Note: Descriptions are shown in the official language in which they were submitted.
2161681
w o 94/25466 PCTrUS94/04681
WATER 501UBIF DERIVATIYES OF CAMPTOTHECIN AND THEIR USE AS ANTITUMOR AGENTS
The present invention relates to water soluble, camptothecin derivatives
substituted in the 7 positionJ their use in the treatment of tumors and methods of
5 their preparation.
BACKGROUND OFTHE INVENTION
Camptothecin, a natural, cytotoxic a~kaloid, is a topoisomerase I inhibitor and
10 potent antitumor agent. It was first isolated from the leaves and bark of the Chinese
plant, Camptotheca accuminata, by Wall, et. al. (J. Am. Chem. Soc., 88 3888
(1 966)).
As depicted, camptothecin is a fused ring system, composed of a quinoline (A
15 and B), fused to a pyrrolidine ring (C), fused to an alpha-pyridone ring (D) which in
turn is fused to a lactone ring (E).
CAMPTOTHECIN
~
20 It has an asymmetric carbon at the 20 position making two enantiomeric forms
possible. However, the natural occurring compound is found in the "S" configura-tion as shown above.
Cytotoxic agents are often employed to control or eradicate tumors i.e., they
25 are chemotherapeutic agents. Camptothecin's cytotoxic activity is thought to be di-
rectly related to camptothecin's potency as a topoisomerase inhibitor. [For detailed
explanations of the topoisomerase function see A. Lehninger, Principles of
Biochemisfry, 813, Worth Publishers, New York (1982); L. F. Liu, "DNA
Topoisomerases," CRC Critical Review in Biochemistry, 1-24,15 (1983) and H
30 Vosberg, ~DNA Topoisomerases: Enzymes that Control DNA Conformation,"
Current Topics in Microbiology and Immunology, 19, Springer-Verlag, Berlin
2 ~ 8 l
WO 94/2~466 PCT/US94/04681
-2 -
(1985).] In particular, camptothecin has been shown to be effective in the treatment
of leukemia (L-1210) and certain solid tumors in laboratory animals, e.g., see
Chem. Rev. 23, 385 (1973) and Cancer Treaf. Rep., 60, 1007 (1967).
Unfortunately, in the ciinic camptothecin's promise as an effective antitumor
agent has not bee~ completely fulfilled. Camptothecin is essentially insoluble in
physiologically compatible, aqueous media, and must be modified to make it suffi-
ciently soluble for parenteral administration, a preferred mode for antitumor treat-
ment. It can be made soluble by forming its sodium salt, that is, by opening thelactone with sodium hydroxide (see F.M. Muggia, et al., Cancer Chemotherapy
Reports, pt. 1, 56, No.4, 515 (1972)). However, M. C. Wani, et al., J. Med. Chem.,
23, 554 (1980), reported that the alpha-hydroxy lactone moiety of ring E is an abso-
lute requirement for antitumor activity.
In the art there are examples of modifications and derivatives of camptothecin
prepared to improve its solubility in water. Although many of these derivatives were
active in vitro and in early animal studies using leukemia (L-1210) models, theywere disappointing in chronic, animal models involving implanted solid tumors.
Miyasaka, et al., U.S. Patent No. 4,399,282, discloses a group of camptothecin
derivatives substituted at the 7 position with, inter alia, hydroxymethyl and
alkoxymethyl. Further, Miyasaka, et. al. in U.S. patent No. 4,399,276 discloses
camptothecin-7-aldehyde and certain related aldehyde derivatives such as acetals,
oximes and hyrazones. More recently, Vishnuvajjala, et al., in U.S. Patent No.
4,943,579, claimed a series of water-soluble camptothecin derivatives with sub-
stituents on the A ring as does Boehm, et al., European Patent Application 0 321122 A2. Other examples of derivatives of camptothecin include Miyasaka, et al.,
U.S. Patent No. 4,473,692 and No. 4,545,880; and W. Kingsbury, et al., J Med.
Chem., 34, 98 (1991). None of these references reported compounds with antitu-
mor activity greater than that of camptothecin itself.
Wani and co-workers reported that 10, 11-methylenedioxycamptothecin is
more potent than unsubstituted camptothecin (see M. C. Wani, et al., J. Med. Chem,
29, 23~8 (1986) and 30, 2317 (1987)). However, its water solubility is as poor as
camptothecin which seriously limits its clinical utility.
We have now found water-soluble analogs of camptothecin with good topoi-
somerase I inhibitory activity in vitro, and impressive, antitumor activity in vivo.
21 61 ~81
~0 94125466 PCT/US94/04681
-3-
SUMMARY OF THE INVENTION
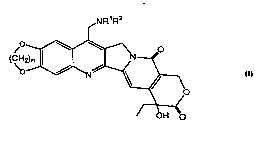
One aspect of the present invention is the water-soluble camptothecin
analogs of formula (I),
5 NR1R2
, ~
(~
OH
wherein:
n represents the integer 1 or 2; and
i) R1 represents:
hydrogen, lower alkyl, (C3 7)cycloalkyl, (C3 7)cycloalkyl
lower alkyl, lower alkenyl, hydroxy lower alkyl, amino lower
alkyl, lower alkylamino lower alkyl, amino lower alkyl,
lower alkoxy lower alkyl or (CH2)tAr
1 5 wherein:
t is 0 to 5 and
Ar represents phenyl, furyl, pyridyl, N-methylpyrrolyl,
imidazolyl; or phenyl, furyl, pyridyl, N-methylpyrrolyl,
imidazolyl, with one or more substituents independently
selected from hydroxy, methoxy, halogen, and amino;
and
R2 represents:
diphenylmethyl or(CH2)tAr; or
ii) R1 and R2 taken together with the linking nitrogen represent;
N-tetrahydroquinolyl or N-tetrahydroisoquinolyl;
and the pharmaceutically acceptable salts and solvates thereof.
Pharmaceutically acceptable salts include, but are not limited to salts with in-organic acids such hydrochloride, sulfate, phosphate, diphosphate, hydrobromide
216168~
WO 94/2~i466 PCT/US94/04681--
and nitrate or salts with an organic acid such as acetate, malate, maleate, fumarate,
tartrate, succinate, citrate, lactate, methanesulfonate, p-toluenesulfonate, palmoate,
salicylate and stearate. Other acids such as oxalic, while not in themselves phar-
maceutically acceptable, may be useful as intermediates in obtaining the com-
5 pounds of the invention and their pharmaceutically acceptable salts.
Another aspect of the invention is a method of inhibiting topoisomerase Type I
in mammalian cells comprising àdministering to a patient a topoisomerase inhibit-
ing amount of a compound of formula (I), and a method of treating a tumor in a
10 mammal comprising administering to a mammal bearing a tumor, an effective anti-
tumor amount of a compound of formula (I). A further aspect comprises pharma-
ceutical formulations containing a compound of formula (I) as an active ingredient.
Methods of preparation of the compounds of formula (I) and the associated novel
chemical intermediates used in the synthesis, as taught herein, are also within the
15 scope of the present invention.
DETAILED DESCRIPTION OF THE INVENTION
Compounds
As used herein the term "lower" in reference to alkyl and alkoxy means 1-6
carbons, especially 1-3 carbons, and in reference to alkenyl means 3-6 carbons
(provided that the double bond is not attached to the carbon which is attached to
the nitrogen). As use herein, the term ~arylU means aromatic ring substituents, e.g.,
25 phenyl, napthyl, furyl, pyridyl, N-methylpyrrolyl or imidazolyl. The group "(CH2)
also includes branched alkylene chains where branching is possible.
As use herein the terms "N-tetrahydroquinolyl" and "N-tetrahydroisoquinolyl"
are defined as follows:
N-tetrahydroquinolyl N-tetrahydroisoquinolyl
The lactone ring, i.e., ring E, of the camptothecin moiety may be opened by
35 alkali metal or alkaline-earth metal bases, for example sodium hydroxide or
calcium hydroxide, to form alkali metal or alkaline-earth metal salts of the
21 6l 68l
~WO 94/2~466 PCTIUS94/04681
-5-
corresponding open E ring form of the compounds of formula (1). Because of its
better solubility in water, the open E ring form may advantageously be purified by
conventional recrystallization techniques. Accordingly, said open E ring form may
then be used as an intermediate to form the compounds of formula (1), for example
5 by treatment with acid, e.g., hydrochloric acid, and thereby produce a purified form
of the compounds of formula (1).
As noted above, the camptothecin moiety has an asymmetric carbon atom at
the 20 position making two enantiomeric forms, i.e., UR" and "S" configurations,possible. This invention includes both enantiomeric forms and any combinations of
these forms. For simplicity, where no specific configuration at the 20 position is
depicted in the structural formulas, it is to be understood that both enantiomeric
forms and mixtures thereof are represented. Unless noted otherwise, the nomen-
clature convention, ''(R,S)U, denotes a racemic (approximately equal portion) mix-
ture of the R and S enantiomers while ~(R)u and "(S)" denote essential opticallypure R and S enantiomers respectively. Also included in the invention are other
forms of the compound of formula (1), such as solvates, hydrates, polymorphs andthe like.
One sub group of compounds of the present invention are the compounds
of formula (I) wherein:
n represents the integer 1 or 2; and
j) R1 represents:
hydrogen, lower alkyl, (C3 7)cycloalkyl, (C3 7)cycloalkyl
lower alkyl, lower alkenyl, hydroxy lower alkyl, amino lower alkyl,
lower alkoxy lower alkyl or (CH2)tAr
wherein:
t is O to 5 and
Ar represents phenyl, furyl, pyridyl, N-methylpyrrolyl,
imidazolyl; or phenyl, furyl, pyridyl, N-methylpyrrolyl,
imidazolyl, with one or more substituents selected from
hydroxy, methoxy, halogen, and amino; or
R2 represents:
diphenylmethyl or(CH2)tAr; and
j;) R1 and R2 taken together with the linking nitrogen represent;
N-tetrahydroquinolyl or N-tetrahydroisoquinolyl;
and the pharmaceutically acceptable salts thereof.
WO 94/25466 21~1~ 8 ~ -6- PCT/US94/04681~
Another sub group of compounds of the present invention are the compounds
of formula (I) wherein:
n represents the integer 1 or 2; and
i) R1 represents:
hydrogen, (C1 3) alkyl or amino (C1 3) alkyl; and
R2 represents:
diphenylmethyl or(CH2)tAr;
1 0 wherein:
tis 1 to3and
Ar represents phenyl, 2-furyl, 2-pyridyl, 4-pyridyl 2-N-
methylpyrrolyl, 4-imidazolyl; or phenyl, 2-furyl, 2-pyridyl, 4-
pyridyl, 2-N-methylpyrrolyl, 4-imidazolyl, with one to two
substituents selected from hydroxy, methoxy, halogen,
and amino; or
ii) R1 and R2 taken together with the linking nitrogen represent
N-tetrahydroisoquinolyl;
and the pharmaceutically acceptable salts thereof.
In one particular group of compounds of formula (I) Ar represents phenyl, furyl,pyridyl, N-methylpyrrolyl, imidazolyl or phenyl substituted with one or two
25 substituents independently selected from hydroxy, methoxy, halogen and amino.Typical Ar groups include phenyl and substituted phenyl, 2-furyl, 2-pyridyl, 4-
pyridyl, 2-N-methylpyrrolyl and 4-imidazolyl. In one particular subgroup of
compounds of formula (I), Ar is substituted phenyl or 4-pyridyl.
In another subgroup of compounds R1 represents hydrogen, lower alkyl, lower
alkylaminolower alkyl or -(CH2)tAr, especially hydrogen or lower alkyl.
In another subgroup of compounds of formula (I), t is 1, 2, or 3, especially 1
and n is 1 or 2, especially 2.
Specific compounds of formula (I) are:
2161681
~0 94/2s466 PCT/US94/04681
-7- - r~
Example
Number Compound Name
1. 7-(N-Ethyl-N-4-pyridylmethyl)- aminomethylene-10, 11 -ethylenedioxy-20(S)-
camptothecin,
2. 7-(4-Aminobenzyl)-aminomethylene-10, 11 -ethylenedioxy-20(S)-
camptothecin,
3. 7-(3-Methoxy-4-hydroxybenzyl)-aminomethylene-10, 11-ethylenedioxy-20(S)-
camptothecin,
10 4. 7-Benzylaminomethylene-10, 11-ethylenedioxy-20(S)-camptothecin,
5. 7-(N-Tetrahydroisoquinolino)methylene-10, 11-ethylenedioxy-20(S)-
camptothecin,
6. 7-Dibenzylaminomethylene-10, 11 -ethylenedioxy-20(S)-camptothecin,
7. 7-(N-Methyl)benzylaminomethylene-10, 11-ethylenedioxy-20(S)-
camptothecin,
8. 7-Furylaminomethylene-10, 11 -ethylenedioxy-20(S)-camptothecin,
9. 7-(3-Phenylpropyl)-aminomethylene-10, 11-ethylenedioxy-20(S)-
camptothecin,
10. 7-(3,4-Dimethoxybenzyl)-aminomethylene-10, 11 -ethylenedioxy-20(S)-
camptothecin,
11. 7-(N-Ethyl, (2-chloro-6-fluorobenzyl))-aminomethylene-10, 11-ethylenedioxy- 20(S)-camptothecin,
12. 7-(3,4-Difluorobenzyl)-aminomethylene-10, 11 -ethylenedioxy-20(S)-
camptothecin,
13. 7-Diphenylmethylaminomethylene-10, 11-ethylenedioxy-20(S)-camptothecin,
14. 7-((R) 1 -Phenylethyl)-aminomethylene-10, 11 -ethylenedioxy-20(S)-
camptothecin,
15. 7-((S) 1-Phenylethyl)-aminomethylene-10, 11-ethylenedioxy-20(S)-
camptothecin,
16. 7-(2(N-Methyl-2-pyrrol)-ethyl)-aminomethylene-10, 11-ethylenedioxy-20(S)-
camptothecin,
17. 7-(N-benzyl-N-(2-Dimethylaminoethyl)-aminomethylene-10, 11 -
" ethylenedioxy-20(S)-camptothecin,
18. 7-(2-(4-lmidazolyl)ethyl)-aminomethylene-10, 11 -ethylenedioxy-20(S)-
camptothecin,
19. 7-(2-Pyridylmethyl)-aminomethylene-10, 11 -ethylenedioxy-20(S)-
camptothecin,
20. 7-Benzylaminomethylene-10, 11-methylenedioxy-20(S)-camptothecin, and
216168~
WO 94/25466 PCT/US94/04681
21. 7-(3,4-Dimethoxybenzyl)-aminomethylene-10, 11-methylenedioxy-20(S)-
camptothecin, and pharmaceutically acceptable salts and solvates thereof.
Particular specific compounds of formula (I) are:
7-(N-Ethyl-N-4-pyridlymethyl)- aminomethylene-10, 11-ethylenedioxy-20(S)-
camptothecin,
7-(4-Aminobenzyl)-aminomethylene-10, 11-ethylenedioxy-20(S)-
1 0 camptothecin,
7-(3-Methoxy-4-hydroxybenzyl)-aminomethylene-10, 11-ethylenedioxy-
20(S)-camptothecin,
and pharmaceutically acceptable salts and solvates thereof.
Prep~ration of Compounds
According to one general process (A), compounds of formula (I) may be
20 prepared by the procedure shown in Step 2 of Scheme l:
~VO 94125466 2161 ~8 1 PCT/US94/04681
\N ~
(CH2)~0 ~ ~
( 11 ) / H 60
/ STEP 1
<
HNR1R2 STEP 2 / H
(V) ~
NR1R2
( ) / OH ~o
SCHEME I
In Step 1 of Scheme 1, a compound of formula (Il), wherein X is a leaving
group (as defined in J. March, Advanced Organic Chemistry, 3rd. Ed., page 179,
5 John Wiley & Sons, New York (1985)), for example, a halogen, e.g., chloro, may be
reacted with a compound of formula (Ill) according to the method taught in US
Patent 4,894,456 (hereinafter, '456), issued January 16, 1990 to Wall et al., incor-
porated herein by reference, to yield a compound of formula (IV).
The reaction of Step 1 is preferably carried out in the presence of an acid or
base catalyst. The acid catalyst is preferably a strong mineral acid, for example
hydrochloric, nitric, sulfuric and phosphoric or a strong organic acid such as C1-8
alkanoic acids and C1.12 arylsulfonic acids, especially p-toluenesulfonic acid. The
base catalyst is preferably an inorganic base, for example sodium and potassium
1~ carbonate and sodium and potassium bicarbonate or an organic base such as a
WO 94/25466 21~ 16 8 1 1o- PCT/US94/04681--
sterically hindered base, for example, triethylamine and diisopropylamine.
This reaction may be carried out neat or in the presence of a polar or non-
polar solvent. Preferred polar solvents are C1.6 alcohols, C1-6 ethers, and
5 dimethylformamide. Preferred non-polar solvents are branched or straight chained
alkyl hydrocarbons having 4-10 carbon atoms and aromatic hydrocarbons having
6-20 carbon atoms especially toluene. The reaction is generally conducted with
heating at reflux.
In Step 2, (General Process A) the compounds of formula (IV) may be
converted to the compounds of formula (I) by displacement of the leaving group, X,
with a compound of formula (V), wherein R1 and R2 are as defined for formula (I).
This displacement reaction may conveniently be carried out in a solvent system, for
example water, a (C1 4) alkanol, a (C2 4) alkylene diol, 1-hydroxy-2-
15 methoxyethane, dimethylacetamide (DMAC), N-methylpyrolidinone, dimethyl for-
mamide (DMF), tetrahydrofuran (THF), dimethyl sulfoxide (DMSO), toluene,
dioxane or a combination of these solvents in the presence of excess amine, i.e.,
excess compound of formula (V), with or without a base, e.g., potassium carbonate,
and/or with Nal as a catalyst.
This method is particularly useful for preparing compounds of formula (I)
wherein R1 is other than hydrogen.
Compounds of formula (V) are commercially available, taught in the chemical
25 literature, or may be readily prepared by one skilled in the art of organic chemistry
using methods and materials known in the art of organic chemistry.
According to another general process (B), compounds of formula (I) may be
prepared by the procedure shown in Step 2a of Scheme IA:
WO 94/2~i466 81 PCT/US94/04681
(CH2)~
b NH2
HNR1R2 ~ ( 11 )
( V ) ~/Step 1a
NR1R2
~ o
(IIA) (1ll)
s~p 2a / OH ~b
y
NR1R2
~-o
OH
SCHEME 1 A
In Step 1a, a compounds of formula (Il) may be converted to a compounds of
formula (IIA) by displacement of the leaving group, X (as defined for Scheme 1), with
5 a compound of formula (V), wherein R1 and R2 are as defined for formula (I). This
displacement reaction may conveniently be carried out in a solvent system, for
example, water, a (C14) alkanol, a (C2.4) alkylene diol, 1-hydroxy-2-meth-
oxyethane, dimethylacetamide (DMAC), N-methylpyrolidinone, dimethyl formamide
(DMF), tetrahydrofuran (THF), dimethyl sulfoxide (DMSO), toluene, dioxane or a
10 combination of these solvents (to the extent of miscibility) in the presence of excess
amine, i.e., excess compound of formula (V), with or without a base, e.g., potassium
carbonate, and/or with Nal as a catalyst.
In Step 2a (General Process B), compound of formula (IIA) is reacted with a
15 compound of formula (Ill) in a similar manner to that taught above in Scheme 1,
Step 1, to yield a compound of formula (I).
WO 94/25466 2 1 6 1 6 8 1 -12- PCT/US94/0468~
Further alternate general method (C), particularly useful for preparing
compounds of Formula (I) wherein R1 is hydrogen, is shown in Step 3b of Scheme
x
/~o
(CH2)~NH2
D~ ~ (11)
(Hal)3C~ NHR ~/ SteP lb
C~ 2 I \N
\~NH~ OH~O
(llb) ~ (1ll)
(Hal)30~0
~NR2
H'B- / OH 6
Step 3b
NR~ B-
O C
(Ib)
SCHEME 1 B
2l~l68l
~VVO 94/25466 PCTfUS94tO4681
- 1 3-
ln Step lb, a compound of formula (Va) (wherein "Hal" is halogen, i.e., fluoroj
chloro, bromo or iodo) e.g., trifluoroacetamide, is reacted with a compound of for-
mula (Il) in a polar, aprotic solvent, e.g., acetonitrile, in the presence of a base sol-
uble in the polar, aprotic solvent, e.g., cesium carbonate if the solvent is acetonitrile,
5 to yield a compound of formula (llb).
In Step 2b, a compound of formula (llb) is reacted with a compound of formula
(Ill) in a similar manner to that taught in Scheme 1, Step 1, to yield a compound of
formula (IVb).
In Step 3b (General Process C), a compound of formula (IVb) is treated with
an acid, H+B-, such as a mineral acid, e.g., hydrochloric acid, to yield a compound
of formula (Ib), i.e. salt of a compound of formula (I). The compound of formula (Ib)
may be treated with a base, such as an alkali metal hydroxide or carbonate, e.g.,
15 sodium hydroxide or potassium carbonate, by standard method of the art to yield
the corresponding free base. For example a compound of formula (Ib) may be
stirred with an aqueous solution of potassium carbonate for about one to about four
hours in the temperature range of from about 5 to about 100C. The free base
can then be converted by conventional means to a pharmaceutically acceptable
20 salt if required.
The compounds of formula (Il) and (Ill) may be prepared according to the
procedure described in EP0 540 099 A1.
The novel, intermediate compounds of formulas (lla), (llb) and (IVb) are within
the scope of this invention.
A compound of formula (I) according to the invention may be converted into
another compound of the invention using conventional procedures.
Thus, for example, a compound of formula (I) wherein R1 represents a
hydrogen atom, may be alkylated using conventional techniques. The reaction
may be effected using a suitable aklylating agent such as an alkyl halide, an alkyl
tosylate or a dialkylsulphate. The alkylation reaction may conveniently be carried
out in an organic solvent such as an amide, e.g. dimethylformamide, or an ether,e.g. tetrahydrofuran, preferably in the presence of a base. Suitable bases include,
for example, alkali metal hydrides, such as sodium hydride, alkali metal
carbonates, such as sodium carbonate, or potassium methoxide, ethoxide or t-
butoxide. The alkylation reaction is conveniently carried out at a temperature of
21616~1
WO 94/25466 PCTIUS94/0468
-14-
from about 25 to about 1 00C.
Alternately, a compound of formula (I) wherein R1 represents a hydrogen
atom may be converted to another compound of formula (I) by reductive alkylation.
5 Reductive alkylation with an appropriate aldehyde or ketone may be effected using
an alkaline earth metal borohydride or cyanoborohydride. The reaction medium,
co~veniently in an alcohol, e.g. methanol or ethanol or an ether, e.g. dioxan ortetrahydrofuran, optionally in the presence of water. The reaction may conveniently
be carried out at a temperature in the range of 0 to 100C, preferably about 5 to
10 about 50C.
Alternatively, a compound of formula (I) wherein R1 represents a lower alkenyl
group may be converted to another compound of formula (I) wherein Rl represents
a lower alkyl group. Reduction may conveniently be effected in the presence of
15 hydrogen and a metal catalyst, for example, Raney nickel or a nobel metal catalyst
such as palladium, platinum, platinum oxide or rhodium, which may be supported,
for example, on charcoal. The reaction may be effected in a solvent such as an
alcohol, for example ethanol and conveniently at a temperature of from about -10 to
about +50C, preferably about 20 to about 30C.
A compound of formula (I) according to the invention, or a salt thereof may br
prepared by subjecting a protected derivative of formula (I) or a salt thereof to reac-
tion to remove the protecting group or groups.
Thus, at an earlier stage in the preparation of a compound of formula (I) or a
salt thereof it may have been necessary and/or desirable to protect one or more
sensitive groups in the molecule to prevent undesirable side reactions.
The protecting groups used in the preparation of compounds of formula (I)
may be used in conventional manner. See for example, "Protective Groups in
Organic Chemistry~ Ed. J.F.W. McOmie (Plenum Press 1973) or "Protective Groups
in Organic Synthesis" by Theodora W. Greene (John Wiley and Sons 1981).
Conventional amino protecting groups may include, for example, aralkyl
3~ groups, such as benzyl, diphenylmethyl or triphenylmethyl groups; and acyl groups
such as N-benzyloxycarbonyl or t-butoxycarbonyl. Thus, compounds of general
formula (I) wherein R1 represents hydrogen may be prepared by deprotection of a
corresponding protected compound.
~vo 94/25466 -15- ~681 PcT/uss4/046s1
Hydroxy groups may be protected, for exampie, by aralkyl groups, such as
benzyl, diphenylmethyl or triphenylmethyl groups, acyl groups, such as acetyl, sili-
con protecting groups, such as trimethylsilyl or t-butyl dimethylsilyl groups or as
tetrahydropyran derivatives.
Removal of any protecting groups present may be achieved by conventional
procedures. Thus, an aralkyl groups such as benzyl, may be cleaved by hy-
drogenolysis in the presence of a catalyst (e.g. palladium on charcoal); an acylgroup such as N-benzyloxycarbonyl may be removed by hydrolysis with, for ex-
ample, hydrogen bromide in acetic acid or by reduction, for example by catalytichydrogenation; silicon protecting groups may be removed, for example, by treat-
ment with fluoride ion or by hydrolysis under acidic conditions; tetrahydropyrangroups may be cleaved by hydrolysis under acidic conditions.
As will be appreciated, in any of the processes described above, it may be
necessary or desired to protect any sensitive groups in the molecule as just de-scribed. Thus, a reaction step involving deprotection of a protected derivative of
general formula (I) or a salt thereof may be carried out subsequent to any of the
above described processes.
Thus, according to a further aspect of the invention, the following reactions
may, if necessary and/or desired by carried out in any appropriate sequence sub-sequent to any of the processes
(i) removal of any protecting groups; and
(ii) conversion of a compound of formula (I) or a salt thereof into a
pharmaceutically acceptable salt thereof.
Where it is desired to isolate a compound of the invention as a salt, for exam-
ple, as an acid addition salt, this may be achieved by treating the free base of gen-
eral formula (I) with any appropriate acid, preferably with an equivalent amount, or
with creatinine sulphate in a suitable solvent (e.g. aqueous ethanol).
As well as being employed as the last main step in the preparative sequence,
the general methods indicated above for the preparation of the compounds of the
invention may also be used of the introduction of the desired groups at an inter-
mediate stage in the preparation of the required compound. It should therefore be
appreciated that in such multi-stage processes, the sequence of reactions shouldbe chosen in order that the reacting conditions do not affect groups present in the
WO 94/25466 2 ~ 6 1 6 81 PCT/US94/04681~
-16-
molecule which are desired in the final product.
The biological activity of the compounds of formula (I) appears to reside in theS enantiomer, and the R enantiomer has little or no activity. Thus, the S enan-
5 tiomer of a compound of formula (I) is generally preferred over a mixture of R and Ssuch as the racemic mixture. However, if the R enantiomer were desired, e.g., for
control studies or synthesis of other compounds, it could be conveniently prepared
by the procedure above using the R enantiomer of the compound of formula (Ill)
prepared according to the teachings of '512.
A compound of formula (I) prepared by reaction Scheme I or Scheme IA may
be purified by conventional methods of the art, e.g., chromatography, distillation or
crystallization.
15 Cle~vable Complex 7n vitro Assay
The data in Table A, below, shows the relative topoisomerase Type I inhibitory
activity of the compounds of Formula (I). This assay performed according to the
method described in Hsiang, Y. et al., J. Blol. Chem., 260:14873-14878 (1985),
20 correlates well with in vivo anti-tumor activity of topoisomerase inhibitors in animal
models of cancer, e.g., camptothecin and its analogs. See Hsiang et al., Cancer
Research, 49:4385-4389 (1989) and Jaxel et al., Cancer Research, 49:146~-1469
(1989).
Those compounds which exhibit observable activity at concentrations greater
25 than 2000 nM ("+~ in Table A) are considered weakly to moderately active, while
those with activity at concentrations less than 500 nM (U++++~ in Table A) are very
active. The term ~ICsoU means the concentration of a compound of formula (I) at
which 50% of the DNA substrate has been captured by topoisomerase 1.
TABLE A
Topoisomerase Inhibitory Activity of
Compounds of Formula (I) in the Cleavable Complex Assay
Example Isomeric Relative
Number form
4 (S) 1 l l l
8 (S) ++++
(S) m 11
14 (S) ~+++
(S) ++++
~0 94/2~466 -17- ~1 PCT/US94/04681
16 (S) ++++
21 (S) ++++
2 (S) ++++
19 (S) ++++
3 (S) ++++
(S) +It I
18 (S) ++++
6 (S) +++
7 (S) +++
(S) +++
12 (S) +++
17 (S) +++
(S) ++
11 (S) ++
13 (S) ++
9 (S) ++
ICso Range Symbol nM
++++ <~500
+++ <~ 1000~~500
++ <~2000>~ 1000
+ >~2000
Utility
In view of such activity, the compounds of formula (I) are active against a widespectrum of mammalian (including human) tumors and cancerous growths such as
cancers of the oral cavity and pharynx (lip, tongue, mouth, pharynx), esophagus,stomach, small intestine, large intestine, rectum, liver and biliary passages, pan-
30 creas, larynx, lung, bone, connective tissue, skin, colon, breast, cervix uteri, corpusendometrium, ovary, prostate, testis, bladder, kidney and other urinary tissues, eye,
brain and central nervous system, thyroid and other endocrine gland, leukemias
(Iymphocytic, granulocytic, monocytic), Hodgkin's disease, non-Hodgkin's Iym-
phomas, multiple myeloma, etc. Hereir~ the terms utumor", Ucancer" and "cancerous
35 growthsU are used synonymously.
The amount of compound of formula (I) required to be effective as an antitu-
mor agent will, of course, vary with the individual mammal being treated and is ul-
timately at the discretion of the medical or veterinary practitioner. The factors to be
2~ 6~ .
W094/25~6 PCT~S94/0~81
. -18-
considered include the condition being treated, the route of administration, the na-
ture of the formulation, the mammal's body weight, surface area, age and generalcondition, and the particular compound to be administered. However, a suitable
effective antitumor dose is in the range of about 0.1 to about 200 mg/kg body
5 weight per day, preferably in the range of about 1 to about 100 mg/kg per day. The
total daily dose may be given as a single dose, multiple doses, e.g., two to six times
per day, or by intravenous infusion for a selected duration. Dosages above or be-
low the range cited above are within the scope of the present invention and may be
administered to the individual patient if desired and necessary.
For example, for a 75 kg mammal, a dose range would be about 75 to about
7500 mg per day, and a typical dose would be about 800 mg per day. If discrete
multiple doses are indic~te~, treatment might typically be 200 mg of a compound of
formula (I) given 4 times per day.
Formul~tions
Formulations of the present invention, for medical use, comprise an active
compound, i.e., a compound of formula (I), together with an acceptable carrier
therefof and optionally other therapeutically active ingredients. The carrier must be
pharmaceutically acceptable in the sense of being compatible with the other in-
gredients of the formulation and not deleterious to the recipient therefor.
The present invention, therefore, further provides a pharmaceutical formula-
tion comprising a compound of formula (I) together with a pharmaceutically accept-
able carrier thereof.
The formulations include those suitable for oral, rectal or parenteral (including
subcutaneous, intramuscular and intravenous) administration. Preferred are those suitable for oral or parenteral administration.
The formulations may conveniently be presented in unit dosage form and may
be prepared by any of the methods well known in the art of pharmacy. All methodsinclude the step of bringing the active compound into association with a carrierwhich constitutes one or more accessory ingredients. In general, the formulations
are prepared by uniformly and intimately bringing the active compound into asso-ciation with a liquid carrier or a finely divided solid carrier and then, if necessary,
shaping the product into desired unit dosage form.
~WO 94/25466 6~7~81 PCT/US94/04681
-19-
Formulations of the present invention suitable for oral administration may be
presented as discrete units such as capsules, cachets, tablets or lozenges, eachcontaining a predetermined amount of the active compound; as a powder or gran-
ules; or a suspension or solution in an aqueous liquid or non-aqueous liquid, e.g.,
6 a syrup, an elixir, an emulsion or a draught.
A tablet may be made by compression or molding, optionally with one or more
accessory ingredients. Compressed tablets may be prepared by compressing in a
suitable machine the active compound in a free-flowing form, e.g., a powder or
10 granules, optionally mixed with accessory ingredients, e.g., binders, lubricants, in-
ert diluents, surface active or dispersing agents. Molded tablets may be made bymolding in a suitable machine, a mixture of the powdered active compound with
any suitable carrier.
A syrup or suspension may be made by adding the active compound to a
concentrated, aqueous solution of a sugar, e.g., sucrose, to which may also be
added any accessory ingredients. Such accessory ingredient(s) may include flavor-
ing, an agent to retard crystallization of the sugar or an agent to increase the solu-
bility of any other ingredient, e.g., as a polyhydric alcohol, for example, glycerol or
20 sorbitol.
Formulations for rectal or vaginal administration may be presented as a sup-
pository with a conventional carrier, e.g., cocoa butter or Witepsol S55 (trademark
of Dynamite Nobel Chemical, Germany, for a suppository base).
For transdermal administration, the compounds according to the invention
may be formulated as creams, gels, ointments or lotions or as a transdermal patch.
Such compositions may, for example, be formulated with an aqueous or oily base
with the addition of suitable thickening, gelling, emulsifying, stabilizing, dispersing,
30 suspending and/or coloring agents.
Formulations suitable for parenteral administration conveniently comprise a
sterile aqueous preparation of the active compound which is preferably isotonic
with the blood of the recipient. Such formulations suitably comprise a solution or
35 suspension of a pharmaceutically and pharmacologically acceptable acid addition
salt of a compound of the formula (I) that is isotonic with the blood of the recipient.
Thus, such formulations may conveniently contain distilled water, 5% dextrose indistilled water or saline and a pharmaceutically and pharmacologically acceptable
acid addition salt of a compound of the formula (I) that has an appropriate solubility
WO 94/2~466 2~ 681 -20- PCTIUS94/04681~
in these solvents, for example the hydrochloride. Useful formulations also com-
prise concentrated solutions or solids containing the compound of formula (I) which
upon dilution with an appropriate solvent give a solution suitable for parental ad-
ministration above.
In addition to the aforementioned ingredients, the formulations of this inven-
tion may further include one or more optional accessory ingredient(s) utilized in the
art of pharmaceutical formulations, e.g., diluents, buffers, flavoring agents, binders,
surface active agents, thickeners, lubricants, suspending agents, preservatives
10 (including antioxidants) and the like.
EXAM PLES
The following examples illustrate aspects of this invention but should not be
15 construed as limitations. The symbols and conventions used in these examples
are consistent with those used in the contemporary chemical literature, for example,
the Journal of the American Chemical Society. As used here in the term "room
temperature" means about 25C.
GENERAL PRO~:EDURE
A flask is charged with (S)-7-chloromethyl-10,11-ethylenedioxycamptothecin
(25-150 mg, 0.06-0.33 mmol), catalytic sodium iodide (1-15 mg, 99.99% Aldrich),
and anhydrous 1,4-dioxane (2-50 mL). The amine is added (2-5 equiv. neat), and
25 the stirred slurry was heated in an oil bath at 75-95 C. The reaction was moni-
tored for disappearance of starting material by thin layer chromatography. The
mixture is worked up by removing the solvent with a rotary evaporator, and triturat-
ing the residue with ether. The solid that is collected by suction filtration is purified
by silica gel chromatography or reverse phase HPLC (Rainin Dynamax 60A col-
30 umn, eluting with 2% trifluoroacetic acid in water (70-80%) and 4:1 acetonitrile THF
(20-30%), monitoring at 254 nm) to afford the product amine as either the free base
or the trifluoroacetic acid salt respectively.
WO 94/25466 `, 61~i681 PCT/US94/04681
F~ample 1
7-(N-Fthyl-N-4-pyridylmethyl)- ~minomethylene-10. 11 -ethylenedioxy-20(S)-
cam~ ptothecin (Compound 1)
(A) 6'-amino-3',4'-ethylenedioxy-2-chloroacetophenone
A 1-L, three-necked, round-bottomed flask was fitted with a magnetic stirring
bar, thermometer, reflux condenser with calcium chloride filled drying tube, and a
nitrogen inlet. The reaction vessel was charged with dry methylene chloride (10010 ml) and 1,4-benzodioxane-6-amine (15.12 9, 100 mmol). The reaction vessel wascooled to 0 C followed by slow addition of 400 ml of a lM solution of boron
trichloride in methylene chloride while maintaining an internal temperature at or
below 10C. Aluminum chloride (13.34 9, 100 mmol) was added quickly in three
portions followed by addition of chloroacetonitrile (7 ml, 110 mmol). The reaction
15 was stirred for 30 min at 0 C then heated to 40C for 16 hours. The reaction was
removed from heat, allowed to cool to room temperature, then quenched into a
mixture of 1 kg of ice/ 500 ml of lN HCI. The mixture was stirred until no solids
were observed. The methylene chloride layer was removed and the aqueous layer
was extracted twice with methylene chloride. The organic layers were combined,
20 washed with brine, dried over magnesium sulfate, filtered, treated with decolorizing
carbon, filtered through a pad of celite, and concentrated to a solid residue. The
solid was recrystallized from ethyl acetate/hexanes to give 8.3 g (36.5%) of 6'-amino-3',4'-ethylenedioxy-2-chloroacetophenone.
MS (Cl): m/z 228 (M+H+).
1 H NMR (CDCI3):â 7.14 (s, lH), 6.15 (s, 1 H), 4.57 (s, 2H), 4.3 (m, 2H), 4.2 (m, 2H),
1.6 (s, broad, 2H).
Anal. (CloHloclNo3)
calc. found
C 52.76 52.66
H 4.43 4.53
7-Chloromethyl-10,11-ethylenedioxy-20(S)-camptothecin. Into a 250 ml round-
30 bottomed flask equipped with a stirring bar, a reflux condeser and a Dean Stark
WO 94/2~466 2 I 6 1 ~ 8 1 PCT/US94/04681--
-22-
trap were added 1.82 9 (8.0 mmol) of 2'-amino-4',5'-ethylenedioxy-2-
chloroacetophenone, 40 ml dry toluene and 2.0 g (7.6 mmol) of (S) tricyclic keto-
lactone. The reaction was stirred under nitrogen and refluxed for 0.5 hours. Thereaction was allowed to cool followed by addition of 100 mg, (0.~3 mmol) of p-
5 toluenesulfonic acid. The reaction was then heated to reflux for 36 hours. Thereaction was cooled, and the solids were collected by filtration and washed withtoluene followed by thorough washings of the solids by anhydrous ethanol. The
remaining greenish-brown solid was dried under vacum at room temperature
yielding 2.67 9 (77.2%) of > than 97% pure materal.
1H NMR (DMSO-d6): ~ 7.47 (s,1H), 7.32 (s, 1H), 6.98 (s, 1H), 5.15 (s, 1H), 5.06 (s,
1H), 4.97 (s, 1H), 4.19 (s, 4H), 2.23 (s, 2H), 1.6 (m, 2H), 0.68 (m, 3H).
MS M+H = 455.
HRMS: Calc. 455.1009; Found 455.1000.
Anal. (C23H1gN2ClO6) C, H
calc. found
C 60.73 60.70
H 4.21 4.30
(C) 7-(N-Ethyl-N-4-pyridylmethyl)- aminomethylene-10, 11 -ethylenedioxy-20(S)-
camptothecin
A flask is charged with (S)-7-chloromethyl-10,11 -ethylenedioxycamptothecin
(0.60 g,1.3 mmol), catalytic sodium iodide (20 mg, 99.99% Aldrich), and anhydrous
20 1,4-dioxane (120 mL). The 4-(ethylaminomethyl)pyridine (0.54 g, 4.0 mmol) is
added and the stirred slurry was heated in an oil bath at 75-95 C. The reactionwas monitored for disappearance of starting material by thin layer chromatography.
The mixture is worked up by removing the solvent with a rotary evaporator, and
triturating the residue with ether. The solid that is collected by suction filtration is
25 purified by reverse phase HPLC (Rainin Dynamax 60A column, eluting with 2%
trifluoro~cetic acid in water (70-80%) and 4:1 acetonitrile:THF (20-30%), monitoring
at 254 nm) to afford the product amine as the trifluoroacetic acid salt.
Mp 178-180C.
W094125466 ~ 681 PCl/US94/04681
FAB MS MH+ 555.
1H NMR (d6-DMSO) ~ 8.58(d, 2H), 7.83(s, lH), 7.58(d, 2H), 7.52(s, 1H), 7.24(s,
1H), 5.42(s, 2H), 5.31(s, 2H), 4.46(s, 4H), 4.15(br s, 2H), 3.79(br s, 2H), 2.65(m, 2H),
1.86(m, 2H), 1.16(t, 3H), 0.87(t, 3H).
FxamDle 2
7-(4-aminobenzyl)aminomethyl-10.11 -ethylenedioxy-(20S)-camptothecin
(ComDound ~)
A flask is charged with 7-chloromethyl-10,11-ethylenedioxy-(20S)-
camptothecin (252 mg, 0.555 mmol), Nal (42 mg, 0.307 mmol) and anhydrous 1,4-
dioxane (2-50 mL). 4-Aminobenzylamine (192 mL, 207 mg, 1.7 mmol), is added
and the stirred slurry is heated in an oil bath at 75-95 C. The reaction is monitored
for disappearance of starting material by thin layer chromatography. The mixture is
15 worked up by removing the solvent with a rotary evaporator, and triturating the
residue with ether. The crude product is collected by suction filtration and purified
by flash chromatography (eluting with 6:5:1 EtOAc/CHCI3/MeOH) to afford the
amine product which was further purified by recrystallizing from t-butylmethyl ether
to afford 100.6 mg (34% yield) of pure product. The TLC salt was formed by
20 dissolving in 2% aqueous trifluoroacetic acid and Iyophylizing to afford the product
TFA salt (141.4 mg) as a bright yellow solid, mp 280 C (dec).
1H NMR (300 MHz, DMSO-d6): ~ 0.85 (t, 3H, J=7.1), 1.78-1.95 (m, 2H), 4.27 (bs,
1H), 4.45 (s, 2H), 4.65 (bs, 1H), 5.42 (s, 2H), 6.63 (d, 1H, J=8.30), 7.21 (d, 1H,
J=8.30), 7.27 (s, 1 H), 7.63 (s, 1 H), 7.75 (s,1 H), 9.05 (bs, 2H).
25 Low resolution ms (M+1): 541.3 (calcd 541).
Anal. Calcd C30H2gN4O8-2TFA-2.5H2O:
calc. found
C 48.23 48.25
H 4.22 4.18
N 6.43 6.39.
wo 94/2~466 ' -24- PCT/US94/04681
Fxample 3
7-(3-Methoxy-4-hydroxyben7yl)-aminomethylene-10. 11-ethylenedioxy-~O(S)-
5 ~rnptothecin (Compound 3)
A flask is charged with (S)-7-chloromethyl-10,11-ethylenedioxycamptothecin ( 250mg, 0. 549 mmol), catalytic sodium iodide ( 20 mg, 99.99% Aldrich), and anhydrous
1,4-dioxane (25 mL). 4-Hydroxy-3-methoxybenzylamine is added ( 254 mg,1.66
mmol neat), and the stirred slurry was heated in an oil bath at 75-95 C for 18h.
10 The reaction was monitored for disappearance of starting material by thin layer
chromatography. The mixture is worked up by removing the solvent with a rotary
evaporator, and triturating the residue with ether. The solid that is collected by
suction filtration is purified by reverse phase HPLC (Rainin Dynamax 60A column,eluting with 2% trifluoroacetic acid in water (70-80%) and 4:1 acetonitrile THF (20-
15 30%), monitoring at 254 nm) to afford the product amine as the trifluoroacetic acidsalt 214 mg (68%).
MS (FAB ) (M+H) +572.3.
1H NMR (DMSO-d6): o 7.74 (s,1H), 7.63 (s,1H), 7.28 (s,1H), 7.24 (br s,1H), 6.95
(d,1H,j=8Hz), 6.82 (d,1H,j=8Hz), 5.42 (s,2H), 4.68 (br s,1H), 4.46 (br s,2H), 4.35 (br
20 s,1H), 3.78 (s,3H), 1.86 (m,2H), 0.85 (t,2H).
Fxamples 4-~1
The following compounds of formula (I) are prepared by the procedure taught
25 in Scheme 1, in an analogous manner to Example 1 using the appropriate
intermediate compounds of formulas (Il), (Ill), and (V).
4. 7-Benzylaminomethylene-10, 11 -ethylenedioxy-20(S)-camptothecin, (as the
trifluoroacetic acid (TFA) salt)
30 Nominal mass spectrum: MH+526,
mp = >300C (decomposition).
5. 7-(N-tetrahydroisoquinolino)methylene- 10, 11 -ethylenedioxy-20(S)-
camptothecin, (as TFA salt)
35 Nominal mass spectrum: MH+552,
mp = 265-270C.
WO 94/25466 6'~1 PCT/US94/04681
6. 7-Dibenzylaminomethylene-10, 11-ethylenedioxy-20(S)-camptothecin, (as
TFA salt)
Nominal mass spectrum: MH+630,
mp = 180C (decomposition).
7. 7-(N-Methyl)benzylaminomethylene-10, 11-ethylenedioxy-20(S)-
camptothecin, (as TFA salt)
Nominal mass spectrum: MH+540,
mp = 205C (decomposition).
8. 7-Furylaminomethylene-10, 11 -ethylenedioxy-20(S)-camptothecin, (as TFA
salt)
Nominal mass spectrum: MH+516,
mp = 185C (decomposition).
9. 7-(3-Phenylpropyl)-aminomethylene-10, 11-ethylenedioxy-20(S)-
camptothecin, (as TFA salt)
Nominal mass spectrum: MH+554,
mp = 280C (decomposition).
10. 7-(3,4-Dimethoxybenzyl)-aminomethylene-10, 11-ethylenedioxy-20(S)-
camptothecin, (as TFA salt)
Nominal mass spectrum: MH+586,
mp = 190C.
11. 7-(N-Ethyl, (2-chloro-6-fluorobenzyl))-aminomethylene-10, 11-ethylenedioxy-
20(S)-camptothecin, (as TFA salt)
Nominal mass spectrum: MH+606,
mp = 172C (decomposition).
12. 7-(3,4-Difluorobenzyl)-aminomethylene-10, 11-ethylenedioxy-20(S)-
camptothecin, (as TFA salt)
Nominal mass spectrum: MH+562,
mp = 274-277C.
13. 7-Diphenylmethylaminomethylene-10, 11-ethylenedioxy-20(S)-camptothecin,
(as TFA salt)
Nominal mass spectrum: MH+602,
mp = 250C (decomposition).
:2~6i~8~
WO 94/25466 -26- PCT/US94/04681
14. 7-((R) 1-Phenylethyl)-aminomethylene-10, 11-ethylenedioxy-20(S)-
camptothecin, (as TFA salt)
Nominal mass spectrum: MH+540,
5 mp = 265-270C.
15. 7-((S) 1 -Phenylethyl)-aminomethylene-10, 11 -ethylenedioxy-20(S)-
camptothecin, (as TFA salt)
Nominal mass spectrum: MH+552,
10 mp = 265-270C.
16. 7-(2(N-Methyl-2-pyrrol)-ethyl)-aminomethylene-10, 11 -ethylenedioxy-20(S)-
camptothecin, (as TFA salt)
Nominal mass spectrum: MH+552,
15 mp = 160C (decomposition).
17. 7-(N-benzyl-N-(2-Diethylaminoethyl)-aminomethylene-10, 11-ethylenedioxy-
20(S)-camptothecin, (as TFA salt)
Nominal mass spectrum: MH+597,
mp = 170-173C.
18. 7-(2-(4-lmidazolyl)ethyl)-aminomethylene-10, 11-ethylenedioxy-20(S)-
camptothecin, (as TFA salt)
Nominal mass spectrum: MH+530,
mp = 190C.
19. 7-(2-Pyridylmethyl)-aminomethylene-10, 11 -ethylenedioxy-20(S)-
camptothecin, (as TFA salt)
Nominal mass spectrum: MH+527,
30 mp = >300C.
20. 7-Benzylaminomethylene-10, 11-methylenedioxy-20(S)-camptothecin, (as the
hydrochloride salt
Nominal mass spectrum: MH+512,
35 mp = >256C (decomposition).
~N0 94/25466 i 2~
21. 7-(3,4-Dimethoxybenzyl)-aminomethylene-10, 11-methylenedioxy-20(S)-
camptothecin, (as the hydrochloride salt)
Nominal mass spectrum: MH+472,
mp = 225C (decomposition).
Fxample 2
Ph~rmaceutic~l formulations
(A) Transdermal System
In~redients Amount
Active compound 600.0 mg
Silicone fluid 450.0 mg
Colloidal silicone dioxide 25.0 mg
15 The silicone fluid and active compound, i.e., a compound of formula (I), are mixed
together and the colloidal silicone dioxide is reacted with to increase viscosity. The
material is then dosed into a subsequently heat sealed polymeric laminate
comprised of the following: polyester release liner, skin contact adhesive
composed of silicone or acrylic polymers, a control membrane which is a polyolefin
20 (e.g. polyethylene),polyvinyl acetate or polyurethane, and an impermeable backing
membrane made of a polyester multilaminate. The system described is a 10 sq. cm
patch.
(B) Oral Tablet
In~redients Amount
Active compound 200.0 mg
Starch 20.0 mg
Magnesium Stearate 1.0 mg
The active compound and the starch are granulated with water and dried.
30 Magnesium stearate is added to the dried granules and the mixture is thoroughly
blended. The blended mixture is compressed into a tablet.
(C) Suppository
In~redients Amount
Active compound 150.0 mg
Theobromine sodium salicylate 250.0 mg
Witepsol S55 1725.0 mg
The inactive ingredients are mixed and melted. The active compound is then
2 ~
WO 94125466 -28- PCT/US94/0468
distributed in the molten mixture, poured into molds and allowed to cool.
(D) Injection
In~redients Amount
Active Compound 20.0 mg
Buffering Agents q.s.
Propylene glycol 0.4
Water for injection 0.6 mL
10 The active compound and buffering agents are dissolved in the propylene glycol at
about 50C. The water for injection is then added with stirring and the resulting
solution is filtered, filled into an ampule, sealed and sterilized by autoclaving.
(E) Capsule
In~redients Amount
Active Compound 200.0 mg
Lactose 450.0 mg
Magnesium stearate 5.0 mg
20 The finely ground active compound is mixed with the lactose and stearate and
packed into a gelatin capsule.