Note: Descriptions are shown in the official language in which they were submitted.
WO 94/00477 213 8 6 ~ 5 PCI/US93/06110
SUBSTITUTED LACTOSE DERIVATIVES
AS CELL ADHESION INHIBITORS
Technical Field
The invention relates to compounds useful in the tre~tment of infl~mm~tion,
allergic reactions, autoimmnnP tli~e~ces~ and related conditions. More specifically,
the invention conr~-rnc substituted lactose that binds to selectin receptors and to
ph~rrn~reutical compositions cont~ining them. The present invention is also
directed to synthetic methods useful in obtaining these analogs and other lactose
derivatives.
Back~round Art
It is now well established that cellular interactions are at least in part
mediated by receptor/ligand interactions. One class of receptors is known to
recognize the peptide sequence "RGD"; other receptors recognize carbohydrate
lig~nrlc
One class of receptors that recognize carbohydrate-based ligands me~ t~s
the adhesion of circulating neutrophils to stimulated vascular endothelium. This is
a primary event of the infl~mm~tQry response and appears to be involved as well in
allergic and autoimmnn~ responses. Several receptors have been implicated in this
interaction, including a family of putative lectins that includes gpgoMEL (Leu8),
ELAM-l, and GMP-140 (PADGEM) and (Gong, J.-G., et al., Nature (1990)
343:757; Johnston, G.I., et al., Cell (1989) 56:1033; Geoffrey, J.S., and Rosen,S.D., J. Cell Biol. (1989) 109:2463; Lasky, L.A., et al., Cell (1989) 56:1045).
These lectins have been termed L-SELECTIN, E-SELECTIN, and P-SELECTIN.
E-SELECTIN is perhaps the best characterized of the three selectins. It is
particularly interesting because of its transient expression on endothelial cells in
response to IL-l or TNF (Bevilacqua, M.P., et al., Science (1989) 243:1160). Thetime course of this induced expression (2-8 hours) suggests a role for this receptor
in initial neutrophil extravasation in response to infection and injury. Furthermore,
Bevilacqua et al. (see Bevilacqua, M.P., et al., Proc. Natl. Acad. Sci. USA (1987)
WO 94/00477 PCr/US93/06110
2~ 3~
84:9238) have demonstrated that human neutrophils or HL-60 cells will adhere to
COS cells transfected with a plasmid cont~ining a CDNA encoding for the
E-SELECTIN receptor. Information regarding the DNA sequences encoding for
endothelial cell-leukocyte adhesion molecules are disclosed within PCT publishedapplication WO90/13300 published November 15, 1990.
Recently, several different groups have published papers regarding the
ligand for E-SELECTIN. Lowe et al., (1990) Cell, 63:475-484 reported a positive
correlation between the E-SELECTIN dependent adhesion of HL-60 cell variants
and tr~nefected cell lines, with their expression of the sialyl Lewis x (sLex)
oligosaccharide, Neu Nac oc2-3Gal-~1-4(Fuc oc1-3)-GlcNAc. By trancfecting cells
with plasmids cont~ining an oc(1,3/1,4) fucosyltransferase, they were able to
convert non-myeloid COS or CHO lines into sLex-positive cells that bind in an
E-SELECTIN dependent m~nn~r Attempts to block E-SELECTIN dependent
adhesion using anti-sLex antibodies were uninterpretable due to the ag~ tin~tiQn of
the test cells by the antibody. They concluded that one or more members of a
family of oligosacch~r~dec consisting of sialylated, fucosylated, lactos~minQglycans
are the ligands for the lectin domain of E-SELECTIN. Phillips et al., (1990)
Science, 250: 1130-1132 used antibodies with reported specificity for sLex to
inhibit the E-SELECTIN dependent adhesion of HL-60 or LEC11 CHO cells to
activated endothelial cells. Liposomes cont~ining difucosylated glycolipids withterminal sLex structures inhibited adhesion, while those cont~ining nonsialylated
Lex structures were partially inhibitory. Walz et al., (1990) Science, 250: 1132-
1135 were able to inhibit the binding of a E-SELECTIN-lgG chimera to HL-60
cells with a monoclonal antibody directed against sLex or by glycoproteins with
the sLex structure, but could not demonctr~te inhibition with CD65 or CD15
antibodies. Both groups concluded that the sLex structure is the ligand for
E-SELECTIN. Patent Application No. W092/02527 assigned to the present
~c.cigne~ and incorporated herein by reference discloses and claims the foregoing
minimum tetr~c;~cçh~ride structure and identi~les the groups putatively interactive
with the ELAM-l receptor.
In contrast to E-SELECTIN, the ~lvpe~lies of the ligands that bind to L-
SELECTIN and P-SELECTIN are not as well worked out. L-SELECTIN appears
WO 94/00477 2 1 3 ~ 6 ~ 5 PCI/US93/06110
to bind a sialic acid bearing ligand based on nellr~minicl~ce treatment of peripheral
lymph node high endothelial venules which inhibits L-SELECTIN recognition.
True et al., 1990, J. Cell Biol. 111, 2757-2764. Further, other studies using soluble
L-SELECTIN in direct binding/inhibition assays suggests that certain carbohydrate
moieties may be important ligand components including m~nnose and fucose,
- particularly when sulfated or phosphorylated. Imai et al., 1990 J. Cell Biol. 111,
1225-1232. More recent studies suggest that L-Selectin binds to sialyl Lewis X.
Foxall, C., et al., (1992) Cell, in press.
The ligand to P-SELECTIN is thought to have an epitope related to sialyl
Lewis x. This con~ sion is based on studies using antibody with this specificitythat block P-SELECTIN me~ t~d ~lh~sic)n of HL-60 cells to activated platelets orCOS cells that express P-SELECTIN. Larsen et al. (1990) Cell 63, 467-474. Other
e~.;.,.çnt.c have shown that the adhesion of HL-60 cells to P-SELECTIN
transfected cells is blocked by the pent~c~rcharide isolated from milk that has the
Lewis~ epitope. Recently, P-Selectin has been shown to bind to slllf~ti~es- Aruffo,
A., et al. (1991) Cell, 67:35-44.
Because of the role of selectins in ~lice~ce, particularly dice~ces involving
unwanted cell-cell adhesion that occurs through selectin-ligand binding on defined
cell types, the ;denti~lc~tiQn and isolation of novel ligands that would permit the
regula~ion of such selectin-ligand binding is sorely n~edetl
WO94/004~ 35~ PCr/US93/06110
Obiects of the Invention
The invention provides agonists and antagonists which bind to selectin
receptors and.thus modulate the course of infl~mm~tion, cancer and related
responses by mocl~ ting cell-cell adhesion events. In this aspect, the invention is
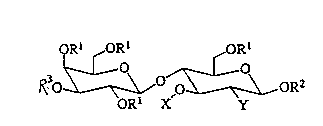
directed to compounds of the formula:
QRI ~ORl ORl
l~o~ \ \~ ~\ oR2
ORl X Y
(1)
whelc;m each Rl is independendy H or lower aLkyl (1-4C);
R2 is H, lower aLkyl(14C), aLkylaryl or one or more additional saccharide
re.ci~ e,s;
R3 is a negatively charged moiety in~luding S04--, P04--, or related group;
Y is H, oHl or lower alkyl(1-4C); and
X is -CHR4(CHoRl)2CHR5ORl wherein R4 and R5 are each independently
H, lower alkyl(14C), or taken together result in a five- or six-membered ring
optionally co,~ g a heteroatom selected from the group concisting of O, S, and
NRI;
said five- or six-membered ring optionally s1lbstit~lted with one substituent
sPlecte~ from the group conci.cting of Rl, CH20Rl, ORI, OOCRI, NRI2, NHCORI,
and SRl with the proviso that if X ~ Gsellts a hexose substituent R4 and R5, taken
together, cannot provide a hexose sllbstitnent
WO 94/00477 2 1 3 8 ~ ~ 5 PCI/US93/06tlO
In another aspect, the invention is directed to a method to synthPsi7e lactose
derivatives which method comprises contacting an intenne~ t~ of the formula
oR6 OR~ ~OR~<
RGO ~_ ~OR7
(2)
wherein each R6 is independ~-ntly H, lower aLkyl (1-4C), or a protecting
group; and
whe.~ l Y' is H, OH~, OOCR6, or SR6;
wherein at least one R6, which is at the position to be s~lbstitllted~ and at
most one ~dj~e.nt R6 is H and all other R6s are protecting groups; and
R' is a protecting group,
with an electrophile-don~ting moiety to obtain a product wherein the
electrophile is sllhstit-lt~.d for the H of the OH at the position to be sl~hstit~t~i
In other aspects, the invention is directed to pharm~e-lti~l compositions
cont~ining the compounds of formula 1 and to methods of treating infl~mm~tion
using these compocitionc~ In other aspects, the invention is directed to compounds
of form~ 2 and ~d~ition~l int~rrnefli~t~s in the synthesis of selectin binding
. ligands or other useful lactosyl residue-cont~ining moieties.
Modes of Carrvin~ Out the Invention
It is int~de.d that all the references cited herein be inco~porated into the
patent application in their entirety.
The invention provides compounds that are useful in the tre~tme~t of
infl~mm~tion by virtue of their ability to bind to selectin receptors. For example,
Figure 1 shows a rli~gr~mm~ti~ view of the role believed to be played by one of
the selectin receptors, ELAM-1, in medi~ting infl~mm~tif~ll Blood vessels are
lined with endothelial cells capable of producing the ELAM-1 surface receptor.
Lymphocytes circulating in the vessel contain on their surf~es carbohydrate
s
WO94/00477~3~6~1 PCr/US93/06110
ligands capable of binding to the ELAM-l receptor. This results in transfer of the
lymphocyte through the vessel wall and into the surrounding tissue. While this
may have a useful effect in some circllmct~n~es~ as in cases when the surrounding
tissue is infected, excessive transfer of the lymphocytes through the vessel wall and
into the tissue may also be excessive and cause unwanted infl~mm~tion. While notwishing to be limited by any particular theory, it is believed that the compounds of
the present invention which bind the ELAM-l receptor, antagonize the action of
the surface ligands on the circ~ ting lymphocytes and thus prevent their transfer
through the blood vessel wall to cause infl~mm~tion in the surrounding tissue.
Assavs to Identify T.i~n-ls
In their most general form assays for identifying lactose derivatives that act
as selectin ligands involve contacting the applup,iate selectin~ L-SELECTIN, E-
SELECTIN, or P-SELECTIN, with a yu~tivc; ligand and measuring its binding
properties.
Several assays are available to measure the capacity of a compound to bind
to L-SELECTIN, E-SELECTIN, or P-SELECTIN, and such assays are well known
in the art. For example, both the selectin and the yu~tivci ligand may be in
solution for a time sufficient for a complex to form consi.cting of the selectin and
ligand, followed by sep~aLing the complex from uncomplexed selectin and ligand,
and measuring the amount of complex formed. ~ltP.rn~tively, the amount of
uncomplexed se.lectin or compound could be measured.
A second and preferred assay format consist of immobilizing either the
selectin or the putative ligand on a solid surface, and forming the selectin-ligand
complex thereon by cont~rting the immobilized reagent with the non-immobilized
reagent. The se11~ctin-ligand complex is separated from uncomplexed reagents, and
the amount of complex formed can be dete~nin~d by m-~c11ring the amount of the
non-immobilized reagent present in the complex. For example, the putative ligandcan be affixed to a microtiter well, followed by adding the desired selectin to the
well and measuring the amount of selectin bound to the ligand.
A variation of the above assay is to genetic~11y en~in~er cells to express
high levels of L-SELECTIN, E-SELECTIN, or P-SELECTIN on their s~ ce, and
wo 94/00477 ~ ~ 3 ~ 6 ~ 5 PCr/US93/06110
.
to use the cells in lieu of purified selectin Radinl~b~le~l COS cells have been used
in this type of assay, and can be transfected with cDNA that encodes for L-
SELECTIN, E-SELECTIN or P-SELECTIN. After the cells have had a sufficient
time to adhere to the ligand coated microtiter well, non-adherent cells are removed
S and the number of adherent cells delc.. lh~ The number of adherent cells
reflects the capacity of the ligand to bind to the selectin
Represe.nt~live of the application of this type of assay is the identification of
E-SELECTIN lig~nflc For eY~mple, a compl~te cDNA for the ELAM-1 receptor
was obtained by PCR starting with total RNA isolated from IL-1 stimulated human
umbilical vein endoth~.linm The r~sl~hing cDNA was inserted into the CDM8
plasmid (see Aruffo, A., and Seed, B., Proc. Natl. Acad. Sci. USA (1987) 84:8573)
and the pl~mi~l amplified in E coli. Plasmid DNA from individual colonies was
isolated and used to transfect COS cells. Positive plasmids were selecte~ by their
ability to gen~o,rat~-. COS cells that support HL-60 cell adhesion. DNA sequencing
positively i(lPntified one of these clones as encoding for ELAM-1 (Bevilacqua,
M.P., et al., Science, (1989) 243:1160; Polte, T., et al., Nucleic Acids Res. (1990)
18:1083; Hession, C., et al., Proc. Natl. Acad. Sci. USA (1990) 87:1673). These
publications are incorporated herein by reference for their disclosure of ELAM-land genetic matt-ri~l coding for its production. The complPte nucleotide sequence
of the ELAM-1 cDNA and predicted amino acid sequence of the ELAM-1 protein
are given in the above cited article by Bevilacqua et al., which DNA and amino
acid sequences are incorporated herein by reference (see also publich~cl PCT patent
application WO90/13300 which was published November 15, 1990, which is
incorporated herein by reference).
A full length cDNA encoding ELAM-1 was obtained by 35 cycles of the
polymerase chain reaction with 1 ,ug of total RNA t;~ d;Led from IL-1 stimulatedhuman nmbilic~l vein endothelial cells, utili7ing primers complementary to the
untranslated fl~nking sequences
(5'-GGTGCGGCCGCGGCCAGAGACCCGAGGAGAG-3' and
5'-GGTGTCGACCCCACCTGAGAGATCCTGTG-3'). The 2Kb insert generated
was gel purified, directionally cloned into the m~mm~ n expression vector,
CDM8 that had been modified by the insertion of a SalI site into the polylinker,
Wo 94/00477 ~ PCr/US93/0611U
and grown in E coli (MC1061/p3). pl~cmitl.c were isolated from individual
colonies and used to tr~ncf~ct COS cells. Putative E-SELECTIN encoding
pl~cmi~l.c were selected based on the ability of these transfected COS cells to
support HL-60 cell adh~sion 72 h posttransfection.
A positive cDNA whose sequence corresponded to the published sequence
of E-SELECTIN with two nucleic acid substitutionc was used in all experim~P.nt.c.
COS cells were tr~ncf~ct~d with 1 ,ug of this pl~cmi~l DNA per 3.5 - 5.0 x 105
cells, with 400 ,ug/ml DEAE-dextran and 100 ~lM chloroquine for 4 h, followed bya brief exposure to 10% DMSO in PBS. Cells were metabolically radiolabeled
overnight with carrier free 32po4 and harvested in PBS supplemPntPd with 0.02%
azide and 2 mM EDTA at 72 h posttr~ncfection for use in cell ~lhPcion studies.
E-SELECTIN transfected COS cells produced by the above method may be
used to assay for glucuronyl glycolipid li~ndc, Similarly, COS cells may be
tr~n.cfec~,d with cDNAs that encode L-SELECTIN and/or P-SELECTIN. The
prodl~ction and char~çteri7~tion of L-SELECTIN IgG chimer~ constructs have been
previously described by Watson S. R. et al., (1990) J. Cell Biol. 110: 2221-2229.
This chimera contains two complemPnt binding dom~inc, con~ci~ct~pnt with its natural
expression. See Watson S. R. et al., (1991) J. Cell Biol. 115:235-243.
P-SELECTIN chimer~ was constructed in a similar manner as described by Walz,
G., et al (1990) Science 250, 1132-1135, and Aruffo, A. et al.(l991) Cell, 67, 35-
44, respectively. The chim~rac may be expressed in a suitable host cell, for
example, 293 cells and purified. Protein A affinity chromatography is the preferred
method of purific~tion. E-SELECTIN and P-SELECTIN may be constructed with
llu~ ed complement binding clom~inc to standardize the size of the chimeras and
to facilitate their secretion. A variation of the above assay is to genetically engineer
cells to express high levels of L-SELECTIN, E-SELECTIN, or P-SELECTIN on
their surface, and to use the cells in lieu of purified selP~tin- Radiolabeled COS
cells have been used in this type of assay, and can be transfected with cDNA that
encodes for L-SELECTIN, E-SELECTIN or P-SELECTIN. After the cells have
had a sufficient time to adhere to the ligand coated microtiter well, non-adherent
cells are removed and the number of adherent cells determined. The number of
adherent cells reflects the capacity of the ligand to bind to the sele~tin
Wo 94/00477 2 1 3 8 64 5 PCr/us93/o6llo
.
Thus, any c~ndid~t~o compound of the formula 1 may be verified to bind
ELAM-l receptors by a positive result in the foregoing assays. These assays
provide a simple screen for def~ g the relative effectiveness of the various
members of the group con.cicting of compounds of formula 1.
Nontherapeutic Uses of ComPounds of Formula 1
In ~f~ fition to their use in treating or pr~;venf llg inflamm~tion as is further
described hereinbelow, the compounds of formula 1 are useful in diagnostic and
p~pd d~ory procedures both in vitro and in vivo.
Compounds of formula 1 may be conjugated to solid substrates and used for
the purifi~tion of sel~ctin receptor protein from biological s~mr~les. This is
conducted most conveniently by ~rranging the coupled substrate as an affinity
chromatography column and applying a sample putatively cont~ining the selectin
receptor protein to the affinity column under conditions wherein the selectin
receptor protein is adsorbed whereas cont~min~ting m~t~.ri~lc are not. The selectin
,eceplor protein is then subsequently eluted, for example, by adjusting the eluent
solution to co~ g comreting amounts of the compound of formula 1 or by
adjusting pH or salt paramPt~r.c. Techniques for affinity p~ri~ tion are well
understood, and routine opl;-"i,i~f;on experimPrt.c will generate the approp-iate
con~ itionc for conduct of the procedure.
The compounds of formula 1 are also useful as detection reagents to
determinP the presence or absence of selectin or related carbohydrate-binding
receptor lig~n~.s. For use in such diagnostic assays, a biological sample suspected
to contain selectin receptor protein or a receptor protein closely related thereto is
treated with the compound of formula 1 under conditions wherein complexation
occurs between the receptor protein and the formula 1 compound, and the
formation of the complex is detected. A wide variety of protocols may be utilized
in such procedures, analogous to protocols applied in immunoassays. Thus, directassay wherein the amount of complex formed is directly measured may be lltili7Pd~ltern~tively~ competition assays may be used wherein labeled selectin receptor
protein is supplied along with, and in competition with, the biological sample. In
some forms of the assay, it is convenient to supply the compounds of formula 1 in
WO 94/00477 ?. ~'3~ ` PCr/US93/06110
labeled form so that the complex is detected dir~;lly, in ~lt~rn~te procedures, the
complex may be detecPd by size sepala~ions, seeondary l~beling re~g~ntc, or other
alt~rn~tce means. Suitable labels are known in the art, and include r~-lioartivelabels, fluorescent labels, enzyme labels, ehromogenic labels, or composites of
these approaehes.
The compounds of formula 1 may also be used as eompetitive diagnostie
reagents to detect the quantity of s~lectin receptor-binding components, such assurface lig~nds, in biological fluids. For the conduct of such assays, thè
compounds of formula 1 are labeled as deserihed above and mixed with the
biological sample and cont~rt~od with the appropliate receptor protein; the
iminlltiQn of binding of the labeled compound of forrnula 1 to seleetin reeeptor in
the presenee of biological sample is then dett-rmin.--l
The eompounds of formula 1 may also be used in im~gining studies in vivo
to determine the loc~tion of seleetin lCC~plOl~i in S . For use in sueh assays, the
eompounds of formula 1 are supplied with labels whieh ean be detected by in vivoim~ging teehniques, sueh as seintigraphic labels incl~ in~ indium 111, teçhn~tillm
99, iodine 131, and the like.
Teehniques for coupling compounds such as those of formula 1 to labels,
chromatographic supports, or other moieties useful in employing the compounds inthe relevant procedures are well nnr~t-.rctçod in the art.
Antibodies may also be prepared to the compounds of formula 1 by
coupling these eompounds to suitable earriers and admini~tering the eoupled
m~tP.ri~lc to m~mm~ n or other vertebrate subjeets in standard immllni~tinn
protocols with proper inclusion of adjuvants. Suitable immnnogenic carriers
include, for example, Keyhole Limpet Hemocyanin (KLH), tetanus toxoid, various
serum albumins such as bovine serum albumin (BSA) and certain viral proteins
sueh as rotaviral VP6 protein. These coupled m~t~-ri~lc are then a(lminictered in
repeated injections to subjeets such as rabbits, rats or mice and antibody titers
monitored by standard immunoassay teehniques. The resulting antisera may be
used per se or the antibody-secreting eells gen.-.r~t~d by the immlmi7~tion may be
immortalized using standard techniques and used as a souree of monoelonal
preparations which are immunoreactive with the compounds of formula 1. The
WO 94/00477 ~ 1 ~ 8 6 ~ 5 PCr/US93/061 l0
.
resulting antibodies are useful in assay systems for dete~mining the presence and/or
amount of the relevant formula 1 compound. Such assays are useful in monitoring
the circul~ting levels of compounds of formula 1 in therapeutic tre~tm~ont.c such as
those descrihed below.
S
Administration in Anti-infl~mm~torv Protocols
The compounds of the invention are ~flminictered to a subject in need
thereof for prophylactically plevt;~ g infl~mm~tion or relieving it after it hasbegun. "Treating" as used herein means pl~vell~ng or aml~linr~ting infl~mm?tion
and/or symptoms associated with infl~mm~tion The compounds are preferably
mini~tered with a ph~rm~rel-tir~lly acceptable carrier, the nature of the carrier
differing with the mode of ~dmini.ctration, for example, oral ~minictration, usually
using a solid carrier and I.V. ~-1minictration using a liquid salt solution carrier.
Typically, inject~hle compositionc are p-~a-cd as liquid solutions or suspensions;
solid forms sllit~hle for solution in, or suspension in, liquid vehicles prior to
injection may also be prepared. The compounds may also be emulsified or the
active ingredient encapsulated in liposome vehicles.
Suitable vehicles are, for example, water, saline, dextrose, glycerol, ethanol,
or the like, and combinations thereof. In ~dr1ition~ if desired, the vehicle maycontain minor amounts of auxiliary su-hst~nres such as wetting or emulsifying
agents or pH burr~ g agents. Actual methods of p~pal.ng such dosage forms are
known, or will be apparent, to those skilled in the art. See, for ~x~mple,
Remin~ton's Pharm~ce~ltiral Sciences, Mack Pnhli.ching Company, Easton, PA,
17th edition, 1985. Formulations may employ a variety of excipients including,
for example, pharm~ceutiç~l grades of m~nnitol, lactose, starch, m~gn~cium
stearate, sodium saccharin cellulose, m~gnlocium carbonate, and the like. Oral
compositions may be taken in the form of solutions, suspensions, tablets, pills,c~rsllles, sllct~in~d release ft~rmlll~tionc, or powders. Particularly useful is the
a~1ministration of the subject ligand molecules directly in tr~ncderm~l formulations
with permeation enhancers such as DMSO. Other topical formulations can be
a~minic~red to treat dermal infl~mm~tion. In addition, transmucosal
atiminictration may be effected using pen.-trantc such as bile salts or fusidic acid
11
WO 94/00477 ~ ,6 4~ PCI/US93/06110
derivatives optionally in combination with a(ldition~l detergent moleeules. These
form1]1~tionc are useful in the preparation of suppositories, for ex~mple, or nasal
sprays. For suppocitoriPs, the vehicle eomposition will inelude trallition~l binders
and c~rners~ sueh as polyaLkylene glyeols, or triglycçrif1Ps. Sueh suppositories may
be formed from mixtures eont~ining the aetive ingredient in the range of about
0.5% to about 10% (w/w), preferably about 1% to about 2%.
Tntr~n~c~l formnl~tionc will usually inelude vehieles that neither cause
irrit~tion to the nasal mueosa nor ci~ni~ ntly disturb eiliary funetion. Diluents
sueh as water, aqueous saline or other known snbst~n-~es ean be employed with the
subjeet invention. The nasal formnl~tiQnc may also eontain preservatives sueh as,
but not limited to, ehlorobutanol and bçn7~1koni1lm ehloride. A surfaetant may be
present to enhanee absorption of the subjeet proteins by the nasal mueosa.
Typically, the eompositions of the instant invention will contain from less
than 1% to about 95% of the active ingredient, preferably about 10% to about
50%. Preferably, between about l0 mg and 50 mg will be ~rlminicte.red to a childand between about 50 mg and 1000 mg will be ~dminictered to an adult. The
frequency of a-lminictr~tion will be detP.rrninPcl by the care given based on patient
responsivelless. Other err~;live doc~ges ean be readily deterrninPd by one of
ordinary skill in the art through routine trials establishing dose response eurves.
In detPrrnining the dose to be ~flmini.ctered, it will be noted that it may not
be decir~hlP. to eompletely bloek all seleetin reeeptors of a particular type. In order
for a normal healing process to proceed, at least some of the white blood cells or
neutrophils must be brought into the tissue in the areas where any wound, infection
or disease state is oeeurring. The amount of the selPctin ligands adminictered as
bloeking agents must be adjusted earefully based on the partieular needs of the
patient while taking into eonsideration a variety of faetors sueh as the type ofdisease that is being treated.
The eompounds of the present invention are useful to treat a wide range of
rlice~ces, for example ~nlo;~ une tlice~cP,c sueh as rheumatoid arthritis and
multiple sclerosis. The compositions of the invention are applicable to treat any
disease state wherein the immune system turns against the body causing the white
WO 94/00477 2 1 3 8 ~ ~ PCr/US93/06110
1-
cells to acc-1m~ t~ in the tissues to the extent that they cause tissue d~m~ge,
swelling, infl~mm~tion and/or pain.
Formulations of the present invention might also be ar1minictered to prevent
the undesirable after effects of tissue damage rçs--lting from heart attacks. When a
heart attack occurs and the patient has been revived, such as by the application of
anticoagulants or thrombolytic (e.g., tPA), the endothelial lining where a clot was
formed has often suffered d~m~ge When the antithrombotic has removed the clot,
the l~m~gecl tissue beneath the clot and other d~maged tissue in the endothelial
lining which has been deprived of oxygen become activated. The activated
endothelial cells then synth~ci~e selectin receptors, for example ELAM-l receptors,
within hours of the cells being ~l~m~ged. The receptors are extended into the blood
vessels where they adhere to glycolipid ligand molecules on the surface of whiteblood cells. Large numbers of white blood cells are quickly captured and broughtinto the tissue surrounding the area of activated endothelial cells, resl-lting in
infl~mm~tion~ swelling and necrosis which thereby decreaces the likelihood of
survival of the patient.
In ~ lition to treating patients ~ur~lhlg from the trauma resulting from
heart attack, patients suffering from actual physical trauma could be treated with
formulations of the invention in order to relieve the amount of infl:lmm~tion and
swelling which normally result after an area of the body is subjected to severe
trauma. Other conditions treatable using formulations of the invention include
various types of arthritis and adult respiratory distress syndrome. After reading the
present disclosure, those skilled in the art will recognize other disease states and/or
symptoms which might be treated and/or mitig~t~l by the adminictration of
formulations of the present invention.
Applications of Compounds of Formula 2
The compounds of formula 2 are inter~nedi~tPs in the preparation of
compounds which contain a lactosyl unit. Notably, the compounds of formula 2
W094/00477 7,~3S~ PCr/US93/06110
are useful in the pl~pa,dLion of compounds of formula 1 whose use is described
hereinabove. In addition to the compounds of formula 1, ~1trrn~tive compounds
cont~ining a lactose residue may also be p~ )ared, such as:
4-0-(3-0-carbonymethyl-~-D-galactopyranosyl)-3-0-[2R,S)-glyceryl]-D-
glucopyranose;
4-0-(3-0-carbonymethyl-~-D-galactopyranosyl)-3-0-[2R,S)-2,3-dideoxy-2,3-
difluoro-propyl]-D-glucopyranose;
4-0-[3-0-{(lR,S)-l-(carboxy)ethyl}-,13-D-gala~;Lopyl~nosyl]-3-0-[(2R,S)-
glycosyl]-D-glucopyldnose;
4-0-[3-O-~(lR,S)-l-(carboxy)ethyl}-,13-D-galac~ )yl~losyl]-3-O-(a-L-
fucopylanosyl)-D-glucopyranose;
4-0-[3-O-(a-Neu5Ac)-~13-D-gala.,-lopyldnosyl]-3-o-[(2R~s)-glyceryl]-D
glucopyldllose;
~0-[3-0-(oc-Neu5Ac)-,~-D-galactopyranosyl]-3-0-[(2R,S)-2,3-dideoxy-2,3-
difluoro-propyl]-D-glucopyranose.
Multivalent Forms of the RecePtor Bindin~ n~s
The affinity of the ligands of the invention for lccep~or can be enh~nced by
providing multiple copies of the ligand in close proximity, preferably using a
scaffolding provided by a carrier moiety. It has been shown that provision of such
multiple valence with optimal spacing between the moieties dr~m~tirally improvesbinding to receptor. For example, Lee, R. et al., Biochem (1984) 23:4255, showedthat providing multivalent forms of lactose inhibited labeled ASOR binding to
m~mm~ n hepatocytes much more effectively when the lactose was supplied as a
multivalent entity; the IC50 dropped from 500 ~lM for a single valent lactose to 9
for a divalent lactosyl compound to 4 for a trivalent lactosyl compound, and with
ideal or optimal spacing between the three lactose moieties to 0.007 ~lM.
The multivalency and spacing can be controlled by selection of a suitable
carrier moiety. Such moieties include molecular supports which contain a
mllltiplicity of functional groups that can be reacted with functional groups
associated with the ligands of the invention. A particularly preferred approach
involves coupling of the lactose-derived ligands of the invention to amino groups
14
W094/00477 213~6~7 PCr/US93/06110
of the carrier through reductive ~min~tion Reductive amination is a particularlyconvGnient way to couple aldehyde moieties to free amino groups by first formingthe Schiff base and then treating the conjugate with a reducing agent, such as ahydride reducing agent. Typically, the amino group-bearing carrier is mixed withS the carbohydrate moiety at about pH 9 and allowed to form the Schiff base; the
solvents are typically evaporated and reducing agent is added at high pH to
compl~t~ the re~-~tion
Particularly convenient carrier moi~ti~s to obtain multivalent forms of the
invention ligands include proteins and peptides, particularly those cont~ining lysyl
residues which have -amino groups available for binclinp- It is also useful to
include in the peptide or protein at least one tyrosine residue, as this offers a
convGnient site for l~heling~ for eY~mr1e with radioactive iodine. A particularly
convGl~ient carrier to obtain a trivalent couple is the peptide Lys-Tyr-Lys.
Comp4t~ reaction of the ligands of the invention with the free amino groups on
this peptide result in a trivalent moiety. Thus, compounds of the invention of the
formula lOH
QRI ~oRl $EI20Rl
~o~_Nll~l~Nn~
QRI ~ORI ~2ORI ~
QRI ~ORI ~R~
'--R~I~/'
wherein X, Y, and Rl, and R3 are as above defined illustrate the multivalent
compounds of the invention. Of course, a variety of carriers can be used, including
proteins such as BSA or HSA, a mu1tip1icity of peptides including, for e~mple,
pentapeptides, decapeptides, pent~lecareptides, and the like. Preferably, the
peptides or proteins contain the desired number of amino acid residues having free
amino groups in their side chains; however, other functional groups, such as
sulnlydlyl groups or hydr~yl groups can also be used to obtain stable link~ges
For example, the carbohydrate ligands of the invention may be oxidized to contain
WO 94/00477 ~)~38~64S PCI/US93/06~10
carboxyl groups at the reducing le~...i~.u~ which can then be derivaLi;~d with either
free amino groups to form amides or with hydroxyl groups to form esters.
P~ ,~a~on of the ComPounds of Formula 1
The compounds of the invention of Formula 1 may be synthPci7Pcl using an
intermçdi~t~ of Formula 2. The int~rmçr~i~t.o. of Forrnula 2, in one embo~lim~nt,
can be prepa~ed directly from D-lactose using standard procedures. In this
conversion, D-lactose is converted to the oct~cet~te in crystalline form, in over
95% yield in the method descrihed by ~u(lson, C., and Kuns, A., J Am Chem Soc
(1925) 47:2052. The oct~et~t~ is, in turn, converted in more than 90% yield by
the method of ~~ con, C. (supra) or of Fischer, E. and Fischer, H., Ber (1910)
43.2521 to the corresponding lactosyl bromide, also a crystalline compound. The
protected lactosyl bromide is con~,~,.~d by the method of Jansson, K., et al., J Or~
Chem (1988) 53.5629, in over 60% yield to the corresponding acylated
trimethylsilyl ethyl lactose, which can be deprotected by deacylation in qu~ntit~tive
yield to obtain 2-(trimethylsilyl)ethyl l~toci~le? 2-(trimethylsilyl)ethyl ~-D-
galactopyranosyl-,B-D glucopy~ o.~i~e. ~ltern~tive protecting groups at position 1
of the ~ cch~ri~lP may also be used.
This precursor of the compounds of Formula 2 is of the formula:
OH 'OH ~OH
~0~ \Q~o~QR1
OH OH
wherein R7 is a protecting group, preferably SE or Bn, wherein SE
represents -CH2CH2SiMe3 and Bn is benzyl.
pce~tion SchPmç 1 outlines the form~tion of one embodiment of the
compounds of Formula 2 from this intermediate, where Bz represents benzoyl:
16
WO 94/00477 2 1 3 g 64 S - Pcr/US93/06110
Reaction Sch~me 1
OH OH OH
Ho~o~oR7
OH OH
~,
S~ep l
. .
H3~'~ ~oR7
OH OH.
.
- 6a R7 = SE
6b R7 = ~n
~p 2
.
H~<~
OBz OBz
7a R7 = SE
7b ~7 = Bn
17
2 ~ 3 ~ PCr/US93/06110
In step l of the reaction scheme, the protected lactose, e.g., the
trimethylsilyl ethyl derivative, is treated with an excess of 2,2-dimetho~yp,opane
and dry c~mphnr sulfonic acid is added to the reaction mixture which is stirred for
2-3 days at about room temperature. A suitable base, such as triethylamine is
added and stirring continued for 10-20 minutes; the mixture is then concentr~tPd to
dryness and the base removed. In the case of benzyl lactoside, the method
employed is that of D. Beith-H~1~hmi et al., Carbohydr. Res., (1967) 5:25, wherein
benzyl lactoside is boiled for 3-4 hours in a large excess of dry açetQnP. cont~ining
4-toluene sulfonic acid. The reaction mixture is worked up using standard
procedures to recover the product 6. This intermerli~tç is then benzoylated under
suitable con~1itic)nc using, for çY~mp1P, benzoyl chloride to obtain the intermediat~
compound shown in reaction scheme as 7.
The intP.rmedi~te 7 may then be further derivatized at the free hydroxyl at
the 3-position of the g1ucosidP residue or this position may be protected and the
compound deprotected at positions 3 and 4 of the galactosyl residue and further
derivatized at position 3. Position 4 of the g~1~rtosyl residue is relatively
unreactive. A typical scheme for utili7~tion of this key intermediate 7 is shown in
Reaction .SchPme 2A. (In this scheme, Bz is benzoyl (PhCO-) and Bn is benzyl
(PhCH2-).
WO 94/00477 ~ f 3 ~ 6 4 ~ ; PCr/US93/061 10
Reaction Scheme 2A
H3C O OBz OBz
H3~0~0~0R7
OBz OBz
CH3 / O~S~3
r ~ 0/ OBn
CH3 0 OBz . ~OBz
CH3><0 ~ \ \~~\ \~.~oR7
OBz 08z
CH
~OBn
¦ OBn
OBn 2 a R7 ~ SE
gb R7 ~ BN
OBz OBz
HO~\ \~o~ \'~R7
OBz OBz
C. / 0--7
~ dBn ~ ~ a R7 = SE
OBn 10 b R7 ~ BN
(C~ J~I on ne~t page)
19
-
wo 94/00477 PCr/US93/061 10
2~38Ç~
Reaction .Sch~.me 2A
3~ (cont;--l,e~)
Aco OBz 08z
Ho~O~OR7
H3C~OBn t~ a ~ = SE
snO
AcO OBz OBz
So3o~o~oR7
H3C~OBn 12b ~ = BN
OBn
snO
OH OH OH
NaS030~,0 ~,~oR7
~13C~OH 13 b R7 = EI(~,B)
WO 94/00477 2 1 3 8 64 5 PC~/US93/06110
As shown in Reaction ,SchPme 2A, the interrnefli~te 7 is converted in two steps to
intermPrli~tP 10 by tre~tmPnt under suitable conditions with protected methyl l-thio-L-fucoside. The reaction is conducted in a nonaqueous aproctic solvent in the
presence of cupric bromide, tetrabutylammonium bromide and molecular sieve. (S.
Sato, et al., Carbohvdr. Res. (1986) 155:C6). The result~nt compound shown as 10- is then selectively acetylated at position 4 of D-gala~lopyldnosyl residue by the
way of its 3,4- orthoester, according to literature procedure, without isolation of the
int~rmedi7,te (R.U. T emiPux and H. Drigwez, J. Amer. Chem. Soc., (1975)
97:4069) to give intP.rmP~i~t~P 11. Sulfation of intermP~ tP 11 produces
intP.rmedi~tP 12 which is deacylated and hydrog~Pn~tPd to yield the final product ~,
a se1Pctin ligand.
In another embodiment of the instant invention, shown in reaction scheme
2B, intermediate ll may be phosphorylated to yield intermP~i~t-e 14 which upon
deacylation and hydrogenation yields the final product 15. This compound would
be expected to act as a selectin ligand.
WO 94/00477 PCr/US93/06110
2~3a6~ ~
Reaction .SchPme 2B
AcO OBz
HO~O~OBn
H3C ~OBIl 1l
OBn
OBn
I
(Ro) ~o~o~p~oBn
H3C ~OBI
OBn
OBn
R=C6Hs or C6HsCH2
HO OH
(NaO)2opo~ j
H3C ~OH
OH
WO 94/00477 ~ 8 6 4 ~ PCr/US93/061 l0
; .
ComPounds of the Invention and Preferred Embo~im~nt.c
As used herein, alkyl (1-6C) refers to a s~t~lr~tecl straight or branched chain
or cyclic hydrocarbyl residue co~ ;.l;l-g 1-6C; lower alkyl is simil~rly defined but
cont~ining only 1-4C.
S As used herein, aL~cylaryl is of the formula (CH2)m-Ar wherein m is 1-10
and Ar is a mono- or bicyclic aromatic residue optionally conlS~ g one or more
heteroatoms. Typical embodim~nt.c of Ar include phenyl, naphthyl, quinolyl,
pyridyl, pyrimidinyl, ben7thi~7oyl, be.n7imi~7oyl, and the like.
R7 is a protecting group suitable for saccharide resid~es Typical protecting
10 groups include benzyl, benzoyl, various silylalkyl groups, such as
trimethylsilylethyl (SE), and the like.
Exemplary compounds of formula 1 of the invention are those wherein R3 is
S04-2, P04-2, or other similar charged moiety.
Additional exemplary compounds of formula 1 include those wherein X is:
6-methyl-3,4,5-trihydroxypyran-2-yl,
6-acetyl-3,4,5,trihydro,~ypyran-2-yl,
6-propylamido-3 ,4,5 ,trihydroxypyran-2-yl,
6-propylamido-2,3,4-trimethoxypyran-2-yl,
6-ethyl-2,3-dihydroxy-4-metho~yL.yl~l-2-yl~
6-N-ethylamino-2-hydroxy-3,4-ethoxypyran-2-yl,
3,4,5-tri-n-propylo~y~yl~l-2-yl,
3 ,4,5-trihydroxypyran-2-yl,
2,3 ,4-trimethoxyfuran-2-yl,
2~3-dihydroxy-4-metho~yru'all-2-yL
2-hydroxy-3,4-ethoxyfuran-2-yl,
3,4,5-tri-n-propyloxyfuran-2-yl, and
3 ,4,5-trihydroxyfuran-2-yl;
or wherein both Rs and R6 are H and all R' in X are H or methyl;
or wherein X is 2,3,4-trihydro~ylJenzoyl.
Thus, particularly preferred compounds of formula 1 are those wherein all
Rl are H or methyl, Y is H, OH, OCH3 or OAc; and/or X is -CH2(CHOH)3H,
3,4,5-trihydro~yl.yl~1-2-yl, 3,4,5-trihydroxy-6-methylpyran-2-yl, 3,4,5-
23
WO 94/00477 c PCr/US93/06~10
~,~3~6~ ~
trimethoxypyran-2-yl, 3,4,5-trimethoxy-6-melhylpyldn-2-yl, 3,4,5-trihydroxyfuran-2-
yl, 3,4,5-trimethoAyfulall-2-yl, 2,3,4-trihydroxybenzoyl, or 2,3,4-
trihydroxynaphthoyl; and R3 is SO4-2, PO4-2, or other similar charged moiety.
Most preferred of the compounds of formula 1 are those wherein all R' are
H, R2 is H, Y is H, ORI, or lower aL~yl.
For those compounds of formula 2 which represent intP.rmP~ tPS preferred
forms are those wherein the protecting groups represented by R6 are benzyl or
benzoyl, the protecting group represented by R7 is trimethylsilylethyl or benzyl, and
wherein yl is H, oR6 wherein 1~6 iS benzyl or benzoyl as set forth above, and
where the free hydroxyl group(s) is at position 3 of the glucosyl moiety or
positions 3 and 4 of the g~l~ctosyl moiety. An additional preferred protecling
group for positions 3 and 4 of the g~l~ctQsyl moiety is isopropylidene.
The following e~r~mpl~s are int~nflecl to ilh-str~t,- but not to limit the
invention.
Example 1
Preparation of 2-(Timethvlsilyl) ethvl 2,6-di-0-benzovl-4-0-(2.6-di-0-
benzoYl-3,4-0-isou,ol,ylidene-~-D-~alactopyranosyl)-~-~lucopyranoside (7a).
2-(Trimethylsilyl) ethyl 4-0-(3,4-0-isopl~,pylidene-,B-D-galactopyranosyl)-~-D-
glucopyranoside (K. Jansson et al., J. Org. Chem. (1988) 53: 5629-5647; 6.6g,
13.75 mmol) was dissolved in dry pyridine (120 mL). The mixture was cooled to -
45C and stirred, while benzoyl rhlori-le (9.07mL, 77.4 mMol.)was added
dropwise, and stirring was contimled for 4h at -45C.
T.l.c. (8.5:1.5 toluene-ethyl acetate) revealed the presence of a major
product, faster-migr~ting than the starting acetal. A small proportion of a still
faster-migrating product (pent~ben70~te) was also revealed by t.l.c. The mixturewas poured into ice-water and extracted with dichlorom~th~n~ The
dichlorometh~ne solution was successively washed with water, aqueous NaHCO3,
and water, dried (Na2S04), and concentrated. The concentr~t~ was applied to a
column of silica gel with 9:1 toluene-ethyl acetate as the eluent and gave a solid
which cryst~lli7~d from m~th~nol to afford 7a (5.2g, 42.3%), [a]D +17.5 (c, 1.1,chloroform). 13C NMR (CDCl3): ~ 167.16, 166.13, 165.87, 165.83 (4xPhCO),
24
WO 94/00477 2 1 3 8 64 5 PCI/US93/06110
.
111.23 ~Me2), 101.50, 100.24 (C-l, C-l'), 82.57, 77.02 (C-3', C-4), 73.65, 73.44,
73.01, 72.96, 72.06, 71.97 (C-5, C-5', C-4', C-3, C-2, C-2'), 67.21 (O_H2CH2Si),63.69, 62.72 (C-6, C-6'), 27.62, 26.28 [C~H3)2], and 17.75 (CH~CH?Si).
Example 2
Preparation of BenzYl 2.6-di-O-benzo~1-4-0-(2,6-di-O-benzoyl-
3,4-O-isopro~,ylidene-~-D-~alactopyranosyl)-~-D-~lucopyranoside (7b).
A stirred and cooled (-45C, bath) solution of benzyl 4-0-(3,4-O-
isoplol)ylidene-,B-D-galactopyranosyl)-~-D-glucopyranocide ( 5g, 10.6 mmol; D.
Beith-~l~hmi et al., Carbohydr Res. (1967) 5: 25) in dry pyridine (120 mL), was
treated with benzoyl chloride (6 mL, 51.8 mmol), dropwise, and the stirring was
contin~-ed for 4h at -45C. T.l.c. (8.5:1.5 toluene- ethyl acetate) revealed thepresence of a major product, faster-migrating than the starting acetal. A small
proportion of a still faster-mi~r~ting product (pent~be.n7o~te) was also revealed by
t.l.c. The ~ u-c; was poured into ice-water and t-xtracted with dichlorometh~ne
The dichlorometh~nt solution was s~cceccively washed with water, aqueous
NaHCO3, and water, dried (Na2SO4), and concentrated. The concentare was then
applied to a column of silica gel and eluted with 9:1 toluene-ethyl acetate. On
concentr~tion, the fractions corresponding to the major product gave a solid residue
which crystalliæd from hot methanol to afford 7b (5.53g, 59%); m.p. 159-161C;
[OC]D -4.2 (c, 1.3, chloroform). lH NMR (CDC13): ~ 8.2-7.00 (m, 25H, arom.),
5.36 (t, lH, J 7.8 Hz, H-2'), 5.30 (dd, lH, J 8.0, and 9.5 Hz, H-2), 4.68 (d, lH, J
8.0Hz,H-1'),4.56(d, lH,J8.1Hz,H-1),3.94(dd, lH,J8.2and9.6Hz,H-3),
3.75 (dd, lH, J 8.2 and 9.7 Hz, H-4), and 1.65 and 1.35 [2s, 3H each, C(CH3)2 ];13C NMR (CDC13): ~ 167.16, 166.17, 165.90, and 165.86 (4xPHCO),111.86
(5~Me2), 102.10, 99.49 (C-l, C-l'), 82.99(C-4), 77.60 (C-3'), 74.02 (C-4'), 73.60,
73.50, 72.66 (C-2, C-2,'C-3, C-5, C-5'), 70.73 (PhCH2), 64.29 and 63.20 ( C-6,
C-6'), and 28.26 and 26.88 [ (CH3)2C]; positive ion LSIMS: 889.7 (M+H)+, 781.6
M-OBn)+, negative ion LSIMS: 934.1 (M+NO2)-, 1041.1 (M+mNBA)~.
WO 94/00477 ~ . ' PCr/US93/06110
6~ --
ExamPle 3
PreParation of Benzyl 2.6-di-0-benzoYl-3-0-(2,3.4-tri-0-benzvl
-oc-L-fucopyranosyl)-4-0-(2,6-di-0-benzoyl-3,4-0-isoplo~ylidene-~-D-
~ala-;lo~ osvl)-~-D-~lucoPv,~ oside (9b).
A ",i~u.e of compound 7b (4g, 4.5 mmol), methyl 2,3,4-tri-0-benzyl-1-thio-
a-L-fucopyranoside 8 (3.6g, 7.75 mmol) and powdered 4 A molec~ r sieves (lOg)
in 5:1 dichloroethane-N,N-dimethylform~mille (120 mL), protected from moisture,
was stirred for 2h at room ~,llpe.~ture. Cupric bromide (2g, 9 mmol) and
tetrabutylammonium bromide (0.29g,0.9 mmol) were added and the stirring was
continued for 35h at room temperature. More of the donor 8 (1.2g, 2.6 mmol, in
14.4 mL of 5:1 dichloroethane-N,N-dimethylform~mi~le), cupric bromide (0.67g,
0.3 mmol), and molecular sieves 4 A (2g) were added, and the stirring was
contimlt-d for 16h at room temperature. T.l.c. (9:1 toluene-ethyl acetate) then
showed the presence of a major product, faster-migrating than 7b; a trace of
unchanged 7b was also revealed by t.l.c. The IllL~lulc; was filtered (a bed of Celite)
and the solids thoroughly washed with chlorofo"". Tne filtrate and washings werecombined and washed with aqueous NaHCO3 and water, dried and concç~
The residue was applied to a column of silica gel and eluted with 9.5:0.5 toluene-
ethyl acetate. ~oncentr7~tion of the fractions corresponding to the major product
fmnicht-d a solid, which cryst~lli7f d from ether to afford 9b (3.68g, 76%), based
on reacted 7b. Compound 9b had m.p. 180-181C; [OC~D -8 (c, 1.1, chloroform).
'H NMR (CDC13): ~ 5.48 ( dd,l H, J 9.3 and 7.9 Hz, H-2'), 5.40 ( d, 1 H, J 3.8
Hz, H-l fuc), 5.22 ( dd, 1 H, J 8.6 and 7.3 Hz, H-2), 4.49 (d, 1 H, J 8.6 Hz, H-1),
4.42 ( d, 1 H, J 7.9 Hz, H-l'), 3.90 ( dd, 1 H, J 10.2 and 3.8 Hz, H-2 fuc), 1.49
and 1.35, ( s, 1 H each, CMe2), and 1.29 ( d, 3 H, J 6.6 Hz, H-6 fuc); l3C,
(CDCl3): o 166.86-165.22 (4xPhCO), 111.44 [~(CH3)21, 100.84, 99.80 (C-l,C-1'),
63.17, 63.01 (C-6,C-6'), 28.35, 26.86 [C(~H3)2], and 17.48 (C-6"); positive ion
LSIMS: 1197.9 (M-OBn)+, negative ion LSIMS: 1350.2 (M+NO2)-, 1 457.3
(M+mNBA)~.
26
WO 94/00477 21 386~S PCl/US93/06110
Example 4
E~cpalalion of 2-(Trimethylsilyl) ethyl 2,6-di-0-benzoyl-3-0-
(2,3,4-tri-0-benzYl-a-L-fucopyranosyl)-4-0-(2,6-di-0-benzoyl-3,4-0-
isop~vlidene-~-D-~alactopyranosyl)-~-D-~lucopyraoside (9a).
S A mixture of compound 7a (5.2g, 5.78 mmol), compound 8 (4.68g,10.17
mmol) and powdered 4A molecular sieves (6g), in 5:1 dichloroethane-N,N-
dimethylform~mide (135 mL), protected from moisture, was stirred for 2h at room
tempelatu~l;. Cupric bromide (2.6g, 11.7 mmol), and tetrabutylammonium bromide
(3.77g, 11.7 mmol) were added, and the stirring was continued for a total of 48hat room tempelatulc;, additional amounts of 8 (2.34g, 5.09 mmol, in 60 mL of 5:1dichloroethane-N,N-dimethylform~mi~le), cupric bromide (1.3g, 5.85 mmol),
tetrabutylammonium bromide (1.9g, 5.85 mmol) and 4A molecular sieves (3g)
being added after 24h. T.l.c. (9:1 toluene- ethyl acetate) revealed the presence of a
major product, faster-migrating tban 7a, Some unreacted 7a was also revealed by
t.l.c. After procçccing as described for 7b (to give 9b), followed by column
chromatography, compound 9a (6.7g, 88%) was obtained as an amorphous solid;
positive ion LSIMS: 1442.6 (M+Na)+, 1340.8 (M-NaSO3)+, negative LSIMS:
1396.2 (M-Na)~ .
Example 5
Preparation of Benzyl 2,6-di-0-benzoyl-3-0-(2,3,4-tri-0-benzyl-a-L-
fucopyranosyl)-4-0-(2,6-di-0-benzoYl-~-D-~alactopyranosyl)
-~-D-~lucoPYranoside (lOb).
Compound 9b (l.Og) in 70% aqueous acetic acid (600 mL), was stirred at
85-90C, the progress of the reaction being monitored by t.l.c.(4:1 toluene - ethyl
acetate). After 2.5h, most of the starting acetal 9b was converted into a slower-
migrating product. T.l.c. also in~ ated some cleavage of the a-L-fucosyl link~ge,
as eviden~ed by the presence of two by-products, one of which was marginally
faster-migr~ting than the product (tribenzyl fucose), and the other slower-nitrating
(disaccharide product). The acetic acid was evaporated under ~liminich~d pressure
( 40C), the last traces being removed by co-evaporation with several added
portions of toluene. The residue so obtained was purified in a column of silica gel
27
wO94/0047~ 3~q~13 PCr/US93/06110
with 9:1 toluene -ethyl acetate as the eluent to give 10b (0.6g. 61.8%), as an
amorphous solid. 13C NMR (CDC13): ~ 167.25, 166.80, 165.23 (4xPhCO), 100.65,
99.85 (C-l, C-l'), 98.18 (C-l fuc),79.55, 79.08 (C-3, C-4), 75.77, 73.20, 72.97,70.30 (4xPhCH2), 63.38, 62.33 (C-6, C-6'), and 17.16 (C-6 fuc); positive ion
S LSIMS: 1263.7 (M+H-2H)+, 1157.7 (M-OBn)+, negative ion LSIMS: 1417.1
(M+mNBA)~, 1310.3 (M+NO2)-, 1263.2 (M-H)- .
ExamPle 6
P~ tion of 2- (Trimethylsilyl) ethYl 2~6-di-O-benzoyl-3-O-
(2,3,4-tri-0-benzyl-a-L-fucoPyranosyl)-4-0-(2,6-di-0-benzoyl-~-D-
~ala.;Lo~v-~losYl)-~-D-~lucopYranoside (lOa).
Compound 9a (3g, 2.3 mmol) was taken in 70% aqueous acetic acid (300
mL) and the mixture was heated, with stirring, for 2h at 85-90 (bath). T.l.c. (4:1
toluene-ethyl acetate) showed the presence of a major product with
chromatographic mobility comparable to that of 10b. Processing as described for
9b (to give 10b), followed by column chromatography, gave trisaccharide diol 10a(2.3g, 79%) as an amorphous solid; [OC]D -20.6 (c, 1.1, chloroform). '3C NMR
(CDCl3): o 167.26, 166.98, 166.78, 165.04 (4 x PhCO), 101.31, 100.55 (C-l, C-l'),
98.19 (C-l fuc), 79.56, 78.98 (C-3, C-4), 75.75, 73.19, 72.98 (3 x PhCH2), 67.84(OCH2CH2Si), 63.48, 62.19 (C-6, C-6'), 18.45 (OCH2CH2Si),and 17.16 (C-6 fuc).
ExamPle 7
Preparation of Benzyl 2,6-di-0-benzoyl-4-0-(4-0-acetYl-2,6-di-0-benzoYl-,~-D-
~alactopyranosvl)-3-0-(2,3.4-tri-O-benzyl-a-L-fucopyranosyl)-
B-D-~lucoPyranoside (1 lb).
Compound 10b (0.56g) was dissolved in a mixture of benzene (30 mL) and
triethyl orthoacetate (30 mL), cont~ining 4-toluenes~-lfonic acid (0.15g), and the
mixture stirred for lh at room temperature. The acid was neutralized with a little
triethylamine, and the mixture evaporated to dryness. It was then taken in 80%
aqueous acetic acid (50 mL) and stirred for 40 min at room temperature. T.l.c. (4:1
toluene-ethyl acetate) showed the presence of a major product,faster-migrating than
diol 10b. The acetic acid was removed under ~liminich~l pressure, and several
28
WO 94/00477 ~1 ~8 6~ ~ PCI/US93/06110
portion of toluene were added to, and evaporated from the residue to furnish 1 lb
(0.56g, 96.6%) as an amorphous solid, [a]D -14.3 (c,l.l, chloroform). lH NMR
(CDCl3): ~ 8.20-7.00 (m, 40 H, arom.), 5.51 (t, 1 H, J 8.0 Hz, H-2'), 5.38 (d, 1 H,
J 3.8 Hz, H-l fuc), 5.30 (d, 1 H, J 3.8 Hz, H-4'), 5.20 (dd, 1 H, J 8.1 and 10.0 Hz,
H-2), 4.62 (d, 1 H, J 8.2 Hz, H-l), 4.44 (d, 1 H, J 7.9 Hz, H-l'), 1.82 (s, 3 H,CH3CO), and 1.34 (d, 3 H, J 6.4 Hz, H-6 fuc). 13C NMR (CDCl3): ~ 170.38
(CH3CO), 166.15, 165.72, 165.57, 164.56 (4xPhCO), 100.45, 99.23 (C-l, C-l'),
97.54 C-l fuc), 79.48 (C-3). 77.57 (C-4), 74.08, 72.94, 72.70, 70.15 (4xPhCH2),
62.54, 60.78 (C-6, C-6'), 20.59 (~H3CO), and 16.88 (C-6 fuc); positive ion
LSIMS: 1307.1 (M+H)+), 1200.8 (M-OBn)+, negative ion LSIMS: 1460.9
(M+mNBA)~, 1353.6 (M+NO2)-, 1306.8 (M-H)- .
Example 8
Preparation of 2-(TrimethYlsilYl) ethvl 2.6-di-0-benzoYl-4-0-(4-0-acetyl-
2,6-di-O-benzoyl-~-D-~alactopyranosyl)-3-0-(2,3,4-tri-O-benzyl-a-L-
fucop~ osvl)-B-D-~alactoPYranoside (l la).
A solution of compound 10a (1.87g), in a mixture of benzene (50 mL) and
triethyl ortho~cet~te (50 mL), col-l .;..;..g 4-tolu~nesulfonic acid (0.25g) was stirred
for lh at room temperature. The acid was then neutralized with a few drops of
triethylamine, and the mixture evaporated to dryness. The residue was mixed with80% aqueous acetic acid (100 mL) and the mixture stirred for 40 min at room
tempe.~lule. Proce~cing as described for 10b ( to give 1 lb), gave the title
compound lla (1.86g,89%); a white amorphous solid; [a]D -2.7 (c, 1.1,
chloroform). 13C NMR (CDCl3): o 171.03 (CH3CO), 166.78, 166.35, 166.18,
165.02 (4 x PhCO), 101.27, 101.09 (C-l, C-l'), 98.17 (C-l fuc), 80.10, 78.20 (C-3,
C-4), 74.67, 73.54, 73.30 (3 x PhCH2), 67.86 (O_H2CH2Si), 63.31, 61.42 (C-6, C-
6'), 21.21 (~H3CO), 18.47 (OCH2_H2Si), and 17.53 (C-6 fuc); negative ion
LSIMS: 1470.8 (M+mNBA)~, 1363.7 (M+NO2)-.
29
Wo 94/00477~S6~ PCI/US93/06110
ExamPle 9
Preparation of Benzvl 2,6-di-O-benzoyl-3-0-(2,3.4-tri-O-benzYl-a-L-
fuco~y~ osvl)-4-0-(sodium 4-0-acetYl-2,6-di-0-benzoYl-~-D-
~alactoPyranosYl 3-sulfate)-~-D-~lucoPvranoci~le (12b).
5A mixture of compound 1 lb (0.6g, 0.46 mmol) and sulfur trioxide-pyridine
complex (0.6g, 6.3 mmol) in dry pyridinè (50 mL) was stirred for 2h at 55-60C
(bath), and then for 16h at room temperature. T.l.c. (6:1 chloroform - methanol)showed the disap~e~r~nre of llb and the presence of a single slower-migrating
product. Methanol (5 mL) was added, and the mixture stirred for 15 min (to
decompose excess reagent). It was then concenLr~led and purified in a column of
silica gel by elution with 10:1, followed by 6:1 chloroform-m~-th~nol. On
concentrAtion, the fractions corresponding to the product gave a solid residue,
which was dissolved in 1:1 chloroform-methanol (30 mL) and treated with
~mberlite IR 120 (Na+) cation-exf~h~nge. resin, and the mixture stirred fo lh atroom temperature. It was then filtered and evaporated to dryness to give 12b
(0.58G, 89%) as an amorphous solid; [a]D -5.1 (c, 1.8, 1:1 chloroform-m~-th~nol);
positive LSIMS: 1433 (M+Na)+, 1411.1 (M+H)+, negative LSIMS: 1563.9
(M+mNBA)-, 1386.7 (M-Na)~.
ExamPle 10
Preparation of 2-(TrimethYlsilyl) ethvl 2,6-di-O-benzovl-3-O-
(2 3,4-tri-O-benzvl-oc-L-fucopyranosvl)-4-O-( sodium 4-O-acetyl-
2,6-di-O-benzoyl-B-D-~alactopvranosyl 3-sulfate)-~-D-~lucoPvranoside (12a).
A mixture of compound 1 la (0.45g, 0.39 mmol ) and sulfur trioxide-
pyridine complex (0.45g, 4.7 mmol) in dry pyridine (25 mL) was stirred for 2h at55-60C, and then overnight at room temperature. After proces~ing and
purification, in a manner similar to the afore described, compound 12a (0.46g,
95.8%) was obtained as an amorphous solid; [CC]D ~2.2 (c,1.5, 1:1 chloroform-
meth~nol); positive ion LSIMS: 1442.6 (M+Na)+, 1341.1 (M-NaSO3)~, negative ion
LSIMS: 1395.5 (M-Na)~.
WO94/00477 ~ 645 PCr/US93/06110
Example 11
u~dLion of O-a-L-fucou~lanosyl-(1~3)-O-rsodium ~-D-
~alactop~ranos~ll 3-sulfate-(1- ~4)l-D-~lucoPYranose (13b).
Compound 12b (0.58g) in m~th~nol (50 mL), conl;.;..il-g a catalytic amount
of sodium msthoxide~ was stirred overnight at 45-50. T.l.c. (13:6:1 chloroform-
mPth~nol-water) showed the presence of a single slower-migrating product. After
cooling to room tem~e~Lulc;, Amberlite IR 120 (H+) cation-exrh~nge resin was
added till the llli~lulc became neutral (pH paper). It was then filtered directly into
a flask co~ g Ambçrlite IR 120 (Na+) cation-exchange resin, and the mixture
stirred for 45 min. It was then conr,entrflt-d and the residue repeatedly extracted
with hexane-ether mixture to remove medlyl ben7O~t~ The partially-protected
intermedi~te so obtained (0.38g,) was sufficiendy pure to be utilized directly in the
next step; negative ion LSIMS: 928.1 (M-Na)~. A portion (0.35g), without furtherpnrifir,atiQn, was taken in 80% aqueous msth~nol (30 mL), cont~ining 10%
p~ (1illm-on-carbon 0.35g). The mixture was stirred overnight at room temperature
under a slight overpressure of H2, when t.l.c. (5:4:1, or 13:6:1 chloroform-
mcth~nQl-water) in(lir~ted the presence of a slower-migr~ting product, together with
traces of some faster-migr~ting cont~min~ntc ( presumably due to incomplete
hydrogenolysis). The mixture was filtered ( Celite bed) directly onto Amberlite IR
120 (Na+) cation-exch~nge resin, and the solids thoroughly washed with aqueous
methanol. After stirring with the resin for lh, the ~ urt; was filtered and
conePntr~ted to a small volume, which was applied to a column of silica gel and
eluted with 5:4:1 chloroform-meth~nQ1-water. Fractions corresponding to the
product were pooled, concentrated to a small volume and treated with Amberlite IR
120 (Na+) cation-exchange resin. The resin was filtered off and washed with water,
and the filtrate and washings combined, refiltered ( 0.2 ~lM CellnlQse acetate
syringe filter), and lyophilized to give 13b, (183 mg, 84.3%; [OC]D -20.5 (c, 0.6,
water). 1 H NMR (D20): ~ 5.45 [d, 1 H, J 4.13 Hz, H-l fuc (~)], 5.39 [d, 1 H, J
3.81 Hz, H-l fuc (oc)], 5.18 (d, 1 H, J 3.81 Hz, H-l), 4.66 (d, 1 H, J 7.93 Hz, H-
1'), 4.55 [d, 1 H, J 7.62 Hz, H-l (,~)]; negative ion LSIMS: 567.5 (M-Na)~, 421
(M-Na-Fuc)~.
-
wo94'4"2~3a64~j PCr/US93/06110
Example 12
Fre~dLion of 2-(TrimethylsilYl ethyl O-a-L-fucopvranosyl-(1~3)-O -r sodium
13-D-~ala~;~o~ osYl 3-sulfate-(1~4)1-~-D-~lucopyranoside (13a).
Compound 12a (0.45g) was O-deacylated in methanolic sodium mrthQxide (50
mL), exactly as described for 10, to afford the corresponding partially benzylated
interme~ te (0.29g), which showed positive ion LSIMS: 983.9 (M+Na)+, 882.1
(M-NaSO3 ), negative ion LSIMS: 938.0(M-Na)~ . This compound (0.24g) without
any further pmifie~tion, was subjected to catalytic hydrogenolysis in 80% aqueous
meth~nQl (30 mL) in the presence of 10% p~ lm-on-carbon (0.24g), and then
processed in a manner analogous to the afore described to afford compound 13a
(125 mg, 72.7%), as a white fluffy m~teri~l; [a]D -49.2 (c,0.6. water).lH NMR
(D2O): ~ S.45 (d, 1 H, J 4.22 Hz, H-1 fuc), 4.55 (d,1 H, J 8.06 Hz, H-1'),4.49 (d, 1
H, J 8,44 Hz,H-1), 4.32 (dd, 1 H, J 3.45 and 9.98 Hz, H-3'); positive ion LSIMS:713.8 (M+Na)+, negative ion LSIMS: 667.6 (M-Na)~ .
Example 13
Preparation of a Multivalent T i~n(l N,6N,6N' Tris (20) LYs-TYr-L~s
Compound 13a or 13b, prepared in Example 127 may be derivatized to the
peptide Lys-Tyr-Lys to obtain the trivalent conjugate derivatized at the two -
amino lysine groups and the a-amino N-sormin~l of the peptide. To obtain this
trivalent compound, 50 111 of 2 mM peptide Lys-Tyr-Lys (100 nmol) in 100 mM
sodium carbonate, pH 9, are placed in a small Eppendorf tube cont~ining 5 ~11 of200 mM 20 (1 mmol), and the sample is evaporated to dryness in a SpeedVac for
about 30 min~lt~s
After evaporation, 50 111 of 800 mM NaCN BH3 (recrystallized, 40 ~lmol) in
100 mM sodium carbonate, pH 9, is added and the mixture is incubated for 48
hours at 55C. The re.s-llting incubated mixture is run on a GPC peptide HPLC
sizing column and fractions are collected and assayed for protein content by BCAprotein assay. Protein-cont~ining fractions are pooled, lyophilized and submitted
for mass spectroscopy.
The results would show the formation of the derivatized peptide as
co~t~ining 1, 2 or 3 moieties of compound 13a or 13b.
32
WO 94/00477 kl 3 86 ~ 5 PCr/US93/06110
The trivalent derivative would be especially effective in inhibiting the
binding of lactose to hepatocytes in an assay conducted as described by Lee, R. et
al., Biochem (1984) 23:4255.
EXAMPLE 14
Selectin Li~and ~ru~)c;lLies of Lactose Derivatives
Compounds 13a and 13b were tested for their capacity to bind to E and L
selectin. The ELISA assay used consists of evaporating 2,3 sLex glycoIipid, at 25
picomoles per well, onto microtiter wells, and then washing the excess off with
water. The wells are blocked with 5% BSA at room temperature for an hour and
then washed with PBS con~i~;..;--g imM Ca. While the plate is being blocked, biotin
labelled goat F(ab')2 IgG (Fc specific) and streptavidin-alkaline phosphatase diluted
1:1500 in 1 % BSA-PBS (lmM Ca) are combined with either the E- or L-Selectin-
IgG chimera (L91-10) at 200 ng/mL and incubated at 37 C for 15 mimltes to allow
a complex to form. This provides a soluble "multivalent" receptor. Compounds
13a and 13b were added at final con~entr~tio~c ranging from 1.5 to 5.0 mM to thesoluble receptor and allowed to react at 37 C for 45 minutes. The solutions were
then placed in the microtiter wells that had been washed after being blocked, and
the plates incubated at 37 C for 45 mim tPs to allow the soluble receptor to bind to
the known natural ligand, 2,3 sLex glycolipid. The positive control was the signal
produced by soluble "multivalent" ,t;ceplor reacted with only the ligand evaporated
to the microtiter well. This was considered "100 % bin~lin~: " The signal produced
by receptor previously reacted with inhibitor is divided by the signal produced by
the positive control, multiplied by 100, to calculate % receptor bound in the
presence of the inhibitor. The reciprocal of this is % inhibition.
It is appa~ t from Table 1 that both compounds 13a and 13b inhibit
binding of E selectin to 2,3 sLex glycolipid. Over the three concentrations tested
13b was the better inhibitor with the greatest difference apparent at 5mM
conrentratiQn. At this concentration 13b showed 82.5% inhibition compared to
48% for 13a.
WO 94/00477 PCr/US93/06110
~6~ ~
2 Table 1
INHIBlTION OF E-SELECTIN BINDING TO sLeX
COMPOUND CONC. (mM) % INHIBITION
s
13a 1.25 30
2.50 34
5.00 48
13b 1.25 48.5
2.50 45.4
5.00 82.5
It is appalt;nt from Table 2 that both compounds 13a and 13b also inhibit
binding of L selectin to 2,3 sLex glycolipid. However, the dirre,ence here was
considerably greater than the difference in % inhibitinn for binding to E selectin. For
example, at 1.25 mM, 13b surprisingly showed 90% inhibition. 100% inhibition wasobserved at 2 mM and S mM. In m~rk.o.~l contrast, 13b displayed only 13% inhihition
at 1.25 mM and a maximum inhibition of 47% at 5 mM.
Table 2
INHIBITION OF L-SELECTIN TO sLex
CONC. (mM) % INHIBITION
13a 1.25 13
2.50 27
5.00 47
13b 1.25 90
2.50 100
5.00 100
34