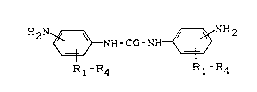Note: Descriptions are shown in the official language in which they were submitted.
21 ~6670
Le A 29 324 - PCT
A method of preparing stabilised aromatic diamines and use
thereof for producing heat-resistant polyurethane-urea
elastomers
The invention relates to a simple industrial method of
producing polyurethane-urea elastomers, in which aromatic
diamines stabilised by reaction with organic and/or
inorganic acid chlorides and in the solid phase are reacted
with isocyanate prepolymers.
It is known to prepare polyurethane-urea elastomers from
polyisocyanates, higher-molecular polyhydroxyl compounds
and aromatic diamines. In order to process reactive
systems of the aforementioned starting components within a
reasonable time, the reactive aromatic isocyanates normally
used in industry are advantageously reacted with diamines,
which are slow to react. In practice in this connection,
the best results have been obtained with aromatic diamines
in which the basicity and consequently the reactivity with
isocyanates have been reduced by introducing halogen or
carboxy substituents. One example is the substance most
frequently used hitherto, i.e. 3,3'-dichloro-4,4'-diamino
diphenyl methane (MOCA).
US-A 3 891 606 describes cross-linking of NCO prepolymers
of polyhydroxyl compounds and an excess of polyisocyanates
with aromatic diamines whose reactivity with isocyanate
groups has been reduced by complexing with certain alkali-
metal salts. The disadvantage of this method is that it is
limited to two particular aromatic diamines. In addition,
2146670
the complex between the aromatic diamine and the alkali-
metal salt must be produced in a separate step.
Another possible method of controlling the rate~-of reaction
between polyisocyanates and aromatic diamines is to carry
out the reaction in an organic solvent. Methods of this
kind are described e.g. in US-A 3 926 922 and in Japanese
Laid-Open Specification 9195/70. The disadvantage of using
organic solvents is obvious. On the one hand the risk of
fire and explosion is increased and on the other hand the
solvent has to be recovered in a complicated industrial
process, for economic and ecological reasons.
At present little is known about production of polyurethane
ureas by reaction of polyisocyanates with aromatic diamines
in the heterogeneous phase. In the prior art, higher-
melting aromatic diamines, which are generally of
particular importance in industry, are used either in
dissolved form, with the aforementioned disadvantages, or
are reacted with polyisocyanates in the melt. Processing
of aromatic diamines in the melt is described e.g. in the
previously-mentioned US-A 3 926 922 or in DE-A 1 122 699.
DE-A 1 122 699 relates to a method of producing poly-
urethane elastomers by cross-linking of liquid isocyanate
prepolymers by reaction with mixtures of primary diamines
and compounds containing a number of hydroxyl groups and
shaping, in which a dispersion of a pulverulent crystalline
diamine in a liquid polyester containing a number of
hydroxyl groups or in a polyether or in castor oil is
introduced into the prepolymer at a temperature below the
melting-point of the diamine, and the material is hardened
in known manner at temperatures above the melting-point of
the diamine in the mixture. In this method also,
therefore, the actual "amine cross-linking" occurs in the
liquid homogeneous phase. The main disadvantage of the
method in DE-AS 1 122 699 is the high temperatures
, 21q6~7D
required, particularly when processing high-melting
diamines such as 1,5-naphthylene diamine (m.p. = 189C) or
4,4'-diamino diphenyl ether (m.p. = 186C), since
experience shows that decomposition reactions a}ready occur
to a considerable extent in polyurethane and uncontrollably
affect the mechanical properties of the products.
US-A 3 105 062 describes a method of producing polyurethane
ureas in which higher-molecular pre-adducts containing
isocyanate groups are reacted in the heterogeneous phase
with diamines, preferably aromatic. The reaction mixtures
are solidified at a temperature at which the "two-phase"
system changes into a "single-phase" system. The
temperature is usually about 100 to 170C.
However the aromatic diamines cited in US-A 3 105 062 are
still soluble, even though slightly, in the reaction medium
(NC0 pre-adduct), so that when the two components are
mixed, uncontrollable preliminary reactions occur even at
room temperature. The result is that the reacting batches
thicken in a very short time and in some cases the
formulations become pasty. They are difficult to process
by normal casting and therefore have to be introduced into
the required mould under pressure before the actual
solidification by heating. According to US-A 3 105 062 the
stability in storage (pot life) of the thickened reaction
mixtures is sufficient for subsequent processing (shaping
under pressure and coating) and amounts to several hours.
The examples, however, show that the reaction mixtures are
preferably those having a maximum pot life of about 1 hour.
They therefore cannot be regarded as long-time systems.
In US-A 3 105 062 it is expressly pointed out that the use
of the cited diamines, in solid form only, in a single-
stage process results in unsatisfactory polyurethanemoulded members. In that case the undesired preliminary
2146670
reaction between the diamine and the diisocyanate is
intensified, and the difficultly-soluble polyurea
precipitates from the reaction mixture and ceases to react.
DE-A 26 35 400 describes another method of producing
polyurethane urea elastomers, in which aromatic diamines
for lengthening the chain are reacted in a single or multi-
stage process. This process is characterised in that the
aromatic diamines are present in solid form in the reaction
mixtures and melt at above 130C. The batches are
solidified by heating at a temperature of 80 to 120C, i.e.
below the melting-point of the aromatic diamine. As a
result of a choice of suitable diamines for lengthening the
chain, no premature reaction and no thickening of the
batches occurs with the NC0-containing pre-adduct (NC0
prepolymer). These systems therefore can also be
efficiently processed by casting. Since the pot life of
these reactive systems is very long, the process is
suitable for many aromatic diamines which were very
difficult to work by the previously-known method.
The examples in DE-A 26 35 400 show that the pot life of
the liquid reaction mixtures, depending on the reactivity
or solubility of the aromatic diamine in a temperature
range, varies from a few minutes to several hours. Under
normal processing conditions, e.g. manual casting, these
reaction batches, particularly those with long pot lives,
can usually be processed without great difficulty.
Problems occur if, as a result of failures of machinery or
other involuntary shut-downs, there is a prolonged pause
between production and reaction batches and the
solidification phase. The requirement for a long
processing time at low temperature and a short processing
time at elevated temperature is therefore becoming
progressively more urgent in practice.
2146670
The finished PUR plastics are stated to have good
mechanical properties in general and often also have
thermal stability suitable for their intended use. In the
prior art, the thermal stability of PUR elastome~rs is
closely dependent on the nature of the chain-lengthening
substances. If for example glycolic chain-lengtheners are
used in the production of elastomers, the resulting PUR
members have lower thermal stability than when compounds
containing amino groups are used. Of course, there are
also considerable differences in thermal stability within
each kind of chain-lengtheners (compounds containing H0 or
NH2 groups).
DE-A 26 35 400 states that numerous diamines varying in
constitution are suitable chain-lengtheners for producing
polyurethane-urea elastomers. The only representative of a
diamino diphenyl urea mentioned is 2,2'-diamino diphenyl
urea.
No experimental example is given.
A check of the application showed that an NC0 pre-adduct
reacted with the aforementioned diamino diphenyl urea
yields a resilient PUR moulded member having perfectly
acceptable mechanical properties. However, the thermal
stability of these elastomers is unexpectedly low. If the
moulded member is subsequently heat-treated at only 120 -
130C, a considerable decrease in mechanical strength
occurs after a short time. At 140 - 150C there is only a
viscous melt, irrespective of whether the test-piece is hot
or cold (example).
DE-A 3 732 728 describes a method of preparing
polyurethane-urea elastomers in which finely-divided
diamino diphenyl ureas having the general formula
21~6670
H2N ~ NH-Co-N~ ~ NH2
Rl-R4 Rl-R4
in which the NH2 groups are in the m and/or p position
relative to the urea group and R1, R2, R3, R4 (which may be
the same or different) denote H or Cl - C6-alkyl radicals,
in combination with NCO pre-adducts yield reaction mixtures
which, at the respective processing temperature, have
processing times of at least several hours, preferably at
least 8 hours (long-time system). The heterogeneous
reaction batches can be solidified at a relatively low
temperature within an economically advantageous time. If
on the other hand the chain-lengtheners according to the
invention are added in dissolved form to the NCO pre-
adducts, they behave like conventional aromatic diamines.After a few seconds the reaction becomes batch cross-linked
and the resulting swollen product can no longer be worked.
It is stated that the pre-adducts comprising NCO groups are
prepared by using polyhydroxyl compounds having a molecular
weight of 400 to 10,000. These include polyester and
polycarbonate polyols particularly suitable for cast
elastomers having excellent mechanical properties, as
described in DE-A 3 732 728 on page 5, lines 51 ff. The
reaction components are reacted by the known single-stage
method or the prepolymer method or the semi-prepolymer
method, often using mechanised equipment, e.g. as described
in US-A 2 764 S65, where performance of the method
according to DE-A 3 732 728 is described on page 8, lines
17 ff.
21~6670
It is specially pointed out that the processing temperature
on the one hand is closely dependent on the nature of the
NC0 pre-adduct and on the other hand should not be too
high, when premature reactions cannot be prevented. This
is particularly critical when highly reactive NC0 pre-
products, which are solid or highly viscous at room
temperature, have to be processed by casting. In that case
the processing temperature has to be raised until the
reaction batch can be properly degassed and cast. The
temperatures are usually up to 130C. In the case of the
polyester polyol-based prepolymers used in examples 1 and 7
in DE-A 3 732 728, the temperature is usually at least
80C. Under these conditions, it is of course more
difficult to meet the requirement for long-time systems
with a processing time of at least several hours,
particularly in view of the already high reactivity of NCO
prepolymers based on polyester or polycarbonate polyols,
compared e.g. with polyether polyols. The stability in
storage of diamino diphenyl ureas in polyester polyol-based
NC0 prepolymers at a temperature of 40 - 50C is therefore
not in reality as described in examples 1 and 7 of DE-A 3
732 728. In the case of 4,4'-diamino diphenyl urea, it
appears possible to increase the storage temperature to the
80C required in practice. In the case of meta-diamino
phenyl urea in example 7, on the other hand, the stability
in storage is relatively low (2 hours) even at 40 to 50C,
showing that processing at 80C is impossible. This is
confirmed by a comparative test (example 1 in this
application). At the normal processing temperature of
about 80C for this system, the reaction batch is found to
thicken so strongly within a very short time (less than 1
minute) that it cannot be degassed and cast to form a high-
quality industrial moulded part.
The aim of the invention therefore is to discover a method
of preparing polyurethane ureas in which the processing
2146670
" " ~
time of the reaction batches, consisting of a combination
of highly-reactive NC0 prepolymers which are highly viscous
or solid at room temperature, based on polyester and/or
polycarbonate polyols and therefore needing to be processed
at elevated temperature, and solid diamino diphenyl ureas
in finely divided form and having the general formula
H2N~t~H-CO-NH~NH2
Rl-R4 Rl-R4
in which the NH2 groups are in the meta position relative
to the urea group and Rl, R2, R3, R4 (which may be the the
same or different) denote H or Cl - C6- alkyl radicals, at
the respective reaction temperature is within a range
enabling the reaction mixture to be efficiently degassed
and cast. It is also desirable that the liquid reaction
batches should solidify at the lowest possible reaction
temperatures within an economically advantageous time.
It has now surprisingly been found that finely-divided
diamino diphenyl ureas having the general formula
H2N ~ NH-C0-NH ~ NH2
Rl-R4 Rl-R4
in which the NH2 groups are in the meta position relative
to the urea group and Rl, R2, R3, R4 (which may be the same
or different) denote H or Cl-C6-alkyl radicals, which can be
stabilised by reaction with organic and/or inorganic acid
chlorides in a proportion of more than 200 ppm relative to
the amount of NC0 pre-product, so that the processing time
of the reaction batches, consisting of the combination of
NC0 pre-polymers, which are highly reactive and/or highly
viscous or solid at room temperature and based on polyester
2t~46670
and/or polycarbonate polyols and the aforementioned
stabilised ureas at the respective reaction temperature is
prolonged so that the mixture can be efficiently degassed
and cast. Solidification is then brought about in
conventional manner by action of heat (120 to 180C)
yielding polyurethane elastomers having very good
mechanical properties and good resistance to heating.
The diamino diphenyl ureas suitable according to the
invention are produced by known methods. For example
nitro-anilines with phosgene or diphenyl carbonate can be
converted into the corresponding dinitro diphenyl ureas and
then converted into the desired diamino diphenyl ureas by
reduction. Another universally applicable method is a
reaction of amino acetanilides with phosgene or diphenyl
carbonate and subsequent alkaline saponification of the
acetamide group to the desired product.
A particularly simple method, which is consequently
preferred for manufacturing the ureas according to the
invention, is to react aromatic m-diamines with urea as
described in US-A 16 17 847 (in an inert solvent or in the
melt) or in US-A 25 03 797 (in sulphuric-acid or neutral
aqueous solution).
As a result of the method of manufacture, however, higher-
molecular, multi-nuclear products having the general
formula II
NH2 11 ~ NH2
[~ ~NH~--NH ~ n > 2
R R R R
~ n
21~6670
are usually formed together with the monomeric diamino
diphenyl ureas I, but within certain limits do not have any
negative influence on the properties of the elastomers.
However the proportion of ureas with n = 1 - 3 must be at
least 60~ by weight relative to the total amount. Ureas
with a maximum proportion of "monomers" (n = 1) are
preferred.
One simple method of producing diamino diphenyl ureas with
a high yield and very low proportions of higher-nuclear
ureas is to react aromatic m-diamines with urea in
chlorobenzene under specific concentration conditions, as
described in EP-A 374 653.
The diamino diphenyl ureas, which are solids, are usually
first finely ground, e.g. in a ball mill, until they have
an average particle size of 1 to 50 ~m, preferably 3 to
10 ~m.
The following are examples of preferred diamines for
preparing the ureas according to the invention: m-phenyl
diamine, 2,4-diaminotoluene, 2,6-diaminotoluene, 1-methyl-
3,5-diethyl-2,6-diaminobenzene and 1,3,5- triethyl-2,4-
diaminobenzene. The following are examples of preferred
diamines for producing the ureas according to the
invention: p-phenylene diamine, m-phenylene diamine, 2,5-
diaminotoluene, 2,4-diaminotoluene, 2,6-diaminotoluene,
l-methyl-3,5-diethyl-2,6-diaminobenzene and 1,3,5-triethyl-
2,4-diaminobenzene. The resulting diamino powders can be
mixed directly with the NCO pre-product or are preferably
applied in suspension form with a small amount of the high-
molecular polyol on which the NCO pre-adduct is based.
Of course, use can also be made of mixtures of the
aforementioned diamino diphenyl ureas with other chain-
lengtheners known in PUR chemistry and comprising at least
2116670
two hydrogen atoms which react with isocyanates and have a
molecular weight of 60 to 400.
The polyhydroxyl compound suitable for the method according
to the invention of producing the pre-adducts comprising
NC0 groups have a molecular weight of about 400 to 10,000,
preferably 600 to 6,000. The polyesters and polycarbonates
in question contain at least 2 and preferably 2 to 4
hydroxyl groups, as known per se for producing homogeneous
and cellular polyurethanes.
The polyesters in question comprising hydroxyl groups are
e.g. reaction products of polyhydric, preferably dihydric
or optionally additionally trihydric alcohols with
polyvalent, preferably divalent carboxylic acids. Instead
of the free polycarboxylic acids, the corresponding poly-
carboxylic acid anhydrides or corresponding polycarboxylic
acid esters of low alcohols or mixtures thereof can be used
to obtain the polyesters. The polycarboxylic acids can be
aliphatic, cycloaliphatic, aromatic and/or heterocyclic and
optionally substituted e.g. by halogen atoms and/or can be
unsaturated. The following are examples: succinic acid,
adipic acid, suberic acid, azelaic acid, sebacic acid,
phthalic acid, isophthalic acid, trimellitic acid, phthalic
acid anhydride, tetrahydrophthalic acid anhydride,
hexahydrophthalic acid anhydride, tetrachlorophthalic acid
anhydride, endomethylene tetrahydrophthalic acid anhydride,
glutaric acid anhydride, maleic acid, maleic acid
anhydride, fumaric acid, dimeric and trimeric fatty acids
such as oleic acid, optionally mixed with monomeric fatty
acids, terephthalic acid dimethyl ester or terephthalic
acid-bis-glycolic ester. The polyhydric alcohols can e.g.
be ethylene glycol, propylene glycol-(1,2) and -(1,3),
butylene glycol-(1,4) and -(2,3), hexanediol-(1,6),
octanediol-(1,8), neopentyl glycol, cyclohexane dimethanol
(1,4-bis-hydroxymethyl cyclohexane), 2-methyl-1,3-
11
2146670
. .
propanediol, glycerol, trimethylol propane, hexanetriol-
(1,2,6), butanetriol-(1,2,4), trimethylol ethane,
pentaerythritol, quinitol, mannitol or sorbitol, methyl
glycoside, or diethylene glycol, triethylene glycol,
tetraethylene glycol, polyethylene glycol, dipropylene
glycol, polypropylene glycols, dibutylene glycol or
polybutylene glycols. The polyesters can contain
proportions of terminal carboxyl groups. Polyesters from
lactones, e.g. ~-caprolactone or hydroxycarboxylic acids
such as ~-hydroxycaproic acid can also be used.
The polycarbonates comprising hydroxyl groups can be those
of known kind, prepared e.g. by reacting diols such as
propanediol-(1,3), butanediol-(1,4) and/or hexanediol-
(1,6), dimethylene glycol, triethylene glycol ortetraethylene glycol with diaryl carbonates, e.g. diphenyl
carbonate, or with phosgene.
Representatives of these compounds for use according to the
invention are described e.g. in High Polymers, Vol. XVI,
"Polyurethanes, Chemistry and Technology", compiled by
Saunders-Frisch, Interscience Publishers, New York, London,
Volume I, 1962, pages 32 - 42 and pages 44 - 54 and Volume
II, 1964, pages 5 - 6 and 198 - 199, and in Kunststoff-
Handbuch, Volume VII, Vieweg- Hochtlen, Carl-Hanser-Verlag,
Munich 1966, e.g. on pages 45 to 71.
Of course, use can be made of mixtures of the afore-
mentioned compounds containing at least two hydrogen atoms
capable of reacting with isocyanates and having a molecular
weight of 400 to 10,000, e.g. mixtures of polyesters and
polycarbonates.
The starting compounds for use according to the invention
can also be aliphatic, cycloaliphatic, araliphatic,
aromatic or heterocyclic polyisocyanates, described e.g. by
12
21~6670
W. Siefken in Justus Liebigs Annalen der Chemie, 562, pages
75 to 136, e.g. ethylene diisocyanate, 1,4-tetramethylene
diisocyanate, 1,6-hexamethylene diisocyanate, 1,12-dodecane
diisocyanate, cyclobutane-1,3-diisocyanate, cyclohexane-1,3
and 1,4-diisocyanate or any mixtures of these isomers,
1-isocyanato-3,3,5-trimethyl-5-isocyanatomethyl cyclohexane
(DAS 1 202 785), 2,4- and 2,6-hexahydrotoluylene
diisocyanate or any mixtures of these isomers, hexahydro-
1,3- and/or -1,4-phenylene diisocyanate, perhydro-2,4'-
and/or 4,4'-diphenylmethane diisocyanate, 1,3- and 1,4-
phenylene diisocyanate, 2,4- and 2,6-toluylene diisocyanate
or any mixtures of these isomers, diphenyl methane-2,4'-
and/or -4,4'-diisocyanate, naphthylene-1,5- diisocyanate,
triphenylmethane-4,4'4''-triisocyanate, polyphenyl
polymethylene polyisocyanates obtained by aniline-
formaldehyde condensation and subsequent phosgenation and
described e.g. in British PSS 874 430 and 848 671,
perchlorinated aryl polyisocyanates as described e.g. in
German laid-open specification 1 157 601, polyisocyanates
containing carbodiimide groups as described in German PS 1
092 007, diisocyanates as described in US-PS 3 492 330,
polyisocyanates containing allophanate groups as described
e.g. in GB-A 994 890, BE-A 761 626 and NL-A 7 102 524,
polyisocyanates containing isocyanate groups as described
e.g. in DE-A 1 022 789, 1 222 067 and 1 027 394 or 1 929
034 and 2 004 048, polyisocyanates containing urethane
groups as described e.g. in BE-A 752 261 or in
US-A 3 394 164, polyisocyanates containing acylated urea
groups according to DE-A 1 230 778, polyisocyanates
containing biuret groups as described e.g. in DE-A 1 101
394, GB-A 889 050 and FR-A 7 017 514, polyisocyanates
produced by telomerisation reactions as described e.g. in
BE-A 723 640, polyisocyanates containing ester groups as
mentioned e.g. in GB-A 965 474 and 1 072 956,
2196670
US-A 3 567 763 and DE-A 1 231 688, or reaction products of
the aforementioned isocyanates with acetals according to
DE--A 1 072 385.
5 Use can also be made of distillation residues, optionally
dissolved in one or more of the aforementioned poly-
isocyanates, and comprising isocyanate groups obtained
during industrial isocyanate production. Any mixtures of
these polyisocyanates can also be used.
Usually special preference is given to polyisocyanates
which are easily obtainable industrially, e.g. 2,4- and
2,6-toluylene diisocyanate or any mixtures of these isomers
("TDI"), polyphenyl polymethylene polyisocyanates obtained
15 by aniline-formaldehyde condensation and subsequent
phosgenisation ("crude MDI") and polyisocyanates comprising
carbodiimide groups, urethane groups, allophanate groups,
isocyanurate groups, urea groups or biuret groups
("modified polyisocyanates").
The polyisocyanates or the isocyanate prepolymers prepared
from the aforementioned polyisocyanates and the afore-
mentioned higher and/or lower-molecular polyols should be
present in liquid form in the reaction with the powdered or
25 suspended aromatic diamine.
If the method according to the invention is used to produce
polyurethane foam, water and/or easily volatile organic
substances are used as the foaming agent. The organic
30 foaming agent can e.g. be acetone, ethyl acetate, methanol,
ethanol, halogen-substituted alkanes such as methylene
chloride, chloroform, ethylidene chloride, vinylidene
chloride, monofluorotrichloromethane, chloro-
difluoromethane, dichlorodifluoromethane, or butane,
35 hexane, heptane or diethyl ether. The foaming effect can
also be obtained by adding compounds, e.g. azo compounds
14
2146670
such as azo-isobutyric acid nitrile, which decompose at
elevated temperatures with evolution of gases, e.g.
nitrogen. Other examples of foaming agents and details
about the use of foaming agents are described in
Kunststoff-Handbuch, Volume VII, published by Vieweg and
Hochtlen, Carl-Hanser-Verlag, Munich 1966, e.g. on pages
108 and 109, 453 and 455 and 507 - 510.
Catalysts can also often be used according to the
invention. The catalysts can be those of known kind, e.g.
tertiary amines such as triethyl amine, tributyl amine,
N-methyl morpholine, N-ethyl morpholine, N,N,N',N'-
tetramethyl ethylene diamine, 1,4-diaza-bicyclo-(2,2,2)-
octane, N-methyl-N'-dimethyl aminoethyl piperazine, N,N-
dimethyl benzylamine, bis-(N,N-diethyl aminoethyl)-adipate,
N,N-diethyl benzylamine, pentamethyl diethylene triamine,
N,N-dimethyl cyclohexyl amine, N,N,N',N'-tetramethyl-1,3-
butanediamine, N,N-dimethyl-~-phenyl ethylamine, 1,2-
dimethyl imidazole or 2-methyl imidazole.
The following are examples of tertiary amines containing
hydrogen atoms which react with isocyanate groups:
triethanolamine, triisopropanolamine, N-methyl-
diethanolamine, N-ethyl-diethanolamine, N,N-dimethyl-
ethanolamine or reaction products thereof with alkyleneoxides such as propylene oxide and/or ethylene oxide.
The catalysts may also be silaamines with carbon-silicon
bonds, as described e.g. in DE-A 1 229 290, e.g. 2,2,4-
trimethyl-2-silamorpholine and 1,3-diethyl aminomethyl
tetramethyl disiloxane.
The catalysts can be nitrogen-containing bases such as
tetraalkyl ammonium hydroxides or alkali-metal hydroxides
such as sodium hydroxide, alkali-metal phenolates such as
sodium phenolate or alkali-metal alcoholates such as sodium
21~6670
"
methylate. Hexahydrotriazines can also be used as
catalysts.
According to the invention other organic metal Gompounds,
more particularly organic tin compounds, can be used as
catalysts.
The organic tin compounds are preferably tin(II) salts of
carboxylic acids such as tin(II) acetate, tin(II) octoate,
tin(II) ethyl hexoate or tin(II) laurate and dialkyl tin
salts of carboxylic acids such as dibutyl tin diacetate,
dibutyl tin dilaurate, dibutyl tin maleate or dioctyl tin
diacetate.
Other examples of catalysts used according to the invention
and details about the operation of the catalysts are
described in Kunststoff-Handbuch, Volume VII, published by
Vieweg and Hochtlen, Carl-Hanser-Verlag, Munich 1966, e.g.
on pages 96 to 102.
The catalysts are normally used in a proportion between
about 0.001 and 10% by weight relative to the quantity of
polyhydroxyl compounds having a molecular weight of 400 to
10, 000.
Surface-active additives (emulsifiers and foam stabilisers)
can also be used according to the invention. The
emulsifiers can e.g. be sodium salts of castor oil
sulphonates or of fatty acids or salts of fatty acids with
amines, such as diethylamine oleate or diethanolamine
stearate. Alkali-metal or ammonium salts of sulphonic
acids, e.g. dodecylbenzene sulphonic acid or dinaphthyl-
methane disulphonic acid or of fatty acids such as
ricinoleic acid or polymeric fatty acids can also be used
as surface-active additives.
2146670
The main foam stabilisers are water-soluble polyether
siloxanes. These compounds are usually constructed by
bonding an ethylene oxide and propylene oxide copolymer
with a polydimethyl siloxane radical. Foam sta~ilisers of
this kind are described e.g. in US-A 2 764 565.
According to the invention, use can also be made of cell
regulators of known kind, such as paraffins or fatty
alcohols or dimethyl polysiloxanes or pigments or dyes and
flame retardants of known kind, e.g. tris-chloroethyl
phosphate or ammonium phosphate or polyphosphate, or
stabilisers against ageing and weathering, or softeners,
fungistatic and bacteriostatic substances, fillers such as
barium sulphate, diatomite, carbon black or whiting.
Other examples of surface-active additives optionally of
use according to the invention, and of foam stabilisers
such as cell regulators, reaction-delaying substances,
stabilisers, flame retardants, plasticisers, dyes, fillers,
fungicidal and bacteriostatic substances and details about
the use and operation of these additives are described in
Kunststoff-Handbuch, Volume VI, published by Vieweg and
Hochtlen, Carl-Hanser-Verlag, Munich 1966, e.g. on pages
103 to 113.
The organic acid chlorides can be compounds having the
general formula
(ClCO)n-R or (ClCOX)-R
where n = 1 - 4, preferably n = 2 - 4, R denotes an
aliphatic, cycloaliphatic or aromatic radical and X = 0
and/or N. The following are examples of compounds
according to the invention: acetic acid chloride, oleic
acid chloride, stearic acid chloride, phthalic acid
chloride, terephthalic acid chloride, or toluylene-2,4-bis
17
2146670
carbomoyl chloride. Use can also be made of inorganic acid
chloride derived from oxo-acids such as sulphuric acid or
phosphoric acid. Thionyl chloride or phosphorus
pentachloride are examples. Hydrochloric acid ~dded to the
NCO prepolymer reacts immediately in situ with free
isocyanate and gives carbamic acid chlorides which can be
classified among the aforementioned organic acid chlorides.
DE-A 3 732 728, page 7, lines 56 ff mentions use of acid-
reacting substances such as hydrochloric acid or organic
acid halides for delaying the reaction. This relates to
the possibility, generally known in the prior art, of using
Brondsted or Lewis acids in ppm proportions for inhibiting
the NCO reaction (Becker/Braun, Kunststoff-Handbuch,
Part 7, Munich, Carl Hanser Verlag, 1983, page 98). This
- method is used mainly for stabilising NCO prepolymers
and/or for forming prepolymers from highly active polyols
or amines. The effect is explained by neutralisation of
traces of bases or inactivation of metals by the acids.
The delaying effect of organic acids in combination with
amines is attributed to salt formation. During the
starting phase, isocyanate is trapped by the acid, thus
delaying the start. The influence of acids and bases on
the production of urethane prepolymers is described in
detail by H. L. Heiss et al in Ind. Eng. Chem., S1 (1959),
pages 929 - 934. One important finding is that addition of
a few ppm of HCl (10 to 20 ppm) is observed to result in a
considerable decrease in exothermy or the speed of the
reaction between toluylene-2,4-diisocyanate with a
polyethylene glycol (MW 400) (see Fig. 1, page 931 in H. L.
Heiss et al in Ind. Eng. Chem., 51 (1959)). If however the
quantity of HCl is considerably increased (> 200 ppm) there
is actually an increase in the rate of reaction. This
shows that inhibitors do not influence the NCO-OH or NH
reaction so much as suppress the side-reaction of
isocyanate to form allophanate, biuret or trimers
18
21~6670
(see Fig. 19, page 214 in H. J. Saunders and K. C. Frisch:
Polyurethanes, Chemistry and Technology, Volume 1, New
York, Interscience, 1962 (High Polymers, Volume 16). From
this prior art, the skilled man would mainly be;inclined to
use organic acids in ppm proportions for delaying the NCO-
NH2 reaction.
In the present case, however, where solid diamino ureas
combine with liquid NCO polymers, no stabilising effect of
organic acids is observed, even if a considerably larger
proportion is used than normally. Consequently there was
no reason to foresee the effect of organic and/or inorganic
acid chlorides which, in a proportion of > 250 ppm relative
to the NCO pre-adduct, i.e. considerably above the normal
proportion, are found to be excellent stabilisers of solid
diamino ureas, without appreciably interfering with the
process of hardening into high-quality end products.
According to the invention, the components are reacted by
the known prepolymer method or the semi-prepolymer method,
often using mechanised equipment, e.g. as described in US-A
2 764 565. Details about processing equipment also used
according to the invention are described in Kunststoff-
Handbuch, Volume VI, published by Vieweg and Hochtlen,
Carl-Hanser-Verlag, Munich 1966, e.g. on pages 121 to 205.
In the method according to the invention, the proportions
of components in the reaction are usually chosen so that
the molar ratio of polyisocyanates to chain-lengtheners
plus the compound with reactive OH groups - depending on
the particular method of processing - is usually between
0.7 and 1.5, preferably between 0.90 and 1.15. The
percentage of NCO in the prepolymer, if the prepolymer
stage is included, can be 1.8 to 6% by weight. The molar
ratio of reactive hydrogen in the chain-lengtheners to
reactive OH groups can vary within wide limits, preferably
19
- 2146670
between 0.4 and 1.5, resulting in soft to hard types of
polyurethane. In addition to the diamines used according
to the invention, the chain-lengtheners can comprise other
diamines or diols, e.g. those mentioned hereinbefore in the
production of polyhydroxyl compounds. However, the molar
fraction of the amine according to the invention in the
chain-lengthener should be between 1 and 0.5, preferably
between 1 and 0.8.
The invention is worked by a simple method. The polyol
component, which has at least two hydroxyl groups and has a
molecular weight of 400 to 10,000, is reacted in known
manner with an excess of diisocyanate to obtain the pre-
adduct containing the NCO groups. The reaction can be
monitored by NCO titration. At the end of the poly-
addition, the stabiliser is added. The temperature during
addition of the stabiliser depends on the solubility
thereof in the NCO pre-adduct. It is simplest to add the
stabiliser at the respective processing temperature of the
batches for casting. In most cases the temperature is
about 80C. Of course, the processing temperature should
not be too high, since in that case it is impossible to
prevent a premature reaction after adding the chain-
lengthener. The diamino diphenyl urea is introduced in the
form of a solid powder (particle size about 5 to 50 ~m)
using a suitable agitator, and the resulting suspension is
intimately mixed. The processing time (Pot Life) of
the system depends on the nature of the diamino diphenyl
urea, the NCO pre-adduct and the proportion of stabiliser.
The proportion of stabiliser is chosen to obtain efficient
degassing and casting under the respective processing
conditions. There should never be a premature reaction
between the NCO pre-adduct and the aromatic diamine, since
any uncontrollable increase in the viscosity of the batch
will increase the difficulty of subsequent processing
during casting. On the other hand, not too much stabiliser
214667~
should be added, since otherwise the solidification process
will suffer, and it will be necessary to accept
uneconomically high solidification temperatures and poorer
mechanical properties. The upper limit of the
concentration of stabiliser is about 5,000 to 10,000 ppm
relative to the prepolymer used. It is simplest to
estimate the optimum proportion of stabiliser for the
respective system and the respective working conditions in
a preliminary test, using a series of concentrations.
In a variant of the process, the solid diamine powder can
first be mixed with the high-molecular liquid polyols on
which the NC0 pre-adduct is based. The batch can then be
degassed, optionally at elevated temperature. In this case
the stabiliser can be added either on the side of the NC0
pre-adduct as already described, or in the polyol-diamine
suspension. The resulting pourable suspension or paste can
then be added to the NC0 pre-adduct. This process has the
advantage of simplicity.
The solidification temperature of the reactive systems
according to the invention varies from 100 to 180C.
When the solidification temperature increases, the
solidification time decreases. The duration of baking,
depending on the temperature, can vary from less than
1 minute to several hours. It is often advantageous to
heat-treat the plastics at lOO~C for a short time after
removal from the mould, to ensure complete and thorough
hardening.
Elastomers produced according to the invention have
numerous uses, e.g. for highly mechanically stressed
moulded members such as tyres, rollers, V-belts or seals
exposed to severe thermal or chemical stress, or for hot-
21~6670
water pipes or motors or for producing sheets, textilecoatings or polyurethane powder.
The chains can also be lengthened in the presen,ce of the
aforementioned foaming agents and additives, preferably in
closed moulds, thus forming foams with a cellular core and
a compact surface.
The resilient, semi-elastic foamed materials obtainable by
the process according to the invention are used e.g. as
cladding materials, mattresses, packing material or, owing
to their resistance to flame, in all sectors where these
properties are particularly important, e.g. in car and
aircraft construction and in the transport sector in
general. The foamed materials can be produced either by
foaming in a mould or by fabrication from slab stock.
The following examples illustrate the method according to
the invention. Unless otherwise stated, numerical values
are proportions or percentages by weight.
2146670
Examples
Prepolymer A: A straight-chain polyester (OH number = 56,
molecular weight = 2,000) prepared from adipic acid and
ethylene glycol was reacted with 2,4-diisocyanato-toluene
in the molar ratio 1:2 at 60 - 80C by the normal process,
yielding a pre-adduct containing NCO groups and having an
NCO content of 3.6 to 3.85% by weight.
Prepolymer B: A straight-chain polyester prepared from
adipic acid with ethylene glycol and butanediol-1,4 was
reacted with 2,4-diisocyanatotoluene in the molar ratio 1:2
at 60 to 80C by the normal process, yielding a pre-adduct
lS containing NCO groups and with an NCO content of 3.3 to
3.5% by weight.
ExamPle 1 Comparative example, not according to the
invention.
This example describes an attempt to cast an NCO polyester
pre-adduct with TDAH without addition of stabiliser.
100 g of prepolymer A were intimately mixed at 70 to 80C
with 26.32 g of 3,3'-diamino-4,4'-dimethyl-diphenyl urea
(prepared from 2,4-diaminotoluylene and urea as per
EP 0 374 653, NH number 390 mg KOH/g). At the
aforementioned temperature, the viscosity increased
considerably in a very short time (< 60 seconds) so that
the batch could neither be degassed nor cast.
23
2146670
Example 2 according to the invention
Use of 1,000 ppm terephthalic acid dichloride
400 g of prepolymer B were heated to 70 to 80C. After
addition of 0.4 g (1,000 ppm) terephthalic acid dichloride,
the mixture was stirred for a further 30 minutes. Next,
45.1 g of 3,3'-diamino-4,4'-dimethyl-diphenyl urea
(prepared according to EP 0 374 653 from 2,4-
diaminotoluylene and urea, NH number 390 mg KOH/g) was
intensively mixed with the prepolymer. The suspension was
thoroughly degassed in a water-jet vacuum.
The time for processing the resulting reactive system was
at least 6.5 hours at 80C. During this time there was no
premature reaction resulting in a considerable increase in
the viscosity of the batch.
The liquid reactive system was poured into a preheated
mould coated with a separating agent, and heated to 140 to
150C.
After 1 to 2 hours the batch solidified and the moulding
could be removed from the mould. It was advantageously
heat-treated at 150C for a further 4 hours.
The product was a highly elastic PUR elastomer having the
following mechanical properties:
Modulus (100%) (MPa) 10.0
Tensile strength (MPa) 31.0
Elongation at break (%) 580
Resistance to tear
propagation (KN/m) 87.9
Elasticity (%) 39
Hardness Shore A 94
2146670
ExamPle 3 according to the invention
Use of terephthalic acid dichloride
400 g of prepolymer B were heated to 70 to 80C. After
addition of 0.2 g (500 ppm) terephthalic acid dichloride,
the mixture was stirred for a further 30 minutes. Next,
45.1 g of 3,3'-diamino-4,4'-dimethyl-diphenyl urea
(prepared according to EP 0 374 653 from 2,4-
diaminotoluylene and urea, NH number 390 mg KOH/g) wasintensively mixed with the prepolymer. The suspension was
thoroughly degassed in a water-jet vacuum.
The time for processing the resulting reactive system was
at least 6.5 hours at 80C. During this time there was no
premature reaction resulting in a considerable increase in
the viscosity of the batch.
The liquid reactive system was poured into a preheated
mould coated with a separating agent, and heated to 140 to
150C.
After 1 to 2 hours the batch solidified and the moulding
could be removed from the mould. It was advantageously
heat-treated at 150C for a further 4 hours.
The product was a highly elastic PUR elastomer having the
following mechanical properties:
Modulus (100%) (MPa) 11.5
Tensile strength (MPa) 38.9
Elongation at break (~) 570
Resistance to tear
propagation (KN/m) 101
35 Elasticity (~) 39
Hardness Shore A 93
2146670
ExamPle 3 not according to the invention
Use of 200 ppm terephthalic acid dichloride
400 g of prepolymer B were heated to 70 to 80C. After
addition of 0.08 g (200 ppm) terephthalic acid dichloride,
the mixture was stirred for a further 30 minutes. Next,
45.1 g of 3,3'-diamino-4,4'-dimethyl-diphenyl urea
(prepared according to EP 0 374 653 from 2,4-
diaminotoluylene and urea, NH number 390 mg KOH/g) wasintensively mixed with the prepolymer. After 10 minutes
the batch had thickened so much as a result of a premature
reaction that degassing and casting to obtain a high-
quality moulded part were impossible.
ExamPle 5 according to the invention
Use of 1,250 ppm oleic acid chloride
200 g of prepolymer A were heated to 70 to 80C. After
addition of 0.25 g (1250 ppm) oleic acid chloride, the
mixture was stirred for a further 30 minutes. Next, 26.3 g
of 3,3'-diamino-4,4'-dimethyl-diphenyl urea (prepared
according to EP 0 374 653 from 2,4-diaminotoluylene and
urea, NH number 390 mg KOH/g) was intensively mixed with
the prepolymer. The suspension was thoroughly degassed in
a water-jet vacuum.
The time for processing the resulting reactive system was
at least 5 hours at 80C. During this time there was no
premature reaction resulting in a considerable increase in
the viscosity of the batch.
26
2146670
The liquid reactive system was poured into a preheated
mould coated with a separating agent, and heated to 140 to
150C.
After 1 to 2 hours the batch solidified and the moulding
could be removed from the mould. It was advantageously
heat-treated at 150C for a further 4 hours.
The product was a highly elastic PUR elastomer having the
following mechanical properties:
Modulus (100%) (MPa) 11.2
Tensile strength (MPa) 37.1
Elongation at break (%) 645
15 Resistance to tear
propagation (KN/m) 111
Elasticity (%) 35
Hardness Shore A 95
Example 6 according to the invention
Use of 833 ppm sebacic acid dichloride
300 g of prepolymer A were heated to 70 to 80C. After
addition of 0.25 g (833 ppm) sebacic acid dichloride, the
mixture was stirred for a further 30 minutes. Next, 39.5 g
of 3,3'-diamino-4,4'-dimethyl-diphenyl urea (prepared
according to EP 0 374 653 from 2,4-diaminotoluylene and
urea, NH number 390 mg KOH/g) was intensively mixed with
the prepolymer. The suspension was thoroughly degassed in
a water-jet vacuum.
The time for processing the resulting reactive system was
at least 4 hours at 80C. During this time there was no
21~6fi70
premature reaction resulting in a considerable increase in
the viscosity of the batch.
The liquid reactive system was poured into a pr~2heated
5 mould coated with a separating agent, and heated to 140 to
150C.
After 1 to 2 hours the batch solidified and the moulding
could be removed from the mould. It was advantageously
10 heat-treated at 150C for a further 4 hours.
The product was a highly elastic PUR elastomer having the
following mechanical properties:
Modulus (100%) (MPa) 10.6
Tensile strength (MPa) 33.5
Elongation at break (%) 640
Resistance to tear
propagation (KN/m) 99.5
Elasticity (%) 30
Hardness Shore A 94
Example 7 according to the invention
Use of 1250 ppm stearic acid dichloride
200 g of prepolymer A were heated to 70 to 80C. After
addition of 0.25 g (1250 ppm) stearic acid dichloride, the
30 mixture was stirred for a further 30 minutes. Next, 26.3 g
of 3,3'-diamino-4,4'-dimethyl-diphenyl urea (prepared
according to EP 0 374 653 from 2,4-diaminotoluylene and
urea, NH number 390 mg KOH/g) was intensively mixed with
the prepolymer. The suspension was thoroughly degassed in
35 a water-jet vacuum.
28
21~6670
The time for processing the resulting reactive system was
at least 5 hours at 80C. During this time there was no
premature reaction resulting in a considerable increase in
the viscosity of the batch.
The liquid reactive system was poured into a preheated
mould coated with a separating agent, and heated to 140 to
150C.
After 1 to 2 hours the batch solidified and the moulding
could be removed from the mould. It was advantageously
heat-treated at 150C for a further 4 hours.
The product was a highly elastic PUR elastomer having the
following mechanical properties:
Modulus (100%) (MPa) 12.3
Tensile strength (MPa) 45.6
Elongation at break (%) 650
20 Resistance to tear
propagation (KN/m) 116
Elasticity (%) 31
Hardness Shore A 94
ExamPle 8 according to the invention
Use of 250 ppm phosphorus pentachloride (PCl5)
200 g of prepolymer B were heated to 70 to 80C. After
addition of 0.05 g (250 ppm) PCl5, the mixture was stirred
for a further 30 minutes. Next, 26.3 g of 3,3'-diamino-
4,4'-dimethyl-diphenyl urea (prepared according to EP 0 374
653 from 2,4-diaminotoluylene and urea, NH number 390 mg
KOH/g) was intensively mixed with the prepolymer. The
suspension was thoroughly degassed in a water-jet vacuum.
21~6670
The time for processing the resulting reactive system was
at least 4 hours at 80C. During this time there was no
premature reaction resulting in a considerable increase in
the viscosity of the batch.
The liquid reactive system was poured into a preheated
mould coated with a separating agent, and heated to 140 to
150C.
After 1 to 2 hours the batch solidified and the moulding
could be removed from the mould. It was advantageously
heat-treated at 150C for a further 4 hours.
The product was a highly elastic PUR elastomer having the
following mechanical properties:
Modulus (100%) (MPa) 10.7
Tensile strength (MPa) 25.7
Elongation at break (%) 580
20 Resistance to tear
propagation (KN/m) 97.8
Elasticity (%) 34
Hardness Shore A 94
Example 9 according to the invention
Use of 2,500 ppm thionyl chloride (SOCl2
200 g of prepolymer A were heated to 70 to 80C. After
addition of 0.5 g (2,500 ppm) SOCl2, the mixture was
stirred for a further 30 minutes. Next, 26.3 g of 3,3'-
diamino-4,4'-dimethyl-diphenyl urea (prepared according to
EP 0 374 653 from 2,4-diaminotoluylene and urea, NH number
390 mg KOH/g) was intensively mixed with the prepolymer.
2146670
The suspension was thoroughly degassed in a water-jet
vacuum.
The time for processing the resulting reactive system was
at least 6 hours at 80C. During this time there was no
premature reaction resulting in a considerable increase in
the viscosity of the batch.
The liquid reactive system was poured into a preheated
mould coated with a separating agent, and heated to 140 to
150C.
After 1 to 2 hours the batch solidified and the moulding
could be removed from the mould. It was advantageously
heat-treated at 150C for a further 4 hours.
The product was a highly elastic PUR elastomer having the
following mechanical properties:
Modulus (100%) (MPa) 10.0
Tensile strength (MPa) 28.1
Elongation at break (%) 600
Resistance to tear
propagation (KN/m) 98.1
25 Elasticity (%) 30
Hardness Shore A 92