Note: Descriptions are shown in the official language in which they were submitted.
21585I 7
HOECHST ARTIENGESELLSCHAFT HOE 94/F 282 Dr. TH/Pl
Description
Aminomethylphosphonic and ~;no~thylphosphonic acid
derivatives and their use for treating degenerative joint
disorders
Osteoarthritis is a degenerative joint disorder with
inflammatory episodes and progressive cartilage destruc-
tion which may lead to impairment of function or even
complete ankylosis. Although to date the concomitant
inflammations and states of pain associated with this
disorder can be treated, there are no available pharma-
ceuticals which have been proven to be able to stop or
cure the progressive cartilage breakdown. Examples of
known therapeutic agents for osteoarthritis are mixtures
of sulfated glucosaminoglycans (Current Therapeutic
Research, 40,6 (1986) 1034) or non-steroidal antiinflamm-
atory drugs, but these are unable to stop the 1088 of
cartilage.
Although the pathogenesis of osteoarthritis is still not
known in detail, it i8 regarded as certain that the
chondrocytes (cartilage cells) are crucially involved in
the increased 1088 of matrix, and that, of the main
constituents of this matrix, in particular the proteo-
glycans (PG) are the first to undergo enzymatic break-
down.
Accordingly, promising approaches to the therapy of
osteoarthritis would be provided by those drugs which,
because of their profile of action, stimulate proteo-
glycan synthesis in chon~rocytes and, furthermore,
counteract a pathologically increased rate of cartilage
breakdown. Moreover, the breakdown of proteoglycans can
be reduced either by inhibiting relevant matrix metallo-
proteinases or else by inactivating reactive oxygen free
radicals.
It has now been found that the aminomethylphosphonic and
~1S8517
-phosphinic acid derivatives according to the invention
~timulate proteoglycan synthesis in cartilage, inhibit
cartilage breakdown and effectively reduce the cartilage
breakdown induced by oxygen free radicals.
Because of their pharmacological properties, the
compounds according to the invention are outstAn~ingly
suitable for the treatment and prophylaxis of
degenerative joint disorders, as well as of disorders of
the rheumatic type in which cartilage breakdown is to be
found, such as for rheumatoid arthritis, joint trauma and
chondrolysis after lengthy immobilization of the joint.
Some of the compounds which can be used according to the
invention have already been described, but nothing is
known about their use for degenerative joint disorders
(DE 2 751 943; H. Oswiecimska et al., J. Prakt. Chem. 318
(1976) pages 403-408; H. Gross et al., J. Prakt. Chem.
317 (1975) pages 890-896; M.G. Pavlichenko et al., Zh
Obshch Khim 56 (1986) pages 2000-2004; R.R. Ismagilov et
al., Zh Obshch Rhim 61 (1991) pages 387-391).
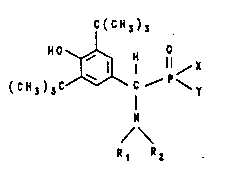
The invention relates to a pharmaceutical containing a
compound of the formula I
C(CH~)5
( C H 3 ), C~ ~
/\ I
Rl R2 ( )
and/or a physiologically tolerated salt of the compound
of the formula I and/or an optionally stereoisomeric form
of the compound of the formula I, where5 Rl and R2 are identical or different and are, indepen-
dently of one another,
215~517
a) a hydrogen atom,
b) (C1-Cl2)-alkyl,
c) (C1-C12)-alkyl, substituted one or more times
independently of one another by
1) phenyl,
2) phenyl, substituted once to three times
independently of one another by
2.1. fluorine, chlorine, bromine or iodine
atom,
2.2. (C1-C3)-alkyl,
2.3. (C1-C3)-alkoxy,
2.4. methylenedioxy,
2.5. nitro,
2.6. amino,
2.7. amidino,
2.8. guanidino,
2.9. hydroxyl,
2.10. (C1-C6)-alkoxycarbonyl or
2.11. cyano,
3) a monocyclic 5- or 6-membered saturated
heterocyclic ring,
4) amino, unsubstituted or substituted once or
twice by (C1-C3)-alkyl, or
5) -C(O)-O-(C1-C3)-alkyl
d) tetralin,
e) phenyl,
f) phenyl, substituted once to three times indepen-
dently of one another by
1) (Cl-C4)-alkyl,
2) (C1-C4)-alkyl, substituted one or more times
by fluorine, chlorine, bromine or iodine
atom,
3) fluorine, chlorine, bromine or iodine atom
4) nitro,
5) aminosulfonyl,
6) amidino,
7) guanidino,
8) (C1-C3)-alkyloxy,
9) methylenedioxy,
21~8517
-- 4
10) hydroxyl,
11) carboxyl,
12) cyano,
13) (C1-C3)-alkoxycarbonyl,
14) -CH=CH-COOH,
o
15) -CH ~ CH-C-O-~C1-C3)-AIkyl,
16) amino or
17) amino, sub~tituted by the radical of the
formula II,
~ OH
- CH
\~X (1l)
o
in which X and Y are, independently of one
another,
17.1. hydrogen atom,
17.2. (C1-C6)-alkyl,
17.3. (C1-C6)-alkoxy,
17.4. (C3-C6)-alkenyloxy,
17.5. phenoxy or
17.6. hydroxyl,
g) naphthyl,
h) naphthyl, substituted one or more time~ as
defined under f)l) to f)17),
i) heteroaryl,
21~i8517
-- 5
k) heteroaryl, substituted one or more times by
phenyl, benzyl or as defined under f)l) to
f)17), or
l) Rl and R2 form, together with the nitrogen atom
to which they are bonded, a 4- to 7-membered
ring,
m) Rl and R2 form, together with the nitrogen atom
to which they are bonded, a 5- to 7-membered
ring in which one carbon atom in the ring is
replaced by an oxygen atom or a nitrogen atom,
or
n) Rl and R2 form, together with the nitrogen atom
to which they are bonded, a 5- to 7-membered
ring in which one carbon atom in the ring is
replaced by a nitrogen atom and this nitrogen
atom is substituted by
1) phenyl or
2) the radical of the formula II in which X and
Y ha~e, independently of one another, the
~n;ng gpecified under f)l7.1. to f)l7.6.
and,
X and Y are identical or different and have, indepen-
dently of one another, the meaning specified under
f)l7.1. to f)l7.6.;
and a physiologically tolerated ~ehicle.
A preferred pharmaceutical contains at least one compound
of the formula I and/or a physiologically tolerated salt
of the compound of the formula I and/or an optionally
stereoisomeric form of the compound of the formula I,
where
Rl and R2 are identical or different and are, indepen-
dently of one another,
a) hydrogen atom,
b) (Cl-Cl2)-alkyl,
c) (Cl-Cl2)-alkyl, substituted one or more times
independently of one another by
1) phenyl,
2) phenyl, substituted once to three times
~1~8~17
-- 6
independently of one another by
2.1. fluorine, chlorine, bromine or
iodine atom,
2.2. (C1-C3)-alkyl,
2.3. (Cl-C3)-alkoxy,
2.4. methylenedioxy,
2.5. nitro,
2.6. amino,
2.7. amidino,
2.8. guanidino,
2.9. hydroxyl,
2.10. (C1-C 6 ) - alkoxycarbonyl or
2.11. cyano,
3) piperazinyl or,
4) morpholinyl,
d) phenyl,
e) phenyl, substituted once to three times indepen-
dently of one another by
1 ) ( Cl - C4 ) - alkyl,
2) (Cl-C4)-alkyl, substituted one or more times
by fluorine, chlorine, bromine or iodine
atom,
3) fluorine, chlorine, bromine or iodine atom
4) nitro,
5) aminosulfonyl,
6) amidino,
7) guanidino,
8) (C1-C3)-alkyloxy,
9) methylenedioxy,
10) hydroxyl,
11) carboxyl,
12) cyano,
13) (C1- C3 ) - alkoxycarbonyl,
14) -CH=CH-COOH,
-CH = CH-C O-(C1-C3l-AIkYI,
15)
21~8517
-- 7
16) amino or
17) amino, substituted by the radical of the
formula II in which X and Y are, indepen-
dently of one another,
17.1. hydrogen atom,
17.2. (C1-C6)-alkyl,
17.3. (Cl-C6)-alkoxy,
17.4. (C3-C6)-alkenyloxy,
17.5. phenoyy or
17.6. hydroxyl,
f) (Cs~ C7) -cyc loalkyl,
g) (C5-C7)-cycloalkyl, substituted one or more
times independently of one another by the radi-
cals mentioned under c)1) to c)4),
h) adamantyl,
i) (Cl-Cl2)-alkyl substituted by amino, unsubsti-
tuted or substituted once or twice by (Cl-C3)-
alkyl,
j) (Cl-Cl2)-alkyl substituted by -C(O)-O-(Cl-C3)-
alkyl,
k) naphthyl,
l) tetralin,
m) pyridyl,
n) pyridyl, substituted one or more times indepen-
dently of one another as defined under e)1) to
e)l7),
o) pyrimidyl
p) pyrimidyl, substituted one or more times
independently of one another as defined under
e)1) to e)17),
q) piperidyl radical, unsubstituted or substituted
once to four times by
1) (Cl-C4)-alkyl,
2) phenyl or
3) benzyl, or
r) Rl and R2 form, together with the nitrogen atom
to which they are bonded, a piperidino radical,
8) Rl and R2 form, together with the nitrogen atom
to which they are bonded, a piperazino or
21~8517
-- 8
morpholino radical, or
t) Rl and R2 form, together with the nitrogen atom
to which they are bonded, a piperazino radical
which is substituted on the nitrogen atom by
1) phenyl or
2) the radical of the formula II in which X and
Y have, independently of one another, the
meAn; ng gpecified under e)17.1. to e)17.6.
and,
X and Y are identical or different and have, indepen-
dently of one another, the me~n;ng specified under
e)17.1. to e)17.6.;
and a physiologically tolerated vehicle.
A particularly preferred pharmaceutical contains at least
one compound of the formula I and/or a physiologically
tolerated salt of the compound of the formula I and/or an
optionally stereoisomeric form of the compound of the
formula I, where
Rl and R2 are identical or different and are, indepen-
dently of one another,
a) hydrogen atom,
b) (Cl-C5)-alkyl,
c) (Cl-C5)-alkyl, substituted one or more times
independently of one another by
1) phenyl,
2) phenyl, substituted once to three times
independently of one another by
2.1. fluorine or chlorine atom,
2.2. methyl, ethyl or propyl,
2.3. methoxy,
2.4. methylenedioxy,
2.5. nitro,
2.6. amino,
2.7. amidino,
2.8. guanidino,
2.9. hydroxyl,
2.10. methoxycarbonyl or
2.11. cyano, or
1 7
g
3) morpholinyl,
d) (C5-C7)-cycloalkyl,
e) (C5-C7)-cycloalkyl, substituted once to three
times independently of one another as defined
under c)l) to c)3),
f) adamantyl,
g) naphthyl,
h) phenyl,
i) phenyl, substituted once to three times indepen-
dently of one another by
1) (Cl-C2)-alkyl,
2) trifluoromethyl,
3) fluorine or chlorine atom,
4) nitro,
5) aminosulfonyl,
6) amidino,
7) guanidino,
8) methoxy,
9) methylenedioxy,
10) hydroxyl,
11) carboxyl,
12) cyano,
13) methoxycarbonyl,
14) -CH=CH-COOH,
0
Il
15) - CH=CH-C-O-CH3,
16) amino or
17) amino, substituted by the radical of the
formula II in which X and Y are, indepen-
dently of one another,
17.1. hydrogen atom,
17.2. methoxy, ethoxy, propoxy or butyloxy,
17.3. allyl,
17.4. phenoYy or,
17.5. hydroxyl,
k) pyridyl,
l) pyrimidyl, or
m) Rl and R2 form, together with the nitrogen atom
2158517
- 10 -
to which they are bonded, a piperidino radical,
n) R1 and R2 form, together with the nitrogen atom
to which they are bonded, a piperazino or
morpholino radical, or
o) Rl and R2 form, together with the nitrogen atom
to which they are bonded, a piperazino radical
which i8 subgtituted on the nitrogen atom by
1) phenyl or
2) the radical of the formula II in which X and
Y have, independently of one another, the
me~n;ng specified under i)17.1. to i)17.5.
and,
X and Y are identical or different and have, indepen-
dently of one another, the meaning specified under
i)17.1. to i)l7.6.;
and a physiologically tolerated vehicle.
A further preferred pharmaceutical contains at least one
compound of the formula I and/or a physiologically
tolerated salt of the compound of the formula I and/or an
optionally stereoisomeric form of the compound of the
formula I, where
R1 and R2 are identical or different and are, indepen-
dently of one another,
a) hydrogen atom,
b) phenyl,
c) phenyl, substituted once to three times indepen-
dently of one another by
1) fluorine or chlorine atom,
2) methylenedioxy or
3) (Cl-C3)-alkoxy, and
X and Y are, independently of one another,
1) hydrogen atom,
2) (C1-C3)-alkoxy, or
3) hydroxyl;
and a physiologically tolerated vehicle.
The term alkyl or alkoxy mean~ radicals whose carbon
chain can be straight-chain, branched or cyclic. Examples
21~ 8 ~1~
of cyclic alkyl radicals are 5- to 7-memhered monocycles
such as cyclopentyl, cyclohexyl or cycloheptyl. The
cyclic alkyl radicals also include polycycles such as
adamantane, twistane or diamantane radicals.
The term "monocyclic 5- or 6-membered saturated,
heterocyclic ring" includes, for example, radicals
derived from pyrrolidine, piperidine, pyrazolidine,
imidazolidine, piperazine, isoxazolidine, morpholine,
isothiazolidine or thiomorpholine.
The term "R1 and R2 form, together with the nitrogen atom
to which they are bonded, a 4- to 7-membered ring"
includes, for example, radicals derived from azetidine,
pyrrolidine, piperidine or azepine.
The term "R1 and R2 form, together with the nitrogen atom
to which they are bonded, a 5- to 7-membered ring in
which one carbon atom in the ring is replaced by an
oxygen atom or nitrogen atom" includes, for example,
radicals derived from isoxazolidine, morpholine,
imidazolidine or piperazine.
The term "heteroaryl" represents aromatic ring systems
which, apart from carbon atoms, also contain hetero atoms
as ring members, for example nitrogen, oxygen or sulfur.
5- or 6-membered monocyclic radicals are preferred, such
as pyrrole, pyridine, furan, thiophene, pyrazole,
imidazole, pyridazine, pyrimidine, pyrazine, oxazole,
isoxazole, thiazole or isothiazole. Also preferred are 9-
to l0-m~hered bicyclic rings such as indole, quinoline,
isoquinoline, indazole, benzimidazole, phthalazine,
quinazoline, ~l;nox~line, purine or pteridine.
Examples of degenerative joint disorders are
osteoarthritis, rheumatic disorders with cartilage
breakdown, rheumatoid arthritis, cho~rolysis after joint
trauma such as after meniscus or patella injuries or
ruptured ligaments, or cho~olysis after immobilization
~1~8~ ~
- 12 -
of joints.
The invention furthermore relates to novel compounds of
the formula I and/or physiologically tolerated salts of
the compound of the formula I and/or an optionally
stereoisomeric form of the compound of the formula I,
where Rl and R2 are identical or different and are,
independently of one another,
a) a hydrogen atom,
b) (Cl-Cl2)-alkyl, substituted one or more times
independently of one another by
1) phenyl, substituted once to three times
independently of one another by
1.1. fluorine, chlorine, bromine or
iodine atom,
1.2. (Cl-C3)-alkyl,
1.3. (Cl-C3)-alkoxy,
1.4. methylenedioxy,
1.5. nitro,
1.6. amino,
1.7. amidino,
1.8. guanidino,
1.9. hydroxyl,
1.10. (Cl-C6)-alkoxycarbonyl or
1.11. cyano,
2) a monocyclic 5- or 6-m~hered saturated
heterocyclic ring,
3) amino, unsubstituted or substituted once or
twice by (Cl-C3)-alkyl, or
4) -C(0)-0-(Cl-C3)-alkyl
c) tetralin,
d) phenyl, substituted once to three times indepen-
dently of one another by
1) (Cl-C4)-alkyl,
2) (Cl-C4)-alkyl, substituted one or more times
by fluorine, chlorine, bromine or iodine
atom,
3) nitro,
4) aminosulfonyl,
21~17
- 13 -
S) amidino,
6) guanidino,
7) (C1-C3)-alkyloxy,
8) methylenedioxy,
9) hydroxyl,
10) carboxyl,
11) cyano,
12) (Cl-C3)-alkoxycarbonyl,
13) -CH=CH-COOH,
Il
14) -CH=CH-C-O-(C1-C3)-alkyl,
15) amino or
16) amino, substituted by the radical of the
formula II, in which X and Y are, indepen-
dently of one another,
16.1. hydrogen atom,
16.2. (C1-C6)-alkyl,
16.3. (C1-C6)-alkoxy,
14.4. (C3-C6)-alkenyloxy,
16.5. phenoxy or
16.6. hydroxyl,
e) naphthyl, substituted one or more times as
defined under d)1) to d)16),
f) heteroaryl,
g) heteroaryl, substituted one or more times by
phenyl, benzyl or as defined under d)1) to
d)16), or
h) Rl and R2 form, together with the nitrogen atom
to which they are bonded, a 5- to 7-membered
ring in which one carbon atom in the ring i6
replaced by an oxygen atom or a nitrogen atom,
or
i) R1 and R2 form, together with the nitrogen atom
to which they are bonded, a 5- to 7-membered
ring in which one carbon atom in the ring is
replaced by a nitrogen atom and thi~ nitrogen
atom is substituted by
1) phenyl or
21~8517
- 14 -
2) the radical of the formula II in which X and
Y have, independently of one another, the
me~n;ng specified under d)16.1. to d)16.6.
and,
X and Y are identical or different and have, indepen-
dently of one another, the me~n;ng Qpecified under
d)16.1. to d)16.6.; or where
R1 is phenyl or phenyl substituted once to three time~
by fluorine, chlorine, bromine or iodine atom,
R2 is hydrogen atom and
X and Y are, independently of one another,
1) hydrogen atom,
2) (Cl-C6)-alkyl,
3) (C3-C6)-alkenyloxy,
4) phenoxy or
5) hydroxyl.
Preferred novel compounds of the formula I are those
where R1 and R2 are identical or different and are,
independently of one another,
a) hydrogen atom,
b) (C1-C12)-alkyl, ~ub~tituted one or more times
independently of one another by
1) phenyl, substituted once to three times
independently of one another by
1.1. fluorine, chlorine, bromine or
iodine atom,
1.2. methylenedioxy or
1.3. cyano,
2) amino, unsub~tituted or substituted once or
twice by (Cl-C3)-alkyl, or
3) -C(O)-O-(Cl-C3)-alkyl
c) phenyl, substituted once to three times indepen-
dently of one another by
1) (Cl-C4)-alkyl, substituted one or more times
by fluorine, chlorine, bromine or iodine
atom,
2) (Cl-C3)-alkyloxy,
3) methylenedioxy,
21~;8~17
- 15 -
4) cyano, or
5) amino, substituted by the radical of the
formula II, in which X and Y are, indepen-
dently of one another,
5.1. hydrogen atom,
5.2. (C1-C6)-alkyl,
5.3. (C1-C6)-alkoxy,
5.4. (C3-C6)-alkenyloxy,
5.5. phenoxy or
5.6. hydroxyl,
d) pyridyl or
e) pyrimidyl, and in which
X and Y have, independently of one another, the meaning
specified under c)5.1. to c)5.6.; or where5 R1 is phenyl or phenyl substituted once to three times
by fluorine or chlorine atom,
R2 is hydrogen atom and
X and Y are, independently of one another,
1) hydrogen atom,
2) (Cl-C2)-alkyl, or
3) hydroxyl.
The invention furthermore relates to processes for
preparing the compound of the formula I, where one
embodiment comprises5 A) reacting a phosphorous diester or phosphorous mono-
ester of the formula III
O
ll/x
\~. (111)
where X and Y are, independently of one another,
a) hydrogen atom,
b) (Cl-C6)-alkyl,
c) (C1-C6)-alkoxy,
d) (C3-C6)-alkenyloxy or
e) phenoxy,
with an azomethine of the formula IV
- 16 - 21~17
H O ~
~H
Il
N ~
~ R , ( I V )
where Rl has the me~n;ng specified under formula I
for b) to k), to give a compound of the formula Ia
~ O
H~l ~P \r
~/ \R, (I~)
where R1 has the meaning stated in formula IV, and
X and Y have the ~e~n;ng stated in formula III, or
B) reacting a compound of the formula NHRlR2, where Rl
and R2 have, independently of one another, the
meaning stated in formula I, in the preRence of
4-hydroxy-3,5-di-tert-butylbenzaldehyde and a com-
pound of the formula III to give a compound of the
formula I, or
C) reacting an addition compound of the formula V
Rl
NH ~ X ~2
R2 (V)
where R1 and R2 have, independently of one another,
the meaning stated in formula I,
in the pre~ence of 4-hydroxy-3,5-di-tert-butylbenz-
aldehyde to give a compound of the formula Ib
21~8~17
H0 ~
Rl R2 (l~)
or
D) hydrolyzing a compound of the formula I where X and
Y are, independently of one another,
a) (C1-C6)-alkyl,
b) (C1-C6)-alkoxy,
c) (C3-C6)-alkenyloxy or
d) phenoxy,
to give a compound of the formula I where X and Y
are, independently of one another, hydroxyl, or0 E) fractionating a compound of the formula I which has
been prepared by process A)-D) and which, by reason
of its chemical structure, occurs in enantiomeric
forms into the pure enantiomers by salt formation
with enantiomerically pure acids or bases, chroma-
tography on chiral stationary phases or derivatiza-
tion using chiral enantiomerically pure compounds
such as amino acids, separation of the diastereomers
obtained in this way, and elimination of the chiral
auxiliary group, or0 F) either isolating in free form the compound of the
formula I prepared by process A)-E) or, in the case
where acidic or basic groups are present, where
appropriate converting it into physiologically
tolerated crystalline salts.
5 The procedure for process variant A) is, for example, to
add the compound of the formula III on to the C=N double
bond of the azomethine compound of the formula IV
(E.K. Fields, J. Am. Chem. Soc. 74 (1952) 1528;
K. Issleib et al., Z. Naturforsch. 36 b (1981) 1392).
2 ~ 17
- 18 -
Equimolar amounts of the compound of the formula III and
formula IV are preferably used in the reaction. The
reaction takes place in the presence of non-polar 801-
ventg such as toluene, xylene, benzene, cyclohexane or
high-boiling hydrocarbons or else in polar solvents such
as alcohols, for example methanol, ethanol, propanol or
butanol, or else in lower aliphatic carboxylic acids such
as acetic acid, dimethylformamide and acetonitrile or
mixtures of the said solvents. The reaction can also be
carried out without addition of solvents, especially when
phosphorous diester such as diethyl phosphite is used in
equimolar amounts or in up to 10-fold excess as phos-
phorus component.
The reaction is carried out in the range from room
temperature to the boiling point of the solvent used in
each case, preferably at 60 to 120C, with reaction
times from 2 to 6 hours. A preferred variant of this
proces6 comprises using a Lewis acid such as aluminum
chloride, which is added in up to equimolar amounts, in
which case the reaction preferably takes place in an
inert solvent such as toluene, expediently at the boiling
point of the solvent.
The azomethines of the formula IV needed for the process
can be prepared by processes known to the skilled worker,
for example by condensation of 4-hydroxy-3,5-di-tert-
butylbenzaldehyde with the appropriate primary amines
H2N-Rl in the presence of a catalyst, for example
p-toluenesulfonic acid in a preferably non-polar solvent
such as toluene or benzene, separating out the water
which is liberated.
The procedure for process variant C) is, for example, to
react the reactants - compound of the formula NHRlR2,
4-hydroxy-3,5-di-tert-butylbenzaldehyde and the compound
of the formula III - in equimolar amounts. The reaction
takes place in a solvent which is immiscible with water,
such as toluene or benzene. To increase the rate of water
elimination, it proves particularly advantageous to add
2~8~17
- 19 -
a Lewis acid such as AlCl3, ZnCl2 or sulfonic acids such
as p-toluene sulfonic acid. The preferred reaction
temperature is from about 50C to the boiling point of
the solvent, in which case the reaction times are 2 to
6 hours.
The reaction conditions for process variant C) are the
same as for process variants A) and B). The compound of
the formula V is prepared by reacting equimolar amounts
of amine and hypophosphorous acid in an inert solvent
such as acetonitrile, an ether, for example diethyl ether
or tert-butyl methyl ether, toluene or methylene chlor-
ide. The reaction takes place at temperatures from 20 to
50C.
The hydrolysis in process ~ariant D) takes place by known
processes. The hydrolysis preferably takes place by
acidolytic cleavage with hydrobromic acid in glacial
acetic acid, preferably at room temperature with reaction
times of up to 8 hours.
The preparation of physiologically tolerated salts from
compounds of the formula I which are capable of salt
formation, including their stereoisomeric forms, takes
place in a manner known per se. Thus, the carboxylic
acids, phosphonic and phosphinic acids and the phosphonic
monoesters form with basic reagents, such as hydroxides,
carbonates, bicarbonates, alcoholates and ~mmo~; a or
organic bases, for example trimethyl- or triethylamine,
ethanolamine or else basic amino acids, for example
lysine, ornithine or arginine, stable alkali metal,
alkaline earth metal or optionally substituted ammonium
salts. Where the compounds of the formula I have basic
groups in the radical R1 or R2, it is also possible to
prepare stable non-toxic acid addition salts with strong
acids. Suitable for this purpose are both inorganic and
organic acids such as hydrochloric, hydrobromic, sul-
3S furic, phosphoric, methanesulfonic, benzene~ulfonic,p-toluenesulfonic, 4-bromobenzenesulfonic,
- 20 -
cyclohexylsulfamic, trifluoromethylsulfonic, acetic,
oxalic, tartaric or trifluoroacetic acid.
Where the compounds of the formula I occur in diastereo-
isomeric or enantiomeric forms, and mixtures thereof are
produced in the chosen synthesis, it is possible to
separate into the pure stereoisomers either by chromato-
graphy on an optionally chiral support material or, where
the racemic compounds of the formula I are able to form
salts, by fractional crystallization of the diastereo-
meric salts formed with an optically active base or acidas auxiliary. Examples of suitable chiral stationary
phases for the separation of enantiomers by thin-layer or
column chromatography are modified silica gel ~upports
(so-called Pirkle phases) and high molecular weight
carbohydrate~ such as triacetylcellulose. For analytical
purposes, gas chromatography methods on chiral stationary
phases can also be used after appropriate derivatization
known to the skilled worker. To separate enantiomers of
the racemic carboxylic acids, phosphonic acids and
pho~phinic acids, an optically active base, which is
usually commercially available, such as (-)-nicotine,
(+)- and (-)-phenylethylamine, quinine bases, L-lysine or
L- and D-arginine are used to form the diastereomeric
salts which have different solubilities, and the less
soluble component is isolated as solid, the more soluble
diastereomer is precipitated from the mother liquor, and
the pure enantiomers are obtained from the diastereomeric
salts obtained in this way. The racemic compounds of the
formula I which contain a basic group such as an amino
group can be converted into the pure enantiomers in the
same way in principle, using optically active acids such
as (+)-camphor-10-sulfonic acid, D- and L-tartaric acid,
D- and L-lactic acid and (+)- and (-)-mandelic acid.
Chiral compounds which contain alcohol or amine func-
tionalities can also be converted with appropriatelyactivated or optionally n-protected enantiomerically pure
amino acids into the correspo~;ng ester~ or amides, or
conversely chiral carboxylic acids can be converted with
21~8~17
- 21 -
carboxyl-protected enantiomerically pure amino acids into
the amides or with enantiomerically pure hydroxy
carboxylic acids such as lactic acid into the corres-
pon~;ng chiral esters. The chirality of the amino acid or
alcohol residue which has been introduced in enantio-
merically pure form can then be utilized for separation
of the isomers by carrying out a separation of the
diastereomers which are now present by crystallization or
chromatography on suitable stationary phases, and then
the chiral moiety included in the molecule can be
eliminated again by suitable methods.
The invention furthermore relates to the use of the
compound of the formula I for the production for
pharmaceuticals for the prophylaxis and therapy of
degenerative joint disorders.
The invention also relates to a process for the produc-
tion of a pharmaceutical, which comprises converting at
least one compound of the formula I into a suitable
administration form with a pharmaceutically suitable and
physiologically tolerated vehicle and, where appropriate,
other suitable active substances, additives or ancillary
substances.
The pharmaceuticals according to the invention are
administered orally, intramuscularly, periarticularly,
intraarticularly, intravenously, intraperitoneally,
subcutaneously or rectally.
Examples of suitable solid or liquid pharmaceutical
formulations are granules, powders, coated tablets,
tablets, (micro)capsules, suppositories, syrups,
solutions, suspensions, emulsions, drops or injectable
solutions, and products with protracted release of active
substance, in whose production conventional auxiliaries
such as excipients, disintegrants, binders, coating
agents, swelling agents, glidants or lubricants, flavor-
ings, sweeteners and solubilizers are used. Examples of
2158517
- 22 -
ancillary substances which are frequently used are
magnesium carbonate, titanium dioxide, lactose, mannitol
and other sugars, talc, lactalbumin, gelatin, starch,
cellulose and derivatives thereof, ~n;~l and vegetable
oils such as fish liver oil, sunflower, arachis or sesame
oil, polyethylene glycol and solvents such as, for
example, sterile water and mono- or polyhydric alcohols,
for example glycerol.
The pharmaceutical products are preferably produced and
administered in dosage units, where each unit contains as
active ingredient a particular dose of the compound of
the formula I according to the invention. This dose can
be up to about 1000 mg, but preferably about 50 to
300 mg, in solid dosage units such as tablets, capsules,
coated tablets or suppositories, and up to about 300 mg,
but preferably about 10 to 100 mg, in injection solutions
in ampoule form.
The daily doses indicated for the treatment of an adult
patient weighing about 70 kg are - depending on the
activity of the compounds of the formula I, about 20 mg
to 1000 mg of active substance, preferably about 100 mg
to 500 mg. However, higher or lower daily doses may also
be appropriate in certain circumstances. The daily dose
may be administered either by a single ~; n; stration in
the form of a single dosage unit or else several smaller
dosage units and by multiple administration of divided
doses at defined intervals.
Example 1
by process variant A)
Diethyl 1-(4-chloroanilino)-1-(3,5-di-tert-butyl-4-
hydroxyphenyl)methanephosphonate
al) N-(3,5-di-tert-butyl-4-hydroxybenzylidene)-4-chloro-
aniline
21~8~17
- 23 -
12.2 g (50 mmol) of 3,5-di-tert-butyl-4-hydroxybenz-
aldehyde semihydrate and 6.4 g (50 mmol) of 4-chloro-
aniline in 130 ml of toluene are heated under reflux
together with 0.2 g of p-toluenesulfonic acid until the
calculated amount of water of reaction has separated out
azeotropically. After concentration of the reaction
mixture under reduced pressure, the residue is
recrystallized from ethanol.
Yield: 15.0 g (87% of theory)
Melting point: 108-110C
a2) Diethyl 1-(4-chloroanilino)-1-(3,5-di-tert-butyl-4-
hydroxyphenyl)methanephosphonate
A solution of 15.0 g (44 mmol) of N-(3,5-di-tert-butyl-4-
hydroxybenzylidene)-4-chloroaniline and 7.0 g (50 mmol)
of diethyl phosphite in 100 ml of toluene is heated under
reflux in the presence of 0.1 g of anhydrous AlCl3 while
stirring with simultaneous azeotropic removal of water
for 6 hours. The re~idue remaining after concentration of
the reaction mixture is recrystallized from diisopropyl
ether.
Yield: 19.5 g (92% of theory)
Melting point: 130-131C
C2sH3sClN4P (MW = 482-0)
Analysi~:
calculated: C 62.30% H 7.74% Cl 7.36% N 2.81% P 6.43%
found: C 61.98% H 7.73% Cl 7.47% N 2.97% P 6.70%
Example 2
by process variant B)
Di-n-propyl 1-(3,4-methylenedioxybenzylamino)-1-(3,5-di-
tert-butyl-4-hydroxyphenyl)methanephosphonate
hydrochloride
2i~8~17
- 24 -
A solution of 12.2 g (50 mmol) of 3,5-di-tert-butyl-4-
hydroxybenzaldehyde semihydrate and 7.5 g (50 mmol) of
3,4-methylenedioxybenzylamine plu8 300 mg of p-toluene-
sulfonic acid in 100 ml of toluene is heated to the
reflux temperature and then 9.1 g (50 mmol) of dipropyl
phosphite and 0.1 g of anhydrous aluminum(III) chloride
are added while stirring. After heating under reflux with
simultaneous azeotropic removal of the water of reaction
for 6 hours, the mixture is filtered hot and, after cool-
ing, methanolic hydrochloric acid is added. The substancewhich precipitates in the form of the hydrochloride is
subsequently recrystallized from acetonitrile.
Yield: 18.1 g (63% of theory)
Melting point: 162-163C
C29H45ClNO6P (MW = 570.11)
Analysis:
calculated: C 61.10% H 7.96% N 2.46% P 5.43%
found: C 59.80% H 8.30% N 2.45% P 4.93%
Example 3
by process variant C)
1-(3,4-Methylenedioxybenzylamino)-1-(3,5-di-tert-butyl-4-
hydroxyphenyl)methanephosphinic acid
cl) 3,4-Methylenedioxyaniline hypophosphite
13.2 g (0.12 mol) of 60% strength hypophosphorous acid
are slowly added dropwise to a solution of 13.8 g
(0.1 mol) of 3,4-methylenedioxyaniline in 20 ml of
acetonitrile while stirring at room temperature. The
precipitate which forms after stirring for 2 hours and
stAn~;ng overnight is filtered off, washed with
acetonitrile and dried under reduced pressure.
Yield: 14.0 g (69% of theory)
Melting point: 132-136C
215~S17
- 25 -
C7HloNO4P (MW = 203.14)
c2) 1-(3,4-Methylenedioxybenzylamino)-1-(3,5-di-tert-
butyl-4-hydroxyphenyl)methanephosphinic acid
A mixture of 11.7 g (50 mmol) of 3,5-di-tert-butyl-4-
hydroxybenzaldehyde and 10.15 g (50 mmol) of 3,4-methy-
lenedioxyaniline hypophosphite in 400 ml of acetonitrile
i8 heated under reflux with stirring for 4 hours. After
cooling, the precipitate is filtered off with suction and
digested several times with hot acetone.
Yield: 19.0 g (91% of theory)
Melting point: 196-197C
C22H30Nosp (MW = 419.46)
Analysis:
calculated: C 63.00% H 7.21% N 3.34% P 7.38%
found: C 62.72% H 6.97% N 3.29% P 7.26%
Example 4
by process D)
1-(4-Choroanilino)-1-(3,5-di-tert-butyl-4-hydroxyphenyl)-
methanephosphonic acid
20 ml of 33% hydrobromic acid in glacial acetic acid are
added to 2 g (4.4 mmol) of dimethyl 1-(4-chloroanilino)-
1-(3,5-di-tert-butyl-4-hydroxyphenyl)methanephosphonate,
and the mixture is then stirred at room temperature for
1.5 hours. Water is added to the mixture, which is then
extracted with ethyl acetate, and the organic phase is
washed with water and dried over Na2SO4. Concentration is
followed by recrystallization from i-propanol/diisopropyl
ether.
Yield: 1.45 g (78% of theory)
Melting point: 162-165C
C21H29ClNO4P (MW = 425.9).
21~8~ 17
- 26 -
Table I: Compound~ of the formula I
E7~ample R1 R2 X Y Melting Process
point
~3 H-OC~H5 -OC2H6130-131 a
2 o~ H-OC3H7 -OC3H7162-163
-Cl12~0
3 o~ H -H -OH 196-197 c
~o
4 ~_ H -OH -OH 162-165 d
-nC4Hg H -OH -OH 183-185 d
6 -nC4Hg H-OC2H5 -OC2H5 93-96
7 {~ H-OCH3 -OCH3 143-145 a
8 {~ H-OC2H5 -OC2H5 104-106
9 --O H-OC3H7 OC3H7 125
{~ H-OC4Hg -OC4Hg 99-l10 a
H-OC2H5 -OC2H5 148-149 a
(HCI)
12 Adamantyl H -OH -OH Subl. from d
230
21~8~17
Rl R2 ~,~ yMelting Process
Example point
13 Adam~n~l H -OC2H6 -OC2H6 132-133
14 -cH2-cH2-Nlc2Hsl2 H -H -OH 213-216 c
/__~ H -OC2H6 -OC2Hs 244~245
-(Cl12)J~ o
16 ~ H -OC2H5-OC2H5112-113 n
-Cll,-CH2-N O
17 ~ H -OC2H5-OC2H5144~145 a
18/~~~ H -OC2H5-OC2H5119-121
~ 2 1
19C H 3 H -OC2H5 -OC2Hs110-111 b
-C~-C02c~ ~HCI~
~ H -OC2H5-OC2H5 135-136 b
-CH-C02CH~
21 -C3H7 -C3H~ -OC2H5 OC2H5 138-139 b
22 ~ -OC2H5 -OC2Hs 128129 b
23 /__~ -OC2H5 -OC2H5 107-109 b
, - o
8~17
- 28 -
Example R1 R2 % Y Melting Process
point
24 /~ h -OC2H5 -OC2H5 62-63 b
\_J ~.2HCI~
~ ~ -OC2Hs -OC2H5 11 ~119 b
\J .
26~0 - P (0 C 2 ~ -OC2H5 -OC2H5 213-214 b
~ -c~
\ ~
~-011
27 -C~2~ H H -OH 223 c
28 o' H H -OH 240-241 c
29 o~ H -OCH3 -OCH3 146-147 b
-CH24~--
o~ ` H -OC2H5 -OC2H5 125 b
- C N 24~--o
21~8~17
- 29 -
Example Rl R2 X Y Melting Process
point
31 o~ H-OC~,Hg -OC~H9162-163 b
-CH~ ~HCI)
32 -CH~OCH~ H -OC2HS-OC2H6 87-88 b
-CH2~C I H -OC2H5-OC2H5 10~107 b
-C~ 3NHz H -OC2H5-OC2H5 77-78 b
~HCII
- C H z~3C N H -OC2H5-OC2H5 98 99 b
(HCI)
36 ~3 H H OH189-190 c
37 ~3 H -OC2H5-OC2H5 125-127 b
38 ~3 H-OC2H5-OC2H5 98 b
~1~8~17
- 30 -
Example Rl R2 X Y MeltingProcess
point
39 ~3 H-OC2Hs -OC2Hs 128-129 ~
~3_ H H -OH 215-217 c
41 ~3 H -CH3 -OH 166-168 8
42 ~c I H -CH3 -OC2H5 185 8
HOCH3 -OCH3 154-156 b
~ H-OC3H7 -OC3H7 120-121 b-
~ H-OC4Hg -OC~,Hg 78-79 b
46 ~3 H-O-Allyl -O-Allyl 9496
21S8~17
- 31 -
Example R1 R2 X Y Melting Process
point
47 ~3~ H -OPh -OPh 178 180 b
48 ~c I H -OC2H5 -OC2H5143-145 b
49 ~ H-OC,,Hg -OC"H9100-102 b
~ H H -OH 187 c .
C~.
~3 H -OC2H5 -OC2H5 154-155
52 ~ H -OC2Hs -C2Hs 121 a
~ ~
cr,
53 ~ -CH3 -OC2H5 -OC2H5 85-86 b
,
_/
54 ~3 -CH3 -OC2H5 -OC2H5 109-111 b
2158517
- 32 -
Example Rt R2 X Y Melting Process
polnt
S5 ~30 H H OH 208-210 c
56 ~30CH, H H OH 231-233 c
~30CHI H-OC2H5 -OC2H5106-107 8
58 ~--OOH, H-OC2H5 -OC2H5117-118 a
OCH,
~oJ -CH3 -C2Hs 230 8
H-OCH3 -OCH3 156-157 8
H-OC2H5 -OC2H512~126
21~17
Example Rl R2 X Y Melting Process
point
62 ~_ H-OC3H7 -OC3H~ 25~252 a
~< J (HCI)
63 ~_ H-OC4Hg -OC4H~ 241-242
~< J (HCI)
64 0c~, H H -OH 16~161 c
~o~,
OC~
~ H-OC2H5 -OC2H5 45-47
COOCH,
66 ~3cooc~1, H -OC2H5 -OC2H5 135136 a-
67 ~ ."1 ",c", H -OC2H5 -OC2H5 184-185
68 {~ H -OC2H5 -0C2HS 223-225 a
2158~17
- 34 -
Example Rl R2 X Y Meltin~ Process
point
69 ~3-02 H -OC2H5 -OC2H5192~193
~3_ CH3-OC2H6 -OC2H5109-110 b
~HCI)
71 ~ N, H -0C2H6 OC2HS 197-199
~HCI~
72 ~3--N, CH3-OC2H5 -0C2H6 68-69 b
~HCI~
73 ~N, "~1 N H -OC2Hs -OC2H5 139-141 b
74 o H -OC2H5 -OC2H5 156-158
~=~ N ~ ( C2 N, J,
~ON
~ H -OC2H5 OC2H6 137-138
76 ~ . H -OC2H5 -OC2H5 128-130
21~517
- 35 -
~ample Rl R2 X Y Melting Process
point
77 ~ H-OC4Hg -OC~H9 135136
~,
78 N H -OH -OC2HS 179-180
~.3
79 N H -OC2Hs -OC2Hs 12~126
~ (HCI)
N H -OC2Hs -OC2Hs 132-133 a
~/~
N--J
21~517
- 36 -
Pharmacological tests
1. Activity in stimulating synthesis of cartilage
matrix, test in cho~rocyte culture
Cells: The hyaline cartilage is removed from the foot
joints of freshly slaughtered cattle, the natural matrix
i8 enzymatically broken down with Pronase (Boehringer
M~nnheim) and collagenase (Sigma), and the cho~ocytes
are plated out in 1% low melting agarose in 24-well
dishes at a cell density of 4 x 106 per well.
Medium: Complete medium contains HAM's F12 (Biochrom RG,
Berlin) and 10% fetal calf serum (Boehringer M~nnheim),
the test substance is dissolved in medium, normally added
in a concentration of 10-5 M and added again at each
change of medium.
Experimental procedure: Treatment takes place from the
third to the tenth day of primary culture and, on day 9,
20 ~Ci/ml (7.4 x 105 Bq) Na235S04 are added to the medium
for 24 h. Dissociative extraction of the proteoglycans
from the agarose layer is carried out with 8M guanidinium
hydrochloride and added proteinase inhibitors (Sigma) by
shaking up at 4C for 24 h. The supernatant after
centrifugation is separated into free and incorporated
sulfate on a PD lO~Sephadex G 25 column, and the activity
thereof is measured on aliquots in a ~ scintillation
counter.
Evaluation: The parameter for matrix production by
c~o~ocytes is the amount of proteoglycans synthesized,
measured as sulfate incorporation in cpm. The mean is
calculated from four wells for each group. This mean is
divided by the mean for the untreated control and thus
yields a stimulation factor which is greater than 1 on
stimulation of matrix synthesis, is less than 1 on
inhibition thereof by the effect of the substance, and is
equal to 1 when matrix synthesis is unchanged.
21~17
- 37 -
Diacerein, which i u~ed a~ therapeutic agent for
osteoarthritis under the proprietary name~ "Artodar" of
Proter and Fi~iodar of Gentili in Italy (Drugs of the
Future 11 (6), 1986) i~ used as ~t~n~rd.
2 1 ~ 7
- 38 -
Table II:
Stimulation of proteoglycan synthesis in agarose cell
culture
Compound of Proteoglycan synthesis
Example stimulation factor
1 1.4
2 1.2
6 1.2
8 1.2
22 1.2
1.4
34 1.2
36 1.2
38 1.7
49 1.2
51 1.6
52 1.7
57 1.5
58 2.5
61 2.9
62 1.2
1.3
78 1.4
79 1.6
Diacerein 1.0
21~851~
- 39 -
2. Activity in cho~rocytic cho~rolysis, test in
chon~rocyte culture
Cells are obtained and plated as in Experiment 1. Medium:
5 ~/ml human rec. interleukin-1 alpha (IL-1, Sigma) are
additionally added to the complete medium as in Test 1
from the start of treatment and is again added, like the
test substance, at each change of medium.
Experimental procedure: Treatment takes place from day 5
to day 13 of primary culture and, on day 12, 20 ~Ci/ml
Na235S04 are added to the medium for 24 h. Dissociative
extraction of the proteoglycans and measurement of
sulfate incorporation are carried out a~ in Test 1.
Evaluation: IL-1 leads to inhibition of synthesis and
increase in degradation of the proteoglycans, which is
reflected by a smaller content of labeled matrix mole-
cules in the agarose layer. The stimulation factor is
therefore related to the mean for the IL-1-treated
control.
Results: The results are listed in Table III.
Table III:
Effect on IL-1 induced chon~rolysis in agarose cell
culture
Compound of Proteoglycan synthesis
Example stimulation factor
1 1.5
2 4.3
3 1.3
6 1.4
7 1.5
1.5
2~58517
- 40 -
Compound of Proteoglycan synthesis
Example stimulation factor
12 2.0
13 2.0
14 1.2
19 4.3
27 1.2
28 5.5
29 1.5
1.5
31 1.7
32 1.4
33 2.2
3.9
36 2.5
37 1.5
38 1.3
2.1
41 3.5
43 1.4
44 1.5
1.2
47 1.8
48 1.7
49 1.5
53 1.7
54 1.4
21~8~17
- 41 -
Compound of Proteoglycan synthesis
Example stimulation factor
57 1.4
58 1.2
1.4
61 1.3
62 1.2
63 1.5
67 1.9
68 1.3
74 3.8
77 1.3
79 1.2
2.4
Diacerein 1.1
3. Inhibition of matrix metalloproteases (MMP)
The compounds according to the invention showed distinct
inhibitory effects on proteolytic enzymes, the so-called
matrix metalloproteases. This is very important because
these enzymes, which are known per se to the skilled
worker, are crucially involved in the proteolytic break-
down of the intact cartilage matrix.
Cell culture:
Rabbit synoviocytes (HIG-82; ATCC, Rockville, Maryland,
USA) are cultivated in HAM's F12 nutrient medium (Sigma,
Deisenhofen, Germany, Catalog No. N-6760) with 10% fetal
bovine serum (Sigma, Deisenhofen, Germany, Catalog No.
F-2442), together with penicillin 100 U/ml and strepto-
mycin 100 ~g/ml. After confluent cell growth, MMP
2~8517
- 42 -
expression is induced in serum-free HAM's F12 by adding
0.3 ~mol/l phorbol 12-myristate 13-acetate. After an
incubation time of 20 h at 37C, the supernatant is
removed.
Activation of MMP:
The supernatant is activated with trypsin (5 ~g/ml).
After 15 minutes (min) at 37C, the activation is stopped
by adding 1 mmol/l phenylmethylsulfonyl fluoride (PMSF)
and incubation is continued for a further 10 min. The
total volume of the mixture is 210 ~1.
Measurement of MMP activity (Lit.: C.G. Rnight et al.,
FEBS Lett. 296, 263 (1992)):
20 ~1 of the abovementioned supernatant are diluted 1:10
and mixed with 240 ~1 of buffer (0.1 Tris/HCl pH 7.5; 0.1
M NaCl; 0.01 M CaCl2; 0.05% Brij). The test substance is
added in the stated concentration (see Table). After an
incubation time of 15 min, the reaction is started by
adding 20 ~mol/l fluorescent substance ((7-
methoxycoumarin-4-yl)acetyl-pro-Leu-Gly-Leu-[3-(2',4'-
dinitrophenyl)-L-2,3-diaminopropionyl~-Ala-Arg-NH2;
Bachem, Heidelberg, Germany, Cat. No.: M-1895). The
reaction is stopped after 30 min by adding 10 mmol/EDTA.
The total volume of the mixture is 320 ~1. The measured
parameters are the fluorescence intensities at ~eX:
328 nm and AeX 393 nm. In order to take account of the
possible intrinsic fluorescence of the test substances,
the fluorescence intensities from parallel measurement6
without substrate are subtracted from the measurements
with substrate. All operations take place at 20C. In the
control experiment without inhibitor, the fluorescence is
equivalent to 0% inhibition, whereas complete quenching
of fluorescence means 100% inhibition.
21S8~;17
- 43 -
Inhibition of MMP
Example 3 83% Inhibition at100 ~M
20% 30 ~M
3% 10 ~M
Example 22 34% 100 ~M
12% 30 ~M
Example 53 25% 100 ~M
11% 30 ~M
Example 59 43% 30 ~M
19% 10 ~M
Example 62 83% 100 ~M
68% 30 ~M
23% 10 ~M
Example 63 60% 30 ~M
26% 10 ~M
Example 74 74% 50 ~M
47% 30 ~M
20% 10 ~M
4. Inhibition of microsomal lipid peroxidation
Oxidative degradation processes are also involved to a
considerable extent in the unwanted breakdown of
cartilage matrix. The compounds according to the
invention showed a strong inhibitory action on biological
oxidation processes and are therefore particularly
suitable for inhibition of oxidative cartilage breakdown.
Isolation of rat liver microsomes:
All steps are carried out at 0C. The liver from a rat is
thoroughly rinsed with 0.9% NaCl solution to remove all
hemoglobin. The coarsely comminuted liver is then treated
in a Potter in 10 mM Tri~/HCl pH 7.4; 250 mM sucrose
(10 ml of buffer/g of liver). Centrifugation is first
21~8517
- 44 -
carried out at 600xg for 5 minutes. The supernatant i8
then centrifuged at 12000xg for 10 min and adjusted
to a concentration of 8 mM with solid CaCl2
(117.6 mg/100 ml). Centrifugation at 25000xg (15 min)
results in a microsome pellet. This pellet is
rehomogenized in the same volume of buffer (10 mM
Tris/HCl pH 7.4; 150 mM KCl) and again centrifuged at
25000xg for 15 min.
Peroxidation by rat liver microsomes:
A test mixture is composed of 20 ~1 of microsomes
(100 mg/ml), see above, dissolved in buffer (250 mM
Tris/HCl pH 6.6, 750 mM gCl), 10 ~1 of 50 mM MgCl2, 10 ~1
of 200 mM isocitric acid, 10 ~1 of 4 mM NADP
(30.028 mg/ml of water), 10 ~1 of 25 mM niacinamide,
10 ~1 of isocitrate dehydrogenase (diluted 1:100) and
20 ~1 of water or test substance. The reaction is started
with 10 ~1 of 0.25 mM FeSO4. Incubation takes place at
37C for 10 minutes. The reaction is stopped with 500 ~1
of ice-cold 20% strength trichloroacetic acid. The
malonaldehyde which has been produced is converted by
addition of 500 ~1 of 0.67% strength thiobarbituric acid
and incubation at 90C for 30 minutes into a pink-colored
compound which is measured at 532 nm. The extinction in
the control mixture without test product is set at 0%
inhibition, while complete disappearance of the signal
means 100% inhibition.
Results: Microsomal lipid peroxidation
Example IC50 (~mol/l)
1 2.43
2 2.30
3 0.79
4 0.71
6 2.25
- 2158~17
- 45 -
Example IC50 (~mol/l)
7 2.40
8 0.73
9 2.10
9 . oo
11 0-97
12 2.70
13 0.83
17 3.18
18 2.03
2.10
21 6.35
22 2.45
23 2.45
2.45
29 0.69
31 2.18
34 0.70
39 2.40
0.49
43 0.79
44 1.05
6.15
46 0.86
48 2.95
49 ~ 10
53 2.03
21~3517
- 46 -
Example IC50 (~mol/l)
54 2.80
0.71
62 0.24
63 0.24
0.73
66 0 75
67 2.22
68 2.37
73 0.67
74 1.25
2.90
76 2.05
77 8.70
79 2.30
2.90
5. Release of interleukins (in particular IL-l~) from
human mo~o~uclear cells
The compounds according to the invention have a strong
inhibitory effect on the release of interleukin~ from
human cells. This is of great pharmacological importance
because interleukins may induce unwanted degradation of
cartilage matrix.
Isolation of mononuclear cells from human blood
10 ml of human blood stabilized with 1 ml of 3.8%
~trength sodium citrate solution are diluted with 10 ml
of PM 16 (Serva, Heidelberg), and a layer of 15 ml of
Lymphoprep (Dr. Molter GmbH, Heidelberg) is introduced
2158~17
- 47 -
underneath. The samples are centrifuged at 400xg (1600
rpm Minifuge 2, Heraeus, Osterode) for 40 minutes (room
temperature). The ~o~uclear cells are visible as a
white ring at the Lymphoprep/plasma boundary. This ring
is carefully removed using a syringe, diluted with the
same volume of PM16 and centrifuged at 400xg for 10
minutes. The precipitate i~ washed with ~ 10 ml of RPMI
1640 (+ 300 mg/l L-gluta_ine, Gibco, Eggenstein). After
resuspension of the cells in ~ 1 ml of RPMI 1640
(+ 300 mg/l L-glutamine, + 25 mM HEPES, + 100 ~g/ml
streptomycin, + 100 ~g/ml penicillin), the cell density
is determined with a Coulter counter JT (Coulter
Diagnostics) and adjusted to 5X106 ml. The cells are
typically composed of 90% lymphocytes and 10% monocytes.
Stimulation of release of cytokines, in particular
interleukin 1~
230 ~l of mo~o~uclear cells are incubated with 10 ~l of
test substance (10 ~M in DMSO/water 1/10) and 10 ~l of
lipopolysaccharides (500 ~g dissolved in 1 ml of dimethyl
sulfoxide (DMSO) and diluted 1/10 with water before start
of test, from Salmonella abortus equi, Sigma,
Deisenhofen) at 37C, 5% CO2 for 20-22 hours. The samples
are cooled to 0C in an ice bath and centrifuged in a
Sigma centrifuge (2 minute~; 2000 rpm). Aliquots of the
supernatant are determined using a commercially available
ELISA (Biermann, Bad Nauheim).
Results:
Inhibition of interleukin release
(all substances from the examples were tested at
10 ~mol/l)
Example % Inhibition Interleukin
7 21 IL-lalpha
22 80 I-lalpha
21~8517
- 48 -
Example % Inhibition Interleukin
22 86 TNF-alpha
23 75 IL-lalpha
23 70 TNF-alpha
IL-lalpha
36 TNF-alpha
31 32 IL-lbeta
22 IL-lalpha
53 53 IL-lbeta
24 TNF-alpha
61 24 IL-lbeta
62 46 IL-lbeta
63 31 IL-lbeta
67 26 IL-lbeta
21 TNF-alpha