Note: Descriptions are shown in the official language in which they were submitted.
WO 94/22852 PCT/US94/03004
to Quinolines as type IV phosphodiesterase inhibitors
BACKGROUND OF THE INVENTION
FIELD OF THE INVENTION
This invention relates to optionally 6,8-substituted quinolines
useful as anti-inflammatory agents, immunosuppressive agents, anti-
allograft rejection agents, anti-graft-vs-host disease agents, anti-
allergic agents (e. g., asthma, rhinitis and atopic dermatitis), broncho-
dilation agents, anti-autoimmune agents or analgetic agents,
to their precursors, to their preparation and to pharmaceutical
compositions using the compounds of the invention.
BACKGROUND INFORMATION
Cyclic 3',5'-adenosine monophosphate (cANlp) modulates a variety of
cellular and physiologic functions in mammals, such as, cell division,
endocrine function, and the immune response. The level of cAN~ is
controlled by a class of enzymes called phosphodiesterases, which
enzymatically deactivate cAMP. There are five general types of phospho-
diesterases, which are categorized according to their function and the type
of cell from which they are isolated. For instance, high-affinity
phosphodiesterase (PDE III) is isolated from human platelet cells and
modulates platelet aggregation. Another type of phosphodiesterase (PDE IV)
is found in various tissues but is the predominant form in human
leukocytes; this enzyme modulates leukocyte activation and function
associated with the immune response and inflammation. Both of these
phosphodiesterases implement their control by modulating the cellular level
of cAN~ in their respective cells. Thus, inhibition of phosphodiesterases
provides a method of modulating any cellular and bodily function that is
controlled by cAN~.
Compounds that are nonspecific phosphodiesterase inhibitors are
known, i.e., these compounds inhibit all or multiple types of
phosphodiesterases. [See, Beavo, J.A. and D.H. Reifsyder, Trends in Pharm.
Science, 11:150-155 (1990); and Nicholson, C.D., R.A.J. Challiss and M.
Shahid, Trends in Phazm. Science, 12:19-27 (1991).] Since CAMP is involved
in so many functions throughout the body, a nonspecific phosphodiesterase
inhibitor has the potential to alter all of the functions modulated by
cAN~, thus nonspecific phosphodiesterase inhibitors are of limited value
because of numerous side-effects.
It has been surprisingly discovered that certain optionally
substituted 6,8-quinolines are potent selective inhibitors of
WO 94/22852 PCT/US94103004
-2_
Phosphodiesterase Type IV (PDE IV). These compounds are well suited for
use as a treatment for any disorder in which PDE IV function plays a role,
such as where leukocyte activation or function is involved.
In particular, these compounds are especially well suited for use as anti-
s inflammatory agents, immunosuppressive agents, anti-allograft rejection
agents, anti-graft-vs-host disease agents, anti-allergic
agents (e. g., asthma, rhinitis and atopic dermatitis), bronchodilation
agents, anti-autoimmune disease agents or analgetic agents.
SUI~IARY OF THE INVENTION
One aspect of the present invention relates to optionally substituted
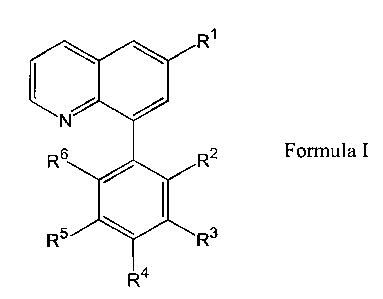
6,8-quinolines, i.e., a compound of Formula I:
R~
\ \
N
R2
Formula I
wherein:
R' is selected from hydrogen; lower alkyl; cycloalkyl; cycloalkyloxy;
cycloalkylamino; cycloalkyl lower alkyl; lower alkoxy; formyl;
hydroxy-lower alkyl; carboxyalkyl; optionally substituted aryl,
aryloxy, arylamino or aryl lower alkyl; optionally substituted
heterocycle, heterocycle-oxy, heterocycle-amino or heterocycle
lower alkyl; and
Rz is optionally substituted phenyl,
or a pharmaceutically acceptable salt or N-oxide thereof.
Preferred aspects of R' are pyridylmethyl, benzyl, cycloalkylmethyl and
lower alkyl.
In another aspect, the invention relates to a pharmaceutical
composition containing a therapeutically effective amount of a compound of
Formula I or a pharmaceutically acceptable salt or N-oxide thereof admixed
with at least one pharmaceutically acceptable excipient.
In still another aspect, the invention relates to a method of use as
an anti-inflammatory agent, immunosuppressive agent, anti-allograft
rejection agent, anti-graft-vs-host disease agent, anti-allergic agent
(e. g., asthma, rhinitis and atopic dermatitis), bronchodilation agents,
2I~96~1
WO 94/22852 PCT/US94/03004
-3-
anti-autoimmune disease agent or analgetic agent, by administering to a
mammal in need of such treatment a therapeutically effective amount of a
compound of Formula I or a pharmaceutically acceptable salt or
N-oxide thereof.
Yet another aspect of the invention relates to the treatment of the
above conditions or diseases by the selective inhibition of PDE IV.
In another aspect, this invention provides compositions useful in the
treatment of inflammatory, allograft rejection, graft-vs-host disease,
allergy, autoimmune or analgetic conditions or diseases in mammals
comprising a therapeutically effective amount of a compound of Formula I or
a pharmaceutically acceptable salt or N-oxide as described above and a
pharmaceutically acceptable excipient.
Another aspect of the invention relates to processes for making the
compounds of Formula I and the pharmaceutically acceptable salts and N-
oxides thereof.
DETAILED DESCRIPTION
DEFINITIONS AND GENERAL PARAMETERS
The following definitions are set forth to illustrate and define the
meaning and scope of the various terms used to describe the invention
herein.
The term "alkyl" refers to a branched or straight chain monovalent
saturated aliphatic hydrocarbon radical of one to twenty carbon atoms.
The term "lower alkyl" refers to a branched or straight chain
monovalent alkyl radical of one to four carbon atoms. This term is further
exemplified by such radicals as methyl, ethyl, n-propyl, isopropyl,
i-butyl, n-butyl and t-butyl.
The term "cycloalkyl" refers to a monovalent carbocyclic radical of
three to six carbon atoms, i.e., cyclopropyl, cyclobutyl, cyclopentyl and
cyclohexyl, which can optionally be substituted, independently, with, e.g.,
hydroxy, amino, imino, lower alkyl, lower alkoxy, carboxy, lower
alkoxycarbonyl, carbamoyl, aryl, aryl, halo, and/or cyano.
The teen "cycloalkyloxy" refers to a cycloalkyl group attached to a
parent structure via an oxy radical, i.e., -O-, such as cyclopentyloxy.
The term "cycloalkylamino" refers to a cycloalkyl group attached to a
parent structure via an imino radical, i.e., -NH-, such as
cyclopropylamino.
The term "cycloalkyl lower alkyl" refers to a cycloalkyl group
attached to a parent structure via a lower alkylene group such as
methylene; e.g., cyclopropylmethyl, cyclopentylethyl, cyclopentylpropyl, or
cyclopentylmethyl.
The term "lower alkylene" refers to a biradical branched or
unbranched saturated hydrocarbon chain containing 1 to 4 carbon atoms, such
WO 94/22852 PCT/US94/03004
~lr~,pbpF~
-4-
as methylene (-CHZ-), ethylene, propylene, isopropylene and butylene.
The term "lower alkoxy" refers to the group -O-R' where R' is lower
alkyl.
The teen "carbonyl" refers to the group -C(O)-.
The term "carboxy" refers to the group -C(O)OH.
The term "carboxyalkyl" refers to the group -alkyl-C(O)OH,
where alkyl is a branched or straight chain monovalent alkyl radical of one
to eight carbon atoms.
The term "lower alkoxycarbonyl" refers to the group -C(O)OR' where R'
is lower alkyl.
The term "aryl" refers to the group -C(O)-R', where R' is lower
alkyl, e.g., acetyl or propionyl; or optionally substituted phenyl or
heterocycle, e.g., phenacyl.
The term "carbamoyl" refers to the group -C(O)NR'R where R and R' are
independently hydrogen or lower alkyl, e.g., where R is hydrogen and R' is
lower-alkyl the group is lower alkylcarbamoyl, where R and R' are lower
alkyl the group is di-lower alkylcarbamoyl.
The term "halo" refers to fluoro, bromo, chloro and iodo.
The term "lower alkylthio" refers to the group R-S-.
The term "lower alkylsulfinyl" refers to the group R-S(O)-.
The term "lower alkylsulfonyl" refers to the group R-S(0)2-.
The term "lower alkoxysulfonyl" refers to the group RO-S(0)2-.
The term "hydroxysulfonyl" refers to the group HO-S(OZ)-.
The term "aryl" refers to a monovalent carbocyclic aromatic radical
(e. g., phenyl), or two condensed carbocyclic rings (e. g., naphthyl) which
can optionally be mono-, di-, or tri-substituted, independently, with
hydroxy, thiol, amino, halo, vitro, lower alkyl, lower alkylthio, lower
alkoxy, mono-lower alkylamino, di-lower alkylamino, carboxy, lower
alkoxycarbonyl, hydroxysulfonyl, lower alkoxysulfonyl, lower alkylsulfonyl,
lower alkylsulfinyl, trifluoromethyl, cyano, tetrazolyl, carbamoyl, lower
alkylcarbamoyl, and di-lower alkylcarbamoyl.
The term "aryloxy" refers to an aryl group as defined above
attached to a parent structure via an oxy radical, i.e., aryl-O-.
The term "arylamino" refers to an aryl group as defined above
attached to a parent structure via an imino radical, i.e., aryl-NH-.
The term "aryl lower alkyl" refers to an aryl group attached to a
parent structure via a lower alkylene group such as methylene; e.g.,
benzyl.
The term "heterocycle" refers to a saturated, unsaturated or aromatic
monovalent cyclic radical having at least one hetero atom (such as
nitrogen, oxygen or sulfur) or a combination thereof, which can optionally
be substituted, independently, with, e.g., hydroxy, amino, imino, lower
alkyl, lower alkoxy, carboxy, lower alkoxycarbonyl, carbamoyl, aryl, aryl,
halo, and/or cyano. Further, the term also includes instances where an atom
WO 94/22852 ~~~~ PCT/US94/03004
-5-
of a heterocycle has been oxidized, e.g., N-oxides, sulfoxides, sulfones,
or oxo. For example, typical heterocycles with one or more nitrogen or
sulfur atoms are pyrrole, imidazole, imidazoline, imidazolidine, pyrazole,
- pyrazine, pyrrolidine, pyrrolidinone, pyrazolidine, piperidine, piperazine,
moxpholine, pyridine, pyridone, triazole, oxazole, oxadiazole, thiazole,
and the like.
The term "heterocycle-oxy" refers to a heterocyclic
group as defined above attached to a parent structure via an oxy radical,
i.e., heterocycle-O-.
The term "heterocycle-amino" refers to a heterocyclic
group as defined above attached to a parent structure via an imino radical,
i.e., heterocycle-NH-.
The term "heterocycle lower alkyl" refers to a heterocyclic
group attached to a parent structure via a lower alkylene group such as
methylene (-CHZ-); e.g., 4-pyridylmethyl.
The term "tetrazolyl" refers to the group
N
-\NH
N-N
The term "optionally substituted phenyl" refers to phenyl and mono-,
di-, tri- or tetra-substituted phenyl, wherein the optional substituents
are lower alkyl, hydroxy, thiol, amino, halo, nitro, cyano, lower alkoxy,
lower alkylthio, mono-lower alkylamino, di-lower-alkylamino, carboxy, lower
alkoxycarbonyl, lower,alkylene-dioxy, hydroxysulfonyl, lower
alkoxysulfonyl, lower alkylsulfonyl, lower-alkylsulfinyl, trifluoromethyl,
trifluoromethyloxy, tetrazolyl, carbamoyl, lower alkylcarbamoyl, and di-
lower alkylcarbamoyl. This term is further exemplified by such radicals as
3-chlorophenyl, 3-nitrophenyl, 4-methoxyphenyl, 3-cyanophenyl, 4-
trifluorophenyl, 3-chloro-4-fluorophenyl and 3,4-methylenedioxyphenyl.
The term "N-oxide" refers to nitrogen heterocycles where a nitrogen
atom in the ring has been oxidized, e.g., 4-pyridyl-N-oxide,
3-pyridyl-N-oxide, or 2-pyridyl-N-oxide.
The term "hydroxy-lower alkyl" refers to a lower alkyl radical
substituted with a hydroxy group, e.g., -CH(OH)CHZCH3 or -C(OH)(CH3)2.
The term "pharmaceutically acceptable salt" refers to any salt
derived from an inorganic or organic acid or base.
The term "pharmaceutically acceptable anion" refers to the anion of
such acid addition salts. The term "pharmaceutically acceptable cation"
refers to the cation of such base addition salts. The salt, anion and/or
the cation are chosen not to be biologically or otherwise undesirable.
The anions are derived from inorganic acids, such as hydrochloric acid,
hydrobromic acid, sulfuric acid (giving the sulfate and bisulfate salts),
nitric acid, phosphoric acid and the like, and organic acids such as acetic
WO 94/22852 2 ~1~~~ ~ ~ ~ ~~ PCTIUS94/03004
-6-
acid, propionic acid, glycolic acid, pyruvic acid, oxalic acid, malic acid,
malonic acid, succinic acid, malefic acid, fumaric acid, tartaric acid,
citric acid, benzoic acid, cinnamic acid, mandelic acid, methanesulfonic
acid, ethanesulfonic acid, salicylic acid, p-toluenesulfonic acid and the
like.
The cations are derived from bases, such as alkaline earth hydroxides,
including calcium hydroxide, potassium hydroxide, sodium hydroxide, lithium
hydroxide and the like.
As used herein, the term "allograft rejection" refers to the humoral
or cellular immune response mounted by the immune system of a mammal after
it has received a histo-incompatible tissue graft from another mammal of
the same species, thereby producing tissue injury to the graft in such a
recipient.
As used herein, the term "graft-vs-host disease" refers to the immune
response that originates from transplanted graft tissue, in particular,
transplanted bone-marrow tissue, and that is directed towards the host
tissue, thereby producing tissue injury in the host.
As used herein, the tezm "autoimmune disease" refers to disorders
wherein the immune system of a mammal mounts a humoral or cellular immune
response to the mammal's own tissue or to antigenic agents that are not
intrinsically harmful to the mammal, thereby producing tissue injury in
such a mammal. Examples of such disorders include, but are not limited to,
systemic lupus erythematosus, rheumatoid arthritis and type I diabetes.
As used herein, the terms "treatment" or "treating" of a condition
and/or a disease in a mammal, means:
(i) preventing the condition or disease, that is, avoiding any
clinical symptoms of the disease;
(ii) inhibiting the condition or disease, that is, arresting the
development or progression of clinical symptoms; and/or
(iii) relieving the condition or disease, that is, causing the
regression of clinical symptoms.
The conditions and diseases treated in the present invention include
inflammation, pain, pyrexia, autoimmune disease, allograft rejection,
graft-vs-host disease, allergies and uveitis.
As used herein, the terns "therapeutically effective amount" refers to
that amount of a compound of Formula I which, when administered to a mammal
in need thereof, is sufficient to effect treatment (as defined above) as an
anti-inflammatory agent, immunosuppressive agent, anti-allograft rejection
agent, anti-graft-vs-host disease agent, anti-allergy agent, autoimmune
disease agent or analgetic agent. The amount that constitutes a
"therapeutically effective amount" will vary depending on the compound, the
condition or disease and its severity, and the mammal to be treated, but
may be determined routinely by one of ordinary skill in the art with regard
to contemporary knowledge and to this disclosure.
As used herein, the term "q.s." means adding a quantity sufficient to
WO 94/22852 ~ PCT/US94I03004
achieve a stated function, e.g., to bring a solution to a desired volume
(e. g., 100 mL).
As used herein, the term "mp" refers to melting point.
All temperatures are given in degrees Celsius (°C).
Unless specified to the contrary, the reactions described herein take
. place at atmospheric pressure over a temperature range from about -
78°C to
about 150°C, more preferably from about 10°C to about
50°C, and moat
preferably at about room (or "ambient"~) temperature, e.g., about 20°C.
Unless specified to the contrary, the ranges of time and temperature
described herein are approximate, e.g., "from 8 to 24 hours at from
10°C to
100°C" means from about 8 to about 24 hours at about 10°C to
about 100°C.
Isolation and purification of the compounds and intermediates
described herein can be effected, if desired, by any suitable separation or
purification procedure such as, for example, filtration, extraction,
crystallization, column chromatography, preparative high pressure liquid
chromatography (preparative HPLC), thin-layer chromatography or thick-layer
chromatography, or a combination of these procedures. Specific
illustrations of suitable separation and isolation procedures can be had by
reference to the examples hereinbelow. However, other equivalent
separation or isolation procedures can also be used.
The following numbering and nomenclature system will be used for
naming the compounds of the invention.
2 5 .a s
s R~
3
2 / ~ 7
a
R2
E.g., the compound of Formula I where R' is 4-pyridylmethyl and
RZ is 3-chloro-4-fluorophenyl can be named 6-(4-pyridylmethyl)-8-(3-
chloro-4-fluorophenyl)quinoline.
The compound of Formula I where R' is isopropyl and RZ is
4-chlorophenyl can be named 6-(isopropyl)-8-(4-chlorophenyl)quinoline.
SYNTHESIS OF THE COMPOUNDS OF FORMULA I
As used in the Reaction Schemes R' and Rz are the same as described in
the Summary of the Invention.
Reaction Scheme A illustrates the preparation of 6,8-(disubstituted)
quinolines, i.e., the compounds of Formula I.
Reaction Scheme A-1 illustrates the preparation of intermediates
c
WO 94/22852 ~ 15 9 ~ 0 ~ PCTIUS94/03004
_g_
of formula 3a which can be converted into compounds of formula I where R'
is cycloalkylmethyl, arylmethyl or heterocycle-methyl by following the
reactions as shown in Reaction Scheme A.
Reaction Scheme 8 illustrates an alternate preparation of 6,8-
(disubstituted) quinolines, i.e., the compounds of Formula I, where the
final two steps in Reaction Scheme A are carried out in reverse order.
Reaction Scheme B-1 illustrates an alternate preparation of
6,8-(disubstituted) quinolines, i.e., the compounds of
Formula I, via the 8-ZnCl-quinoline intermediate.
Reaction Scheme B-2 illustrates the preparation of
6-substituted-8-haloquinolines, in particular, the compounds of Formula 5
where X is halo and R' is cycloalkylaminomethyl, arylaminomethyl,
heterocycle-aminomethyl or heterocycle-methyl, i.e., where the methylene
group is attached to a nitrogen atom of a heterocycle.
Reaction Scheme B-3 illustrates the preparation of
6,8-(disubstituted) quinolines, in particular, the compounds of Formula I
where R' is cycloalkyloxy, aryloxy or heterocycle-oxy.
Reaction Scheme C illustrates the preparation of
6-[lower alkyl-(hydroxy)methyl]-8-quinolines, and 6-formyl-8-quinolines,
i.e., the compounds of Formula I where R' is formyl, or lower alkyl-
(hydroxy)methyl.
Reaction Schemes C-1 and C-2 illustrate alternate preparations of
6,8-(disubstituted)quinolines, in particular, the compounds of Formula I
where R' is alkyl, cycloalkylmethyl, cycloalkenylmethyl, arylmethyl or
heterocycle-methyl.
Reaction Scheme C-3 illustrates an alternate preparation of 6,8-
(disubstituted)quinolines, in particular, the compounds of Formula I where
R' is mono- or di-alkylaminomethyl, cycloalkylaminomethyl, arylaminomethyl,
heterocycle-aminomethyl or heterocycle-methyl, i.e., where the methylene
group is attached to a nitrogen atom of a heterocycle.
REACTION SCHEME A
R1 R1 R'I
/ / /
X
N02 NH2 NHz
C ~ 7 C 2J ~ 3]
where X is chloro,
bromo or iodo
WO 94122852 , ~~~j PCT/US94/03004
-9-
R~
' C3~ + R2-B[OH]z
_R2
[38~ NHz
where R2 is optionally [9]
substituted phenyl
R~
OH
[ 4 ] + ~ --
OH OH N
R2
Formula I
REACTION SCHEME A-1
R / R R R
CHO
\ \
\ ~ I \
i i
x
NOZ N02 NH2 NHz NHZ
(~a] [2a] [2b] [3a]
where R is alkyl, cycloalkyl,
where X is
aryl lower alkyl or chloro or bromo
heterocycle lower alkyl
STARTING MATERIALS
Referring to the Reaction Schemes, the compounds of Formula 1 (i.e.,
4-optionally substituted nitrobenzene) and Formula 2 (i.e., 4-optionally
substituted aniline) are commercially available from Aldrich Chemicals Co.,
Inc., Fluka Chemical Corporation, Lancaster Synthesis Ltd., Karl
Industries, Maybridge Chemical Co. Ltd. or Tokyo Kasai International. The
compounds of Formula 3A, i.e., optionally substituted benzene boronic acid,
are commercially available from Lancaster Synthesis Ltd., or alternatively
can be prepared following the procedures in Organic Synthesis, Coll Vol 4.
WO 94/22852 PCTIUS94/03004
~~59Gt~
-10-
Those compounds that are not commercially available can be prepared by one
of ordinary skill in the art following procedures set forth in references
such as, "Fieser and Fieser's Reagents for Organic Synthesis", Volumes 1-
15, John Wiley and Sons, 1991; "Rodd's Chemistry of Carbon Compounds",
Volumes 1-5 and Supplementals, Elservier Science Publishers, 1989; and
"Organic Reactions", Volumes 1-40, John Wiley and Sons, 1991.
PREPARATION OF FORMULA 2
An optionally substituted p-nitrobenzene is combined with about 5
molar equivalents of a reducing agent, such as SnClz~H20, Fe/acetic acid, or
palladium on carbon/Hz, preferably SnC12~H20, in a solvent such as ethanol,
or ethyl acetate, preferably ethanol. The solution is heated. The
temperature and duration will vary according to the reagent and solvent
used, e.g., using ethanol and SnCl~-H20, the solution is heated at a
temperature in the range of about 50°C to 90°C, preferably about
70°C, for
a period of about 1 hour to 3 hours, preferably about 2 hours. The
progress of the reaction is monitored by TLC (thin layer chromatography).
When the reaction is substantially complete, the product is isolated and
purified by conventional means yielding the desired optionally substituted
p-aminobenzene compound (i.e., a compound of Formula 2).
PREPARATION OF FORMULA 3
A solution of about 1 molar equivalent of a halogenating agent, such
as N-bromosuccinimide (NBS), or N-chlorosuccinimide (NCS), preferably N-
bromosuccinimide, in a solvent, preferably DMF is added in a gradual manner
to a solution of an optionally substituted p-aminobenzene compound (Formula
2) dissolved in a solvent (such as DMF, preferably DMF). The reaction
mixture is stirred at about room temperature for a period of about 1 to 5
hours, preferably about 3 hours. When the reaction is substantially
complete (by TLC), the product is isolated and purified by conventional
means yielding the desired optionally substituted p-amino-m-halobenzene
compound (i.e., a compound of Formula 3 where X is chloro or bromo).
PREPARATION OF FORMULA 4
An optionally substituted p-amino-m-halobenzene compound (Formula 3)
is combined with about 1 to 5 molar equivalent, preferably about 2 molar
equivalent, of an optionally substituted benzene boronic acid, i.e.,
Formula 3B where RZ is optionally substituted phenyl, 2 M Na=C03 (about
4 molar equivalent), methanol or ethanol, preferably ethanol, and benzene
or toluene, preferably benzene. To this solution is added about 0.01 to 0.1
molar equivalent, preferably about 0.035 molar equivalent, of palladium
tetrakis triphenylphosphine. The reaction mixture is heated at about
reflux for a period of about 3 to 9 hours, preferably about 6 hours. When
the reaction is substantially complete (by TLC), the product is isolated
WO 94/22852 PCT/US94/03004
-11-
and purified by conventional means yielding .he desired optionally
substituted p-amino-m-arylbenzene compound (e.g., a compound of Formula 4).
PREPARATION OF FORMULA I
An optionally substituted p-amino-m-arylbenzene compound (Formula 4)
is combined with about 1 molar equivalent of an oxidant, such as ferric
oxide, m-nitrobenzenesulphonic acid, nitrobenzene, iron(II) sulfate
(heptahydrate)/nitrobenzene or arsenic pentoxide, preferably arsenic
pentoxide, and 3 molar equivalent of glycerol under an inert atmosphere.
The mixture is heated at a temperature in the range of about 75°C to
125°C,
preferably about 100°C, for a period of about 15 minutes to 45 minutes,
preferably about 30 minutes. About 12 molar equivalent of a concentrated
acid (preferably concentrated H~SO,) is added to the mixture in a gradual
manner, and the mixture is heated at a temperature in the range of about
100°C to 200°C, preferably about 150°C for a period of
about 1 to 3 hours,
preferably about 2 hours. The progress of the reaction is monitored by TLC
(9:1 hexane:ethyl acetate). When the reaction is substantially complete (by
TLC), the product is isolated and purified by conventional means yielding
the desired 6-optionally substituted-S-arylquinoline compound (i.e., a
compound of Formula I).
REACTION SCHEME B
R~
OH
\ ~ ~ \ \
~ /
OH OH X N
NHZ X
C37 C5~
where X is chloro,
bromo or iodo
R'
+ Rz-B[ OH] 2
4o CS]
N
[38] R
2
Formula I
WO 94/22852 ~ . PCT/US94/03004
-12-
REACTION SCHEME B-1
R~ R~ R~-OTf \ \ R
\ \ \ \ C3C]
i /
i / / /
N
N N ~ or Rz-X
X ZnCI R2
15
C5] Formula I
where X is chloro, where R2 is
bromo or iodo optionally substituted phenyl
REACTION SCHEME H-2
\ \ \X NH2R \ \ NHR \ \ NR2
i / ~
N or NHRZ ~ N~ / or ~ Ni
X X X
CS] CS] C5]
where X is where R is lower where R2 Ts
chloro, alkyl, cycloalkyl, independently
bromo, or aryl or hetero- lower alley or aryl;
iodo cycle or NR2 is heterocycle
REACTION SCHEME B-3
OMe OMe OMe
\ \ \ \ ~ \ \
I i / I i / ~ /
N ~ N ~ N
NO2 NH2 Br
C s7
45
w WO 94/22852 ' PCTIUS94/03004
-13-
OMe ~H R-X \ \ ~~R
\ \ \ \
i
i / i / N /
N N
Br Br er
C57 C 57
where R is lower nlkyl,
cycloalkyl, aryl or heterocycle,
and X is chloro, bromo or iodo
PREPARATION OF FORMULA 5
An optionally substituted p-amino-m-halobenzene compound (Formula 3)
combined with about 1 molar equivalent of arsenic pentoxide and 3 molar
equivalent of glycerol under an inert atmosphere. The mixture is heated at
a temperature in the range of about 75°C to 125°C, preferably
about 100°C,
for a period of about 15 minutes to 45 minutes, preferably about
30 minutes. About 12 molar equivalent of a concentrated acid (preferably
concentrated HzSO,) is added to the mixture in a gradual manner, and the
mixture is heated at a temperature in the range of about 100°C to
200°C,
preferably about 150°C for a period of about 1 to 3 hours, preferably
about
2 hours. When the reaction is substantially complete (by TLC), the product
is isolated and purified by conventional means yielding the desired
6-optionally substituted-8-haloquinoline compound (i.e., a compound of
Formula 5).
PREPARATION OF FORMULA 3B
About 2 molar equivalents of trimethylborate is dissolved in an
apolar solvent (such as diethyl ether, or tetrahydrofuran, preferably
diethyl ether) and cooled at a temperature in the range of about -50 to
-80°C, preferably about -65°C.
An optionally substituted phenyl Grignard reagent is added to the solution
in a gradual (e. g., dropwise) manner over a period of about
20 minutes/molar equivalent. The mixture is then stirred at a temperature
in the range of about -50°C to -80°C for about 15 to 45 minutes,
preferably
about 30 minutes. The mixture is allowed to warm to about -10°C to
10°C,
preferably about 0°C and stirred for a period of about 1 hour. Water is
added to the reaction mixture and the mixture is stirred for a period of
about 1 hour. The organic layer is removed and the residue is extracted
(e. g., 3 x ethyl acetate).
The organic layers are combined and dried over a drying agent (e. g.,
MgSO,). The solution is concentrated, hexanes are added to the solution and
the solution is stirred for a period of about 1 hour until a free flowing
suspension forms. The suspension is filtered and air dried. The resultant
optionally substituted benzene boronic acid (i.e., a compound of Formula
WO 94/22852 ~ PCT/US94/03004
-14-
3B) is carried on to the next step of the process.
PREPARATION OF FORMULA 3C
An optionally substituted phenol is dissolved in an aprotic solvent (such
as CHzClz or THF, preferably CH2Clz) and cooled to 0 C. About 5 molar
equivalents of triethylamine are added, followed by about 1.5 molar
equivalents of trifluoromethanesulfonic anhydride (triflic anhydride)
(e.g., via addition funnel) under nitrogen over a period of 15 min to 60
min, preferably 30 min. The solution is poured into a saturated NaHC03
solution and extracted with an aprotic solvent such as CHZC12. The organic
layer is washed with water (2 x), and brine (100 mL), dried over a drying
agent (e. g., MgSO,), filtered, and concentrated. Flash chromatography using
10-50%, preferably 20%, ethyl acetate/hexanes provides a slightly impure
brown oil Which can be distilled under vacuum to give the
trifluoromethanesulfonyloxy derivative (triflate) as a yellow liquid.
PREPARATION OF FORMULA I
An 6-optionally substituted-8-haloquinoline compound (i.e., a
compound of Formula 5) is combined with about 2 molar equivalent of an
optionally substituted benzene boronic acid, i.e., Formula 3A, 2 M Na~C03
(about 4 molar equivalent), methanol, and benzene. To this solution is
added about 0.1 molar equivalent of palladium tetrakis triphenylphosphine.
The reaction mixture is heated at about reflux for a period of about 3 to 9
hours, preferably about 6 hours. The progress of the reaction is monitored
by TLC. Upon completion of the reaction, the mixture is allowed to cool to
about room temperature, and the solvents are removed. About 40 mL/molar
equivalent of ethyl acetate is added to the residue and filtered through a
drying agent (e. g., NazSO,). The product is isolated by chromatography,
preferably preparative thin layer chromatography, yielding the desired
6-optionally substituted-8-aryl-quinoline compound (i.e., a compound of
Formula I).
REACTION SCHEME C
X
CH3
\ \ \ \ ~X
\ \ ~X
/ and/or
N
N
Rz
R
R
Formu I o I Formu I a I
where RZ is optionally where X is chloro,
substituted phenyl bromo or iodo
zy~
WO 94/22852 ~O~ PCT/US94/03004
-15-
\ \ X \ \ X ~ \ \ ~H
~ i / and/or ~ i / ~ N~
N ~ N ~ Rz
Formu I a I Formu I a I
where X is chloro, where R1 is formyl
bromo or iodo
OH
Formu I a I \ \ \R
where R~ is formyl ~ ~
N
R2
Formula I
where R~ is -CCOH]-R
and R is lower alkyl
REACTION SCHEME C-1
\ \ ~X R'BC~H]2 \ \ ~R
/ C3B] ~ i /
N ~ N
C 17 C I]
where X is chloro, where R is cycloalkenyl,
bromo or iodo aryl or heterocycle
45
WO 94/22852 PCT/US94/03004
-16-
REACTION SCHEME C-2
CN
\ \ x \ \ cN I \ \ ~R
i / -~. ~ i / ~ ~
N N N
~2
~I] CI] CI]
where X is chloro, where R is alkyl,
cycloalkyl,
bromo or iodo
or heterocycle
CN
\ \ ~R \ \ 'R
i / ~ / /
N ~ N
Rz R2
Formula f
where Rz is optionally where R is alkyl,
substituted phenyl cycloalkyl or heterocycle
REACTION SCHEME C-3
\ \ x
NH2R \ \ ~NHR \ \ ~NR2
/
N or NHRZ ~ N~ / or ~ N~ /
R2 R2 R~
CI] CI] CI]
40
where X is where R is lower where R2 is
chloro or alkyl, cycloalkyl, independently
bromo aryl or hetero- lower alkyl or aryl;
cycle or NRZ is heterocycle
PREPARATION OF FORMULA I iaHERE R1 IS -CHz-B AND/OR -CH-%
A 6-methyl-8-optionally substituted phenyl-quinoline (i.e., a
compound of Formula I where R' is methyl, prepared according procedures
described in Reaction Scheme A or B) is dissolved in a solvent such as
WO 94/22852 PCT/US94103004
-17-
carbon tetrachloride and heated under reflux. About 1 molar equivalent of
a halogenating reagent, such as N-bromosuccinamide (NBS) or N-
chlorosuccinamide (NCS), preferably N-bromosuccinamide and about 0.2 molar
equivalent of 2,2'-azobis(2-methylpropionitrile) are added to the refluxing
solution. The reaction mixture is optionally exposed to light (e.g., 250 W
light bulb) for a period of about 30 minutes to 90 minutes, preferably
about 1 hour. The reaction mixture is then stirred for a period of about 1
to 3 hours, preferably about 2 hours. The progress of the reaction is
monitored by TLC. Upon conversion of greater than about 90% of the starting
material, the reaction solution is cooled to about 0°C, then poured
through
a drying agent, such as Na2S0,. The solution is concentrated yielding the
mono- and di-halogenated products (i.e., Formula I, where R' is CHZ-X and -
CH-Xz) which are taken on to the next step without further purification or
isolation.
PREPARATION OF FORMULA I WHERE R' IS FORMYL
The mono and dihalogenated quinoline mixture from the previous step
is dissolved in a solvent, such as methylene chloride or chloroform,
preferably chloroform, and added in a gradual manner to an oxidizing
reagent, such as about 5 molar equivalents of tetra n-butylammonium
dichromate in a solvent, such as methylene chloride or chloroform,
preferably chloroform. The reaction mixture is heated, preferably at
reflux temperature, and refluxed for about 2 to 6 hours, preferably about 4
hours. The reaction mixture is allowed to cooled to about room temperature
and filtered, preferably through a silica gel pad.
The residue is eluted with an ether, preferably diethyl ether, and isolated
and purified by chromatography to obtain the desired 6-formyl-8-(optionally
substituted phenyl)quinoline (i.e., Formula I where R' if formyl).
Alternatively, the mono and di-halogenated quinoline mixture can be
oxidized by the Kornblum oxidation [Chew Rev, Vol 67, No. 3, (1967), p 247-
260] or the Sommelet oxidation [Org. React., Vol 8, p 197-217 (1954)].
PREPARATION OF FORMULA I WHERE R1 IS -CH(OH)-(LOWER ALKYL)
A 6-formyl-8-(optionally substituted phenyl)quinoline is dissolved in
a nonpolar solvent, preferably tetrahydrofuran, and cooled to a temperature
in the range of about -50°C to -100°C, preferably about -
78°C. To this
solution is added an alkylation reagent, such as a lower-alkyl Grignard
reagent, or a lower-alkyl lithium reagent (about 1 to 3 molar equivalent,
preferably about 2 molar equivalent) in a gradual manner. The reaction
mixture is stirred for a period of about 10 to 30 minutes, preferably about
20 minutes. The reaction mixture is then quenched with an aqueous salt
solution, preferably saturated ammonium chloride. The desired product is
purified by extraction (preferably With ethyl acetate), and drying
(preferably over MgSO,). The residue is further purified and isolated by
WO 94/22852 PCTIUS94103004
-18-
chromatography to yield the desired optionally substituted 6-[(lower
alkyl(hydroxy)methyl]-8-(optionally substituted phenyl)quinoline, i.e.,
Formula I where R' is lower alkyl (hydroxy)methyl.
PREPARATION OF THE SALT OF FORMULA I
The pharmaceutically acceptable salts of Formula I are prepared by
dissolving a compound of Formula I in a suitable solvent (such as methanol,
dioxane or diethyl ether) adding 1 to 3 molar equivalents (preferably about
two molar equivalent) of an appropriate acid (such as hydrogen chloride
gas) or base (such as an alkaline earth hydroxide, e.g., lithium hydroxide,
calcium hydroxide, potassium hydroxide, sodium hydroxide or the like;
preferably sodium hydroxide) and stirring.
The salt is isolated by lyophilization or by precipitation, using
techniques that will be apparent to~those skilled in the art.
PREPARATION OF N-OXIDES OF FORMULA I
A compound of Fozmula I, Where R' is pyridylmethyl (e.g., 4-
pyridylmethyl, 3-pyridylmethyl or 2-pyridylmethyl), is converted to the
corresponding N-oxide derivative by treating it with an oxidizing agent
(e. g., m-chloroperoxybenzoic acid) in a solvent (e. g., methylene chloride).
The solution is stirred for a period of about 30 to 90 minutes, preferably
about 60 minutes at a temperature in the range of about 0°C to
50°C,
preferably about room temperature. Following the reaction, the desired
product is isolated and purified by preparative thin-layer chromatography.
PREFERRED COMPOUNDS
Presently preferred are the compounds of Formula I where RZ is
3-chlorophenyl, 3-nitrophenyl, 3-cyanophenyl or 3-chloro-4-fluoro- phenyl.
Of the compounds where RZ is 3-chlorophenyl, 3-nitrophenyl or 3
cyanophenyl most preferred are the compounds of Formula I where and R' is
4-pyridylmethyl, benzyl, ethyl, n-propyl, isopropyl, n-butyl or 1-hydroxy-
1-methylethyl.
Of the compounds where RZ is 3-chloro-4-fluorophenyl most preferred
are the compounds of Formula I where R' is 4-pyridylmethyl, benzyl, ethyl,
n-propyl, isopropyl, n-butyl or 1-hydroxy-1-methylethyl.
PREFERRED PROCESSES
A preferred process for making 6-optionally substituted 8-aryl-
quinolines is combining the corresponding optionally substituted benzene
boronic acid with the corresponding 6-optionally substituted quinoline.
Another preferred process for making 6-optionally substituted 8-aryl-
quinolines is combining the corresponding optionally substituted p-amino-m-
benzene with glycerol and arsenic pentoxide.
Other preferred processes for making 6-optionally substituted 8-azyl-
WO 94/22852 ~~~ PCT/US94/03004
-19-
quinolines are shown in Reaction Schemes C, C-1, C-2, and C-3.
LAST STEPS
The compounds of this invention are prepared according to the following
processes:
A process for the preparation of a compound of the formula
R~
\ \
N
R2
Formula I
wherein:
R' is selected from hydrogen; lower alkyl; cycloalkyl; cycloalkyloxy;
cycloalkylamino; cycloalkyl lower alkyl; lower alkoxy; formyl;
hydroxy-lower alkyl; carboxyalkyl; optionally substituted aryl,
aryloxy, arylamino or aryl lower alkyl; optionally substituted
heterocycle, heterocycle-oxy, heterocycle-amino or heterocycle
lower alkyl; and
Rz is optionally substituted phenyl,
provided that when R' is methoxy, Rz is not 4-nitrophenyl or 4-
aminophenyl;
or a pharmaceutically acceptable salt or N-oxide thereof,
which comprises
(a) reacting a compound of the formula (4)
R~
R2
NH2
where
R' and R= are as defined above, with glycerol in the presence of an
oxidant; or
WO 94/22852 PCTIUS94/03004
-20-
(b) reacting a compound of the formula (5)
R ~'
\ \
N
X
where
R' is as defined above and X is chloro, bromo or iodo, with a boronic
acid of the formula (3B)
RZ-B (OH),
where RZ is optionally substituted phenyl; or
(c) reacting a compound of the formula
-
R~
\ \
i / i
N
ZnCI
where
R' is as defined above, with a trifluoromethane-
sulfonyloxy benzene of the formula (3C)
RZ-OTf
where Rz is optionally substituted phenyl, and -OTf is trifluoro-
methanesulfonyloxy; or
(d) reacting a compound of the formula (5)
0
\ \ \R
N
Br
where
R is lower alkyl, cycloalkyl, aryl, or heterocycle, with a boronic
acid of the formula (3B)
RZ-B (OH)
where R'- is optionally substituted phenyl; or
WO 94/22852 ~~~, PCT/US94/03004
-21-
(e) reacting a compound of the formula (I)
0
\ \ ~H
s ~ i /
N
Rz
where
R' is optionally substituted phenyl, with a lower alkyl Grignard
reagent or a lower alkyl lithium reagent; or
(f) reacting a compound of the formula
\ \ ~x
N
Rz
where
RZ is optionally substituted phenyl, with a boronic acid of the
formula (3B)
R-B (OH) Z
where R is cycloalkenyl, aryl or heterocycle; or
(g) reacting a compound of the formula
CN
\ \ ~R
N
Rz
where
R' is optionally substituted phenyl, and R is alkyl, cycloalkyl or
heterocycle, with a concentrated acid; or
(h) reacting a compound of the formula
\ \ ~x
N
Rz
where
RZ is optionally substituted phenyl and X is chloro, bromo or iodo,
WO 94/22852 PCT/US94/03004
2159643
-22-
with a compound of the formula
NFizR or NHR2,
where R is lower alkyl, cycloalkyl, aryl or heterocycle; and RZ is
independently lower alkyl or aryl; or NR2 is heterocycle; or
(i) reacting the free base of a compound of Formula I with an acid
to give a pharmaceutically acceptable acid addition salt; or
(j) reacting an acid addition salt of a compound of Formula I with
a base to give the corresponding free base; or
(k) converting an acid addition salt of a compound of Formula I to
another pharmaceutically acceptable acid addition salt of Formula I.
GENERAL UTILITY
UTILITY, TESTING AND ADMINISTRATION
The compounds of this invention, including the pharmaceutically
acceptable salts and N-oxides thereof, and the compositions containing them
are particularly useful as anti-inflammatory, immunosuppressive, anti-
allograft rejection, anti-graft-vs-host disease, anti-allergic agents
(e. g., asthma, rhinitis and atopic dermatitis), bronchodilation agents,
anti-autoimmune disease or analgetic agents. The compounds of this
invention act as PDE IV selective inhibitors, thereby modulating cANlP
levels. Thus, these compounds are of use for the treatment of cAMP related
conditions or diseases, particularly those that are modulated by leukocyte
cAMP.
For example, inflammation, autoimmune diseases, graft-vs-host disease
and allograft rejection are conditions that are manifested by the
proliferation of lymphocytes. The proliferation is triggered by the
presence of cAMP at specific levels. Inhibition of lymphocyte
proliferation is accomplished by increasing levels of cAN~ resulting from
the inhibition of lymphocyte phosphodiesterase.
TESTING
Potency and selectivity of compounds as inhibitors of PDE IV is
determined by following, for example, the procedures described in Example
22, or modifications thereof.
The immunomodulatory and anti-inflammatory activity of the compounds
of the invention can be determined by a variety of assays utilizing both in
vitro and in vivo procedures.
Inhibition of the proliferation of lymphocytes in response to
mitogenic stimulation is determined by the procedures described by Greaves,
et al. [~~Activation of human T and B lymphocytes by polyclonal mitogens,~~
WO 94122852 ~ , ~ ~~'~ ~ PCT/US94/03004
-23-
Nature, 248, 698-701 (1974)], or modifications thereof
(Example 23).
Inhibition of lymphocyte activation in response to antigenic
challenge is determined in vitro by inhibition of a cytolytic T-cell assay
(CTL) as described by Wunderlich, et al., Nature (1970), Vol. 228, p. 62,
or a modification thereof.
Immune modulation is determined by in vivo procedures utilizing the
Jerne Hemolytic Plaque Assay, [Jerne, et al., °The agar plaque
technique
for recognizing antibody producing cells,~~ Cell-bound Antibodies, Amos, B.
and Kaprowski, H. editors (Wistar Institute Press, Philadelphia) 1963,
p. 109] or a modification thereof (Example 24).
Anti-inflammatory activity is determined by the Arachidonic
Acid-Induced Mouse Ear Edema Assay [Young, et al., J. Invest. Derm., 82:
367-371 (1984)] (Example 25).
Anti-inflammatory activity is also determined by the Adjuvant
Arthritis assay [Pearson, C.M., Proc. Soc. Exp. Biol. Med., 91:95-101
(1956)], or modifications thereof (Example 26).
Anti-autoimmune activity in treating autoimnnune disease can be
determined utilizing the survivability of MR.L/lpr mice described by
Theofilopoulos, et al., Advances in Immunology, Vol 37, pages 269-390
(1985) on pages 274-276, or a modification thereof (Example 27).
Analgetic activity is determined by the Phenylquinone-induced Mouse
Writhing Assay [Hendershot, et al., J. Pharmacol. Exp. Ther., 125: 237-240
(1959)] (Example 28).
ADMINISTRATION
The compounds of this invention are administered at a therapeutically
effective dosage, i.e., that amount which, when administered to a manunal in
need thereof, is sufficient to effect treatment, as described above (for
example, to reduce or otherwise treat inflammation, pain and/or pyrexia in
the mammal). Administration of the active compounds and salts described
herein can be via any of the accepted modes of administration for agents
that serve similar utilities.
The level of the drug in a formulation can vary within the full range
employed by those skilled in the art, e.g., from about 0.01 percent Weight
(%w) to about 99.99%w of the drug based on the total formulation and about
.O1%w to 99.99%w excipient. Preferably the drug is present at a level of
about 10%w to about 70%w.
Generally, an acceptable daily dose is of about 0.001 to 50 mg per
kilogram body weight of the recipient per day, preferably about 0.05 to 25
mg per kilogram body weight per day, and most preferably about 0.01 to 10
mg per kilogram body weight per day. Thus, for administration to a 70 kg
person, the dosage range would be about 0.07 mg to 3.5 g per day,
preferably about 3.5mg to 1.75 g per day, and most preferably about 0.7 mg
to 0.7 g per day depending upon the individuals and disease state being
WO 94/22852 PCT/US94/03004
-24-
treated. Such use optimization is well within the ambit of those of
ordinary skill in the art.
Administration can be via any accepted systemic or local route, for
example, via parenteral, oral (particularly for infant formulations),
intravenous, nasal, bronchial inhalation (i.e., aerosol formulation),
transdermal or topical routes, in the form of solid, semi-solid or liquid
dosage forms, such as for example, tablets, suppositories, pills, capsules,
powders, solutions, suspensions, aerosols, emulsions or the like,
preferably in unit dosage forms suitable for simple administration of
precise dosages. The compositions will include a conventional
pharmaceutical carrier or excipient and an active compound of Formula I
and, in addition, may include other medicinal agents, pharmaceutical
agents, carriers, adjuvants, etc. Carriers can be selected from the
various oils, including those of petroleum, animal, vegetable or synthetic
origin, for example, peanut oil, soybean oil, mineral oil, sesame oil, and
the like. Water, saline, aqueous dextrose, and glycols are preferred
liquid carriers, particularly for injectable solutions. Suitable
pharmaceutical carriers include starch, cellulose, talc, glucose, lactose,
sucrose, gelatin, malt, rice, flour, chalk, silica gel, magnesium stearate,
sodium stearate, glycerol monostearate, sodium chloride, dried skim milk,
glycerol, propylene glycol, water, ethanol, and the like. Other suitable
pharmaceutical carriers and their formulations are described in
"Remington's Phazmaceutical Sciences" by E. W. Martin.
If desired, the pharmaceutical composition to be administered may
also contain minor amounts of non-toxic auxiliary substances such as
wetting or emulsifying agents, pH buffering agents and the like, such as
for example, sodium acetate, sorbitan monolaurate, triethanolamine oleate,
etc.
The compounds of this invention are generally administered as a
pharmaceutical composition which comprises a pharmaceutical excipient in
combination with a compound of Formula I. The level of the drug in a
formulation can vary within the full range employed by those skilled in the
art, e.g., from about .O1 percent weight (%w) to about 99.99%w of the drug
based on the total formulation and about .O1%w to 99.99%w excipient.
Preferably, the formulation will be about 3.5 to 60% by weight of the
pharmaceutically active compound, with the rest being suitable
pharmaceutical excipients.
INTRAVENOUS ADMINISTRATION
Intravenous injection has proven to be an important route of
administration for therapeutic agents. The compounds of the present
invention can be administered via this route, for example, by dissolving
the compound, ester, ether, N-oxide or salt in a suitable solvent (such as
water or saline) or incorporation in a liposomal formulation followed, by
dispersal into an acceptable infusion fluid. A typical daily dose of a
WO 94/22852 ~ PCT/US94103004
-25-
compound of the invention can be administered by one infusion, or by a
series of infusions spaced over periodic intervals.
ORAL ADMINISTRATION
Oral administration can be used to deliver the compound of Formula I
using a convenient daily dosage regimen which can be adjusted according to
the degree of affliction or for renal impairment, or to compensate for the
toxic effects of other medications administered contemporaneously. For
such oral administration, a pharmaceutically acceptable, non-toxic
composition is formed by the incorporation of any of the normally employed
excipients, such as, for example, pharmaceutical grades of mannitol,
lactose, starch, magnesium stearate, sodium saccharine, talcum, cellulose,
glucose, gelatin, sucrose, magnesium carbonate, and the like. Such
compositions take the form of solutions, suspensions, tablets, pills,
capsules, powders, sustained release formulations and the like. Such
compositions may contain between .01 wt/wt% and 99.99 wt/wt% of the
compound of Formula I, but preferably such compositions will contain
between 25 wt/wt% and about 80 wt/wt%.
Preferably the compositions will take the fornl of a capsule, pill or
tablet and thus the composition will contain, along with the active
ingredient, a diluent such as lactose, sucrose, dicalcium phosphate, and
the like; a disintegrant such as starch or derivatives thereof; a lubricant
such as magnesium stearate and the like; and a binder such as a starch,
polyvinylpyrrolidone, gum acacia, gelatin, cellulose and derivatives
thereof, and the like. For oral administration to infants, a liquid
formulation (such as a syrup or suspension) is preferred.
AEROSOL ADMINISTRATION
Aerosol administration is an effective means for delivering a
therapeutic agent directly to the respiratory tract. Some of the
advantages of this method are: 1) it circumvents the effects of enzymatic
degradation, poor absorption from the gastrointestinal tract, or loss of
the therapeutic agent to the hepatic first-pass effect;
2) it administers therapeutic agents which would otherwise fail to reach
their target sites in the respiratory tract due to their molecular size,
charge or affinity to extra-pulmonary sites; 3) it provides for fast
absorption into the body via the alveoli of the lungs; and 4) it avoids
exposing other organ systems to the therapeutic agent, which is important
where exposure might cause undesirable side effects. For these reasons,
aerosol administration is particularly advantageous for treatment of
asthma, local infections of the lung, and other diseases or disease
conditions of the lung and respiratory tract.
There are three types of pharmaceutical inhalation devices,
nebulizers inhalers, metered-dose inhalers (MDI) and dry powder inhalers
(DPI). Nebulizer devices produce a stream of high velocity air that causes
WO 94122852 ~ PCT/US94/03004
-26-
the therapeutic agent (which has been formulated in a liquid form) to spray
as a mist which is carried into the patient's respiratory tract. MDIs
typically have the formulation packaged with a compressed gas. Upon
actuation, the device discharges a measured amount of therapeutic agent by
compressed gas, thus affording a reliable method of administering a set
amount of agent.
DPIs administer therapeutic agents in the form of a free flowing
powder that can be dispersed in the patient's inspiratory air-stream during
breathing by the device. In order to achieve a free flowing powder, the
therapeutic agent is formulated with an excipient, such as lactose. A
measured amount of the therapeutic agent is stored in a capsule form and is
dispensed with each actuation. Examples of DPIs being used are Spinhaler
(for the administration of disodium cromoglycate), Rotahaler' (for
albuterol) and Turbuhale= (for terbutaline sulfate). All of the above
methods can be used for administering the present invention, particularly
for the treatment of asthma and other similar or related respiratory tract
disorders.
LIPOSOMAL FORMULATIONS
Pharmaceutical formulations based on liposomes have recently reached
human clinical trials. Their benefits are believed related to favorable
changes in tissue distribution and pharmacokinetic parameters that result
from liposome entrapment of drugs, and may be applied to the compounds of
the present invention by those skilled in the art.
The formulations can be designed to either target drug to disease
sites [see: Lopez-Berestein et al., J. Infect. Dis., 151: 704-710 (1985);
Gotfredsen et al., Biochemical Pharmacology, 32: 3389-3396 (1983)]; or to
the reticuloendothelial system [see Eppstein et al., Int. J. Immunotherapy,
2: 115-126 (1986)], to increase duration of drug action [see: Gabizon et
al., Cancer Res., 42: 4734 (1982); Eppstein et al., Delivery Systems for
Peptide Drugs, Eds. S.S. Davis, L. Illum and E. Tomlinson, Plenum Pub.
Corp., New York, pp. 277-283; C.A. Hunt, Biochemica et Biophysica Acta.,
719: 450-463 (1982); and Senior et al., Biochemica et Biophysica Acta.,
839: 1-8 (1985)], or to divert a drug away from organs that are
particularly sensitive to its toxic effects [see: Weinstein et al.,
Pharmac. Ther., 24: 207-233 (1983); Olson et al., Eur. J. Cancer Clin.
Oncol., 18: 167-176 (1982); and Gabzion et al., supra.].
Controlled release liposomal liquid pharmaceutical formulations for
injection or oral administration are described in U.S. Patent No.
4,016,100. Liposomal applications for oral drug delivery of a lyophilized
liposome/peptide drug mixture filled into intestine capsules have also been
suggested, see U.S. Patent No. 4,348,384.
WO 94/22852 PCTIUS94/03004
'~~0~
-27-
SUPPOSITORIES
For systemic administration via suppository, traditional binders and
carriers include, for example, polyalkaline glycol or triglycerides fe.g.,
PEG 1000 (96%) and PEG 4000 (4%)]. Such suppositories may be formed from
mixtures containing active ingredients in the range of from about 0.5
wt/wt% to about 10 wt/wt%; preferably from about 1 wt/wt% to about
2 wt/wt%.
LIQUIDS
Liquid pharmaceutically administrable compositions can, for example,
be prepared by dissolving, dispersing, etc. an active compound (about 0.5%
to about 20%), as described above, and optional pharmaceutical adjuvants in
a carrier, such as, for example, Water, saline, aqueous dextrose, glycerol,
ethanol and the like, to thereby form a solution or suspension.
Actual methods of preparing such dosage forms are known, or will be
apparent, to those skilled in this art; for example, see Remington~s
Pharmaceutical Sciences, Mack Publishing Company, Easton, Pennsylvania,
16th Ed., 1980. The composition to be administered will, in any event,
contain a quantity of the active compounds) in a pharmaceutically
effective amount for relief of the particular condition being treated in
accordance with the teachings of this invention.
EgAMPLES
The following examples are given to enable those skilled in the art
to more clearly understand and to practice the present invention. They
should not be considered as limiting the scope of the invention, but merely
as being illustrative and representative thereof.
EgAMPLE 1
PREPARATION OF 4-(4-AMINOHENZYL)PYRIDINE
lA. Formula 2, Whare R' is 4-Pyridylmethyl
4-(4-Nitrobenzyl)pyridine (214.22 mg) was combined with absolute
ethanol (10 mL) and SnCl2-2H20 (225.63 mg) . The reaction mixture was heated
to 70°C under nitrogen for 2 hours. The progress of the reaction was
monitored by thin-layer chromatography (e.g., an aliquot of the mixture was
collected, neutralized with saturated NaHC03 to pH 7-8, extracted into
ethyl acetate). When all of the starting material had been converted, the
reaction mixture was allowed to cool to room temperature, neutralized with
saturated NaHC03 to pH 7-8 and extracted With ethyl acetate. The organic
layers Were collected and dried over MgSO,. The solvents were removed and
the residue was triturated with ethyl ether. The desired product, i.e., 4-
(4-aminobenzyl)pyridine, was filtered out of the solution as cream colored
crystals and air dried, mp 159.1°C - 160.3°C; elemental
analysis:
WO 94/22852 PCT/US94/03004
;:. :~~596a3
-28-
[calc(found)] C: 78.2 (78.12), H: 6.56 (6.45), and N: 15.20 (15.33).
1B. Compounds of Formula 2, Where R' Is Varied
Other desired compounds of Formula 2 can be prepared by following the
procedures described in Example lA, and using different starting compounds.
For example: 4-isopropylnitrobenzene, 4-methylnitrobenzene, 4-
benzylnitrobenzene, 4-ethylnitrobenzene, 4-(3-propenyl)nitrobenzene, 4-
propylnitrobenzene, 4-butylnitrobenzene, 4-pentylnitrobenzene,
4-hexylnitrobenzene, 4-methoxynitrobenzene, 4-ethoxynitrobenzene,
4-(trifluoromethyl)nitrobenzene, 4-(3-pyridylmethyl)nitrobenze,
4-(2-pyridylmethyl)nitrobenzene, 4-(cyclopentylmethyl)nitrobenzene,
4-(cyclopropylmethyl)nitrobenzene, 4-(thiomethyl)nitrobenzene, and
4-(methylsulfonylmethyl)nitrobenzene can be converted to the corresponding
(substituted) anilines.
EgAMPLE 2
PREPARATION OF 4-(4-AMINO-3-BROMOHENZYL)PYRIDINE
2A. Formula 3, Where R' is 4-Pyridylmethyl
A solution of N-bromosuccinimide (1.4 gm) in 10 mL dimethylformamide
was added in a dropwise manner to a solution of
4-(4-aminobenzyl)pyridine (1.5 gm) in 10 mL of dimethylformamide.
The reaction flask was wrapped in aluminum foil to prevent exposure
of the reagents to light. The reaction mixture was stirred at room
temperature for 3 hours. The progress of the reaction was monitored
by thin-layer chromatography. When the reaction was completed, the
reaction mixture was combined with 100 mL of H20 with stirring.
A reddish brown precipitate was formed, filtered out of solution, washed
with H20, and air dried. The solid Was dissolved in ethyl acetate, back
washed with saturated NaCl, dried over MgSO, and triturated with ethyl
ether. The solid was filtered from the solution and air dried yielding
1.6 gm of 4-(4-amino-3-bromobenzyl)pyridine as reddish/tan crystals.
mp 102.7°C-103.9°C;
2H. Other Prepared Compounds of Formula 3~ i4here R' Is Varied
Other compounds of Formula 3 were also prepared following the
procedures described in Example 2A, and substituting 4-(4-aminobenzyl)-
pyridine with different starting compounds. For example, following is a
list of starting compounds and the corresponding desired compounds that
were obtained by following the above-referenced procedure:
4-isopropylaniline was converted to 2-bromo-4-isopropylaniline,
4-methylaniline was converted to 2-bromo-4-methylaniline,
4-benzylaniline Was converted to 4-benzyl-2-bromoaniline, and
4-(4-pyridylmethyl)aniline was converted to 2-bromo-4-(4-
pyridylmethyl)aniline.
WO 94/22852 PCT/US94/03004
-29-
2C. Compounds of Formula 3, Where R' Is Varied
In addition, other desired compounds of Forniula 3 can be prepared by
following the procedures described in Example 2A, and using different
starting compounds.
For example, 4-ethylaniline, 4-(3-propenyl)aniline,
4-propylaniline, 4-butylaniline, 4-pentylaniline, 4-hexylaniline,
4-trifluoromethylaniline, 4-(3-pyridylmethyl)aniline,
4-(2-pyridylmethyl)aniline, 4-(cyclopentylmethyl)aniline,
4-(cyclopropylmethyl)aniline, 4-(methylthiomethyl)aniline, and
4-(methylsulfonylmethyl)aniline can be converted to the corresponding
2-bromo-4-(substituted)-anilines.
E%AMPLE 3
PREPARATION OF
3-CHLOROBENZSNE BORONIC ACID
3A. Formula 3B Where RZ is 3-chlorophenyl
A solution of trimethylborate in 200 mL of ethyl acetate was cooled
to -65°C. 3-Chlorobenzene magnesium chloride (0.8 M, 60 mL), i.e., a
Grignard reagent, was added to the solution in a dropwise manner over
20 minutes. The mixture was kept in the temperature range of -60°C to
-70°C and stirred. After 30 minutes, the mixture was allowed to warm to
0°C and stirred for 1 hour. The mixture was quenched With H20 (25 mL)
and
stirred at room temperature for 1 hour. The solvent was removed and the
remaining mass was extracted with ethyl ether (3 x 100 mL). The organic
layers were combined and washed with H20 (2 x 50 mL), dilute HCl (2 x 100
mL), H20 (2 x 50 mL) and brine (1 x 50 mL). The organic layer was dried
over MgSO" concentrated and allowed to stand. 100 mL of hexanes Was added
and the solution was stirred for 1 hour. The solution was filtered and
allowed to air dry yielding 4.6 g.of 3-chlorobenzene boronic acid as a
white solid.
3H. Compounds of Formula 3B where Rz is optionally substituted
Other desired compounds of Formula 3B can be prepared by following
the procedures described in Example 3A, and using different starting
compounds. For example, 3-chloro-4-fluorobenzene magnesium chloride, 4-
chlorobenzene magnesium chloride, benzene magnesium bromide, 3,4-
dichlorobenzene magnesium bromide, 3-bromobenzene magnesium bromide, and
3-(trifluoromethyl)benzene magnesium bromide can be converted to the
corresponding (substituted) benzene boronic acids.
EBAMPLE 4
PREPARATION OF 4-[4-AMINO-3-(3-NITROPHENYL)-
HENZYL]PYRIDINE
4A. Formula 4, tahere R' is 4-Pyridylmethyl, RZ is 3-Nitrophenyl.
WO 94/22852 PCT/US94103004
2 ~ 5~g'~ ~ 3
-30-
4-(4-Amino-3-bromobenzyl)pyridine (1.0 gm), 3-nitrobenzene boronic
acid (0.63 gm), palladium tetrakis triphenylphosphine (0.47 gm), methanol
(6.5 mL), 2.0 M NaZC03 (1.9 mL) and benzene (32 mL) were combined in a
reaction flask that was wrapped in aluminum foil (to prevent exposure of
the reagents to light). The reaction mixture was heated under reflex for 6
hours. The progress of the reaction was monitored by thin-layer
chromatography (9:1 hexane:ethyl acetate). When the starting material was
converted, the reaction mixture was allowed to cool and the solvents were
removed. Ethyl acetate was added to the residue, the resultant solution
was filtered through a pad of Na2S04, and concentrated. The product was
isolated by preparative thin-layer chromatography (9:1 hexane: ethyl
acetate) yielding 983 mg of 4-[4-amino-3-(3-nitrophenyl)benzyl]pyridine as
an orange oil.
Characteristic analytical data: ms m/e 305(M+); 'H NMR (CDC13) b 3.74
(bs, 2H), 3.92 (s, 2H), 6.77 (d, 1H, J=8.1 Hz), 6.95 (d, 1H, J=2.1 Hz),
7.03 (dd, 1H, J=2.1 Hz, J=8.1 Hz), 7.14 (d, 1H, J=5.7 Hz), 7.62 (dd, 1H,
J=8.2 Hz, J=7.7 Hz), 7.82 (ddd, 1H, J=7.7 Hz, J=1.9 Hz, J=2.5Hz), 8.21
(ddd, 1H, J=8.2 Hz, J=2.5 Hz, J=l.9Hz), 8.34 (dd, 1H, J=1.9 Hz, J=1.9 Hz),
and 8.5 (dd, 1H, J=5.7 Hz).
4H. Other Compounds of Formula 4, ~4here R' and RZ Are Varied.
Other compounds of Formula 4 were also prepared following the
procedures described in Example 4A, and substituting for 4-(4-amino-3-
bromobenzyl)pyridine and 3-nitrobenzene boronic acid with different
starting compounds. For example, following is a list of starting compounds
and the corresponding desired compounds that were obtained by following the
above-referenced procedure:
2-bromo-4-isopropylaniline and benzene boronic acid were combined to
form 2-phenyl-4-isopropylaniline,
2-bromo-4-isopropylaniline and 3-nitrobenzene boronic acid were
combined to form 2-(3-nitrophenyl)-4-isopropylaniline,
4-benzyl-2-bromo-aniline and 3-nitrobenzene boronic acid were
combined to form 4-benzyl-2-(3-nitrophenyl)-aniline,
2-bromo-4-methylaniline and 3-chloro-4-fluorobenzene boronic acid
were combined to form 2-(3-chloro-4-fluorophenyl)-4-
methylaniline,
2-bromo-4-isopropylaniline and 3-chloro-4-fluorobenzene boronic acid
were combined to form 2-(3-chloro-4-fluorophenyl)-4-
isopropylaniline,
4-benzyl-2-bromo-aniline and 3-chloro-4-fluorobenzene boronic acid
were combined to form 4-benzyl-2-(3-chloro-4-fluorophenyl)-
aniline,
2-bromo-4-(4-pyridylmethyl)aniline and 3-chloro-4-fluorobenzene
boronic acid were combined to form 2-(3-chloro-4-fluorophenyl)-
WO 94/22852 ~ ~~0~ PCT/US94/03004
-31-
4-(4-pyridylmethyl)aniline,
2-bromo-4-methylaniline and 4-chlorobenzene boronic acid were
combined to form 2-(4-chlorophenyl)-4-methylaniline,
2-bromo-4-isopropylaniline and 4-chlorobenzene boronic acid were
combined to form 2-(4-chlorophenyl)-4-isopropyl aniline, and
2-bromo-4-benzylaniline and 4-chlorobenzene boronic acid were
combined to form 2-(4-chlorophenyl)-4-benzyl- aniline.
4C. Compounds of Formula 4, Where R1 and R= Are Varied
In addition, other desired compounds of Formula 4 can be prepared by
following the procedures described in Example 4A, and using different
starting compounds. For example,
2-bromo-4-ethylaniline and 3-(trifluoromethyl)benzene boronic acid
can be combined to form 2-(3-trifluoromethylphenyl)-4
ethylaniline,
2-bromo-4-isopropylaniline and 3,4-dichlorobenzene boronic acid can
be combined to form 2-(3,4-dichlorophenyl)-4-isopropylaniline,
2-bromo-4-(3-propenyl)aniline and 3-nitrobenzene boronic acid can be
combined to form 2-(3-nitrophenyl)-3-propPnylaniline,
2-bromo-4-propylaniline and 3-nitrobenzene boronic acid can be
combined to form 2-(3-nitrophenyl)-4-propylaniline,
2-bromo-4-(n-butyl)aniline and 3-nitrobenzene boronic acid can be
combined to form 2-(3-nitrophenyl)-4-(n-butyl)aniline,
2-bromo-4-(n-hexyl)aniline and benzene boronic acid can be combined
to form 2-phenyl-4-(n-hexyl)aniline,
2-bromo-4-pentylaniline and 3-nitrobenzene boronic acid can be
combined to form 2-(3-nitrophenyl)-4-pentylaniline,
2-bromo-4-benzylaniline and 3,4-dichlorobenzene boronic acid can be
combined to fozm 2-(3,4-dichlorophenyl)-4-benzylaniline,
2-bromo-4-cyclopentylmethylaniline and 3-chlorobenzene boronic acid
can be combined to form 2-(3-chlorophenyl)-4-
cyclopentylmethylaniline,
2-bromo-4-cyclopropylmethylaniline and 3-nitrobenzene boronic acid
can be combined to form 2-bromo-4-cyclopropyl- methylaniline,
2-bromo-4-(methylthiomethyl)aniline and 3-chlorobenzene boronic acid
can be combined to form 2-(3-chlorophenyl)-4-
(methylthiomethyl)aniline,
2-bromo-4-(methylsulfonylmethyl)aniline and benzene boronic acid can
be combined to form 2-phenyl-4-(methylsulfonyl- methyl)aniline,
2-bromo-4-(cyclopentylmethyl)aniline and 2,3-dichlorobenzene boronic
acid can be combined to form 2-(2,3-dichlorophenyl)-4-
(cyclopentylmethyl)aniline, and
2-bromo-4-(cyclopentylmethyl)aniline and 3-nitrobenzene boronic acid
can be combined to form 2-(3-nitrophenyl)-4-
(cyclopentylmethyl)aniline.
F
WO 94122852 ~5 9 6. ~ 3 PCT/US94/03004
-32-
EXAMPLE 5
PREPARATION OF 6-(4-PYRIDYLMETHYL)-8-
(3-NITROPHENYL)QUINOLINE
5A. Formula I, Where R' is 4-Pyridylmethyl, R2 is 3-Nitrophenyl.
4-[4-Amino-3-(3-nitrophenyl)benzyl]pyridine (600 mg) was combined
with glycerol (489 mg) and arsenic pentoxide (325 mg) under nitrogen. The
reaction mixture was heated to 100°C for 30 minutes. Concentrated
sulfuric
acid (406 mg) was added in a dropwise manner and the reaction mixture was
further heated to 150°C for 2 hours. The reaction was monitored by TLC
(9:1, hexane:ethyl acetate). When TLC indicated that greater than 90% of
the starting material had been converted, the reaction mixture was removed
from the heat, ice (approximately 1 gm) was added, and NH40H was added to
basify the mixture. A precipitate was filtered out of the mixture, washed
with HZO and air dried.
The resulting solid was suspended in hot ethyl acetate and filtered. The
filtrate was concentrated and chromatographed by preparative thin-layer
chromatography (4:1, hexane:ethyl acetate), the band with the higher RE
value was isolated yielding 181 mg of 6-(4-pyridylmethyl)-8-(3-
nitrophenyl)quinoline as a viscous yellow oil. The product was
recrystallized from diethyl ether as a light yellow solid.
Characteristic analytical data: mp 131°C-143°C; elemental
analysis,
calc(found) C: 73.89 (73.90), H: 4.43 (4.50), and N: 12.31 (12.30); 'H NMR
CDC13 b 4.21 (s, 2H), 7.21 (d, 2H, J=5.9 Hz), 7.48 (dd, 1H, J=8.3 Hz, J=4.2
Hz), 7.6 (d, 1H, J=2 Hz), 7.66 (dd, 1H, J=8 Hz, J=7.7 Hz), 7.7 (d, 1H,
J=2Hz), 8.03 (ddd, 1H, J=7.7 Hz, J=1.4 Hz, J=1.2 Hz), 8.2 (dd, 1H, J=8.3
Hz, J=1.7 Hz, J=1.2 Hz), 8.28 (ddd, 1H J=8 Hz, J=1.44 Hz, J=1.2 Hz),
8.55(m, 3H), 8.93 (dd, 1H, J=4.2 Hz, J=1.7 Hz).
5H. Other Compounds of Formula I, Where R1 and R2 Are Varied.
Other compounds of Formula I were also prepared following the
procedures described in Example 5A, and substituting 4-[4-amino-3-(3-
nitrophenyl)benzyl]pyridine with different starting compounds. For
example, following is a list of starting compounds and the corresponding
desired compounds that were obtained by following the above-referenced
procedure:
2-phenyl-4-isopropylaniline Was converted to 6-isopropyl-8-
phenylquinoline as an oil,
2-(3-nitrophenyl)-4-isopropylaniline was converted to 6-isopropyl-8-
(3-nitrophenyl)quinoline as an oil,
4-benzyl-2-(3-nitrophenyl)aniline was converted to 6-benzyl-8-(3-
nitrophenyl)quinoline, mp 101°C-103°C,
2-(3-chloro-4-fluorophenyl)-4-methylaniline was converted to 6-
methyl-8-(3-chloro-4-fluorophenyl)quinoline, mp 96°C-114°C,
2-(3-chloro-4-fluoro)-4-isopropylaniline was converted to
WO 94122852
PCT/US94/03004
~~e~
-33-
6-isopropyl-8-(3-chloro-4-fluorophenyl)quinoline as an oil,
4-benzyl-2-(3-chloro-4-fluorophenyl)aniline was converted to 6-
benzyl-8-(3-chloro-4-fluorophenyl)quinoline as an oil,
2-(3-chloro-4-fluorophenyl)-4-(4-pyridylmethyl)aniline was converted
to 6-(4-pyridylmethyl)-8-(3-chloro-4-fluorophenyl)quinoline as
an oil,
2-(4-chlorophenyl)-4-methylaniline was converted to 6-methyl-8-(4-
chlorophenyl)quinoline, mp 96°C-98°C,
2-(4-chlorophenyl)-4-isopropylaniline was converted to 6-isopropyl-8-
(4-chlorophenyl)quinoline as an oil, and
4-benzyl-2-(4-chlorophenyl)aniline was converted to 6-benzyl-8-(4-
chlorophenyl)quinoline as an oil.
5C. Formula I, Where R' and R= Are Varied
In addition, other desired compounds of Formula I can be prepared by
following the procedures described in Example 5A, and using different
starting compounds. For example,
2-(3-trifluoromethylphenyl)-4-ethylaniline can be converted to
6-ethyl-8-(3-trifluoromethylphenyl)quinoline,
2-(3,4-dichlorophenyl)-4-isopropylaniline can be converted to
6-isopropyl-8-(3,4-dichlorophenyl)quinoline,
2-(3-nitrophenyl)-4-(3-propenyl)aniline can be converted to
6-(3-propenyl)-8-(3-nitrophenyl)quinoline,
2-(3-nitrophenyl)-4-propylaniline can be converted to
6-propyl-8-(3-nitrophenyl)quinoline,
2-(3-nitrophenyl)-4-butylaniline can be converted to
6-butyl-8-(3-nitrophenyl)quinoline,
2-(phenyl)-4-hexylaniline can be converted to
6-hexyl-8-(phenyl)quinoline,
2-(3-nitrophenyl)-4-pentylaniline can be converted to
6-pentyl-8-(3-nitrophenyl)quinoline,
2-(3,4-dichlorophenyl)-4-benzylaniline can be converted to
6-benzyl-8-(3,4-dichlorophenyl)quinoline,
2-(3-methoxycarbonylphenyl)-4-(3-pyridylmethyl)aniline can be
converted to 6-(3-pyridylmethyl)-8-(3-methoxycarbonylphenyl)-
quinoline,
2-(3-methoxycarbonylphenyl)-4-(2-pyridylmethyl)aniline can be
converted to 6-(2-pyridylmethyl)-8-(3-methoxycarbonylphenyl)-
quinoline,
2-(3-chlorophenyl)-6-cyclopentylmethylaniline can be converted to 6-
cyclopentylmethyl-8-(3-chlorophenyl)quinoline,
2-(3-nitrophenyl)-4-cyclopropylmethylaniline can be converted to 6-
cyclopropylmethyl-8-(3-nitrophenyl)quinoline,
2-(3-chlorophenyl)-4-(methylthiomethyl)aniline can be converted to 6-
methylthiomethyl-8-(3-chlorophenyl)quinoline,
WO 94/22852 ~- . PCTIUS94/03004
-34-
2-phenyl-4-(methylsulfonylmethyl)aniline can be converted to 6-
methylsulfonylmethyl-8-(phenyl)quinoline,
2-(3,4-dichlorophenyl)-4-(cyclopentylmethyl)aniline can be converted
to 6-cyclopentylmethyl-8-(3,4-dichlorophenyl)quinoline, and
2-(3-nitrophenyl)-4-(cyclohexylmethyl)aniline can be converted to 6-
cyclohexylmethyl-8-(3-nitrophenyl)quinoline.
EBAMPLE 6
PREPARATION OF 6-ISOPROPYL-8-BROMOQUINOLINE
6A. Fozmula 5 Where R' Is Isopropyl
2-Bromo-4-isopropylaniline (3.0 gm) [obtained from Aldrich Chemical
Co.J, glycerol (2.8 M1) and arsenic pentoxide (3.22 g) Were combined and
heated to 100°C. HzSO, (concentrated, 1.9 M1) was added to the reaction
mixture in a dropwise manner. The mixture was then heated to 150°C for
2.5
hours. The resulting black oil was added in a dropwise manner to a
stirring mixture of saturated NaHC03 (300 M1) and ethyl acetate (100 M1).
After completion of the addition, the reaction mixture was stirred for 30
minutes. The reaction mixture was then extracted with ethyl acetate (2 x
200 M1). The organic layers were washed with brine, dried over MgSO"
filtered and concentrated. The resulting material was further purified and
isolated by chromatography on silica gel (30:70 ethyl acetate/hexanes)
which yielded 2.5 g of 6-isopropyl-8-bromoquinoline as a brown oil.
Characteristic analytical data: elemental analysis calc.(found): C
57.62 (57.65), H 4.84 (4.78), and N 5.60 (5.57); 'H NMR (CDC13) b 1.34 (d,
6H, J=6.9 Hz), 3.05 (m, 1H, J=6.9 Hz), 7.42 ((dd, 1H, J=4.2 Hz, J=8.3 Hz),
7.57 (d, 1H, J=1.8 Hz), 7.97 (d, 1H, J=1.8 Hz), 8.1 (dd, 1H, J=8.3 Hz,
J=1.7 Hz), and 8.9 (dd, 1H, J=4.2 Hz, J=1.7 Hz).
E%AMPLE 7
PREPARATION OF 6-ISOPROPYL-
8- (3 -NITROPHENYL) QUINOLINE HYDROCFILORIDE
7A. Formula I Where R' Is Isopropyl and R2 Is 3-Nitropheayl.
6-Isopropyl-8-bromoquinoline (1.15 g) was dissolved in 46 M1 of
ethanol/benzene (1:1). 3-Nitrobenzene boronic acid (1.4 g), Na2C03
(2M, 9.2 M1) and tetrakis(triphenylphosphine)palladium (0.23 g) were added
successively. The reaction mixture was refluxed for 6 hours and then
cooled and concentrated. The resulting residue was partitioned between 75
mL H20 and 100 mL of ethyl acetate, and extracted with ethyl acetate (2 x
100 mL). The extracts were then dried over MgS04, filtered, concentrated
and chromatographed on silica gel (20:80 ethyl acetate:hexane) yielding 1.2
g of 6-isopropyl-8-(3-nitrophenyl)- quinoline as a slightly impure yellow
oil.
The desired product was obtained in its pure form by
reczystallization of its hydrochloride salt. The oil was dissolved in 25
WO 94/22852 , ~~~ PCT/US94103004
-35-
mL of 10% methanol:methylene chloride, to which a solution of 1M HC1/Et20
(100 mL) was added. The solution was stirred for 10 minutes and then
concentrated. The resulting white solid was recrystallized from ethyl
acetate: ethanol yielding 0.74 g of 6-isopropyl-8-(3-nitrophenyl)quinoline
as pale yellow crystals.
Characteristic analytical data: elem. anal. calc. (found):
C 65.75 (65.92), H 5.21 (5.21), and N 8.52 (8.67); and 'H NMR (DMSO)
b 1.37 (d, 6H, J=6.9 Hz), 3.2 (m, 1H, J'=6.9 Hz), 7.81 (m, 2H), 7.92
(d, 1H, J=1.9 Hz), 8.09 (d, 1H, J=1.9 Hz), 8.11 (m, 1H), 8.43 (m, 1H),
8.53 (m, 1H), 8.7 (dd, 1H, J=1.5 Hz, J=8.3 Hz), 8.9 (dd, 1H, J=4.7 Hz,
J=8.3 Hz).
EXAMPLE 8
PREPARATION OF
6-METHYL-8-BROMOQUINOLINE
8A. Formula 5 inhere R' is Methyl and X is Hromo
A slurry of 2-bromo-4-methylaniline (5 g), glycerol (6.7 g),
and arsenic pentoxide (3.9 g) was formed and heated to 100°C for
30 minutes. Concentrated HZS04 (4.9 g) was added dropwise and the mixture
was heated to 150°C for two hours. The progress of the reaction was
monitored by thin-layer chromatography (9:1 hexane: ethyl acetate). Upon
completion, the reaction mixture was worked up by adding water, basifying
with saturated NaHC03, extracting with ethyl acetate, and drying the
organic layer over MgS04. The desired product was isolated and purified by
column chromatography (9:1 hexane:ethyl acetate), yielding 6-methyl-8-
bromoquinoline (1.8 g) as a yellow liquid.
Characteristic analytical data: 'H NMR (CDC13) b 2.5 (s, 3H), 7.4 (dd,
1H, J=8.3 Hz, J=4.3 Hz), 7.52 (bs, 1H), 7.9 (d, 1H, J=1.8 Hz), 8.07 (dd,
1H, J=8.3 Hz, J=1.7 Hz), 8.88 (dd, 1H, J=4.3 Hz, J=1.7 Hz).
8H. Formula 5 Where R' Is Varied
Following the procedures described in Example 8A the following
desired compounds of Formula 5 can be obtained from the indicated starting
compounds. For example,
2-bromo-4-isopropylaniline and glycerol can be combined to form 6-
isopropyl-8-bromoquinoline,
2-bromo-4-methylaniline and glycerol can be combined to form
6-methyl-8-bromoquinoline,
2-bromo-4-benzylaniline and glycerol can be combined to form
6-benzyl-8-bromoquinoline,
2-bromo-4-(4-pyridylmethyl)aniline and glycerol can be combined to
form 6-(4-pyridylmethyl)-8-bromoquinoline,
2-bromo-4-ethylaniline and glycerol can be combined to form
6-ethyl-8-bromoquinoline,
WO 94/22852 PCT/US94/03004
-36-
2-bromo-4-(3-propenyl)aniline and glycerol can be combined to form 6-
(3-propenyl)-8-bromoquinoline,
2-bromo-4-propylaniline and glycerol can be combined to form
6-propyl-8-bromoquinoline,
2-bromo-4-(n-butyl)aniline and glycerol can be combined to form 6-(n-
butyl)-8-bromoquinoline,
2-bromo-4-pentylaniline and glycerol can be combined to form
6-pentyl-8-bromoquinoline,
2-bromo-4-hexylaniline and glycerol can be combined to form
6-hexyl-8-bromoquinoline,
2-bromo-4-(3-pyridylmethyl)aniline and glycerol can be combined to
form 6-(3-pyridylmethyl)-8-bromoquinoline,
2-bromo-4-(2-pyridylmethyl)aniline and glycerol can be combined to
form 6-(2-pyridylmethyl)-8-bromoquinoline,
2-bromo-4-(cyclopentylmethyl)aniline and glycerol can be combined to
form 6-(cyclopentylmethyl)-8-bromoquinoline,
2-bromo-4-(cyclopropylmethyl)aniline and glycerol can be combined to
form 6-(cyclopropylmethyl)-8-bromoquinoline,
2-bromo-4-(methylthiomethyl)aniline and glycerol can be combined to
form 6-(methylthiomethyl)-8-bromoquinoline, and
2-bromo-4-(methylsulfonylmethyl)aniline and glycerol can be combined
to form 6-(methylsulfonylmethyl)-8-bromoquinoline.
EXAMPLE 9
PREPARATION OF
6-METHYL-8-(3-NITROPHENYL)QUINOLINE
9A. Formula I, Where R' is Methyl and RZ is 3-Nitrophenyl.
To a mixture of 6-methyl-8-bromoquinoline (1.0 g), 3-nitrobenzene
boronic acid (0.63 g), purchased from Lancaster Chemicals, 2M NaC03 (1.9
mL), methanol (6.5 mL) and benzene 32 mL) was added palladium tetrakis
triphenylphosphine (0.47 g). The mixture was heated under reflux for 6
hours. The progress of the reaction was monitored by thin-layer
chromatography. Upon completion, the reaction mixture was cooled and the
solvents removed. Ethyl acetate was added to the residue and the solution
was filtered through a pad of NazSO,. The solution was concentrated and the
desired product was purified and isolated by preparative thin-layer
chromatography yielding 6-methyl-8-(3-nitrophenyl)quinoline (983 mg) as a
yellow oil. Elemental analysis [calc.(found)] C: 72.72 (72.63), H: 4.58
(4.32), and N: 10.60 (10.72).
9B. Formula I, Where R' and RZ Are Varied
The compounds of Formula I that were prepared following the
procedures described in Examples 1-5 (i.e., following Reaction Scheme A)
can also be prepared by following the procedures described in Examples 6,
WO 94122852 PCT/LJS94103004
-37-
7, 8 and 9 (i.e., following Reaction Scheme B).
For example,
6-isopropyl-8-bromoquinoline combined with benzene boronic acid can
be converted to 6-isopropyl-8-phenylquinoline,
6-isopropyl-8-bromoquinoline combined with 3-nitrobenzene boronic
acid can be converted to 6-isopropyl-8-(3-nitrophenyl)-
quinoline,
6-benzyl-8-bromoquinoline combined with 3-nitrobenzene boronic acid
can be converted to 6-benzyl-8-(3-nitrophenyl)quinoline,
6-methyl-8-bromoquinoline combined with 3-chloro-4-fluorobenzene
boronic acid can be converted to 6-methyl-8-(3-chloro-4-fluoro-
phenyl)quinoline,
6-isopropyl-8-bromoquinoline combined with 3-chloro-4-fluoro- benzene
boronic acid can be converted to 6-isopropyl-8-
(3-chloro-4-fluorophenyl)quinoline,
6-benzyl-8-bromoquinoline combined with 3-chloro-4-fluorobenzene
boronic acid can be converted to 6-benzyl-8-(3-chloro-4-
fluorophenyl)quinoline,
6-(4-pyridylmethyl)-8-bromoquinoline combined with 3-chloro-4-
fluorobenzene boronic acid can be converted to 6-(4-
pyridylmethyl)-8-(3-chloro-4-fluorophenyl)quinoline,
6-methyl-8-bromoquinoline combined with 4-chlorobenzene boronic acid
can be converted to 6-methyl-8-(4-chlorophenyl)- quinoline,
6-isopropyl-8-bromoquinoline combined with 4-chlorobenzene boronic
acid can be converted to 6-isopropyl-8-(4-
chlorophenyl)quinoline, and
6-benzyl-8-bromoquinoline combined with 4-chlorobenzene boronic acid
can be converted to 6-benzyl-8-(4-chlorophenyl)- quinoline.
9C. Formula I, Where R1 and Rz Are Varied
In addition, other desired compounds of Formula I can be prepared by
following the procedures described in Example 9A, and using different
starting compounds. For example,
6-ethyl-8-bromoquinoline and 3,4 dichlorobenzene boronic acid can be
combined to form 6-ethyl-8-(3,4-dichlorophenyl)quinoline,
6-(3-propenyl)-8-bromoquinoline and 3-(trifluoromethyl)benzene
boronic acid can be combined to form 6-(3-propenyl)-
8-(3-trifluoromethylphenyl)quinoline,
6-propyl-8-bromoquinoline and 3-nitrobenzene boronic acid can be
combined to form 6-propyl-8-(3-nitrophenyl)quinoline,
6-(n-butyl)-8-bromoquinoline and 3-chlorobenzene boronic acid can be
combined to form 6-(n-butyl)-8-(3-chlorophenyl)quinoline,
6-pentyl-8-bromoquinoline and 3,4-dichlorobenzene boronic acid can be
combined to form 6-pentyl-8-(3,4-dichlorophenyl)quinoline,
6-hexyl-8-bromoquinoline and 3-nitrobenzene boronic acid can be
WO 94122852 , PCT/US94/03004
.~ .~r~9603
-38-
combined to form 6-hexyl-8-(3-nitrophenyl)quinoline,
6-(3-pyridylmethyl)-8-bromoquinoline and 3-nitrobenzene boronic acid
can be combined to form 6-(3-pyridylmethyl)-8-
(3-nitrophenyl)quinoline,
6-(2-pyridylmethyl)-8-bromoquinoline and 3-chlorobenzene boronic acid
can be combined to form 6-(2-pyridylmethyl)-
8-(3-chlorophenyl)quinoline,
6-(cyclopentylmethyl)-8-bromoquinoline and 3-chloro-4-fluorobenzene
boronic acid can be combined to form 6-(cyclopentylmethyl)-8-
(3-chloro-4-fluorophenyl)quinoline,
6-(cyclopropylmethyl)-8-bromoquinoline and benzene boronic acid can
be combined to form 6-(cyclopropylmethyl)-8-phenyl- quinoline,
6-(methylthiomethyl)-8-bromoquinoline and benzene boronic acid can be
combined to form 6-(methylthiomethyl)-8-phenylquinoline, and
6-(methylsulfonylmethyl)-8-bromoquinoline and 3-nitrobenzene boronic
acid can be combined to form 6-(methylsulfonyl- methyl)-8-(3-
nitrophenyl)quinoline.
E%AMPLE 10
PREPARATION OF
8-(3-CHLOROPHENYL)-6-QUINOLINECARBOBALDEHYDE
10A. Formula I Where R' is Bromomethyl
6-Methyl-8-(3-chlorophenyl)quinoline (prepared following the
procedures described in Example 9, Reaction Scheme B) (1.64 g) was combined
with 30 mL of carbon tetrachloride and heated to reflux.
N-Bromosuccinamide (1.34 g) and 2,2'-azobis(2-methylpropionitrile) (0.025
g) was added and the reaction mixture was exposed to light from a 250 watt
light bulb for 1 hour. The reaction mixture Was then stirred for an
additional 2 hours. The progress of the reaction was monitored by thin-
layer chromatography (9:1 hexane/ethyl acetate). Upon completion of the
reaction, the reaction mixture was cooled to 0°C and poured through a
2cm pad of NazS04 on lcm pad on silica gel. The filtrate was concentrated
yielding 2.26 g of a yellow oil as a mixture of the monobrominated compound
as the major product as well as the dibrominated compound as the minor
product. The crude oil (1.84 g) Was dissolved in 25 mL of chloroform and
added dropwise to a solution of tetra n-butylammonium dichromate (19.4 g)
in methylene chloride (30 mL) and refluxed for 4 hours. The reaction
mixture was then cooled to room temperature, filtered through silica gel,
eluted with ether and concentrated. The residue was chromatographed in
hexane:ethyl acetate (70:30) to obtain 8-(3-chlorophenyl)-6-
quinolinecarboxaldehyde
(0.93 g) .
Characteristic analytical data: ~H NMR (CDC13) b 7.41 (m, 2H) 7.52
(dd, 1H, J=5.2 Hz, J=4.2 Hz), 7.6 (m, 1H), 7.71 (m, 1H), 8.19 (d, 1H, J=1.9
WO 94/22852 ~ PCT/US94/03004
-39-
Hz), 8.38 (d, 1H, J=1.9 Hz), 8.4 (dd, 1H, J=5.2 Hz, J=1.8 Hz), 9.6 (dd, 1H,
J=4.2 Hz, J=1.8 Hz) , 10.22 (s, 1H) ; and elemental analysis for C,~I-i,oClNO,
calc(found) C, 71.78 (71.79) ; H, 3.77 (3.81) ; N, 5.23 (5.39) .
lOH. Formula I, Where R' Is Hromomethyl and RZ Is varied.
Following the procedures described in Example l0A the following
desired compounds of Formula I where R'~ is bromomethyl can be obtained from
the indicated starting compounds. For example,
6-methyl-8-(3-nitrophenyl)quinoline can be converted to
8-(3-nitrophenyl)-6-quinolinecarboxaldehyde,
6-methyl-8-phenylquinoline can be converted to
8-phenyl-6-quinolinecarboxaldehyde,
6-methyl-8-(3-chlorophenyl)quinoline can be converted to
8-(3- chlorophenyl)-6-quinolinecarboxaldehyde,
6-methyl-8-(3-trifluoromethylphenyl)quinoline can be converted to 8-
(3-trifluoromethylphenyl)-6-quinolinecarboxaldehyde,
6-methyl-8-(3-chlorophenyl)quinoline can be converted to
8-(3-chlorophenyl)-6-quinolinecarboxaldehyde,
6-methyl-8-(3-chloro-4-fluoro-phenyl)quinoline can be converted to 8-
(3-chloro-4-fluorophenyl)-6-quinolinecarboxaldehyde,
6-methyl-8-(3,4-dichlorophenyl)quinoline can be converted to
8-(3,4-dichlorophenyl)-6-quinolinecarboxaldehyde, and
6-methyl-8-(4-chlorophenyl)quinoline can be converted to
8-(4-chlorophenyl)-6-quinolinecarboxaldehyde.
EBAMPLE 11
PREPARATION OF
6-(1-HYDROgMETHYL)-8-(3-CHLOROPHENYL)QUINOLINE
11A. Formula I Where R' Is 1-hydroxyethyl
8-(3-Chlorophenyl)-6-quinolinecarboxaldehyde (0.8 g) was dissolved in
20 mL of tetrahydrofuran and cooled to -78°C. Methyllithium (4.5 mL,
1.4M)
in diethyl ether was added dropwise to the solution over 5 minutes. The
solution was stirred for 20 minutes, poured into 50 mL of saturated
ammonium chloride, stirred for 5 minutes and extracted with ethyl acetate
(2 x 75 mL). The extracts were combined and dried over MgSO" and
concentrated. The residue was chromatographed on silica gel (30:70 ethyl
acetate:hexanes) to obtain 0.72 g of 6-(1-hydroxyethyl)-8-(3-chlorophenyl)-
quinoline as a yellow oil.
Characteristic analytical data: 'H NMR (CDC13) 5 1.6 (d, 3H, J=6.5
Hz), 5.1 (q, 1H, J=6.5 Hz), 7.4 (m, 3H), 7.55 (m, 1H), 7.67 (m, 1H), 7.7
(d, 1H, J=1.9 Hz), 7.8 (d, 1H, J=1.9 Hz), 8.18 (dd, 1H, J=8.3 Hz, J=1.8
Hz), 8.9 (dd, 1H, J=4.2 Hz, J=1.8 Hz).
WO 94122852 PCT/US94/03004
~1~9~a~
-40-
11B. Formula I, 'there R' Is Formyl and R2 Is Varied
Following the procedures described in Example 11A the following
desired compounds of Formula I where R' is formyl can be obtained from the
indicated starting compounds. For example,
8-(3-nitrophenyl)-6-quinolinecarboxaldehyde and methyllithium can be
converted to 6-(1-hydroxyethyl)-8-(3-nitrophenyl)- quinoline,
8-phenyl-6-quinolinecarboxaldehyde and n-butyl magnesium chloride can
be converted to 6-(1-hydroxypentyl)-8-phenylquinoline,
8-(3-chlorophenyl)-6-quinolinecarboxaldehyde and ethyllithium can be
converted to 6-(1-hydroxypropyl)-8-(3-chlorophenyl)- quinoline,
8-(3-trifluoromethylphenyl)-6-quinolinecarboxaldehyde and hexyl
magnesium chloride can be converted to 6-(1-hydroxy- heptyl)-8-
(3-trifluoromethylphenyl)quinoline,
8-(3-chlorophenyl)-6-quinolinecarboxaldehyde and methyl magnesium
chloride can be converted to 6-(1-hydroxyethyl)-8-(3-
chlorophenyl)quinoline,
8-(3-chloro-4-fluorophenyl)-6-quinolinecarboxaldehyde and
methyllithium can be converted to 6-(1-hydroxyethyl)-8-(3-
chloro-4-fluorophenyl)quinoline,
8-(3,4-dichlorophenyl)-6-quinolinecarboxaldehyde and propyllithium
can be converted to 6-(1-hydroxybutyl)-8-(3,4-
dichlorophenyl)quinoline, and
8-(4-chlorophenyl)-6-quinolinecarboxaldehyde and hexyllithium can be
converted to 6-(1-hydroxyheptyl)-8-(4-chlorophenyl)-
quinoline.
EBAMPLE 12
PREPARATION OF
6-(4-PYRIDYL-N-OgIDE-METHYL)-8-(3-NITROPHENYL)QUINOLINE
To a solution of 6-(4-pyridylmethyl)-8-(3-nitrophenyl)quinoline
(100 mg) in methylene chloride (10 mL) under nitrogen at room temperature
was added m-chloroperoxybenzoic acid (59 mg). The reaction mixture was
stirred at room temperature for 3~ hours. The progress of the reaction Was
monitored by TLC. Upon completion of the reaction, the reaction mixture
was worked up by preparative thin-layer chromatography yielding 6-(4-
pyridyl-N-oxide-methyl)-8-(3-nitrophenyl)quinoline (87 mg) as creme colored
crystals.
Characteristic analytical data: 'H (CDC13) b 4.21 (s, 2H), 7.13 (d,
2H, J=5.2 Hz), 7.43 (dd, 1H, J=8.2 Hz, J=4.2 Hz), 7.5 (d, 1H, J=1.9 Hz),
7.6 (dd, 1H, J=8 Hz, J=8 Hz), 7.7 (d, 1H, J=1.9 Hz), 7.95 (ddd, 1H
J=7.7 Hz, J=1.4 Hz, J=1.3 Hz), 8.13 (d, 2H, J=5.2 Hz), 8.18 (dd, 1H, J=8.2
Hz, J=1.7 Hz), 8.71 (ddd, 1H, J=8.2 Hz, J=J.14, J=1.3 Hz), 8.55 (dd, 1H,
J=1.3 Hz, J=1.3 Hz), 8.92 (dd, 1H, J=4.2 Hz, J=1.7 Hz).
WO 94/22852 PCT/US94/03004
-41-
EXAMPLE 13
(REACTION SCHEMES H-1, H)
PREPARATION OF
6-HROMOMETHYL-8-HROMOQUINOLINE
Formula 5. Where R' Is Hromomethyl and R~ Is Bromo
3.7g (16.66 mmol) of 6-Methyl-8-bromoquinoline Was dissolved in 85mL of
CC1, and 3g of N-bromosuccinamide (16.66 mmol) was added followed by a
catalytic amount of azobisisobutyronitrile (AIBN). The reaction mixture was
heated at reflux under nitrogen for 1.5 hours and irradiated with a lamp,
the cooled and pour into water (100mL). The organic layer was separated and
washed with H20 (100mL) and brine (100mL), dried over MgSO" filtered and
concentrated to give a brown solid. Crystallization from ethyl
acetate/hexane yielded 2.848 (9.44 mmol) of light brown crystals. The
filtrate was concentrated and the residue crystallized from the same
solvent system. A second crop of light brown crystals (0.778 2.56 mmol) was
obtained. Column chromatography on silica gel of the concentrated filtrate
in 20% ethyl acetate/hexanes gave 0.598 (1.96 mmol) of 6-bromomethyl-8-
bromoquinoline as a brown solid (4.2g total, 13.95 mmol, 84%), mp 156.2-
157.8°.
PREPARATION OF
6-(N-PYRROLIDINYLMETHYL-8-BROMOQOINOLINE
Formula 5, Where R' Is Pyrrolidinylmethyl and Ri Is Hromo
0.628 (2.1 mmol) of 6-bromomethyl-8-bromoquinoline was slurried in 15 mL of
THF and pyrrolidine (0.38 mL, 4.6 mmol) was added dropwise under nitrogen.
The reaction mixture was stirred for 1.5h, and then poured into half
saturated NaHC03 (50mL) and 25 mL EtOAc. The organic layer was washed with
H~O (20mL), and brine (25mL), dried over MgSO" filtered and concentrated.
Flash chromatography of the residue on silica gel using 5% MeOH/CHzCl.,
yielded 0.448 (1.51 mmol, 72%) of 6-(N-pyrrolidinylmethyl)-8-bromoquinoline
as a dark oil.
PREPARATION OF
6-(N-PYRROLIDINYLMETHYL-8-(3-NITROPHENYL)QUINOLINE
Formula 5. Where R' Is Pyrrolidinylmethyl and RZ Is 3-Nitrophenyl
0.328 (1.1 mmol) of 6-(N-pyrrolidinylmethyl)-8-bromoquinoline was dissolved
in 10 mL of 1:1 EtOH/benzene under nitrogen. 3-nitrophenyl boronic acid
(0.378, 2.2 mmol), 2M NaZC03 (2.2 mL, 4.4 mmol), and tetrakis(triphenyl-
phosphine)palladium Pd(PPh3), (0.05g, 0.044 mmol) Were added successively
and the reaction mixture was refluxed for 4h, then cooled and poured into
water (lSmL) and extracted with EtOAc (15 mL). The organic layer Was washed
with water (10 mL) and brine (lOmL), dried over MgSO" filtered and
concentrated. The residue was chromatographed using 10% methanol/ CH=C1~.
WO 94/22852 PCT1US94/03004
215964
-42-
0.298 (0.87 mmol) of 6-(N-pyrrolidinylmethyl)-8-(3-nitrophenyl)quinoline
was obtained as a yellow oil. The oil was dissolved in 15 ml of EtOAc, and
the HCL salt was formed with a saturated solution of HC1/EtOAc. The residue
was concentrated and crystallized from ethanol/ethyl acetate/ether/hexane
to give 0.298 (0.71 mmol, 65%) of 6-(N-pyrrolidinylmethyl)-8-(3-nitro-
phenyl)quinoline dihydrochloride as yellow crystals, mp 223-225°C.
PREPARATION OF
6-(N-PYRROLIDINONYLMETHYL)-8-BROMOQUINOLINE
Formula 5, Where R' Is Pyrrolidinonylmethyl and RZ Is Hromo
8-bromo-6-bromomethylquinoline (1.33 mmol) was dissolved in THF/[1,3-
dimethyl-3,4,5,6-tetrahydro-2(1H)pyrimidinone (DMPU) and reacted With a
solution of NaH (1.6 mmol) and pyrrolidinone (1.6 mmol) at reflux for 24 h
to obtain 8-bromo-6-(N-pyrrolidinonylmethyl) quinoline as a yellow oil that
was used directly in the next step.
PREPARATION OF
6-(N-PYRROLIDINONYLMETHYL-8-(3-NITROPHENYL)QUINOLINE
Formula I, HThere R' Is Pyrrolidinonylmethyl and Rz Is 3-Nitrophenyl
8-bromo-6-(N-pyrrolidinonylmethyl) quinoline (1.05 mmol) Was reacted with
3-nitroboronic acid (2.1 mmol), 2M Na2C03 (4.2 mmol), and Pd(PPh3)4 (0.04
mmol) to obtain 8-(3-nitrophenyl)-6-(N-pyrrolidinonylmethyl) quinoline as
white crystals (0.55 mmol, 52%), mp 127.4-128.3°C.
EBAMPLE 14
(REACTION SCHEME C-3)
PREPARATION OF
6-PYRROLYLMETHYL-B-(3-NITROPHENYL)QUINOLINE
Formula I, 'Vhere R' Is Pyrrolylmethyl and RZ Is 3-Nitropheayl
0.68 (1.75 mmol) of freshly distilled pyrrole was added to a slurry of NaH
(0.0468, 1.9 mmol) in 5 mL of dzy DMF under nitrogen. The reaction mixture
was stirred for 10 min and then 0.68 (1.75 mmol) of 6-bromomethyl-8-(3-
nitrophenyl)quinoline, dissolved in 5 mL of DMF, was added to forth a dark
solution. The soh.tion was stirred at room temperature for 2h, then heated
at 80°C for 2h. The reaction mixture was cooled and poured into 150 mL
of
water, extracted with CHZC12 (4 x 25 mL). The combined organic layers were
washed with water (50 mL), brine (50mL), and dried over MgSO,. Filtration
and concentration provided a solid which was purified by flash
chromatography on silica gel using 30% ethyl acetate/hexane.
Yield: 0.38 of a yellow oil.
The oil was dissolved in 15 mL of ethyl acetate and the HC1 salt was formed
from ethyl acetate/HC1. The solution was concentrated and the resulting oil
was crystallized from ethanol/ether . Yield: 0.268 (0.64 mmol, 37%) of
WO 94/22852 PCT/US94/03004
~~3
-43
6-pyrrolylmethyl-8-(3-nitrophenyl)quinoline dihydrochloride as yellow
crystals, mp 183.2-184.8°C.
PREPARATION OF
6-(PHENYLAMINOMETHYL)-8-(3-NITROPHENYL)QUINOLINE
Formula I, Where R' Is Phenylaminomethyl and RZ Is 3-Nitrophenyl
0.5g (1.46 mmol) of 6-bromomethyl-8-(3-nitrophenyl)quinoline was dissolved
in lOmL of ethanol. 0.15g (1.61 mmol) of aniline and 0.3g
(2.2 mmol) of IC2C03 was added. The reaction mixture was refluxed under
nitrogen for 24h, then poured into water (50 mL) and ethyl acetate (50 mL).
The organic layer was separated and washed with water (30 mL) and brine
(30 mL), dried over MgS04, filtered, concentrated and purified by flash
chromatography using 30% ethyl acetate/hexane. An impure yellow oil was
obtained which was further purified by preparative TLC using the same
solvent system to obtain 0.2g of yellow oil. Crystallization from ethyl
acetate/hexane gave O.lg (0.28 mmol, 19%) of 6-(phenylaminomethyl)-8-(3-
nitrophenyl)quinoline as yellow crystals, mp 149.9-150.5°C.
PREPARATION OF
6-(1,2,4-TRIAZOLYLMETHYL)-8-(3-NITROPHENYL)QUINOLINE
Formula I, Where R' Ia 1,2,4-Triazolylmethyl and RZ Is 3-Nitropheayl
6-Bromomethyl-8-(~-nitrophenyl)quinoline (1.75 mmol) was reacted with
1,2,4-triazole (3.5 mmol), ICzC03 (4.38 mmol), and tetrabutylammonium iodide
(0.18 mmol) in refluxing ethyl acetate to obtain 6-(1,2,4-
triazolylmethyl)-8-(3-nitrophenyl)quinoline (0.91 mmol, 57%) as yellow
czystals, mp 160.7-161.5°C.
PREPARATION OF
6-(1,2,4-TRIAZOLYhMETHYL)-8-(3-CHLOROPHENYL)QUINOLINE
Formula I, Where R' Is 1,2,4-Triazolylmethyl and RZ Ia 3-Chlorophenyl
6-Bromomethyl-8-(3-chlorophenyl)quinoline (1.26 mmol) was reacted with a
solution of NaH (1.89 mmol) and 1,2,4-triazole (1.89 mmol), in THF for 24h
to give 6-(1,2,4-triazolylmethyl)-8-(3-chlorophenyl)quinoline
dihydrochloride (0.22 mmol, 18%) as light brown crystals, mp 208.1-
208.5°C.
PREPARATION OF
6-(IMIDAZOLYLMETHYL)-8-(3-CHLOROPHENYL)QUINOLINE
Formula I, Where R' Ia Imidazolylmethyl and R2 Is 3-Chlorophenyl
6-Bromomethyl-8-(3-chlorophenyl)quinoline (1.7 mmol) was reacted with a
solution of s-BuLi (1.7 mmol) and imidazole (1.87 mmol) in THF at
-78°C, then room temperature for lh to obtain 6-(imidazolylmethyl)-8-(3-
chlorophenyl)quinoline dihydrochloride (1.35 mmol, 79 %),
mp 65-65.4°C.
WO 94122852 PCT/US94/03004
44
PREPARATION OF
6-(IMIDAZOLYLMETHYL)-8-(3-NITROPHENYL)QUINOLINE
Formula I, Where R' Is Imidazolylmethyl and Rz Is 3-Nitrophenyl
6-Bromomethyl-8-(3-nitrophenyl)quinoline (1.28 mmol) was reacted with a
solution of n-BuLi (1.92 mmol) and imidazole (1.92 mmol) in THF at -78°
C,
then room temperature for 24 h to give 6-(imidazolylmethyl)-8-(3-
nitrophenyl)quinoline (0.52 mmol, 40%), mp 129.5-130.5°C.
EBAMPLE 15
(REACTION SCHEME C-1)
PREPARATION OF
6-(4-FLUOROBENZYL)-8-(3-NITROPHENYL)QUINOLINE
Formula I, Where R' Is 4-Fluorobenzyl and R= Is 3-Nitrophenyl
0.5g (1.46 mmol) of 6-bromomethyl-8-(3-nitrophenyl)quinoline was dissolved
in 15 mL of 1:1 ethanol/benzene. 0.41g ( 2.9 mmol) of 4-fluorophenyl
boronic acid, 3 mL (5.8 mL) 2M NazC03, and 0.07g (0.06 mmol) Pd(PPh3), were
added. The reaction mixture was refluxed under nitrogen for 20 min, cooled
and poured into water (30 mL) and extracted with ethyl acetate (30 mL). The
organic layer was washed with water (25 mL) and brine (25 mL), dried over
MgSO, , filtered and concentrated. The residue was purified by flash
chromatography using 30% ethyl acetate/ hexane to obtain 0.41g of an impure
yellow oil which was further purified by preparative TLC using the same
solvent system. 0.33g of a clear oil was obtained which was crystallized
from ethyl acetate/hexanes at 0° C to give 0.2g (0.56 mmol, 36%) of 6-
(4-
fluorobenzyl)-8-(3-nitrophenyl)quinoline as white crystals, mp 97.7-
98.5°C.
PREPARATION OF
6-(4-METHOXYHENZYL)-8-(3-NITROPHENYL)QUINOLINE
Formula I, Where R' Is 4-Methoxybenzyl and Ri Is 3-Nitrophenyl
6-Bromomethyl-8-(3-nitrophenyl)quinoline (1.17 mmol) was reacted with 4-
methoxyboronic acid (2.34 mmol), 2M NazC03 (4.68 mmol), and Pd(PPh3)4 (0.05
mmol) to obtain 6-(4-methoxybenzyl)-8-(3-nitrophenyl)quinoline (0.49 mmol,
42%) as tan crystals, mp 117.5-119.1°C.
EXAMPLE 16
(REACTION SCHEME H-1)
PREPARATION OF
6-ISOPROPYL-8-(3,4-METHYLENEDIOXYPHENYL)QUINOLINE
Formula I, Where R' Is Isopropyl and RZ Is 3,4-Methylenedioxyphenyl
0.848 (3.36 mmol) of 6-isopropyl-8-bromoquinoline was dissolved in 15 mL of
dzy THF and cooled to -78°C. s-BuLi ( 2.7 mL, 3.53mmo1,
(1.3M/cyclohexane))
WO 94/22852 ~ ,~;~ PCT/US94103004
-45-
was added dropwise under nitrogen. The reaction mixture was stirred for 5
min and then ZnClz (7 mL, 3.53 mmol, (0.5M/TFiF)) was added dropwise. The
reaction mixture was warmed to room temperature over a period of 1 h.
0.44mL (3.7 mmol) of 1-Bromo-3,4-methylenedioxybenzene was added dropwise
followed by 0.198 ( 0.17mmo1) Pd(PPh3),. The reaction mixture was refluxed
for 2.5 h, cooled and poured into water (25 mL) and ethyl acetate (25 mL).
The organic layer was Washed with water (20 mL) and brine (20 mL), dried
over MgSO" filtered and concentrated. The resulting brown oil was purified
by flash chromatography using 15% ethyl acetate /hexanes to obtain 0.478 of
a pure, clear oil which Was crystallized from ethyl acetate/hexanes. Yield:
0.258 (0.85 mmol, 25%) of 6-isopropyl-8-(3,4-methylenedioxy-
phenyl)quinoline as white platelets, mp 86.6-88.2°C.
PREPARATION OF
6-ISOPROPYL-8-(3-CYANOPHENYL)QUINOLINE
Formula I, Where R' Is Isopropyl and Ri Is 3-Cyanophenyl
6-Isopropyl-8-bromoquinoline (3.8 mmol) was reacted with 1.3M s-BuLi (3.99
mmol) , ZnClz (3.8 mmol) , 3-cyanobromobenzene (3.8 mmol) , and Pd(PPh3)4
(0.19 mmol) to obtain 6-isopropyl-8-(3-cyanophenyl)quinoline
dihydrochloride (1.1 mmol, 29%) as white crystals, mp 194.8-197.6°C.
PREPARATION OF
6-ISOPROPYL-8-(4-METHO%YPHENYL)QUINOLINE
Formula I, Where R' Is Isopropyl and Ri Is 4-Methoxypheayl
6-Isopropyl-8-bromoquinoline (3 mmol) was reacted with 1.3M s-BuLi (3.03
mmol) , ZnClz (3 mmol) , 4-methoxyiodobenzene (3.6 mmol) , and Pd(PPh3)4 (0.15
mmol). 6-isopropyl-8-(4-methoxyphenyl)quinoline (0.67 mmol, 23%) was
obtained as a clear oil.
PREPARATION OF
6-ISOPROPYL-8-(4-TRIFLUOROMETHYLPHENYL)QUINOLINE
Formula I, Where R' Is Isopropyl and RZ Is 4-Trifluoromethylphenyl
6-Isopropyl-8-bromoquinoline (2.7 mmol) was reacted with 1.3M s-BuLi (2.83
mmol), ZnCl,, (2.83 mmol), 4-trifluoromethyliodobenzene (4.05- mmol), and
Pd(PPh3)4 (0.14 mmol) to obtain 6-isopropyl-8-(4-
trifluoromethylphenyl)quinoline hydrochloride (0.97 mmol, 36 %) as off-
white crystals, mp 167.5-168.4°C.
EBAMPLE 17
(REACTION SCHEME B)
PREPARATION OF
METHYL 3-TRIFLUOROMETHANESULFOgYBENZOATE
Formula 3C, HThere R2 Is Trifluoromethanesulfonyloxy
WO 94/22852 PCT/US94/03004
-46-
108 (65.73 mmol) of Methyl 3-hydroxybenzoate was dissolved in 330 mL of
CHZC12 and cooled to 0°C. 45.8 mL (328.65 mmol) of triethylamine was
added
to form a light brown solution. Trifluoromethanesulfonic anhydride (triflic
anhydride) ( 16.6 mL, 98.6 mmol) was added via addition funnel under
nitrogen over 0.5h to form a dark brown solution. The reaction mixture was
poured into a saturated solution of NaIiC03 (200 mL) and extracted with
CHzCl2 (200 mL). The organic layer was washed with water (2 x 100mL) and
brine (100 mL), dried over MgS04, filtered, and concentrated. Flash
chromatography using 20% ethyl acetate/ hexanes provided a slightly impure
brown oil which was distilled (1.2 torr, 92-94.5°C) to yield 13.798 (
48.52
mmol, 74%) of 3-(trifluoromethanesulfonyloxy)benzoic acid methyl ester
(methyl 3-trifluoromethanesulfoxybenzoate) as a yellow liquid.
PREPARATION OF
6-ISOPROPYL-8-(3-CARBOMETHOXYPHENYL)QUINOLINE
Formula I, Where R' Is Isopropyl and RZ Is 3-Methylbenzoate
6-Isopropyl-8-bromoquinoline (4.12 mmol) was reacted with 1.3M s-BuLi (4.33
mmol), ZnClz (4.33 mmol), methyl 3-trifluorosulfoxybenzoate (4.33 mmol),
and Pd(PPh3)4 (0.21 mmol) to obtain 6-isopropyl-8-(3-carbomethoxy-
phenyl)quinoline hydrochloride (0.568, 1.64 mmol, 40%) which was
crystallized from ethanol/ether/hexanes to yield light yellow crystals,
mp 166.5-167.6°C.
EXAMPLE 18
(REACTION SCHEME C-1)
PREPARATION OF
6-CYANOMETHYL-8-(3-NITROPHENYL)QUINOLINE
Formula I, Where R' Is Cyanomethyl and RZ Is 3-Nitrophenyl
lg (2.91 mmol) of 6-Bromomethyl-8-(3-nitrophenyl)quinoline was dissolved in
5mL of CHZC12. To this solution Was added dropwise 0.788 (2.91 mmol) of
tetrabutylammonium cyanide dissolved in 5 ml of CHZC12 over 5 min. The
reaction mixture was stirred for lh at room temperature then refluxed
overnight. An additional 0.398 (1.45 mmol) of tetrabutylammoniun cyanide
was added and the reaction refluxed for 3 more hours. The reaction was then
concentrated and ethyl acetate (30 mL) and water (30 mL) Were added. The
organic layer was washed with water (2 x 30 mL), and brine (30 mL). CH,Clz
(50 mL) was added to the organic layer to solubilize the suspended solids
and the solution was then dried over MgS04, filtered and concentrated. The
residue was dissolved in a minimal amount of CH2Clz, absorbed on silica gel,
and placed on top of a silica gel column and eluted With 50% ethyl acetate/
hexanes. Yield 0.68 (2.07 mmol, 71%) of 6-cyanomethyl-8-(3-
nitrophenyl)quinoline as a yellow solid, mp 178.0-180.5°C.
WO 94/22852 PCT/US94/03004
_47_
PREPARATION OF
6-[1-(4-CHLOROPYRIDAZINYL)-1-CYANOMETHYL]-8-(3-NITROPHENYL)QUINOLINE
Formula I, Where R' Is (1-(4-Chloropyridazinyl)-1-cyanomethyl] and RZ Is 3-
Nitrophenyl
0.458 (1.56 mmol) of 6-cyanomethyl-8-(3-nitrophenyl)quinoline and 0.49
(3.28 mmol) of 1,4-dichloropyridazine were dissolved in 15 mL of dry DMF
under nitrogen and cooled to 0 C. NaH (0.0788 3.28 mmol) was added
portionwise and the reaction stirred at 0 C for 45 min and then 30 min at
room temperature. The reaction mixture was poured into CHZClz (150 mL) and
150 mL of half saturated NH,C1 was added. The organic layer was separated
and the aqueous layer was washed with CH=C12 (100 mL). The combined organic
layers were washed with water (2 x 100 mL), and brine (100 mL), dried over
MgS04, filtered and concentrated. Chromatography using 50% ethyl acetate/
hexanes yielded 0.518 (1.27 mmol, 81%) of 6-(1-(4-chloropyridazinyl)-1-
cyanomethyl)-8-(3-nitrophenyl)quinoline as an orange foam.
PREPARATION OF
6-PYRIDAZINONYLMETHYL)-8-(3-NITROPHENYL)QUINOLINE
Formula I, Where R' Is Pyridazinonylmethyl] and R~ Is 3-Nitrophenyl
0.48 (1 mmol) of 6-(1-(4-chloropyridazine)-1-cyanomethyl)-8-(3-
nitrophenyl)quinoline was dissolved in 5 mL acetic acid, 5 mL water, and 10
mL cone. hydrochloric acid. The reaction mixture was refluxed for 24h, and
then concentrated. The residue was dissolved in water (25 mL), saturated
NaHC03 (150 mL) and ethyl acetate (100 mL) were added. The organic layer
was washed with Water (100 mL) and brine (100 mL), dried over MgSO"
filtered and concentrated to obtain 0.278 of a brown solid. The residue
was purified by preparative TLC using 5% MeOH/ CH~Clz. Yield: 0.258 (0.7
mmol, 70%) of 6-(4-pyridazinonylmethyl)-8-(3-nitrophenyl)quinoline as an
off-white solid, mp 199.5-202.0° C.
EXAMPLE 19
(REACTION SCHEMES A-l, B)
PREPARATION OF
4-NITRO-1-CYCLOPENTYLIDINYLBENZENE
Formula la, Where R Is Cyclopentyl
5g (33.42 mmol) of Cyclopentyltriphenylphosphine bromide was slurried in
100mL of dry THF and 18.4 mL of 1.8M PhLi ( 33.09 mmol) was added over 5
min. The reaction mixture was stirred for ih at room temperature, then
refluxed for 15 min, and cooled to room temperature. 5g (33.09 mmol) of 4-
nitrophenyl carboxaldehyde in 40 mL of dzy THF was added dropwise. The
reaction mixture was stirred at 40° C for 24 h, then poured into water
(100
mL) and ethyl acetate (100 mL). The aqueous layer was extracted with ethyl
acetate (3 x 50 mL). The combined organic layers were dried over MgSOa,
WO 94/22852 PCTIUS94/03004
-48-
filtered, and concentrated. Flash chromatography using 50% ethyl
acetate/hexanes yielded 4.7 g (23.13 mmol) of 4-vitro-1-cyclo-
pentylidinylbenzene as an impure black oil.
PREPARATION OF
CYCLOPENTYLIDINYLANILINE
Formula lb, Where R Is Cyclopentyl
4.7g of 4-vitro-1-cyclopentylidinylbenzene Was dissolved in ethanol (100
mL) and catalytically hydrogenated (Pd/C) under 1 atm of hydrogen for 24h.
The reaction mixture was filtered through Celite, concentrated to an orange
semi-solid, and chromatographed using 10% ethyl acetate to obtain 2.14g
(12.35 mmol) of 4-cyclopentylidinylaniline as an yellow semi-solid.
PREPARATION OF
4-CYCLOPENTYLMETHYLANILINE
Formula 2a, Where R Is Cyclopentyl
1.83g (10.32 mmol) of 4-cyclopentylidinylaniline was dissolved in ethanol
(50 mL) and catalytically hydrogenated (Pt02) under 1 atm of hydrogen for
30 min. The reaction mixture was filtered through Celite and concentrated.
The residue was chromatographed using 20% ethyl acetate/ hexanes to afford
1.52 g of a yellow oil as a mixture of starting material and product. The
yellow oil was dissolved in ethanol (25 mL) and catalytically hydrogenated
(PtOz) under 1 atm of hydrogen for 5h. Filtration through Celite,
concentration, and chromatography using 20% ethyl acetate/ hexanes gave
1.52g (8.67 mmol 84%) of 4-cyclopentylmethylaniline as a clear oil.
PREPARATION OF
2-BROMO-4-CYCLOPENTYLMETHYLANILINE
Formula 3a, Where R Is Cyclopentyl and $ is Bromo
1.078 (6.1 mmol) of 4-cyclopentylmethylaniline was dissolved in DMF (2.5
mL) and placed under nitrogen. 1.098 (6.1 mmol) of N-bromosuccinamide,
dissolved in DMF (2.5 mL), was added dropwise.
The reaction mixture was stirred for 5h, then poured into water (100 mL),
and extracted with ether (3 x 25 mL). The combined ether extracts were
dried over MgS04 and filtered. Concentration and flash chromatography on
silica gel using 10% ethyl acetate/hexanes provided 0.98g (3.86 mmol, 63%)
of 2-bromo-4-cyclopentylmethylaniline as a light brown oil.
PREPARATION OF
6-CYCLOPENTYLMETHYL-8-BROMOQUINOLINE
Formula 5, Where R Is Cyclopentylmethyl and ~ is Hromo
0.668 (2.6 mmol) of 2-bromo-4-cyclopentylmethylaniline was slurried in
0.094g (0.34 mmol) Iron (II) sulfate heptahydrate, 0.17 mL (1.66mmo1)
nitrobenzene, and lg (10.4 mmol) of glycerol, and heated to 115°C under
WO 94/22852 ~~~ ; PCTIUS94103004
-49-
nitrogen. HzS04 (0.55 mL) was added dropwise and the reaction mixture was
heated to 165°C, stirred for 2h and then poured into saturated NaHC03
(300
mL) and ethyl acetate (50 mL). The solution was filtered through glass wool
and the filtrate was extracted with ethyl acetate (2 x 50mL). The combined
extracts were dried over MgSO, and filtered. Concentration and flash
chromatography (10% EtOAc/hexanes) yielded 0.588 (2 mmol, 77%) of 6-
cyclopentylmethyl-8-bromoquinoline as a yellow oil.
PREPARATION OF
6-CYCLOPENTYLMETHYL-8-(3-CHLOROPHENYL)QUINOLINE
Formula I, Where R' Is Cyclopentylmethyl aad RZ is 3-Chlorophenyl
0.19g (0.65 mmol) of 6-Cyclopentylmethyl-8-bromoquinoline was dissolved in
6.5 mL of 1:1 ethanol/ benzene under nitrogen. 0.2g (1.3 mmol) of 3-
chloroboronic acid, 1.3 mL (2.6 mmol) of 2M Na2C03, and 0.038 (0.026 mmol)
of Pd(PPh3)4 were added successively. The reaction mixture was refluxed for
3h, then cooled and poured into water (25 mL) and ethyl acetate (25 mL).
The organic extract was dried over MgS04 and filtered. Flash
chromatography on silica gel using 20% ethyl acetate/hexanes yielded 0.178
of impure product which was purified further by preparative TLC in the same
solvent system to obtain 0.168 yellow oil. Formation of the HC1 salt with
saturated HC1/EtOAc and crystallization of the resulting residue from
EtOH/EtOAc/ether yielded 0.14 g (0.4 mmol, 60%) of 6-cyclopentylmethyl-8-
(3-chlorophenyl)quinoline hydrochloride as white czystals, mp 179.3-
183.7°C.
PREPARATION OF
6-CYCLOPENTYLMETHYL-8-(3-NITROPHENYL)QUINOLINE
Formula I, Where R' Is Cyclopentylmethyl and RZ is 3-Nitrophenyl
Similarly, 6-Cyclopentylmethyl-8-bromoquinoline (0.55 mmol) was reacted
With 3-chloroboronic acid (1.1 mmol), 2M NazC03 (2.2 mmol), and Pd(PPh3)4
(0.02 mmol) to obtain 6-cyclopentylmethyl-8-(3-nitrophenyl)quinoline
hydrochloride (0.42 mmol, 77 %), as tan czystals from EtOH/EtOAc/EtzO, mp
185.8-190.4°C.
EBAMPLE 20
(REACTION SCHEMES H-3, B)
PREPARATION OF
6-METH0g7C-8-AMINOQUINOLINE
Formula 5, Where R' Is Methoxy aad 8 is Amino
4 g (19.6 mmol) of commercially available 6-methoxy-8-nitroquinoline was
dissolved in ethanol (100 mL) and THF (25 mL) and catalytically
hydrogenated (Pd/C, 1 atm H2) for 5h. The reaction mixture was filtered
through Celite and concentrated. Flash chromatography using 5% MeOH/ CH2C1=
WO 94/22852 r ' PCT/US94/03004
-50-
yielded an impure product which Was re-chromatographed in 50% EtOAc/hexanes
to give 2.768 (15.84 mmol, 81%) of 6-methoxy-8-aminoquinoline as a yellow
oil.
PREPARATION OF
6-METHOX7t-8-HROMOQUINOLINE
Formula 5, There R' Is Methoxy and x is Bromo
1.25 g (7.18 mmol) of 6-methoxy-8-aminoquinoline was mixed with 48% HBr (6
mL) and water (6 mL) to form an orange slurry. The mixture Was then cooled
to OC and a solution of 5 g of NaNOz (9.3 mmol) in 5 mL of water was added
dropwise. The resulting brown solution was stirred for 15 min and then
added to a solution of 1.24 g CuBr (8.6 mmol) in 15 mL of 48% HBr with
stirring at 75°C. The reaction mixture was stirred at 75°C for
5.5h and Was
then made basic with 10% NaOH and filtered through filter paper. The
filtrate was partitioned between water (50 mL) and EtOAc (50 mL) and
extracted with EtOAc (2 x 50 mL). The combined organic extracts were dried
over MgSO" filtered, and concentrated to give 0.6 g of a dark oil which
was purified by preparative TLC (20% EtOAc/hexanes) to afford 0.368 (1.5
mmol, 21%) of 6-methoxy-8-bromoquinoline as a tan solid, mp 64.5-
65.6°C.
PREPARATION OF
6-HYDROXY-8-HROMOQUINOLINE
Formula 5. Where R' Is Hydroxy and X is Bromo
0.268 ( 1.1 mmol) of 6-methoxy-8-bromoquinoline was dissolved in CHzCl2(11
mL) and cooled to -78°C. 2.7 mL (2.75 mmol) of BBr3 (1M in CHZC12) was
added
dropwise to form a yellow slurry. The reaction mixture was stirred for 10
min at -78°C, the bath was removed and the reaction mixture was allowed
to
warm to room temperature, stirred for 1.5 h and then MeOH (20 mL) was added
to quench the reaction. The reaction mixture was concentrated under reduced
pressure, 20 mL of MeOH was added and again concentrated (repeated 2x). The
residue Was partitioned between EtOAc and water. The organic layer was
washed 2x with NaOH (0.5 N). The base extracts were neutralized with
saturated NH4C1 and extracted with EtOAc (2 x 25 mL). The organic layers
were washed with brine, dried over MgS04, filtered, and concentrated to
yield O.llg (0.49 mmol, 45 %) of 6-hydroxy-8-bromoquinoline as an off-White
solid, mp 249.5-250.6°C.
PREPARATION OF
6-CYCLOPENTYLOgSC-8-BROMOQOINOLINE
Formula 5, Where R' Is Cyclopentyloxy and X is Bromo
0.2g (0.89 mmol) of 6-hydroxy-8-bromoquinoline was dissolved in DMF
(5 mL) . 0.25g (1.78 mmol) of powdered FCzC03 and 0.2 mL(1.78 mmol)
cyclopentylbromide were added, and reaction mixture was heated at 75°C
for
1.5h. The reaction mixture was then cooled and poured into water (20 mL)
WO 94122852 PCT/US94/03004
-51-
and extracted with ether (20 mL). The ether layer was washed with water (30
mL) and brine (30 mL). The aqueous layers were re-extracted with ether (20
mL) and the combined organic layers were dried over MgS04, filtered and
concentrated to obtain 0.268 (0.89 mmol, 100%) of 6-cyclopentyloxy 8-
bromoquinoline as a crude yellow solid, mp 76.2-79.2°C.
PREPARATION OF
6-CYCLOPENTYLOBY-8-(3-NITROPHENYL)QUINOLINE
Formula I, Where R' Is Cyclopentyloxy and RZ is 3-Nitrophenyl
0.228 (0.75 mmol) of 6-cyclopentyloxy-8-bromoquinoline was dissolved in 7.5
mL of 1:1 EtOH/benzene and 0:258 (1.5 mmol) 3-nitroboronic acid, 1.5 mL (3
mmol) 2M Na2C03, and 0.0358 (0.3 mmol) Pd(PPh3), were added successively. The
reaction mixture was refluxed for 2h under nitrogen, then partitioned
between water/EtOAc. The organic layer was washed with water, brine, dried
over MgSO" and filtered. Concentration and purification by preparative TLC
using 15% EtOAc/hexanes provided 0.28 of a yellow oil. The hydrochloride
salt was formed with saturated HC1/EtOAc and crystallized from
EtOH/EtOAc/ether. Yield: 0.218 (0.56 mmol, 75%) of 6-cyclopentyloxy-8-(3-
nitrophenyl)quinoline as white crystals, mp 190-191.4°C.
EBAMPLE 21
By following the procedures in Examples 1-20, other 6,8-disubstituted
quinolines within the scope of this invention can be prepared.
For example, the following 6,8-disubstituted quinoline compounds of formula
I were prepared by following the above described procedures:
6-isopropyl-8-(3-nitrophenyl)quinoline, (hydrochloride),
mp 204.5-216 °C;
4-pyridylmethyl-8-(3-nitrophenyl)quinoline, (dihydrochloride),
mp 238-240°C;
6-[1-(1,2,4-triazolyl))-8-(3-nitrophenyl)quinoline, mp 160.7-162.5°C;
6-anilinomethyl-8-(3-nitrophenyl)quinoline, mp 149.9-150.5°C;
6-(4-pyridazinonylmethyl)-8-(3-nitrophenyl)quinoline, mp 199.5-202°C;
6-(2-pyrrolidinonylmethyl)-8-(3-nitrophenyl)quinoline,mp 127.4-128.3°C.
6-cyclopentylmethyl-8-(3-nitrophenyl)quinoline, (hydrochloride),
mp 186.3-191.2°C.
6-imidazolylmethyl-8-(3-nitrophenyl)quinoline, mp 129.5-130.5°C.
6-cyclohexyl-8-(3-nitrophenyl)quinoline, (hydrochloride),
mp 188.5-194.8°C.
6-cyclopentyloxy-8-(3-nitrophenyl)quinoline, (hydrochloride),
mp 190-191.4°C.
6-ethyl-8-(3-nitrophenyl)quinoline, (hydrochloride),
mp 193.1-193.6°C.
WO 94/22852 PCT/US94103004
-52-
6-n-propyl-8-(3-nitrophenyl)quinoline, (hydrochloride),
mp 208.5-209.7°C.
6-n-butyl-8-(3-nitrophenyl)quinoline, (hydrochloride),
mp 194.1-198.5°C.
6-t-butyl-8-(3-nitrophenyl)quinoline, (hydrochloride),
mp 214-214.8°C;
6-n-pentyl-8-(3-nitrophenyl)quinoline, (hydrochloride),
mp 175.9-177.9°C;
6-(4-fluorobenzyl)-8-(3-nitrophenyl)quinoline, mp 97.7-98.5°C.
6-pyrrolidinyl-8-(3-nitrophenyl)quinoline, (dihydrochloride)
mp 223-225°C.
4-pyridylmethyl-8-(3-chlorophenyl)quinoline, (dihydrochloride),
mp 243.0-246.4°C;
6-[1-(1,2,4-triazolyl)]-8-(3-chlorophenyl)quinoline, (dihydrochloride),
mp 208.1-208.5°C;
6-imidazolylmethyl-8-(3-chlorophenyl)quinoline, (dihydrochloride),
mp 64-64.5°C.
6-ethyl-8-(3-chlorophenyl)quinoline, (hydrochloride),
mp 184.8-185.9°C.
6-n-propyl-8-(3-chlorophenyl)quinoline, (hydrochloride),
mp 206.4-210.4°C.
6-n-butyl-8-(3-chlorophenyl)quinoline, (hydrochloride),
mp 202.3-208.7°C.
6-t-butyl-8-(3-chlorophenyl)quinoline, (hydrochloride),
mp 197.5-198.5°C;
6-n-pentyl-8-(3-chlorophenyl)quinoline, (hydrochloride),
mp 182.2-183.6°C;
6-cyclohexyl-8-(3-chlorophenyl)quinoline, (hydrochloride),
mp 197.7-203.7°C.
6-(1-hydroxy-1-methylethyl)-8-(3-chlorophenyl)quinoline, (hydrochloride),
mp 171.2-171.9°C.
6-isopropyl-8-(3,4-methylenedioxyphenyl)quinoline, (hydrochloride),
mp 86.6-88.2°C.
6-isopropyl-8-(3-carbomethoxyphenyl)quinoline, (hydrochloride),
mp 166.5-167.6°C.
6-n-propyl-8-phenylquinoline, (hydrochloride), mp 168-169°C.
6-isopropyl-8-phenylquinoline, (hydrochloride), mp 155-158.9°C.
6-n-pentyl-8-phenylquinoline, (hydrochloride), mp 151.5-152.3°C.
6-isopropyl-8-(4-trifluoromethylphenyl)quinoline, (hydrochloride),
mp 167.5-168.6°C.
6-isopropyl-8-(3-cyanophenyl)quinoline, (hydrochloride),
mp 206.5-207.6°C.
WO 94/22852 ~~~ PCT/US94/03004
EXAMPLE 22
Determination of Potency and Selectivity
of Inhibitors for PDE IV
Preparation of Human Platelet Phosphodiesterase (PDE III)
Platelet high-affinity cAMP PDE (PDE III) was obtained from human
blood in accordance with previously described procedures described in Mol.
Phara~acol. 20:302-309, Alvarez, R., Taylor, A., Fazarri, J.J., and Jacobs,
J.R. (1981).
Blood was collected into evacuated tubes containing EDTA (7.7 mM,
final concentration). PRP was obtained by centrifuging the blood in
polycarbonate tubes at 200 x g for 15 min at 4°C. A platelet pellet was
resuspended in a volume of buffer A (0.137 M NaCl, 12.3 mM Tris-HC1 buffer,
pH 7.7, containing 1 mM MgCh. The hypotonically-lysed platelet suspension
was centrifuged at 48,000 x g for 15 min and the supernatant was saved.
The pellets were frozen on dry ice and briefly thawed at 22°C. The
supernatant was combined with the pellet fraction and the resulting
suspension was centrifuged at 48,000 x g for 30 min. The supernatant
fraction was stored in 0.5 mL aliquots at -20°C and used as the soluble
PDE. Enzyme activity was adjusted to 10-20% hydrolysis after 10 minutes of
incubation by dilution with lOmM cold Tris-HC1 buffer, pH7.7.
Preparation of Human Lymphocyte Phosphodieaterase (PDE Iv)
Human B cell line (43D) were cultured at 37°C in 7% COz in RPMI
1640
with L-glutamine and 10% Nu-Serum. Prior to the assay -1.5x108 cells were
centrifuged at 1000 xpm for 10 minutes in a table top clinical centrifuge.
The pellet was resuspended in 2-3 mL of 45 mM Tris-HC1 buffer, pH 7.4. The
suspension was homogenized and centrifuged at 12,000 x g at 4°C for 10
minutes. The supernatant was diluted to 28 mL with Tris-HC1 buffer and
used directly in the assay or stored at -20°C. The final concentration
of
DMSO in the PDE incubation medium Was 1%. Nitraquazone was included in each
assay (10 and 100~M) as a reference standard.
Human Platelet cAM.P Phosphodiesterase Assay
The phosphodiesterase incubation medium contained 10 mM Tris-HC1
buffer, pH 7.7, 10 mM MgSOd, 0.1-1~M [3Fi]-AMP (0.2 ~Ci) in a total volume of
1.0 mL. Following addition of the enzyme, the contents were mixed and
incubated for 10 min at 30°C. The reaction was terminated by immersing
the
tubes in a boiling-water bath for 90 sec. After the tubes were cooled in
an ice-water bath, 0.1 mL (100~g) of 5'-nucleotidase from snake venom
(Crotalus atrox, Sigma V-7000) was added to each tube. The contents were
mixed and incubated for 30 min at 30°C. The nucleotidase reaction was
terminated by immersing the tubes in a boiling water bath for 60 sec.
Labeled adenosine was isolated from alumina columns according to the method
WO 94/22852 PCTIUS94/03004
-54-
described in Anal. Biochem., 52:505-516 (1973), Filburn, C.R., and Karn,
J.. Assays were performed in triplicate. Hydrolysis of cAMP ranged from
10-20%. Test compounds were dissolved in DMSO. The final concentration of
DMSO in the phosphodiesterase assay was 1% when tested with compounds up to
0.1 mM. When tested at 1 mM the DMSO concentration was 10% and this
activity was compared to control PDE activity in the presence of 10% DMSO.
PDE III Activity
Compound I ICs: 53 N,M
Compound I is 6-(4-pyridylmethyl)-8-(3-nitrophenyl)quinoline.
Human Lymphocyte CAMP Phosphodiesterase Assay
The phosphodiesterase incubation medium contained 40 mM Tris-HC1
buffer, pH 7.7, 0.1 mM MgSO" 3.75 mM mercaptoethanol, and 0.1-1.0 ~M [3Fi]
cANtp (0.2 ~Ci) in a total volume of 1.0 mL. The reaction was performed and
processed according to the procedure used (above) for human platelet PDE.
The final concentration of DMSO was 1%.
The representative compounds of the present invention exhibit potency
and selectivity as inhibitors of PDE IV When tested by the human platelet
cAMP phosphodiesterase assay and the human lymphocyte cANlp
phosphodiesterase assay.
Inhibition of Human Lymphocyte PDE IV
Compound I ICs: 0.023 nM
Compound II ICs: 0.11 nM
Compound III ICs: 0.063 nM
Compound I is the compound of Formula I where R' is
4-pyridylmethyl and Rz is 3-nitrophenyl, namely 6-(4-pyridylmethyl)-8-
(3-nitrophenyl)quinoline.
Compound II is the compound of Formula I where R' is
4-pyridylmethyl and RZ is 3-chlorophenyl, namely 6-(4-pyridylmethyl)-8-(3-
chlorophenyl)quinoline.
Compound III is the compound of Formula I where R' is
isopropyl and RZ is 3-nitrophenyl, namely 6-isopropyl-8-(3
nitrophenyl)quinoline.
EXAMPLE 23
Determination of Immunosuppreasive Activity
Btilizing Responses of Human Peripheral Hlood
Lymphocytes to Mitogen
This procedure is a modification of a procedure initially described
by Greaves, et al. ["Activation of human T and B lymphocytes by polyclonal
mitogens," Nature, 248, 698-701 (1974)].
WO 94122852 ~~~ PCT/US94/03004
-55-
Human mononuclear cells (PBL) were separated from heparinized whole
blood by density gradient centrifugation in Ficoll-Paque (Pharmacia).
After washing, 5 x 10' cells/well were cultured in microtiter plates with
minimal essential media supplemented with 1% human serum, gentamicin,
sodium bicarbonate, 2-mercaptoethanol, glutamine, non-essential amino
acids, and sodium pyruvate. The mitogen concanavalin A (Sigma) Was used at
a.concentration of 2 ~g/ml. Test materials were tested at concentrations
between 10' and 10''° M, by addition to the culture at time 0. Cultures
were
set up in quadruplicate and incubated at 37~C in a humidified atmosphere
with 5% COZ for 48 hours. A pulse of 1.0 ~Ci/well of 3fi-thymidine was added
for the last 4 hours. Cells were collected on glass fiber filters with an
automatic harvester and radioactivity was measured by standard
scintillation procedures. The 50% inhibitory concentration ("ICS") for
mitogenic stimulation was determined graphically.
The representative compounds of the present invention showed
immunosuppressive activity when tested by this method.
EBAMPLE 24
Determination of Immunosuppressive Activity
Utilizing The Hemolytic Plaque Forming Cell Assay
This procedure is a modification of "The agar plaque technique for
recognizing antibody producing cells," a procedure initially described by
Jerne, et al. (Cellbound Antibodies, Amos and Kaprowski editors (Wistar
Institute Press, Philadelphia, 1963) p. 109].
Groups of 5-6 adult C3H female mice were sensitized with 1.25 x 108
sheep red blood cells (SRBC) and simultaneously treated with an oral dosage
form of the test material in an aqueous vehicle. Animals in a control
group received the same volume of vehicle. Four days after SRBC
inoculation, spleens were dispersed in glass homogenizers. The number of
nucleated cells (WBC) was determined and the spleen cell suspension was
mixed with SRBC, guinea pig complement and agar solution at 0.5%
concentration. Aliquots of the above mixture (0.1 mL) Were dropped on four
separate quadrants of a Petri dish and were covered with cover slips.
After two hours incubation at 37°C, areas of hemolysis around
plaque-forming cells (PFC) were counted with a dissecting microscope.
Total WBC/spleen, PFC/spleen and PFC/106 WBC (PPM) were calculated for each
mouse spleen. Geometric means of each treatment group were then compared
with the vehicle-treated control group.
The representative compounds of the present invention showed
immunosuppressive activity when tested by this method.
WO 94/22852 , PCT/US94I03004
~l~9bb~
-56-
EXAMPLE 25
Determination of Anti-Inflammatory Activity Utilizing
Arachidonic Acid-Induced Ear Edema (AAEE) in the Mouse
This procedure is a modification of a procedure described by Young et
al., J. Invest. Derm., 82:367-371 (1984).
Female Charles River ICR mice 23-27 grams were administered 0.2 mL of
test material. The mice were challenged with 20 ~1 of arachidonic acid
applied topically to the ear. One hour after challenge, the weight of an
8 mm disc was determined. The mean increase in ear plug weight Was
calculated.
Materials with anti-inflammatory activity inhibit the increase in ear
plug weight.
The representative compounds of the present invention exhibited
anti-inflammatory activity when tested by this method.
Anti-inflammatory Activity (AAEE)
Compound I EDT O.Olmg/kg
Compound II EDT 0.2mg/kg
Compound III EDT 4.5mg/kg
EBAMPLE 26
Determination of Anti-Inflammatory
Activity Utilizing Adjuvant-Induced
Arthritis In The Rat
This procedure is a modification of a procedure initially described
by Pearson, C.M., Proc. Soc. Exp. Biol. Med., 91:95-101 (1956).
Female Charles River albino rats weighing 160-180 g received
0.1 mL of a suspension in paraffin oil of heat-killed Mycobacterium
butyricum (10 mg/ml) by means of an intradermal injection into the proximal
1/4 of the tail on day 0. Beginning on day 1, the test material was
administered orally in an aqueous vehicle (0.5 mL/dose) once each day for
17 days. On day 18 the intensity of the swelling of the four foot pads and
tail was determined utilizing a scoring system in which the swelling in the
four paws was scored 0-4 for each paw and the tail swelling was scored 0-3,
such that the total maximum score was 19.
The representative compounds of the present invention exhibited
anti-inflammatory activity when tested by this method.
EBAMPLE 27
Determination of Activity Towards
Autoimmune Disease Utilizing Survival of MRL/lpr Mice
MRL/lpr mice develop a multisystemic disease characterized by
glomerulonephritis, arthritis, arteritis, lymphoid hyperplasia. The length
WO 94/22852 ~~~ PCT/US94/03004
-57-
of survival of mice with this disease is approximately one-third that of
non-disease developing MRL/n mice. These mice have a high incidence of
autoantibodies and the disease process is considered autoimmune in nature
as described by Theofilopoulos, et al., Advances in Immunology, 37:269-390
(1985) .
The representative compounds of the present invention significantly
extended the lifespan of the MRL/lpr mice.
EXAMPLE 28
Determination of Analgetic Activity
Utilizing Phenylquinone-Induced Stretching in the Mouse
This procedure is a modification of a procedure described by
Hendershot, et al. J. Pharmacol. Exp. Then., 125:237-240 (1959).
Groups of 8 Female CD-1 mice were administered test materials orally
in an aqueous vehicle. At various times following administration of test
materials, 0.25 mL of a 0.02% solution of phenylquinone was administered
intraperitoneally. The number of stretches for each animal was enumerated
over a ten minute period following the phenylquinone administration.
Analgetic activity was determined by inhibition of the mean number of
stretches.
The representative compounds of the present invention showed
analgetic activity when tested by this method.
EBAMPLE 29
Capsule Formulation
This example illustrates the preparation of a representative
pharmaceutical formulation for oral administration containing an active
compound of Formula I, e.g., 6-isopropyl-8-(3-nitrophenyl)quinoline.
Ingredients Quantity (mg/capsule)
Active compound 200
lactose, spray-dried 148
magnesium stearate 2
The above ingredients are mixed and introduced into a hard-shell
gelatin capsule.
Other compounds of Formula I, such as those prepared in accordance
with Examples 1-20 can be used as the active compound in the preparation of
the orally adminiEtrable formulations of this example.
EXAMPLE 30
Oral Formulation
This example illustrates the preparation of a representative
pharmaceutical formulation containing an active compound of Formula I,
e.g., 6-isopropyl-8-(3-nitrophenyl)quinoline.
9~~~
WO 94/22852 PCT/US94/03004
-58-
An suspension for oral administration is prepared having the
following composition:
Ingredients Quantity
Active compound 1.0 g
fumaric acid 0.5 g
sodium chloride ' 2.0 g
methyl paraben 0.1 g
granulated sugar 25.5 g
sorbitol (70% solution) 12.85 g
Veegum K (Vanderbilt Co.) 1.0 g
flavoring 0.035 mL
colorings 0.5 mg
distilled water q.s. to 100 mL
Other compounds of Formula I, such as those prepared in accordance
with Examples 1-20 can be used as the active compound in the preparation of
the orally administrable formulations of this example.
EBAMPLE 31
Tablet Formulation
This example illustrates the preparation of a representative
pharmaceutical formulation containing an active compound of Formula I,
e.g., 6-isopropyl-8-(3-nitrophenyl)quinoline.
A tablet for oral administration is prepared having the following
composition:
Ingredients Quantity (mg/tablet)
Active compound 400
corn starch 50
lactose 145
magnesium stearate 5
The above ingredients are mixed intimately and pressed into single
scored tablets.
Other compounds of Formula I, such as those prepared in accordance
with Examples 1-20 can be used as the active compound in the preparation of
the tablet formulations of this example.
EXAMPLE 32
Injectable Formulation
This example illustrates the preparation of a representative
pharmaceutical formulation containing an active compound of Formula I,
e.g., 6-isopropyl-8-(3-nitrophenyl)quinoline.
WO 94/22852 ~ ~ ~ PCTIUS94/0300::-
-59-
An injectable preparation is prepared having the following
composition:
Ingredients Quantity
Active compound 0.2 g
water (distilled, sterile) q.s. to 20.0 mL
Other compounds of Formula I, such as those prepared in accordance
with Examples 1-20 can be used as the active compound in the preparation of
the injection administrable formulations of this example.
E%AMPLE 33
Suppository Formulation
This example illustrates the preparation of a representative
pharmaceutical formulation containing an active compound of Formula I,
e.g., 6-isopropyl-8-(3-nitrophenyl)quinoline.
. A suppository totalling 2.5 grams is prepared having the following
composition:
Ingredients Quantity
Active compound 500 mg
Witepsol H-15- q.s. to 2.5 g
(*triglycerides of saturated vegetable fatty acid; a product of Riches-
Nelson, Inc., New York, N.Y.).
Other compounds of Formula I, such as those prepared in accordance
with Examples 1-20 can be used as the active compound in the preparation of
the suppository formulations of this example.
TO%ICOLOGY
No serious toxicological effects were observed in the above described
assays.