Note: Descriptions are shown in the official language in which they were submitted.
WO 95/06479 PCT/NL94/00208
METHOD TO REDUCE MYOCARDIAL INJURY DURING ACUTE MYOCARDIAL
INFARCTION
Rield of the Invention
This invention is in the field of immunology/cardiology/
biochemistry, and describes more particularly a method to reduce
myocardial cell injury during acute myocardial infarction.
Background Of the Invention
Mechanical failure of heart muscle is one of the leading
causes of in-hospital deaths in patients with acute myocardial
infarction (AMI). Friedberg C.K., 1968, Circulation 39 suppl.IV:
252. An important determinant in the development of such failure
is the amount of necrotic tissue in the jeopardized myocardium.
Page D.L. et al., 1971, New Encx J Med 285: 133. Experimental
studies in animals have shown that irreversible myocardial cell
injury starts about 30 minutes after occlusion of coronary
vessels and proceeds for hours. Maroko P.R. et al., 1973, Ann
Jnt Med 7~: 720. However, even interventions given as late as
6 hours after c~~_onary occlusion are able to reduce infarction
size after AMI .by about 35~ compared to untreated control
animals. Libby r', et al., 1973, J Clin Invest ~: 599. Electro-
cardiographic studies indicate that also in humans a substantial
amount of myocardial tissue may not become irreversibly injured
until hours or even days after occlusion of the coronary vessel,
i.e. at a time that most patients will have been admitted to a
hospital. Reid P.R. et al., 1974, New Engl J Med ~Q: 123. A
number of experimental and clinical studies have attempted to
minimize infarction size by reducing the myocardial necrosis
that occurs in the later stages of AMI. Maroko P.R. et al.,
1973, Ann Int Med 7~: 720.
The later phase of myocardial cell injury likely results
from an ensuing acute inflammatory reaction characterized by
infiltration of neutrophilic granulocytes (neutrophils). Entman
WO 95/06479 ~ PCT/NL94/00208
2
M.L. et al., 1991, FASEB J ~: 2529. Initially, the importance of
an inflammatory reaction in mediating myocardial cell injury
during AMI was recognized in animal studies which showed that
corticosteroids could reduce infarction size by 20 to 35~. Libby
P. et al., 1973, J Clin Invest ~: 599; Maclean D. et al., 1978,
J Clin Invest ~: 541. However, clinical application of methyl-
prednisolone in AMI to minimize myocardial necrosis, was not
successful mainly because this treatment interfered with scar
formation and healing, leading in some patients to the develop-
ment of aneurysm and rupture of the ventricle wall. Roberts R.
et al., 1976, Circulation 53 Suppl-II: 204. A similar effect has
been observed in long-term experiments in rats. Maclean D. et
al., 1978, J Clin Invest ~: 541. These disappointing results
tuned down further clinical studies that aimed at reducing
infarction size by attenuating the inflammatory reaction
following AMI.
The inflammatory reaction which occurs in the course of AMI
comprises some important events: the local production of chemo-
tactic factors, the infiltration and activation of neutrophils,
the local production of cytokines (such as tumor necrosis
factor-a and interleukin-6) to enhance adherence of neutrophils
to cardiac myocytes, and the local activation of the complement
system. Entman M.L. et al., 1991, FASEB J ~: 2529.
A role of complement activation in AMI was initially
suggested by Hill and Ward who provided evidence that complement
activation products generated in the infarcted myocardium were
responsible for the infiltration of neutrophils. Hill J.H. et
al., 1970, J Ex~ ~: 885. Later studies showed that plasma
levels of activated complement components are increased in
patients with AMI and that several complement components become
localized in the infarcted area during the course of AMI, as has
been demonstrated both in animals as well as in patients.
Pinckard R.N. et al., 1975, J Clin Invest ~: 740; Langlois P.F.
et al., 1988, Atherosclerosis ~: 95; Yasuda M. et al., 1990,
Circulation $~: 156; Pinckard R.N. et al., 1980, J Clin Invest
1050; McManus L.M. et al., 1983, Lab Invest ~$: 436; Schafer
H. et al., 1986, J Immunol ~7: 1945; Hugo F. et al., 1990, ~n
Ex8 Immunol $~: 132.
WO 95/06479 PCT/NL94/00208
3
Furthermore, a number of studies have demonstrated that
complement activation products such as the anaphylatoxins and
TCC may have deleterious effects on the myocardium by mechanisms
dependent and independent of neutrophils, such as the local
productian of thromboxane A2 and peptidoleukotrienes LTC4 and
LTD4, the release of histamine, plasmalemmal disruption and the
activatian of neutrophils in the coronary circulation with
subsequent plugging of capillary vessels, formation of toxic
oxygen radicals and the release of proteolytic enzymes. These
mechanisms may lead to vasoconstriction, impaired micro-
circulation, an increase in coronary perfusion pressure, and
result in ischaemia, contractile failure of the myocardium,
tachycardia and impairment of atrioventricular conduction. Del
Balzo U. et al., 1984, Proc Natl Acad Sci USA $~: 886; Martin
S.E. et al., 1988, Circ Res ~: 483; Ito B.R. et al., 1989, it
Res ,~: 1220; Del Bazzo U. et al., 1989, Circ Res ~: 847; Ito
B.R. et al., 1990, Circ Res ~: 596; Stahl G.L. et al., 1990,
Circ Res ~: 1103; Engler R.L. et al., 1991, FASEB J ,~: 2983;
Homeister J.W. et al., 1992, Circ Res ,Z,~: 303.
The molecular mechanism of the observed activation of
complement during AMI is not clear, although released
mitochondrial constituents, presumably membranes, have been
frequently claimed to induce the activation. Pinckard R.N. et
al., 1973, J Immunol 110: 1376; Pinckard R.N. et al., 1975,
Clin Invest ~: 740; Giclas P.C. et al., 1979, J Immunol 122:
146; Storrs S.B. et al., 1981, J Biol Chem 256: 10924; Rossen
R.D. et al., 1988, Circ Res ~: 572; Kagiyama A. et al., 1989,
Circ Res ~: 607. However, it should be noted that most of these
studies dealt with activation of complement following
reperfusion of ischaemic myocardium rather then that following
ischaemia due to permanent occlusion of coronary vessels. The
deleterious effects of complement activation products on the
myocardium have been substantiated by observations that in
animal models complement depletion prior to or shortly after
permanent occlusion of a coronary vessel significantly reduces
the amount of myocardial necrosis. Maroko P.R. et al., 1978,
Clin Invest ~: 661; Maclean D. et al., 1978, J Clin Invest ~1:
541; Pinckard R.N. et al., 1980, J Clin Invest ~: 1050;
WO 95/06479 . ' PCTIIVL94/00208
~~ ~ 4
Crawford H.R. et al., 1988, Circulation 7$: 1449. None of the
studies investigating the effect of complement inhibition on
myocardial damage after permanent occlusion of a coronary vessel
have used a true inhibitor of the complement cascade, all these ,
studies were done with an agent, i.e. Cobra Venom Factor, that
intravascularly activates and depletes the system. However, this "
method of complement inhibition/depletion cannot be used in
clinical situations considering the inherent dangers of
intravascular complement activation such as the development of
adult respiratory distress. Goldstein IM, 1992, In: Gallin JI,
Goldstein IM, Snyderman R (eds): Inflammation: Basic Principles
and Clinical Correlates, New York, Raven Press, p.63; Craddock
P.R. et al., 1977, New Enq J Med 296: 769; Stimler N.P. et al.,
1980, Am J Pathol 100: 327; Hosea S.F. et al., 1980, J Clin
Invest ,~: 375; Ward P.A. et al., 1985, ~,Clin Invest 7~: 517.
The discussion above deals with activation of the
complement system during permanent occlusion of coronary
vessels. However, it is now generally accepted to treat patients
with AMI with thrombolytic therapy or coronary angioplasty to
reperfuse the jeopardized myocardium. The sooner after the
occlusion this therapy is given, the more effective it is to
salvage the ischaemic myocardium. Clinical studies indicate that
more than half of the effect of thrombolytic therapy is lost
when treatment is delayed 60-75 minutes. Hermens WTh et al.,
1992, Lancet ~Q: 1297. However, more than 90~ of the patients
with AMI do not reach the hospital within 75 minutes after the
occlusion of a coronary artery and, therefore, will hardly
benefit from thrombolytic therapy.
There is ample evidence that reperfusion of ischaemic
myocardium itself may induce an inflammatory reaction, also
known as ischaemic-reperfusion injury, which is caused by
activation of complement and neutrophils and the generation of
oxygen radicals. Rossen R.D. et al., 1985, Circ Res ~: 119; '
Rossen R.D. et al., 1988, Circ Res ~: 572; Dreyer W.J., 1989,
Circ Res ~5: 1751; Dreyer W.J. et al., 1992, sirs Res 71: 1518; "
Zucchesi BR et al., 1989, J Mol Cell Cardiol ~: 1271; Engler R
et al., 1989, Circulation 7~: 1137. This ischaemic-reperfusion
injury may damage the cardiac tissue and limit the beneficial
WO 95/06479 PCT/NI94/00208
,
effects of a restored circulation. Herdson PB et al., 1965, A~
Pathol ~: 367. Reperfusion therapy in AMI can, therefore, be
regarded as a "double edged sword" and is better not applied as
late as 2 hours or more after the onset of AMI.
5 Until now there are no clinical studies in patients with
AMI showing activation of complement due to reperfusion of the
infarcted myocardium. However, inhibition of complement in these
patients may be beneficial since in rats undergoing reperfusion
of ischaemic myocardium, treatment with a recombinant soluble
form of the human complement receptor type 1 has been shown to
reduce myocardial infarction size considerably. Weisman H.F. et
al., 1990, Science ~: 146.
To date there is no report in literature describing a
beneficial effect of a complement inhibitor (i.e., a protein or
substance which inhibits a [activated] complement protein rather
then depletes it by activation) on the myocardial infarction
size after permanent occlusion of a coronary vessel. The present
invention describes a simple method to reduce myocardial
infarction size by administering a naturally occuring inhibitor
of the activated first component of complement.
Summary of the Invention
It has now been found that administration of the serine-
proteinase inhibitor C1-esterase inhibitor reduces the size of
myocardi;-__1 infarction when given as late a~ 2 hours following
occlusion of a coronary w-essel.
Therefore, the present invention contemplates a therapeutic
or prophylactic treatment method of AMI, which method comprises
administering exogenous C1-esterase inhibitor, alone or in
combination with other drugs, to a patient with acute myocardial
infarctian or to a patient at risk for acute myocardial
infarctian.
The treatment is applicable, independently from whether or
not the patient is receiving medical or surgical treatment to
V
restore blood flow to the jeopardized myocardium. In particular,
the invention is also applicable in patients who, because of the
time elapsed between the onset of AMI and the admission to a
hospital, may not (or not anymore) be treated by reperfusion
WO 95/06479 . PCT/NL94/00208
6
therapy. Thus, the invention provides an alternative method to
treat these patients (more than 90~ of all patients with AMI!),
since it is still effective in reducing myocardial tissue injury
when given as late as 2 hours after the occlusion of a coronary
vessel.
Exemplary C1-esterase inhibitor may be derived from human ,
plasma or any other biological source, or recombinant C1-
esterase inhibitor, or mutants derived therefrom, or recombinant
proteinase inhibitor with specificity for the activated form of
the first component of complement.
Examples of other drugs which may be administered in
combination with C1-esterase inhibitor are substances which
improve the blood flow to the myocardium, such as tissue
plasminogen activator, urokinase and streptokinase, and
substances having anti-inflammatory properties, such as oxygen
radical scavengers and cytokine antagonists.
The invention will be more fully understood after a
consideration of the following description of the invention.
Brief Description of the Drawings
The effect of administering plasma-derived human C1-
esterase inhibitor on myocardial damage of AMI was studied in
dogs that underwent a permanent ligation of the left anterior
descending coronary artery (LAD). Forty-eight hours after
occlusion the animals were killed and the hearts excised. The
ischaemic myocardium was cut into pieces of about 1 gram.
Myocardial damage was assessed by measuring the residual
a-hydroxybutyrate dehydrogenase (HBDH) activity in each piece of
myocardial tissue. The blood flow in each piece of myocardium at
15 minutes and at 48 hours was measured by injecting labeled
microspheres intravenously 15 min and 48 hours after occlusion
and counting radio-activity of the pieces. The results were
expressed as ~, 100 being the value observed for normal
myocardium. The results shown were obtained from 4 dogs that
received C1-esterase inhibitor at 2 and 8 hours after occlusion
(treated) and 4 dogs that did not receive C1-esterase inhibitor
(control).
WO 95/06479 PCT/NL94/00208
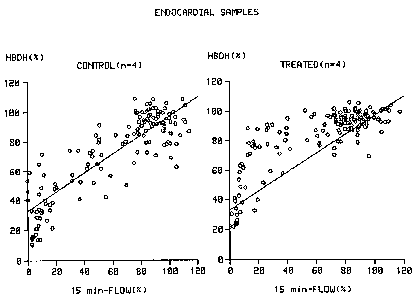
Figure 1 shows that for the same degree of initial
ischaemia (flow at 15 minutes) the hearts of the animals treated
with C1-esterase inhibitor had lost about 50~ less muscle enzyme
than the controls.
a
Figure 2 shows the same as Figure 1 except that the results
shown were obtained with epicardial tissue specimens.
Figure 3 shows that the reduction of myocardial enzyme
depletion in animals treated with C1-esterase inhibitor runs
parallel to about 50~ recovery of blood flow in the infarcted
area during the first 48 hours, in spite of continued LAD
ligation.
Figure 4 shows the same as Figure 3 except that the results
shown were obtained with epicardial tissue specimens.
Detailed D scr,'_~t,'--on of the Invention
The invention provides a method to inhibit the inflammatory
reaction, more specifically the activation of the complement
system, which occurs in the course of acute myocardial
infarction. The preferred inhibitor is C1-esterase inhibitor,
which may include C1-esterase inhibitor purified from plasma, or
recombinant C1-esterase inhibitor, or recombinant variants
derived therefrom, or recombinant constructs of other inhibitors
having a specificity similar to C1-esterase inhibitor.
Several patent documents and scientific articles are
referred to below that discuss various aspects of the materials
and methods used to realize the invention. It is intended that
all of the references be entirely incorporated by reference.
Basic for the present invention is the realization that
activation of complement causes substantial myocardial damage
during AMI, indepedently from whether or not medical treatment,
such as thrombolytic therapy, is instituted to re-open the
occluded coronary vessels, and that inhibition of this
activation would reduce or prevent this myocardial damage. The
preferred method to inhibit complement activation in persons
' 35 suffering from AMI would be administering plasma-derived Cl-
esterase inhibitor, but by no means should this invention be
constructed so narrowly. Virtually every method to inhibit
complement activation by inhibiting the activity of the first
WO 95/46479 ~ , PCT/NL94100208
8
component of complement is intended to come into the scope of
this invention.
To more clearly define the present invention, it will be
described in three sections. The first section contains ,
definitions of particular terms as they will be employed herein.
These definitions are generally consistent with their usage in
the art. The second section descibes more specifically the
various C1-esterase inhibitor molecules which are intended to
come into the scope of the invention. The third section
describes the method to administer exogenous C1-esterase
inhibitor to a patient with AMI.
I. Defin~t~ons
As used herein, the phrase "complement system" refers to a
set of proteins, most of which circulate in blood as inactive
precursor proteins, also known as factors. During activation of
the system, one factor activates the subsequent one by limited
proteolysis and so on. This activation process resembles a
cascade and, therefore, the complement system is also considered
as one of the major plasma cascade systems, the other being the
coagulation, the fibrinolytic and the contact systems. The
physiological role of the complement system is to defend the
body against invading micro-organisms. The system may become
activated via two pathways, a classical and an alternative
pathway, which both can activate a common terminal pathway.
Cooper N.R., 1985, Adv Immunol ~: 151; Muller-Eberhard H.J, et
al., 1980, Adv Immunol ~: 1; Muller-Eberhard H.J., 1992, In:
Gallin JI, Goldstein IM, Snyderman R (eds): Inflammation: Basic
Principles and Clinical Correlates, New York, Raven Press, p.33.
A number of biologically active peptides, also known as the
anaphylatoxins, are generated during activation of complement.
Vogt W., 1986, Complement ~: 177. The anaphylatoxins, in
particular C3a and CSa, are chemotactic for neutrophils and able
to aggregate, activate and degranulate these cells. Vogt w.,
1986, Complement ~: 177; Goldstein IM, 1992, In: Gallin JI, '
Goldstein IM, Snyderman R (eds): Inflammation: Basic Principles
and Clinical Correlates, New York, Raven Press, p.63.
Furthermore they may enhance vasopermeability, stimulate
WO 95!06479 PCT/NL94/00208
9
adhesion of neutrophils to endothelium, activate platelets and
induce the production of vasoactive eicosanoids, thromboxane A2
and peptidoleukotrienes such as LTC4, LTD4 and LTE4. Also the
so-called terminal complement complexes (TCC), formed upon
activatian of the common pathway, have important effects such as
the capacity to kill cells. Muller-Eberhard H.J., 1986, Ann Rev
9.: 503. Activation of the classical pathway of
complement starts with activation of the first component, which
consists of a macromolecular complex of 5 proteins, one Clq, two
Clr and two C1s proteins. The C1q protein of the C1 complex
binds to an activator, for example immune complexes, which leads
to activation of both Clr and both Cls subcomponents. Cooper
N.R., 1985, Adv Immunol ~: 151. During activation the C1r and
C1s proteins are converted from a single peptide-chain inactive
protein to a two-chain active serine proteinase. Cooper N.R.,
1985, Adv Immunol ~7: 151. The activated C1 complex then
activates the complement factors ~::' and C2, which both form a
complex that can activate C3, the complement factor. Several
plasma proteins can inhibit activation of the classical pathway
of. complement, notably, C1-esterase inhibitor, C4-binding
protein and the serine-proteinase factor I. Muller-Eberhard
H.J., 1992, In: Gallin JI, Goldstein IM, Snyderman R (eds):
Inflammation: Basic Principles and Clinical Correlates, New
York, Raven Press, p.33; Cooper N.R., 1985, Adv Immunol ~: 151.
As used herein, the phrase "contact system" refers to a set
of proteins, which circulate in blood as inactive precursor
proteins, and which is also known as the contact system of
coagulation or the kallikrein-kinin system. Colman R.W., 1984, ,~
Clin Invest 7~: 1249; Kaplan A.P, et al., 1987, Blood 7~: 1;
Kozin F., et al, 1992, In: Gallin JI, Goldstein IM, Snyderman R
(eds) : Inflammation: Basic Principles and Clinical Correlates,
New York, Raven Press, p.103. The contact system also belongs to
the major plasma cascade systems, and is often regarded as one
of the two pathways via which the coagulation system can be
activated, the so-called extrinsic pathway of coagulation being
the other. The physiological role of the contact system is not
precisely known, although it is known that this system may
WO 95/06479 PCTINL94/00208
~~ 10
become activated in inflammatory conditions. Colman R.W., 1984,
J Clin Invest ~: 1249; Kaplan A.P. et al., 1987, _Blood 7~.: 1;
Kozin F., et al, 1992, In: Gallin JI, Goldstein IM, Snyderman R
(eds): Inflammation: Basic Principles and Clinical Correlates, ,
New York, Raven Press, p.103.. Activation of the contact system
starts with binding of factor XII, also known as Hageman factor, ,
to an activator. Subsequently, bound factor XII may become
activated, during which process it is converted from a single-
chain inactive to a two-chain active serine proteinase. Tans G.
et al., 1987, Sem Thromb Hemost ,1,~: 1. Activated factor XII then
activates prekallikrein, that together with its cofactor high
molecular weight kininogen is bound to the activator, into the
active serine proteinase kallikrein. Kallikrein in turn may
activate bound but not yet activated factor XII (reciprocal
activation), and/or factor XI, which in turn can activate factor
IX to start activation of coagulation. Cochrane C.G. et al.,
1982, Adv Immunol ,~: 290; Colman R.W., 1984, J Clin Invest 7~:
1249; Kaplan A.P. et al., 1987, Blood 7Q: 1; Kozin F., et al,
1992, In: Gallin JI, Goldstein IM, Snyderman R (eds):
Inflammation: Basic Principles and Clinical Correlates, New
York, Raven Press, p.103. Activation of the contact system is
controlled by the same protein which also inhibits the classical
complement pathway, C1-esterase inhibitor. During activation of
the contact system several biologically active components are
formed such as bradykinin, kallikrein and activated factor XII,
which may enhance activation and degranulation of neutrophils,
increase vasopermeability and decrease vascular tonus. Colman
R.W., 1984, J Clin Invest 7~,: 1249; Kozin F., et al, 1992, In:
Gallin JI, Goldstein IM, Snyderman R (eds): Inflammation: Basic
Principles and Clinical Correlates, New York, Raven Press,
p.103.
As used herein, the phrase "C1-esterase inhibitor" refers
to a protein that is present in blood and is the main inhibitor
of the classical pathway of complement and of the contact '
system. Cl-esterase inhibitor can inhibit the activated form of
the first component of complement and activated factor XII, and
it is also a major inhibitor of kallikrein. Schapira M. et al.,
WO 95/06479 PCT/NL94/00208
11 .
1985, omplement ~: 111; Davis A.E., 1988, Ann Rev Immunol ~:
595; Sim R.B. et al., 1979, FEBS Lett ~7: 111; De Agostini A. et
al., 1984, J Glin Inv ~t ~,; 1542; Pixley R.A. et al., 1985,
Biol Chem ~Q: 1723; Schapira M. et al., 1982, J Clin Invest
462; Van der Graaf F. et al., 1983, J Cl~n Invest ~: 149;
Harpel P.C. et al., 1975, J Clin Inves ,~: 593. In addition,
C1-esterase inhibitor may inhibit activated factor XI, tissue-
type plasminogen activator and plasmin. Meijers J.C.M. et al.,
1988, Biochem~s~; 959; Harpel P.C. et al., 1975, J Clin
Invest ,~,~; 149; Booth N.A. et al., 1987, Blood ~,9: 1600.
C1-esterase inhibitor inhibits proteinases by forming
stable complexes with these proteinases, which are rapidly
cleared from the circulation. De Smet B.J.G.L. et al., 1993,
Blood .$1.: 56. Full-length genomic and cDNA coding for C1-
esterase inhibitor has been cloned. Bock S.C. et al., 1986,
B?ochemistr~ ~; 4292; Carter P.E. et al., 1988, Eur J Biochem
173: 163. Functional recombinant Cl-esterase inhibitor protein
has been expressed in COS cells and found to be similar to the
plasma protein. Eldering E. et al., 1988, J Biol Chem 263:
11776. In addition, several variants of recombinant C1-esterase
inhibitor with amino acid mutations at the P1 and the P3 and/or
P5 position of the reactive centre have been expressed in the
same system. Eldering E. et al., 1988, J Bio1 Chem 263: 11776;
Eldering E. et al., 1993, J Biol Chem 267: 7013; Eldering E. et
al., 1993, J Glin Invest ~; 1035; Patent Cetus Corp, US617920.
Moreover, some variants isolated from patients with hereditary
angioedema have been cloned and expressed in the same system.
Davis A.E, et al., Na ~r n t-ir~ ~; 354.
C1-esterase inhibitor belongs to a superfamily of
homologous proteins which together are known as the serine-
proteinase inhibitors, also called serpins. Travis J. et al.,
1983, Ann Rev BlOChPm ~; 655; Carrel R.W. et al., 1985, '/rends
' Bioch Sci ,~Q: 20. The serpins share a similar mechanism of
inhibition, which is characterized by forming stable bi-
' 35 molecular complexes with the proteinase to be inhibited. In
these complexes the active site of the proteinase is bound to
the so-called reactive centre of the serpin and hence rendered
inactive. Travis J. et al., 1983, Ann Rev Biochem ~: 655.
WO 95/06479 PCT/1VI94/00208
12
Serpins have specificity for certain proteinases and this
specificity is in part due to the amino acid sequence of the
reactive centre. A number of studies have shown that it is
possible to alter the specificity of a serpin by changing the ,
amino acid sequence of the reactive centre and/or other parts of
the inhibitor, for example by site-directed mutagenesis. Owen _
M.C. et al., 1983, New Eng J Med 309: 694; Carrel R.W. et al.,
1985, Trends Bioch Sci ,Z,Q: 20; Courtney M. et al., 1985, Nature
77; George P.M. et al., 1989, Blood 7~: 490; Rubin H. et
al., 1990, J Biol Chem ~: 1199; Holmes W.E. et al., 1987,
Biochemistry ~: 5133. Although a recombinant variant of serpins
other than C1-esterase inhibitor with a specificity for
activated C1 has not been described, it is reasonable to expect
that such inhibitors can be constructed.
As used herein the phrase "acute myocardial infarction"
("AMI") refers to a common clinical condition caused by necrosis
of myocardial tissue. This condition is well-known in the art
and is characterized by the occurrence of pain, in most cases
precordial, characteristic electrocardiographic changes and an
increase in plasma levels of intracellular enzymes released by
the necrotic cardiac tissue such as creatinine phosphokinase and
a-hydroxybutyrate dehydrogenase. AMI may be accompanied by
hypotension, circulatory failure, pulmonary edema and
arhythmias. In most cases, but not exclusively, AMI results from
vascular injury and thrombosis in the coronary vessels, which
causes these vessels to become occluded with subsequent impaired
blood flow to the jeopardized myocardium. Fuster V. et al.,
1992, New Engl J Med 326: 242 and 310. In most cases the time of
the occlusion of the coronary vessel can be estimated from the
medical history, the course of plasma levels of intracellular
heart muscle enzymes and electrocardiographic changes.
II. C1-esterase inhibitor reparations
The activity of C1-esterase inhibitor in plasma or in
purified preparations can be measured with several assays
including chromogenic and esterolytic assays, in which the
inhibition of conversion of substrates by active C1s is
WO 95/06479 ~ ~ PCT/NL94/00208
13
monitored. These assays are well-known in the art. Also, a
radioimmunoassay, in which the binding of Cl-esterase inhibitor
to solid-phase bound active C1s is assessed, can be used to
measure levels of functional C1-esterase inhibitor. Nuijens J.H.
et al., 1989, J Clin Invest $Q: 443. Levels of functional C1-
esterase inhibitor can be expressed in various ways. Here Units
per milliliter (U/ml) will be used where one U/ml is the
concentration of functional C1-esterase inhibitor present in
pooled normal plasma, which is approximately 270 ~.g per ml of
plasma. Nuijens J.H. et al., 1989, J Clin Invest $~: 443.
Intended to come into the scope of the invention is the
application of the following forms of Cl-esterase inhibitor
molecules: native C1-esterase inhibitor purified from human or
animal plasma or any other biological source, or fragments
derived therefrom that maintain biological activity; recombinant
native C1-esterase inhibitor, human or animal, or variants or
fragments therefrom that maintain biological activity; or
recombinant inhibitors manipulated to inhibit the activated form
of the first component of complement.
The C1-esterase inhibitor preparation is dissolved into a
pharmaceutically acceptable vehicle, and in the preferred
embodiment of this invention, given by intravenous injection.
Such vehicles are well-known in the art and examples include
water, saline, dextrose solution, Ringer's solution and
solutions containing small amounts of human serum albumin. It
will, of course, be understood that intended to come within the
scope of this invention is virtually any method of administering
C1-esterase inhibitor to yield sufficient concentrations of this
inhibitor in the jeopardized myocardium.
III Treatment of acute myocardial infa ction with C1-esterase
inhibitor
The C1-esterase inhibitor preparation described herein,
alone or in combination, may be used to treat a host organism
either suffering from AMI, or at risk for developing this
condition.
In the preferred embodiment of the invention C1-esterase
inhibitor is administered intravenously to a patient with AMI to
WO 95/06479 ~ ~ PC'~YNL94/00208
14
yield sufficient amounts of C1-esterase inhibitor in the
jeopardized myocardium during the first 24 hours after occlusion
of the coronary vessel. In most cases this can be achieved by
administering C1-esterase inhibitor until plasma levels of ,
functional C1-esterase inhibitor are within the range of 2 to
2.5 U per ml. For most patients, two intravenous injections of
30 to 40 U of C1-esterase inhibitor per kg of body weight each,
for example at the time the patient is admitted to the hospital
and 6 hours later, will yield this plasma level. In case of
clinical and biochemical evidence for further ongoing necrosis
of myocardial tissue, as manifested by the course of intra-
cellular heart muscle enzymes or electrocardiographic changes,
additional gifts of C1-esterase inhibitor can be given. C1-
esterase inhibitor therapy can be given to patients who, because
of the time elapsed between the onset of AMI and admission to
the hospital, may not benefit anymore from reperfusion therapy.
Activation of the complement system not only contributes to
the myocardial tissue damage due to permanent occlusion of a
coronary vessel, but also to that caused by reperfusion of the
ischaemic tissues following coronary angioplasty or treatment
with thrombolytic agents. Therefore, patients with AMI may be
treated with C1-esterase inhibitor independent from whether they
do or do not receive medical or surgical treatment to re-open
coronary vessels. Typically, a patient is admitted to a hospital
because of AMI. When appropriate, the patient may receive
thrombolytic therapy or acute percutaneous transluminal coronary
angioplasty, which procedures are well-known in the art, and at
the same time C1-esterase inhibitor may be injected intravenous-
ly at a dose of 30 to 40 U per kg of body weight. This may be
followed by a second gift of C1-esterase inhibitor at a similar
concentration 6 hours thereafter. In another embodiment of the
invention C1-esterase inhibitor administration treatment is
instituted in a patient with AMI, who does not receive angio-
plasty or thrombolytic therapy. Typically, such a patient may
receive an intravenous injection of C1-esterase inhibitor at a '
dose of 30 to 40 U per kg of body weight upon admission to the
hospital, which administration may be repeated 6 hours
thereafter.
CA 02170739 2004-08-06
' 15
Patients with partial occlusions of the coronary vessel:>
may receive medical therapy such as percutaneous transluminal
coronary angioplasty, which are accompanied by temporary
- complete occlusion of the coronary vessels. Reperfusion of the
jeopardized myocardium may be accompanied by inflammatory
., . reactions which may affect the function of the myocardial
vessels. Intended to be within the scope of this invention is
the prophylactic treatment of these patients by administering
them a single intravenous injection of C1-esterase inhibitor at
a dose of 30 to 40 U per kg of body weight.
Having described what the applicants believe their
invention to be, the following examples will be presented to
illustrate the invention. The examples are intended as
illustrative of the present invention and not limiting.
Examy le 1
In the preferred embodiment of the invention, the
therapeutic composition contains plasma-derived C1-esterase
inhibitor as the active ingradient, prepared according to
Voogelaar E.F. et al., 1974, Vox Sana. ~: 118. The virus safety
of this preparation is guaranteed by the addition of hepatitis
B-immunoglobulin and a heat treatment of the freezed-dried
preparation in the final container. Brummelhuis H.G.J. et al.,,
1983, Vox Sang. g~: 205, Tersmette et al., 1986, Vox Sancr.
239. C1-esterase inhibitor is prepared from human plasma,
depleted of vitamin K-dependent coagulation factors, according
to a procedure which involves the following purification steps:
1) the starting plasma is 1 to 10 diluted with sterile destilled
water; 2) the diluted plasma is incubated with DEAF-Sephadex~;A50
(Pharmacia Fine Chemicals, Uppsala, Sweden) at a concentration
of 2 g/kg, for 60 minutes at 8-10 oC; 3) the DEAF-Sephadex is
collected and washed with 150 mM sodium chloride, pH 7.0, and
eluted with 10 mM trisodium citrate, 2 M sodium chloride, pH
7.0; 4) ammonium sulphate is added to the eluate to yield a
final concentration of 50%, v/v; 5) after centrifugation at
13,000 rpm, ammonium sulphate is added to the supernatant to
yield a final concentration of 65%, v/v; 6) the precipitate is
collected by centrifugation and dissolved in 10 mM trisodium
*Trade-mark
CA 02170739 2004-08-06
16
citrate, pH 7.0; 7) a diafiltration is performed to remove the
ammonium sulphate and to concentrate the solution to a protein
concentration of 40-50 mg/ml; 8) after the addition of Hepatitis
B immunoglobulin (0.4 IU/ml), the solution is filtered through a
0.22 Etm filter, dispensed in vials and freeze-dried: 9) the _
freeze-dried product is heat-treated for 72 hours at 60 °C.
In the preferred embodiment of the invention, C1-esterase
inhibitor will be administered by intravenous injection. For
parenteral administration, freeze-dried heat-treated C1-esterase
inhibitor is dissolved into water for injections. The final
concentration of functional C1-esterase inhibitor in the
preparation to be administered may be about 50 U per ml.
Example 2
The usage of animal models for AMI is well-known in the
art. A model frequently used by investigators is to induce Aril
in dogs by occluding the left anterior descending coronary
artery (LAD). The usefulness of administering C1-esterase
inhibitor as a therapeutic treatment for AMI will be illustrated
using this model. Experimental details of the model are given
elsewhere. Hermens W.Th et al., 1990, Circulation ~,: 649.
Thoracotomy was performed in Mongrel dogs of either sex,
after appropriate premedication and anaesthesia. Then; a snare
(Mersilene; 3 metric) was placed around the LAD, which had been
dissected, distal to the first diagonal branch, and fixed into a
polyethylene tube in such a way that an outward pull over a
distance of 2 cm caused complete ligation of the LAD. In
addition, a catheter (Tygon~S 50 HL1) was inserted through the
auricle of the left atrium for microsphere injection, collection
of blood samples and administration of medication. The thorax
was closed and the dogs were allowed to recover. After one week
the LAD was occluded after appropriate medication of the dogs,
by pulling the snare 2 cm out of the polyethylene tube.
Occlusion was verified by electrocardiography and by inspection
of the site of occlusion at the end of the experiment. At
various time intervals regional myocardial flow was determined
by injecting 3-5x106 tracer microspheres (New England Nucear
Corp.; diameter 15 dun) labeled with different isotopes. After
*Trade-mark
WO 95!06479 PCT/NL94/00208
17
48 hours the animals were killed with an overdose of sodium
pentobarbital. The hearts were excised, rinsed and further
processed. The right ventricle was removed, the left was cut
from base to apex into 5 slices 1-2 cm thick. These slices were
then cut into 108 pieces totally. Each piece was weighed.
Residual activity of the intracellular enzyme a-hydroxybutyrate
dehydrogenase as well as the radioactivity of the tracer micro-
spheres were measured in each piece of tissue. The relation
between impaired regional myocardial flow after the occlusion
and the myocardial cell damage can be visualized by plotting the
activity of the tracer microsphere~ injected 15 minutes after
the occlusion versus residual ~i-hydroxybutyrate for each tissue
piece. In non-treated dogs that undergo a permanent occlusion of
the LAD, a linear correlation between regional myocardial flow
at 15 minutes after the occlusion and myocardial cell damage at
48 hours, exists (Figs 1 and 2, left panel). This correlation is
altered in dogs that undergo a permanent occlusion of the LAD
and who are treated with intravenous injections of C1-esterase
inhibitor at a dose of 35 U per kg of body weight at 2 and 8
hours after the occlusion. In these latter animals significantly
more residual ~i-hydroxybutyrate activity is found in the
myocardial tissues pieces, in particular in the endocardial
tissue pieces, than is expected based on the regional myocardial
flow at 15 minutes after the occlusion (Figs 1 and 2, right
panels), illustrating that administration of C1-esterase
inhibitor to the dogs significantly reduces myocardial cell
damage after permanent occlusion of the LAD. Similarly, in non-
treated animals with permanent occlusion of the LAD, there is a
linear correlation between regional myocardial blood flow at 15
minutes and that at 48 hours after occlusion (Figs 3 and 4, left
panels). C1-esterase inhibitor treatment of dogs with permanent
occlusion of the LAD is accompanied by a better flow at 48 hours
than is expected based on the flow at 15 minutes after the
occlusion (Figs 3 and 4, right panels), which suggests that the
treatment with C1-esterase inhibitor is able to partially
restore regional blood flow in the jeopardized myocardium.