol) was
coupled with 3(S),2(R)-3-amino-2-hydroxy-4-phenylbutyramide (USP 4 668 769,
164
mg, 0.710 mmol) to yield crude product in unexpectedly low yield (30 mg,
9°~6). The
aqueous layers from the extractions were brought to pH 7 and saturated with
NaCI, and
the resulting solution was repeatedly extracted with chloroform. These
extracts were
combined, dried and concentrated giving 102 mg of a yellow foam which was
chromatographed on silica eluted with 2°0, 6°~6, and 1096
ethanol in dichloromethane
giving 26 mg of the title substance.
B. N-(4-oxo~peridine-1-carbonyl)-L-phenylalanyl-N-f3-amino-2,3-dioxo-
~S)-(phenylmethyl)propel-L-prolinamide
The compound was prepared in a manner according to the procedure described
as General Procedure C. The product of the preceding Example (22 mg, 0.036
mmol)
was treated with the periodinane, and the crude product, isolated by
chloroform
extraction, was purified on silica eluted with 296, 496, 690, 10°ro,
and 2096 ethanol in
dichloromethane giving the title substance (7 mg, 32°~) LSIMS 564
(30°~, M++H), 292
(10096).