Note: Descriptions are shown in the official language in which they were submitted.
WO94/18224 PCT~W4/003~
21S5~3,~
STEROIDS FOR TREATING MENOPAUSi~L COMPLAINTS
The invention relates to the use of steroids for the
manufacture of a medicament for treating or preventing
menopausal complaints, especially for treating or
preventing osteoporosis.
Many of the steroids according to this invention, which
are used for the treatment of menopausal complaints, are
known per se.
For instance, ll~-alkyl steroid are known from USP
3,983,144. These steroids are described to have
interesting anti-fertility activity. Other ll~-alkyl
steroids having anabolic, androgenic, and progestational
activity are disclosed in USP 3,325,520. Related
progestational compounds for menstrual and ovulation
control are further known from i~ustralian patent
application AL 6614974, European patent application
0,145,493, and USP 3,465,010. Steroids having unsatu-
rated hydrocarbon groups at position ll of the steroid
skeleton are known from USP 4,292,'251, which patent
discloses uterotropic and ovulation inhibiting activity,
and from above-mentioned EP 0,145,493.
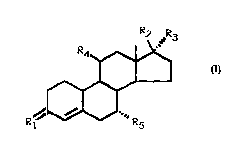
It has now been found that compounds having the general
formula:
~ ~R3
R4~
R~' ~ I
WO94/182~ PCT~4/003~
Z
2~S5~2..,'~
wherein
R1 represents O, (H,OH), or two hydrogens atoms;
R2 is hydroxy, optionally etherified or esterified;
R3 is (2-6 C) alkynyl, optionally substituted with
hydroxy;
R4 is CN or one of the hydrocarbon groups selected from
(1-6 C) alkyl, (1-6 C) alkoxy, (2-6 C) alkenyl, (2-6 C)
alkynyl, and (2-6 C) alkylidene, each of said hydro-
carbon groups may optionally be substituted with
halogen, hydroxy or (1-6 C) alkoxy; and
R5 is hydrogen or (1-6 C) alkyl, can be used for
treating or preventing postmenopausal complaints, and
especially osteoporosis.
One of the most serious menopausal complaints is bone
loss (osteoporosis), which characteristically affects
women. The aim of this invention is to provide a
medicament which is able to prevent bone loss, and
possibly to increase bone mass, and further to treat
climacteric complaints. Preferably the active ingre-
dients of these medicaments have strong estrogen and
weak or non-existing androgenic activity. Preferred
medicaments further have favourable bleeding properties,
do not induce endometrium proliferation, and have a
favourable HDL/LDL (high/low density lipid) ratio.
In a preferred embodiment the compounds have the general
formula I wherein Rl represents O or two hydrogens
atoms; R2 is hydroxy; R3 is ethynyl; R4 is selected from
the group consisting of methyl, (2-6 C) alkynyl, (2-6 C)
alkylidene, and one of (2-6 C) alkyl, (2-7 C) alkoxy-
alkyl, (1-6 C) alkoxy, or (2-6 C) alkenyl, which may
optionally be substituted with halogen; and R5 is hydro-
gen or (1-6 C) alkyl.
More preferred is the use of stero ds having the general
formula I wherein: Rl is O; R2 is hydroxy; R3 is ethyn-
WO94/18224 PCT~4/003~
~ 3 21 SSS~2
y'; R4 is ethyl, 2-fluoroethyl, ethynyl, (2-6 C) alkenyl
optionally substituted with fluorine, o;l (2-6 C) alkyli-
dene optionally substituted with fluorine; and R5 is
hydrogen or methyl. In the preferred embodiment R5 is
hydrogen.
Even more preferred is the use of steroids having the
general formula I wherein: R1 is O; R2 :Ls hydroxy; R3 is
ethynyl; R4 is ethyl or ethynyl; and R5 is hydrogen.
The invention further relates to new steroids having the
general formula I wherein:
R1 represents O;
R2 is hydroxy, optionally etherified or esterified;
R3 is (2-6 C) alkynyl, optionally substituted with
hydroxy;
R4 is CN, (2-6 C) alkyl optionally subst:ituted with
halogen, or (2-6 C) alkenyl substituted with halogen;
and
R5 is hydrogen or (1-6 C) alkyl;
or wherein
Rl represents two hydrogens atoms;
R2 is hydroxy, optionally etherified or esterified;
R3 is (2-6 C) alkynyl, optionally substi.tuted with
hydroxy;
R4 is CN or one of the hydrocarbon groups selected from
(2-6 C) alkyl, (1-6 C) alkoxy, (2-6 C) alkenyl, (2-6 C)
alkynyl, and (2-6 C) alkylidene, each oi-` said hydro-
carbon groups may optionally be substituted with
halogen, hydroxy or (1-6 C) alkoxy; and
R5 is (1-6 C) alkyl.
The steroids of general formula I wherein R1 is O; R2 is
hydroxy; R3 is ethynyl; R4 is 2-fluoroet:hyl or 2-fluoro-
ethenyl; and R5 is hydrogen are preferred steroids.
WO94/18224 PCT~4/003~
~l$5~3~
Other preferred steroids are steroids of general formula
I wherein Rl represents two hydrogens atoms; R2 is
hydroxy; R3 is ethynyl; R4 is (2-6 C) alkyl, (2-6 C)
alkylidene or (2-6 C) alkenyl, each of which groups may
be substituted with fluorine; and R5 is methyl.
In the definition of R2 the OH group may be etherified
or esterified. The term etherified means that the
hydroxy group is etherified with a lower alkyl group,
preferably having 1-6 carbon atoms, such as methyl,
ethyl, propyl, sec-butyl and the like. The term
esterified means that the hydroxy group is esterified
with a lower alkanoyl group, preferably having 2-6
carbon atoms, like acetyl, propionyl, and the like. In
principle any ester suffices as long as the ester group
cleaves when the compound is administered in vivo.
The (1-6 C) alkyl group in the definition of formula I
is a branched or unbranched alkyl group having 1-6
carbon atoms, such as methyl, ethyl, propyl, butyl,
tert-butyl, pentyl and hexyl. Preferably the alkyl group
is methyl (especially for R5) and ethyl (especially for
R4). The term (2-6 C) alkyl has the same meaning with
the exception of methyl.
The (2-6 C) alkenyl group is a branched or unbranched
alkenyl group having 2-6 carbon atoms, like vinyl, 2-
propenyl, and 1,3-butadienyl.
The (2-6 C) alkynyl group is a branched or unbranched
alkynyl group having 2-6 carbon atoms, like ethynyl,
propynyl, butynyl, and the like.
The (2-6 C) alkylidene group is a branched or unbranched
alkylidene group having 2-6 carbon atoms, like
ethylidene, propylidene, 2-methylpropylidene, and the
like.
WO94/18224 PCT~P94/00348
5 21~1SS
The term halogen used in the definition of formula I
means fluorine, chlorine, bromine or iodine. Fluorine is
the preferred halogen.
5 The term (1-6 C) alkoxy means an alkoxy group, the alkyl
moiety of which is a (1-6 C) alkyl group as previously
defined.
The term (2-7 C) alkoxyalkyl means a (1-6 C) alkyl group
as previously defined substituted by a (1-6 C) alkoxy
group as previously defined, the total number of carbon
atoms being between 2 and 7.
The new compounds of this invention ca,n be prepared by
condensation of 11-keto steroids of the general formula:
~ 3
R ~ "'R5 II
wherein
R1 represents O;
R2 is hydroxy, optionally etherified or esterified;
R3 is (2-6 C) alkynyl, optionally substituted with
hydroxy; and
R5 is hydrogen or (1-6 C) alkyl;
or wherein
R1 represents two hydrogens atoms;
R2 is hydroxy, optionally etherified or esterified;
R3 is (2-6 C) alkynyl, optionally substituted with
hydroxy; and
R5 is (1-6 C) alkyl;
the active groups of which are optiona,:Lly protected, is
condensed with a Wittig(-like) compound of the formula
R4'R4''CH-W, wherein R4'R4''C forms the group R4 which
is the optionally halogen, hydroxy, or(1-6 C) alkoxy
-
WO94/18224 215~ 5 PCT~ ~4/003~
~.
substituted (2-6 C) alkylidene group as previously
defined, or R4'''Li, wherein R4''' is (2 6 C) alkyl or
(2-6 C) alkenyl optionally substituted with halogen,
hydroxy, or (1-6 C) alkoxy, in which reactive groups can
be protected by protective groups which are known in the
art (see for example T.W. Green: Protec~ive Groups in
Organic Synthesis, Wiley, NY, 1981), and W is a Wittig,
Wittig-Horner or Peterson-like moiety, optionally
followed by halogenation and dehydration or by
hydration, after which the compound obtained is
converted into a nitrile or condensed with a Wittig(-
like) compound of the formula R6W, wherein W has the
previously given meaning and R6 is independently
hydrogen, halogen or (1-6 C) alkyl, followed by
hydroboration, optionally followed by alkylation,
halogenation, or halogenation and dehydrohalogenation,
or (partial) hydrogenation, after which optionally
present protective groups are removed.
Suitable reagents are triphenylphosphoranes such as
R4'R4''CH-P(EIal)Ph3 and the like, and suitable Peterson
reagents are, for example, trimethylsilane reagents like
R4'R4''C(MgHal)Si(CH3)3, wherein Hal denotes a halogen
like chlorine or bromine.
The steroids of the invention can be used to prevent and
treat estrogen deficiency induced disorders such as
menopausal complaints, as is demonstrated in the
estrogen-induced bone loss assay. In this assay young
mature female Wistar rats are ovariectomized and treated
with the test compound for 1 month. After 1 month blood
is collected and in the lithium-heparin plasma the bone
turnover parameter (osteocalcin) is determined according
to the method of Verhaeghe et al., J. Endrocrinol., 120,
143-151. At autopsy the right femur is dissected and the
bone density of the distal part of the metaphyse is
measured using an X-ray densitometric method. The bone
density, in mm aluminum equivalent, is expressed in a
W094118-~4 ' ~ ~T~P94/00348
percentage relative to the intact control group which is
100 ~ by definition, and to the ovariectomized control
group which is 0 % by definition.
The value for osteocalcin is defined as 100 % for the
ovariectomized group and 0 % for the intact group.
Active compounds inhibit the bone turnover, and thus
have an osteocalcin value lower than loO %.
Table I gives the results of this assay.
Table 1
dose bone osteo-
Rl R2 R3 R4 R5density calcin
(~g) (%) (%)
reference:
O OH C-CH H H 1000 24 55
this invention:
O OH C-CH C2H5 H 64 94 -117
O OH C-CH CH2Cl H 10 57 -78
O OH C-CHCH2OCH3 H 250 66 -45
O OH C--CHCH=CH2 H 100 48 -52
O OH C-CH C-CH H 250 115 -117
O OH C--CH C--N HlD00 113 -35
O OH C--CHCH2 CH CH3lD00 90 -80
O OH C5CH OCH3 H1000 73 -46
O OH C--CH CH3 H 125 81
O OH C--CHCH2CH2F H 500 93 -59
O OH C--CH (E)=CHCH3 H 64 86
O OH C--CH C--N H 125 119 -66
O OH C-CH C2H5 CH3250 115
- 30 H2 OH CeCH CH3 H1000 80 -54
H2 OH C-CH CH2Cl H 10 43 -72
H2 OH C-CH CH2Cl H 100 81 -112
H2 OH C--CHCH=CH2 CH31000 124 -80
H2 OH C--CH C-CH H 32 66 -17
WO94/182~ 2 ~S ~ ~ 3 PCT~4/003~
The compoùnds of the invention may be administered
enterally or parenterally, and for humans preferably in
a daily dosage of O,OO1-lO mg per kg body weight. Mixed
with pharmaceutically suitable auxiliaries, e.g. as
described in the standard reference, Gennaro et al.,
Remington's Pharmaceutical Sciences, (18th ed., Mack
Publishing Company, 1990, see especially Part 8:
Pharmaceutical Preparations and Their Manufacture) the
compounds may be compressed into solid dosage units,
such as pills, tablets, or be processed into capsules or
suppositories. By means of pharmaceutically suitable
liquids the compounds can also be applied as an
injection preparation in the form of a solution,
suspension, emulsion, or as a spray, e.g. a nasal spray.
For making dosage units, e.g. tablets, the use of
conventional additives such as fillers, colorants, poly-
meric binders and the like is contemplated. In general
any pharmaceutically acceptable additive which does not
interfere with the function of the active compounds can
be used.
Suitable carriers with which the compositions can be
administered include lactose, starch, cellulose
derivatives and the like, or mixtures thereof, used in
suitable amounts.
The invention is further illustrated by the following
examples.
Example 1
To a solution of 115 g of (7~,11~)-11-hydroxy-7-methyl-
estr-4-en-3,17-dione in 5 1 of acetone were added drop-
wise at 0.5 C llO ml of 8 N chromic acid. After
stirring for 1 h 50 ml of propanol-2 were added and 15
min later the mixture was concentrated partially and
diluted with 3 1 of water. After stirring for several
W094/l~2U ~555 CT~ ~4/0~3
hours, the precipitate was filtered, taken up in a small
volume of dichloromethane and dried wit:h sodium sulfate,
to give after evaporation of the organic solvent 107 g
of triketone (7~)-7-methylestr-4-en-3,~1,17-trione. Rf =
0.54 (toluene-ethyl acetate 4-6 v/v).
A mixture of 115 g of (7~)-7-methylestr-4-en-3,11,17-
trione, 8 g of p-toluenesulfonic acid and 40 ml of
ethanedithiol in 1 l of absolute ethanol was refluxed
for 1 h. After cooling the mixture was diluted with 1 l
of water and stirred in the cold for several hours. The
precipitate was filtered and washed with 1 N NaOH, water
and cold methanol. After drying 134 g of (7a)-3,3-
ethanedithio-7-methylestr-4-en-11,17-dione were obtain-
ed. Rf = 0.62 (toluene-ethyl acetate 8~2).
A solution consisting of 36 g of (7~)-3,3-ethanedithio-
7-methylestr-4-en-11,17-dione, 300 m~l of dichloro-
methane, 85 ml of triethylorthoform,ate, 70 ml of
ethylene glycol and 2 g of p-toluenesulfonic acid was
stirred for 7 h. Then the mixture wa~ washed with 10%
sodium carbonate solution and dried, concentrated and
passed through a silica gel column to provide 44 g of
product (7~)-3,3-ethanedithio-17,17-ethylenedioxy-7-
methylestr-4-en. Rf = 0.65 (toluene-ethyl acetate 8/2).
A mixture of 9.2 g of potassium tert-butoxide, 34.8 g of
methyltriphenylphosphonium bromide in :260 ml of toluene
was refluxed for 1 h. Then 7.8 g of (7~)-3,3-ethanedi-
thio-17,17-ethylenedioxy-7-methylestr-4-en were added
and the mixture was boiled for another 2 h. Then the
reaction was cooled, washed with water, dried and
concentrated. The residue was chromatographed to give
6.1 g of (7~)-3,3-ethanedithio-17,17-ethylenedioxy-7-
methyl-11-methylene-estr-4-en. Rf = 0.77 (toluene-ethyl
acetate 9/1).
WO941182~ . ~ PCT~4/003~
~ 2~S32 1 0 ~
A mixture of 6.1 g of (7~)-3,3-ethanedithio-17,17-
ethylenedioxy-7-methyl-11-methylene-estr-4-en, 90 ml of
acetone, 50 ml of tetrahydrofuran and 3 ml of 6 N
hydrochloric acid was stirred at ambient temperature.
After 1 h the mixture was diluted by addi~ion of 700 ml
of 5~ sodium carbonate and stirred for ~ h. The
precipitate was filtered and dried to provide 5.4 g of
(7~)-3,3-ethanedithio-7-methyl-11-methylene-estr-4-en-
17-one. Rf = 0.74 (toluene-ethyl acetate 9/1).
Acetylene gas was passed through a solution of 50 g of
potassium tert-butoxide in 500 ml of dry tetrahydro-
furan. After 2 h a solution of 30 g of (7~)-3,3-
ethanedithio-7-methyl-11-methylene-estr-4-en-17-one in
400 ml of tetrahydrofuran was introduced dropwise at 0
C. After stirring for an additional 1 h the mixture was
poured into 7 l of water and stirred for an additional ~
h. The precipitates were filtered and dried to give 31 g
of (7~,17~)-3,3-ethanedithio-17-hydroxy-7-methyl-11-
methylene-19-norpregn-4-en-20-yne. Rf = 0.64 (toluene-
ethyl acetate 9/1).
To a solution of 8 g of sodium in 300 ml of liquid
ammonia at -50 C was added dropwise a solution of 16 g
of (7~,17~)-3,3-ethanedithio-7-methyl-11-methylene-19-
norpregn-4-en-20-yn-17-ol in 75 ml of tetrahydrofuran.
After stirring for 1 h, excess sodium was destroyed by
addition of 10 ml of ethanol. After evaporation of
ammonia, the residue was partioned between dichloro-
methane and water. The organic layer was separated,
washed and dried. The material which left after
evaporation of the solvent was chromatographed to
provide 5.4 g of (7~,17~)-7-methyl-11-methylene-l9-nor-
pregn-4-en-20-yn-17-ol. Rf = 0.59 (toluene-ethyl acetate
9/l).
094J1~ZJ ~ CT~W4/003
Example 2
~ A mixture of 15 g of (7~,17~)-3,3-ethanedithio-7-methyl-
11-methylene-19-norpregn-4-en-20-yn-17--ol, 300 ml of
methanol, 20 ml of water, 6 g of calcium carbonate and
51 ml of methyl iodide was refluxed for 6 h. After
filtration over Hy-flow, the filtrate was concentrated
taken up in dichloromethane, washed, dried and eva-
porated. The r~; n; ng residue was chromatographed to
provide 6.5 g of (7a,17~)-17-hydroxy-7--methyl-11-methyl-
ene-19-norpregn-4-en-20-yn-3-one. Rf = 0.40 (toluene-
ethyl acetate 9/1).
Example 3
To a solution of 134 g of (7~)-3,3-ethanedithio-7-
methylestr-4-en-11,17-dione in 5 1 of dry tetrahydro-
furan were added in portions 110 g of lithium tri-tert-
butoxy aluminium hydride at 0-5 C. Aft:er stirring for 3
h the mixture was poured into 10 1 of ice water and
acidified slightly by addition of 1 1 of 2 N
hydrochloric acid. The product was extracted with ethyl
acetate. After drying with sodium su]fate the organic
material was treated with diethyl ether, to provide 133
g of essentially pure (7~,17B)-3,3-ethanedithio-17-
hydroxy-7-methylestr-4-en-11-one. Rf = 0.45 (toluene-
ethyl acetate 6/4).
To a solution of 132 g of (7~,17~)-3,3-ethanedithio-17-
hydroxy-7-methylestr-4-en-11-one in 800 ml of pyridine
were added at 0 C 182 ml of trimethylsilyl chloride.
After stirring for 1 h the mixture was poured into ice
water and the product was extracted with ethyl acetate.
After washing, drying and evaporation of the solvent,
the residue was co-evaporated with 1toluene and then
treated with hexane to provide 137 g of (7~,i7~)-3,3-
WO94/18224 '`
2 1~ S 5 3 2 1 2 PCT~W4/003~ ~
e_hanedithio-7-methyl-17-trimethylsilyloxy-estr-4-en-11-
one. Rf = 0.63 (toluene-ethyl acetate 6/4).
To a suspension of 334 g of methoxymethyltriphenyl-
phosphonium chloride in 6 1 of dry ether were added
dropwise 600 ml of 1.6 M ~utyl lithium at 0.5 C. After
stirring for 1 h a solution of 44.6 g of (7Q,17B)-3,3-
ethanedithio-7-methyl-17-trimethylsilyloxy-estr-4-en-11-
one in 1.5 1 of ~iethyl ether were added and the mixture
was stirred fo~ 24 h. The organic solution was then
washed with water and dried. The residue which remained
after concentration of the organic phase was partitioned
between hexane-methanol-water (1/0.7/0.3 v/v/v), and
stirred for lS min. The hexane phase was dried and
concentrated to ?rovide 47 g of (7Q,17B)-3,3-ethanedi-
thio-ll-methoxyme~nylene-7-methyl-17-trimethylsilyloxy-
estr-4-en. Rf = 0.50 (hexane-ethyl acetate 3/1).
To a solution of 265 g of (7Q,17B)-3,3-ethanedithio-11-
methoxymethylene-7-methyl-17-trimethylsilyloxy-estr-4-en
in 800 ml of acetone were added 80 ml of concentrated
hydrochloric acid and the mixture was stirred at room
temperature. After 1 h the mixture was poured into water
and extracted with ethyl acetate. After washing, drying
and evaporation of the organic solvent, the remaining
residue was passed through a silica qel column and
eluted with dichloromethane-acetone 9/1 to provide 63 g
of (7Q,llB,17B)-3,3-ethanedithio-17-hydroxy-7-methyl-
estr-4-en-11-carboxaldehyde. Rf = 0.38 (toluene-ethyl
acetate 7/3).
To a solution of 55 g of (7Q,11~,17B)-3,3-ethanedithio-
17-hydroxy-7-methylestr-4-en-11-carboxaldehyde and 165
ml of dihydropyrane in 1100 ml of dry tetrahydrofuran
were added 1.3 g of p-toluenesulfonic acid. After
stirring for 2 h the mixture was poured into 5 1 of 5~
sodium hydrogencarbonate solution and the product was
WO94/182
1 3 ~ PCT~4/003
extracted with ethyl acetate. After concentration of the
organic phase (7~,11B,17~)-3,3-ethanedithio-17-tetra-
hydropyranyloxy-7-methyl~str-4-en-11-carboxaldehyde was
isolated.
A mixture of 1 g of (7~,11B,17B)-3,3-ethanedithio-17-
tetrahydropyranyloxy-7-methylestr-4-en-~1-carboxaldehyde
and 2 g of hydroxylamine hydrochlori~e in 12 ml of
pyridine was stirred at 80 C for 1 h. Then it was
cooled, poured into water and extracted with ethyl
acetate. After washing, drying and concentration O.9 g
of amorphous (7Q,llB,17B)-3,3-ethanedithio-17-tetra-
hydropyranyloxy-7-methylestr-4-en-11-carboxaldehyde
oxime were obtained. Rf = 0.60 (toluene-ethyl acetate
8/2).
Dehydration of the oxime was effected by reacting 0.8 g
of (7~,11B,17B)-3,3-ethanedithio-17-tetrahydropyranyl-
oxy-7-methylestr-4-en-11-carboxaldehyde oxime in 8 ml of
acetic anhydride during 45 min. Concomitant replacement
of the 17-tetrahydropyran ether by an acetate group was
observed. The reaction mixture was poured into 50 ml of
ice water and stirred for 30 min. After neutralization
with 2 N sodium hydroxide and extraction with ethyl
acetate 0.75 g of (7~,11B,17B)-3,3-ethanedithio-17-
acetyloxy-7-methylestr-4-en-11-carbonitrile was
obtained. Rf = 0.58 (toluene-ethyl acetate 9/1).
The acetate functionality was saponi:Eied by stirring
0.75 g of (7~,1lB,17B)-3,3-ethanedithio-17-acetyloxy-7-
methylestr-4-en-11-carbonitrile for 30 min in a mixture
of 20 ml of tetrahydrofuran and lO ml of water
containing 1 g of sodium hydroxide. The mixture was
diluted and extracted with ethyl acetate. After drying
and evaporation 0.50 g of (7~,11B,17B)-3,3-ethanedithio-
17-hydroxy-7-methylestr-4-en-11-carbonitrile was
obtained. Rf = 0.34 (toluene-ethyl acetate 9/l).
WO94/182~ PCT~4/003
2~ss~32
To a solution of 20 g of (7~,11B,17B)-3,3-ethanedithio-
17-hydroxy-7-methylestr-4-en-11-carbonitrile in 600 ml
of dry dichloromethane were added 20 g of sodium
acetate, followed by 85 g of pyridinium chlorochromate.
After stirring for 3 h the reaction turned out to be
complete. Excess oxidant was removed by addition of 40
ml of propanol-2. The mixture was filtered over Hy-Flow,
concentrated and chromatographed to provide 13 g of
(7~,11B)-3,3-ethanedithio-17-keto-7-methylestr-4-en-11-
carbonitrile. Rf = 0.75 (toluene-ethyl acetate 8/2).
Acetylene gas was passed through a solution of 10.5 g of
potassium tert-butoxide in 60 ml of dry tetrahydrofuran
for 1 h at 0 C. Then a solution of 9.3 g of (7~,11B)-
3,3-ethanedithio-17-oxo-7-methylestr-4-en-11-carbo-
nitrile in 100 ml of tetrahydrofuran were added drop-
wise. After stirring for 1 h at 0-5 C the mixture was
poured into 500 ml of saturated ammonium chloride
solution and the product was extracted with ethyl
acetate. After washing, drying and concentration 9.5 g
of (7~,11B,17~)-3,3-ethanedithio-17-hydroxy-7-methyl-19-
norpregn-4-en-20-yne-11-carbonitril was obtained. Rf =
0.28 (toluene-ethyl acetate 9/1).
A mixture of 6.5 g of (7~,11B,17a)-3,3-ethanedithio-17-
hydroxy-7-methyl-19-norpregn-4-en-20-yne-11-carbo-
nitrile, 200 ml of methanol, 100 ml of tetrahydrofuran,
2.4 g of calcium carbonate, 8.5 ml of water, and 35 ml
of methanol was refluxed for several hours, additional
methanol was added from time to time and after dis-
appearance of starting material the mixture was cooled,
filtered and concentrated. The residue was passed
through a silica column and provided 2.5 g of pure
(7~,1lB,17~)-17-hydroxy-7-methyl-3-oxo-19-norpregn-4-en-
20-yne-11-carbonitrile. M.p. 234 C. Rf = 0.33 (toluene-
ethyl acetate 7/3).
~094/I82
PCT~4/003
1 5 ~ss3~
~xample 4
Hydroboration of the 11-methylene function in 3,3,17,17-
bis(ethylenedioxy)-ll-methylene-estr-5-en was accom-
plished as follows:
To a solution of 2.18 ml of 10 ~[ boron hydride-
dimethylsulfoxide complex in 10 ml of dry tetrahydro-
furan were added at 0 C 2.7 ml of cyclo-octadiene.
After additional reflux for 1 h a solution of 2.7 g of
3,3,17,17-bis(ethylenedioxy)-11-methylene-estr-5-en in
30 ml of tetrahydrofuran were added. The mixture was
stirred for 16 h and then treated with 10 ml of 10~
sodium hydroxide, followed by 10 ml of 30~ hydrogen
peroxide. After stirring for an additional 4 h the
mixture was poured into water and the product extracted
with dichloromethane. Final purification was achieved by
chromatography to provide 2 g of (l~B)-3,3,17,17-bis-
(ethylenedioxy)-11-(hydroxymethyl)-estr-5-en. Rf = 0.25
(toluene-ethyl acetate 1/1).
To a suspension of 20 g of pyridinium chlorochromate in
200 ml of dichloromethane were added 9.3 g of (llB)-
3,3,17,17-bis(ethylenedioxy)-11-(hydroxymethyl)-estr-5-
en in 100 ml of dichloromethane. After stirring for 1 h,
excess oxidant was destroyed by addition of 40 g of
sodium hydrogensulfite in 200 ml of water, followed by
extraction of the product with ethyl acetate. After
drying and concentration of the organic phase the
residue was purified by chromatography to provide 5.4 g
of (11~)-3,3,17,17-bis(ethylenedioxy)-estr-5-en-11-
carboxaldehyde. Rf = 0.50 (hexane-ethyl acetate 1/1).
A solution of 1.6 M butyl lithium in hexane (44 ml) was
added dropwise to a suspension of ~4.4 g of chloro-
methyltriphenylphosphonium chloride in 500 ml of ether.
After stirring for 15 min a solution of 5.4 g of (11~)-
3,3,17,17-bis(ethylenedioxy)-estr-5-en-11-carboxaldehyde
.
WO9~/182~ ~5~3 PCT~/003
1 6
iII 30 ml of tetrahydrofuran were added clropwise. After
12 h the mixture was poured into 0.5 l of water and the
organic phase was separa'ed, washed, dried and concen-
trated. The residue was chromatographed to provide 3.7 g
of E/Z (llB)-3,3,17,17-bis(ethylenedioxy)-11-(2-chloro-
ethenyl)-estr-5-en. Rf = 0.4 (hexane-ethyl acetate 7/3).
To a suspension of lithium amide, prepared from 920 mg
of lithium in 130 ml of ammonia (liq.), were added at
-45 C a solution of 3.6 g of (11~)-3,3,17,17-bis-
(ethylenedioxy)-11-(2-chloroethenyl)-estr 5-en in 30 ml
of tetrahydrofuran. After stirring for 1 h the excess
reagent was destroyed by addition of 15 g of ammonium
chloride, followed by evaporation of ammonia. The
residue was partioned between dichlorometl~ane and water;
the organic phase was dried and concentrated and
chromatographed to provide 1.8 g of (11~)~3,3,17,17-bis-
(ethylenedioxy)-11-ethynyl-estr-5-en; M.p. 200 C. Rf =
0.45 (hexane-ethyl acetate 7/3).
A mixture of 8 g of (llB)-3,3,17,17-bis(ethylenedioxy)-
11-ethynyl-estr-5-en, 200 ml of acetone, 100 ml of
methanol and 100 ml of tetrahydrofuran was treated with
5 ml of 6 N hydrochloric acid and stirred overnight.
After treatment with sodium hydrogencarbonate and
concentration, the residue was chromatographed to give
5.1 g of (11~)-11-ethynyl-estr-4-en-3,17-dione. Rf =
0.48 (hexane-ethyl acetate 1/1).
A mixture of 2.7 g of potassium tert-butoxide in 12 ml
of tetrahydrofuran and 5 ml of tert-butanol was placed
under nitrogen and acetylene gas was bubbled through for
1.5 h at 0 C. Then a suspension of 1.85 g of (11~)-11-
ethynyl-estr-4-en-3,17-dione in 5 ml of tetrahydrofuran
was introduced, and stirring was prolonged for 1 h. The
mixture was diluted with water (200 ml), neutralized by
addition of 2 N hydrochloric acid and extracted with
W0941182~ ''
PCT~4/003
I 7 S~3?
e.hyl acetate, dried and concentrated, The residue was
chr~matographed to provide 1.6 g of (llB,17~
et~yny1-17-hydroxy-19-norpregn-4-en-20-yn-3-one; M.p.
~68 C. Rf = 0.60 (hexane-ethyl acetate 1/1).
Example 5
To a solution of 4 ml of borane methylsulfide (lOM in
tetrahydrofuran) in 20 ml of tetrahydrofuran were added
dropwise 5 ml of 1,5-cyclo-octadiene. ~fter stirring for
1 h a solution of 5 g of 11~-vinyl-3,3,17,17-bisethyl-
enedioxy-estr-5-ene (obtained by selective Lindlar
hydrogenation from (11~)-3,3,17,17-bi,s(ethylenedioxy)-
11-ethynyl-estr-5-en) in 25 ml of tetrahydrofuran was
added dropwise. After stirring for an additional hour 20
ml of 15~ aq sodium hydroxide and 20 ml of 30% hydrogen
peroxide were added. After stirrin,g overnight the
product was extracted with ethyl acetal:e and the organic
material thus obtained was purified by chromatography to
give 3.4 g of llr,-(2-hydroxyethyl)-3,3,17,17-bisethyl-
enedioxy-estr-5-ene; M.p. 190 C.
To a solution of 5 g of 11~-(2-hydroxyethyl)-3,3,17,1~-
bisethylenedioxyestr-5-ene in 7 ml of tetrahydrofuran
were added at -30 C 500 mg of 2,6-di-t-butylpyridine
followed by 800 mg of trifluoromethanesulfonic
anhydride. After stirring for an additional 15 min at
-30 C, 10 ml of a lM solution of tetrabutylammonium
fluoride in tetrahydrofuran were added and the mixture
was stirred for 2 h at room temperature and then poured
into 30 ml of 10~ sodium hydrogencarbonate solution.
Then the mixture was extracted with ethyl acetate and
the organic material was purified by chromatography to
give 600 mg of 11~-(2-fluoroethyl)-3,3,17,17-bisethyl-
enedioxy-estr-5-ene; M.p. 196 C.
W09~/182~ ~55~3' ~
PCT/EP94/00348 ~
1 8
To a solution of 580 mg of 11~-(2-fluoroethyl)-
3,3,17,17-bisethylenedioxy-estr-5-ene in a mjxture of 3
~l of acetone and 3 ml of tetrahydrofuran were added 6
ml of 3M hydrcchloric acid. After stirring for 2 h
sodium hydrogencarbonate and water were added and the
product was extracted with ethyl acetate. The residue
obtained after evaporation of the solvent was treated
with diisopropyl ether to give 400 mg of llB-(2-fluoro-
ethyl)-estr-4-er.-3,17-dione; M.p. 85 C.
Acetylene gas was passed into a solution of 0.7 g of
potassium tert-butoxide in 5 ml of tetrahydrofuran and 1
ml of tert-butanol. After 15 min a solution of 400 mg of
11~-(2-fluoroethyl)-estr-4-en-3,17-dione in 5 ml of
tetrahydrofuran was added. After stirring for an
additional 15 min the solution was poured into water and
the product was extracted with ethyl acetate. The
organic material thus isolated was purified by column
chromatography and treated with ether, to provide 320 mg
of (llB,17~)-11-(2-fluoroethyl)-17-hydroxy-19-norpregn-
4-en-20-yn-3-one; M.p. 212 C.
Example 6
To a solution of lithium diisopropylamide (prepared from
250 mg of diisopropylamine and 1.6 ml of a 1.6M butyl
lithium-hexane solution) in 3 ml of tetrahydrofuran was
added a solution of 600 mg of difluoromethyldiphenyl-
phosphinoxide in 2 ml of tetrahydrofuran at -50 C.
After stirring for 15 min a solution of 800 mg of (11
3,3,17,17-bis-(ethylenedioxy)-estr-5-en-11-carboxalde-
hyde in 3 ml of tetrahydrofuran was added and the
mixture was stirred overnight at room temperature. The
reaction products were poured into water and extracted
with ethyl acetate. Chromatography of the organic
material provided 465 mg of llB-(2,2-difluoroethenyl)-
~094/18~ ~ ~ T~4/0~3
3,3,17,17-bis-(ethylenedioxy)-estr-5-en; M.p. 180-181
o~
A solution of 430 mg of above-mentioned product in a
mixture of 3 ml of acetone and 2 ml of tetrahydrofuran
was treated with 2 ml of 4N hydrochloric acid. After
stirring for 2 h the mixture was neutralized with solid
sodium hydrogencarbonate and the product was extracted
with ethyl acetate. The organic material thus obtained
was crystallised from diisopropyl ether to provide 270
mg of 11~-(2,2-difluoroethylene)-estr-4-en-3,17-dione;
M.p. 150 C.
Acetylene gas was bubbled into a solution of 0.48 g of
potassium tert-butoxide in a mixture of 5 ml of
tetrahydrofuran and 0.5 ml of t-butanol. After 15 min a
solution of 250 mg of llB-(2,2-difluoroethylene)-estr-4-
en-3,17-dione in 3 ml of tetrahydrofuran was added and
15 min later the mixture was quenched with water and the
product extracted with ethyl acetate.
The organic material thus isolated was treated with
diisopropyl ether to provide 160 mg of (11~,17~)-11-
(2,2-difluoroethenyl)-17-hydroxy-19-norpregn-4-ene-20-
yn-3-one; M.p. 196 C.
Example 7
A mixture of E/Z 11~,3,3,17,17-bis-(ethylenedioxy)-11-
(2-chloroethenyl)-estr-4-en was separat:ed on a silica
column to provide the pure E-isomer (m.p. 143 C) and
- the Z-isomer (m.p. 182 C). Treatment of a solution of 3
g of (E)11~,3,3,17,17-bis-(ethylenedioxy)-11-~2-chloro-
ethenyl)-estr-5-en in 20 ml of acetone with 3 ml of
conc. hydrochloric acid during 1 h follcwed by neutrali-
zation with sodium hydrogencarbonate solution and ethyl
acetate extraction provided the desired diketone
WO9~/182~ 2 `~ PCT~4/003~ ~
(llBtE)-11-(2-chloroethenyl)-estr-4-ene-3,17-dione (2.3
g~
This product was dissolved in 10 ml of tetrahydrofuran
and added dropwise to a solution of potassium acetylide
in a mixture of 14 ml of tetrahydrofuran and 3 ml of
tert-butanol (potassium acetylide was generated by
passing acetylene gas into a solution of 2.9 g of
potassium tert-butoxide in above-mentioned t-butanol-
tetrahydrofuran mixture). After stirring for ~ h the
mixture was quenched with water and the product was
extracted with ethyl acetate. The organic material was
purified by chromatography to provide 1.9 g of
(ll~,E,17~)-ll-(2-chloroethenyl-17-hydroxy-19-norpregn-
4-en-20-yn-3-one; M.p. 180 C.
~5
Example 8
Similarly as above-described were prepared:
Rl R2 R3 R4 R5 M.p.( C)
O OH C-CH C2H5 H 217
O OH C-CH (E)CHCH3 H 213
O OH C-CH C-CH CH3 268
O OH CsCH (E)CHC4Hg CH3 230
0 OH C3CH C2H5 CH3 222
O OH C-CH CH3 CH3 219
O OH C-CH CN CH3 234
O OH C-CH CH20CH3 CH3 amorphous
O OH C-CH CH3 H 221
o OH C-CH CsCH H 165
O OH C=CH CH=CH2 CH3 223
O OH CsCH CN H 208
O OH C-CH CH=C(CH3)2 H 212
O OH C--CH (Z)CH=CHF H 215
0 OH C-CH CH2CH2~ CH3 151
O OH C-CH n C3H7 H 208
~O 94/18224 ~ . P~T/EP91/00348
2 1 ~5
Rl R2 R3 R4 R5M.p. ( C)
O OH CeCH CH=CF2 CH3169
O OH C--CH ( E ) CH=CHCH3 H 168
O OH C--CH ( Z ) CH=CHCH3 H 169
O OH C--CH (E)=CH--CH3 CH3 235
O OH C--CH CH2CH2F H 212
O OH C----CH CH=CF2 H 196
O OH C--CH ( E ) CH=CHC l H 180
O OH C--CH ( Z ) CH=CHCl H 186
O OH C--CH (E)=CH-CH2F H
O OH C----CH ( E ) =CH--CH2F CH3
H 2 OH C----CH C--CH CH 3 116
H2 OH C--CH ( E ) CHC4Hg CH3 120
H 2 OH C--CH CN CH 3 172
H2 OH C----CH C2H5 CH3 120
H2 OH C--CH CH3 CH3 119
H2 OH C----CH CH2OCH3 CH3 134
H2 OH CSCH CH3 H 107
H2 OH C--CH C--CH H 98
H2 OH C--CH C=CH2 CH3 135
~3OH OH C3CH ( E ) CHCH 3 H 145
130H OH C----CH (E)CHC4H9 H 172
130H OH C--CH C----CH CH 3 217
~3OH OH C----CH C2H5 CH3 133
BOH OH C--CH CH3 CH3 151
I~OH OH C--CH CN CH 3 243
J3OH OH C----CH CH3 H 183
13OH OH C--CH CH20CH3 CH3 amorphous
J3OH OH CsCH CH2 CH3 194
30 ~3OH OH C3CH CH=C(CH3)2 H 164
~30H OH CsCH ( Z ) CH=CHF H 79
J3OH OH C--CH CH2CH2F CH3 185
J3OH OH C--CH n C3H7 H 109
~3OH OH C----CH CH=CF2 CH3 158
~3OH OH C--CH ( E ) CH=CHCH 3 H 112
~3OH OH C--CH ( Z ) CH=CHCH3 H 9 O
WO94/182~ 2 ~$5~3~ PCT~4/003~ ~
~ 22
Rl R2 R3 R4 R5 M.p.( C)
BO~ OH C--CH (E)=CH-CH3 CH3 124
~o~ o~ C=CH CH2CH2F H 95
~OH OH C--CH CH=CF2 H 135
~OH OH C--CH (E)CH=CHCl H amorphous
BOH OH C-CH (Z)CH=CHCl H amorphous
BOH OH C--CH (E)=CH-CH2F H
~o~ OH C-CH (E)=CH-CH2F CH3
1~
Example g
A tablet having the following composition was prepared:
(11~,17~)-17-hydroxy-11-ethyl-
l9-norpregn-4-en-20-yn-3-one2.5 mg
starch 10 mg
ascorbyl palmitate 0.2 mg
magnesium stearate 0.5 mg
lactoseto make up to 100 mg
Base granules were prepared by mixing the lactose with a
portion of starch. The remainder of the starch was mixed
to a slurry with water and added to the mixture. The
whole was granulated and dried. These base granules were
mixed with ascorbyl palmitate and the active ingredient,
sieved, finely mixed with magnesium stearate and then
tabletted.
Example 10
A tablet having the same composition as in Example 9 was
prepared with (11~,17~)-11-ethynyl-17-hydroxy-19-nor-
pregn-4-en-20-yn-3-one as active ingredient.
~094/18224
_ PCT~4/003
~5~3~ 23 ~ .
E~mple 11
A tablet having the same composition as in Example 9 was
prepared with (7~ ,17~)-11-methylene-17-hydroxy-7-
m~thyl-19-norpregn-4-en-20-yn as active ingredient, by
first mixing the active ingredient with 10 % of the
lactose and the ascorbyl palmitate and then mixing this
mixture with the lactose, starch and st:arch slurry. The
mixture was dried, finely mixed with magnesium stearate
and tabletted.
Ex~mple 12
A capsule having the following composition was prepared:
(11~,17~)-11-ethyl-17-hydroxy-19-
norpregn-4-en-20-yn-3-one 2.5 mg
starch 10 mg
ascorbyl palmitate 0.2 mg
magnesium stearate 0.5 mg
Avicel to make up to 100 mg
The components were mixed with one another in the manner
described in Example 9, granulated and filled into
gelatin capsules.