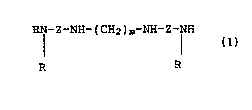Note: Descriptions are shown in the official language in which they were submitted.
W094/07480 2 1 ~ ~ 3 1~ PCT/US93/08517
POLYAMINE DERIVATIVES AS ANTI-CYTOMEGALOVIRAL AGENTS
BACRGROUND OF T~E lNY~NlION
Human cytomegalovirus (CMV) infections occur in a range
of severity, from a passive silent infection without
consequences to diseases that are manifested by fever,
hepatitis, pneumonitis, and after congenital or neonatal
infection may result in severe brain damage, stillbirth, or
perinatal death.
Cytomegalovirus infections frequently occur through out
life, with the incidence of seropositivity most frequent in
the elderly. Intrauterine infection with CMV has a high
frequency of infection, with CMV the leading cause of
congenital viral infection, occurring in about 0.4 to 2.2~
of all births. CMV infections can have serious effects in
diseases in immunocompromised patients with a high
frequency of morbidity and mortality, and is the major
cause of sight-threatening infections in AIDS patients.
CMV is ubiquitously transmitted in all populations.
= However, risk to CMV infections varies considerably in
different geographic areas. Infected individuals may
transmit the virus in urine, saliva, cervical secretions,
semen, feces, milk, or infected blood and organs.
W094/07480 21 ~ ~ 3 ~ ~ PCT/US93/08517
Molecular analysis of the DNA of CMV isolates reveals
minor strain-specific differences that are useful markers
in epidemiological investigation. CMV is highly host-
specific and cannot be propagated in laboratory animals or
in most non-human cell cultures. Therefore, experiments
designed to be predictive of CMV activity generally are
restricted to human cell lines that have resident the CMV
virus or have become infected with CMV.
It is well known that aliphatic polyamines, such as
spermine and spermidine, play a role in cell growth and
proliferation. These naturally occurring polyamines are
found in animal cells and are produced in a biosynthetic
pathway involving putrescine as a precursor. Putrescine is
formed by a decarboxylation of ornithine by ornithine
decarboxylase (ODC) and this is a highly regulated stage in
the biosynthesis of spermidine and spermine.
The first indication that polyamines may have a role in
certain viral function was discovered when putrescine and
spermidine were found to be present in the bacteriophage
T2. Later the polyamines, putrescine and spermidine, were
found in other viruses, including human cytomegalovirus,
poxvirus, vaccinia virus, and human type 5 adenovirus to
name a few. However, the fact that polyamines are present
in several animal viruses does not directly indicate a role
for polyamines in viral replication. Unusually, CMV
infection of cells in vitro is antagonised by inhibition of
ODC by specific inhibitors of the enzyme (Tyms et al., J.
Antimicrobial Chemotherapy 22, 403-427 1988).
The human cytomegaloviruses are a subgroup of agents
within the herpes group of viruses, all of which have the
propensity for remaining latent in man. No specific
therapy is generally available for CMV infections.
Although ganciclovir and foscovir are both licensed for use
against some infections in the immunocompromised host, both
~ W O 94/07480 2 1 4 6 3 1 9 PC~r/US93/08517
compounds can induce major side effects. It is a feature
of the present invention that the CMV agents afford less
cell and/or tissue toxicity compared to other agents in
use.
CMV, compared to other herpes viruses, herpes simplex
type 1 and type 11 and varicella-zoster virus, is
relatively resistant to the action of acyclovir. Research
in this area is primarily focused toward identifying agents
which would be therapeutically effective in humans. A
further feature of the present invention is that the
compounds described herein provide a CMV agent that
selectively acts against CMV compared to other Herpes
viruses.
It has now been found that certain polyamine
derivatives are effective therapeutic agents when used
against cell cultures infected with CMV. An object of the
present invention is the use of the describe polyamine
derivatives as therapeutic agents against CMV.
SUMMARY OF THE INVENTION AND DETAILED DESCRIPTION
Synthesis of compounds of the present invention as
anticancer agents has been described in part in U.S. Patent
Application 07/602,530 which is herein incorporated by
reference. Synthesis of compounds of the present invention
as antiprotozoal agents has been described in part in U.S.
Patent Application 07/840,575 which is herein incorporated
by reference. Synthesis of compounds of the present
invention as potentiating cell-mediated immunity has been
described in part in U.S. Patent Application 07/856,818
- which is herein incorporated by reference.
This invention relates to methods o use of certain
polyamine derivatives in the treatment of patients
21~3~9 ~
W O 94/07480 PC~r/US93/08517
suffering from CMV disease states and to pharmaceutical
compositions containing these polyamine derivatives.
More specifically, this invention relates to a method
for the treatment of patients suffering from CMV infections
which comprises administering a therapeutically effective
amount of a compound of the formula (I): wherein Z is a
HN-Z-NH-(CH2)m-NH-Z-NH
R R
( C2-C6 ) saturated or branched chain alkyl moiety; m is 7 or
8; each R group independently is hydrogen, a Cl-C6 saturated
or unsaturated hydrocarbyl, or -(CH2)x-(Ar)-x wherein Ar is
phenyl or napthyl, X is H, Cl-C6 alkoxy, halogen Cl-C4
alkyl, wherein x is an integer 0, 1, or 2; with the proviso
that both R groups cannot be hydrogen; or said compounds of
formula I can be a pharmaceutically acceptable acid
addition salt thereof.
As a further aspect of the present invention, it has
been found that compounds of formulae (I) provide an anti-
CMV effect in patients in need thereof. Compounds of
formuale I generally produce effective treatment without
similar delayed toxicity effects produced by other agents.
The present invention relates to the use of novel
compounds of the formula (I) for CMV infections, or more
specifically to the novel compounds of formula (Ia) and
(Ib).
W094/07480 2 1 ~ 6 3 1 9 PCT/US93/08517
The novel compounds of formula (Ia) are of the formula:
HN~Zl~NH~(CH2)m~NH~Zl~NH
1 (la)
R R
wherein Zl is a branched chain (C2-C6) alkyl moiety; m is 7
or 8; and each R group independently is hydrogen, a Cl-C6
saturated or unsaturated hydrocarbyl, or -(CH2)x-(Ar)-X
wherein Ar is phenyl or napthyl, X is H, Cl-C6 alkoxy,
halogen Cl-C4 alkyl, wherein x is an integer 0, l, or 2;
with the proviso that both R groups cannot be hydrogen; or
said compounds of formula Ia can be a pharmaceutically
acceptable acid addition salt thereof.
The novel compounds of formula (Ib) are of the formula:
HN-Z2-NH-(CH2)m-NH-Z2-NH
l I (Ib)
R R
wherein Z2 is a straight chain (C2-C6) alkyl moiety; m is 7
or 8; each R group independently is hydrogen, a Cl-C6
saturated or unsaturated hydrocarbyl, or -(CH2)x-(Ar)-X
wherein Ar is phenyl or napthyl, X is H, Cl-C6 alkoxy,
halogen Cl-C4 alkyl, wherein x is an integer 0, l, or 2;
with the proviso that both R groups cannot be hydrogen; or
said compounds of formula Ib can be a pharmaceutically
acceptable acid addition salt thereof.
The center alkylene moiety of compounds of the formula
(I) is a saturated, straight-chain hydrocarbyl radical
comprising 7 or 8 carbon atoms, i.e., "(CH2)7" or "(CH2)8".
As used herein, the term "Z" is understood to mean a
saturated hydrocarbylene radical of straight (Z2) or
branched-chain configuration (Zl) comprising 2 to 6 carbon
W O 94/07480 2 1 ~ ~ 3 1 ~ P~r/US93/08517
atoms including, but not limited to, -cH2cH2-~ -CH2CH2CH2-~
-CH2 ( CH2 ) 2cH2-, -cH2 ( CH2 ) 3cH2-, -CH2 ( CH2 ) 4CH2- ~
-CH(CH3)CH2-, -CH(CH3)CH2CH2-, -CH2CH(CH3)CH2- and the like.
In those instances where R is an unsaturated hydrocarbyl
moiety, such compounds include straight, branched, or
cyclized hydrocarbyl moieties such as -CH2CH=CH2, -
CH2CH2CH=CH2, -CH2CH-CH, and -CH2CH=C=CH2.
Compounds of the formula (I) can be used according to
the present invention as pharmaceutically acceptable acid
addition salts thereof. The term "pharmaceutically
acceptable acid addition salt" encompasses both organic and
inorganic acid addition salts including, for example, those
prepared from acids such as hydrochloric, hydrofluoric,
sulfuric, sulfonic, tartaric, fumaric, hydrobromic,
glycolic, citric, maleic, phosphoric, succinic, acetic,
nitric, benzoic, ascorbic, p-toluenesulfonic,
benzenesulfonic, naphthalenesulfonic, propionic, and the
like. The hydrochloric acid addition salts are preferred.
The selection and preparation of pharmaceutically
acceptable non-toxic acid addition salts are within the
ability of one of ordinary skill in the art utilizing
procedures and techniques well known and appreciated in the
art.
In general, the compounds of formula (I) may be
prepared by chemical reactions analogously known in the
art. The choice of any specific route of preparation is
dependent upon a variety of factors. For example, general
availability and cost of the reactants, applicability of
certain generalized reactions to specific compounds, and so
forth, are all factors which are fully understood by those
of ordinary skill in the art and all contribute to the
choice of synthesis in the preparation of any specific
compound embraced by formula (I).
W094/07480 21~631~ PCT/US93/08517
A preferred route for the synthesis of compounds of the
formula (I) wherein Z is -CH2CH2CH2-, but also applicable by
analogy for other compounds of formula (I) wherein Z is an
alkyl-substituted propyl group (such as -CH(CH3)CH2CH2-), is
presented in Reaction Scheme A.
Reaction Scheme A
H2N ( CH2 ) mNH2
EtOH ~ Nc(cH2)2NH(cH2)mNH(cH2)2cN
H2C=CHCN 2
PtO2 H2N(CH2)3NH(CH2)mNH(CH2)3NH2-4HCl
2 + H2
HCl/AcOH
~ NaOH/H2O HN(cH2)3N-(cH2)mlN-(cH2)3lNH
2 + O-C-O-C-O THF Boc Boc Boc Boc
20 1 1
Y Y 4
4 + R-halide Kt-BuO ~ RN(CH2)3N(CH2)mN(CH2)3NR
25 DMF Boc Boc Boc Boc
Et20
5 + HCl ~ RNH(CH2)3NH(CH2)mNH(CH2)3NHR 4HCl
EtOH
W O 94/07480 2 ~ ~ ~ 3 1 ~ PC~r/US93/08517
wherein m is 7 or 8, R is as defined in formula (I) except
when R is X-(Ar)-(CH)x, x cannot be zero. In reaction
scheme A Boc is the t-butoxycarbonyl protecting group, and
Y is tert-butyl.
The initial step of this process entails an N-
alkylation of the appropriate diamine with 2 equivalents of
acrylonitrile by heating reactants, either in a suitable
solvent or neat, according to standard conditions well
known in the art. The resulting cyano derivatives (2) are
chemically reduced by reaction with hydrogen in the
presence of a catalyst (PtO2) in a suitable solvent, such as
acetic acid containing 8 equivalents of hydrochloric or
hydrobromic acid, to produce the resulting hydrohalic salts
according to standard procedures well known in the art. Of
course, other reducing systems, e.g., reduction with
lithium aluminum hydride, may also be utilized to produce
compounds of formula (3). Following the preparation of
these compounds the hydrohalic salts are neutralized with
base and the nitrogen atoms are protected, preferably with
di-t-butyldicarbonate according to standard operating
conditions well known in the art. The tetra N-protected
amines (4) are alkylated by reacting (4) with the
appropriate alkyl halides (chloro or bromo) in the presence
of potassium butoxide according to standard alkylation
procedures well known in the art. When it is desired to
provide compounds of the formula (I) wherein both R groups
are the same, about 3 equivalents of the alkyl halide is
reacted. When it is desired to provide compounds of the
formula (I) wherein the R groups are not the same,
monosubstitution of compounds of formula (4) is effected by
reacting about 1 to about 1.5 equivalents of the alkyl
halide with subsequent isolation of the monosubstituted
compound according to standard procedures well known in the
art and optionally further reacting the monosubstituted
W094/07480 2 1 4 6 3 1 9 PCT/US93/08517
compound with the desired different alkyl halide.
Following alkylation the N-protective groups of compound
(5) are removed by standard procedures, e.g., treatment
with acid, preferably HCl, in the presence of a suitable
solvent or solvent system, e.g., diethyloxide in ethanol,
to obtain the desired products t6).
Alternatively, compounds of formula (3) and their
otherwise prepared homologs may be subjected to a reductive
alkylation using an appropriate aldehyde. The reduction is
effected by hydrogenation in the presence of PtO2 or sodium
cyanoborohydride according to well known procedures. This
procedure does not generally require protection of the
nitrogen atoms of the intermediates.
A preferred route for the preparation of compounds of
formula (I) wherein Z is -CH2(CH2)2CH2-, but which is also
applicable by analogy to those compounds wherein Z is any
straight chain, is presented in Reaction Scheme B.
W094/07480 2~ 4~3~ PCT/US93/08517
--10--
Reaction Scheme B
H2N(cH2)noH Rl-CHO ~ RHN(CH2)nOH
PtO2/H2 8 N-Protect
RN(CH2)nOH MsCl RN-(CH2)nOMs
C
Boc pyridine Boc
NH(CH2)mNH
9 10
Boc Boc
R-N-(CH2)n-N-(CH2)m-N-(CH2)n-N-R
Boc Boc Boc Boc
wherein m is 7 or 8, n is an integer 2 to 6 describing a
straight chain alkylene moiety, Boc is the t-butoxycarbonyl
protecting group, R is as defined in formula (I), Ms is
mesyl and Rl is hydrogen, methyl or ethyl.
This synthesis is initiated by reductive alkylation
techniques well known in the art using an amino alcohol (7)
and an appropriate aldehyde to form R- substituted amino
alcohols (8). The nitrogen atom is protected, preferably
with di-t-butyldicarbonate, according to standard operating
conditions well known in the art, to yield the N-protected
amino alcohols (9) which are converted to their mesylates
(10) by known reaction conditions, e.g., reaction with
mesylchloride in the presence of pyridine, preferably in a
solvent such as CH2C12.
W094/07480 2 1 4 6 31 9 PCT/US93/08517
The mesylate is subjected to alkylation with an N-
protected diamine (i.e., BocNH(CH2)mNHBoc) in the presence
- of potassium t-butoxide in a solvent such as DMF. The so-
produced tetra N-protected tetramines (11) are deprotected
as in Scheme A. In essence the foregoing reductive
alkylation, N-protection, mesylation, alkylation and
deprotection procedures all employ techniques and reaction
conditions which are well known in the art.
In those instances wherein it is desired to prepare
compounds of formula (I) wherein Z is -CH2-CH2-, it is
preferred to employ Reaction Scheme C to obtain the
necessary intermediates (14) which could be subjected to
the alkylation procedures discussed above in Scheme A
wherein m is 7 or 8.
Reaction Scheme C
H2NCH2CH2NH2+ Br(CH2)mBr H2N(CH2)NH(CH2)mNH(CH2)2NH2
12 13 14
The foregoing N-alkylation entails the reaction of an
appropriate dihaloalkane (13) with excess quantities (lOx)
of ethylene diamine (12) by heating the reactants at reflux
temperatures in a suitable solvent, e.g., ethanol.
Preparation of the final products bearing the desired R
substituents on the terminal nitrogen atoms of the
intermediates (14) may be effected by N-protection,
alkylation with the appropriate alkyl halide, and
deprotection in an analogous manner to that described for
Reaction Scheme A. Preferably, the alkylation can be
carried out by the reductive alkylation procedures without
N-protection as alternatively described for Reaction
Scheme A.
W094/07480 2 1~ 6 3 ~ 9 PCT/US93/08~17
-12-
In general, compounds of the formula (Ia) can be
prepared in an analogous manner to that described in
Reaction Scheme D.
Reaction Scheme D
H2N-Zl-OH Rl-CHO ~ R-N-Zl-OH
Ptoz/H2 H
l6
15 or 16 N-Protect~ R-N-Zl-OH
Boc
l7
17 + MsCl pyridine R-N-Zl-OMs
Boc
18 + HN-(CH2)m-NH ~ R-N-Zl-NH-(CH2)m-NH-Zl-N-R
_ 1 1 1 1 1 1
Boc Boc Boc Boc Boc Boc
19 + HCl Et2O ~ NH-Zl-NH-(CH2)m-NH-Zl-NH 4HCl
EtOH
R R
W094/07480 ~ 4 6 3 19 PCT/US93/08517
wherein m is 7 or 8, R and Zl are as generically defined for
formula (Ia), and Rl is hydrogen.
The appropriate primary amino alcohol (15) containing a
branched chain hydrocarbyl moiety (i.e., Zl) is prepared by
standard procedures well known in the art. If desired, the
primary amine can at this point be converted to a secondary
amine (16), by a reductive alkylation with the appropriate
aldehyde. The amino alcohol is reacted as described in
Reaction Scheme A by standard conditions well known in the
art to effect protection of the amines with an appropriate
N-protecting group such as Boc (17). The mesylates of the
N-protected amino alcohols (18) are prepared and are
alkylated with the appropriate N-protected diamine (i.e.,
BocNH(CH2)mNHBoc) using standard procedures well known in
the art as discussed for Reaction Scheme B. The so-
produced tetra N-protected tetramines (19) are deprotected
as in Scheme A to yield compounds of the formula (Ia). In
essence, the foregoing reductive alkylation, N-protection,
mesylation, alkylation and deprotective procedures all
employ techniques and reaction conditions which are well
known in the art.
Where it is desired to provide a compound of the
formula (Ia) wherein each R group is not the same, the
substituted mesylates (18) are prepared separately and
monoalkylation of the appropriate N-protected diamine
(i.e., BocNH(CH2)mNHBoc) is effected by reacting the diamine
with about 1.0 to 1.5 equivalents of one of the mesylates
(18) with subsequent isolation of the monosubstituted
compound and optionally further reacting the
monosubstituted compound with the desired differently
substituted mesylate (18).
W094/07480 PCT/US93/08517
~4~3~9
-14-
In those instances in which it is desired to prepare
compounds of the formula (Ia) wherein Zl is an alkyl-
substituted propylene group such as *-CH(Q)CH2CH2- wherein Q
is a saturated alkyl radical comprising 1 to 3 carbon atoms
of straight or branched chain configuration, Reaction
Scheme E can be used to obtain intermediates of the formula
(25) which can be subjected to alkylation of the N-terminal
groups in a manner analogous to that described in Reaction
Scheme A prior to de-protection,
W094/07480 ~ PCT/US93/08~17
Reaction Scheme E
H2N~(CH2)m~NH2 H2/PtO2 I HN(CH2)m-NH
EtOH CIH2 ICH2
Il 0 0
0--C
H 21
0
Il 11 11
21 + H2C=CH-C MeOH C~(CH2)2~lN~(CH2)m~N~(CH2)2~C
Q Q f H2 CH2 Q
0 0
22
_ 23
N-OH N-OH
23 NH2OH HCl 11 11
C~(CH2)2~N~(CH2)m~N~(CH2)2~c
NaOH
Q CH2 CH2 Q
0 0
24LAH/AlC13H2N-CH-(CH2)2-N-(CH2) m~ I ~ ( CH2 ) 2-CI H-NH2
THF Q CH2 CH2 Q
0 0 25
wherein m is 7 or 8, 0 is phenyl, and Q is as defined
above.
The initial step of the process entails a reductive
alkylation wherein the appropriate diamine is reacted with
hydrogen gas and 2 equivalents of benzaldehyde in the
presence of a catalyst such as PtO2 to yield the N-protected
W O 94/07480 PC~r/US93/08517
~4~319
-16-
diamine (21) under standard conditions well known in the
art. The N-protected diamine (21) is then alkylated with 2
equivalents of the appropriate vinyl ketone (22) in a
suitable solvent such as methanol using standard
techniques. The resulting N-substituted diamine (23) is
further reacted under standard conditions with
hydroxylamine hydrochloride in the presence of base such as
NaOH in a suitable solvent such as ethanol/water. The
resulting oximes (24) are reduced to the corresponding N-
protected di-primary amines (25) by reaction with lithium
aluminum hydride (LAH) in the presence of AlC13 in a
suitable solvent such as THF according to standard
procedures. The N-protected di-primary amines (25) can be
further alkylated with an appropriate aldehyde prior to
deprotection in a manner analogous to that described for
Reaction Scheme A.
Compounds of formula (Ia) wherein Zl is
*-CH(CH3)CH2CH2- or *-CH(C2H5)CH2CH2- are generally preferred
in their end-use application. Compounds of formula (Ia)
wherein each R group is the same moiety are also preferred.
Compounds of formula (Ia) wherein each R group is methyl or
ethyl are particularly preferred.
Of course, it is appreciated that in those instances
wherein a compound of formula (Ia) possesses one or more
chiral centers, the individual stereoisomers as well as
mixtures of stereoisomers are included within the scope of
the present invention. For example, the following
compounds are specifically included within the scope of
formula (I) and (Ia):
(R,R)-N,N'-bis[3-(methylamino)butyl]-1,7-diaminoheptane
(S,S)-N,N'-bis[3-(methylamino)butyl]-1,7-diaminoheptane
(R,S)-N,N'-bis[3-(methylamino)butyl]-1,7-diaminoheptane.
A preferred method for preparing compounds of formula [I]
wherein -(CH)x-(Ar)-X represents phenethyl or
W094/07480 214 ~ 319 PCT/US93/08517
naphthylethyl, particularly wherein Z is 3 and m is 8, is
the reaction of an aroylchloride according to the method
depicted in Reaction Scheme F wherein depicted in Scheme F
- Bn is benzyl, ~ is phenyl, and LAH is lithium aluminum
hydride.
REACTION SCHEME F
H2N(CH2)3N-Bn O
+
(cH2)8 ~cH2
H2N(CH2)3N-Bn
127] 128]
~pCH2C-CNH(CH2)3N-Bn LAH ~pCH2CH2NH(CH2)31~-Bn
(CH2)8 Reduction (CH2)s
l l
~CH2C-C-CNH(CHz)3N-Bn ~CH2CH2NH(CH2)3N-Bn
Il l30]
O [29]
~CH2CH2NH(CH2)3~H
H2
Pd/C (CH2)8
3 5 cpCH2CH2N H (CH 2)3 N H
131]
W094/07480 ~ 3 PCT/US93/08517
-18-
As stated above, the foregoing reaction is a preferred
method for the preparation of one particular compound which
entails N-alkylation of a partially protected intermediate
[27] with an arylacetyl chloride [28] in the presence of
triethylamine, using an inert solvent, to form an amide
[29] which is chemically reduced, preferably with LAH, and
the resulting product [30] is catalytically de-benzylated
(H2Pd/C) to form the desired end product [31]. These steps
entail reaction techniques and procedures well known in the
art. Of course the same reaction scheme can be applied for
the preparation of other compounds of formula [I]; adoption
of the technique being with the usual caveats well
understood by those of ordinary skill in the art.
In those instances wherein -(CH)x-(Ar)-X represents an
aromatic moiety (X-phenyl or X-naphthyl) which is attached
directly to the terminal nitrogen atoms (i.e., x is zero)
then such compounds may be prepared according to the
general reactions of Reaction Scheme G.
~ 214~9
W O 94/07480 PC~r/US93/08517
--19--
REACTION SCHEME G
cpNH(cH2)2cN LAH (pNH(CH2)3NH N-Protect
or H2
[32] [33]
~tpN(CH2)3NH I(CH2)n Iq)N(CH2)3~ -Boc
Boc Boc NDaMHFI ~oc (CH2)m
~34] ~pN(CH2)3N-Boc
Boc
[35]
The foregoing reaction scheme depicts the preparation
of compounds wherein Ar is phenyl, the first step of which
is a LAH reduction effected according to procedures
published is the art (Bul. Soc. Chim. Fr., Part 2, 165-7
(1979)). Of course this reaction scheme can be expanded to
include napthyl and X-substituted intermediates which will
not be adversely affected by the reaction conditions.
Preferably the N-protection uses the t-butoxycarbonyl
protecting groups which are put on and taken off according
to standard techniques already mentioned hereinabove. The
N-protected compounds are alkylated by reaction with an
appropriate dihaloalkane using standard and well known
procedures.
W094/07480 ` PCT/US93/08517 ~
2 1 ~
-20-
In those instances wherein it is desired to prepare
compounds of formula [I] which contain an unsaturated
hydrocarbyl moiety, i.e., acetylenic, allenic, or allylic
moiety-containing compounds, it is preferred to use the
techniques of Reaction Scheme H wherein as shown R2 is an
appropriate unsaturated hydrocarbyl moiety, Bn is benzyl,
MsCl is methanesulfonyl chloride, and Boc is the t-
butoxycarbonyl protecting group.
WO 94/07480 ~1 4 ~3 1 9 PCI/US93/08517
--21--
REACTION SCHEME H
BnNH(CH2)mNHBn Cl-Z-OH HO-Z-N(CH2)ml\-Z-OH
nBnOH
NaI Bn Bn
[36]
[37l
DebenzylateHO-Z-NH(CH2)mNH-Z-OH
138]
138] N-ProtectHO-Z-N(CH2)mNH-Z-OH
Boc Boc
[39]
[39] MsCIMsO-Z-N(CH2)m~-Z-OMs
CH 2Cl2
Pyridine
Boc Boc
l40]
2 5 R2NHBoc
[40] NaH, NaIRzN-Z-l\(CH2)m~~Z~~R2
DMF
Boc Boc Boc Boc
41] HCI/EtOH [41]
R2NH-z-NH(cH2)mNH-z-NHR2
142]
W094/07480 ~ 3 ~ 9 - PCT/US93/08517
-22-
In the foregoing reaction a dibenzylated diamine [36]
is N-alkylated by a simple displacement reaction to form
compounds [37] which are sequentially benzylated [38] and
N-protected. These steps are effected according to well
known and standard procedures. The resulting
bishydroxyamino-alkanes [39] are mesylated and the
mesylates [40] are alkylated with two equivalents of an N-
protected amine bearing an appropriate unsaturated
hydrocarbyl moiety, e.g., N-(t-butoxycarbonyl)-2,3-
butadienylamine. A so-obtained tetra protected tetramine
[41] is then readily de-protected to produce the desired
compounds [42].
In those instances wherein it is desired to convert an
alkylthio substituent to one of its higher oxidation states
the alkyl thioether is treated with a peracid according to
known conditions. Suitable oxidizing agents are H2O2 and
NaIO4, but meta-chloroperoxybenzoic acid is preferred. In
effecting the oxidation to a sulfinyl derivative l molar
equivalent (per alkylthioether moiety) is used and 2 molar
equivalents of the peracid will yield the sulfonyl
derivatives. The oxidations are effected at temperatures
of about OoC to room temperature in solvents which
themselves are not susceptible to oxidation. Preferred
solvents are CH2Cl2, CHCl3, acetic acid, and ethyl acetate.
The foregoing reaction scheme I depicts a method of
preparation of the compounds of formula I. In scheme I the
following abbreviations are used: Ts is a tolunesulfonyl
substituent, DEAD is diethyl azodicarboxylate, THF is
tetrahydrofuran, TFA is trifluoroacetic acid, and Boc is
the t-butoxycarbonyl protecting group. It is intended that
the reaction scheme is illustrative, and not limiting, of
the chemistry depicted, wherein it is understood the
reaction scheme can be expanded to include variations of m,
Z, and R as defined in formula I. For instance, the
reaction scheme can be expanded to include the variations
~ W094/07480 ~14~ 3.1 9 PCT/US93/08~17
of Ts protecting group by various other arylsulfonyl groups
beside toluenesulfonyl, such as mesitylenesulfonyl,
benzylsulfonyl and the like.
Initial step (a), of scheme I, is the formation of
compound [44] by reaction of N-t-butyloxycarbonyl-p-
tolunesulfonamide with a chloroalkyl alcohol in the
presence of triphenylphosphine (TPP) and DEAD to form the
compound of [44]. Reactions involving step (a) is
especially preferred to step (g) when Z=4.
Step (b) involves the reaction of two equivalents of
Compound [44] with a suitably protected diaminoalkane,
e.g., as shown a di-tolsyl protected alkylydiamine
(TsNH(CH2)mNHTs) can be used to form intermediate [45].
An alternative route (step (g)) for forming
intermediate [45] can be accomplished by reaction [43] with
the approximately l equivalent of the alkyldiol shown in
the presence of TPP and DEAD in a suitable solvent, such as
THF to form intermediate [49]. Intermediate [49] can then
be reacted with a protected diaminoalkane, e.g., as shown,
the ditosyl protected alkyldiamine (TsNH(CH2)mNHTs), to form
intermediate [45]. This route of synthesis, step (g), is
suitable in those instance when Z is not 4.
Further reaction of compound [45] can occur by
deprotection o~ the Boc protecting groups with a suitable
acid, as for example with trifluoroacetic acid in an
aprotic solvent to form intermediate [46]. Further
deprotection with a stronger acid, e.g. HBr, can be used to
form structures of compound [48] where in the substituted R
group is hydrogen. Rather than a two step treatment of
acid by steps (c) and (d), the more general route of
deprotection would be to directly treat [45] with a strong
acid, e.g. HBr, to directly form [48] wherein R is
hydrogen.
W094/07480 ~1~ 6 3 lg PCT/US93/08~17
-24-
Optional substitution of compound [48] wherein R is
desired to be other than hydrogen, can be accomplished in
either of two ways. First, compounds of formula [45] can
be reacted with a R-alkyl alcohol in the presence of TPP
and DEAD in an appropriate solvent (step e).
Alternatively, intermediate [46] can be reacted with a R-
alkyl halide in the presence of hydride ion and sodium
iodide to give the compound of formula [47]. Compounds of
formula [47] can then be deprotected upon treatment with
acid, to form the compounds of formula [48], wherein R is
other than hydrogen.
219~319
WO 94/07480 PCI/US93/08517
--25--
E m
T ¦ E
Z Z ,~ D) û
Z U I ~I ~ n \
U U \
E Z u
~ O,~, I E
Z-- _ ~
1~ ~ I I
U
U ~1 1 1
f~ z _
O
1--~ I 111 ,
U
ov O
Z ~
W094/07480 ~ PCT/US93/08517
2~
-26-
REACTION SCHEME J
(a)
HCHO
H2N NH-(CH)m-NH NH2
10~J 509 ~J
(b)
H N N-(CH 2)m-N --~H Ac20
15 ~ 51 ~
(C)
~\ ~ LAH
~ !N N-(CH2)m-N N~ ~ I
(d)
CH3CH2-N N-(CH2)m-N N-CH2CH3 HCI
53
~0
CH3CHz-NH NH-(CH2)m-NH NH-CH2CH3
~ 54
The foregoing reaction scheme J depicts the preparation
of N,N'-Bis[3-(ethylamino)propyl]1,7-heptanediamine
~ 214~3~9
W094/07480 PCT/US93/08517
tetrahydrochloride. Of course, certain parts of the
reaction scheme can be expanded to include the variations
of m and R o formula I, however, Z is generally limited to
~ being propyl, wherein Z is 3. The R group introduced in
scheme J is shown as ethyl, but the reaction scheme is not
limited only to ethyl derivatives but may include those R
groups of formula I to form corresponding R substituted
derivatives.
The initial step (a) of scheme J is a protection of the
nitrogens of formula [50] by reaction with aqueous HCHO
using standard and well known procedures to give the
corresponding diaza ring systems in compound [51].
In step (b) the N-protected amines [51] are alkylated
by reacting with the appropriate acid anhydride according
to standard alkylation procedures known to those skilled in
the art. When it is desired to provide compounds of the
formula (I) wherein both R groups are the same, about 3-6
equivalents of the acid anhydride is reacted. When it is
desired to provide compounds of the formula (I) wherein the
R groups are not the same, monosubstitution of compounds of
formula [52] is effected by reacting about 1 to about 1.5
equivalents of the acid anhydride with subsequent isolation
of the monosubstituted compound according to standard
procedures well known in the art and optionally further
reacting the monosubstituted compound with the desired
different acid anhydride or alkyl or aryl halide.
The resulting acetylated derivatives of [52] are then
chemically reduced (step c) by reaction with lithium
aluminum hydride in anhydrous conditions according to
standard procedures well known in the art. Of course,
other reducing systems, e.g., reduction with H2 and PtO2,
may also be utilized under appropriate conditions to
produce compounds of formula [53].
W094/07480 ~1 4 ~ 3 19 PCT/US93/08517
-28-
Following reduction, the N-protective groups of
compound [53] are removed by standard procedures, e.g.,
treatment with acid, preferably HC1, in the presence of a
suitable solvent or solvent system, to obtain the desired
products of formula [54].
As is well known in the art of pharmaceutical
inventions wherein generic classes of compounds are
involved, certain specific compounds are more effective in
their end use applications than other members of the
generic class. The following compounds are preferred in the
method of use described by the present invention:
1,18-Bis[(phenyl)methyl]-1,5,14,18-tetraazaoctadecane-4HCl,
1,20-Bis[(phenyl)methyl]-1,16,15,20-tetraazaeicosane-4HCl,
N,N'-Bis(3-aminobutyl)-1,8-octanediamine,
N,N'-Bis(3-ethylamino)butyl]-1,7-diaminoheptane
tetrahydrochloride,
1,4,13,16-tetra(t-butoxycarbonyl)-
1,4,13,16tetraazahexadecane,1,18-Bis[(2-phenyl)ethyl]-1,5,14,18-
tetraazaoctadecane-4HCl,
1,18-Bis(phenyl)-1,5,14,18-tetraazaoctadecane,
1,18-Bis(2,3-butadienyl)-1,5,14,18-tetraazaoctadecane
tetrahydrochloride.
Especially preferred are the following compounds:
3,7,15,19-tetraazaheneicosane tetrahydrochloride,
3,17-dimethyl-2,6,14,18-tetraazanonadecane
tetrahydrochloride, and
4,16-dimethyl-2,6,14,18-tetraazanonadecane
tetrahydrochloride.
W094/07480 ~ 3 1~ PCT/US93/08517
-29-
Based upon standard laboratory experimental techniques
and procedures well known and appreciated by those skilled
in the art, as well as upon comparisons with compounds of
known usefulness, the compounds of formula (I) can be used
in the treatment of patients suffering disease states for
CMV infections.
Of course, one skilled in the art will recognize that
not every compound of formula (I) will be effective against
each of the CMV disease states, and that selection of the
most appropriate compound is within the ability of one of
ordinary skill in the art and will depend on a variety of
factors including assessment of results obtained in
standard animal tumor models.
As used herein, the term "patient" refers to a warm-
blooded animal such as a mammal which is afflicted with a
neoplastic disease state. It is understood that dogs,
cats, rats, mice, horses, bovine cattle, sheep, and humans
are examples of animals within the scope of the meaning of
the term.
The term "cytomegaloviral infection" as used herein
refers to an abnormal state or condition characterized by
an active or latent CMV infection or state whenever the
patient has virus residing active or dormant in the
patients tissues, fluids, or body cavities.
Treatment of a patient afflicted with a CMV
disease state comprises administering to such patient an
amount of a compound of the formula (I) which is
therapeutically effective in controlling the symptomatic
and/or associated viral states of CMV beyond that expected
in the absence of such treatment.
W094/07480 ~ PCT/US93/08517 ~
'~14~319
-30-
As used herein, "controlling CMV infection" refers to
slowing, interrupting, arresting or stopping a CMV
infection and does not necessarily indicate a total
elimination of the virus. It is believed that the
prolonged survivability of a patient with the instant
compounds indicates control of a CMV infection.
In effecting treatment of a patient afflicted with a
CMV infection or prophylatic treatment to prevent infection
a compound of formula (I) can be administered parenterally
in any manner which makes (I) bioavailable in effective
amounts including for example, by intraperitoneal (i.p.),
subcutaneous (s.c.), or intravenous (i.v.) injection.
Administration by intravenous injection is preferred.
A therapeutically effective dose or amount can readily
be determined by the attending diagnostician and is a
function of a number of factors including, but not limited
to, the species of mammal, its size, age and general
health, the specific CMV infection involved, the stage of
the CMV infection, the compound selected and mode of
administration, the bioavailability characteristics of the
preparation administered, the dose regimen selected, and
use of concomitant medication. The correct amount for any
specific situation can be readily determined by those
skilled in the art using conventional range finding
techniques and analogous results observed under other
circumstances. A therapeutically effective amount of (I)
will vary from about 1 milligram per kilogram of body
weight per day (mg/kg/day) to about 500 mg/kg/day and
preferably will be about 5 mg/kg/day to about 50 mg/kg/day.
It is believed that compounds of the formula (I)
administered at the above doses to a patient suffering from
a CMV infection are therapeutically effective in
con-trolling the growth of one or more disease states a
patient may have so as to prolong the survivability of the
W094/07480 ~1 4 ~ 3 1 g PCT/US93/08517
-31-
patient beyond that expected in the absence of such
treatment.
Another embodiment of the present invention relates to
pharmaceutical compositions for parenteral administration
~ for compounds of the formula (I). These pharmaceutical
compositions comprise a therapeutically effective amount of
one or more compounds of the formula (I) in an admixture
with one or more pharmaceutically acceptable excipients,
Such compositions are prepared in conventional manner well
known in the art of pharmaceutical science. The amounts of
the active ingredient(s) in a unit dosage form and the
dosage regimen are adjusted to provide a sustained
pharmacologic effect at the dose regimen selected.
Pharmaceutically acceptable excipients are substances
that are chemically inert to the active compound(s) and
have no detrimental side effects or toxicity to mammals
under the conditions of use. Suitable excipients include
solvents such as water, alcohol, and propylene glycol,
surface active agents, suspending agents, lubricants,
flavors, colorants, and the like. Such carriers and
excipients are known to those in the art and are disclosed,
for example, in texts such as Reminqton's Pharmaceutical
Manufacturinq, 13th Edition, Mack Publishing Co., Easton,
PA (1965).
Injectable dosage forms of a solution or suspension of
(I) can be prepared, for example, in a physiologically
acceptable diluent with a pharmaceutical carrier which can
be a sterile liquid such as water and oils with or without
the addition of a surfactant and other pharmaceutically
acceptable adjuvants. Illustrative of oils which can be
employed in these preparations are those of petroleum,
animal, vegetable or synthetic origin, for example, peanut
oil, soybean oil and mineral oil. In general, water,
saline, aqueous dextrose and related sugar solution
W094/07480 2 1 4 ~ 3 1 9 PCT/US93/08517 ~
-32-
ethanols and glycols such as propylene glycol or
polyethylene glycol are preferred liquid carriers,
particularly for injectable solutions.
As is well known in the art of pharmaceutical inven-
tions wherein generic classes of compounds are involved,
certain subgeneric and certain specific compounds are more
efficient in their end-use applications than other members
of the generic class. In this invention, those compounds
wherein Z is -CH2CH2CH2- or -CH(CH~)CH2CH2- are most
preferred. In all instances it has been shown that the
symmetrical compounds are preferred. Compounds for which
each R is independently methyl or ethyl are preferred for
this method of use and compounds for which both R groups
are methyl or both R groups are ethyl are preferred.
Compounds for which both R groups are the same moiety are
generally preferred. Further compounds of formula I may
have m = to 7 or 8
The following compounds are preferred in the method of
use described by the present invention:
N,N'-bis[3-(methylamino)butyl]-1,7-diaminoheptane;
N,N'-bis[3-(ethylamino)propyl]-1,7-diaminoheptane
(MDL28,314);
N,N'-bis[3-(methylamino)propyl]-1,7-diaminoheptane;
N,N'-bis[3-(methylamino)-2-(methyl)propyl]-1,7-
diaminoheptane.
Amongst the listed compounds, and the respective
substituent which are structurally part their of may be
used to form a more preferred grouping of compounds.
For instance, of the compounds listed, N,N'-bis[3-
(ethylamino)propyl]-1,7-diaminooctane and N,N'-bis[3-
(ethylamino)propyl]-1,7-diaminoheptane are most preferred.
W094/07480 ~ 3 1 9 PCT/US93/08517
In order to illustrate the preparation of compounds of
formulas (Ia) and (Ib), the following examples are
provided. The examples are illustrative only and are not
intended to limit the invention in any way. All
temperatures are in degrees Celsius and the following
abbreviations are used: (g) is grams, (mol) is moles, (ml)
is milliliters, (1) is liters, (lb/in2) is pounds per square
inch, (TLC) is thin layer chromatography, (THF) is
tetrahydrofuran, (DMF) is dimethylformamide, (mp) is
melting point, (mm/Hg) is pressure expressed as millimeters
of mercury, (bp) is boiling point.
C~EMICAL EXAMPLES
EXAMPLE 1
N,N-Bis((3-methylamino)propyl)-1,8-octanediamine
tetrahydrochloride
Step A: N,N'-Bis(2-(cyano)ethyl)-1,8-octanediamine
Dissolve 14.4 g (0.1 mol) of 1,8-diaminooctane and 14.5 ml
(0.22 mol) of acrylonitrile in 100 ml of ethanol and reflux
overnight. Remove the solvent at reduced pressure.
Analysis showed the title compound to be >98% pure.
Step B: N,N'-Bis(3-(amino)propyl)1,8-octanediamine
tetrahydrochloride
Combine 14.4 g (0.057 mol) of the product of Step A, 200 ml
of acetic acid, 30 ml of conc. HCl, and 1.2 g PtO2 and treat
the mixture with H2 at 45 lbs/in2 in a shaker flask until H2
is no longer being reacted. Filter the mixture and remove
the solvent at reduced pressure. 22.5 g of the title
compound is obtained after purification. (Rf is 0.17 for
TLC on silica gel developed with 40~ conc.
ammonia/methanol).
W094/07480 p~&3~9 PCT/US93/0~S17 ~
Step C: 1,5,14,18-Tetra(t-butoxycarbonyl)-1,5,14,18-
tetraazaoctadecane
Combine 22.5 g (0.052 mol) of the product of Step B with
8.83 g (0.22 mol) of NaOH, 100 ml H2O and 500 ml THF and
stir until a homogenous solution is obtained. To this
solution add 48.13 g (0.22 mol) of di-t-butyldicarbonate
and stir the resulting mixture overnight. Pour the mixture
into 1 1. of ethyl acetate, separate the organic layer, and
dry over anhydrous MgSO4. Remove the solvent at reduced
pressure. Purify the residue by flash chromatography
(silica gel), eluting with 25% ethyl acetate/hexane to
yield 13.5 g of the title compound (Rf is 0.28 for TLC on
silica gel developed with 25% ethyl acetate/hexane).
Step D: 1,18-Bis(methyl)-1,5,14,18-tetra(t-butoxy-
carbonyl)-1,5,14,18-tetraazoctadecane
Combine 4.3 g (0.0068 mol) of the product of Step C, 0.94
ml (0.015 mol) of iodomethane, 1.69 g (0.015 mol) of
potassium t-butoxide, and 15 ml DMF and stir overnight.
Remove the solvent at reduced pressure and dissolve the
residue in 500 ml ethyl acetate and 200 ml of H2O. Wash the
organic layer with 100 ml H2O (2x) and dry over anhydrous
MgSO4. Remove the solvent at reduced pressure and purify
the residue by flash chromatography (silica gel), eluting
with 20% ethyl acetate/hexane to yield 4.4 g of the title
compound (Rf is 0.20 for TLC on silica gel developed with
20% ethyl acetate/hexane.)
W O 94/07480 - PC~r/US93/08517 3 1 9
Step E: N,N'-Bis(3-(methylamino)propyl)-1,8-octanediamine
tetrahydrochloride
Dissolve 4.4 g (0.0065 mol) of the product of Step D in 3
ml ethanol and treat the solution with 50 ml of 2N HCl in
diethyl ether stirring overnight. Filter the resulting
mixture and crystallize the residue from
methanol/isopropanol/water (20/60/20,v/v/v) at reduced
temperature. Filter and dry the product at 79C over P2O5
at 0.1 mmHg to yield 2.08 g of the title compound (mp
>300C). Elemental analysis: calculated, C-44.44, H-9.79,
N-12.86, C1-32.80; Found C-44.44, H-9.82, N-12.95,.
EXAMPLE 2
N,N'-Bis(3-(ethylamino)propyl)-1,8-octanediamine
tetrahydrochloride
Step ~: 1,18-Bis(ethyl)-1,5,14,18-tetra(t-butoxy-
carbonyl)-1,5,14-18-tetraazaoctadecane
Combine 9.5 g (0.0144 mol) of the product of Step C in
Example 1, 2.91 g (0.026 mol) of potassium t-butoxide, and
45 ml of DMF and cool to 0C. Add 2.1 ml (0.026 mol) of
iodoethane and stir at 0C for 4 hours. Allow the mixture
to warm slowly to room temperature and stir overnight.
Remove the solvent at reduced pressure and partition the
residue between 1400 ml ethyl acetate and 200 ml H20. Wash
the organic layer with 100 ml H2O (2x) and dry over
anhydrous MgSO4. Remove the solvent under reduced pressure
and purify the residue by flash chromatography (silica gel)
eluting with 20% ethyl acetate/hexane to yield 3.3 g of the
title compound (Rf is 0.26 for TLC on silica gel developed
with 20~ ethyl acetate/hexane.)
W O 94/07480 PC~r/US93/08517 ~146319
-36-
Step B: N,N'-Bis(3-(ethylamino)propyl)-1,8-octanediamine
tetrahydrochloride hemihydrate
Dissolve 3.3 g (0.0046 mol) of the product of Step A in 7
ml ethanol and treat with 70 ml of 2 N HCl in diethyl ether
stirring overnight. Filter the mixture and dry the residue
at 70C at reduced pressure to yield 1.95 g of the title
compound, mp >300C. Elemental analysis: Calculated, C-
46.09, H-10.10, N-11.95, Cl-30.24; Found C-46.23, H-9.94,
N-12.11, Cl-29.99.
EXAMPLE 3
N-(3-Aminopropyl)-N'-(3-(ethylamino)propyl)-1,8-
octanediamine tetrahydrochloride
Step A: l-Ethyl-1,5,14,18-tetra-(t-butoxycarbonyl)-
1,5,14,18-tetraazaoctadecane
Follow the procedure described in Step A of Example 2 to
yield 2.5 g of the title compound after flash
chromatography (Rf is 0.17 for TLC on silica gel developed
with 20% ethyl acetate/hexane).
Step ~: N-(3-Aminopropyl)-N'-(3-(ethylamino)propyl)-1,8-
octanediamine tetrahydrochloride
Dissolve 2.5 g (0.0036 mol) of the product of Step A in 5
ml of ethanol and treat with 60 ml of 2 N HCl in diethyl
ether stirring overnight. Filter the mixture and dry the
residue to yield 1.35 g of the title compound, mp >300C.
Elemental analysis: Calculated, C-43.54, H-9.82, N-12.69,
Cl-32.13; Found, C-43.43, H-9.60, 9.55; N-12.60, 12.62; Cl-
32.30.
W094/07480 ~1 4 63 ~ 9 PCT/US93/08517
-37-
EXAMPL~ 4
N,N'-Bis[3-(methylamino)butyl]-1,7-diaminoheptane
tetrahydrochloride
Step A: N,N'-Bis[(phenyl)methyl]-1,7-heptanediamine
Combine 1,7-diaminoheptane (65.0 g, 0.5 mol), benzaldehyde
(106 gm, 1 mol) and platinum oxide (PtO2)[2.0 g] in ethanol
(800 ml) and treat the mixture with hydrogen gas (45 lb/in2)
until the uptake of gas ceases. Remove the catalyst by
filtration and remove the solvent in vacuo. Purify the
residue by bulb to bulb distillation to yield 99.4 g of the
title compound (bp 191-195C @ 1.0 mm/Hg).
Step B: N,N'-Bis~(3-oxo)butyl]-N,N'-bis[(phenyl)methyl] -
1,7-diaminoheptane
Dissolve N,N'-bis[(phenyl)methyl]-1,7-heptanediamine (9.3
g, 0.03 mol) in methanol (120 ml) and while stirring the
mixture introduce methyl vinyl ketone (5.6 ml, 0.066 mol)
in a stream of nitrogen gas. Stir the mixture for 18 hours
to yield the title compound.
Step C: N,N'-Bis[(3-hydroxyimino)butyl]-N,N'-bis[(phenyl)
methyl]-1,7-diaminoheptane
Cool the reaction mixture obtained in step B to 0C and to
this mixture add a solution of hydroxylamine hydrochloride
(4.38 g, 0.063 mol) and sodium bicarbonate (5.54 g, 0.066
mol) in water (40 ml). Stir the mixture at 0C for 30
minutes and then stir at ambient temperature for 2 hours.
Remove the solvent in vacuo and partition the residue
between water (200 ml) and dichloromethane (200 ml). Wash
the aqueous layer 3 times with 200 ml of dichloromethane
each time. Combine the organic layers and dry over
- anhydrous MgSO4. Remove the solvent in vacuo to yield 14.4
g of the title compound. Rf is 0.53 for TLC on silica gel
developed with ethyl acetate.
W094/07480 PCT/US93/08517
~1~63 1~ -38-
Step D: N,N'-Bis[3-(amino)butyl]-N,N'-bis[(~henyl)
methyl]-1,7-diaminoheptane
Add a solution of N,N'-bis[(3-hydroxyimino)butyl]-N,N'-
bis[(phenyl)methyl]-1,7-diaminoheptane (14.4 g, 0.03 mol)
in THF (70 ml) to a mixture of lithium aluminum hydride
(5.8 g, 0.15 mol) in THF (250 ml) and reflux the mixture
overnight. Cool the mixture and ~uench slowly with water
(5.8 ml), followed by 15~ NaOH (5.8 ml), followed by water
(17.4 ml). Filter the mixture and wash the filtrate 3
times with 100 ml of THF each time. Combine the organic
layers and remove the solvent in vacuo to obtain 13.4 g of
the title compound as a clear viscous oil. Rf is 0.33 for
TLC on silica gel developed with 4% conc. ammonia in
methanol.
Step E: 2,16-Bis(methyl)-1,5,13,17-tetra(t-
butoxycarbonyl)-1,5,13,17-tetraazaheptadecane
Combine N,N'-bis[3-(amino)butyl]-N,N'-bis[(phenyl)methyl]-
1,7-diaminoheptane (13.4 g, 0.029 mol), Pearlman's Catalyst
(2.0 g) and ethanol (90 ml) and treat the mixture with
hydrogen gas at 45 lb/in2 until gas uptake ceases. Remove
the catalyst by filtration and remove the solvent in vacuo
to obtain 7.7 g of N,N'-bis[3-(amino)butyl]-1,7-
diaminoheptane (Rf is 0.37 for TLC on silica gel developed
with 40% conc. ammonia in methanol). Dissolve the residue
in dichloromethane (90 ml) and treat the mixture with di-t-
butyldicarbonate (26.2 g, 0.12 mol) for 3 hours. Remove
the solvent in vacuo and purify the residue by flash
chromatography on silica gel eluting with 25% ethyl acetate
in hexane to yield 17.1 g of the title compound as a clear
oil. Rf is 0.35 for TLC on silica gel developed with 25%
ethyl acetate in hexane.
3 1 9
W094/07480 PCT/US93/08517
-39-
Step F: 1,2,16,17-Tetramethyl-1,5,13,17-tetra(t-
butoxycarbonyl)-1,5,13,17-tetraazaheptadecane
Combine 2,16-bistmethyl)-1,5,13,17-tetra(t-butoxycarbonyl)-
1,5,13,17-tetraazaheptadecane (8.5 g, 0.0126 mol) and
sodium hydride (60% in oil)[1.21 g, 0.03 mol] in DMF (75
ml) and stir until hydrogen evolution ceases. To this
mixture add methyl iodide (1.88 g, 0.03 mol) and stir for 2
hours. Remove the solvent in vacuo and partition the
residue between ethyl acetate (400 ml) and water (200 ml).
Dry the organic layer over anhydrous MgSO4 and remove the
solvent in vacuo. Purify the residue by flash
chromatography on silica gel eluting with 22% ethyl acetate
in hexane to yield 3.8 g of the title compound as a clear
oil. Rf is 0.22 for TLC on silica gel developed with 20%
ethyl acetate in hexane.
Step G: N,N'-Bis[3-(methylamino)butyl]-1,7-diaminoheptane
tetrahydrochloride
Add lN HCl in methanol (50 ml) to 1,2,16,17-tetramethyl-
1,5,13,17-tetra(t-butoxycarbonyl)-1,5,13,17-
tetraazaheptadecane (3.8 g, 0.0054 mol) and stir overnight.
Remove the solvent in vacuo and recrystallize the residue
two times from methanol/acetonitrile (40/60, v/v) to yield
0.74 g of the title compound as a white solid (mp 238-9
C). Rf is 0.31 for TLC on silica gel developed with 40%
conc. ammonia in methanol.
W094/07480 ~g ~ 3 PCT/US93/08~17
-40-
EXAMPLE 5
N,N'-Bis[3-(ethylamino)butyl]-1,7-diaminoheptane
tetrahydrochloride
Step A: 1,17-Diethyl-2,16-dimethyl-1,5,13,17-tetra(t-
butoxycarbonyl)-1,5,13,17-tetraazaheptadecane
Combine 2,16-bis(methyl)-1,5,13,17-tetra(t-butoxycarbonyl)-
1,5,13,17-tetraazaheptadecane (8.5 g, 0.0126 mol), made as
described in Example 5, and sodium hydride (60% in
oil)[1.21 g, 0.03 mol] in DMF (75 ml) and stir until
hydrogen evolution ceases. To this mixture add ethyl
iodide (4.68 g, 0.03 mol) and stir for 2 hours. Remove the
solvent in vacuo and partition the residue between ethyl
acetate (400 ml) and water (200 ml). Dry the organic layer
over anhydrous MgSO4 and remove the solvent in vacuo.
Purify the residue by flash chromatography on silica gel
eluting with 22% ethyl acetate in hexane to yield 3.9 g of
the title compound as a clear oil. Rf is 0.31 for TLC on
silica gel developed with 20% ethyl acetate in hexane.
Step B: N,N'-Bis[3-(ethylamino)butyl]-1,7-diaminoheptane
tetrahydrochloride
Add lN HCl in methanol (50 ml) to 1,17-diethyl-2,16-
dimethyl-1,5,13,17-tetra(t-butoxycarbonyl)-1,5,13,17-
tetraazaheptadecane (3.9 g, 0.0054 mol) and stir overnight.
Remove the solvent in vacuo and recrystallize the residue
two times from methanol/acetonitrile (40/60, v/v) to yield
O.90g of the title compound as a white solid (mp 249-50
C). Rf is 0.56 for TLC on silica gel developed with 40%
conc. ammonia in methanol.
W094/07480 ~ 3 1 9 PCT/US93/08517
-41-
~XAMPLE 6
L N,N'-Bis[3-(ethylamino)propyl]-1,7-heptanediamine
Steps A and B; 1,5,13,17-Tetraazaheptadecane
tetrahydrochloride
Prepare the title compound by the method of Israel et al.,
J. Med. Chem. 7, 710 (1964).
Step C: 1,5,13,17-Tetra(t-butoxycarbonyl)-1,5,13,17-
tetraazaheptadecane
Combine_1,5,13,17-tetraazaheptadecane tetrahydrochloride
(3.9 gm, 0.01 mol) and sodium hydroxide (1.76 gm, 0.44 mol)
in water (44 ml) and stir until homogeneous. To this
mixture add di-t-butyldicarbonate (9.6 gm, 0.044 mol) in
THF (88 ml) and stir for 3 hours. Dilute the mixture with
ethyl acetate (EtOAc) [300 ml] and separate the organic
layer. Dry the organic layer over anhydrous MgSO4 and
evaporate in vacuo to obtain a viscous oil. Purify the
residue by flash chromatography (silica gel) eluting with
25% EtOAc/hexane to yield 3.0 gm of the title compound. Rf
is 0.20 on silica gel plates eluted with 25% EtOAc/hexane.
Step D: 3,7,15,19-Tetra(t-butoxycarbonyl)-3,7,15,19-
tetraazaheneicosane
Combine 1,5,13,17-tetra(t-butoxycarbonyl)-1,5,13,17-
tetraazaheptade~-ane (3.0 gm, 0.0046 mol) and sodium hydride
(50~ in oil) [0.45 gm, 0.011 mol] in DMF (9 ml) and stir
the mixture until hydrogen evolution ceases. Add ethyl
iodide (0.9 ml, 0.011 mol) and stir the mixture for 18
hours. Evaporate the DMF in vacuo and partition the residue
between ethyl acetate (600 ml) and water (200 ml).
Separate the organic layer, dry the organic layer over
W094/07480 ~4~3 i9 PCT/US93/08517 ~
-42-
anhydrous MgSO4 and evaporate in vacuo. Purify the residue by
flash chromatography (silica gel) eluting with 20%
EtOAc/hexane to yield 1.68 gm of the title compound. Rf is
0.5 on silica gel plates eluted with 25~ EtOAc/hexane.
Step E: N,N'-Bis[3-(ethylamino)propyl]-1,7-heptanediamine
Treat 3,7,15,19-tetra(t-butoxycarbonyl)-3,7,15,19-
tetraazaheneicosane (1.68 gm, 0.0024 mol) with HCl in
methanol (50 ml, 1.0 N) and stir overnight. Filter the
mixture and recrystallize the title compound from
methanol/water (20:80, v/v) to yield 0.5 gm of the title
compound. Rf is 0.39 on silica gel plates eluted with 40%
ammonia (concentrated) in methanol; mp 322-23C with
degradation.
EXAMPLE 7
1,18-Bis[(phenyl)methyl]1,5,14,18-tetraazaoctadecane-4HCl
Step A: N,N'-Bis-[2,2'-bis(cyano)ethyl]-1,8-diamino-octane
Dissolve 28.8 gm (0.2 mol) of 1,8 diaminooctane in 250 ml
of EtOH. Add 27 ml (0.41 mol) of acrylonitrile and gently
reflux the mixture overnight. Remove the solvent at
reduced pressure. Analysis shows desired material to be
>95% pure.
Step B: 1,5,14,18-Tetraazaoctadecane tetrahydrochloride
Combine 50.0 gm of the product of Example 1, 2.0 gm PtO2,
133 ml of conc. HCl at 45 lbs./sq. in. in a shaker flask
until hydrogen is no longer taken up. Filter the resulting
mixture, evaporate the solvent and triturate the product
with 1 liter of EtOH~ Filter and dry the product to obtain
51.6 gm of the title compound, Rf is 0.17 (silica gel
plates eluted with 40% conc. NH3/CH30H).
W094/07480 ~1463 1~ PCT/US93/08517
-43-
Step C: 1,5,14,18-Tetra(t-butoxycarbonyl)-1,5,14,18-
tetraazaoctadecane
- Treat 28.0 gm (0.069 mol) of the product of Step B with
10.99 gm (0.274 mol) of NaOH in 120 ml H2O. When a
homogenous solution is obtained add 65.7 gm (0.307 mol) of
di-t-butyldicarbonate in 750 ml of THF and stir the
resulting mixture for 16 hours. Separate the layers,
remove and wash (2x) the aqueous layer with 500 ml CH2Cl2.
Combine and dry (MgS04) the organics, filter and evaporate
(invacuo) the solvents and flash chromatograph the residue
(silica gel), eluting with 25% EtOAc/hexane to yield 30.2 g
of the desired product. Rf is 0.33 on silica gel plates
eluted with 25% EtOAc/hexane).
Step D: 1,18,-Bis[(phenyl)methyl]-1,5,14,18-tetra(t-
butoxycarbonyl)-1,5,14,18-tetraazaoctadecane
Dissolve 20.0 gm (0.03 mol) of the product from Step C in
30 ml DMF and treat with 7.5 gm (0.067 mol) KtBuO and 7.96
ml (0.067 mol) BnBr, with stirring for 18 hours. Evaporate
the volatiles (0.5 mm and 45C) and take up the resulting
residue in 1400 ml of EtOAc and water-wash (2x, 500 ml).
The organic layer is then dried (MgSO4) and the solvent is
evaporated (in vacuo). Flash chromatography on silica gel
eluted with 20% EtOAc/hexane yields 12.4 gm (50%) of
desired product as a clear viscous oil. Rf is .42 (silica
gel plates eluted with 25% EtOAc/hexane).
Step E: 1,18-Bis-[(phenyl)methyl]-1,5,14,18-tetraaza-
octadecane-4HCl
Dissolve 12.4 g (0.0147 mol) of the product of Step D in
14.7 ml of anhydrous EtOH and treat with 160 ml of 2N HCl
in Et2O with stirring overnight. Filter, wash the filter
cake with Et20, and dry to obtain 7.2 gm of the desired
compound, mp >300C. Rf is 0.24 (from silica gel eluted
with 10% conc. NH3/CH30H).
W094/07480 PCT/US93/08517 ~
i g
-44-
EXAMPLE 8
1,20-Bis[(phenyl)methyl]-1,16,15,20-tetraazaeicosane-4HC1
Step A: N,N'-Bis(t-butoxycarbonyl)-1,8-octanediamine
Dissolve 10.8 gm (0.075) of diaminooctane in 200 ml CH2C12
and 100 ml CH30H, add 32.7 gm (0.156 mol) of di-t-
butyldicarbonate and stir the mixture overnight. Evaporate,
invacuo, and crystallize the residue from hexane to obtain
20.2 gm of the desired compound, mp 96-97C.
Step B: 4-[[(Phenyl)methyl]amino]-butan-l-ol
Combine 4-amino-butan-1-ol (8.9 gm - 0.1 mol), benzaldehyde
(10.6 gm - 0.1 mol), EtOH (100 ml) and PtO2 (0.3 gm), and
hydrogenate the mixture at 45 lbs./sq.in. until H2 is no
longer taken up. Filter, evaporate the solvent ( in vacuo) to
yield 17.7 gm of the desired compound. Rf is 0.70 (eluted
from silica gel with 10~ conc. NH3/CH30H).
Step C: 4-[N-(t-butoxycarbonyl)-N-[(phenyl)methyl]amino]
butan-l-ol
Combine the butanol of Step B (17.7 g - 0.1 mol) and di-
tbutyldicarbonate in 100 ml of CH2C12 and stir the mixture
overnight. Evaporate off the solvents, in vacuo, and flash
chromatography of the residue, eluting from silica gel with
25% EtOAc/hexane to obtain the desired compound. Rf is .27
(silica gel plates eluted with 20% EtOAc/hexane).
Step D: 4-[N-(t-butoxycarbonyl)-N-[(phenyl)methyl]-amino]-
lnethansulfonyl butane
Cool (ice-bath) a mixture containing the product of Step C
(21.8 gm - 0.078 mol), 250 ml CH2C12 and 9.7 ml pyridine
(0.12 mol), add in a dropwise fashion (20 minutes)
mesylchloride (6.65 ml - 0.086 mol) in 6.6 ml CH2Cl2 and
allow the mixture to warm to room temperature, stirring the
mixture for 2 hours. Pour the resulting mixture into 200
ml CH2Cl2, wash with 500 ml 0.5 N HCl, saturated NaHCO3, dry
over MgS04, evaporate (in vacuo) and flash chromatograph
3 1 ~
W094/07480 PCT/US93/08517
: ':
-45-
eluting from the silica gel with 25% EtOAc/hexane to obtain
10.7 g of desired product, Rf is ).36 (silica gel plates
eluted with 25% EtOAc/hexane).
Step E: 1,20-Bis[(phenyl)methyl]-1,16,15,20-tetra-(t-
butoxycarbonyl)-1,6,15,20-tetraazaeicosane
Admix the products of Step A (5.16 gm - 0.015 mol) and of
Step D of this example (10.7 g - 0.032 mol), Kt-BuO (3.92
gm), NaI 0.2 gm), and 60 ml DMF and stir the mixture for
72 hours at room temperature. Evaporate the solvent ( in
voc~o), take up the residue in 600 ml EtOAc and wash (2x)
with 200 ml water. Dry the organic layer (MgS04), evaporate
the solvents, and flash chromatograph the viscous residue
on silica gel eluting with 20% EtOAc/hexane to obtain the
desired product, Rf is 0.22 (silica gel plates eluted with
20~ EtOAc/hexane).
Step F: 1,20-Bis[(phenyl)methyl]-1,6,15,20-tetraeicosane-4
HCl
Dissolve the product of Step E (4.7 gm) (0.0054 mol) in 5
ml EtOH and treat with 54 ml of 2N HCl in EtO2, stir the
mixture overnight, filter and recrystallize to so-obtained
solids from isopropanol/water. Cool, filter and dry the
desired product, mp >300C, Rf is 0.47 (eluted from silica
with 10% conc. NH3/CH30H).
EXAMPLE 9
N,N'-Bis(3-aminobutyl)-1,8-Octanediamine
Step A: N,N'-Bis((phenyl)methyl)-1,8-octanediamine
Combine 14.4 g (0.1 mol) of 1,8-octanediamine, 20.3 ml (0.2
mol) of benzaldehyde, 0.3 g PtO2 and 150 ml ethanol and
treat the mixture with H2 at 45 lb/in2 in a shaker flask
until no more gas is taken up. Remove the catalyst by
filtration and remove the solvent at reduced pressure to
yield the title compound.
W094/07480 - PCT/US93/08517
21~6319
-46-
Step B: N,N'Bis((3-oxo)butyl)-N,N'-bis((phenyl)methyl)-
1,8Octanediamine
Dissolve the product obtained in Step A in 1400 ml of
methanol and introduce 21.6 of methyl vinyl ketone on a
stream of N2 gas. Stir for 16 hours to yield the title
compound.
Step C: N,N'-Bis((3-hydroxyimino)butyl)-N,N'-Bis-
((phenyl)methyl)-1,8-octanediamine
Combine 18.07 g hydroxylamine hydrochloride, 10.4 g of NaOH
and 40 ml of H20 and add to the solution obtained in Step B.
Reflux the mixture for 3 hours, then cool and evaporate the
solvent. Pour the reaction mixture into 300 ml of ethyl
acetate and wash with 300 ml H20. Wash the aqueous layer
with 300 ml of ethyl acetate (2x). Combine the organic
layers and dry over anhydrous MgS04. Remove the solvent at
reduced pressure. Purify the product by flash
chromatography (silica gel), eluting with ethyl acetate to
yield 34.8 g of the title compound (Rf is 0.42 for TLC on
silica gel developed with ethyl acetate).
Step D: N,N'-Bis((3-amino)butyl)-N,N'-Bis((phenyl)-methyl)-
1,8-octanediamine
Add 34.8 g of the product of Step C in 100 ml THF to 12.10
g (0.310 mol) of lithium aluminum hydride in 540 ml THF
and reflux the mixture while stirring overnight. Cool the
mixture and slowly add 15 ml H20 followed by 45 ml lN NaOH
and stir the mixture for 6 hours. Filter the mixture to
remove a white granular precipitate and remove the solvent
at reduced pressure. Subject the residue to short path
distillation to yield 17.0 g of the title compound (bp 230-
235C at 0.1 mmHg).
Step E: N,N'-Bis((3-amino)butyl)-1,8-octanediamine
Combine 5.0 g (0.01 mol) of the product of Step D, 0.5 g of
20% Pd(OH)2 on carbon (Pearlman's Catalyst), and 50 ml or
ethanol and treat the mixture with H2 at 45 lb/in2 in a
W094/07480 2 1 ~ 6 3 1 9 PCT/US93/08517
-47-
shaker flask until no more gas is taken up. Remove the
catalyst by filtration and remove the solvent at reduced
pressure. Subject the residue to short path distillation
- to yield 1.59 g of the title compound (bp 145-148C at 0.012
mmHg).
~ XAMPLE 10
N,N'-Bis[3-(methylamino)butyl]-1,7-diaminoheptane
tetrahydrochloride
Step A: N,N'-Bis[(phenyl)methyl]-1,7-heptanediamine
Combine 1,7-diaminoheptane (65.0 g, 0.5 mol), benzaldehyde
(106 gm, 1 mol) and platinum oxide (PtO2)[2.0 g] in ethanol
(800 ml) and treat the mixture with hydrogen gas (45 lb/in2)
until the uptake of gas ceases. Remove the catalyst by
filtration and remove the solvent in vacuo. Purify the
residue by bulb to bulb distillation to yield 99.4 g of the
title compound (bp 191-195C @ 1.0 mm/Hg).
Step B: N,N'-Bis[(3-oxo)butyl]-N,N'-bis[(phenyl)methyl]-
1,7- diaminoheptane
Dissolve N,N'-bis[(phenyl)methyl]-1,7-heptanediamine (9.3
g, 0.03 mol) in methanol (120 ml) and while stirring the
mixture introduce methyl vinyl ketone (5.6 ml, 0.066 mol)
in a stream of nitrogen gas. Stir the mixture for 18 hours
to yield the title compound.
Step C: N,N'-Bis[(3-hydroxyimino)butyl]-N,N'-bis[(phenyl)
methyl]-1,7-diaminoheptane
Cool the reaction mixture obtained in step B to 0C and to
this mixture add a solution of hydroxylamine hydrochloride
(4.38 g, 0.063 mol) and sodium bicarbonate (5.54 g, 0.066
mol) in water (40 ml). Stir the mixture at 0C for 30
minutes and then stir at ambient temperature for 2 hours.
Remove the solvent in vacuo and partition the residue
between water (200 ml) and dichloromethane (200 ml). Wash
the aqueous layer 3 times with 200 ml of dichloromethane
W094/07480 ~146319 PCT/US93/08517
-48-
each time. Combine the organic layers and dry over
anhydrous MgS04. Remove the solvent in vacuo to yield 14.4
g of the title compound. Rf is 0.53 for TLC on silica gel
developed with ethyl acetate.
Step D: N,N'-Bis[3-[amino)butyl]-N~N'-bis[(phenyl) methyl]-
1,7-diaminoheptane
Add a solution of N,N'-bis[(3-hydroxyimino)butyl]-
N,N'bis[(phenyl)methyl]-1,7-diaminoheptane (14.4 g, 0.03
mol) in THF (70 ml) to a mixture of lithium aluminum
hydride (5.8 g, 0.15 mol) in THF (250 ml) and reflux the
mixture overnight. Cool the mixture and quench slowly with
water (5.8 ml), followed by 15% NaOH (5.8 ml), followed by
water (17.4 ml). Filter the mixture and wash the filtrate
3 times with 100 ml of THF each time. Combine the organic
layers and remove the solvent in vacuo to obtain 13.4 g of
the title compound as a clear viscous oil. Rf is 0.33 for
TLC on silica gel developed with 4% conc. ammonia in
methanol.
Step E: 2,16-Bis(methyl)-1,5,13,17-tetra(t-butoxycarbonyl)-
1,5,13,17-tetraazaheptadecane
Combine N,N'-bis[3-(amino)butyl]-N,N'-bis[(phenyl)methyl]-
1,7diaminoheptane (13.4 g, 0.029 mol), Pearlman's Catalyst
(2.0 g) and ethanol (90 ml) and treat the mixture with
hydrogen gas at 45 lb/in2 until gas uptake ceases. Remove
the catalyst by filtration and remove the solvent in vacuo
to obtain 7.7 g of N,N'-bis[3-(amino)butyl]-1,7-
diaminoheptane (Rf is 0.37 for TLC on silica gel developed
with 40% conc. ammonia in methanol). Dissolve the residue
in dichloromethane (90 ml) and treat the mixture with di-t-
butyldicarbonate (26.2 g, 0.12 mol) for 3 hours. Remove
the solvent in vacuo and purify the residue by flash
chromatography on silica gel eluting with 25% ethyl acetate
in hexane to yield 17.1 g of the title compound as a clear
oil. Rf is 0.35 for TLC on silica gel developed with 25%
ethyl acetate in hexane.
W094/07480 ~ 31~i ``` PCT/US93/08517
-49-
Step F: l~2~l6~l7-Tetramethy~ 5~l3~l7-tetra(t-but
carbonyl)-1,5,13,17-tetraazaheptadecane
- Combine 2,16-bis(methyl)-1,5,13,17-tetra(t-butoxycarbonyl)
1,5,13,17-tetraazaheptadecane (8.5 g, 0.0126 mol) and
sodium hydride (60% in oil)[1.21 g, 0.03 mol] in DMF (75
ml) and stir until hydrogen evolution ceases. To this
mixture add methyl iodide (1.88 g, 0.03 mol) and stir for 2
hours. Remove the solvent in vacuo and partition the
residue between ethyl acetate (400 ml) and water (200 ml).
Dry the organic layer over anhydrous MgS04 and remove the
solvent in vacuo. Purify the residue by flash
chromatography on silica gel eluting with 22% ethyl acetate
in hexane to yield 3.8 g of the title compound as a clear
oil. Rf is 0.22 for TLC on silica gel developed with 20%
ethyl acetate in hexane.
Step G: N,N'-Bis[3-(methylamino)butyl]-1,7-diaminoheptane
tetrahydrochloride
Add lN HCl in methanol (S0 ml) to 1,2,16,17-tetramethyl-
1,5,13,17-tetra(t-butoxycarbonyl)-1,5,13,17-tetraazahepta-
decane (3.8 g, 0.0054 mol) and stir overnight. Remove the
solvent in vacuo and recrystallize the residue two times
from methanol/acetonitrile (40/60, v/v) to yield 0.74 9 of
the title compound as a white solid (mp 238-9 oc). Rf is
0.31 for TLC on silica gel developed with 40% conc. ammonia
in methanol.
EXAMPLE 11
N,N'-Bis[3-(ethylamino)butyl]-1,7-diaminoheptane
tetrahydrochloride
Step A: 1,17-Diethyl-2,16-dimethyl-1,5,13,17-tetra(t-
butoxycarbonyl)-1,5,13,17-tetraazaheptadecane
Combine 2,16-bis(methyl)-1,5,13,17-tetra(t-butoxycarbonyl)-
1,5,13,17-tetraazaheptadecane (8.5 g, 0.0126 mol), made as
described in Example 5, and sodium hydride (60% in
W094/07480 2~ ~ 6 3 1 9 PCT/US93/08517
-50-
oil)[1.21 g, 0.03 mol] in DMF (75 ml) and stir until
hydrogen evolution ceases. To this mixture add ethyl
iodide (4.68 g, 0.03 mol) and stir for 2 hours. Remove the
solvent in vacuo and partition the residue between ethyl
acetate (400 ml) and water (200 ml). Dry the organic layer
over anhydrous MgSO4 and remove the solvent in vacuo. Purify
the residue by flash chromatography on silica gel eluting
with 22% ethyl acetate in hexane to yield 3.9 g of the
title compound as a clear oil. Rf is 0.31 for TLC on
silica gel developed with 20% ethyl acetate in hexane.
Step B: N,N'-Bis[3-(ethylamino)butyl]-1,7-diaminoheptane
tetrahydrochloride
Add lN HCl in methanol (50 ml) to 1,17-diethyl-2,16-
dimethyl-1,5,13,17-tetra(t-butoxycarbonyl)-1,5,13,17-
tetraazaheptadecane (3.9 g, 0.0054 mol) and stir overnight.
Remove the solvent in vacuo and recrystallize the residue two
times from methanol/acetonitrile (40/60, v/v) to yield
O.90g of the title compound as a white solid (mp 249-50
C). Rf is 0.56 for TLC on silica gel developed with 40%
conc. ammonia in methanol.
3 1 9
W094/07480 PCT/US93/08517
EXAMPLE 12
1,4,13,16-Tetra(t-butoxycarbonyl)-1,4-13,16-tetraazahexa-
- decane
Combine 4.75 gm 1,8-dibromooctane (0.017 mol), 20 ml EtOH
and 9.32 ml of ethylene diamine and reflux the mixture
overnight. Cool and treat the mixture with 1.4 gm NaOH.
Evaporate off the solvent and triturate the residue with
CH2Cl2 (200 ml 2x), filter. Treat the filtrate with 66.6 gm
of di-t-butyldicarbonate and stir the mixture overnight.
Remove the solvent and subject the residue to flash
chromatography, eluted with 25% EtOAc/hexane to yield the
desired product. Rf is 0.64 eluted from silica gel with
50~ EtOAc/hexane.
The foregoing may be bis-N-alkylated and the product
deprotected by methods analogous to Steps D and E of
Example 7 to produce desired compounds of the Formula
R'HN(CH2)2N(CH2)gN(CH2)2NHR', e.g., 1,16--
Bis[(phenyl)methyl]-1,4,13,16-tetraazahexadecane-4 HCl.
EXAMPLE 13
1,18-Bis[(2-phenyl~ethyl]-1,5,14,18-tetraazaoctadecane-4HCl
Step A: 1,18-Bis[[(phenyl~methyl]carbonyl]-5,14-bis-
[(phenyl)methyl]-1,5,14,18-tetraazaoctadecane
Chill a solution of 5,14-bis[(phenyl)methyl]-1,5,14,18-
tetraazaoctadecane (2.2 g, 5 mmole) and triethylamine (2 g,
20 mmole) in chloroform (100 ml) in an ice bath. Add a
solution of phenylacetyl chloride (2.3 g, 15 mmole) in
chloroform (10 ml) dropwise. Remove the ice bath and stir
the mixture at ambient temperature for 18 hours. Extract
the reaction mixture with aqueous sodium bicarbonate, dry
the organic layer and evaporate. Chromatograph the residue
on a flash silica gel column (ethyl acetate) to give 3 g of
the desired product as a thick oil.
W094/07480 ~1 4 ~ 3 i~ PCT/US93/08517
-52-
Step B: Add a solution of the product of Step A in THF (150
ml) dropwise to a suspension of LAH (0.5 g) in THF (500
ml). Stir the mixture for 48 hours at ambient temperature.
Decompose the excess reducing agent by dropwise addition of
1 ml of water, 1 ml of 15% NaOH then 3 ml of water. Filter
the mixture and evaporate the filtrate. Take the residue
up in ethanol (100 ml) and add anhydrous HCl gas to convert
the product, 1,18-bis[(phenyl)ethyl]-5,14-bis-
[(phenyl)methyl]1,5,14,18-tetraazaoctadecane, to its
tetrahydrochloride salt. Hydrogenate this product in
ethanol (150 ml) in the presence of Pearlman's catalyst
(0.3 g) at 43 psi on a Parr hydrogenation apparatus for 24
hours. Filter off the catalyst and evaporate the filtrate.
Crystallize the residue from 2-propanol to give the product
1,18-bis-[(phenyl)ethyl]-1,5,14,18-tetraazaoctadecane
tetrahydrochloride salt hemihydrate, mp 228-231C.
EXAMPLE 14
1,18-Bis(phenyl)-1,5,14,18-tetraazaoctadecane
Step A: N-(Phenyl-N,N'-bis(t-butoxycarbonyl)propanediamine
Cool 200 ml of anhydrous Et20 in an ice bath and add lithium
aluminum hydride (8.74 gm -0.23 mol). Add, in a dropwise
fashion over 30 minutes, 3-anilinopropionitrile (14.6 gm)
in 50 ml of Et20, remove the ice bath, and reflux the
resulting mixture overnight. Sequentially add 8.7 ml of
water, 1.5 g of NaOH (in 10 ml of water) and 25 ml of
water. Filter the resulting ppt, rinse with 200 ml of Et20
and remove the solvent, in vacuo, and treat the resulting
N-(phenyl) propanediamine with 43.6 g of di-t-
butyldicarbonate in 600 ml of CH2C12. After stirring
overnight, evaporate off the solvent and subject the
residue to flash chromatography from silica gel eluting
with 17% EtOAc/hexane to produce the desired compound. Rf
is 0.50 (eluted from silica gel with 25% EtOAc/hexane).
214~31~
W094/07480 PCT/US93/08517
-53-
Step B: 1,18-Bis(phenyl)-1,5,14,18-tetra(t-butoxycarbonyl)-
1,5,14,18-tetraazaoctadecane
Stir a mixture containing the product of Step A (13.0 gm),
diiodooctane 3.70 gm) and 4.14 g of potassium t-butoxide in
200 ml of DMF for about 16 hours. Evaporate the solvent at
0.5 mm and 45C, take up the residue in 800 ml of EtOAc.
Wash (2x) with 300 ml of water, dry (MgS04) and remove the
solvent in vacuo. Subject the so-obtained viscous oil to
flash chromatography, eluting with 15% EtOAc from silica
gel to yield 5.7 g of the desired product. Rf of 0.36
(eluted from silica gel with EtOAc/hexane). Remove the N-
boc protecting groups according to the procedure of Step E
of Example 7 to produce the title compound of this example
as its hydrochloride salt. mp 264-2670C.
EXAMPLE 15
1,18-Bis(2,3-butadienyl)-1,5,14,18-tetraazaoctadecane
tetrahydrochloride
Step A: N-(t-Butoxycarbonyl)proparqylamine
In a dropwise fashion, add propargylamine (25 gm) in 25 ml
of CH2Cl2 to a stirring mixture of di-t-butyldicarbonate
(99.18 gm) in 900 ml of CH2Cl2. After 2 hours, remove the
solvent, in vacuo, to obtain 70 gm of the desired N-
protected propargylamine.
Step B: N-(t-Butoxycarbonyl)-2,3-butadienylamine
Reflux a mixture containing N-(t-butoxycarbonyl)-
propargylamine (70 gm), 93.5 ml of 32% formaldehyde, 76.4
ml of diisopropylamine, 19.66 gm of cuprous bromide and 860
ml of p-dioxane for 12 hours. Cool and dilute the
resulting mixture with 3000 ml of Et20, wash with 500 ml of
water, 1000 ml acetic acid, 500 ml of water (2x), 200 ml
sat'd. sodium chloride, dry (MgS04) and evaporate in vacuo.
Flash chromatograph the residue eluting from silica gel
with 10~ Et20/hexane to yield 40.8 g of the desired
W094/07480 PCT/US93/08517
~14~319
-54-
compound. Rf is 0.31 (eluted from silica gel with 10%
EtOAc/hexane).
Step C: N,N-Bis[(phenyl)methyl]-1,8-diaminooctane
Combine 14.4 gm of diaminooctane, 20.3 ml of benzaldehyde
and 0.66 gm of Pt20 in 100 ml of ethanol. Treat the
resulting mixture with hydrogen at 45 lbs./sq.in. until no
further hydrogen is taken up. Filter, evaporate the
solvent (in vacuo), and distill the rendered material to
obtain 25.5 gm of the desired product, bp 185-190C at 0.1
mm.
Step D: 1,18-Bis(hydroxy)-5,14-bis[(phenvl)methyl]-5,14-
diazaoctadecane
Reflux a mixture containing 25.5 g of the product of Step
C, 13.2 ml of 3-chloro-1-hydroxy-propane, 50.4 gm of Na2C03
and 1.19 gm of sodium iodide in 40 ml of n-butanol for 18
hours. Cool the mixture and pour into 700 ml of
ethylacetate, wash with water, dry over MgSO4 and remove
the solvent (in vacuo) to obtain a residue which upon
distillation yields 30.0 gm of the desired product, bp 250-
252C at 0.1 mm.
Step E: 1,18-Bis(hydroxy)-5,14-diazaoctadecane
Hydrogenate a mixture containing 3.0 gm of the product of
Step D, 30 ml of AcOH and 0.6 gm of palladium oxide at 45
lbs./sq.in. until no further hydrogen is taken up. Filter
and remove the solvent (in vacuo) to yield 1.77 gm of the
desired product, Rf is 0.37 (eluted from silica gel with
10% conc. NH3/CH30H)-.
W094/07480 ~ 3 ~ 9 PCT/US93/08517
Step F: 1,18-Bis(hydroxy)-5,14-bis-(t-butoxycarbonyl)-5,14-
diazaoctadecane
Stir a mixture containing 1.77 gm of the product of Step E,
2.97 gm (0.0136 mol) of di-t-butyldicarbonate, 3 ml of
triethylamine and 50 ml of CH2Cl2 overnight. Dilute the
mixture with 200 ml of CH2C12, wash with 200 ml of 0.5N HCl,
and then 100 ml of sat'd NaCl, dry (over MgS04) and remove
the solvent (in vacuo). Flash chromatograph the residue,
eluting from silica gel with 75% EtOAc to obtain the
desired product, Rf 0.29, (eluted from silica gel with 75%
EtOAc/hexane).
Step G: 1,18-Bis(methanesulfonyl)-5,14-bis(t-
butoxycarbonyl)- 5,14-diazaoctadecane
Cool to 0C a mixture containing 3.0 gm of the product of
Step F, 3.3 ml of triethylamine and 70 ml of CH2Cl2. In a
dropwise fashion add 1.22 ml of mesylchloride in 10 ml of
CH2Cl2 and stir the resulting mixture at 0C for 1~ hours.
Pour the mixture into 100 ml of CH2Cl2, wash with 200 ml of
lN AcOH, 100 ml of water, 100 ml of sat'd sodium
bicarbonate, dry over MgS04 and remove the solvent in vacuo.
Flash chromatograph the residue, eluting from silica gel
with 60% EtOAc/hexane to obtain 3.5 gm of the desired
product. Rf is 0.39.
Step ~: 1,18-Bis(2,3-butadienyl)-1,5,14,18-tetra-(t-
butoxycarbonyl)-1,5,14,18-tetraazaoctadecane
Combine a mixture containing 3.5 gm of the product of Step
G, 1.74 gm of sodium iodide, 0.51 gm of hexane washed
sodium hydride (60% in oil) in 12 ml of DMF with 2.16 gm of
N-(tbutoxycarbonyl)-2,3-butandienylamine (i.e., the product
of Step B) and allow the resulting mixture to stand for 2
; hours. Remove the solvent (in vacuo), add 350 ml of ethyl
acetate to the residue, wash with 50 ml of water (4x), 100
ml sat'd sodium chloride and dry over MgS04. Remove the
solvents (in vacuo) and flash chromatograph the residue
3l-D
W094/07480 PCT/US93/08517
-56-
from silica gel eluting with 30% EtOAc/hexane to yield 0.5
gm of the desired product, as a viscous oil. Rf is 0.39
(eluting from silica gel with 25% EtOAc/hexane).
Step I: 1,18-Bis(2,3-butadienyl)-1,5,14,18-tetraazaocta-
decane-4HCl
Dissolve 0.5 gm of the product of Step H in 2 ml of EtOH
and while stirring treat the mixture with 10 ml of 2N HCl
in Et20. Stir the resulting mixture overnight, filter and
dry the solids (in vacuo) to obtain 0.22 gm of the desired
product, mp 283-284C dec.
EXAMPLE 16
N,N'-Bis[3-(ethylamino)propyl]-1,7-heptanediamine
tetrahydrochloride (MDL 28314QA)
Step A: 1,7-Bis(hexahydropyrimidin-l-yl)heptane (MDL
102533)
A solution of N,N'-bis(3-aminopropyl)-1,7-heptanediamine
4HCI (MDL 26752QA, 10.0 g, 25.6 mmol)4 in 1 N NaOH (103 mL,
4 eq) was cooled to 5C (ice-water bath) and aqueous HCHO
(37 wt%, 3.8 mL, 50.8 mmol) was added in one portion. The
solution was stirred for 1 h at 0-5C and then 1 h at room
temperature. The reaction solution was extracted with CH2CI2
(3 x 100 mL). The extracts were combined, dried (K2C03)
and concentrated to give crude MDL 102533 (6.9 g, 100%) as
a light yellow solid. This material was used without
further purification. Purification of a sample by flash
chromatography using eluant A gave analytically pure MDL
102533 as a white solid: mp 52-55C; TLC (eluant B) Rf
0.46;1H NMR (CDCI3) ~ 1.3 (m, 6), 1.46 (br pentet, 4), 1.61
(pentet, 4, J=5.5 Hz), 1.7 (br s, 2, NH), 2.21 (dd, 4,
J=7.6, 8.6 Hz), 2.56 (br t, 4, J=4.8 Hz), 2.81 (t, 4, J=5.5
Hz), 3.37 (s, 4); 13C NMR (CDCI3) ~ 26.79, 27.05, 27.58,
29.39, 45.07, 53.12, 55.66, 69.84; mass spectrum (Cl/CH4),
W094/07480 2 1 ~ 6 3 1 9 PCT/US93/08517
m/z(rel intensity) 270(21), 269(100j, 97(18), 83(19),
79(14).
Anal. Calcd for C15H32N4 (268.45): C, 67.11; H, 12.02; N,
20.87. Found: C, 66.65; H, 12.85; N, 20.69.
i
Step B: 1,7-Bis(3-acetylhexahydropyrimidin-1-yl)heptane
(MDL 44868)
A stirred solution of crude bis(hexahydropyrimidine) MDL
102533 (5.0 g, 18.7 mmol) in EtOAc (40 mL), Et3N (7.6 g, 75
mmol) and Ac2O (7.6 g, 75 mmol) was heated at reflux for 8 h
under N2. The reaction solution was cooled and concentrated
(50/10 mm). The concentrate was partitioned in CH2CI2 (75
mL) and 1 N NaOH (40 mL), the layers were separated and the
aqueous layer was extracted with CH2CI2 (2 x 75 mL). The
CH2CI2 layers were combined, dried (K2CO3) and concentrated
to give crude MDL 44868 (7.2 g, 110%) as an orange oil.
This material was used without further purification.
Purification of a sample by flash chromatography using
eluant C gave analytically pure MDL 44868 as a colorless
oil: TLC (eluant C) Rf, 0.35; lH NMR5 (CDCI3) ~ 1.3 (m, 6),
1.5 (m, 4), 1.63/1.70 (overlapping, complex pentets, 4,
J=5.6/5.6 Hz), 2.09/2.11 (s, 6), 2.4 (m, 4), 2.72/2.73 (two
overlapping t, 4, J=6.3/6.3 Hz), 3.48/3.60 (t, 4, J=5.6/5.6
Hz), 4.04/4.23 (s, 4); 13C NMR (CDCI3) ~ 20.99, 21.12,
23.00, 23.06, 23.40, 26.99, 27.21, 29.16, 41.45, 45.96,
51.71, 51.82, 52.21, 53.37, 53.46, 62.46, 62.54, 67.78;
mass spectrum (Cl/CH4), m/z(rel intensity) 381(21), 354(28),
353(100), 351(10).
Anal. Calcd for Cl9H36N402 (352.52): C, 64.74; H, 10.29; N,
15.89. Found: C, 63.13; H, 10.59; N, 15.52.
Step C: 1,7-Bis(3-ethylhexahydropyrlmldln-1-yl)heptane (MDL
45692)
W094/07480 ~46319 PCT/US93/08517
-58-
A solution of crude bis(acetylhexahydropyrimidine) MDL
44868 (2.0 g, 5.7 mmol) in anhydrous THF (40 mL) was added
to a stirred suspension of LAH (0.85 g, 22.7 mmol) in
anhydrous THF (60 mL) under N2. The reaction mixture was
heated at reflux for 16 h and then allowed to cool to room
temperature. The reaction mixture was stirred vigorously
and quenched by the cautious addition of saturated aqueous
Na2SO4 (5 mL) at room temperature. It was necessary to stir
the mixture for 16 h to ensure total quench. The mixture
was filtered (Celite) and the filter cake was washed with
THF (3 x 10 mL). The filtrate and washings were combined
and concentrated, the residue was dissolved in CH3CN (50 mL)
and was concentrated again to give crude MDL 45692(1.69,87%)
as a yellow oil. This material was used without further
purification. Purification of a sample by flash
chromatography using eluant D gave analytically pure MDL
45692 as a light yellow oil: TLC (eluant D) Rf 0.56; 1 H
NMR (CDCI3) ~ 1.07 (t, 6, J=7.2 Hz), 1.3 (m 6), 1.46 (br
pentet, 4, J=6.3 Hz), l.Ç7 (pentet, 4, J=5.6 Hz), 2.30
(overlapping dd, 4, J=7.6, 7.6 Hz), 2.39 (q, 4, J=7.2 Hz),
2.4-2.5 (m, 8), 3.08 (br s, 4); 13C NMR (CDC13) ~ 12.23,
23.67, 27.10, 27.42, 29.39, 49.00, 52.03, 52.44, 55.38,
76.13; mass spectrum (Cl/CH4), m/z(rel intensity) 353(16),
326(22), 325(100), 324(57), 323(94).
Anal. Calcd for ClgH40N4 (324.56): C, 70.31; H, 12.42; N,
17.26. Found: C, 69.50; H, 12.75; N, 16.70.
W094/07480 ~lg ~ 31~ PCT/US93/08517
Step D: N,N'-Bis[3-(ethylamino)propyl]-1,7-heptanediamine
tetrahydrochloride (MDL 28314QA)
To a stirred solution of crude
- bis(ethylhexahydropyrimidine) MDL 45692 (0.50 g, 1.5 mmol)
in MeOH (20 mL), conc. HCI (5 mL) was added in one portion.
The reaction solution was heated at reflux for 3 h, using a
nitrogen sweep to remove HCHO/MeOH distillate (MDL 28314QA
precipitated from the reaction solution shortly after
heatin~ began). The loss in reaction volume was
periodically adjusted to the original level by addition of
MeOH. The reaction mixture was allowed to cool to room
temperature and then was filtered. The solid was washed
with MeOH (2 x 5 mL) and air-dried to give MDL 28314QA
(0.40 g, 58%) as an off-white solid. Recrystallization
from H20 (0.8 mL) and i-PrOH (2.9 mL) gave pure MDL 28314QA
(0.32 g, 80~ recovery) as a white solid: mp 313C (dec);
TLC (eluant E) Rf 0.39; lH NMR (D20) ~ 1.30 (t, 6, J=7.3
Hz), 1.4 (m, 6), 1.70 (br pentet, 4, J=7.3 Hz), 2.1 (m, 4),
3.1 (m, 4), 3.13 (q, 4, J=7.3 Hz), 3.15 (m, 8); 13C NMR
(D2O) ~ 13.30, 25.50, 28.21, 28.25, 30.48, 45.87, 46.71,
47.17, 50.61; mass spectrum (Cl/CH4), m/z(rel intensity)
329(17), 302(21), 301(100), 300(22), 299(30), 197(8),
159(6), 75(10).
Anal. Calcd for Cl7H40N4 4HCI (446.38): C, 45.74; H, 9.94;
N, 12.55; Cl, 31.77. Found: C, 45.49; H, 10.48; N, 12.33;
Cl, 31.20.
W094/07480 PCT/US93/08517
~ 4~3~ ~
-60-
~XAMPLE 17
1,6,14,19-Tetraazanonadecane Tetrahydrobromide
Step A: N-t-Butyloxycarbonyl-N-4-chlorobutyl-p-
toluenesulfonamide
A solution of DEAD (17.4 g, 0.1 mole) in THF (20 ml) was
added dropwise to a solution of 25 (27.1 g, 01. mole),
triphenylphosphine (26.2 g, 0.1 mole) and 4-chloro-1-
butanol (10.8 g, 0.1 mole) distilled to remove HCl) in THF(600 ml). After 4 hours at ambient temperature the mixture
was evaporated and the residue was chromatographed
(toluene) to give the product (29.4 g, 81%) as an oil. IR
(film) 2980, 1728, 1456, 1394, 1356, 1292, 1256, 1186,
1156, 1088, 10000, 814, 772, 722, 674, 598, 576 and 546 cm-
1. NMR (CDC13) 1.35 (s, 9H), 1.8-2.0 (m, 4H), 2.45 (s,
3H), 3.6 (t, J=7.5 Hz, 2H), 3.86 (t, J=7.5 Hz, 2E), 7.33
(J=7.5 Hz) and 7.78 (d, J=7.5 Hz, 2H). MS (CI/CH4) 362 (M
+ H). Anal Calcd for Cl6H24ClN04S 1/8 toluene: C, 54.28;
H, 6.75; N, 3.75. Found: C, 54.39; H. 694; N, 3.70.
Step B: 1,7-bis-p-Toluenesulfonamidoheptane
A mixture of dichloromethane (600 ml), aqueous sodium
bicarbonate (600 ml) and 1,7-diaminoheptane (25 g, 0.19
mole) was stirred while p-toluenesulfonyl chloride (109 g,
0.57 mole) was added in portions over 1 hour. The layers
were separated, the organic layer was extracted with lN HC1
and evaporated. The residue was triurated with toluene to
give the product (58g, 70%) as a white solid mp. IR (KBr)
3256, 2942, 2864, 1430, 1328, 1306, 1160, 1094, 1078, 808,
706, 668, 578, 552 and 522 cm-l. NMR (DMSO-d6) 1.1-1.2 (m,
6H), 1.25-1.3 (m, 4H), 2.4 (s, 6H), 2.6-2.7 (m, 4H), 7.37
(d, J=7.5 Hz, 4H), 7.45 (t, J=7.5 Hz, 2H) and 7.75 (d,
J=7.5 Hz, 4H). MS (CI/CH4) 439 (M + H). Anal. Calcd for
C21H30N204S2: C, 57.50; H, 6.89; N, 6.39. Found: C, 57.87;
H, 7.13; N, 6.20.
W094/07480 ~ 6 3 ~ ~ PCT/US93/08517
-61-
Step C: 1,6,14,19-Tetraaza-1,19-bis-t-butyloxycarbonyl-
1,6,14,19-tetra-p-tol-uenesulfonylnonadecane
Sodium hydride (0.8 g, 16.8 mmol, 50% mineral oil
dispersion) was added in portions to a solution of 27 (3 g,
8.4 mmol) in DMF (300 mL) and the mixture was stirred for 1
h at ambient temperature. Sodium iodide (0.1) and a
solution of 26 (5.2 g, 16.8 mmol) in DMF (50 mL) were added
and the mixture was heated at 55 C for 18 h. The mixture
was cooled, methanol (10 mL) was added to decompose any
unreacted sodium hydride, and the mixture was evaporated to
dryness. Dichloromethane (300 mL) and water (300 mL) were
added, the organic layer was separated, dried and
evaporated. Chromatography (toluene/ethyl acetate 6/1)
gave the product (4.4 g, 51~) as a thick oil. IR ( film)
2980, 2934, 1726, 1458, 1394, 1348, 1306, 1286, 1258, 1186,
1156, 1090, 1006, 814, 756, 722, 674, 654, 598, 576 and 548
cm-l. NMR (CDCL3) 1.25 (s, 6H), 1.32 (s, 18H), 1.42-1.153
(m, 4H), 1.55-1.65 (m, 4H), 1.7-1.8 (m, 4H), 2.55 and 2.56
(s,together 12H), 3.02-3.18 (m, 8H), 4.82 (t, J=7.5 Hz,
4H), 7.25-7.35 (m, 8H), 7.68 (d, J=7.5 Hz, 4H) and 7.77 (d,
J=7.5 Hz, 4H). MS (FAB) 1089 (M+). Anal: Calcd for
C53H76N40l2S4, C, 58.42; H, 7.03; N, 5.14. Found: C,60.50:
H,7.25; N, 5.08.
Step D: 1,6,14,19-Tetraaza-1,6,14,19-tetra-p-
toluenesulfonylnonadecane
A solution of 28 (20 g, 18 mmol) in a mixture of
dichloremethane (500 mL) and TFA (20 mL) was stirred
overnight at ambient temperature. The mixture was
evaporated, toluene (400 mL) was added and the mixture was
evaporated to dryness. The residue was chromatographed
(toluene/ethyl acetate 3/1) to give the product (14.3 g,
89~) as an oil. IR (film) 3286, 2936, 2864, 1598, 1454,
1426, 1330, 1306, 1290, 1268, 1158, 1092, 1040, 816, 736,
704, 656, 570 and 550 cm-l. NMR (CDCL3) 1.25 (s, 6H),
1.45-1.6 (m, 12H), 2.42 (s, 12H), 2.86-2.95 (m, 4H), 3.0-
3.1 (m, 8H), 4.88 (t, J=7.5 Hz, 2H), 7.15-7.3 (m, 8H), 7.43
W094/07480 2 1 ~ 6 3 ~ ~ PCT/US93/08517 ~
-62-
(D, J=7.5 Hz, 4H) and 7.72 (d, J=7.5 Hz, 4H). MS (CI/CH4)
889 (M + H). Anal: Calcd for C43H60N40gS4 3/4 toluene: C,
60.38; H, 7.15; N, 5.70. Found: C, 60.48; H, 7.17; N,
5.57.
Step E: 1,6,14,19-Tetraazanonadecane Tetrahydrobromide
A solution of 29 (12.2 g, 13.7 mmol) in 48% aq HBr (500 mL)
was heated in a 100 C oil bath for 24 h, cooled and
evaporated to dryness. The residue was recrystallized
(ethanol/H20) to give the product (5.2 g, 63%) as a white
solid, mp 303 C. IR (KBr) 2952, 2874 and 2810 cm-l. NMR
(D20) 1.4 (s, 6H), 1.6-1.85 (m, 12H) and 3.0-3.1 (m, 12H).
MS (CI/CH4) 273 (M + H). Anal: Calcd for C15H36N4 4HBr:C,
30.22; H, 6.76; N, 9.40. Found: C, 30.60; H, 6.90; N,
9.09.
Test compound numbers relate to the following
compounds:
MDLl9,190 = NH=C(NH2)-N=C(CH3)-CH=N-NH-C(NH2)=NH
2,2'-(1-methyl-1,2-ethanediylidene)-
bis[hydrazinecarboximidamide], (methyl-glyoxal
bis(guanylhydrazone))
MDL27,393 = CH3CH2-NH-(CH2)3-NH-(CH2)8-NH-(CH2)3-NH-
CH2CH3
N,N'-Bis(3-(ethylamino)propyl)-1,8-octanediamine
MDL27,616 = ~CH2-NH-(CH2)3-NH-(CH2)3-NH-CH2~
N-(phenylmetnyl)-N'[3-[(phenylmethylamino)propyl]-1,3-
propanediamine
21~31~
W094/07480 PCT/US93/08517
-63-
MDL27,695 =~CH2-NH-(CH2)3-NH-(CH2)7-NH-(CH2)3-NH-
CH2~
1,18-Bis[(phenyl)methyl]1,5,14,18-
tetraazaoctadecane
MDL28,314 =CH3CH2-NH-(CH2)3-NH-(CH2)7-NH-(CH2)3-NH-
CH2CH3
N,N'-Bis(3-(ethylamino)propyl)-1,7-heptanediamine
MDL28,454 =CH3CH2CH2-NH-(CH2)3-NH-(CH2)7-NH-(CH2)3-
NH-CH2CH2CH
N,N'-bis[3-(propylamino)propyl]-1,7-diaminoheptane
gaciclovir = 9-[(2-ydroxy-1-hydroxymethylethoxy)-
methyl]guanine
BIOLOGICAL EXAMPLES
EXAMPLE 18: Al.~ lvlKAL ACl lYl 1 Y
The antiviral effects of, polyamine analogues was
determined by a standard plaque reduction assay as
described by Tyms et al. (J. Antimicrobial Chemotherapy 8,
6S-72, 1981). Monolayers of human embryo fibroblasts (MR~-
5 strain) were formed in the presence or absence of varying
concentrations of compound and infected with CMV strain
AD169 or Towne (lOO pfu/105 cells) in the presence or
absence of compound and incubated in the presence or
absence of compound for ten days at 37C. For incubation
post infection, cell monolayers were overlaid in
maintenance medium (Eagles MEM with 2% bovine fetal calf
serum) which contained 0.5% agarose, and fixed and stained
with methylene blue at the appropriate time (7-10 days post
infection). From the percent reduction in plaque formation
compared to untreated controls, a dose res~onse plot was
made and the 50% inhibitory concentration (IC50)
determined. A range of IC50 values for various polyamine
analogues are shown in Table A. The preferred compound,
W094/07480 ~ 1 4 63 1~ PCT/US93/08517
-64-
MDL27,393, consistently gave IC50 values about 10
pmoles/litre when cells were also pre-exposed to compound
prior to infection. Using these protocols, the IC50 values
for MDL 27,393 were about 100,000 lower than those r
5 recorded for the current therapies for CMV infections,
ganciclovir and foscovir (Table B).
With similar protocols using MRC-5 cells (Table C)
infected with HSV type 1 (17i strain) but using a three day
post-infection incubation period, IC50 values for MDL27,393
(TABLE Cl) and MDL28,314 (TABLE C2) were between 1-10
~moles/litre with the pre-post treatment protocol. No
antiviral activity was observed in MRC-5 cells even at 100
~M with treatment post-infection only. In vero cells
(simian origin) treatment with MDL 27,393 (<100 ~M), pre-
post or post-only, failed to inhibit the growth of HSV-l
(strain 17i). This was evidence that the activity of the
analogues was related to cellular or virus specific events
involved in CMV but not HSV replication.
Natural polyamines putrescine, spermidine or spermine
when presented to infected cells, as described in the
above protocol, failed to inhibit CMV growth at
concentrations < lO~M moles/litre.
Similar experiments were carried out with the same
pretreatment protocol as above but at high multiplicity
infection (approximately 105/pfu/105 cells): monolayers were
maintained in the absence of agarose in the maintenance
medium. Production of progeny virus and the synthesis of
"late" viral proteins were measured as previously described
Tyms et al (J. Gen. Virol, 68, 1563-1573, 1987). Virus
production was inhibited by the preferred compound,
MDL27,393, by 90% at 1 nmoles/litre and 95~ at
lOnmoles/litre with a corresponding reduction in the
synthesis of the three major "late" viral proteins the
major capsid protein ( 153KD) tegument protein ( 69KD) and a
W094/07480 ~1 4 6 3 1 9 PCT/US93/08517
-65-
DNA-binding protein ( 51KD). This was confirmed in
experiments in which numbers of CMV infected cells
expressing the "late" antigens under these conditions,
identified by a specific monoclonal antibody to a late
5 protein, were shown to be reduced. These results were
L interpreted to mean that the production of infectious
progeny virus in infected cells was curtailed which limited
the spread of infection in cell monolayers when infected at
low multiplicity of infection. This was considered due to
10 an interference with the synthesis of CMV "late" viral
proteins by the preferred compound.
EXAMPLE 19: ~Yl~l~XICITY
MRC-5 cells were seeded at low plating density and
15 grown in the presence or absence of the preferred compound,
MDL 27,393, or MDL 28,314 and cell numbers were determined
at five days postseeding. The results showed that growth of
these cells was not inhibited at concentrations of 0.5
~moles/litre or less although the time to confluency of the
20 monolayers was longer compared to untreated controls at
concentrations between 1 and 5 ~moles/litre (data not
shown).
On the basis of these results the therapeutic window,
for the preferred compound MDL 27,393, established by the
amount of compound required to inhibit virus growth by 50%
compared to the concentration required to inhibit cell
growth by 50%, was greater than 10,000.
.
WO 94/07480 PCr/US93/08517
3~9
66--
TABLE A
lVl~OF NAT~RAL POLYN~lN~;~; A~:AT~l~T CMV
lN~J~ ION
S REF: PA219
Mean Mean Mean
C~JMC Spermine %Spermidine % DHPG VC
100 45,52,0 47% 99,91 93% 0,0 0%
10 106,107,0 100% 99,92 96% 10,17 12%
- As VC - 100% - As VC - 100% 68,67 57%
0.1 -ASVC- 100% -AsVC- 100% 106,95 91%
0.01 -AsVC- 100% -AsVC- 100% -AsVC- 100%
VC 103,99, 100% 103,99, 100% 106,129, 100%
105 105 99
P aque reduction assa~ - I owne stra n grown in MR~-5 c~lls
TABLE: B
ANTIVIRAL EFFECT OF POLYAMINE ANALOGUES AGAINST CMV INFECTION
ICso Values
Compound Pre/Post Post only
MDL27,393 <O.lnM <100nM
MDL 28,314 10nM 100nM
MDL 27,484 < 1 nM 100nM
MDL29,587 <lOOnM 11~M
MDL27,666 1nM <1~JM
MDL27,667 10nM ~1,uM
MDL 27,394 < 1 On M < 1 ~M
Ganciclovir 1 ~M < 10~JM
Plaque reduction assay - Towne strain grown in MRC-5 cells .
WO 94/07480 ~ 1 ~ 6 ~ 1 ~ PCI/US93/08517
--67--
TABLE C
EFFECT OF POLYAMINE ANALOGUES AGAINST HSV TYPE ll INFECTION:
REF: PA241
MDL 27, 393: TABLE C1
Conc Pre/Post Mean % Post Mean %
100 0,0,0 0% 68,71,71 77%
16,17, 20 19% 91,90,92 100%
30,27,30 32% As VC 100%
0.1 49,50, 61 59% AsVC 100%
1X10-2 75,71, 60 76% As VC 100%
1 x10-3 81, 63,70 78% As VC 100%
lS 1x10-4 65,80,80 81% AsVC 100%
VC 95,87,91 100% 90, 89,92 100%
20 MDL 28, 314: TABLE CZ
C~oMc Pre/Post Mean % Post Mean %
100 0,0,0 0% 90,76,82 81%
24, 20,15* 22% AsVC 100%
58,65, 51 64% As VC 100%
0.1 63, 65,59 69% AsVC 100%
1x10-2 78,71,60 77% AsVC 100%
1 x10-3 - As VC - 100% As VC 100%
1x10-4 - AsVC- 100% AsVC 100%
VC 87,97, 89 100% 103,99,105 100%