Note: Descriptions are shown in the official language in which they were submitted.
~10 95102585 ~ PCT/IB94/00062
~1672~7
-1 -
ENANTHIOSELECTIVE PREPARATION OF THIAZOLE DERIVATIYES
Back~round of the Invention
This invention relates to a particularly useful process for preparing the
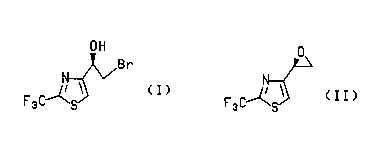
5 compounds of formulas (I) and (Il),
~Br
(I) (II)
in subsl~"lially enantiomerically pure form by enar,lios~l~ctively reducing the prochiral
ketone of formula (Ill)
o
J~Br
F3C ~ ~
~III)
with a borane reducing agent in the plesence of an oxazaborolidine catalyst. Theoptically pure compounds of formulas (I) and (lI) are useful intermeri;-tes in the
synthesis of useful ar,li-liabetic compounds of formula (IV),
-
WO95/02585 '?,~.6~ 2- PcT/Issllono6~
OH H
5F3 Cl~ O~C 0 0 H
( I V )
10which are ~isclosed in U.S. 4,886,814, and which is incorporated herein by reference.
Summarv of the Invention
The pl`~Selll invention provides a process for enat,liosele~ ely prepari~g the
compound of formula (I)
OH
15N~Br
F3C~S~
(I)
in suL sL~nlially enar,lioselectively pure form. The process of this invention CG" ",, i~es
reacting the procl,i,~l ketone of formula (Ill),
o
J~B r
F3C~
( I I I )
V0 95102585 ~ 2 ~ 7 PCT/IB94/00062
with a borane reducing agent in the presence of a chiral oxazaborolidine catalyst of the
- formula (~/),
R2 R3
)~
`B' 7
~1
(V~
wherein R' is hydrogen, (C,-C8)alkyl, benzyl, heterocyclyl or phenyl optionally
substituted independently with up to three (C1-C8)alkyl, (C1-C8)alkoxy or halo groups;
R2 and R3 are syn, are taken separately, and are each independeully (C1-C8)alkyl,
benzyl, heterocyclyl or phenyl optionally sl~hstitl~ted with up to three (C1-C8)alkyl, (C1-
5 C8)alkoxy or halo groups, provided that when R2 is CH3 and R3 is phenyl, R' is H, orwith a chiral oxazaborolidine catalyst of the formula (Vl),
)
~\
H ~B~0
1 1
(VI )
25 wherein R' is as defined above and D is a cis-fused 4-6 membered c~rbo",oi)ocyclic
ring oplioll&lly substituted indepel,derllly with up to three (C1-C8)alkyl, heterocyclyl or
phenyl o,,lio, lal!y sl Ihstit~ Iteci independer,lly with up to three (C1-C8)alkyl, (C,-C8)alkoxy
or halo groups; a cis-fused 6-9 membered carLobicyclic system optionally substituted
i"dep~ndently with up to three (C,-C8)alkyl, heterocyclyl or phenyl o,,lionally substituted
30 indeper,de, Illy with up to three (C,-C8)alkyl, (C,-C8)alkoxy or halo groups; or a cis-fused
WO 95/0258~ PCT/1~394/0006~
5 system hrlving the structure ~ wherein R~ and R' are each independently
H, (C,-C8)alkyl, (C,-C8)alkoxy or halo, in a reaction inert solvent under a reaction inert
,osphere.
A pr~r~r,ed process of this invention is the above process wherein said
oxazaborolidine catalyst is
~"~A~ C",H"3 ~
H N~B~o o r `B'
H H
(VII) (VIII)
A particularly pr~"ed pl~cess within the pr~fe,.ed process is the process
wherein said reaction inert solvent is tetrahydrofuran, dioxane, diethyl ether, toluene or
25 ber,~ene; said reaction inert atmospher~ is nitrogen and said borane reducing agent
is borane methylsulfide complex.
A more particularly prefer,ed process within the particularly pr~:~"~:d process
is the process wherein said chiral oxazaborolidine catalyst is ,ur~par~d in situ prior to
the ad.litiGn of said procl~i,al ketone of formula (Ill).
~O 95/02585 ~ 6 72~ ~ PCT/IBg4/00062
The present invention also provides a process for enantioselectively prepari"g
the compound of the formula (Il),
r 5 /~
F3 C~S~
(II)
which co" ,pri~es prepari"g the compound of formula (1) as described l~erei. ,above and
reacting said compound of formula (I) with a base to form the compound of formula (Il)
in suL,sldnlidlly er,ar,liG",erically pure form.
Detailed Descli~,lion of the Invention
The presenl invention provides a process for preparing the optically active
compounds of formulas (I) and (Il) hereinabove in subslar,lidlly enarl~iGmerically pure
form. The scheme for this process is shown in Scheme 1, below.
W O 95/0258~ PCT~B94/0006
-6-
chiral 1,2-disubstituted + borane reducing ( I I I )
aminoethanol agent
O OH
)~Br Borane reduc i ng agen t N--A~Br
F3C--~ c h i ral c atal y~ t F3C--~S~
(III) (I)
0
Ba~e ,~
1~
F3C--~S~
(II)
SCHEME I
The process of this invention is readily carried out. The con,pound of formula
(I) is prepared in suL,~I~r,lially enantiomerically pure form via the reduction of the
prochiral ketone, 4-bromoacetyl-2-trifluoromethylthia_ole (Ill).
In the reduction process of this invention, the precursor to the chiral
30 oxA~Ahor~,lidi"e catalyst, in the form of a chiral 1 ,2-dis~ ~hstituted aminoethanol
derivative, is dissolved in a reaction inert solvent under a reaction inert atmosphere at
ambient temperature. The chiral 1,2-~isl~hstih~ted amino-ethanol derivative can be
chosen from among any of the 1 ,2-disl~hstih~ted aminoethanol derivatives which give
rise to the chiral oxazaborolidine catalysts which are used in the process of this
vo gs/02s85 21~ 728 7 PCT/IB94/00062
. ~ . ~
invention. However, pr~er,ed 1,2-disubstituted amino-ethanol derivatives are (lS, 2R)-
(+)-2-amino-1,2-diphenylethanol and (lS, 2R)-(+)-r~orephedrine. The reaction inert
solvents which are particularly pr~fer,ed include but are not limited to dioxane,
5 tetrahydrofuran, diethyl ether, toluene and benzene. More particularly pr~fened
solvents are tetrahydrofuran and toluene. A suitable borane reducing agent is added
to the reaction mixture and the reaction mixture is left at amL i~nt temperature for 2 to
24 hours. The borane reducing agent may be selectecl from borane methylsulfide
complex and borane tetrahydrofuran complex, but most prerer"ad is borane
10 methylsulfide complex. Generally the chiral oxazaborolidine catalyst will have formed
within 10-16 hours.
After the chiral oxazaborolidine catalyst has formed, the prochiral ketone of
formula (Ill) is added to the reac~ion mixture at ambient ter"perdlure. The reaction
reducing the ketone to the alcohol is yener~lly colrrleti within 10-15 minutes after
15 addition is cor, Ir'~te. I lo~Jer, occasionally a longer amount of time may be required
to ensure complete reaction depending upon a variety of factors including the particular
solvents chosen or amounts of materials used and so on. The reaction mixture is then
cooled, generally to about 0C, and quenched by the careful addition of a protonsource, generally methanol. The compound of formula (I) is isolated accGI~Jing to the
20 slandard methods of organic chelr,i~,y.
Altematively, the reduction process of this invention can be carried out by
reacting a prochiral ketone of the formula R4R5Co, wherein R4 and R5 are definedhereinbelow with a borane reducing agent in the presence of a chiral oxazaborolidine
catalyst accor.li.,g to formula (\/) or formula (\/I). Said process results in the
25 enantioselective reduction of said prochiral ketone, such that only one of two possible
alcohol enantiomers is formed in pl~fer~nce to the corlespond;ng enantiomer. Thedegree of enantio sele~,ti~rity which is obtained will vary depenJi"g upon the size of the
R4 and R5 groups attached to the carbonyl group forming the prochiral ketone. When
the R~ and Rs groups are similar in size, the degree of enanlioselection will be lower.
30 As the R4 and R5 groups become i"cleasi"yly ~ parate in size, the degree of enantio-
selection will be greater. However, it should be under~lood that the size of the R4 and
R5 groups is not the sole deler~"i"i"g factor affecting the degree of enantioselectivity
achieved. Oldi.,arily, with prochiral ketones wherein R4 and R5 are at least moderately
WO 95/02~85 ~ PCT/IB94/0006
-8-
dillerer,l in size, at least 90% of the desired enarlLio",er will be obtained. However,
typically greater than 90% of the desired enantiomer is Gbta;"ed
The prochiral ketone is dissolved in a suitable reaction inert solvent such as
5 toluene, diethyl ether, dioxane, tetrahydrofuran or the like. r, ~t " ed is tetrahydrofuran.
A catalytically effective amount of a chiral oxazaborolidine compound of formula (V) or
formula ~/I) is added to the reaction mixture at from about -78C to about room
ter"peraLure, ~l~r~lerably at room temperature; howevcr, the pl~er.ed temperature will
vary depending upon the particular borane reducing agent being used. The pr~lel.ed
10 amount of said catalyst is about 5-10 mole % with r espect to said ketone. The reaction
mixture is then treated slowly with about 4.2 hydride equivalents of a borane reducing
agent such as borane dimethylsulfide complex, borane tetrahydrofuran complex,
catecholborane or the like. When the prochiral ketone cor,Lui,,s an R4 or R5 group
which bears a borane-coordinating functionality, addiLiol,al hydride equivalents of
15 reducing agent are necess~y. Generally pr~far,ed for its ease of use is borane
dimethylsulfide complex. Generally the reducing agent is added at a rate which
mod~ t~s the rate of the catalytic reduction. The ~eacLiGn is sometillles cGr, ~!~ e as
soon as all of the reducing agent has been added, as can be determined by monit~"i"g
the course of the reaction via thin layer c:hr~,l"alography according to the sLand.~.d
20 pra~,~Lice of organic ch~ l, y. I low_vcr, occasionally it will be desi, al~le to allow the
reaction mixture to stir for longer periods of time such as overnight, or to heat the
Fea~,1iGn mixture to temperatures of up to 40C to 65C in order to ensure completion
of the reaction. Additionally, with some sub~L,ales and reducing agents, it may be
n ecessA~ y to stir the reaction mixture at -78C for a lengthy period of time such as 16
25 hours. Ordinarily the reaction mixture is stirred at about room temper~lure for about
fifteen minutes. The ter"pe,aLure of reactiGI- mixture is then ~lju~ted to 0C and
quenched with a proton source. Said proton source, usually a lower alkanol such as
",~Lt,anol, is added slowly to control the exothermic rea~LiGn. The product is isGl~ d
by removing the solvent in vacuo fcl'~wed by pa,liliol,i.,g between an ~g~,ic solvent
30 andan~qlleousacidf~llcvl~dbysepar~LionoflayersandplJ,ilicaLionaccordingtothe
~Lal.dar:l tecl,n, les of or~anic chelni~LIy.
The compound of formula (Il) of this plocess is also readily prepared. The
compound of formula (I) is di~solved in aqueous base and vigorously stirred. The,ur~,,ed base is sodium hydroxide, however other bases such as potassium hydroxide
~0 9S/0~585 , ,, ~7
and potassium t-butoxide may also be utilized. The debr-""il,ation and cyclization of
the compound of formula (I) to the epoxide of formula (Il) is ~r~uled rapidly and without
racer"i~alion of the chiral center. Generally the reaction is CG~ lele within 5 to 10
5 minutes, however the reaction may require longer periods depending upon a variety of
factors including sl.el,ylll of base, nature of base, amount of ~"~lle,ials used and so on.
After the epoxide is formed, the epoxide is isolaled from the re&ctiGI~ mixture utilizing
well-known methods of organic cher"i~l~y.
The prochiral ketone starting mcl~:,ial for the process of this reaction is prepared
by the method disclosed in U.S. Patent 4,886,814. The utility of these compounds as
intermediates in the process for the preparclion of the antidiabetic cGi"pound of
formula (IV) is also described therein.
The chiral 1 ,2-~isl ~hstihlted aminoethanol derivatives are gener_"y readily
available from commercial sources such as Aldrich or Sigma. Where the chiral 1,2-
15 ~iSI ~hstit~ ~ted aminoethanol derivative is not readily a\~ 1 e, said erythro amir,oeth&r,olderivatives are pl~pared by methods well known to those of ordinary skill in the art,
such as provided by Reetz et al., Angew. Chemie Int. Ed. Eng., 26, 1987, 114143 and
Matsunaga et al., T~t-cll)e.l~on Letters, 32, 1991, 7715-18.
The fcllo/,l:.,g terms and ph,ases, when used herein and in the appendant
20 claims, are defined as follows:
1. "Alkyl" means a br~nched or unbranched saturated hydrocarbon group
containing the specified number of carbon atoms, e.g., C,-C8. Examples include, but
are not limited to methyl, ethyl, isopropyl, n-butyl, t-butyl and the like.
2. "Alkoxy" means a br~nched or ~" Ibran.~hed saturated hydrocarbon
25 containing the specified number of carbon atoms and a single oxygen atom by which
said hydrocarbon is ~,llL~ched to a central backL,ol-e. Exa",r'es include, but are not
limited to methoxy, ethoxy and the like.
3. H~lel~cyclylR means a 5- or 6-membered aro",~lic group containing up
to three heteroatoms, each of said h~lel~loms selected from N, 0 and S and which30 may be optionally benzo-fused, said heterocyclyl group being optionally s~hstihlted
independently with up to three (C,-C8)alkyl, (C,-C8)alkoxy or halo groups.
4. A '`,~rochi.~l ketone", denoted by R~R5Co, is a ketone in which R4 and R5
are non-identical, so that the secondary alcohol recl~ction product R~R5CHoH has a
chiral center at the alcohol carbon. For cyclic pl~Jchi.~l ketones, it is u"der-~lood that
WO 95/02585 c~ PCT/IB94/0006~
-10-
R4 and R5 are taken together, forming a ring including the ketone, and that the ring so
formed has no plane of symmetry across a plane drawn perpendicular to the plane
CGI llni. Iil l9 the carbonyl group and the two carbon atoms allached directly thereto, said
5 plane containing both the carbon and oxygen atoms of the carbonyl group as points
therein.
7. Reaction inert solvent means a solvent which does not interact with the
reaclnl lls, il llt:r, I ,e~ s or products in such a way that adversely affects the yield of the
desired products.
8. "Syn" means that the s~hstitllents sllhstitllted on adjacent ring carbon
atoms are located on the same side of a plane which enco"~p~cses the bond between
said carbon atoms and the bonds by which each of said carbon atoms are attached
to the ring.
9. ~n6riliGIneric excess", or e.e., is the excess of one of two ent~l,lio",~
over the other, usually ex~ ssed as a percentage, i.e., a 90% e.e. reflects the presence
of 95% of one enar,liol"er and 5% of the other in the ",~l~rial in question.
10. ~Ambient tel "peral.lre" means the temperature of the immediate eAI~ al
environment surrounding the reaction flask. This tel"pelalure is usually room
temperature (20-25C).
11. In situ is the reaction cGr,dit;on v~;,erei., the chiral oxa~aborvlidines of
formula (\/) or formula (\/I) of the invention are formed from the precursor aminc~'c ~ hol
and borane. The prochiral ketone is added after the o.Y~ hor~ '.dine is generated. The
chiral o.~ horolidines of the invention are not isol-~ed under these cor,diliol ,s.
12. "ne&~,tion inert al",osphere" means a gas which does not illlernct with
25 the r~actn"l~ r,nel l; '.es or products in such a way that adversely affects th2 yield
of the desired products.
The presel,l invention is illustrated by the f~ v.;.lg eAtll~l; 18s. I low_vcr, it
should be understood that the invention is not limited to th~ spe~ c details of these
examples. All reactions are conducted under an inert ~I",osphe,t:, such as nil,ogen
30 or argon, unless otherwise specified. All solvents are anhydrous, i.e., contain such a
small amount of water that said water does not interact with the r eage, ll~, intermediates
or products in such a way that adversely affects the yield of the desired products.
Where used herein, '~HF" means tetrahydrofuran.
0 95/02585 PCT/IB94/00062
~?1 6 7~8 7
Example One
(S)-Oxiranvl-2-trifluorol "~thylthiazole.
A. (S)-4-(2-Bromo-1 -hydroxyethyl)-2-trifluorol "ethyllhia~cl~. To atetrahydrofuran
(55mL) solution of (1S, 2R)-(+)-2-amino-1,2-diphenylethanol (586mg, 2.75mmol, 0.05
equivalents based on bromoketone) under a nitrogen atmosphere was added neat
borane methylsulfide complex (Aldrich, ~lOM, 7.70mL, 77mmol, 1.4 equivalents based
on bromoketone) all at once and allowed to stand 16hrs. (EIMS) M+ 223.1159 (Calcd
223.1168). Blu~oketone lll, (15.0g, 54.7mmol, 1.0 equivalent) in tetrahydrofuran(28mL) was added dropwise over 1 hr at 25C. Fifteen minutes after the addiliGn was
complete the starting ketone was consumed, the raa 1ion was cooled to 0C, quenched
with r"~tl,anol (55mL), and stirred overnight. The solvents were removed under
vacuum, toluene (140mL) was added to the residue, and the organic phase was
washed successively with pH4 phosphate buffer (140mL), water (140mL) and treated15 with magnesium sulfate. Eiltlalion and removal of the solvent under vacuum a~urded
the chiral alcohol as a pale yellow oil (13.58g, 90% mass balance, 94%ee).
B. (S)-Oxiranyl-2-trifluoromethylll _~le. A~ueo~-s sodium hydroxide (4N,
12.52mL) was added to the neat oil (13.42g) obl~i"ed ih A. above with vigorous stirring.
After 9 min. methylene chloride (lOOmL) and water (lOOmL) were added, the phases20 were sep~ ~led, and the ~queous phase washed with water (3 x 100mL) and dried with
magnesium sulfate. rill~alion and removal of the solvent under vacuum ar~u,~Jed the
crude chiral epoxide as an off white liquid, 9.35g. Distillation of 8.83g of the crude
product at 4mmHg/boiling point 42~4C yielded 6.99g (70% overall from the
bromoketone) of the chiral epoxide as a cc!orless oil.
Example Two
(S)-4-(2-Bromo-1 -hvdroxvethyl)-2-trifluoromethylll ,' - le .
To a tetrahydrofuran (lOmL) solution of (lS, 2R)-(+)-nGrephedli"e (76mg,
0.5mmol) under a nil~oyen al",osphere was added neat borane methylsulfide complex
(1 OM,1.4mL,14mmole) at ambient temperature (20-25C) and the reaction was allowed
to stand for 16 hrs. Bromoketone lll, (2.74g, 10mmol) in tetrahydrofuran (5mL) was
added dlùpJl:se over 1 hr, the reaction stirred for 15 min after the addition was
cGm,~le~ts, then cooled to 0C and quenched with ",~tl,anol (lOmL). The quenchedI ~a~,1ion was stirred for 18hr, the solvents were removed under vacuum and methylene
chloride (25mL) was added. The Glyanic phase was washed s~ccessiv01y with pH4
phospl)ate buffer (25mL), water (25mL), and dried with rllayllesium sulfate. After
WO 95/02585 PCT/IB94/0006~
~6'~ 12-
filtering, the solvent was removed under vacuum to afford the (S) alcohol 2.24g as a
yellow oil (83%, 84%ee).
Example Three
(Sl-Oxiranyl-2-trifluoro" ,~lh~,lll, ~ - le
A. (S)-4-(2-Bromo-1 -hydroxyethyl)-2-trifluor~,l "~thylll ,i~zoie. Reduction intoluene
and cycli~lion to the chiral epoxicle To a toluene (41 mL) solution of (1 S, 2R)-2-amino
1,2-diphenyl~tl,anol (438mg, 2mmol, 0.05 equivalents based on bromol~etone) under
a nitrogen atmosphere was added neat borane methylsulfide cc mr\le: (Aldrich, ~lOM,
5.80mL, 58mmol, 1.4 equivalents based on Lru,,,oketone) all at once and allowed to
stand 16 hrs. Blu~ol~etone ((Ill), 11.319, 41mmol, 1.0 equivalent) in toluene (21mL)
was added dl opv.~; ~ over 1 hr at 25C. Fifteen minutes after the addiliol, was co" ,, iet
the ~ .li"g ketone was consumed, the rea~,1ion was cooled to 0C, quenched with
methanol (41mL), and stirred overnight. The solvents were removed under vacuum,
toluene (100mL) added to the residue, the organic phase washed successively withueous sulfuric acid (1 M, 50mL), water (100mL), and treated with magnesium sulfate.
Removal of the solvent under vacuum a~urded the chiral alcohol as a pale yellow oil
(10.849, 95% mass balance, 90%ee).
B. (S)-Oxiranyl-2-trifluoromethyl lhi~ol~. Aq~eo~s sodium hyd~ùxic-ie (4N,
10.53mL) was added to the neat oil (10.84g) with vigorous stirring. After 10 min.,
toluene (84mL) and water (84mL) were added, the phases separ~led, the ~ql~eous
phase washed with water (3 X 84mL) and dried with magnesium sulfate. Removai of
the solvent under vacuum &~orded the crude chiral epoxide as a off orange liquid,
6.509. Distillation of 6.079 of the crude product at 4mmHg/boiling point 42-44Cyielded 4.659 (61% overall from the bromoketone) of the chiral epoxide as a ~colerless
oil.