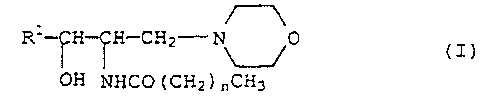Note: Descriptions are shown in the official language in which they were submitted.
- 1 -
FP-2097 (PCT}
SPECIFICATION
AGENT FOR CURING NEURONAL DISEASES
(TECHNICAL FIELD)
This invention relates to an agent for curing neuronal
diseases, which comprises a substance for accelerating
biosynthesis of glycosphingolipids as an effective ingredi-
ent .
(BACKGROUND}
It has been known that glycosphingolipids (hereinafter
referred to as GSL) exist as a constitutional component of
cell surface membranes of mammal cells and they are closely
related to a cellular function such as generation, growth,
differentiation, transformation, immunoreaction, etc.
through a receptor function of a physiologically active
substance, an intercellular mutual recognition function,
intercellular interaction, etc.
Among them, ganglioside is GSL containing sialic acid,
and it is said that it has activity to recoveries from
injury of peripheral nerves and a disorder of central
nerves, i.e., acceleration of regeneration of nerves and a
process of neurotransmission. Heretofore, effectiveness of
exogenous ganglioside to various neurotic disease models
has been investigated. A medicine named CronassialR has
already been put on the market in Italy as a medicine uti-
lizing this, and a patent application pertinent thereto was
filed (Japanese Provisional Patent Publication No.
34912/1977). However, there have been observed clinical
cases that an anti-ganglioside antibody is generated
because of administration of this medicine, i.e., the
exogenous ganglioside, whereby various neurotic symptoms
are caused. For example, there have been reported amyo-
trophic lateral sclerosis in which an anti-GM2 antibody is
generated (Lancet, 337, 1109-1110, 1991) and Guillain-Barre
syndrome in which. an anti-~Ml antibody (_Lancet, 338, 757,1991) .
-2- 21677
At present, a means for searching a function of
ganglioside which has been used most frequently is a
means of a type in which ganglioside is added to an
experiment system from outside. In that case, a relation
to endogenous ganglioside becomes a problem. That is, it
is considered that a result obtained by further adding
ganglioside to a system in which endogenous ganglioside
existing in cell membrane has already formed a complex
with various cell surface receptors, etc. does not always
reflect actual cytophysiological significance of
endogenous ganglioside. Thus, in order to know an
inherent role of ganglioside in cytophysiology, a method
of specifically changing biosynthesis of endogenous GSL
is required. The present inventors have previously
synthesized 1-phenyl-decanoylamino-3-morpholino-1-
propanol (PDMP) which is an analogue of ceramide and
proved that D-threo-PDMP specifically inhibits a
glucosylceramide biosynthesizing enzyme and extremely
decreases an intracellular content of all GSL using
glucosyl-ceramide as a starting substance (J. Lipid.
Res., vol. 28, 565-571, 1987) Further, it has been
reported that a GSL content is lowered by D-threo-PDMP,
whereby extension of neurites is suppressed (J. Biochem.,
110, 96-103, 1991).
On the other hand, we have found that L-threo PDMP
which is an optical enantiomer of D-threo PDMP has
possibility of accelerating biosynthesis of GSL (J. Cell.
Physiol., 141, 573-583 (1989)). However, whether or not
L-threo-PDMP increases an endogenous ganglioside level in
neurocytes and whether or not increase of endogenous
ganglioside activates a function of neurocytes are
unknown issues and have not been investigated.
An object of an aspect of the present invention is
to provide an agent for curing various diseases caused by
21 6~7 7
_3_
disorders of central nervous system and peripheral
nervous system, using a medicine which accelerates
biosynthesis of endogenous CSL in neurocytes,
particularly ganglioside.
(SUMMARY OF THE INVENTION)
The present inventors have studied variously in
order to develop an agent for curing neuronal diseases
based on a new mechanism and consequently found that a
specific 2-acylaminopropanol derivative accelerates
biosynthesis of GSL and significantly accelerates neurite
extension and synapse formation, to accomplish the
present invention.
According to an aspect of the invention, there is
provided, an agent for curing neuronal diseases caused by
disorders of peripheral nervous system or central nervous
system, which comprises a 2-acylaminopropanol derivative
represented by the formula ( I )
Rl-~~-I~H---CH2- ~0
OH ~NHCC7 (CH2) ~,CHg
[wherein Rl represents a phenyl group or cyclohexyl group
each of which may be substituted by 1 to 3 same or
different substituents selected from the group consisting
of alkyl, alkoxy, hydroxy and nitro, or represents an
alkyl group, and n represents an integer of 0 to 16] or a
pharmaceutically acceptable salt thereof as an effective
ingredient.
Also, the present invention relates to a method of
curing neuronal diseases, which comprises administering
the 2-acylaminopropanol derivative represented by the
above formula (I) or a pharmaceutically acceptable salt
,.
1 7 7
-4-
thereof in an effective amount for accelerating
biosynthesis of glycosphingolipids, accelerating neurite
extension and/or accelerating synapse formation to
mammals which suffer from neuronal diseases caused by
disorders of peripheral nervous system or central nervous
system.
According to another aspect of the invention, there
is provided, use of a medical composition which comprises
a 2-acylaminopropanol derivative represented by the
formula (I)
RI ~Fi--~Fi CH2-- N ,0 ( ~ )
~H NHG~ (CHZ) ~CH~3
[wherein R1 represents a phenyl group or cyclohexyl group
each of which maybe substituted by 1 to 3 same or
different substituents selected from the group consisting
of alkyl, alkoxy, hydroxy and nitro, or represents an
alkyl group, and n represents an integer of 0 to 16], a
pharmaceutically acceptable salt thereof or both of them,
for preparing an agent for curing neuronal diseases
caused by disorders of peripheral nervous system or
central nervous system.
According to another aspect of the invention, there
is provided, a liposome preparation composition for
curing neuronal diseases, which comprises at least a 2-
acylaminopropanol derivative represented by the formula
CA 02168877 2005-03-23
- 4 a-
R1~H~H~CH2., ~0 ( I )
0H NHC~ (CHZ) nCH3
[wherein R1 represents a phenyl group or cyclohexyl group
each of which may be substituted by 1 to 3 same or
different substituents selected from the group consisting
of alkyl, alkoxy, hydroxy and nitro, or represents an
alkyl group, and n represents an integer of 0 to 16] or a
pharmaceutically acceptable salt thereof and a
phospholipid.
According to a further aspect of the invention,
there is provided, use, as an agent for treating neuronal
diseases of a liposome preparation composition comprising
at least one 2-acylaminopropanol derivative of the
formula
R1~~H~~H~CH2~ ~C ( I )
OH NHCCf (CH2) nCHg
[wherein R1 represents a phenyl group or cyclohexyl group
each of which may be substituted by 1 to 3 same or
different substituents selected from the group consisting
of alkyl, alkoxy, hydroxy and nitro, or represents an
alkyl group, and n represents an integer of 0 to 16],
said derivative has efficacy for accelerating
biosynthesis of glycosphingolipids, accelerating neurite
extension and/or accelerating synapse formation, or a
pharmaceutically acceptable salt thereof and a
phospholipid.
-4b-
(DETAILED DESCRIPTION OF THE
PREFERRED EMBODIMENTS OF THE INVENTION)
In the following, the present invention is explained
in detail.
In the above formula (I), the carbon number of alkyl
or alkoxy as a substituent of the phenyl group or
cyclohexyl group of R1 is preferably 1 to 4. The carbon
number of the alkyl group of Rl is preferably 6 to 15,
most preferably 10 to 15. The alkyl group is exemplified
by hexyl, heptyl, octyl, nonyl, decyl, undecyl, dodecyl
(lauryl), tridecyl, tetradecyl (myristolyl), pentadecyl,
etc. n is an integer of 0 to 16, preferably an integer of
4 to 16, most preferably 6 to 12.
Among the compounds represented by the above formula
(I), a particularly preferred one is 1-phenyl-2-
acylamino-3-morpholino-1-propanol in which n is 6 to 12,
and a most preferred one is 1-phenyl-2-decanoylamino-3-
morpholino-1-propanol (hereinafter referred to as PDMP).
In the compound of the present invention, stereoisomers
exist, and either of the stereoisomers can be used. Also,
a mixture of isomers such as a racemic mixture, etc. can
be used. There may be specifically mentioned a D-threo
isomer (1R, 2R), a L-threo isomer (1S, 2S), a DL-threo
isomer, a D-erythro isomer (1S, 2R), a L-erythro isomer
(1R, 2S) and a DL-erythro isomer. The L-threo isomer (1S,
2S) is particularly preferred from the point that it has
a glycolipid biosynthesis-accelerating effect.
The compound represented by the above formula (I) is
a known substance (U. S. Patent No. 5,041,441 and Japanese
.aAw:
~~:
- 5 -
Provisional Patent Publication No. 254623/1989) and can be
synthesized by, for example, the following method described
in J. Lipid. Res., 28, 565-571, (1987) and J. Biochem.,
111, 191-196, ( 1992 ) .
CH20, HN O
R1-CO-~g2 ~ R1-CO-~ H-CH2-
NHCO (CHZ ) nCH3 NHCO (CHZ ) nCH3
NaBH4 Rl-CH-CH-CHZ- ~0
OH NHCO ( CHZ ) nCH3
A mixture of the resulting 4 isomers can be separated
by fractional crystallization using chloroform/ether to
obtain a DL-threo isomer and a DL-erythro isomer, respec-
tively. Further, the DL-threo isomer can be also crystal-
lized as a salt of dibenzoyl-D-tartaric acid or dibenzoyl-
L-tartaric acid to obtain a D-threo isomer or a L-threo
isomer, respectively.
As a pharmaceutically acceptable salt of the compound
represented by the above formula (I), there may be men-
tioned a salt of an inorganic acid such as hydrochloric
acid, phosphoric acid, sulfuric acid, nitric acid, formic
acid, etc.; and a salt of an organic acid such as acetic
acid, citric acid, lactic acid, malic acid, oxalic acid,
malefic acid, fumaric acid, succinic acid, trifluoroacetic
acid, methanesulfonic acid, p-toluenesulfonic acid, etc.
By administering an effective amount of the 2-acyl-
aminopropanol derivative represented by the formula (I) or
a pharmaceutically acceptable salt thereof to mammals
including human which suffer from neuronal diseases caused
by disorders of peripheral nervous system or central
nervous system, said animals can be treated. As a repre-
sentative disease, there may be mentioned various central
nervous system diseases which are expected to be cured by
regenerating nerve fibers, for example apoplexy, cerebral
- 6 -
infarction, cerebral hemorrhage, cerebral injury, dysm-
nesia, senile dementia, Alzheimer's disease, parkinsonism,
etc.; and various peripheral nervous system diseases, for
example, polyneuropathy caused by cacochymia, mechanical
neuropathy, toxic neuropathy, etc.
Pharmaceutical preparation
A pharmaceutical preparation to be administered orally
or parenterally can be obtained by using the compound of
the present invention with a carrier, an excipient, a dilu-
ent and other additives. Further, the compound of the
present invention can pass a blood-brain barrier (J. Lipid
Res., 32, 713-722 (1991)) so that effectiveness to cerebral
neuronal diseases as an injection and an oral agent can be
expected. In particular, a liposome preparation having a
fine size such as lipid nanosphere, etc. or a lipid emul-
sion preparation on which the compound of the present
invention is carried can pass a blood-brain barrier by
about 10 times as compared with a physiological saline
solution so that it is effective when the agent of the
present invention is used for curing cerebral neuronal
diseases.
As an oral preparation, there may be mentioned a solid
preparation such as a powder, a granule, a capsule, a
tablet, etc.; and a liquid preparation such as a syrup, an
elixir, an emulsion, etc.
The powder can be obtained by, for example, mixing
with an excipient such as lactose, starch, crystalline
cellulose, calcium lactate, calcium hydrogen phosphate,
magnesium aluminometasilicate, silicic acid anhydride, etc.
The granule can be obtained by adding the above excipient
and, if necessary, for example, a binder such as saccha-
rose, hydroxypropyl cellulose, polyvinylpyrrolidone, etc.
or a disintegrator such as carboxymethyl cellulose, calcium
carboxymethyl cellulose, etc. and granulating the mixture
by a wet method or a dry method. The tablet can be obtain-
ed by tableting the above pocader or granule as such or with
a lubricant such as magnesium stearate, talc, etc. Fur-
ther, the above tablet or granule can be made an enteric or
sustained action preparation by covering it with an enteric
base such as hydroxypropylmethyl cellulose phthalate, a
methyl methacrylate copolymer, etc. or covering it with
ethyl cellulose, carnauba wax, hardened oil, etc. A hard
capsule can be obtained by filling a hard capsule with the
above powder or granule. Further, a soft capsule can be
obtained by dissolving the compound of the present inven-
tion in glycerin, polyethylene glycol, sesame oil, olive
oil, etc. and covering the mixture with a gelatin film.
The syrup can be obtained by dissolving a sweetener such as
saccharose, sorbitol, glycerin, etc. and the compound
represented by the above formula or a salt thereof in
water. In addition to the sweetener and water, essential
oil, ethanol, etc. may be added to prepare an elixir, or
gum arabic, tragacanth, polysorbate 80, sodium carboxy-
methyl cellulose, etc. may be added to prepare an emulsion
or a suspension. Further, a corrective, a coloring agent,
a preservative, etc. may be. added to these liquid prepara-
tions, if necessary.
As a parenteral preparation, there may be mentioned an
injection, an intrarectal administration agent, a pessary,
an endermic agent, an inhalant, an aerosol, an ophthalmic
agent, etc.
The injection can be obtained by adding a pH-adjusting
agent such as hydrochloric acid, sodium hydroxide, lactic
acid, sodium lactate, sodium monohydrogen phosphate, sodium
dihydrogen phosphate, etc.; an isotonizing agent such as
sodium chloride, glucose, etc.; and distilled water for
injection to the compound of the present invention, steri-
lizing and filtering the mixture and then filling an
ampoule, etc. with the mixture. Further, an injection
which is dissolved when it is used can be obtained by
adding mannitol, dextrin, cyclodextrin, gelatin, etc. and
lyophilizing the mixture under vacuum. Also, an emulsion
- g _
for injection can be made by adding an emulsifier such as
lecithin, polysorbate 80, polyoxyethylene hardened castor
oil, etc. to the compound of the present invention and then
emulsifying the mixture in water.
Further, as an injection, there may be mentioned a
liposome preparation which enables improvements of solubil-
ity and a transition rate to a target organ. In particu-
lar, nanosphere liposome (lipid nanosphere) can not only
heighten a concentration in blood without being taken into
reticuloendothelial tissues and lower a minimum effective
dose required for exhibiting a pharmaceutical effect, but
also pass a blood-brain barrier easily so that it is
suitable when it is used for curing cerebral neuronal
diseases. The liposome preparation can be prepared
according to a known liposome preparation method (C. G.
,Knight, Ziposomes: From Physical Structure to Therapeutic
Applications, pp. 51-82, Elsevier, Amsterdam, 1981; Proc.
Nat,. Acad. Sci., USA, 75, 4194, 1978).
That is, as an amphipathic substance forming a lipo
some membrane, there may be used a phospholipid such as a
natural phospholipid (yolk lecithin, soybean lecithin,
sphingomyelin, phosphatidylserine, phosphatidylglycerol,
phosphatidylinositol, diphosphatidylglycerol, phospha-
tidylethanolamine, sphingomyelin,- cardiolipin, etc.), a
synthetic phospholipid (distearoyl phosphatidylcholine,
dipalmitoyl phosphatidylcholine, dipalmitoyl phosphatidyl-
ethanolamine, etc.) and others. Further, in order to
improve membrane stability, fluidity and membrane perme-
ability of the medicine, there may be added known various
additives such as cholesterols (cholesterol, ergosterol,
phytosterol, sitosterol, stigmasterol, etc.), a substance
which is known to impart negative charge to liposome
(phosphatidic acid, dicetyl phosphate, etc.), a substance
which is known to impart positive charge (stearylamine and
stearylamine acetate), an antioxidant (tocopherol, etc.),
- 9 -
an oily substance (soybean oil, cottonseed oil, sesame oil,
cod-liver oil, etc.) and others.
Preparation of liposome can be carried out by, for
example, the following method. The above amphipathic sub-
s stance and additives, and the compound of the present
invention are dissolved in an organic solvent (a single
solvent such as chloroform, dichloromethane, ethanol,
methanol, hexane, etc. or a mixed solvent thereof), respec-
tively, both solutions are mixed, the organic solvent is
removed in a vessel such as a flask, etc. in the presence
of an inert gas (a nitrogen gas, an argon gas, etc.), and a
thin membrane is attached to a vessel wall. Then, this
thin membrane is added to a suitable aqueous medium (phys-
iological saline, a buffer, a phosphate buffered physio-
logical saline, etc.), and the mixture was stirred by a
stirrer. In order to obtain liposome having a small parti-
cle size, the mixture was further dispersed by using an
ultrasonic emulsifier, a pressurization type emulsifier, a
French press cell pulverizer, etc. As described above,
preparation of liposome proceeds by treating, with a mem-
brane filter, a liquid in which the amphipathic substance,
etc. required for preparation of liposome and the compound
of the present invention are dispersed in the aqueous
medium to obtain nanosphere liposome (lipid nanosphere; a
particle size of about 25 to 50 nm) in which a particle
size distribution is controlled. Further, liposome may be
subjected to fractionation treatment such as ultrafiltra-
tion, centrifugation, gel filtration, etc. to remove the
medicine which is not carried.
Further, by making the compound of the present inven-
tion to be carried on liposome having, on a membrane
thereof, a glucose residue, a tyrosine residue, a mannose
residue or sulfatide obtained by adding ~-octylglucoside,
L-tyrosin-7-amido-4-methylcoumarin, phenylaminomannoside or
sulfatide as a membrane-forming substance in addition to
the above amphipathic substance and additives, the liposome
- 10 -
can be made to permeate a blood-brain barrier easily (as to
a method itself, see Japanese Provisional Patent Publica-
tion No. 69332/1992).
The intrarectal administration agent can be obtained
by adding a base for a suppository such as mono-, di- or
triglyceride of cacao aliphatic acid, polyethylene glycol,
etc. to the compound of the present invention, then melting
the mixture by heating, pouring it into a mold and cooling
it, or dissolving the compound of the present invention in
polyethylene glycol, soybean oil, etc. and then covering
the mixture with a gelatin film.
The endermic agent can be obtained by adding white
petrolatum, beeswax, liquid paraffin, polyethylene glycol,
etc. to the compound of the present invention, heating the
mixture, if necessary, and kneading it. A tape agent can
be obtained by kneading the compound of the present inven-
tion with an adhesive such as rosin, an alkyl acrylate
polymer, etc. and spreading the mixture on non-woven fab-
ric, etc. The inhalation can be obtained by, for example,
dissolving or dispersing the compound of the present inven-
tion in a propellant such as a pharmaceutically acceptable
inert gas, etc. and filling a pressure container with the
mixture.
Administration method
The administration method of a medicine containing the
compound of the present invention as an effective ingredi-
ent is not particularly limited, but when it is used for
curing neuronal diseases caused by disorders of central
nervous system, an intramuscular injection, an intravenous
injection, a hypodermic injection or an intraperitoneal
injection is preferred. Particularly when it is used for
curing cerebral neuronal diseases, a method of injecting
the liposome preparation or the lipid emulsion preparation.
Dose
The dose may be suitably determined depending on
administration method, age, health condition, weight, etc.
- 11 -
of a patient, but it is generally 0.25 to 200 mg/kg,
preferably 0.5 to 100 mg/kg by one dose or divided doses
per day.
Acute toxicity
A solution of L-threo-PDMP hydrochloride dissolved in
a nonionic surfactant (Myrj 52) was administered intraperi-
toneally to ICR mice (male, 6 weeks old, weight: about 28
to 30 g). The ZD~p value was 350 mg/kg.
A solution of DL-threo-PDMP hydrochloride or DL-ery-
thro-PDMP hydrochloride dissolved in DMSO (125 mg/ml) was
administered intraperitoneally to ICR mice (male, 8 weeks
old, weight: about 38 to 40 g). The LDSp value of DL-
threo-PDMP was about 250 mg/kg, and that of DL-erythro-PDMP
was about 700 mg/kg.
General toxicity
Solutions of L-threo-PDMP hydrochloride and DL-threo-
PDMP hydrochloride dissolved in a nonionic surfactant (Myrj
52), respectively, were administered (intraperitoneally) at
a rate of 100 mg/kg/day (calculated on the above PDMP
hydrochloride) to ICR mice continuously for 10 days. As to
either of the compounds, neither decrease in weight nor
decrease in neutrophile, acidocyte, etc. due to suppression
of bone marrow was observed, and as a result of observation
for 3 months, no abnormality was observed.
{BRIEF DESCRIPTION OF THE DRAWINGS)
Fig. 1 is a graph showing glycolipid biosynthesis-
accelerating actions of L-threo-PDMP and analogues thereof
having different aryl chain lengths.
Fig. 2 is phase contrast microphotographs of biologi-
cal morphologies showing a neurite extension-accelerating
effect of L-threo-PDMP on primary cultured rat cerebral
neurocytes (solid lines in the photographs show scales of
the cells, and the total length shows 100 nm).
A: control
B: S~.M L-threo-PDMP added
C: 20~.M L-threo-PDMP added
~~g~~$
- 12 -
D: 40~M L-threo-PDMP added
Fig. 3 is a graph showing an accelerating effect of L-
threo-PDMP on neurite extension of primary cultured rat
cerebral neurocytes.
Fig. 4 is a graph showing influences of L-threo-PDMP
and D-threo PDMP on frequency (o) of fluctuation of intra-
cellular Ca2+, reflecting the number of synapse formation
of primary cultured rat cerebral neurocytes.
(BEST MODE FOR PRACTICING THE INVENTION)
In the following, Examples of the present invention
are shown, but the present invention is not limited
thereby. All of D-threo-PDMP or L-threo PDMP used the fol-
lowing Examples are hydrochlorides, but the same results
can be also obtained when other pharmaceutically acceptable
salts are used.
Example 1
Rat fetuses at the 18th day of pregnancy were taken
out, and forebrain base portions of the fetuses were
extracted aseptically and cut into small pieces by scis-
sors. After the pieces were treated with papain (in a
phosphate buffer containing 0.02 o L-cysteine containing
180U papain, 0.02 o bovine serum albumin and 0.5 o glucose,
pH 7.4, at 37 °C for 30 minutes), they were washed with a
mixed solution (DF medium) of 1 . 1 of a Dulbecco modified
Eagle's medium and a Ham's F12 medium and suspended in a DF
medium containing 5 o bovine fetal serum and 5 o equine
serum. This neurocyte suspension was charged into a cul-
ture plate with 24 wells (Falcon, Primaria) in an amount of
1 x 106 cells/well. Subsequently, 20~M D-threo-PDMP or L-
threo-PDMP was added, and the primary culture of the neuro-
cytes was carried out for 3 days. After completion of the
culture, the cells in the respective wells were washed with
a phosphate buffer and recovered by rubber policeman. Fur-
ther, a ganglioside fraction was extracted by adding
chloroform/methanol/water (1 . 2 . 0.8) and shaking the
mixture for 5 minutes. This ganglioside fraction was dis-
- 13 -
solved in 50 ~.1 of water, and the amount of ganglioside-
binding sialic acid was measured by the method of Hara et
al. (Anal. Biochem. 179, 162-166, 1989) to determine a
ganglioside content. The no addition group (control) which
was defined as 100 o was compared with the test groups, and
the results~are shown in Table 1.
Table 1 Influences of D-threo-PDMP and L-threo-PDMP
on ganglioside content of primary cultured
rat cerebral neurocytes
Ganqlioside content
Control 100 0
D-threo-PDMP 63.4 0
L-threo-PDMP 127.1 0
In the treatment with 20~.M D-threo-PDMP, as expected,
decrease in the ganglioside level based on inhibition of
the glucosylceramide biosynthesizing enzyme was clearly
observed. On the other hand, in the neurocytes treated
with 20~.M L-threo-PDMP for 3 days, the ganglioside level
was increased by about 30 o as compared with that of the
control. Thus, it was found that L-threo-PDMP has a GSL
biosynthesis-accelerating effect on rat cerebral neurocytes
and elevates a level of endogenous ganglioside.
Example 2
B16 melanoma cells derived from neuroectoderm were
charged into a culture plate with 12 wells in an amount of
1 x 105 cells/well and cultured for 24 hours in a Dulbecco
modified Eagle's medium containing 10 o bovine fetal serum.
Thereafter, the medium was exchanged with the same culture
solution containing Su.M L-threo-PDMP or analogues thereof
in which acyl chain lengths are changed, and 3H-galactose
was added. After 24 hours, the medium was removed, the
cells were washed with 1 ml of a phosphate buffer contain-
ing 0.1 o EDTA, 1 ml of 0.25 o trypsin was added, and the
mixture was left to stand at 37 °C for 5 minutes. When the
- 14 -
cells were recovered, non-labeled B15 melanoma cells (1.4 x
10~ cells) were added, the mixture was washed with a phos-
phate buffer, and then a mass of the cells was obtained. 4
ml of methanol and chloroform were added successively to
the mass of the cells to extract all GSL, followed by
evaporation to dryness. The residue was subjected to
alkali hydrolysis with chloroform . methanol (1 . 1) con-
taining 0.2N sodium hydroxide, neutralized with acetic acid
and then evaporated to dryness. The product evaporated to
dryness was dissolved in water and then desalted with a
Sep-Pak cartridge C18 (Waters). The adsorbed GSL was dis-
solved out by 1 ml of methanol and 4 ml of chloroform .
methanol (1 . 1) and then evaporated to dryness under a
nitrogen stream. Next, the GSL was purified by a conven-
tional method using acetylation by pyridine-acetic anhydr-
ide, Florisil column and deacetylation (J. Lipid Res., 12,
257-259, 1971), then desalted and evaporated to dryness.
This purified GSL fraction was dissolved in 50 ~.1 of
chloroform . methanol (1 . 1), and 40 ~l of the solution
was applied to silica gel plate (TLC) and developed with
chloroform . methanol . H20 (60 . 35 . 8). Thereafter, the
positions of glucosylceramide, lactosylceramide and gangli-
oside GM3 were confirmed by iodine coloration, they were
scratched off from TLC; respectively, and radioactivities
thereof were measured by a liquid scintillation counter.
The results are shown in Fig. 1. As can be clearly seen
from Fig. 1, it was found that among L-threo-PDMP and the
analogues thereof in which acyl chain lengths are changed,
the compounds of the formula (I) in which n is 6, 8, 10 and
12 significantly accelerate biosynthesis of ganglioside.
Example 3
Hippocampi of rat brains at the 0th day after birth
were extracted aseptically, subjected to enzyme treatment
with papain in the same manner as in Example 1 and then
washed with a DF medium. Neurocytes were sowed in a plate
with 96 wells which had been covered with poly-L-lysine, at
~ .. a '~ "~
- 15 -
a density of 2 x 104 cells/cm2, and at the same time, 5).!.M,
20~.M and 40~M L-threo-PDMP were added, respectively. After
culture for 24 hours, the neurocytes were fixed by para-
formaldehyde, and a degree of neurite extension was
recorded by microphotographs and then observed. As can be
clearly seen from the microphotographs of Fig. 2, L-threo-
PDMP exhibits a significant neurite-extending action in the
primary culture of the rat cerebral neurocytes.
Further, as shown in Fig. 3, when the lengths of the
neurites were measured by using four photographs of the
respective test groups and statistical processing was con-
ducted, it was apparent that L-threo-PDMP accelerates neu-
rite extension with a significant different of P c 0.005
over a wide concentration range (1 to 40 ~M).
Thus, from Examples 1 to 3, it was strongly suggested
that L-threo-PDMP of the present invention and the ana-
logues thereof in which acyl chain lengths are changed have
remarkable neurite extension-accelerating actions, i.e.,
differentiation-accelerating actions to neurocytes by an
action of increasing an endogenous ganglioside level of the
neurocytes.
Example 4
(1) The present inventors have recently advocated a
"tracing circuit" model that a dynamic change of synapse
binding occurs even in human mature brain, and selective
removal of synapse outside a circuit and increase in the
number of synapse binding of synapse within the circuit are
a mechanism of human long-term memory {Kuroda Y., Neuro-
chem. Intern., 14, 309-319, 1989).
Further, as a method of measuring synapse formation
relatively simply and easily, there has been developed a
multipoint observation system of Ca2+ in neurocytes using
Fura-2, and it has been confirmed that the number of
synapse formation is in direct proportion to frequency of
intracellular Ca2+ fluctuation in synchronization therewith
{Br. J. Pharmacol., 89, 191-198, 1986; Neurosci. Lett., 78,
- 16 -
69-74, 1987). We investigated influences of L-threo-PDMP
and D-threo-PDMP exerted on synapse formation between rat
cerebral cortex neurocytes by using this method, and the
results are shown below.
Neurocytes of cerebral cortexes of rat fetuses at the
18th to 19th day of pregnancy were isolated by enzyme
treatment with papain and cultured in the same manner as in
Example 1. By adding L-threo-PDMP or D-threo-PDMP and mea-
suring frequency of intracellular Ca2+ fluctuation from the
first day of the culture, a degree of synapse formation was
examined. The results are shown in Fig. 4, and increase in
the number of synapse formation depending on a dose by L-
threo-PDMP was clearly observed. Thus, L-threo-PDMP not
only has an action of accelerating extension of neurites,
but also accelerates formation of synapse so that possibil-
ity of being effective for curing various neuronal diseases
was strongly suggested. Further, D-threo-PDMP having a GSL
biosynthesis-inhibiting action suppressedly acted on
synapse formation so that it was found that endogenous GSL,
particularly ganglioside fulfills an important function in
the process of synapse formation.
(2) By using cerebral cortexes of rat fetuses at the 15th
day of pregnancy similarly as in (1), explant culture was
carried out by the following method to confirm the effect
of the agent for curing neuronal diseases of the present
invention.
That is, according to the method described in J. Neu-
rochem., 61, 2155-2163 (1993), the above cerebral cortexes
were cut into small pieces, and when culture of tissues
including cerebral cortex neurons was conducted by cultur-
ing the pieces in a medium containing no serum, as groups
to which the medicine was administered, L-threo-PDMP or D-
threo-PDMP was added to the medium so that the concentra-
tion became 5 to 20E1.M, and the mixtures were cultured for 2
days. The effect of neurite extention by administering the
medicine after the culture was judged by measuring the num-
- 17 -
ber of neurocytes having neurites existing in the explants
(the cultured tissues).
As a result, the number of the above neurocytes in the
group to which 10~.M L-threo-PDMP was administered was about
one and half times of that of the group to which no medi
cine was administered. On the other hand, in the group to
which 10~.t,M D-threo-PDMP was administered, the neurite
extension-accelerating effect observed in the case of L-
threo-PDMP was not observed.
As described above, even in the tissue culture of
cerebral cortexes, effectiveness of L-threo-PDMP to neurite
extension was exhibited so that it was strongly suggested
that by exhibiting the similar action in vivo when the
agent for curing neuronal diseases of the present invention
was administered to mammals including human, it is effec-
tive for curing neuronal diseases accompanied with a
regressive change of cerebral cortex neurons such as
Alzheimer's disease, etc.
Example 5
Preparation example of capsule
100 mg of L-threo-PDMP hydrochloride, 150 mg of potato
starch, 50 mg of light silicic acid anhydride, 10 mg of
magnesium stearate and 765 mg of lactose were mixed
uniformly and 200 mg of this mixture was apportioned and
charged into a hard capsule.
Preparation example of tablet
100 mg of L-threo-PDMP hydrochloride, 670 mg of lac-
tose, 150 mg of potato starch, 60 mg of crystalline cellu-
lose and 50 mg of light silicic acid anhydride were mixed
and to the mixture was added a solution of 30 mg of
hydroxypropyl cellulose dissolved in methanol (10 o by
weight of hydroxypropyl cellulose). The mixture was
kneaded and then granulated. Next, the granules were
extruded through a screen with a size of 0.8 mm to obtain
fine granules. After the granules were dried, 15 mg of
- 18 -
magnesium stearate was added thereto and each 200 mg of the
mixture was tableted.
Preparation example of inj-ection
Propylene glycol was added to 100 mg of L-threo-PDMP
hydrochloride so that the total amount was 10 ml to dis-
solve L-threo-PDMP hydrochloride. After this solution was
sterilized and filtered, each 0.2 ml was apportioned to
ampoules and the ampoules were sealed.
In the respective preparation examples, 100 mg of L-
erythro-PDMP hydrochloride was used in place of L-threo-
PDMP hydrochloride to obtain the respective preparations of
capsules, tablets and injections.
Example 6
Liposome~re.paration and kinetic in vivo
As shown in Examples 3 and 4, L-threo-PDMP exhibits an
action to central nervous system tissues (cerebral cortex
and around hippocampus of brain) so that it is required
that it permeates a blood-brain barrier and elevates a con-
centration in brain by intravenous administration. L-
threo-PDMP is a fat-soluble substance, and its solubility
in physiological saline is maximally 0.5 mg/ml so that when
it is solubilized by using a surfactant in order to enable
intravenous administration, it is taken into reticuloendo-
thelial tissues before reaching a target organ, whereby it
cannot be expected to obtain an effective pharmaceutically
effective concentration at an action site.
In view of the above situations, the present inventors
have studied a method of elevating transition from blood to
central nervous tissues (particularly cerebral cortex and
around hippocampus of brain) and consequently found that
the above problem can be solved by incorporating L-threo-
PDMP into nanosphere liposome as shown in the following
experiment.
That is, they have found that by incorporating L-
threo-PDMP into nanosphere liposome and intravenously
administering the mixture to rats, an effective concentra-
- 19 -
tion for exhibiting a pharmaceutical effect in brain can be
maintained.
(1) Preparation of liposome preparation
A chloroform solution containing phosphatidylcholine
(18 N.mol), phosphatidylserine (3 ~mol) and cholesterol (9
~imol ) and a /chloroform . methanol ( 2 . 1 ) solution of
[ 14C ] -L-threo-PDMP ( 0 . 5 mg, 5 . 5 ~,Ci ) were mixed, and the
mixture was condensed and evaporated to dryness in a flask
under a nitrogen stream. 1 ml of physiological saline was
added thereto, and the mixture was stirred, subjected to
ultrasonic treatment for 10 minutes and then passed through
a membrane filter (CORNING Disposable Sterile Syringe
Filter, 25 mm, 0.2 ~,1,, Cellulose Acetate Membrane) to pre-
pare a nanosphere liposome liquid containing L-threo-PDMP.
The method itself of preparing the nanosphere liposome is a
known method (see Japan Medicine Society, the 114th Annual
Meeting, Summaries of Lectures 4, p. 32, Subject No. 13-127
(1994) ) .
(2) Kinetics in vivo
The liposome solution (4.54 ~LCi) of [14C]-L-threo-PDMP
prepared in the method of the above (1) and a physiological
saline solution (4.54 ~.Ci) of 0.5 mg/ml of [14C]-L-threo-
PDMP were intravenously administered to blister strain male
rats (10 weeds old) from femoral arteries over 30 seconds,
respectively, and blood was collected with a lapse of time.
From blood samples, L-threo-PDMP was quantitated according
to a conventional method (J. Lipid. Res., vol. 32, 713-722,
1991). As a result of examining a change of a concentra-
tion in blood with a lapse of time, the results which could
be analyzed by a typical 2-compartment model were obtained.
By a simplex method of non-linear least square method pro-
gram MULTI, pharmaceutically kinetic parameters were calcu-
lated. As a result, the half-times of disappear were 2.56
minutes (liposome) and 2.77 minutes (physiological saline)
in the first phase and 25.2 minutes (liposome) and 27.5
minutes (physiological saline) in the second phase.
- 20 -
From the above results, it was observed that the area
under the concentration time curve of the medicine in blood
(AUC; area under the concentration curve) is increased by
slightly less than 9 times by preparation of liposome, and
the residual property of L-threo-PDMP in blood is elevated
by preparation of liposome. As a result of determining, by
simulation, a time when the peripheral compartment concen-
tration of the 2-compartment model was maximum, it was 10
minutes so that it was estimated that the concentration in
brain is also maximized in 10 minutes. Therefore, the
concentration in brain and the concentration in blood were
measured at 10 minutes and 60 minutes when an equilibrium
state of the compartment was established. Also the concen-
tration in brain, L-threo-PDMP was quantitated according to
a conventional method (J. Lipid. Res., vol. 32, 713-722,
1991).. The results are shown in Table 2.
Table 2 Concentration in blood and concentration
in brain after intravenously administering
L-threo-PDMP to rats
Liposome Physiological
saline solution
10 min 60 min 10 min 60 min
Concentration 1789.1 170.5 451
in 2 118
0
blood (pmol/ml) .
.
Concentration
in
25.7 1.0 2.5 ND
brain ().1,M)
Blood/brain con- 0,0144 0.0060 0
0056 ND
centration ratio .
From the results of Table 2, it became apparent that
by preparation of liposome, the concentration in brain is
also elevated by about 10 times as compared with admini-
stration of the physiological saline solution.
As a result of carrying out simulation of a continuous
intravenous injection (intravenous injection) of a periph-
eral compartment by using the ratio of concentration in
brain/concentration in blood in a steady state (60 minutes)
- 21 -
of liposome administration, it became apparent that it is
possible to reach within a 1 % limit of the concentration
in brain in a steady state by the continuous intravenous
injection for 4 hours.
From an in vitro experiment, an pharmaceutical effect
has been observed by culture at 25~.M for 24 hours in B16
melanoma cells derived from neuroectoderm, culture at 40~.1.M
for 8 hours in culture of neurocytes of cerebral cortexes
of rat fetuses and in the range of 5 to 20~.M in culture of
cerebral cortex pieces of rat fetuses. By applying these
conditions to an in vivo experiment, simulation was carried
out. The results obtained by calculating, by simulation, a
rate and a liquid amount of continuous intravenous injec-
tion required for reaching a predetermined steady concen-
tration in brain are shown in Table 3.
- 22 -
m
~ ~r o ~
W uo co0
o ~ cn ~ o
O ~
-G-y 00 M r-I
~ ~
-1 ~ N tn
r ~-1
f~S ~..
* O
M N N N
'd '-i S-1O tf)Wit'~~
.ri
V~
-r-I O f~W -Iu~
O .t-)
-,-1
,~.InN r-1O
-r-i
U ra t~ ~ U
tn
U
-n
G p I~M ~ M ~1->
-~ ~ N r1 ~ Wit' cn
tn +.) ~ crt~ N r1
Q ~
~ -' c-~O O O
~ -n
~-i
~
~ ~ ~ ctS U
-O O
~ r1
+~
rti ~ cn
-rl bi ~
_
O ~ O O O O O
~
U O O -~ -
O
~ -r1 \ O O O O
cn
V-I U ~ O O V~N -r1
cn
O ~ ~t O N r-iV'N
N
ftS .~ ~ N LO ~1'C
C'.
N ~-I ~ M M ~ -~I
f-~
-r1
+~ .1-1 ~ ~ M rl
r-I
td ~
4-1
rti
U' -'-1
O ~
U
~
O ~ W
O o M O o
U ~
U t~N ~!'N G
" '1J '~ ~ N r-iCOc~' O
~
4-1
O ~ ~ ~ ~i'N O O
(d
N
v N !LS
-t~ ~
.1-5
cd N
O
cd -~ ~ S
i
- O O N ~ U
td ~ lflM r-iO
~
~
-~-I " O O O O U
-r-I ''
'~ '~
~ ~' a~
U
r-I ~ ~ ~ N LO O tl~
S-I
~ l~ M ~-I cN
'CS -r-i ~ '~O O O O
~-I O
~
O O O O O oho
O
N U ctf
~ ~
~4-~ S-I O O O O O
O N
.J~ O \ O O V'N
-.-I r-1N ~-iN ~
U ~
U N O O ~ a0 ~ f~- -t->
.7-~ ~ t~~ u7C~ -rl
c~ -n
,
M ~d ~ t~00 M r-i 3
G -~I
c~ O ~i
-r-I
r-i
N b~
G
G
c~i O N
H -~ ~ U
~
-~i cd rd
U
Sa ~ ~ o m o m
cn
N .-i
..
N S-I *
'~
U .Q *
cd
U
O
~
U -~ *
cn
~1~~~'~'~
- 23 -
From the results of the above simulation, in the phys-
iological saline solution, it is 39.956 m1/28 hrs in the
case of the steady concentration in brain being 50~.M and
19.964 m1/28 hrs in the case of 25~.M so that it can be seen
that the liquid amount of continuous intravenous injection
is too large relative to the total rat humor amount. On
the other hand, in the liposome solution, it is 4.256 m1/28
hrs in the case of the steady concentration in brain being
50~.M and 2.128 m1/28 hrs in the case of 25~M so that it can
be said that this liquid amount is a liquid amount within a
physiological range to be administrated.
From the above results, it is apparent that by con-
verting L-threo-PDMP into liposome, an effective pharmaceu-
tical effect is exhibited even to mammals including human
by intravenously injecting a suitable liquid amount contin-
uously.
(UTILIZABILITY IN INDUSTRY)
The agent for curing neuronal diseases of the present
invention accelerates neurite extension and synapse forma
tion by elevating biosynthesis of endogenous GSLs of neuro
cytes, particularly ganglioside, whereby it is effective
for curing various diseases of central nervous system and
peripheral nervous system. It is effective for various
central nervous system diseases which are expected to be
cured by regenerating nerve fibers, for example, cerebral
apoplexy, cerebral infarction, cerebral hemorrhage, cere-
bral injury, dysmnesia, senile dementia, Alzheimer's dis-
ease, parkinsonism, etc. Also, it is effective for various
peripheral nervous system diseases, for example, polyneuro-
pathy caused by cacochymia, mechanical neuropathy, toxic
neuropathy, etc.
The agent for curing neuronal diseases of the present
invention can pass a blood-brain barrier (J. Lipid Res.,
32, 713-722 (1991)) so that it is effective for cerebral
neuronal diseases as an injection or an oral agent. In
particular, nanosphere liposome (lipid nanosphere) on which
- 24 -
the agent for curing neuronal diseases of the present
invention is carried can not only heighten a concentration
in blood without being taken into reticuloendothelial tis-
sues and lower a minimum effective dose required for
exhibiting a pharmaceutical effect, but also pass a blood-
brain barrier by about 10 times as compared with a physio-
logical saline solution so that it is extremely effective
when the agent for curing neuronal diseases of the present
invention is used for curing cerebral neuronal diseases.