Note: Descriptions are shown in the official language in which they were submitted.
-
WO 94/18981 PCTtUS94/01881
~15~12~
TITLE OF THE INVENTION
FIBRINOGEN RECEPTOR ANTAGONISTS
BA(~KGROIJN~ OF THE IN~ENTION
The invention relates generally to mod~ ting cell adhesion
and to inhibiting the binding of fibrinogen and other proteins to blood
platelets, and inhibiting the aggregation of blood platelets specifically to
the IIb/IIIa fibrinogen receptor site. Fibrinogen is a glycoprotein
present in blood plasma that participates in platelet aggregation and in
fibrin formation. Platelets are cell-like anucleated fragments, found in
the blood of all m~mm~lc, that also participate in blood coagulation.
Interaction of fibrinogen with the IIb/IIIa receptor site is known to be
essential for normal platelet function.
When a blood vessel is ~l~m~ged by an injury or other
5 causative factor, platelets adhere to the disrupted subendothethial
surface. The adherent platelets subsequently release biologically active
constituents and aggregate. Aggregation is initi~ted by the binding of
agonists, such as thrombin, epinephrine, or ADP to speci~lc platelet
membrane receptors. Stimulation by agonists results in exposure of
20 latent fibrinogen receptors on the platelet surface, and binding of
fibrinogen to the glycoprotein IIb/IIIa receptor complex.
Attempts have been made to use natural products and
synthetic peptides to determine the mech~ni.cm of adhesion and platelet
aggregation. For example, Rouslahti and Pierschbacher in Science, 23~,
491-497 (19P~7), describe adhesive proteins such as fibronectin,
vitronectin, osteopontin, collagens, thrombospondin, fibrinogen, and
von Willebrand factor that are present in extracellular matrices and in
blood. The proteins contain the tripeptide arginine-glycine-aspartic acid
(RGD) as their glycoprotein IIb/IIIa recognition site. These arginine-
glycine-aspartic acid cont~ining tripeptides are recognized by at least
one member of a family of structurally related receptors, integrins,
which are heterodimeric proteins with two membrane-spanning
subunits. The authors state that the conformation of the tripeptide
WO 94tl8981 PCT/US94/01881
~ ,~s~ 3 - 2 -
sequence in the individual proteins may be critical to recognition
specificity.
Cheresh in Proc. Nat'l Acad. Sci. U.S.A.. 84, 6471-6475,
(1987), describes an Arg-Gly-Asp directed adhesion receptor expressed
by hl-m~n endothelial cells that is structurally similar to the IIb/~Ia
complex on platelets but is antigenically and functionally distinct. This
receptor is directly involved in endothelial cell attachment to
fibrinogen, von Willebrand factor, and vitronectin.
Pierschbacher and Rouslahti, in J. of Biol. Chem., 262,
(36), 17294-17298 (19P~7) hypothesized that the Arg-Gly-Asp se~uence
alone would be a sufficient signal for receptor recognition and binding
and that, therefore, the conformation of the tri-peptide sequence would
be determinative. Various synthetic peptides were produced and the
authors concluded that the stereochemical conformLation of Arg-Gly-Asp
as influenced by enantiomeric substitutions or additions to this sequence
significantly influenced receptor-ligand interaction. The authors
further showed that cyclization of a decapeptide by forming a disulfide
bridge between non-terminal residues Pen and Cys, rendered the peptide
much less effective at inhibiting attachment to fibronectin.
In Proc. Nat'l Acad. Sci. U.S.A., ~1, 59~5-59~8 (19~S4), the
same authors describe tetrapeptide variants of the cell recognition site of
fibronectin that retain attachment-promoting activity. Peptides having a
tetrapeptide recognition site are described in U.S. Pat. Nos. 4,5~9,~ 1
and 4,614,517. A number of large polypeptide fragments in the cell-
binding domain of fibronectin have cell-attachment activity. For
example, see U.S. Pat. Nos. 4,517,686, 4,661,111 and U.S. Pat. No.
4,57~s,079.
Ruggeri et al., Proc. Nat'l Acad. Sci. U.S.A., ~3, 570P~-
5712 (19~6) explore a series of synthetic peptides designed in lengths to
3 16 residues, that contain RGD and a valine attached to the aspartic acid
residue of RGD that inhibit fibrinogen binding to platelets. See also
Koczewiak et al., Biochem. 23, 1767-1774 (19~4); Ginsberg et al., L
Biol. Chem. 260(7), 3931-3936 (1985); and Haverstick et ah, Blood
WO 94/18981 2 ~ S~ 12 3 PCT/US94/01881
66(4), 946-952 (1985). Other inhibitors are disclosed in Eur. Pat. App.
Nos. 275,748 and 29~,820.
A number of low molecular weight polypeptide factors
have been isolated from snake venom. These factors apparently have
high affinity for the gpIIb/lIIa complex. For example, Huang et ah, J.
Biol Chem., 262, 16157-16163 (19~7); Huang et al., Biochemistry 28,
661-666 (19~9) describe the primary structure of the venom trigramin
which is a 72 amino acid polypeptide that contains the RGD subunit.
Echistatin is another venom which has high affinity for the gpIIb/IIIa
complex. This polypeptide contains 49 amino acids and has the RGD
subunit and various disulfide bridges. Gan et ah, J. Biol. Chem., 263,
19827-19832 (1988). See also, Dennis et ah, Proc. Nat'l Acad. Sci.
USA. 87, 2471-2475 (1989). However, these snake venom factors also
have high affinity for other members of the adhesive protein receptor
farnily including the vitronectin and fibronectin receptors so are not
selective for the gpIIb/IIla complex.
While it is known that the tripeptide sequence Arg-Gly-Asp
is present in certain polypeptides that can duplicate or inhibit the cell
attachment-promoting effects of fibronectin and vitronectin, the tri-
peptide Arg-Gly-Asp has low activity. At present, there is little
understanding of how other amino acids coupled to this sequence
influence binding specificity. U.S. Pat. No 5,023,233, assigned to
Merck & Co., Inc., discloses small cyclic hexapeptides which contain the
sequence Arg-Gly-Asp and are useful platelet aggregation inhibitors.
U.S. Pat. No. 5,037,808 discloses the use of indolyl platelet-aggregation
inhibitors which are believed to act by antagonizing interactions
between fibrinogen and/or extracellular matrix proteins and the platelet
gpIIb/IIIa receptor. U.S. Pat. No. 5,037,~0~s discloses guanidino
peptide mimetic compounds that retain an Asp residue which inhibit
platelet aggregation. The application PCT/US90/02746 describes the
use of antibody-poly-peptide conjugates wherein said polypeptides
contain the Arg-Gly-Asp (RGD) sequence.
The application PCT/US91/00564 discloses the use of large
cyclic peptides cont~inin~ RGD flanked by proline residues which are
W O 94/18981 ~CTrUS94101881
4 -
platelet aggregation inhibitors. The application PCT/US90/037~g
discloses small cyclic platelet aggregation inhibitors which are synthetic
cyclic pentapeptides containin g the tripeptide sequence Arg-Gly-Asp and
a thioether linkage in the cycle. The application PCTIUS90/05367
5 published May 2, 1991, also discloses the use of peptides and
pseudopeptides such as N-amidino-piperidine-3-carboxylglycyl-L- -
aspartyl-L-valine that inhibit platelet aggregation and thrombus
formation in m ~ m m ~ n blood. The application lCur. Pat. App. No.
91103462.7 discloses linear compounds which can include internal
piperazinyl or piperidinyl derivatives. Eur. Pat. App. No. 91300179.8,
assigned to Merck & Co., Inc., and published on July 17, 1991, discloses
linear polypeptide fibrinogen receptor antagonists. Eur. Pat. App. No.
90101404.3 discloses compounds of the R1-A-(W)a-X-(CH2)b-(Y)C-B-
Z-COOR wherein Rl is a guanidino or amidino moiety and A and B are
chosen from specific monosubstituted aryl or heterocyclic moieties.
While a multitude of compounds or peptide analogs
believed to inhibit platelet aggregation by inhibiting binding to a blood
platelet by fibrinogen are known, the present invention provides novel
20 fibrinogen receptor antagonists that have significant binding activity and
are, therefore, useful for the reasons stated herein. A number of very
serious diseases and disorders involve hyperthrombotic complications
which lead to intravascular thrombi and emboli. Myocardial infarction,
stroke, phlebitis and a number of other serious conditions create the
25 need for novel and effective fibrinagen receptor antagonists.
WO 94/18981 PCT/US94/OI881
~ 51~
- 5 -
SUMMARY OF THE INVENTION
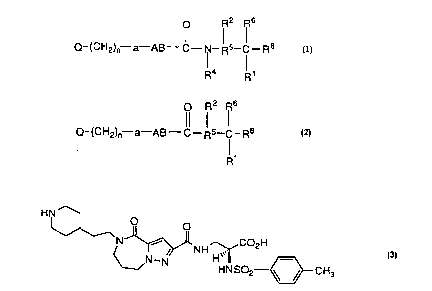
Compounds of the invention have the formula
Q-(CH2)n--a--AB--C--IN--Rs--C--Rr' or
R4 :~1
O R2 R6
Q- (CH2)n--a--AB--C--R5--C - R8
for exarnple
HN~ COzH CH3
. . The compounds have fibrinogen receptor antagonist
actlvlty.
.,
WO 94/18981 PCT/US94/01881
., i ~
S~ 6-
DETAILED DESCRIPTION OF THE rNVENTION
o R2 R6
Il l I
Q- (CH2)n--a--AB--C~ R~--C--R8 or
R4
O R2 R6
Il l I
Q- (C H2)n--a--AB--C--R5--C - R8
wherem
Qis
H2N--C-- ; H2N--C--NH-- ; R7HN ; or
Q is a 4-9 membered mono- or bi-cyclic ring system cont~ining 1, 2 or
3 heteroatoms chosen from N, O or S and either unsubstituted or
substituted with R8;
25 AB is a fused ring system sharing adjacent carbon and nitrogen atoms,
wherem
A is a 5, 6 or 7 membered saturated or lm~ lrated ring
cont~ining 1, 2 or 3 heteroatoms selected from O, S or N;
B is a 5, 6 or 7 membered salulated or~ s~lula~ed^ nng
cont~inin,e 1, 2 or 3 heteroatoms selected from O, S or N;
W O 94/18981 . PCT~US94/01881
_ 7 _
Rl is H, Cl 4 alkyl, N(R8)2, -N(R8)So2R7~ NR8Co2R7~ NR8C(o)R7,
NR8C(o)N(R7)R8, N(R~)So2N(R7)R~S, N(R~)So2N(R~)C(o)oR7,
C(o)N(R7)2, or a cyclic group with R6 as defined below;
R2 is H, Cl 4 alkyl, Cl 4 branched alkyl, Cl 4 alkyl aryl, or aryl;
R4 is H, Cl 4 alkyl, Cl 4 branched alkyl, cyclic Cl 4 alkyl or Cl 4
alkenyl;
R5 i.s CH, -CH(CH2)n, a bond, or when R5 is adjacent N(R4),
-C(CH2)n;
- o
R6 is COOH, CH2OH, C(o)NR7)2, CO2R9, tetrazole, acylsulfonamide,
or
Il (OH)2
O , or a cyclic group with R1 as defined below;
wherein the cyclic group of R1 with R6 is
~ R8 O Rs O
R O"S~O y
wherein y = O or S;
R7 is H, branched or straight chain Cl 4 substituted or unsubstituted
alkyl, branched or straight chain lower alkenyl, Cl 4 alkylaryl,
substituted aryl, or S or 6 membered heteroaryl cont~ining 1, 2, or 3 N,
S, or O heteroatoms
W O 94/18981 PCTnUS94/01881
2~ - 8 -
wherein substituted àlkyl is hydroxy substituted or Cl 4 alkoxy
substituted aIkyl, and wherein substituted aryl is substituted by
one, two or three of the following groups: halogen, Cl 4 alkoxy,
hydroxy, or Cl 4 alkyl;
R8 is H, branched or straight chain Cl 4 alkyl;
R9 is H, Cl 4 alkyl or aryl;
n is O-7;
n' is 0-3; and
ais
F~7
N ll or a bond,
and pharrnaceutically acceptable salts.
In one embodiment, the compounds have the formula
o R2 R6
Il I I .
Q- (CH2)n--a--AB--C--1~1--R5--C R8 or
R4
Q-(CH2)n--a--AB--C--R5--C-R8
PCT/USg4/01881
wo 94/18981 2 1 ~Sl 2 3
g
wherein
Qis
~IH
H N ~ ; H N J~N--~; R7NI ~;
H H
(CH2)n--NyNH ~
~N ;R N ~ ~ ;
R8 ~(CH2)n'
(CH2)2
/3~ ; R8N N- ~; O N--~;
~0 ~0
R8N N--~; R8N~N--~; N~;
WO 94/18981 PCT/US94/01881
R8
H2N~N~ =H~N~N~
H N~{ ~ ~ ; H2N )~N/~)n
H2N (C H2)n H
H2N N H~ (R8)2N
NH
H2N)~; or ~ ;
~,~
n = 0-7;
n' = 0-3;
25 R4 = H, Cl 4 alkyl, Cl 4 branched aLkyl, cyclic Cl 4 alkyl or Cl 4
alkenyl;
RS = CH, -CH(CH2)n, or a bond;
WO 94/18981 Z 15 ~12 3 PCT/US94/01881
R2 is H, Cl 4 alkyl, Cl 4 branched alkyl, Cl 4 alkyl aryl, or aryl;
Rl = H, Cl 4 alkyl, N(R~)2, -N(R~)So2R7, NR8Co2R7~ C(o)R7,
NR8C(o)N(R7)Rg~ N(R8)so2N(R7)R~ N(Rg)So2N(R~)C(o)oR7
C(o)N(R7)2~ or a cyclic group with R6;
R6 = COOH, CH2OH, C(o)NR7)2~ CO2R9, tetrazole, acylsulfonamide,
or
1 0 i(OH)2
, or a cyclic group with Rl;
wherein the cyclic group of Rl with R6 is
R7~ R o
whereiny=OorS;
R7 = H, branched or straight chain Cl 4 substituted or unsubstituted
alkyl, branched or straight chain lower alkenyl, Cl 4 alkylaryl,
substituted aryl, or S or 6 membered heteroaryl containing 1, 2, or 3 N,
25 S, or O heteroatoms
wherein substituted alkyl is hydroxy substituted or Cl 4 alkoxy
substituted alkyl, and wherein substituted aryl is substituted by
one two or three of the following groups: halogen, Cl 4 alkoxy,
hydroxy, or Cl 4 alkyl;
Rg = H, branched or straight chain Cl 4 alkyl;
R9 = H, Cl 4 alkyl or aryl;
WO 94/18981 PCT/US94/01881
2-
a=
O
J~
N
R7 or a bond;
A = a 5, 6 or 7 membered saturated, partially saturated, or unsaturated
o ring containing 1, 2 or 3 heteroatoms selected from O, S or N;
B = a 5, 6 or 7 membered saturated, partially saturated, or unsaturated
ring cont~ining 1, 2 or 3 heteroatoms selected from O, S or N;
wherein A and B form a fused ring system sharing adjacent carbon and
nitrogen atoms.
In the compounds of the present invention, the components
having asymmetric centers occur as racemates, racemic mixtures, and as
20 individual enantiomers and/or diastereomers. All isomeric forms are
included in the present invention.
In one class of this embodiment, the compounds have the
formula,
R2 R6
Q-(CH2)n--a--AB--C--~--R5--C--R8
R4 R
WO 94/18981 l S~ S 123 PCT/US94/01881
- 13 -
AB is selected from the group of
5~--N~
D~N V wherein V is N or CR7,
V~sSs
and D is CH2, CH2-CH2,
lo CH2C(R7)2CH2, or
1 5 H,~H2
(CH2)n' (cH2)n
O ~
~ ~ wherein X = N or CR3,
wherein R3 = CN, C(o)N(R7)R8,
O ~
~,,~N~O ,
o r(CH2)n'
~N~ , or
~ NAN--R3;
WO 94/18981 PCT/US94/01881
.~
14 -
~s--N~
O
~`N~N'~
~NJ ; and
N ~ N
< ,NJ
(H2C)n
In a subclass of this class of this embodiment compounds
are those having the forrnula
o R2 R6
Q-(CH2)n--a--AB--C--N--R5--1--R8
where AB is
_~ wherein V is N or CH,
D`N~ and D is CH2-CH2 or
V=~SsS CH2c(R4)2cH2
wo 94~18981 2 1 5 5 1 2 S PCT/US94/01881
- 15 - ,
In another class of this embodiment the compounds have
the formula
Q-(cH2)n--a--AB--C--R5--I--R
o AB is selected from the group of
55SyV~O
V-N ,N--55s wherein V is N or C R7
and D is CH2, CH2-CH2,
CH2C(R7)2CH2, or
~r
H~H2
(CH2)n (CH2)n
~XN ~ wherein X = N or CR3,
wherein R3= CN, C(o)N(R7)R8,
0 ~ ~
~,~N O, or
r(CH2)n'
;
W O 94/18981 P~CTrJS94/01881
16 -
~`NJ~N'~
~NJ
o
~N~N ~
,NJ ; and
o
~s--N~ wherein y3is 0 or H2.
y3
In a subclass of this class of this embodiment, the
compounds have the formula
o R2 R6
Q - (CH2)n--a--AB--C--R5--C--R8
o
~--NJ~
a = R7 ; and
AB is selected from
WO 94/18981 ~ PCT/US94/01881
~155I23
V-N~D,N--s55 wherein V is N or CR7
and D is CH2, CH2-CH2.
CH2C(R7)2CH2, or
~\
H~H2
(CH2)n. (cH2)n
;and
\~N ~ wherein X= N or CR3,
wherein R3 = CN, C(o)N(R7)R8,
O ~
~L,~N O, or
O,~ r~CH2)n
WO 94/18981 PCTIUS94/01881
- 18 -
In another subclass of this class of this embodiment, the
compounds have the formula
R2 R6
Q-(CH2)n--a--AB--C--R5--C--R8
R1
a = a bond;
and AB is selected from
~5~ N J~ N
~NJ
o
5~NJ--N'~
sss--N~ wherein y3 is O or H2.
y3
WO 94/18981 PCT/US94/01881
215~12~
- 19 -
Specific examples of compounds of the invention are those
selected from the following group of compounds and their
pharmaceutically acceptable salts:
O O
O~ N OH
HN~/\,N N N H H NHS02NHCO2CH2Ph,
HN~\,N~N-N H NHso2NH27
HN~ N ~CH
2s HN~ ,~N COOH
H HN ~CH3
HN~ ~N COOH
H HNSO2C4Hg
E~CTrJS94/01881
W O 94/18981
~5~ 20 -
H N ~ o
~ ~ N ~ C O2C H3
H H N S O 2C4Hg
H N ~ ~ N ~ C O O H
N - N H H N S 2 ~ C H3
HN~ N~COOH Cl
HN~ ~ o
N ~COOH
- N H H N S O2-N H C4Hg
N~ ~ N~COO CH3
N~ N~N~/Co2c2H5
H H N S 2 ~ C H3
3C H N ~ ~ ~ ~ S 2 ~ C H3
WO 94/18981 PCT/US94/01881
~ 3
HN'~ O
~, ~NI ~C02H
H HNSOZC4Hg
HN~l
~ ~ IN--'C2H
H
HN '--1
~J O
~ H HNSO2~CH3
~ ~N O
(~ HNSO2~CH3
HN~ ~N~COOH
H
HN~ ,N~COOH
H HNSO2~CH3,
HN~ ,~N, CO2H
PCT/US94/01881
WO 94/18981
22 -
HN~ N~` CO2H
~,N~ N I H `
H HNS02~CH3
HN~ ~ NI ~CO2H
H NH2
HN~ N ~'~N~co2H
l~,N - N
H
HO2C~N~ H NH
o
HN~ ~ ~N ,CO2H
`N H
HN~ N~N CO2H
H HNSO2C4Hg
HN~ N~ 2
NEt2
WO 94/18981 PCT/US94101881
~55~23
HN~ O
~ ~--N~f N CO2H
,,~ I HNSO2C4Hg
NEt2 o
HN O
--N ~--N~co2H
~ I
NC H
C 1--NH
HO2C ~N~~J
NC
HN O H
--N~--Nl--, CO2H
H H
W O 94/18981 P~CTrUS94/01881
24 -
HN~ `N~ N~CO2H
- ~ HNSO2G4Hg .
s H
H2N~ HNSO2~CH3
~N
H2N NH N~N~co2H
H HN S02~CH3
~N~ 0
NH H N~-J` Nl ~co2~
H HNS02~CH3
O ( ,~ C02H
H HN SO2~ , and
wo 94/18981 ~ I 5 ~ 1 ~ 3 PCTIUS94/01881
.
HN'~ 0
I~N _~N~co2H
H HNS02~CH3
Additional examples of compounds of the invention are
HN~ N~COOH CH3
HN~ N~COO Cl
HN~ `N CO2H
H HNS02~CH3
2s
HN~ 0
( ,~N CO,H CH3 ~,d
WO 94118981 . PCT/US94/01881
.
~,s'j~3 26-
HN'~ O
~N R~ ~CO2H
H HNSO2~CH3
The term "pharmaceutically acceptable salts" shall mean
o non-toxic salts of the compounds of this invention which are generally
prepared by reacting the free base with a suitable organic or inorganic
acid. Representative salts include the following salts: Acetate,
benzenesulfonate, benzoate, bicarbonate, bisulfate, bitartrate, borate,
bromide, calcium edetate, camsylate, carbonate, chloride, clavulanate,
citrate, dihydrochloride, edetate, edisylate, estolate, esylate, fumarate,
gluceptate, gluconate, glllt~m~te, glycollylars~nil~e, hexylresorcinate,
hydrabamine, hydrobromide, hydrochloride, hydroxynapthoate, iodide,
isothionate, lactate, lactobionate, laurate, m~l~te, maleate, m~ndelate,
mesylate, methylbromide, methylnitrate, methylsulfate, mucate,
20 napsylate, nitrate, oleate, oxalate, pamaote, palmitate, panthothenate,
phosphate/diphosphate, polygalacturonate, salicylate, stearate,
subacetate, succinate, t~nn~te, tartrate, teoclate, tosylate, triethiodide,
valerate.
The term "pharmaceutically effective amount" shall mean
25 that amount of a drug or pharmaceutical agent that will elicit the
biological or medical response of a tissue, system or ~nim~l that is being
sought by a researcher or clinician. The term "anti-coagulant" shall
include heparin, and warfarin. The term "thrombolytic agent" shall
include streptokinase and tissue pl~minogen activator. The term
30 "platelet anti-aggregation agent" includes, for example, aspirin,
ticlopidine, and dipyridamole.
The term "alkyl" means straight or branched alkane, alkene
or alkyne.
The term "aryl" means a S-10 membered lln~tllrated
mono- or bicyclic ring group.
WO 94/18981 ~ 1 ~ 5 ~ 2 3 PCT/US94/01881
- 27 -
The term "heteroaryl" means aryl cont~ining l, 2, 3 or 4
heteroatoms.
The term "heteroatom" means N, O, or S.
The terrn~"~yclic," unless otherwisermore specifically
5 defined, means mono- or bicyclic saturated ring groups having S-lO
members.
The term "heterocyclic" means cyclic cont~ining l, 2, 3 or
4 heteroatoms.
In the compounds of the invention, heteroaryl groups and
heterocyclic groups contain no more than 2 0 atoms or 2 S atoms.
The term "alkoxy" includes an alkyl portion where alkyl is
as defined above.
The term.s "arylalkyl" and "alkylaryl" include an alkyl
portion where alkyl is as defined above and to include an aryl portion
5 where aryl is as defined above. The Co-n or Cl n designation where n
may be an integer from l-lO or 2-lO respectively refers to the alkyl
component of the arylalkyl or alkylaryl unit.
The term "halogen" includes fluorine, chlorine, iodine and
bromine.
The term "oxy" means an oxygen (O) atom. The term
"oxo" means (= O). The term "thio" means a sulfur (S) atom. Under
standard nomenclature used throughout this disclosure, the terminal
portion of the designated side chain is described first followed by the
adjacent functionality toward the point of attachment. For example, a
25 Cl 5 alkyl substituted with Cl 6 aIkylcarbonylamino is equivalent to
O
C1 5alkyl --N--C C1 6alkyl
In the schemes and examples below, various reagent
symbols have the following me~nings:
W O 94/18981 PCTrUS94/01881
28-
BOC(Boc): t-butyloxycarbonyl.
Pd-C: palladium on activated carbon catalyst.
DMF: dimethylformamide.
0: ~ dimethylsulfoxide. -
CBZ: carbobenzyloxy.
CH2Cl2: methylene chloride.
CHC13: chloroform.
EtOH: ethanol.
- MeOH: methanol.
EtOAc: ethyl acetate.
HOAc: acetic acid.
BOP: benzotriazol- 1 -yloxytris(dimethylamino)-
phosphonium, hexafluorophosphate.
EDC: 1-(3-Dimethylaminopropyl)-3-ethyl-
carbodiimide
Oxone: potassium peroxymonosulfate
LDA: lithium diisopropylamide
DMA: N,N-Dimethyl~niline
HOBT: Hydroxybenzotriazole
20 Therapeutic Treatment
Compounds of the invention may be used for inhibiting
integrin protein-complex function relating to cell attachment activity.
They may be ~1mini~tered to patients where inhibition of hllm~n or
m~mm~ n platelet aggregation or adhesion is desired.
2s Certain compounds of the invention are elimin~ted from
circulation rapidly and are particularly useful in inhibiting platelet
aggregation. Thus, these compounds may find utility in surgery on
peripheral arteries (arterial grafts, carotid endaterectomy) and in
cardiovascular surgery where manipulation of arteries and organs,
and/or the interaction of platelets with artificial surfaces, leads to
platelet aggregation and consumption. The aggregated platelets may
form thrombi and thromboemboli. They may be ~ministered to these
surgical patients to prevent the formation of thrombi and
thromboemboli.
WO 94/18981 PCT/US94/01881
~ 2~ 3
- 29 -
The compounds of the present invention can be
a-lministered in such oral forms as tablets, capsules (each of which
includes sustained release or timed release formulations), pills, powders,
granules; elixers, tinctures, suspensi~ns, syrups, and emulsions.
Likewise, they may be ~lministered in intravenous (bolus or infusion),
intraperitoneal, subcutaneous, sublingual, intranasal or intramuscular
form, all using forms well known to those of ordinary skill in the
pharmaceutical arts. An effective but non-toxic amount of the
compound desired can be employed as an anti-aggregation agent.
Compounds of the invention may be a~lministered to
patients where prevention of thrombosis by inhibiting binding of
fibrinogen to the platelet membrane glycoprotein complex IIb/IIIa
receptor is desired. They are useful in surgery on peripheral arteries
(arterial grafts, carotid endarterectomy) and in cardiovascular surgery
where manipulation of arteries and organs, and/or the interaction of
platelets with artificial surfaces, leads to platelet aggregation and
consumption. The aggregated platelets may form thrombi and
thromboemboli. They may be ~lmi~i~tered to these surgical patients to
prevent the formation of thrombi and thromboemboli.
Extracorporeal circulation is routinely used for
cardiovascular surgery in order to oxygenate blood. Platelets adhere to
surfaces of the extracorporeal circuit. Adhesion is dependent on the
interaction between gpIIb/IIIa on the platelet membranes and fibrinogen
adsorbed to the surface of the circuit. (Gluszko et ah, Amer. J.
Physiol., 252(H), 615-621 (1987)). Platelets released from artificial
surfaces show impaired hemostatic function. Compounds of the
invention may be a~lmini~tered to prevent adhesion.
Other applications of these compounds include prevention
of platelet thrombosis, thromboembolism and reocclusion during and
after thrombolytic therapy and prevention of platelet thrombosis,
thromboembolism and reocclusion after angioplasty or coronary and
other arteries and after coronary artery bypass procedures. They may
also be used to prevent myocardial infarction.
WO 94/18981 PCT/US94/01881
3 30-
The dosage regimen utilizing the compounds of the present
invention is selected in accordance with a variety of factors including
type, species, age, weight, sex and medical condition of the patient; the
severity of the condition to be treated; the route of ~lministration~ the
5 renal and hepatic function of the patient; and the particular compound
or salt thereof employed. An ordinarily skilled physician or
veterinarian can readily determine and prescribe the effective amount of
the drug required to prevent, counter, or arrest the progress of the
condition.
Oral dosages of the present invention, when used for the
indicated effects, will range between about 0.01 mg per kg of body
weight per day (mg/kg/day) to about 100 mg/kg/day and preferably
0.05-100 mg/kg/day and most preferably 0.1-20 mg/kg/day.
Intravenously, the most preferred doses will range from about 1 to
about 10 ,ug/kg/minute during a constant rate infusion. Advantageously,
compounds of the present invention may be ~lmini~tered in divided
doses of two, three, or four times daily. Furthernnore, preferred
compounds for the present invention can be ~lmini~tered in intranasal
form via topical use of suitable intranasal vehicles, or via transdermal
20 routes, using those forms of transdermal skin patches well known to
those of ordinary skill in that art. To be ~lmini~tered in the form of a
transdermal delivery system, the dosage ~lmini~tration will, of course,
be continuous rather that intelmittent throughout the dosage regime.
In the methods of the present invention, the compounds
25 herein described in detail can form the active ingredient, and are
typically ~lmini.~tered in admixture with suitable pharmaceutical
diluents, excipients or carriers (collectively referred to herein as
"carrier" materials) suitably selected with respect to the intended form
of ~ mini~tration, that is, oral tablets, capsules, elixers, syrups and the
3 like, and consis~nt with convention pharmaceutical practices.
For instance, for oral ~lmini~tration in the form of a tablet
or capsule, the active drug component can be combined with an oral,
non-toxic, pharmaceutically acceptable, inert carrier such as lactose,
starch, sucrose, glucose, methyl cellulose, magnesium stearate,
WO 94/18981 PCT/US94/01881
2~S~2~
- 31 -
dicalcium phosphate, calcium sulfate, mannitol, sorbitol and the like; for
oral a~lministration in liquid form, the oral drug components can be
combined with any oral, non-toxic, pharmaceutically acceptable inert
carrier~such as ethanolj~glycerol, water and the like. ~oreover, when
desired or necessary, suitable binders, lubricants, disintegrating agents
and coloring agents can also be incorporated into the mixture. Suitable
binders include starch, gelatin, natural sugars such as glucose or beta-
lactose, corn-sweeteners, natural and synthetic gums such as acacia,
tragacanth or sodium alginate, carboxymethylcellulose, polyethylene
o glycol, waxes and the like. Lubricants used in these dosage forms
include sodium oleate, sodium stearate, magnesium stearate, sodium
benzoate, sodium acetate, sodium chloride and the like. Disintegrators
include, without limitation, starch methyl cellulose, agar, bentonite,
xanthan gum and the like.
The compounds of the present invention can also be
~lmini~tered in the form of liposome delivery ,systems, such a,s small
unilamellar vesicles, large llnil~mellar vesicles and multilamellar
vesicles. Liposomes can be formed from a variety of phospholipids,
.such as cholesterol, stearylamine or phosphatidylcholines.
Compounds of the present invention may also be delivered
by the use of monoclonal antibodies as individual carriers to which the
compound molecules are coupled. The compounds of the present
invention may also be coupled with soluble polymers as targetable drug
carriers. Such polymers can include polyvinlypyrrolidone, pyran
25 copolymer, polyhydroxy-propyl-methacrylamide-phenol, polyhydroxy-
ethyl-aspartamide-phenol, or polyethyleneoxide-polylysine substituted
with palmitoyl residues. Furthermore, the compounds of the present
invention may be coupled to a class of biodegradable polymers useful in
achieving controlled release of a drug, for example, polylactic acid,
30 polyglycolic acid, copolymers of polylactic and polyglycolic acid,
polyepsilon caprolactone, polyhydroxy butyric acid, polyorthoesters,
polyacetals, polydihydropyrans, polycyanoacrylates and cross linked or
amphipathic block copolymers of hydrogels.
WO 94/18981 PCT/US94/01881
~ . --
~55~3
The compounds of the present invention can al.so be co-
administered with suitable anticoagulants, including antiplatelet agent,s
such as heparin, aspirin, warfarin, dipyridamole and other compounds
and 2[gents kT~wrr t~ inhibit blood clot formation, a~nd ~hrombolytic
5 agents such as pl~minogen activators or streptokinase, to achieve
beneficial effects in the treatment of various vascular pathologies.
The novel compounds of the present invention were
prepared according to the procedure of the following examples. The
most preferred compounds of the invention are any or all of those
specifically set forth in these examples. These compounds are not,
however, to be construed as forming the only genus that is considered as
the invention, and any combination of the compounds or their moieties
may itself form a genus. The following examples further illustrate
details for the preparation of the compounds of the present invention.
Those skilled in the art will readily understand that known variations of
the conditions and processes of the following preparative procedures
can be used to prepare these compounds. All temperatures are degrees
Celsius unless otherwise noted.
In addition to the following preparative procedures, several
examples of in vitro bioactivity of compounds within the scope of the
present invention are indicated. To illustrate, one test which is used to
evaluate fibrinogen receptor antagonist activity is based on evaluation of
inhibition of ADP-stimulated platelets. Aggregation requires that
fibrinogen bind to and occupy the platelet fibrinogen receptor site.
Inhibitors of fibrinogen binding inhibit aggregation. In the ADP-
stirnulated platelet aggregation assay used to determine inhibition
associated with the compounds claimed in the instant invention, hllm~n
platelets are isolated from fresh blood, collected into acid
30 citrate/dextrose by differential centrifugation followed by gel filtration
on Sepharose 2B in divalent ion-free Tyrode's buffer (pH 7.4)
cont~ining 2% bovine serum albumin.
Platelet aggregation is measured at 37C in a Chronolog
aggregometer. The reaction mixture contains gel filtered human
platelets (2 x 108 per ml), fibrinogen (100 micrograms per ml (ug/ml))~
wo s4tl8s81 ~ ~ 5 51~ 3 PCT/US94/01881
Ca2+ (l mM), and the compound to be tested. The aggregation is
initiated by adding 10 mM ADP 1 minute after the other components
are added. The reaction is then allowed to proceed for at least 2
minutes. The extent of inhibition of aggregation i.s expreR,sed a~, the
percentage of the rate of aggregation observed in the absence of
inhibitor. The ICso is the dose of a particular compound inhibiting
aggregation by 50% relative to a control lacking the compound.
In the following examples, all temperatures are in degrees
Celsius, unless otherwise indicated.
SCHEME A
Preparation of sulfonamide intermediate compounds
~CONH2 NaOH
H20/dioxane
H2N~_c02H n-C4HgSO2CI
~9~
~CONH2
(C4H9)so2NH ~- C02H
1. Br, NaOH
2. Boc20, NaOH NH Boc
(C4H9)so2NH H C02H
A-3
L-Asparagine-a-butanesulfonamide (also N-(n-Butyl-sulfonyl)-L-
aspara~ine) A-2
O 94/18981 PCT/US94/01881
,; ' '
5~ ~3 34 -
A solution cont~inin~ L-asparagine (~.45 g, 4~.9 mmol)
and NaOH (2.0 g, 50.0 mmol) in 100 ml of 50% aqueous dioxane was
cooled to 0 in an ice bath. To this rapidly stirred mixture, a solution
of NaOH (2.2 g, 55.0 mmol) in 50 ml of water and neat butane sulfonyl
5 chloride (7~0 ml, 53.9 mmol) were added alternately over a period of
30 min. The reaction solution was concentrated to a volume of 50 ml at
reduced pressure and aqueous residue was cooled, acidified with
concentrated HCl, and extracted into ethyl acetate (3 x 100 ml). The
organic extracts were dried over Na2SO4 and concentrated to a volume
of approximately 50 ml, anhydrous ether (50 ml) was added and the
re.sulting white precipitate was isolated by vacuum filtration yielding
A-2, mp. 154-155.
-
L-,~ Boc-a-butane sulfonamido-,~ amino alanine (also 2(S)-(n-Butyl-
sulfonvlamino)-3-(N-Boc-aminopropionic acid) A-3
A solution cont~inin~ NaOH (6.04 g, 151 mmol) in 50 ml
H2O was cooled to 0 and bromine (1.40 ml, 26.~ mmol) was added.
The resulting solution was stirred at 0 for 5 min. Next, a cooled
solution of A-2 (5.23 g, 20.7 mmol) and NaOH (1.66 g, 41.4 mmol) in
15 ml of H2O was added at once and mixture stirred at 0 for 5 min
then heated to ~s0 for 15 min. The solution was then cooled to 25 and
s acidified with 12N HCl (11 ml) and stirred until gas evolution ceased.
The solution was then made basic by the addition of 2N NaOH and 20
ml of THF was added along with di-t-butyldicarbonate (9.0 g, 41.4
25 mmol). After stirring overnight at 25 the THF was removed at
reduced pressure and the basic aqueous phase extracted with ethyl
acetate (2 x 50 ml). The aqueous phase was ~en made acidic with 10%
KHSO4 and extracted with ethyl acetate (3 x 100 ml). The pooled
acidic extracts were dried over Na2S04 filtered and evaporated giving
3 A-3 as a white solid, mp 111-112.
WO 94/18981 PCT/US94/01881
t 2 ~;
HCI
NH Boc~ NH2
(C4H9)so2NH~- C02H (C4H9)SO2NH~- C02H
~ A-4
2(S)-(n-Butylsulfonvlamino)-3-aminopropionic acid (A-4)
A solution of A-3 (3.83 g, 11.8 mmol) in 200 ml of ethyl
acetate was cooled to 0, HCI gas was bubbled through the solution for 5
min. The solution was then warmed to 25 and stirred for 30 min then
~ concentrated at reduced pressure to 50% of its volume and diluted with
100 ml of ether. The resulting white solid was collected by vacuum
5 filtration giving A-4 as a solid.
Ethyl 2(S)-(n-Butylsulfonylamino)-3-amino propionate (A-5)
A solution of A-4 (1.0 g, 3.8 mrnol) in 50 ml of anhydrous
ethanol was saturated with HCl gas then heated at reflux for 3.0 h. The
20 solvent was evaporated to afford pure A-5 as a white solid.
1H NMR (300 MHz, CD30D) ~ 4.38 (m, 2H); 4,32 (m, lH); 4.23 (m,
2H); 2.85 (m, 2H); 1.65-1.45 (m, 4H); 1.3 (t, 2H); 0.96 (t, 3H).
WO 94/18981 PCTIUS94/01881
i ' . l ~;
., _
2,~$5~23 - 36 -
NaOH
H2N CO2H H20/dioxane
~- NH2 CH3~SO~CI
H2N ~H NHSO2~=~ CH3
9~
Dioxane ~CO2H ~CH3
2 H~NH-S02~ CH3
~
N-Tosyl-L-Asparagine (A-6): L-Asparagine (10.0 g, 75.7
mmol) was placed in a 500 ml round bottom flask equipped with a
30 magnetic stir bar and an addition funnel. lN Sodium hydroxide (85 ml,
1.1 eq.) was added. p-Toluenesulfonyl chloride (15.88g, 83.27 mrnol)
was dissolved in ethyl acetate (100 ml). This solution was added to the
reaction flask with vigorous stirring. lN Sodium hydroxide (85 ml,
1. leq.) was placed in the addition funnel, then added dropwise with
vigorous stirring over a 2 h period. The reaction mixture was stirred
WO 94/18981 2 I 5 51 2 3 PCTIUS94/01881
r
- 37 -
an additional 2 h, at room temperature. The organic and aqueous layers
were separated and the aqueous layer was washed with ethyl acetate
(2xS0 ml). The aqueous liquid was cooled to 0 then acidified with
hydrochloric acid (conc.). A white crystalline solid was obt:~ine-l
5 Recrystallization from hot water yielded A-6.
lH NMR (300 MHz DMSO-d6) o 7.91 (d, J=8.79 Hz, lH); 7.64 (d,
J=g.06 Hz, 2H); 7.32 (s, d (overlapping), J=~.06 Hz, 3H); 6.87 (s, br,
lH); 4.03 (m, lH); 3.32 (s, H2O); 2.49 (m, lH); 2.43 (d, d, J=7.08,
15.3~ Hz, lH); 2.35 (s, 3H); 2.21 (d, d, J=6.11, 15.3~ Hz, lH).
2(S)-To.sylamino-3-aminopropionic acid (A-7)
A solution cont~inin~ NaOH (22.0 g, 550 mmol) in 100 ml
H2O was cooled to 0 and bromine (5.03 ml, 97.5 mmol) was added
5 dropwise. The resulting solution was stirred at 0 for 10 min., then a
cooled (0) solution of A-6 (21.5 g, 75.0 mmol) and NaOH (6.68 g, 161
mmol) in H2~ (50 ml) was added in a single portion. After stirring at
0 for 20 min, the reaction was heated to ~s0 for 30 min, then cooled.
The cooled solution was adjusted to pH=7 with concentrated HCI and the
20 resulting white solid filtered to give A-7.
lH NMR (300 MHz, DMSO-d6) ~ 8.2-7.2 (br, 2H, (NH,COOH)); 7.70
(d, J=8.18 Hz, 2H); 7.38 (J=8.18 Hz, 2H); 3.7-3.0 (br, 2H, (NH2~
H2O)); 3.12 (q, J=4.76 Hz, lH); 2.99 (d, d, J=4.64, 11.96 Hz, lH); 2.79
(d, d, J=9.52, 11.96 Hz, lH); 2.36 (s, 3H).
tert-Butyl-2(S)-(Toluenesulfonylamino)-3-amino propionic acid (A-~)
A-7 (5.0 g, 19.4 mmol) was suspended in Dioxane (100 ml)
in a 1 liter pressure bottle. The bottle was cooled to -15C and
3 isobutylene (100 ml) was condensed into the dioxane. Concentrated
H2SO4 (5 ml) was added and the bottle sealed and stirred at room
temperature for 36 h. The bottle was opened, and the excess
isobutylene carefully vented. The solution was diluted with ethyl acetate
(200 ml) and washed with lN NaOH, (200 ml). The organic layer was
WO 94/18981 PCT/US94/01881
5~?*3
dried (Na2S04), filtered and evaporated to give A-~s a~ a white
crystaline solid.
1H NMR (300 MHz, CDC13) ~ 7.68 (d, J = 8.18 Hz, 2H); 7.35 (d, J =
8.18 Hz, 2H); 3.85 (m, H); 2.93-2.79 (m, 2H); 2.32 (s, 3H); 1.38 (s,
9H).
S~HEME 1
tBu02C~ tBu02C~¢~ Cl3COCOCCI3
N Cl toluene
1-1 1-2
tBu02C~
NJ~ N
o~N~H
1 -3
tert-Butvl 2-Hydrazinopvridine-3-carboxylate (1-2
A solution of tert-butyl 6-chloro-nicotinate (735 mg, 3.44
mmol) in ethanol (5 mL) was cooled to 0 and treated with anhydrous
30 hydrazine (2.75 g, 86 mmol) dissolved in ethanol (5 mL). This mixture
was allowed to reach 25 and stirred 20 h and then warmed to 60 for
2 h. The mixture was dissolved in water and extracted with.ethyl
acetate. The organic portion was washed with water and brine, dried
(Na2SO4), and concentrated to give 1 2 as an oil that was used directly
in the next step.
WO 94/18981 PCT/US94/01881
2~55~
- 39 -
tert-Butyl [2,3-Dihydro-3-oxo-1,4-triazolo-[4,3-a]pyridin-6-yl]-
carboxvlate ( 1-3)
1 2 was dissolved in 35 mL of toluene and slowly added to
a refluxing solution of bis(trichloromethyl)carbonate (1.15 g, 3.9
5 mmol) in toluene (35 mL). This was further refluxed for 1.2 h and
cooled, added to water and extracted with ethyl acetate. The organic
portion was dried (Na2S04), concentrated, and flash chromatographed
on silica gel to yield 1 3.
o lH NMR (DMSO-d6, 300 MHz): ~ = 1.58 (s,9H); 7.25 (d, lH); 7.45
(d,lH); g.25 (s, lH); 12.40 (s, lH).
ICH2CH2
1.
N tBuO2C ~
tBuO2C~¢~ 1-4 CBZl~N~l N
N N 2.TFA ,~N,
o~N~H (CH2)2 ~?
1-3 CBZ
1 -5
wo 94/18981 Pcr/uss4/olss1
, . . --
5~ ~3 - 40 -
o
HN3 CH2CH2oH CICOCH2Ph CB 3 CH2CH2OH
NaHCO3, CH2CI2
H2O
12, Ph3P
imidazole
benzene
r~
CBZ-N ~CH2CH21
20 2(N-CBZPiperidin-4-yl)ethyl iodide ( 1-4)
A mixture of [4-(2-hydroxyethyl)piperidine] (Aldrich)
(5.0 g, 3~.7 mmol), sat. NaHCO3 (50 ml) and CH2C12 (150 ml) was
treated with benzyl chloroformate (6.05 ml, 42.5 mmol). After stirring
at room temperature for 2.5 h, the organic layer was removed and
25 washed wi~ H20 and brine, dried (Na2S04), ~en filtered and
concentrated to give the protected alcohol as a colorless oil.
lH NMR (300 MHz, CDC13) ~ 7.26 (m, 5H); 5.13 (s, 2H); 4.23 (d, 2H);
3.63 (t, 2H); 2.85 (t, 2H); 1.83 (d, 2H); 1.68 (m, 2H); 1.43 (m, lH);
30 1-03 (m, 2H);
This protected alcohol (6.3 g, 23.9 mmol) was combined
with Ph3P (7.0 g, 23.9 mmol), iodine (6.06 g, 23.9 mrnol), and
imidazole (1.9~ g, 28.7 mmol) in benzene (100 ml) and refluxed for
WO 94/18981 PCT/US94/01881
~ ~155~ ~3
- 41 -
2.5 h. The solution was cooled, filtered and then concentrated. The
residue was chromatographed on silica gel (1:1 ethyl acetate/Hexane) to
give 1 4 as a white solid.
lH NMR (300 MHz, CDC13) â 7.26 (m, 5H); 5.14 (s, 2H); 4.23 (d, 2H);
3.86 (m, 4H); 1.85 (d, 2H); 1.68 (m, 2H); 1.45 (m, lH); 1.03 (m, 2H).
3-[[(2,3-dihydro-3-oxo-[2-(N-CBZ-Piperidin-4-yl)ethyl] - 1,2,4-
triazolor4.3-al pvridin-6-yllcarboxylic acid (1-5)
o 1-3 (350 mg, 1.6 mmol) dissolved in 20 ml of acetonitrile
was treated with powdered potassium carbonate (630 mg) and heated to
60 for 20 h. The mixture was cooled to 25 added to water and
extracted with ethyl acetate. The organic portion was dried (Na2SO4),
concentrated, and flash chromatographed on silica gel (35% ethyl
5 acetate in hexane) to give the desired ester as a yellow oil.
The crude tert-butyl ester was converted to the acid by
treating with 15 mL of methylene chloride and 15 mL of trifluoro-
acetic acid at 0 and then warming to 25 for 1.2 h. The mixture was
concentrated to dryness under vacuum, added to water and extracted
20 with ethyl acetate. The organic portion was dried (Na2SO4),
concentrated, and crystallized from ethylacetate/ether (25/1) to give 1 5
as a pale yellow powder.
lH NMR (DMSO-d6, 300 MHz): ~ = 1.05 (m, 2H); 1.45 (m, lH); 1.75
25 (m, 4H); 2.79 (m,2H); 3.95 (q, 4H); 5.05 (s, 2H); 7.30 (m, 6H); 7.50 (d,
lH); ~.25 (s, lH).
wo 94/18981 PCT~S94/01881
,
42 -
O
HOOC ~ ,
HO2C 1. HOBT, DIEA, EDC NH \~
2. LiOH N N
,~N THF/H20 (CH2)2
O (CH2)2 /
N
N CBZ
CBZ
1-~ 1-6
3-[[[(2,3-Dihydro-3-oxo-2[2-(N-CBZ-Piperidin-4-yl)ethyl]- 1 ,2,4-
15 triazolor4.3-alPyridin-6-yllcarbonyllaminolpropionic acid (1-6)
A solution of 1-5 (75 mgs, 0.1~ mmol) in dimethyl-
formamide (1 mL) was treated sequentially with hydroxybenztriazole
(40 mgs, 0.26 mmol), diisopropyl ethyl amine (87 ,ul, 0.5 mmol), ethyl
2-aminopropionate hydrochloride (40 mgs, 0.26 mmol), and EDC (50
20 mgs, 0.26 mmol). This mixture was stirred at 25 for 15 h. The
mixture was dissolved in water and extracted with ethyl acetate. The
organic portion was washed with water and brine, dried (Na2SO4), and
concentrated to provide the desired ester.
~his crude ethyl ester was dissolved in 5 mL of TH~, 5 mL
2s of water and treated with 0.37 mL of lN aqueous LiOH solution. This
was stirred 3 h at 25. The mixture was dissolved in water and
extracted with ethyl acetate. The organic portion was washed with
water and brine, dried (Na2SO4), and concentrated to a provide 1-6,
mp 201-203 (dec).
WO g4/18981 PCT/US94/01881
~ ~$`~ ~3
- 43 -
.
HO2C~ ~N ~ N-(CH2)2~NCBZ Me3Sil, CH3CN
s 1-6 ~
HO2C NH~ r-(CH2)2~NH
o 1-7
3-[[[(2,3-Dihydro-3-oxo-2-(2-(piperidin-4-yl)ethyl]- 1 ,2,4-triazolo-
r4.3-alpyridin-6-yllcarbonyllaminolpropionic acid (1-7)
A solution of 1 6 (64 mg, 0.129 mmole) in acetonitrile
(15 ml) at 0 was treated with iodotrimethylsilane (107.0 mg, 0.533
mmole) and the reaction stirred 0.5 h. The reaction was quenched into
water, extracted with diethyl ether and chromatographed on silica using
EtOH/NH40H/H20 (10/1/1) to give upon concentration a white foam.
20 Cryst~11i7~tion from ethanol gave 1 7 as a white solid, mp 267-269.
WO 94/18981 PCT/US94/01881
~S1 ~3
ClCO2i-Bu, Et3N
O CH2CI2
CBZN~,CH2)2-N~N - - ~co2H
1-5 A-4
o O Me3Sil
CBZN3(CH2)2-N N ~ ~6CO2H CH3CN
1-8
)~N ~ NH ~SCO2H
HN3(CH2)2-N~N - - ~d H- NHSO2C4Hg
1 -9
2s 2(S)-[6-Butylsulfonyl)amino]-3[[[2,3-dihydro-3-o~co-2-[2-(N-CBZ-
Piperidin4-yl)ethyl] - 1,2,4-triazolo[4,3-a]Pyridin-6-yl]carbonyl[amino
Propionic acid (l-f~,)
1 5 was coupled to A-4 as described for 1 6 to provide 1 8.
30 lH NMR (300 MHz, DMSO d6) ~ 8.26 (t, lH), 8.50 (s, lH); 7.63 (d,
lH); 7.54 (d, lH); 7.4-7.31 (m, 6H); 5.01 (s, 2H); 4.10 (m, lH); 3.96
(m, 4H); 3.60 (m, lH); 3.46 (m, lH); 2.95 (t, 2H); 2.73 (brm, 2H); 1.71
(m, 2H); 1.53 (m, 2H); 1.46 (m, lH); 1.01 (m, 2H); 0.~3 (t, 3H).
WO 94tl8981 PCT/US94/01881
~ 2~
- 45 - I i
2(S)-[(n-Butylsulfonyl)amino]-3[[[2,3-dihydro-3-oxo-2-[2-(piperidin-4-
yl)ethyl]-1,2,4-triazolo[4,3-a]Pyridin-6-yl]carbonyl]amino propionic
acid (1-9)
l-~s was treated with trimethylsilyl iodide in CH3CN as
5 described for 1-7 to afford 1 9.
lH NMR (300 MHz, D20) o ~.10 (s, lH); 7.43 (d, lH); 7.15 (d, lH);
4.00 (m, 4H); 3.60 (d, 2H); 3.31 (m, 2H); 3.23 (d, 2H); 3.93 (m, 2H);
3.~S1 (t, 2H), 1.93 (d, 2H); 1.~0 (m, 2H); 1.6-1.23 (m, 7H); 0.~5 (t, 3H).
SCHEME 2
HO2C~ CH30H CH,02C CH3
NBS; BP0 CH302C~
CCI4 N~ Br
Methyl 6-methylpyridine-3-carboxylate (2-2)
A solution of 6-methyl nicotinic acid, 2-1 (5 g, 36.5 mmol)
in 100 ml of anhydrous methanol was placed in a 250 ml three neck
30 flask equipped with a dro~pi.lg funnel vertical condenser and CaC12
drying tube. The reaction solution was cooled to -15 in an ice acetone
bath and SOC12 (5 ml, 69.1 mmol) was added dropwise. The solution
was then heated at reflux for 3 h then cooled and the solvent removed at
reduced pressure. The resulting white solid was treated with 60 ml of
saturated NaHCO3 and extracted into CH2Cl2 (3xS0 ml). The pooled
WO 94/18981 PCT/US94/01881
2~1f55~2~ - 46 -
extracts were dryed (Na2S04 ), filtered and evaporated at reduced
pressure. The resulting oil crystallized on standing giving 2-2 as a
white solid.
lH NMR (300 MHz, CDCl3) ~ 9.05 (d, J = 1.4 Hz, lH); 8.0R (dd, J=1.4
and 6.8 Hz); 7.19 (d, J=6.8 Hz, lH); 3.98 (s, 3H); 2.63 (s, 3H).
Methyl 6-Bromoethylpvridine-3-carboxylate (2-3)
2-2 (10.6 g, 71.5 mmol) was combined with NBS (12.73 g,
71.5 mmol), 100 mg benzoyl peroxide and 200 ml CCl4 and refluxed
under an inert atmosphere for 18 h. The reaction solution was cooled,
filtered, and concentrated to a viscous orange oil which was flash
chromatographed on silica gel using 20% ethyl acetate in hexane giving
the desired pyridyl bromide 2-3.
lH NMR (300 MHz, CDC13) ~ 9.05 (d, J=1.4 Hz" lH); 8.08 (dd, J=1.4
and 6.8 Hz); 7.19(d, J=6.8 Hz, lH); 5.38 (s, 2H); 3.9~ (s, 3H).
2s
WO 94/18981 PCT/US94/01881
- ~ 2'~51~3
/--\
BOCN~ CH2CH2NH2
CH302C~,~ . ~L
NJ~ Br CH3CN
N~T/C2CH3 triphosgene
BOCN~-(CH2)2-NH ~ J~J DMA, toiuene
~
BocN ~ o
~ ~CO2CH3
o
Methyl 6-[2-(N-Boc-Piperidin-4-yl)ethylamino]-methylpyridine-3-
carboxylate (2-5)
A mixture of ~ (1.0 g, 4.34 mmol), 2-4 (2.16 g, 10.0
30 mmol) and K2CO3 (0.66 g, 4.4 mmol) in 100 ml of anhydrous CH3CN
was placed in a 250 ml flask and- refluxed for 3 h then cooled and
filtered. The filtrate was concentrated at reduced pressure and
chromatographed on silica gel using 10% CH30H/EtOAc as eluent to
afford ;~ as a yellow residue.
WO 94/18981 PCT/US~4/0188I
;
48 -
lH NMR (CDC13) ~ 9.18 (d, J=1.4Hz, lH); ~..15 (dd, J=1.4 and 6.~S Hz,
lH); 7.39 (d, J=6.8 Hz, lH); 4.08 (br d, J=12 Hz, 2H); 3.98 (s, 2 H);
3.95 (s, 3H); 2.75 (overlapping m, 6H); 1.78 (d, J=12.Hz, 2H); 1.5
(overlapping m, 4H); 1.4 (s, 9H); 0.98 (m, 2H).
Methyl l-(Chlorocarbonyl)-2,3-dihydro-3-oxo-2-~[2-(N-Boc-
piperidin-4-vl)ethyllimidazo~l,S-alpyridin-6-yllcarboxylate (2-6)
2-S (480 mg, 1.27 mmol) was dissolved in 50 ml of
0 toluene, N,N- dime~yl aniline (645 ml, 4.08 mmol) was added and the
solution cooled to 0. To this, a solution of triphosgene (1.13 g, 3.18
mmol) in 15 ml toluene was added dropwise over 30 min. The
solution was then heated to 100 for 1.5 h then cooled, washed twice
wi~ lN HCI, water and brine (50 ml of each), dried over Na2S04 and
evaporated giving 515 mg of a yellow crystalline, solid 2-6.
lH NMR (CDC13) ~ 8.82 (d, J=1.4Hz, lH); 8.25(d, J=6.8 Hz, lH); 7.91
(d, J=6.~. Hz, lH) (dd, J=1.4 and 6.8 Hz,l H; 4.08 (br d, J=12 Hz, 2H);
3.9~. (s, 2 H); 3.95 (s, 3H); 2.75 (overlapping m, 6H); 1.78 (d, J=12.Hz,
20 2H); l.S (over lapping m, 4H); 1.4(s, 9H); 0.98 (m, 2H).
BcN~L O~N CO2CH3 Et2NH-HCI
(CH2)2-N~ CH2CI2, NaHCO3
Cl~
0
BocN~L Q~N CO2CH3
(CH2)2-N,~
(Et)2
o
2-7
WO 94/18981 21~51 ~3 PCT/US94/01881
- 49 - ~ ,
Methyl l-[(N,N-Diethylamino)carbonyl]-2,3-dihydro-3-oxo-2-[[2-(N-
Boc-piperidin-4-yl)ethyl]-imidazo[1,5-a]pyridin-6-yl]carboxylate (2-7)
2-6 (0.3 g, 0.64 mmol) was dissolved in 100 ml of CH2C12
and diethylamine hydrochloride (105 mg, 0.97 mmol) was added along
with 50 ml of saturated NaHCO3 solution. This biphasic mixture was
stirred for 1 h, then the organic layer was separated and washed with
10% citric acid then brine (50 ml), dried over Na2SO4 and evaporated
giving 2-7.
H NMR (CDC13) ~ 8.45 (d, J=1.4Hz, lH); 7.15 (dd, J=1.4 and 6.8 Hz,
lH); 6.78 (d, J=6.8 Hz, lH); 4.08 (m, 4H); 3.95 (s, 3H); 3.51 (m, 4H);
2.75 (m, 3H); 1.78 (d, J=12.Hz, 2H); 1.5 (over lapping m, 4H); 1.4
(s, 9H); 1.1 (t, 6H); 0.98 (m, 2H).
tert-Butyl- 1 [[(N,N-diethylamino)carbonyl]-2,3-dihydro-3-oxo-2[2-(N-
Boc-piperidin-4-yl)ethyl]imidazo[1,5-a]Pyridin-6-yl]carbonyl]amino
propionate (2-8)
~ (315 mg, 0.64 mmol) was dissolved in 10 ml of
CH30H, 15 ml H2O and 0.725 ml lN NaOH added and mixture stirred
at room temperature for 3.5 h. The organic solvent was removed at
reduced pressure and the aqueous residue acidified with citric acid and
extracted with CH2C12. The organic extracts were washed with H20,
and brine then dried over Na2SO4 filtered and evaporated to give the
25 desired acid.
1H NMR (CDC13) ~ 8.32 (d, J=1.4Hz, lH); 7.10 (dd, =1.4 and 6.8 Hz,
lH); 6.78 (d, J=6.8 Hz, lH); 4.08 (m, 4H); 3.51 (m, 4H); 2.75 (m, 3H);
1.78 (d, J=12.Hz, 2H); 1.5 (overlapping m, 4H); 1.4 (s, 9H); 1.1
(t, 6H); 0.98 (m, 2H).
wo 94/18981 PcT/uss4/0188
50 -
BocN ~ ~N~co2cH3
~ (CH2)2-N~
s (Et)2N~ 1. NaOH, CH30H, H20,
2. ClCO2-i-Bu, Et3N
HCI-H2N ~ CO2tBu
2-7 CH2CI
~N N~CO2tBu HCI
BocN3(CH2)2-N>~ EtOAc
0~
~ NEt2
HN~(CH2)2- ~N, CO2H
(Et)2N
o
;2~
This acid (105 mg, 0.215 mmol) was dissolved in 10 ml of
CH2C12, Et3N (48ml, 0.47 mmol) was added and solution cooled to
-10. Ne~t, isobutyl chloroformate (30 ml, 2.36 mmol) was added and
the mixture stirred at -10 for 30 min. To this a solution of ,B-alanine
tert-butyl ester hydrochloride (58.7 mg, 323 mmol) and Et3N (32 ml,
0.323 mmol) in 10 ml of CH2C12 was added and solution warmed to
room temperature. Reaction solution washed with 10% citric acid, H20
WO 94118981 . PCT/US94/01881
~ 5~23
- 51 -
and brine (10 ml each) and dried over Na2SO4, concentrated and
chromatographed giving 2-~s.
lH NMR (CDC13) o ~S.16 (s, lH~; 7.02 (d~ J=6.~s Hz, lH); 6.7~ (d, J=6.8
Hz, lH); 6.63 (t, 5.6Hz, lH); 4.08 (m, 4H); 3.51 (m, 6H); 2.75 (m, 2H);
2.45 (m, 2H); 1.78 (d, J=12.Hz, 2H); 1.5 (overlapping m, 6H); 1.4 (s,
9H); 1.37 (s, 9H); 1.1 (t, 6H); 0.98 (m, 2H).
l -[(N ,N-Diethylamino)carbonyl] -2,3-dihydro-3-oxo-2[2-(piperidin-4-
yl)ethyllimidazo~l.5-alpvridin-6-yllcarbonYllpropionic acid (2-9)
2-g (100 mg, 0.16 mmol) was dissolved in 20 ml of ethyl
acetate anhydrous HCl was passed through the solution at 0C for 5 rnin
then the mixture was stirred at room temperature for 1 h. The solvent
was evaporated at reduced pressure and the residue triturated with ethyl
acetate and filtered to give 2-9.
lH NMR (DMSO-d6) ~ 8.90 (br s, lH); 8.60 (br s, lH); 8.3 (s, lH);
7.6 (t, 3H); 7.1 (d, lH); 6.89 (d, lH); 4.08 (m, 4H); 3.51 (m, 6H); 2.75
(m, 2H); 2.45 (m, 2H); 1.7~ (d, J=12.Hz, 2H); 1.5 (overlapping m, 6H);
1.1 (t, 6H); 0.9~ (m, 2H).
HN3CH2CH2oH BC2. BOCN3CH2CH2OH 2 3.
NaOH, imidazole
dioxane benzene
2-10 2-1 1
BOCN/~CH2CH21 1. NaN3, Dl~/ISO /~\
BOCN ~ CH2CHzNH2
2. Ph3P, THF
2-12
WO 94/18981 PCT/US94/018gl
~'' ''
52 -
2-(N-Boc-Piperidin-4-vl)ethanol (2- 11)
4-Piperidine-2-ethanol (2- 10) (Aldrich) (130 g, 1.0 mole)
was dissolved in 700 mL dioxane, cooled to 0 and treated with 3 N
NaOH (336 mL, 1.0 mole), and di-t-butyldicarbonate (221.8 g, 1.0
mole). The ice bath was removed and the reaction stirred overnight.
The reaction was concentrated, diluted with water and extracted with
ether. The ether layers were combined, washed with brine, dried over
MgSO4, filtered and evaporated to give 2-11.
Rf = 0.37 in 1:1 EtOAc/Hexances, ninhydrin stain~
lH NMR (300 MHz, CDC13) ~ 4.07 (bs, 2H); 3.7 (bs, 2H); 2.7 (t, J =
12.5 Hz, 2H); 1.8-1.6 (m, 6H); 1.51 (s, 9H); 1.1 (ddd, J = 4.3, 12.5, 12
~ Hz, 2H).
5 2-(N-Boc-Piperidin-4-yl)ethyl iodide (2-12)
2-11 (10.42 g, 0.048 mol) was dissolved in 400 ml benzene,
imidazole (4.66 g, 0.068 mol), triphenylphosphine (15.24 g, 0.05 mol)
and iodine (0.048 mol) were added at room temperature. After 6 hours
20 the reaction mixture was filtered and the filtrate was evaporated to give
a dark residue. This was purified by flash chromatography on silica gel
eluting with 10% EtOAc-hexanes to give 2-12 as a yellow oil.
2-(N-Boc-Piperidin-4-yl)ethyl amine (2-4)
To 2-12 (27.9 g, 0.0~s2 moles) dissolved in DMSO (400 ml)
was added sodium azide (5.01 g, 0.086 moles) as room temperature and
the resulting solution was heated at 65 for 2 h. The cooled reaction
mixture was ~ te-l with 250 ml EtOAc, extracted with 2 x 100 ml
portions of water 2 x 50 ml portions of brine and then dried (MgS04).
Solvent removal provided the desired azide as a pale yellow oil, Rf 0.5
(silica gel, 70% acetone/hexane).
This azide (19.3 g, 0.076 moles) in THF (400 ml)lH2o
(195 ml) was added triphenylphosphine (~0.0 g, 0.305 moles) in one
portion at room temperature. This was stirred at room temperature 3
WO 94/18981 . PCT/US94/01881
~ 2~5~
-- 5 3 5
hours and the organic solvents were then removed in vacuo. The
residue was acidified to pH 2 with 10% KHS04 solution and this was
extracted 4 x 100 ml portions of EtOAc. The organic extract was
extracted with 2 x 100 mol portions of 10% KHS04 and the aqueous
5 phases were combined and the pH was adjusted to 10 with 2N NaOH.
This solution was extracted with 4 x 200 ml portions of CH2C12. These
were combined, dried (MgS04) and the solvent was removed to give
2-4 as an oil. Rf 0.3 (silica gel, eluting with 10% CH30H in
CHC13/NH3).
1 H NMR (300 MHz, CDC13) ~ 4.05 (broad, 2H); 2.72 (t, J = 7.2 Hz,
2H); 2.62 (m, 2H); 1.64 (d, J = 12.2 Hz, 2H); 1.43 (s, 9H); 1.42-1.32
(m, 5H); 1.09 (m, 2H).
SCHE~E 3
CO2H
~,N 1. HCI, CH30H (3-1a) C02CH3
20HO2C Nl 2. K2CO3, CH3CN ,¢~;
H Br2cH2cH2Br H3C02C N
3-1 3-2
Dimethvl 1-(2-Bromoethvl)pvrazole-3.5-dicarboxylate (3-2)
A solution of pyrazole-3,5-dicarboxylic acid (75 g, 431
30 mmol) in 11 of anhydrous methanol was treated with anhydrous HCI
gas. The HCI addition was continued for 30 min after which, the
solution was allowed to cool to room temperature and allowed to stand
for 16 h. The solution was then heated at reflux for 3 h then cooled and
the solvent removed at reduced pressure. The resulting white solid was
WO 94/18981 PCT/US94/01881
,
~ 3 c,4
treated with 600 ml of saturated NaHCO3 and extracted into CH2C12
(3xS00 ml). The pooled extracts were dried (Na2S04), filtered and
evaporated at reduced pressure. The resulting white solid was
recrystalized from methanol with the addition of anhydrous ether to
5 give dimethyl pyrazole-3,5-dicarboxylate (3- 1 a).
H NMR (CDC13) ~ 7.38 (s, lH); 3.98 (s, 3H); 3.93 (s, 3H).
A solution of this ester (5.0 g, 27.2 mmol) in 150 ml of
anhydrous acetonitrile was treated with K2co3 (5.2 g, 40.0 mmol) and
1,2-dibromoethane (25.0 ml, 291 mmol). The resulting mixture was
heated to reflux under argon. After 25 min the reaction suspension was
cooled, filtered and the filtrate evaporated to dryness at reduced
pressure and placed on a high vacuum line for 12 h. The resulting
white solid was reclystalized from hexane to give 3-2 as a white solid.
lH NMR (CDC13) ~ 7.38 (s, lH); 5.03 (t, J=8.2 Hz, 2H); 3.98 (s, 3H);
3.93 (s, 3H); 3.75 (t, J=8.5 Hz, 2H).
CO2CH3
~N r~
H3CO2C ~N BocN~ CH2CH2NH2
2-4
2s ) DIEA, Kl
Br CH3CN
3-2
0
BocN~ (CH2)2-N ~ CO2H
WO 94/18981 PCT/US94tO1881
. . ~
215512~
- 55 -
, ,~,, .
Methyl-[4,5,6,7-Tetrahydro-4-oxo-5-[2(N-Boc-Piperidin-4-yl)ethyl] -
pyrazolo~l.S-alpyrazin-2-yllcarboxylate (3-3)
A solution of ~ (14.0 g, 48.0 mrnol), diisopropylethyl
amine (25 ml, 144 mmol), Boc-4-aminoethylpiperidine ~12.0 g~ 52.6
mrnol), and potassium iodide (2.39 g, 0.3 mrnol) in 250 ml CH3CN was
refluxed under N2 for 4.5 h then cooled, filtered and evaporated at
reduced pressure. The resulting yellow residue was chromatographed
on silica gel using EtOAc as eluent to give 3-3 as an off-white
crystalline solid.
lH NMR (300 MHz, CDC13) o 7.15 (s, lH); 4.29 (t, J=7.0 Hz, 2H);
3.93 (br d, J=12 Hz, 2H); 3.76 (s, 3H); 3.61 (t, J=5.3 Hz, 2H); 3.42 (t,
J=7.3 Hz, 2H); 2.65 (t, J=7.6 Hz, 2H); 1.55 (d, J=12.5 Hz, 2H); 1.38 (m,
2H); 1.33-1.25 (m, lH); 1.27 (s, 9H); 1.01 (m, 2H).
A solution cont~ining LiOH (14.05 mg, 0.335 mmol) in 10
ml H2O was added to a solution of 3-3 (90.81 mg, 0.223 mmol) in 10
ml CH30H and the mixture was heated to 60 for 2.5 h then cooled, and
the CH30H removed at reduced pressure. The rem~ining aqueous phase
was acidified with 10% aqueous citric acid and extracted with CH2C12
(2 x 50 ml). The pooled organic extracts were dried over Na2SO4 then
evaporated to give the desired acid as a white solid.
H NMR (300 MHz, CDC13) ~ 7.43 (s, lH); 4.48 (t, J=7.0 Hz, 2H); 4.01
(br d, J=12 Hz, 2H); 3.77 (t, J=5.3 Hz, 2H); 3.51 (t, J=7.3 Hz, 2H); 2.71
(t, J=8.3Hz, 2H); 1.72 (d, J=12.5Hz, 2H); 1.53 (m, 2H); 1.42-1.37 (m,
lH); 1.35 (s, 9H); 1.10 (m, 2H).
WO 94/18981 PCT/US94/01881
t
.
~s~?~3 - 56 -
BocN 1. LiOH, CH30H, H20
<~ 2. ClCO2-i -Bu, NMM, THF
(cH2)2 H2N~co2H
\ ~o H NHSO2C4Hg
~ N~40
3-3 OH
BocN ~
<
(C~H~
NH
CO2H
N HSO2C4Hg
3-4
2(S)-[(n-Butylsulfonyl)amino-3[[[4,5,6,7-tetrahydro-4-oxo-5-[2-(N-Boc-
piperidin-4-yl)ethyl]pyrazolo-[ 1 ,S-a]pyrazin-2-yl]carbonyl]amino
30 propionic acid (3-4)
Isobutyl chloroformate (1.75 ml, 13.35 mmol) was added
to a cooled solution (0) cont~ining this acid (4.98 g, 12.72 mmol) and
N-methyl morpholine (1.53 ml, 14.00 mmol) in 100 ml THF. This
mixture was stirred under an atmosphere of dry nitrogen. After
reacting for 1 h, HPLC analysis of an aliquot indicated that the reaction
WO 94/18981 PCT/US94/01881
~l$512~-
- 57 -
was >90% complete. The N-methyl morpholine-HCI was removed by
filtration and the filtrate poured into a solution cont~ining A-4 (4.30 g,
16.54 mmol), diisopropylethylamine (4.27 ml, 33.10 mmol) THF (60
ml) a-nd H20 (20 ml). The T~IF was then removedlfrom the reaction
5 solution at reduced pressure and the rem~ining aqueous portion
acidified with sat. KHSO4 and extracted with ethyl acetate (3 x 200 ml).
Pooled extracts were dried over Na2SO4, filtered, and concentrated
giving a red colored oil from which 3-4 formed as a white solid.
H NMR (DMSO-d6) ~ 8.31 (t, J=6Hz, lH); 7.62 (d, J=8.5 Hz, lH);
7.01 (s, lH); 4.43 (t, J=6.6 Hz, 2H); 4.11 (m, lH); 3.92 (d, J=12 Hz,
2H); 3.80 (t, J=6.6 Hz, 2H); 3.51 (t, J=7.3 Hz, 2H); 3.65 (m, 2H); 3.51
(t, J 6.8 Hz, 2H); 2.96 (t, J=7.2 Hz, 2~I); 1.70 (d, J=l l Hz, 2H); 1.53 (m,
2H); 1.60-1.49 (overlapping m, SH); 1.40 (s, 9H); 1.28 (q, J=7.1Hz,
2H); 1.05 (m, 2H); 0.79 (t, J=7.1Hz, 3H).
2(S)-[(n-Butylsulfonyl)amino]-3-[[[4,5,6,7-tetrahydro-4-oxo-5-{2-
(piperidin-4-yl)ethyl]pyrazolo[1,5-a]pyrazin-2-yl]carbonyl]amino
propionic acid monohydrochloride (3-5)
25HN~(CH2)2-N~H HNSO2C4Hg
3-~
30A solution of ~ (278 mg, 0.437 mmol) in 30 ml ethyl
acetate was cooled to 0 and HCl gas bubbled through for 3 min. The
reaction mixture was warmed to room temperature, stirred for 30 min
then taken to dryness on a rotary evaporator. The rem~ining white
solid was recrystalized from ethanol/water (90:10) filtered and vacuum
dried over P2O5 giving ~ as a white solid.
WO 94/18981 PCT/US94/01881
~3
lH NMR (DMSO-d6) ~ 8.95 (br s, lH); 8.33 (t, J=5.7 Hz, lH); 7.64 (d,
J=9 Hz, lH); 7.02 (,s, lH); 4.35 (t, J=5.1 Hz, 2H); 4.10 (m, lH); 3.~1 (t,
J=5.2 Hz, 2H); 3.6-3.4 (m, 4H); 3.21 (d, J=10.5 Hz, 2H); 2.95 (t, J=7.
Hz, 2H); 2.~1 (br m, 2H); 1.96 (d, J=l lHz, 2H); 1.62-1.2 (overlapping
multiplets, 9H); 0.80 (t, J=7.3Hz, 2H).
SCHEME 4
--6NHCBZ CHqOH HCI-H2N~6NHCBZ
H HCI
4-1 4-1 a
Methyl 2(S)-N-Benzyloxycarbonylamino-3-aminopropionate
hydrochloride (4-1 a)
Commercially available 2(S)-N-benzyloxycarbonylamino-
20 3-aminopropionic acid (Fluka) was refluxed in methanolic HCl for 2.5 h
then evaporated and the residue crystalized from methanol/ether to give
4-la as a white solid.
1H NMR (300 MHz, DMSO-d6) ~ 7.63 (m, SH); S.93 (d, lH); 5.15 (s,
25 2H); 4.56 (m, lH); 3.95-3.~3 (m, 2H); 3.73 (s, 2H).
WO 94/18981 PCT/US94101881
2~.5~12~
59
BocN~ (C H2)2-N ~ C02H H2N ~C02C H3
N-N NHCBZ
3-3
O O
BocN~ (CH2)2-N ~ NH ~ z H2, Pd/C
CH30H
4-2
lS
BocN ~
(CH2)2-N~NH/~cO2cH~
4-3
2s
Methyl-2(S)-[(CBZ)amino]-3[[[4,5,6,7-tetrahydro-4-oxo-5-[2(N-Boc-
piperidin-4-yl)ethyl]pyrazolo[ 1 ,5-a]pyrazin-2-yl]carbonyl]amino]-
propionate (4-2)
A solution of 3-3 (5.6 g, 14.0 mmol), Na-Cbz-L-2,3-
30 ~ minopropionic acid methyl ester hydrochloride (4-la) (4.5 g, 15.5
mmol), HOBT (2.37 g, 15.5 mmol), and Et3N (4.1 ml, 29.5 mmol~ in
65 ml anhydrous DMF was stirred under N2 for 48 h at room
temperature. The DMF removed at reduced pressure and the residue
dissolved in 700 ml ethyl acetate and washed successively with saturated
NaHCO3 solution, H2O, 10% citric acid, H2O and brine (1 x 100 ml
W O 94/18981 P~CTrUS94/01881
60 -
each), dried over Na2SO4, filtered and evaporated. The resulting clear
glass was chromatographed on silica gel using 3% CH3OH/CH2C12 as
eluent to yield pure 4-2 as a white solid.
lH NMR (CDC13) ~ 7.43 (m, SH); 7.35 (s, lH); 7.18 (t, J=6.5 Hz, lH);
5.98 (d, J=6.8 Hz, lH); 5.09 (s, 2H); 4.59 (m, lH); 4.38 (m, 2H); 4.10
(br d, J=12 Hz, 2H); 3.8 (s, 3H); 3.73 (t, J=5.3 Hz, 2H); 2.71 (t, J=8.3
Hz, 2H); 1.72 (d, J=12.5 Hz, 2H); 1.53 (m, 2H); 1.42-1.37 (m, lH); 1.35
o (s, 9H); 1.10 (m, 2H)
Methyl-2(S)amino-3-~[[4,5,6,7-tetrahydro-4-oxo-5 -[2(N-Boc-piperidin-
4-vl)ethyllpyrazolorl.5-alpyrazin-2-yllcarbonyllaminolpropionate (4-3)
To 4-2 (6.3 g, 10.26 mmol) in 700 ml CH30H was added
650 mg 10% Pd on C and the resulting mixture stirred under 1 atm of
H2 for 48 h. The catalyst was removed by filtration through celite and
the filtrate concentrated to give a colorless glass which was trihlrated
with Et2O and filtered to afford 4-3 as a white solid.
1H NMR (300 MHz, CDC13) ~ 7.43 (m, 5H); 7.3S (s, lH); 7.18 (t, J=6.5
Hz, lH); 5.98 (d, J=6.8 Hz, lH); 5.09 (s, 2H); 4.59 (m, lH); 4.38 (m,
2H); 4.10 (br d, J=12 Hz, 2H); 3.81 (s, 3H); 3.73 (t, J=5.3 Hz, 2H); 2.71
(t, J=8.3 Hz, 2H); 1.72 (d, J=12.5 Hz, 2H); 1.53 (l[n, 2H); 1.42-1.37 (m,
lH); 1.35 (s, 9H); 1.10 (m, 2H).
WO 94/18981 2 3~51 2~ PCTIUS94/01881
.
- 61 -
BocN ~
BocN~\
Ac20 ~_(
(C~H2)2... ~ ~
~ N~ ~CO CH
lo H NH2 ll
o
4-4
4-3
5 Methyl-2(S)-(Acetylamino)-3-[[[4,5,6,7-tetrahydro-4-oxo-5-[2-(N-Boc-
piperidin-4-yl)ethyl]pyrazolo[ 1 ,5-a]pyrazin-2-yl]carbonyl]amino]-
propionate (4-4)
Acetic anhydride (70 ml, 0.76 mrnol), was added to a
cooled (0) solution of 4-3 (350 mg, 0.69 mmol) in 10 ml THF. The
20 resulting solution was allowed to warm to room temperature and stirred
for 1~ h, then concentrated, and the residue was dissolved in 50 ml ethyl
acetate and washed successively with NaHCO3, H2O, 10% KHSO4,
H20, and brine (25 ml each). The organic layer was dried over
Na2SO4 and evaporated giving a colorless residue which was
25 chromatographed on silica gel with 3% CH3OH/CH2C12 to yield 4-4 as
a white solid.
lH NMR (CDC13) ~ 7.29 (s, lH) 7.24 (t, J=6.4 Hz, lH); 6.gl (d, J=7.6
Hz, lH); 4.79 (m, lH); 4.3~ (m, 2H); 4.10 (br d, J=12 Hz, 2H); 3.~1
30 (s, 3H); 3.~0 (m, 2H); 3.73 (t, J=5.3 Hz, 2H); 2.71 (t, J=~S.3Hz, 2H);
2.01 (s, 3H); 1.72 (d, J=12.5 Hz, 2H); 1.57 (m, 2H); 1.42-1.37 (m, lH);
1.37 (s, 9H); 1.09 (m, 2H).
WO 94/18981 `. . - . PCT/US94/01881
.
5~3 - 62-
2-(S)-(Acetylamino)-3-~[[4,5 ,6,7-tetrahydro-4-oxo-5 -[2-(piperidin-4-
yl)ethyl]pyrazolo[l,5-a]pyrazin-2-yl]carbonyl]amino propionic acid
(4-5)
A solution of 4-4 (203 mg, 0.38 mmol), 1 N LiOH (0.76
ml, 0.76 mmol), H20, CH30H, and THF (5 ml each) was stirred
overnight at room temperature. The organic solvents were removed at
reduced pressure and the rem~ining solution was diluted with 25 ml
H2O, made acidic with 10% KHSO4, and extracted into ethyl acetate.
The e~yl acetate was washed with H20 and brine, dried over Na2S04,
filtered and evaporated to provide the desired acid.
lH NMR (CDC13) ~ 7.93 (br, 1 H); 7.81 (br, 1 H); 7.29 (s, lH); 4.79
(m, lH); 4.348 (m, 2H); 4.10 (br d, J=12 Hz, 2H); 3.80 (br m, 2H);
3.73 (br, t, 2H); 2.71 (t, J=8.3Hz, 2H); 2.11 (s, 3H); 1.72 (d, J=12.5 Hz,
2H); 1.57 (m, 2H); 1.42-1.37 (m, lH); 1.37 (s, 9H); 1.12 (m, 2H).
H~
BocN~\ 1. LiOH,CH30H/H20~ 0
<~ 2 HCI, EtOAc ~_~ ~
2s ~N~O C H H NH IClCH3
NH ~ 02C 3 4-5
H NHlclcH3
3 o 4-4
This acid (169 mg, 32.~s mmol) was dissolved in 50 ml
ethyl acetate was cooled to 0 and treated with dry HCl for 30 min.
wo 94/18981 2 1 ~ ~ 2 3 PCT/USg4/01881
. ~
- 63 -
, f
The solvent was removed in vacuo and the residue triturated with
anhydrous ether, filtered and dried over P2O5, to give 4 5 as a white
solid, mp 150-156.
H~N ~6C2H
H NHcBz
4-1
BocN3 (CH2)2-N~ /~6NHCBZ
4-6
O
1. H2, Pd/C~ ,JI
HN ~(CH2)2-N ~\ J~NH ~,CO2H
2. HCI/EtOAc\~ ~N_N/~ H NH2
4-7
2(S)-[(Cbz-Amino)]-3-[[[4,5,6,7-tetrahydro-4-oxo-5-[2(N-Cbz-
piperidin-4-yl)ethyl]pyrazolo[ 1 ,5-a]pyrazin-2-yl]carbonyl]amino
25 propionic acid (4-6)
4-3 was coupled to Na-CBZ-L-2,3-~ mino-propionic acid
(Fluka) (~) using the procedure described for 3-4 to provide 4-6, the
doubly protected adduct.
30 2(S)-Amino-3-[[[4,5,6,7-tetrahydro-4-oxo-5-[2-(piperidin-4-yl)-
ethyllpyrazolorl~5-alpvrazin-2-vllcarbonvllamino propionic acid
Treatment of 4-6 with H2 in the presence of Pd/C gave the
desired acid, mp. 157. The Boc group was then removed with
HCl/EtOAc in standard fashion to give pure 4-7, mp. 195-19~s.
WO 94/18981 PCT/US94/01881
3 - 64 -
BocN~ (CH2kN~ NH /~CO2cH3 CISO2C4Hg
~N-N H NH2 NMM, THF
BocN3(CH2)2-N~NH ~C:02CH3
~N-N H NHSO2c4Hs
lS 4~~
Methyl 2(S)-[(n-Butylsulfonylamino)]-3-[[[4,5,6,7-tetrahydro-4-oxo-5-
[2-(N-CBZ-piperidin-4-yl)ethyl]pyrazolo[1,5-a]pyrazin-2-yl]carbonyl]-
20 aminopropionate (4 8)
A solution of 4-3 (0.30 g, 0.61 mmol), n-butyl sulfonyl
chloride (0.16 g, 0.91 mmol), and N-methyl morpholine in 50 ml of
THF was stirred at room temperature for 12 h. The solvent was
evaporated at reduced pressure and ~e resulting oil was dissolved in
25 CH2C12 (50 ml) washed with 10% KHSO3 (50 ml) then dried over
Na2S04, filtered and evaporated. The resulting residue was
chromatographed on silica gel giving 4-Ps as a colorless glass.
lH NMR (CDC13) ~ 8.31 (t, J = 6Hz, lH); 7.62 (d, J = 8.5 Hz, lH); 7.01
30 (s, lH); 4.43 (t, J = 6.6 Hz, 2H); 4.11 (m, lH); 3.92 (d, J = 12 Hz, 2H);
3.~s3 (s, 3H); 3.~S0 (t, J = 6.6 Hz, 2H); 3.51 (t, J = 7.3 Hz, 2H); 3.65 (m,
2H); 3.51 (t, J = 6.8 Hz, 2H); 2.96 (t, J = 7.2 Hz, 2H); 1.70 (d, J = 11
Hz, 2H); 1.53 (m, 2H); 1.60-1.49 (overlapping m, SH); 1.40 (s, 9H);
1.2~ (q, J = 7.1 Hz, 2H); 1.05 (m, 2H); 0.79 (t, J = 7.1 Hz, 3H).
WO 94/18g81 PCTJUS94/01881
2i~` ~;5 ~
- 65 -
SCHEME S
CH302C~BrCH2CH2CH2Br ~ CO2CH3
N~ N
Br
3-1 a 5-1
l oNaN3, DMSO ~ CO2CH3
25, 5 hr ,J-- 1. H2, Pd/C, EtOH
N3 2. C6H6, reflux
5-2
HN ~ CO2CH3
20(~N--N// BocN~ CH2CH2i
5-4
5-3 NaH, DMF
O
BocN3(cH2)2-N~co2cH3 NaOH, CH30H, H20
~N--N
5-5
~CTrUS94101881
W O 94/18981
- 66 -
~S~3
SCHEME 5 (CONT'D)
O o HOBT, Et3N, CH2C12
BocN3 (CH2)2-~ H N ~6CO2C2H6
H N HSO2C4Hg
5-6 A-5
O O
~ocN~(CH2)2~~ NH /~CO2C2Hs
1. NaOH, CH30H, H20
2. HCI, EtOAc
O O
HN~}(CH2)2-N~D~ ~6NHSO2C4Hg
5-8
WO 94/18981 PCT/US94/01881
~l~5123
- 67 -
Dirnethyl 1-(3-Bromopropyl)pvrazole-3.5-dicarboxylate (5-1)
Compound 5-1 was obtained as a white crystalline solid
using 1,3-dibromopropane in the procedure described for 3-2.
lH NMR (CDC13) o 7.38 (s, lH); 4.95 (t, J=8.2Hz, 2H); 3.95 (s, 3H);
3.92 (s, 3H); 3.75 (t, J=8.5 Hz, 2H) 2.51 (m, 2H).
Dimethyl 1-(3-Azidopropyl)pyrazole-3~5-dicarboxvlate (5-2)
A solution of 5-1 (1.0 g, 3.45 mmol) in 10 ml DMSO was
treated with NaN3 (0.883 g, 13.~ mmol) and mixture stirred at 25C
for 5 h. Next, the reaction mixture was diluted with 100 ml of H2O and
then extracted with ethyl acetate (3x100 ml). The combined organic
extracts were washed with water (2x100 ml) and brine (1 x100 ml),
dried over Na2SO4 and evaporated to give 5-2 as a colorless oil.
Methyl-5,6,7,8-tetrahydro-4-oxo-4H-pyrazolo[1,5-a] [1,4]diazepin-2-yl]-
carboxylate (5-3)
A solution of 5-2 (851 mg, 3.25 mmol) in 100 ml absolute
EtOH was treated with 100 mg 10% Pd on C and the mixture was
20 shaken on a Parr hydrogenator at 45 Psi for 5 h. The catalyst was
removed by filtration through celite and the filtrate was evaporated to
give 800 mg of a colorless oil. NMR analysis showed ~is material to a
mixture of 1-(3-aminopropyl) Dimethylpyrazole-3,5-dicarboxylate and
the cyclic diazapineone. This mixture was dissolved in 50 ml of
benzene and refluxed for 15 h then evaporated. The resulting tan solid
was recrystalized from CH2C12/hexane to afford 5-3 as a white solid.
m.p. = 220-221C. 1H NMR (CDC13) o 7.36 (s,lH); 6.42 (br t, lH);
4.58 (t, J=8.0 Hz, 2H); 3.95 (s, 3H); 3.39 (dt, J=7.2 Hz, 2H); 2.31 (m,
30 2H).
WO 94/18981 PCT/US94/01881
3S~ 6~-
Methyl-5,6,7,~-tetrahydro-4-oxo-5-[2-(N-Boc-piperidin-4-yl)ethyl]-4H-
pyrazolor 1.5-al~ 1.41diazepin-2-yllcarboxylate (X-S)
To a solution of 5-3 (175 mg, 0.83 nnmol), in 50 ml DMF
was, added 60~Q NaH (36 mg, 0.91 mmol~, the mixt~re was stirred
under N2 at -15 for 30 min. To this mixture a solution of 2-(N-Boc-
Piperidin-4-yl)ethyl iodide (~) (283 mg, 083 ~lmol) in 25 ml DMF
was added dropwise over 20 min. The resulting solution was stirred for
30 min at -15 then warmed to room temperature and allowed to stir
overnight. The DMF was evaporated at reduced pressure and the
residue redissolved in ethyl acetate, filtered and chromatographed on
silica gel using ethyl acetate as eluent to afford pure 5-5 as a glass.
1H NMR (CDCl3) o 7.24 (s, lH); 4.50 (t, J=7.0 Hz, 2H); 3.93 (br d,
J=12 Hz, 2H); 3.94 (s, 3H); 3.61 (t, J=5.3 Hz, 2H); 3.42 (t, J=7.3 Hz,
2H); 2.7 (br t, J=6.3 Hz, 2H); 2.3 (m, 2H); 1.55 (d, J=12.5 Hz, 2H);
1.38 (m, 2H); 1.33-1.25 (m, lH); 1.27 (s, 9H); 1.01 (m, 2H).
5,6,7,~s-Tetrahydro-4-oxo-5-[2-(N-Boc-piperidin 4-yl)ethyl-4H-
pyrazolorl~5-al~1.41diazepin-2-yll-carboxylic acid (5-6)
2 A solution of 5-5 (166 mg, 0.395 mmol) in 10 ml CH30H,
was treated with lN NaOH (0.435 ml, 0.43 mmol). The resulting
solution was stirred at room temperature for 1~ h and then CH30H
removed at reduced pressure. The rem~ining aqueous phase was
acidified with 10% aqueous citric acid and extracted with CH2C12 (2 x
2s 50 ml). The pooled organic extracts were dried over Na2S04 then
concentrated to give 5-6 as a white solid.
lH NMR (CDCl3) ~ 7.29 (s, lH); 4.52 (t, J=7.0 Hz, 2H); 4.12 (br d,
J=12 Hz, 2H); 3.94 (s, 3H); 3.61 (t, J=5.3 Hz, 2H); 3.42 (t, J=7.3 Hz,
3 2H); 2.7 (br t, J=6.3 Hz, 2H); 2.3 (m, 2H); 1.55 (d, J=12.5 Hz, 2H);
1.38 (m, 2H); 1.33-1.25 (m, lH); 1.27 (s, 9H); 1.01 (m, 2H).
WO 94/18981 PCT/US94/01881
2i5~i23
- 6 9 - , -} s
Ethyl-2(S)-[(n-Butylsulfonyl)amino]-3-[[[5,6,7,~-tetrahydro-4-oxo-5-[2-
(N-Boc-piperidin-4-yl)ethyl]-4H-pyrazolo[ 1 ,5-a][ 1 ,4]-diazepin-2-
yllcarbonyllamino propionoate (5-7)
5-6 (147 mg, 0.36 mmol) in CH2cl2/s ml was treated with
ethyl 2(S)-n-butanesulfonamido-3-amino-propionate (A-5) (115 mg,
0.40 mmol), HOBT (49 mg, 0.36 mmol), and Et3N (0.10 ml, 0.724
mmol) in 50 ml CH2C12 and this solution was stirred under N2 for 18 h
at room temperature. The reaction solution was washed successively
with sat. NaHCO3, H2O, 10% citric acid, H2O and brine (1 x 20 ml
each), dried over Na2SO4, filtered and evaporated. The resulting clear
glass was chromatographed on silica gel using 5% CH30H/EtOAc as
eluent giving pure 5-7.
H NMR (CDC13) ~ 7.32 (s, lH); 7.24 (t, J=6.~ Hz, lH); 5.54 (t, J=7.2
Hz, lH; 4.43(t J=7.8 Hz, 2H); 4.32 (m, lH); 4.2~s (q, J=7.1 Hz, 2H);
4.10 ( br d, J=12 Hz, 2H); 3.85 (m, 2H); 3.61 (t, J=5.3 Hz, 2H); 3.42 (t,
J=7.3 Hz, 2H); 3.03 (t, J=7.1 Hz, 2H); 2.7 (br t, J=6.3 Hz, 2H); 2.3 (m,
2H); 1.65 -1.45 (overlapping m, 7H); 1.3~ (m, 2H); 1.33-1.25 (m, lH);
1.37 (s, 9H); 1.30 (t, J=7.4 Hz, 3H); 1.01 (m, 2H); 0.96 (t, J=7.3 Hz,
3H).
2(S)-[(n-Butylsulfonyl)amino]-3-[[[5 ,6,7,~-tetrahydro-4-oxo-5-[2-
(piperidin-4-yl)ethyl]-4H-pyrazolo[ 1 ,5-a] [ 1 ,4]diazepin-2-yl]carbonyl]-
amino propionic acid (5-~)
To a solution of ~ (100 mg, 0.156 mmol) in 10 ml
CH30H, was added lN NaOH (160 ml, 0.16 mmol) and H2O, 10 ml.
The resulhng solution was stirred at room temperature for 3.5 h then
CH30H removed at reduced pressure. The rem~inin~ aqueous phase
was acidified with 10% aqueous citric acid and extracted with CH2C12
(2 x 50 ml). The pooled organic extracts were dried over Na2SO4 then
evaporated to give the desired acid.
lH NMR (CDC13) ~ 17.24 (t, J=6.~ Hz,lH); 7.2~s (s, lH); 6.0 (d, J=7.2
Hz, lH); 4.43 (t, J=7.~ Hz, 2H); 4.32 (m, lH); 4.10 (br d, J=12 Hz, 2H);
WO 94/18981 PCT/US94/01881
70-
3.~5 (m, 2H); 3.61 (t, J=5.3 Hz, 2H); 3.42 (t, J=7.3 Hz, 2H); 3.03 (t,
J=7.1 Hz, 2H); 2.7 (br t, J=6.3 Hz, 2H); 2.3 (m, 2H); 1.65-1.45
(overlapping m, 7H); 1.38 (m, 2H); 1.33-1.25 (m, lH); 1.37 (s, 9H);
1.01 (m, 2H); 0.96 (t, J=7.3 Hz, 3H).
This acid (89 mg) in 15 ml ethyl acetate was cooled to 0
and HCI; gas bubbled through for 3 min. The reaction mixture was
warmed to room temperature, stirred for 30 min then taken to dryness
on a rotary evaporator. The rem~ining white solid was triturated with
o ether, filtered and vacuum dried over P2O5 to give 5-8 as a white solid.
lH NMR (DMSO-d6) ~ 8.95 (br s, lH); 8.33 (t, J=5.7 Hz, lH); 7.64 (d,
J=9 Hz, lH); 7.02 (s, lH); 4.35 (t, J=S.l Hz, 2H); 4.10 ( m, lH); 3.~s1 (t,
J=5.2 Hz, 2H); 3.6-3.4 (m, 4H); 3.21 (d, J=lO.S Hz, 2H); 2.95 (t, J=7.8
5 Hz, 2H); 2.~1 (br m, 2H); 1.96 (d, J=l lHz, 2H); 1.62-1.2 (overlapping
multiplets, 9H); 0.80 (t, J=7.3Hz, 2H).
WO 94tl8981 PCT/US94/01881
~ 21~5~2~
- 7 1
SCHEME 6
O
)~N~CO2CH3 1. acetone, H20
B'ocN3 (CH2)2-N>~ EtOH
Cl
)~N~\s,,CO2CH3
BocN3(CH2)2-N~
H
6-1A, cis
6-1 B, trans
H 1. NaOH, CH30H, H20
BocN3 (CH2)2-N ~OCH3 HCI
H H2N ~CO2tBu
Ç~
)~N~ NH ~ CO2tBu CH2C12
BocN3(CH2)2-N~ ~
H 6-2
)~ ~ NH~--C2H
HN3 (CH2)2-N - ~
6-3
WO 94tl8981 PCTIUS94/01881
5~ ,3 - 72 -
Ethyl-3-oxo-2[2-(N-Boc-piperidin-4-yl)ethyl]octahydroimidazo-
rl.S-alpyridin-6-vllcarboxylate (6-lA~ 6-lB)
2-6, (514 mg, 1.1 mmol) was dissolved in 25 ml of acetone
10 ml of H2Q was added and the mixture heated to 60C for 3.5 h. The
5 acetone was removed at reduced pressure and the resulting yellow
precipitate filtered. This crude material was dissolved in 100 ml of
toluene and refluxed for 3 h. The toluene was evaporated giving a
yellow solid.
This solid (500 mg, 1.11 mmol) was dissolved in 100 ml of
ethanol 75 mg of 10% Pd on C was added and mi~ture shaken on Parr
hydrogenator at 55 psi for 13 h. The catalyst was removed by filtration
~rough celite and the solvent evaporated. The resulting colorless oil
was chromatographed on silica gel using 70% ethyl acetate/30% hexane
to give 268 mg of the cis reduction product 6-lA along with 132 mg of
the trans product 6- 1 B .
Isomer 6- 1 A
lH NMR (CDC13) ~ 4.15 (m, lH); 4.06 (m, 2H); 3.6~ (s, 3H); 3.41 (m,
2H); 3.22 (m, lH); 2.90 (m, lH); 2.80 (m, 2H); 2.68 (m, 2H); 2.43 (m,
lH); 2.16 (m, lH); 1.81 (m, lH); 1.69 (m, 2H); 1.60 (m, lH); 1.45 (s,
9H); 1.43 (m, 3H); 1.39 (m, lH); 1.11 (m, 2H).
Isomer 6-lB
lH NMR (CDCl3) ~ 4.34 (m, lH); 4.06 (m, 2H); 3.69 (s, 3H); 3.40 (m,
3H); 3.36 (m, lH); 3.30 (m, lH); 3.11 (m, lH); 2.89 (m, lH); 2.84 (m,
lH); 2.67 (m, 2H); 2.63 (m, lH); 2.30 (m, lH); 1.69 (m, 2H); 1.67 (m,
2H); 1.60 (m, lH); 1.45 (s, 9H); 1.41 (m, lH); l.û9 (m, 2H).
WO 94/18981 PCT/US94/01881
1Z3~
tert-Butyl (~)-cis-[[3-oxo-2[2(N-Boc-piperidin-4-yl)ethyl]octahydro-
imidazo~l.S-alpyridin-6-vllcarbonyllaminolpropionate (6-2)
6-lA was hydrolyzed with lN NaOH in CH30H/H20 as
described for 1 6 to give the desired acid. This acid was coupled with
5 ,B-alanine t~-butyl ester as described for 2-8 to provide 6-2.
lH NMR (CDC13) ~ 6.32 (t, lH); 4.06 (m, 2H); 3.98 (m, lH); 3.56 (m,
5H); 3.21 (m, 2H); 2.91 (m, lH); 2.92 (m, lH); 2.80 (m, 2H); 2.40 (t,
2H); 2.23 (m, lH); 2.09 (m, lH); 1.81 (m, 3H); 1.69 (m, 2H); 1.60 (m,
lH); 1.45 (s, 18H); 1.43 (m, 3H); 1.11 (m, 2H).
(:t)cis-3-Oxo-2[2-(piperidin-4-yl)ethyl]octahydroimidazo[1,5-a]pyridin-
~ 6-yllcarbonvllaminopropionic acid (6-3)
6-2 (65.3 mg, 0.13 mmol) was dissolved in 10 ml of
5 anhydrous CH2C12 and cooled to 0C. Trifluoroacetic acid (0.200 ml)
was added and solution stirred for 1 h then evaporated at reduced
pressure to give pure 6-3.
lH NMR (CD30D) ~ 4.06 (m, 2H); 3.91 (m, lH); 3.56 (m, SH); 3.21
20 (m, 2H); 2.91 (m, lH); 2.92 (m, lH); 2.80 (m, 2H); 2.40 (t, 2H); 2.23
(m, lH); 2.09 (m, lH); 1.81 (m, 3H); 1.68 (m, 2H); 1.60 (m, lH); 1.28
(m, 3H); 0.91 (m, 2H).
WO 94118981 PCT/US94/01881
$5~ 3 ~ 74 -
SCHEME 7
E~ocN3(CH2)z-N~NH/~co2cH3
ClSO2NHCO2CH2Ph
CH2cl2~ Et3N
BocN3(CH2)2~N J~ NH~CO2CH3
~N-N HNHso2NHco2cH2ph
7 1 1. LiOH, THF
2. HCI, EtOAc
o
HN3(CH2)2-N ~ NH~co2H
l~N_N H NHso2NHco2cH2ph
7-2 1. H2, Pd/C
2. HCI, EtOAc
HN3(CH2)2-N~NH/~co2H
WO 94/18981 Zi~ 551~3 PCT/US94/01881
- 75 -
Methyl-2(S)-[(N-CBZ-Aminosulfonyl)amino] -3-[[[4,5,6,7-tetrahydro-
4-oxo-5-[2(N-BOC-piperidin-4-yl)ethyl]pyrazolo[1,5-a]pyrizine-2-
yllcarbonyllaminolpropionate (7- 1)
To a 0 solution of chlorosulfamylisocyanate (45.1 ,ul,
0.508 mmol) in methylene chloride was added benzyl alcohol (53 ml,
0.508 mmol). The reaction was aged 90 min at 0 and a solution of
4-3 (250 mg., 0.50~ mmol) in methylene chloride cont~ining
triethylamine (142 ml, 1.02 mmol) added. The reaction was allowed to
warm to room temperature and stirred overnight (18 hr). The reaction
was adjusted to a pH=3.0 with aqueous sodium bisulfate and the product
was extracted with methylene chloride(3xlO ml). The organic extracts
were combined, concentrated and chromatographed on silica (eluent
95% methylene chloride, 5% methanol) to give 7-1.
lH NMR (300 MHz, CDC13) o 1.40 (s, 9H), 2.65 (t, 2H), 3.65 (s, 3H),
5.10 (s, 2H), 6.65 (d, lH), 7.2-7.5 (m, 6H), 8.90 (s, lH)
2(S)-[(N-CBZ-Aminosulfonyl)amino]-3-[[[4,5,6,7-tetrahydro-4-oxo-5-
[2-(N-Boc-piperidin-4-yl)ethyl)pyrazolo[1,5-a]pyrazine-2-yl]carboxyl]-
amino propionic acid (7-2)
To a solution of ~ (100 mg) in THF (5 ml) was added lN
LiOH (0.6 ml) and the mixture stirred at room temperature for 18 h.
The reaction was quenched by addition of aq. sodium bisulfate (pH=3.0)
and product extracted into ethyl acetate (2 x 15 ml). Concentration of
25 the extracts gave the desired acid.
H NMR 1.4 (s, 9H), 2.6 (br, t, 2H), 7.1-7.2 (br.m, 5H), 7.3 (s, lH).
This acid was dissolved in EtOAc, cooled to -5, and
30 treated with HCl (gas). The reaction mixture was concentrated and
flushed with ethyl acetate to give 7-2, mp >200 (dec.)
CHN analysis Calc. C, 44.86; H, ~.92; N, 14.20
Found: C, 44.50, H, 6.09; N, 13.fs0
WO 94/18981 r ~ PCT/`US94/(~1881
'
76 -
2(S)-(Aminosulfonyl)amino-3-[[[4,5,6,7-tetrahydro-4-oxo-5-[2-
(piperidin-4-yl)ethyl]pyrazolo[ 1 ,5-a]Pyrazin-2-yl]carbonyl]amino- r
propionic acid (7-3)
A solution of 7-2 (70 mg) in methanol (20 ml) was treated
with 1 0%Pd/C (35 mg) and the mixture hydrogenated ( 1 atm.)
overnight (18 hrs). The mixture was filtered and concentrated to give
46 mg of an oil. The oil was dissolved in ethyl acetate, cooled to 0 and
HCI gas bubbled in over 30 min. Concentration of the reaction gave 7-3
as a white solid, mp >200, FAB MS, M+1=458.
PCT/US94/01881
WO 94118981
2~5~1 23
- 77 -
SCHEME ~
C2H5O2C~ NaH, TI~IF
HN_~CO2C2H5 BrCH2CH2Br
8-1
C2H5O2C BocN~CH2CH2NH2
~ CO2C2Hs 2-4
Br Kl, DIEA
8-2
0
Jl 1. LiOH, CH30H, H20
BocN3(CH2)2 N~CO2CH3 2. EDC, HOBT, TEA
8-3 HCI H2N ~ C02tBu
,¦¦ o HCI, EtOAc
BocN~(CH2)2-N~ NH~ CO2tBu
8-4
HN3(CH2)2-N~ NH/ CO2H
~-5
W O 94/18981 PCTrUS94/01881
~5~ 7~-
Diethyl 1-(2-Bromoethyl)pylTole-2~4-dicarboxylate (~-2)
A solution of diethyl pyrrole-2,4-dicarboxylate (5.50 g,
29.4 mmol) in tetrahydrofuran (200 ml) was cooled in an ice bath and a
suspension of NaH (60%) (6.5 g, 6~s.6 mmol) in tetrahydro-furan (50
ml) was added in a stream. The reaction flask was warmed to room
temperature. After stirring 1 h at room temperature 1,2-
dibromoethane (25.2 ml, 294 mmol) was added and the mixture was
refluxed for 24 h. Water (50 ml) was added to the reaction flask. The
mixture was rotary evaporated to remove tetrahydrofuran. Saturated
o sodium bicarbonate solution (100 ml) was added to the residue and the
resulting solution was extracted with methylene chloride (4 x 50 ml).
The combined organic extracts were dried with anhydrous sodium
sulfate. The drying agent was removed by filtration, and the filtrate
was rotary evaporated to give a yellow solid. This material was
recrystalized from hexane ethyl acetate 80:20 to give ~-2 as a yellow
solid.
1H NMR (DMSO-d6) ~ 7.~3 (d, lH); 7.15 (d, lH); 4.69 (t, 2H); 4.25
(m, 4H); 3.7~ (t, 2H); 3.31 (H2O); 1.29-1.22 (m, 6H).
Ethyl [4,5,6,7]-Tetrahydro-4-oxo-5-[2-(N-Boc-Piperidin-4-yl)ethyl]-
pvITolorl.5-alpyrazin-2-vllcarboxylate (~s-3)
The alkyl bromide ~-2 (3.30 g, 10.4 mmol., 1.0 eq.),
(3.52 g, 15.~ mmol., 1.5 eq.), potassium iodide (5.18 g, 31.2 mmol),
diisopropylethyl~mine (5.42 ml, 31.2 mmol., 3.0 eq.), and acetonitrile
(50 ml) were combined. The suspension was heated to reflux for 24 h,
and then rotary evaporated to remove acetonitrile. Saturated sodium
bicarbonate solution (100 ml) was added, and the solution was extracted
with ethyl acetate (5 x 50 ml). The combined organic extract.s were
3 dried with anhydrous sodium sulfate and concentrated to a brown oil.
The crude product was subjected to column chronnatography using
silica. The colurnn was eluted with methylene ch~oride then methylene
chloride con~ining 1% methanol to give pure ~s-3 as a white solid.
WO 94/18981 PCT/US94/01881
~ 3
- 79 -
lH NMR (CDC13) ~ 7.32 (d, lH); 7.29 (d, lH); 4.28 (q, 2H); 4.19-4.00
(m, 4H); 3.7-3.6 (t, 2H); 3.6-3.5 (t, 2H); 2.68 (t, 2H); 1.8-1.7 (m, br,
2H, H2O); 1.6-1.4 (s, m, 1 lH); 1.33 (t, 3H); 1.2-1.1 (m, 2H).
tert-Butyl [4,5,6,7]-tetrahydro-4-oxo-5[2-(N-Boc-piperidin-4-
yl)ethyllpyrrolo~l~5-alpyrazin-2-vllamino propionate (8-4)
~ -3 (620 mg, 1.54 mmol) lithium hydroxide monohydrate
(160 mg, 4.00 mmol., 2.6 eq.), water (15 ml), and methanol (10 ml)
o were combined in a 50 ml round bottom flask equipped with a magnetic
stir bar. The solution was stirred for 4 h at room temperature then
heated to 90 for 1 h. Any rem~ining methanol was removed by rotary
evaporation and the aqueous residue was acidified with 10% K2SO4
then extracted with ethyl acetate (4xS0 ml). The combined organic
15 extracts were dried with anhydrous sodium sulfate, filtered, and
evaporated giving the desired acid as a white solid.
lH NMR (D~SO-d6) o 7.51 (d, 2H); 6.84 (d, lH); 4.19 (m, 2H); 3.90
(d, br, 2H); 3.63 (m, 2H); 3.43 (m, 2H); 3.4-3.2 (H2O); 2.6-2.8 (br,
20 2H); 1.66 (d, 2H); 1.43 (m, 2H); 1.36 (s, 9H); 1.1-0.9 (m, 2H).
This acid (150 mg, 554 mmol), EDC (117 mg, 0.609
mmol), l-hydroxybenzotriazole (82.2 mg, 0.609 mmol), triethylamine
(0.300 ml, 1.11 mmol., 4.0 e4.), ~-alanine-t-butyl ester (111 mg, 0.609
25 mmol), and methylene chloride (10 ml) were combined in a 100 ml
round bottom flask equipped with a m~netic stir bar. The resulting
solution was stirred at room temperature overnight. Solvent was rotary
evaporated from the reaction flask and the resulting residue was
subjected to column chromatography using silica. The column was
30 eluted with methylene chloride, methylene chloride with 2% methanol
then 4% methanol. Fractions cont~inin~; product were pooled to
provide ~s-4 as a white solid.
lH NMR (CDC13) ~ 7.32 (d, lH); 7.04 (d, lH); 6.5~ (m, lH); 4.2-4.0
(m, 4H); 3.7-3.5 (m, 6H); 2.68 (t, 2H); 2.51 (t, 2H); 1-70 (m, 3H, H2O);
1.53 (m, 2H); 1.45 (s, 9H); 1.21-1.0 (m, 2H).
WO 94/18981 PCT/US94/01881
80 -
4,5,6,7-Tetrahydro-4-oxo-5-~2-(piperidin-4-yl)ethyl] [ 1 ,5-a~pyrazine-2-
vllcarbonyllamino propionic acid (8-5)
The ester ~-4 (18a mg, 0.347 mmol) and ethyl acetate (10
5 ml) were cpmbined in a 50 ml round bottom flask. The suspension was
cooled in an ice bath. Hydrogen chloride was bubbled through the
.suspension for 1.5 min. The reaction flask was warmed to room
temperature, then solvent was removed by vacuum filtration giVillg 8-5
as a white solid, mp 248-249.
lH NMR (DMSO-d6) ~ 9.0-8.5 (br, 2H); 8.05 (m, lH); 7.42 (d, lH);
7.05 (d, lH); 4.15 (m, 2H); 3.62 (m, 2H); 3.5-3.3 (m, 4H, H2O), 3.25-
3.15 (d, br, 2H); 2.85-2.7 (br, 2H); 2.5-2.4 (m, 4H); 1.81 (d, 2H); 1.6-
1.4 (m, 3H); 1.4-1.2 (m, 2H).
WO 94/18981 PCT/US94/01881
21~
- 81 -
SCHEME 9
.
BOCNH ~ 6CO2C2H5BOCNH ~ 6CO2C2H5
H NH2 CISO2NHC4H9 H NHSO2 NHC4Hg
9-1py, CH2cl2 9-2
HCI,H2N~6NHSO2 NHC4Hs
EtOAc DMF, 3-3
9-3
BocN3 (CH2)2 N J~ NH~CO2C2Hs
~N-N H NHSO2 NHC4Hg
9~4
1. NaOMe, MeOH
2. HCI, EtOAc
HN3(CH2)2 N~ N~co2H
~N-N H~ NHSO2 NHC4Hg
Ethyl 2(S)-Amino-3-(N-Boc-amino)propionate (9-1)
Commerically available 2(S)-3-~ minopropanoic acid
(Fluka) (10 g, 96.2 mmol) was dissolved in absolute ethanol (200 ml)
and the solution was saturated with anhydrous HCI gas then heated at
reflux for 2.5 h. The solvent was removed and the residue
WO 94/18981 PCT/US94/01881
~?,3 - 82 -
recrystallized from EtOH/Et20 to afford the ethyl/ester dihydro-
chloride as a hygroscopic white solid.
This material (5 g, 24.3 mmol) was suspended in CH2C12
(200 ml) cooled to -50. Next, triethyl~mine (7.0 ml, 51 mmol) was
5 added and the mixture stirred for 5 min. A solution of di-tert-butyl
dicarbonate (5.30 g, 24.3 mmol) in 100 ml CH2C12 was added dropwise
over a 30 min period and the mixture stirred at -50 for 1.5 h, then
warmed to room temperature. The solution was washed with water (2 x
100 ml), then dried (Na2S04) and evaporated. The resulting residue
was chromatographed silica gel (~0:20 CH2C12/CH3OH) to afford pure
9-1 .
lH NMR (300 MHz, CDC13) 6.03 (brt, lH); 4.35 (m, lH); 3.~5 (m,
2H); 3.23 (m, 2H); 1.21 (t, 3H).
Ethyl 2(S)-n-Butylaminosulfonylamino-3-(N-BOC-amino)propionate
(9-2)
A solution of ~ (500 mg, 2.15 mmol) in CH2C12 (10 ml)
was treated with pyridine (261 ml, 3.23 mmol) and n-butylsulfoamoyl
chloride (406 mg, 2.37 mmol). The solution was stirred at room
temperature for 3 h, ~en poured onto silica gel and eluted with 30%
acetone/hexane to give pure 9-2 as a white solid.
1H NMR (300 MHz, CDC13) ~ 5.43 (d, lH); 4.9~ (t, lH); 4.40 (brs,
lH); 4.10 (q, 2H); 4.05 (m, lH); 3.56 (m, 2H); 3.0~ (m, 2H); 1.~-1.2
(overlaping multipets, 16H); 0.90 (t, 3H).
Ethyl 2(S)-(n-Butylaminosulfonylamino)-3-aminopropionate (9-3)
A solution of 9-2 (612 mg, 1.65 mmol) in ethyl acetate
(50 ml) was cooled to -5 and anhydrous HCl was bubbled in for 30
min. The reaction was concentrated and the product isolated by
filtration to give 9-3 as a white solid.
WO 94/18981 PCT/US94/01881
2~5123l
- 83 -
lH NMR (300 MHz, DMSO-d6) ~ 5.82 (d, lH); 4.56 (brs, lH); 4.20
(q, 2H); 4.02 (m, lH); 3.45 (m, 2H); 3.01 (m, 2H); 1.9-1.36 (m, 7H);
0.93 (t, 3H).
Ethyl 2(S)-[(n-Butylaminosulfonyl)amino]-3-[[4,5,6,7-tetrahydro-4-oxo-
5-[2-(piperidin-4-yl)ethyl]pyrazolo[1,5-a]pyrazin-2-yl]carbonyl]amine
propionate (9-4)
Coupling or 9-3 with 3-3 with EDC and HOBT in DMF as
described for 1 6 provided 9-4.
lH NMR (300 MHz, CDC13) o 7.29 (s, lH); 7.19 (t, lH); $.43 (d, lH);
4.41 (t, 2H); 4.26 (q, 2H); 4.20 (m, lH); 4.0~s (d, 2H); 3.86 (m, 2H);
3.76 (m, 2H); 3.60 (t, 2H); 3.08 (t, 2H); 2.68 (t, 2H); 1.78s (d, 2H); 1.6-
1.08 (m, 8H); 1.43 (s, 9H); 0.92 (t, 3H).
2(S)-[(n-Butylaminosulfonylamino]-3-[(4,5,6,7-tetrahydro-4-oxo-5-
(piperidin-4-yl)ethyl]pyrazolo[1,5-a]pyrazin-2-yl]carbonyl]-
aminopropionic acid (9-5)
Hydrolysis of 9-4 with NaOMe, isolation of the crude acid,
and subsequent treatment with HCl in EtOAc as described for 5-7
provided 9-5 as a white solid, mp. 155-160.
w o 94/18981 . F~rrus94/01881
~5~ - 84 -
S C H E M ~E 10
HCI
5GH302C~ H N ~CO2tBu
CH2Br K2CO3, CH3CN
2-3
CH302C~, NH~ triphosgene
2t u toluene, DMA
10-1
1. NH40H. CH2C12
CH3O2C~N--4 ~ CO2tBu 2. MeO2C-SO(3NEt3
~< CH2CI2
~CI 3. 1 N NaOH
10-2
0 1. EDC, HOBT, Et3N
2s HO2C ~[~<N~ CO2tBu 2-4
CN
10-3 2. TFA
3 0 HN~} (C H2)2 N H ~N~ CO2H
CN
10-4
WO 94/18981 PCT/US94/01881
St23
- 85 -
.
N-r2-(5-Carbomethoxy)pyridylmethvll-~-alanine tert-butyl ester (10-1)
A mixture of 2-3 (871 mg, 3.79 mmol)"~-alanine tert-
butyl ester-HCI (2.7 g, 15 mmol), and K2C03 (4.5 g, 30 mmol) in 100
ml of anhydrous CH3CN was placed in a 250 ml flask and refluxed for
5 3 h, then cooled and filtered. The filtrate was concentrated at reduced
pressure and chromatographed on silica gel using EtOAc as eluent to
afford 10-1 a.s a colorless glass.
H NMR (CDC13) ~ 9.18 (d, J=1.4Hz, lH); ~.l(dd, J=1.4 and 6.8 Hz,
lH); 7.39 (d, J=6.~ Hz, lH); 4.08 (s, 2 H); 3.95 (s, 3H); 3.04 (t, 2H);
2.60 (t, 2H); 1.4 (s, 9H).
tert-Butyl-2-(2-carboxyethyl)- 1 -chlorocarbonyl-3-oxo-2,3-dihydro-
imidazor l .S-alpyridine-6-carboxylate (10-2)
10-l (~00 mg, 2.71 mmol) was dissolved in 50 ml of
toluene. N,N-dimethyl aniline (2.0 ml, 16.7 mmol) was added an
solution cooled to 0. To this, a solution of triphosgene (1.7 g, 5.7
mmol) in 15 ml toluene was added dropwise over 30 min. The solution
was then warmed to 25 and stirred for 3.0 h then washed twice with
20 lN HCl, water and brine (50 ml of each), dried over Na2SO4 and
evaporated giving 10-2 as a yellow crystalline solid.
1H NMR (CDC13) ~ 8.83 (d, J=1.4Hz, lH); 8.25 (d, J=6.g Hz; lH); 7.82
(dd, J=1.4 and 6.8 Hz,lH); 4.43 (t, J=7.2 Hz, 2H); 3.98 (s, 3 H); 2.75
5 (t, J=7.2 H~, 2H); 1.4 (s, 9H).
tert-Butyl-2-(2-carboxyethyl)- 1 -cyano-3-oxo-2,3-dihydroimidazo-
r l .S-alpvridine-6-carboxvlate (10-3)
10-2 (250 mg, 0.65 mmol) was dissolved in 100 ml of
30 CH2C12 10 ml of ammonium hydroxide was added and this biphasic
mixture was stirred for 1 h then the organic layer separated and washed
with 10% citric acid then brine (50 ml), dried over Na2SO4 and
evaporated.
WO 94/18981 . PCT/US94/01881
. t
$~3 - 86 -
This residue (150 mg, 0.42 mmol) was dissolved in 100 ml
of CH2C12, 355.7 mg of Methoxycarbonylsulfamoyl-triethyl-
ammonium hydroxide, inner salt (Burgess reagent, 1.48 mmol) was
added in three portions over a 2 h period. The resulting solution wa~
5 stirred at room temperature for an additional hour and then
concentrated and chromatographed on silica gel using 1:1 hexane/ethyl
acetate as eluent giving the nitrile in ql-~ntit~tive yield. This material
was subjected to saponification using lN LiOH to give the desired
carboxylic acid 10-3.
lH NMR (CD~13) ~ 8.75 (s, lH); 7.43 (d, lH); 7.1~ (d, lH); 4.21
(t, 2H); 2.85 (t, 3H); 1.4 (s, 9H).
3-[ 1 -cyano-3-oxo-6-2-(piperidin-4-yl)ethylcarbamoyl)-2,3-dihydro-
5 imidazorl.5-alpyridin-2-yllpropionic acid (10-4)
10-3 (170 mg, 0.51 mmol) was dissolved in 10 ml of
CH2C12, Et3N ~71 ml, 0.51 mrnol) was added along wi~ HOBT (69.3
mg, 0.51 mmol), EDC (98.6 mg, 0.52 mmol) and ~ (117.2 mg, 0.51
mmol). The mixture was stirred under N2 for 18 h then washed with
20 10% citric acid, H20 and brine (10 ml each) and dried over Na2S04,
concentrated and chromatographed giving a yellow solid (215 mg, 0.43
mmol). This material was deprotected using trifluoroacetic acid in
CH2C12 to give the 10-4 TFA salt as a yellow solid, m.p.= 173.
2s lH NMR (300 MHz, DMSO d6) ~ 1.75 (s, lH); 7.43 (t, lH); 7.31
(d, lH); 7.15 (d, lH); 4.23 (t, 2H); 3.41 (t, 2H); 3.23 (d, 2H); 2.~S3 (m,
2H); 1.85 (d, 2H); 1.53 (m, 2H); 1.41 (m, lH); 1.32 (m, 2H).
WO 94/18981 PCT/US94/01881
2~ 3
- 87 -
SCHEME 1 1
,~CO2CH3 HCI-H2N / CO2tBu
CH302C l~N' K2CO3, CH3CN
3-2
o 1. LiOH, H20, CH30H
CH302C~N~CO2tBu 2. BocN~} CH2CH2NH2
N--N 2-4
EDC, HOBT
11-1
3. HCI/EtOAc
~N~co2H
HN3CH2CH2-NH N_N~J
11-2
Methyl [4,5,6,7-tetrahydro-4-oxo-5-[3(tertbutyl propionyl)]pyrazolo
30 rl~5-alpvrazin-2-yllcarboxylate (11-1)
3-2 (1.4 g, 4.8 mmol), ~-alanine tert-butyl ester-HCI
(0.90 g, 5 mmol), and potassium carbonate (0.7~ g, 5.2~ mmol) in 150
ml CH3CN was refluxed under N2 for 4.5 h then cooled, filtered and
evaporated at reduced pressure. The resulting yellow residue was
WO 94/18981 PCT/US94/01881
8P~ -
chromatographed on silica gel using 2% CH3OH/CH2C12 giving the
diester 11-1 as a colorless glass.
lH NMR (CDC13) o 7.31 (s, lH); 4.48 (t, 2H), 3.93 (s, 3H), 3.61
(t, 2H); 2.71 (t, 2H); 2.35 (t, 2H); 1.23 (s, 9H).
3-[4,5,6,7-Tetrahydro-4-oxo-2-(2-(piperidin-4-yl)ethylcarbamoyl)-
pyrazolorl.5-alpyrazin-5-yllpropionic acid (11-2)
A solution cont~ining LiOH (145 mg, 3.41 mmol) in 10 ml
H2O was added to a solution of the ester 11-1 (1.0 g, 3.1 mmol) in 10
ml CH30H and the mixture was heated to 60C for 2.5 h then cooled
and the solvent removed at reduced pressure. The rem~ining residue
was acidi~led with 10% citric acid and extracted CH2C12 (2 x 100 ml).
The pooled organic extracts were washed with H20, dried and
evaporated to afford the desired acid as a colorless glass.
lH NMR (CDC13) ~ 7.21 (s, lH); 4.48 (t, 2H); 3.63 (t, 2H); 2.71
(t, 2H); 2.32 (t, 2H); 1.23 (s, 9H).
This acid (500 mg, 1.62 mmol) was dissolved in 10 ml of
CH2C12, HOBt (220 mg, 1.62 mmol) was added along with EDC (309
mg, 1.62 mmol), and 2-4 (356 mg, 1.63 mmol). The mixture was
stirred under N2 for 16 h then washed with 10% citric acid, H20 and
brine (10 ml each) and dried over Na2S04, concel,tl~ted and
chromatographed giving a colorless foam. This material was
deprotected using HCl in ethyl acetate, to give the HCl salt of 11-2 as a
white solid. MP= 192- 194C.
lH NMR (300 MHz, DMSO d6) o 9.0 (br, s, lH); 8.35 (m, lH); 6.99 (s,
lH); 4.38 (t, J = 6.0 Hz, 2H); 3.83 (t, J = 5.5 Ht, 2H); 3.64 (t, J = 7.1
Hz, 2H); 3.30-3.1 (m, 4H); 2.85-2.65 (q, 2H); 2.54 (t, J = 7.1 Hz, 2H);
1.85-1.75 (s, br, 2H); 1.60-1.20 (overlapping m, 5H).
wo 94/18981 PCT/USg4/01881
~ 21~5~
- 89 -
SCHEME 12
r CH3O2C~CO2CH3 N~CH2CH2NH2
Br~_N_N i 12-1
K2CO3, CH3CN
3-2
O
10 ~ (cH2)2-N~J~co2cH3
-N 1. LiOH, H20, CH30H
H N~CO~tBu CH3
N~ (CH2)2-N~ NH /~6CO2tBu
-N H NHSO2~CH3
12-3
TFA, CH2CI2
2s
~(CH2)z~N~NH~6CO2H
-N H NHSO2~CH3
12-4
WO 94118981 . PCT/US94101881
.... _
90 -
2-(4-Pyridyl)ethylamine (12- 1)
A solution of NH4CI in 200 ml of H20 was placed in a lL
Flask. 4-Vinyl pyridine (56.4 ml, 0.52 mol) was added along with 150
ml CH30H and on the mixture heated at 60 for 18 h. The reaction
5 solution was cooled to 0 in an ice bath and made Ibasic by the addition
of 30% NaOH. The basic solution was extracted with CH2C12 (5 x 100
ml) and the pooled extracts dried, then evaporated. Vacuum distillation
of the residue afforded 12 1 as a colorless liquid.
HNMR(300MHz,CDC13)~.53(d,3=6.1 H~,2H);7.25(d,J=6.1
H2, 2H), 3.02 (t, 2H); 2.77 (t, 2H); 1.4 (br.s, 2H).
~ Methyl [4,5,6,7-tetrahydro-4-oxo-5-[2-(pyridin~ yl)ethyl]pyrazolo-
S-alpyrazin-2-yllcarboxylate (12-2)
A solution of 3-2 (1.4 g, 4.~ mmol), 4-(2-aminoethyl-
pyridine) (0.645 g, 5.28 mrnol), and potassium carbonate (0.78 g, 5.28
mmol) in 150 ml CH3CN was refluxed under N2 for 4.5 h then cooled,
filtered and evaporated at reduced pressure. The resulting yellow
residue was redissolved in 50 ml of DMF and treated with NaH (200 mg
20 of a 60% oil dispersion) and heated at 90 for 3 h then concentrated at
reduced pressure and chromatographed on silica gel using 20%
CH30H/CH2C12 giving the ester a 12-2 as a pale yellow glass (0.9lg,
3.0 mmol, 68%).
2~ lH NMR (CDC13) ~ 8.32 (d, 2H); 7.52 (d, 2H); 7.34 (s, lH); 4.4
(t, 2H); 3.91 (s, 3H); 3.61 (t, 2H), 2.71 (t, 2H) 2.35 (t, 2H).
tert-Butyl 2(S)-[(p-toluenesulfonyl)amino]-3-[[[4,5,6,7-tetrahydro-4-
oxo-5-[2-(4-pyridyl)ethyl]pyrazolo[ l ,S-a]pyrazin-2-yl]carboxylate
3 (12-3)
A solution cont~inin~ LiOH (130 mg, 3.05 mmol) in 10 ml
H2O was added to a solution of 12-2 (910 mg, 3.0 mmol) in 10 ml
CH30H and the mixture was heated to 60 for 2.5 h then cooled and the
solvent removed at reduced pressure. The renl~inin~ residue purified
WO 94/18981 PCT/US94/0188I
2~1S~
- 91 -
by ion exchange chromatography on Dowex-50W re.sin to affording the
desired acid as an off-white solid, mp 187.
This acid (300 mg, 0.78 mmol) was suspended in 50 ml of
anhydrous DMF, A-8 (293 mg, 81 mmol), EDC (150 mg, 0.78 mmol),
HOBt (105 mg, 0.78 mmol) and N-methyl morpholine (87 ml, 0.78
mmol) were added and the resulting clear solution was stirred at 25C
for 19 h. The solution was diluted with 100 ml of EtOAc, washed
successively with sat. NaHCO3, H2O, and brine (25 ml), dried over
Na2SO4 and evaporated to provide 12-3.
lH NMR (300 MHz, DMSO-d6) ~ 8.63 (d, 2H); 7.80 (d, 2H); 7.58 (d,
2H); 7.29 (t, lH); 6.93 (s, lH); 5.95 (d, 2H); 4.40 (t, 2H); 4.08 (m, lH);
~ 3.86-3.74 (m, 4H); 3.35-3.20 (m, 2H); 3.10 (t, 2H); 1.30 (s, 9H);
2(S)-[(p-Toluenesulfonyl)amino]-3-[[[4,5,6,7-tetrahydro-4-oxo-5-[2-(4-
pyridyl)ethyl]pyrazolo[ l ,5-a]pyrazin-2-yl]carboxylic acid-TFA salt
(12-4)
12-3 was deprotected using TFA in CH2C12 and purified by
reverse phase chromatography to give 12-4 as its TFA salt, mp 182-
185.
lH NMR (300 MHz, DMSO-d6) ~ 8.72 (br d, 2H); 8.19 (t, lH); 8.00
(d, lH); 7.81 (d, 2H); 7.58 (d, 2H); 7.19 (d, 2H); 6.85 (s, lH); 4.40
(t, 2H); 4.01 (m, lH); 3.83-3.76 (m, 4H); 3.48 (m, lH); 3.26 (m, lH);
3.10 (t, 2H).
.
PCT/aS941018gl -
wo 94/18981
~35~,3 92-
SCHEME 13
O
H, ll NaH, DMF
~OCH3 NC~ Br
5-3 CN o
,¢OCH3
13-1
1. LiOH, H20/rHF
2 H N~CO2tBu ~CH3
= HOBt. EDC, CH2CI2
CN o
~ ~NHSO2~CH3
13-2
TFA
CH2CI2
WO 94/18981 PCT/US94101881
2 1 5~
- 93 -
SCHEME 13 (CONT'D.)
CN
NHSO2~CH3
13-3
o 1 . SH2, Pyridine/Et3N
2. CH31, acetone
3. NH4CI, CH30H
HN 0
H2N~?~ NHSO2~CH3
13-4
2s
Methyl-5 ,6,7 ,8-tetrahydro-4-oxo-5-[3(cyanophenyl)methyl-4-H-
pyrazolorl.5-alrl~41diazepin-2-vllcarboxylate (13-1)
A solution of 5-3 (3.02 g, 14.5 mmol) in 60 ml anhydrous
DMF was cooled to 0C and treated with NaH (60% in oil) (636 mg,
30 15.98 mmol). The resulting mixture was stirred at 0 for 1.5 h, then a
solution of 3-cyanobenzyl bromide (3.11 g, 15.89 mmol) in 50 ml of
DMF was added dropwise. The resulting mixture was stirred at 25 for
1 8h ~en diluted with 200 ml EtOAc and washed with H2O (3 x 100 ml)
and brine (100 ml). The organic layer was dried (NaSO4), filtered and
WO 94/18981 PCT/US94/01881
~ 5~3 94-
evaporated. The resulting solid was recrystallized from
CH2C12/CH30H to give 13-1 as a white solid.
lH NMR (CDC13) ~ 7.85 (s, lH); 7.7~s (d, J = ~ Hz, lH); 7.59 (d, J = ~
Hz, lH); 7.46 (m, lH); 7.30 (s, lH); 4.80 (s, lH); 4.43 (t, J = 8 Hz, 2H);
3.89 (s, 3H); 3.38 (t, J = 8 Hz, 2H); 2.09 (m, 2H).
tert-Butyl 2(S)-[(p-Toluenesulfonyl)amino]-3-[[[5,6,7,8-tetrahydro-4-
oxo-5-[(3-cyanophenyl)methyl]-4H-pyrazolo[ 1 ,5-a] [ 1 ,4]diazapin-2-yl]-
carbonyllaminolpropanoate (13-2)
A solution of ester 13 1 (1.5 g, 4.04 mmol) in 100 ml THF
was treated with lN LiOH (5.1 ml, 5.1 mmol) and 100 ml H2O and
stirred at 25 for 1.5 h. The THF was removed at reduced pressure and
the aqueous residue acidified with lN HCl. The resulting precipitate
5 was filtered and dried in vacuo to give the desired product as a white
solid.
lH NMR (CDC13) ~ 7.95 (s, lH); 7.73 (d, lH); 7.S3 (d, lH); 7.43
(m, lH); 7.30 (s, lH); 4.85 (s, 2H); 4.43 (t, 2H); 3.31 (t, 2H); 2.0P~ (m,
20 2H).
The above acid (1.0 g, 3.23 mmol) was combined with A-9
(1.24 g, 3.54 mmol), HOBt (480 mg, 3.54 mmol); EDC (641 mg, 3.54
mmol) in 100 ml CH2C12. N-methyl morpholine ~403 ,ul, 3.83 mmol)
25 was added and the resulting solution stirred at room temperature for
16 h, then was washed successively with sat. NaHCO3, 10% KHSO3 and
brine (100 ml each), then dried over Na2SO4, filtered and evaporated.
The residue was chromatographed on silica gel (EtOAc) to give 13-2 as
a white solid.
lH NMR (300 MHz, CDC13) o 7.73 (d, J = 6.8 Hz~ 2H); 7.68 (s, lH);
7.65 (d, lH); 7.51 (m, lH); 7.37 (d, J = 6.8 Hz, 2H); 7.20 (d,-lH); 7.18
(t, lH); 5.63 (d, J = 6.5 Hz, lH); 4.~0 (s, 2H); 4.78 (m, lH); 4.45 (t,
WO 94/18981 PCT/US94/01881
~ t ~ 5 1 2 3
- 95 -
2H); 3.85 (m, lH); 3.0~ (m, lH); 3.40 (t, 2H); 2.43 (s, 3H); 2.19 (m,
2H); 1.65 (s, 9H).
2(S)-[(p-toluensulfonyl)aminol-3-L[[5,6,7,~-tetrahydro-4-oxo-5-[(3-
cyanophenyl)methyl]-4H-pyrazolo[1,5-a][1,4]diazapin-2-yl]carbonyl]-
aminolpropanoate (13-3)
A solution of 13-2 in CH2C12 (15 ml) was treated with 5
ml of TFA. The solution was stirred at 0 for 2.5 h then evaporated
giving 13-3 as a colorless solid.
lH NMR (300 MHz, CDC13) o 7.73 (d, J = 6.8 Hz, 12H); 7.65 (s, lH);
7.65 (d, lH); 7.50 (m, lH); 7.31 (d, J = 6.8 Hz, 2H); 7.3 (d, lH); 7.28
(t, lH); 6.15 (d, J = 6.5 Hz, lH); 4.80 (s, 2H); 4.63 (m, lH); 4.43 (t,
2H); 3.~2 (m, lH); 3.65 (m, lH); 3.48 (m, 2H); 2.43 (s, 3H); 2.19 (m,
2H).
2(S)-[(p-Toluenesulfonyl)amino] -3-[[[5,6,7,~-tetrahydro-4-oxo-5-[(3-
amidinophenyl)methyl]-4H-pyrazolo[1,5-a][1,4]diazapin-2-yl]-
carbonyllaminolpropanoic acid (13-4)
13-3 (400 mg, 0.73 mmol) was dissolved in 10 ml of a 4:1
mixture of pyridine and Et3N. The solution was saturated with SH2 and
stirred until the nitrile could no longer be detected by HPLC (2.5h).
The excess SH2 was removed by passing a stream of nitrogen through
the solution. The rem~ining solution was then evaporated and the
residue triturated with lN HCl and filtered giving a yellow solid. This
material was dissoved in 15 ml of acetone and treated with CH3I
(250 ,ul) and then heated to 50 until the thioamide could no longer be
detected by HPLC (2 h). The solvent and excess CH3I were evaporated
and the residue redissolved in CH30H cont~ining (NH4)2C03 (144 mg,
1.14 mmol). The solution was heated at 50 for 12.5 h then evaporated,
and l3-4 was isolated by preparative reverse phase chromatography.
WO 94/18981 PCT/tJSg4/01881`
,
'-
96-
lH NMR (300 MHz, DMSO-d6) ~ 9.38 (s,2H); 9.17 (s,2H); 8.19
(t, lH); 8.16 (d,2H); 7.78 (s, lH); 7.75 (m,2H); 7.63 (m, lH); 7.60
(d,2H); 7.21 (d,2H); 6.93 (s, lH); 4.81 (s,2H); 4.40 (t,2H); 3.93 (m,
lH); 3.40 (m,2H); 3.35 (m,2H); 2.23 (s,3H); 2.13 (m,2H).
WO 94/18981 2 ~ S~ ¦ ~ 3 PCT/US94/01881
- 97 -
~CHEME 14
o O
H-~ ~OCH3 NaH, DMF Cl-(CH2)3 N~OCH3
OBr(CH2)3-CI ~N-N
5-3 1 4-1
1. NaN3, DMSO
2. LiOH, H20/THF
N3CH2-(CH2)2--N J~OH 14-2
~N--N
1. EDC, HOBt, DMF NMM
H N ~6CO2tBu CH3
A-9
2. 10% Pd on C, H2
H2NCH2-(CH2)2--N ~ N ~6C02tBu
~N--N H NHSO2~cH3
14-3
WO 94/18981 PCT/US94/01881
98 -
SCHEME 14 (CONT'D.)
1.
CH3
H \~N)~;~`CH3
DMF, H20, DIPEA
r 2. TFA, CH2cl2
H~ \~N--(CH2)3--N~` N ~`` 6CO2H
H2N H (~ N--N l~ H NHSO2~CH3
14-4
H2N--(CH2)3--N~ N~6C02tBU
~ o NHSO2~ CH3
14-3 L~ S-cH3-H
N
DMF, Et3N
2. TFA, CH2CI2
N
\~N (CH2)--N~ NH~,CO2H
NH H ~ N--N ~f H ~NHSO2~CH3
14-5
WO 94/18981 PCT/US94/01881
~f ~I;2~
99
Methyl-5,6,7,8-tetrahydro-4-oxo-5-(3 -chloropropyl)-4H-pyrazolo-
1.5-al ~ 1.41diazepin-2-yllcarboxylate (14- 1)
~ (2.0 g, 9.5 mmol) was alkylated with 1-chloro-3-bromo
propane (1.5 ml, 10.5 mmol) was described for 13-1 to give 14-1 as a
white solid.
lH NMR (300 MHz, CDC13) o 7.28 (s, lH); 4.58 (t, 2H); 3.93 (s, 3H);
2.78 (t, 2H); 2.68 (t, 2H); 2.46 (t, 2H); 3.27 (m, 2H); 2.18 (m, 2H).
o 5,6,7,8-Tetrahydro-4-oxo-5-(3-azidopropyl)-4H-pyrazolo[1,5-a] [1,4] -
diazepin-2-yllcarboxylic acid (14-2)
A solution of this chloride (909 mg, 3.2 mmol) and NaN3
(620 mg, 9.5 mmol) in DMF (1~ ml) was stirred at room temperature
for 36 h. The solution was diluted with ethyl acetate (50 ml) ~e washed
with H20 (3 x 50 ml), then dried (Na2S04), filtered and evaporated to
give the azide as a white solid. This material was hydrolyzed in the
usual m~nner to afford 14-2 as a white solid.
lH NMR (300 MHz, DMSO-d6) ~ 7.00 (s, lH); 4.41 (t, 2H); 3.52
(t, 2H); 3.41 (t, 2H); 3.26 (t, 2H); 2.20 (m, 2H); 1.80 (m, 2H).
tert-Butyl 2(S)-(p-Toluenesulfonylamino)-3-[5,6.7,8-tetrahydro-4-oxo-
5-(3-aminopropyl)-4H-pyrazolo[1,5-a][1,4]diazepin-2-yl]carboxyl)-
amino)propionate (14-3)
The acid 14-2 was coupled with A-9 as described for 13-2
to give the desired product as a white solid. This material was dissolved
in ethanol and residual over 10% Pd on C under a H2 atmosphere to
give l4-3 as a white solid.
30 lH NMR (300 MHz, DMSO -d6) 8.18 (t, lH); 7.6~S (d, 2H); 7.23 (d,
2H); 6.98 (s, lH); 4.4 (m, 3H); 3.93 (t, 2H); 3.48-3.2 (m, 6H); 2.78 (t,
2H); 2.43 (s, 3H); 2.69 (m, 2H); 1.86 (m, 2H); 1.08 (s, 9H).
WO 94/18981 ~;~ 4/OI881
~5~3 - loo-
2(S)-(p-Toluensulfonylamino)-3-[5,6,7,~-tetrahydro-4-oxo-5-(3-guan-
idinopropyl~-4H-pyrazolo[1,5-a] [1,4]diazepin-2-yl]carbonyl)amino)-
propionic acid (14-4)
A solution of 14-3 (60 mg, 0.1 mrnol) in DMF (5 ml) was
treated with DIPEA (90 ,ul, 0.5 ml) and 3,5-dimethylpyrazole-1-
carboxamidine (30 mg, 0.5 mmol) and heated at 80C for 12 h. The
solution was evaporated and the residue purified by chromatography on
neutral al--mini~ (CH2C12/CH30H/NH40H, gO/20/1) to give the desired
product as a white solid. ~his material was deprotected with TFA in the
usual manner and purified by preperative rever.se-phase chromato-
graphy to give (14-4) as a white solid.
IH NMR (300 MHz, D20) ~ 8.2 (t, lH); 7.58 (s, 2H); 7.1~ (d, 2H); 4.3~s
(t, 2H); 3.51 (m, SH); 3.45 (t, 2H); 3.2 (m, lH); 3.18 (m, 2H); 2.10
(S, 3H); 2.08 (m, 2H); 1.8 (m, 2H).
2(S)-(p-Toluenesulfonylamino)-3-[5,6,7,8-tetrahydro-4-oxo-5-[3[N-
(imidazolin-2-yl)amino]propyl]-4H-pyrazolo[1,5-a] [1 -4]diazepin-2-
yllcarboxyllaminolpropionic acid (14-5)
A solution of 14-3 was reacted with 2-methylthio-2-
imidazoline hydroiodide using the procedure described in 14-4. The
crude material was deprotected with TFA and 14-5 isolated by
preperative reverse-phase chromatography.
lH NMR (300 MHz, DMSO-d6) 8.20 (t, lH); 8.10 (d, 2H); 7.60 (d, 2H);
7.21 (d, 3H); 6.86 (s, lH); 4.46 (t, 2H); 4.01 (m, 2H); 3.8-3.5
(overlapping m, 8H); 2.23 (s, 3H); 2.10 (t, 2H); 1.80 (t, 2H).
W O 94/18981 215~ 7 2 3 ~CTrUS94/01881
- 1 0 1 -
SCHEME 15
1. Isobutylchloroformate
NMM, THF
5-6
~6NHSO2~ 2. Et3N, THF, DIEA
A-7
O O
BocN~ (CH2)2-lN ~ NH /~6C2H
~ N--N NHSO2~cH3
15-1
C H2CI2
O O
HN~(CH2)2 ,NJ~NH~6CO2H
--N NHSO2~CH3
1 5-2
WO 94/18981 , PCT/USg4/01881
L~s~j3~3 102-
2(S)-[(p-Toluenesulfonyl)amino]-3-[[[5,6,7,8-tetrahydro-4-oxo-5-[2-(N-
BOC-piperidin-4-yl)ethyl]-4H-pyrazolo[1,5-a]-[1,4]diazepin-2-yl]-
carbonvllaminolpropionic acid (15-1)
A solution of 5-6 (5.0 g, 12.3 mmol) in THF (150 ml) was
cooled to 0-10 and N-methylmorpholine (2.11 ml, 19.2 mmol) was
added via syringe. After mixing 20 min., isobutyl chloroformate (2.38
ml, 18.2 mmol) was added dropwise via syringe, ~nd the resulting
solution was stirred for 0.5 h to afford the desired mixed anhydride.
~:Z (7.00 g, 27.1 mmol), THF (125 lml), and
diisopropylethylamine (4.71 ml, 27.1 mmol) were combined in a 500
ml round bottom flask with a magnetic stir bar. ~ater was added in
small portions until a clear solution resulted. The resulting solution was
cooled in an ice bath. The mixed anhydride suspension was added in a
single portion to the solution of 9 with vigorous mixing. After 20 min.
stirring the reaction solution was concentrated to remove THF. The
rem~inin~ aqueous material was acidified with 10% potassium bisulfate
and the resulting precipitate was filtered to give white solid.
This material was subjected to flash column
chromatography using silica (EM Science, 230-400 mesh, 10 x 20 cm).
The column was eluted with methylene chloride:methanol:ammonium
hydroxide 98:2:0.2, 95:5:0.5, 90:10:1, then 85:15:1.5 to give the pure
15-1 as a white solid.
lH NMR (DMSO-d6) ~ 8.23 (q, J = 3.40 Hz, lH); 7.64 (d, l = 8.20 Hz,
25 2H); 7.32 (d, J = 8.20 Hz); 7.2-7.0 (br, lH); 6.86 (s, lH); 4.36 (t, J =
6.70 Hz, 2H); 3.89 (d, br, J = 12.21 Hz, 2H); 3.59 (m, lH); 3.47 (t, J =
7.08 Hz, 2H); 3.5-3.1 (m, br, SH, H2O); 2.8-2.6 (br, 2H); 2.33 (s, 3H);
2.17 (t, J = 6.47 Hz, 2H); 1.66 (d, br, J = 11.97 Hz, 2H); 1.55-1.45 (m,
br, 3H); 1.37 (s, 9H); 1.1-0.9 (m, br, 2H).
WO 94/18981 ~ L 23 PCT/US94/01881
- 103-
..
2(S)-[(p-Toluenesulfonyl)amino]-3-[[[5 ,6,7,8-tetrahydro-4-oxo-5-[2-
(piperidin-4-yl)ethyl]-4H-pyrazolo-[ 1 ,5-a] [ 1 ,4]diazepin-2-yl]carbonyl]-
aminolpropionic acid (15-2)
15-1 (7.42 g, 1 1.48 mmol) was placed in a lL round
5 bottom flask equipped with a magnetic stir bar. Methylene chloride was
added and the reaction mixture was cooled to 0-5. Hydrogen chloride
was bubbled through the suspension with stirring. After about 2 min.
the solid went into solution, and soon afterward a second precipitate
formed. After bubbling gas through the suspension for an additional 5
o min. the reaction flask was warmed to room temperature. After 30
min. the contents of the reaction flask were concentrated. The resulting
white solid was the hydrochloride salt of 15-2 and by HPLC analysis
was of >99% purity.
This hydrochloride salt of 15-2 was subjected to ion
5 exchange chromatography using Dowex SOX8-200 ion exchange resin
(1 10 g, 4.1 1 meq/g). The resin was prepared by washing with water,
methanol, water, 6N hydrochloric acid, and water (500 ml each). At
this time the eluent was pH7. The hydrochloride was dissolved in
water (30 ml) and then applied to the top of the column. The column
was eluted with water. The pH of the eluant became strongly acidic.
When the pH of eluant returned to 7, the column was eluted with
ammoniun hydroxide:acetonitrile:water 50:25:25(1.5L). Portions
containing U.V. active material were combined then concentrated at
high vacuum. The resulting white foam was dried for ~ h on the high
2s vacuum to provide 15-2.
lH NMR(DMSO-d6)~9.0-8.5 (br, lH);8.19-8.16 (m, lH); 7.67 (d, J
=8.18Hz,2H);7.32 (d, J = 8.18Hz,2H);6.89(s,1H);4.38 (t, J = 6.84
Hz,2H);3.75-3.65 (m, br, lH);3.46 (t, br, 2H);3.5-3.1 (m, br, 8H,
H20);2.77 (t, br, J = 11.36,2H);2.35(s,3H);2.17(t, J = 6.47Hz,
2H);l.~0 (d, br, J = 12.7 Hz,2H);1.53-1.42 (m, br, 3H);1.33-1.24 (m,
br, 2H).
WO 94/18981 ~1~U~47018f~1
- 104-
a3
Using the methods set forth previously, particularly in
Schemes 3 and 4, the following compounds of Table 1 were prepared.
TABLE 1
HN~-N 9~`X--
R mp (C) s~lt form
SO2~OCH3 115-120 TFA
110-1 20 TFA
SO2~CH 160-165 zwiterion
H 195-198 zwiterion
So2~_43 180-188 HCI
O
150-156 HCI
CH3
~NH~ 3 195-200 zwiterion
N O 121-124 TFA
0~
WO 94/18981 PCT/US94/01881
- 105 21~5123
TABLE l (CONT'D)
R mp(C) salt form
s SO2/~ 210-212 TFA
SO2- CH3 85-95 TFA
SO2~CI 175-180 TFA
SO2~ 115-120 HCI
~N 78-80 TFA
SO ~NH HCI
SO ,NH~O~ph 200 (dec) HCI
SO ,NH2 210 (dec) HCI
Additional compounds, prepared according to procedures
analogous to those of the exemplary procedures described above, are
shown in the following tables:
~ ; l /U~4/0188
wo 94/18981
,, ~
3 - 106-
TABLE 2
HN~ N~co2H
R A B mp(C)
,C2H5
C2H~ H H 145-152
~N,CH3
H H H 168-170
~ N,CH3
H CH3 H 164-167
~N~
H H H 110-135
-CN H H 180-188
PCTlUS94/01881
WO 94/18981
- 107 - ~ $5
TABLE 2 (CONT'D)
R A B mp(C
o
L~ N,C2H5
C2H5 HNHSO2C4Hg 156-160
l O ~ N,C H3
o H HNHSO2C4Hg 134-140
~ N~CH3
lS CH3 HNHSO2C4Hg 110-125
I~N--
HNHSO2C4H9 130-135
N~J HNHSO2C4Hg 128-135
~N,C2H5
C2H5 HS2~CH3 185-190
-
E~CTrUS94/01881
W O 94/18981
1 0~ -
TABLE 3
HN~ ~N ~ NH-(C Hz)m-C H2CO2H
n m mp (C)
2 0 110-115
2 1 115-120
121-123
0 2 135-141
0 1 140-145
WO g4118981 . PCT/US94/01881
- 109- ~ 3
TABLE 4
~N~ NH~co2H
~/ R
amide stereochemistry N H~
R relative to H6 chemical shift (PPM)
H cis 6.35
H trans 7.38
lS NHSO2C4Hg cis 6.31
NHSO2C4Hg trans 7.29
"H6" refers the hydrogen group at position 6 of the bicyclic structure
o
2 s N~
W O 94/18981 P~CTtUS94tO1881
1 1 0
TABLE S
HN~ ~NH~
R1 R~ M P (C)
H SO2C4Hs 110-116
H SO2~CH3 186
CH3 SO2~CH3 164-171
H SO2~ Cl 164-170
O
~,OH 135-1 40
SO2~ not determined
Br
H S O2 ~ CH3 173-1 74
3 0 (~N 185-189
H H 178-179
WO 94/18981 ~2 I 5~1 2 3 PCT/US94/01881
- 111 -
TABLE 6
HN~ ~n R2
n R1 R2 MP ( C)
1 H H 248-249
1 H NHSO2~CH3 175-180
1 CH3 H 118-122
1 CH3 NHSO2~CH3 155-160
2 Ph H 137-139
2 Ph NHSO2~CH3 185-188
2 H NHSO2~CH3 192-194
2 H NHCO2 - Ph 176-178
H NHSO2C3H7 168-170
2 H NHSO2C2H5 173-174