Note: Descriptions are shown in the official language in which they were submitted.
r
_
21 7~3 1 3
PATENT CASE AL0386
~RICYCLIC DERIVAT~ . COMPOSITIONS
AND MET~ODS OF USE
F~r.n OF T~F. INVFNTION
The present invention relates to tricyclic derivadves,
ph~ çeutical compositions and methods of using such derivatives.
The compounds of the present invention inhibit tumor necrosis factor a
(nTNF-a" )
RACKGRouND OF T~lF INVENTION
Tumor necrosis factor ~ ("TNF-an) is a polypeptide cytokine
known to induce a variety of inflamm~tory and metabolic processes in
25 vivo. See, e.g.,Ann. Rev. Immunol. 7:625 (1989). However,
overproduction or inappropriate production of TNF-~ has been shown to
be involved in several pathoIogical conditions, including septic shock
and various allergic diseases and infl~mm~tory conditions. See, e.g.,
Immunol Res. 10:122 (1991), Science 229:869 (1985) and Proc. Natl.
30 Acad. Sci. 89:7375 (1992). Thus, compounds that could inhibit TNF-a
would be quite valuable in treating these conditions.
In view of the substantial interest in agents that inhibit
TNF-~, the identification of compounds having anti-TNF-~ activity
35 would be a valuable contribution to the art. This invention provides
just such a contribution by providing novel compounds having anti-
TNF-~ activity. In addition, this invention provides methods of using
such compounds.
21 7531 3
- 2 -
SUMMARY OF T~ INVENTION
We have now unexpectedly found that compounds having
t~e general fo~mula I (set forth below) provide surprisingly good
ac~vity ~ inhibitors of tumor necrosis factor ~ (TNF-a). More
specifically, we believe that the compounds of formula I provide this
activity by inhibiting the biosynthesis of TNF-a. In view of this
surprising anti-TNF-a activi~,r, it is believed that compounds of formnl~
I are useful in the relief of septic shock, allergic diseases, and
infl~mm~tory conditions.
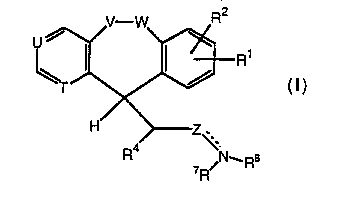
Folmula I is as follows:
~r~ n'
R ~N_Rs
or a pharmaceutically acceptable salt or solvate thereof, wherein:
one of T and U represents N and the other represents =CH-; or each of T
and U represents =CH-;
one of V and W represents oxygen and the other represents -CH2-; or each
of V and W represents -CH2-;
R1 and R2 are each independently selected from the group consisting of H
and halogen;
R4 is alkenyl, alkoxy, or-OH;
- - -
3 21 7531 3
Z N represents an optional double bond;
when Z N is a double bond, Z represents -CH=, or-CH2C(R5)=,
wherein R5 is H or lower alkyl; and R7 and R8 together represent OR9;
when Z N represents a single bond, Z represents -CH2-,
5 -CH=CH-, or -CH2C(R5)(R6)-, wherein R~ and R6 are inde~,endently H or lower
alkyl; and R7 and R8 are independently H, alkyl, alkenyl, alkynyl, aryl, alkaryl,
aralkyl, cycloalkyl, -OR9; -C(O)OR1 ~; -CH2C(O)OR9; -C(O)R1 ~; -SO2R1 ~;
-CO-4-pyridyl-N-oxide; -(CH2)n-N(CH3)2, where n is 2 to 4; -(CH2)mO(CH2)J~H,
c(o)~
where m and j are independently 2 or 3; C(O)OR9; or R7 and R8
10 together fomm either a five-membered or a six-membered ring optionally substituted
with COOR9; a six-membered ring containing NR10; or a five-membered ring fused
to a benzene ring;
R9 is H or lower alkyl; and
R10 is alkyl or aryl.
More preferred compounds of this invention are represented by
Formula l wherein R4 is alkoxy, and more preferably wherein R4 is ethoxy.
More preferred compounds also include those of Formula l wherein
20 R4 is alkenyl.
More preferred compounds also include those of Formula I
wherein each of T and U represents =CH-.
More preferred compounds also include those of Formula l wherein
R7 and R8 are independently H, alkyl, alkenyl, alkynyl, aryl, alkaryl, or aralkyl.
25 Further, when R7 and R8 and N taken together form either a five-membered ring, a
six-membered ring, or a five-membered ring fused to a benzene ring, the portion of
the 5- or 6-membered ring represented by R7 and R8 is preferably carbocyclic
optionally having a nitrogen atom substituted for one of the carbon atoms.
- ~ - - -
4 21 75:~1 3
Representative compounds of this invention include, but are not
limited to:
f ~ H5C20
~ NHCH3 ~(CH3)2
(IA) (IB)
h' N(CH3)2 ~(CH3)2
(IC) (IF)
H5C2 HCH3 N(CH3)2
(IG) (IH)
2 1 753 1 3
- 5 -
N(CH3)C(O)CH3 ~
~ N(cH3)c(o)oczH5
(IJ) (IK)
~CI ~c
N(CH3)C(O)cH3 N(CH3)z
(IL) (IM)
h Cl
NH(CH3)
(IN)
t
2 1 753 1 3
-6 -
This invention also provides a pharmaceutical composition
comprising an effective amount of a compound of Formula I in
combination with a ph~rrnAceutically acceptable carrier.
In addition, this invention provides a method for inhibiting
TNF-~ in a mAmm~l complising A~imini~stering to the m~mmAl an
amount of a compound of Formnl~ I effective to inhibit TNF-a.
In view of the surprising anti-TNF-a activity of compounds
of formlllA I, this invention provides the following methods of
treatment:
- a method for treating inflAmmation in a m~mmAl
comprising Atlministering to the mAmm~l an effective anti-
inflAmmAtory amount of a compound of For_ula I;
- a method for t,reating septic shock in a mAmmAl
comprising Atimini.~tering to the mAmmAl an effective anti-septic shock
amount of a compound of Porml-lA I; and
- a method for treating allergic reaction in a mAmm~l
comprising ~-lministering to the mAmmAl an effective anti-allergic
amount of a compound of Pormula I.
It also has been surprisingly found that the following
compounds, which are either known (Kl) or generically covered by a
known generic formula (Kl-K7), have anti-TNF-a activity:
H C N(CH3)2
~HCH3
(K l ) (K2)
7 2175313
H3C N H C H3 H3C N(CH3)C(O)Oc2H5
(K3) (K4)
~ ~CI
H3C N(CH3)C(O)CH3 H3C N(CH3)2
tK5) (K6)
,~ Cl
H3C NH(CH3)
(K7)
-- ----
-
-8- 21 7531 3
With regard to the above-listed compounds, prior to t~e
present application compound Kl was only disclosed as having activity
as an anti-depressant. See Pro~va et al, J. Med. Pharm. Chem. ~L, 411
5 (1961); Winter et al, German Patent Publication 2335943 (1975). The
genenc folmula cove~ing compounds K2-K7 (as well as Kl) has only
been disclosed as having cardiac and circulatory activity. See U.S.
Patent 4,070,373 (1978) to Winter eJ al.
Thus, in view of tlle surprising anti-TNF activity of the
above-mentioned compounds Kl-K7, this invention also provides
me~ods of using these compounds to treat a m~mm~l for infl~mm~tion,
septic shock, and allergic reaction. Accordingly, the present invention
also provides methods of using compounds of fo~mula K (set forth
15 below) for inhibidng TNF-a and for treating a m~mm~l for
infl~mm~tion, septic shock, and allergic reaction.
FoImula K is as follows:
Ul ~ X ~R2 R1
(~
H ~_Z;;;~
R N_ R~
20 or a pharmaceutically acceptable salt or solvate thereof, wherein:
one of T and U represents N and the other represents =CH-; or each of T
and U represents =CH-;
one of V and W represents oxygen and the other represents -CH2-; or each
of V and W represents -CH2-;
R1 and R2 are each independently selected from the group consisting of H
and halogen;
~1 7531 3
R4 is H or lower alkyl;
Z N represents an optional double bond;
when Z N is a double bond, Z represents -CH-, or ~H2C(R5)-,
wherein R5 is H or lower alkyl; and R7 and R8 together represent OR9;
when Z N f~l)f~se~-ts a single bond, Z represents -CH2-,
~H=CH-, or-CH2C(R5)(R6)-, wherein R5 and R6 are independently H or lower
alkyl; and R7 and R8 are independently H, alkyl, alkenyl, alkynyl, aryl, alkaryl,
aralkyl, cycloalkyl, -OR9; -C(O)ORl~; -CH2C(O)OR9; -C(O)R10; -SO2R1~;
-C~4-pyridyl-N-oxide; -(CH2)n-N(CH3)2, where n is 2 to 4; -(CH2)mO(CH2)jOH,
c(o)~
where m and j are independently 2 or 3; C(O)OR9; or R7 and R8
together form either a five-membered or a six-membered ring optionally substituted
with COOR9; a six-membered ring containing NR10; or a five-membered ring fused
to a benzene ring;
R9 is H or lower alkyl; and
R10 is alkyl or aryl.
In a more preferred embodiment for FormuIa K,
Z N represents a single bond and R7 and R8 are independently H or
alkyl.
The present invention will be described in detail below in
connection with several preferred embodiments. However, additional
embodiments of the present invention will be apparent to those having
ordinary skill in the art.
nETALED l)F.SCRIPrION OF THF INVFNTION
As used herein, the following terms are used as defined
below unless otherwise indicated:
- - i
2175313
- 10-
alkyl - (including the alkyl portions of alko~y and
cycloalkyl) - represents straight and branched carbon chains and
contains from one to twenty carbon atoms, preferably one to six carbon
atoms;
alkenyl - (including the alkenyl portions of cycloalkenyl)
represents straight and branched carbon chains having at least one
C~bOn to carbon double bond and cont~inin~ from 2 to 12 carbon
atoms, preferably from 2 to 6 carbon atoms;
alkynyl - represents straight and branched carbon chains
having at least one carbon to carbon triple bond and containing from 2
to 12 carbon atoms, preferably from 2 to 6 carbon atoms;
aryl - represents a carbocyclic group (preferably phenyl or
substituted phenyl) containing from 6 to 14 carbon atoms and having at
least one phenyl or fused phenylene ring, with all available
substitutable carbon atoms of the carbocyclic group being intended as
possible points of attachment, said carbocyclic group being optionally
substituted with one or more of halo, alkyl, hydro~y, alkoxy, phenoxy,
cyano, cycloalkyl, alkenyloxy, alkynyloxy, -SH, ~S(O)eR12 (wherein e is 1
or 2 and R12 is alkyl or aryl), -CF3, ~mino, alkylamino, dialkylamino,
12
-COOR or-NO2;
acyl - (including the acyl portions of acyloxy) represents
-C(O)-alkyl, -C(O)-alkenyl, -C(O)-alkynyl, -C(O)-cycloalkyl, -C(O)-
cycloalkenyl or -C(O)-cycloalkynyl;
alkaryl - represents an aryl group, as defined above, in
which an alkyl group, as defined above, is substituted for one of ~e
aryl H atoms;
alkoxy - represents an alkyl group, as defined above,
attached to a molecule through an oxygen molecule (-O-alkyl);
alkoxymethyl - represents an alkoxy group as de~lned above
attached to a molecule through a methylene group;
aralkyl - represents an alkyl group, as defined above, in
which an aryl group, as defined above, is substituted for one of the
alkyl H atoms;
and halo - represents fluoro, chloro, bromo and iodo.
2 1 7 5 3 1 3
Certain compounds of the invention may exist in different
isomeric (e.g., enantiomers and diastereoisomers) as well as
conformational forms. The invention contemplates all such isomers
both in pure form and in admixture, including racemic mixtures.
5 Tautomeric forms are also included.
The compounds of Formula I can exist in unsolvated as well
as solvated forms, including hydrated forms, e.g., hemi-hydrate. In
general, the solvated forms, with pharmaceutically acceptable solvents
10 such as water, ethanol and the like are equivalent to the unsolvated
forms for purposes of the invention.
Lines drawn into the ring systems indicate that the
indicated bond may be attached to any of the substitutable ring carbon
1 5 atoms.
Certain compounds of the invention will be acidic in nature,
e.g. those compounds which possess a carboxyl or phenolic hydro~yl
group. These compounds may form pharmaceutically acceptable salts.
20 F~ mples of such salts may include sodium, potassium, calcium,
alurninum, gold and silver salts. Also contemplated are salts formed
with pharmaceutically acceptable amines such as ammonia, alkyl
amines, hydroxyalkylamines, N-methylglucamine and the like.
Certain basic compounds of the invention also form
pharmaceutically acceptable salts, e.g., acid addition salts. For example,
the pyrido-nitrogen atoms may form salts with strong acid, while
compounds having basic substituents such as amino groups also form
salts with weaker acids. F.~mples of suitable acids for salt formation
are hydrochloric, sulfuric, phosphoric, acetic, citric, oxalic, malonic,
salicylic, malic, fumaric, succinic, ascorbic, maleic, methanesulfonic and
other mineral and carboxylic acids well known to those skilled in the
art. The salts are prepared by contacting the free base form with a
suf~lcient amount of the desired acid to produce a salt in the
con~ entional manner. The free base forms may be regenerated by
treating the salt with a suitable dilute aqueous base solution such as
dilute aqueous sodium hydroxide, potassium carbonate, ammonia and
-12 2175313
sodium bicarbonate. The free base forms differ from their respective
salt forms somewhat in certain physical properties, such as solubility in
polar solvents, but the acid and base salts are otherwise equivalent to
their respective free base forms for purposes of the invention.
All such acid and base salts are intended to be
pharmaceutically acceptable salts within the scope of the invention and
all acid and base salts are considered equivalent to the free forms of
the corresponding compounds for purposes of the invention.
The invention disclosed herein is exemplified by the
following preparative examples, which should not be construed to limit
the scope of the disclosure. Alternative synthetic pathways and
analogous structures within the scope of the invention may be apparent
15 to those of ordinary skill in the art. Further, those skilled in the art will recognize that the reacdons are conducted under conditions, e.g.,
temperature, that will allow the reaction to proceed at a reasonable rate
to completion. Unless indicated otherwise, the substituents for the
formulas given hereinafter have the same definition as those of
20 Formula I.
-13- 21 7531 3
PREPARATIVE METHODS AND REACTION SCHEMES
Scheme 1
V--W R2 V--W R2
U~~ ~ step 1 U~ ~ step 2
~ , /~ Rl ~--T~ Rl
O
COOMe
V--W R2 V--W R2
step4 U/~ ~ step3 U/~
Q_ ~,' Rl ~Q_ '~ R
R4/~OH R4 COOMe
V--W R2 V--W R2
U~( ~ step 5
Q_T/~'R1 ~_ ~/~R
N R
R4 CHO R4 \ R8
Step 1: This step is preferably carried out with the reagent
trimethyl phosphono~cetate and sodium hydride in a polar aprotic solvent (e.g.
N,N-dimethylformamide, N,N-dimethylacetamide, or dimethylsulfoxide) under an
inert atmosphere (nitrogen or argon). Preferred temperature range is 25~C to
80~C.
-14- 21 7531 3
Step 2: This step is preferably carried out with a suitably
substituted R4 silyl reagent and tetra-n-butyl ammonium fluoride in an inert solvent
such as a polar aprotic solvent (e.g. N,N-dimethylformamide or N,N-
dimethylacetamide) with a cosolvent (e.g. 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-
5 pyrimidinone or hexamethylphosphoramide) under an inert atmosphere (nitrogenor argon). Preferred temperature range is between 25~C and 80~C.
Step 3: This step is preferably carried out with any suitable
reducing agent (e.g. diisobutylaluminum hydride, aluminum hydride, or lithium
10 trimethoxyaluminum hydride) in an inert solvent such as ether (e.g. diethyl ether,
tetrahydrofuran, or dioxane) at temperatures preferably between -78~C and 25~C
under an inert atmosphere (nitrogen or argon).
Step 4: This step is preferably carried out with a suitable
15 oxidizing agent (e.g. pyridinium chlorochromate, chromium trioxide-pyridine,
pyridinium dichromate, oxalyl chloride-dimethylsulfoxide, acetic anhydride-
dimethylsulfoxide, dicyclohexylcarbodiimide-dimethylsulfoxide, or periodinane) in
an inert solvent such as chlorinated hydrocarbons (e.g. dichloromethane, 1,2-
dichloroethane, or chloroform). Preferred temperature range is between -78~C and20 25~C.
Step 5: This step is preferably carried out with a suitably
substituted amine (usually as its acid salt e.g. hydrochloride or maleate) and
sodium cyanoborohydride in a solvent mixture of ether (e.g. diethyl ether, tetra-
25 hydrofuran, or dioxane) and protic solvent (e.g. methanol or ethanol) with 3Amolecular sieves. Preferred temperature range is 25~C to 70~C.
- -
1~- 21 7531 3
Scheme 2
step6 u~ ~ step7
OH O
I B
step9 ~ ,step8 U~¦~'R
~ HO~
~N\ ~N\
R8 R7 R8 R7
R90
~N \
Step 6: This step is preferably carried out by first adding a base
(e.g. sodium hydride, potassium hydride, or potassium bis(trimethylsilyl)amide) in
an inert solvent (e.g. ether such as diethyl ether, tetrahydrofuran, or dioxane) under
an inert atmosphere (nitrogen or argon). Subsequently, the alkylating reagent 2-10 chloro-N,N-dialkylacetamide is added, and the preferred temperature range is
25~C to 65~C.
21 7531 3
- 16-
Step 7: This step is preferably carried out with a strong base
(e.g. sodiurn hydride, potassium hydride, potassium bis(trimethylsilyl)amide, orlithium diisopropylamide) in an inert solvent (e.g. benzene or toluene) between
5 80~C and 110~C under an inert atmosphere (nitrogen or argon).
Step 8: This step is preferably carried out by first adding a base
(e.g. sodium hydride or potassium hydride) in an inert solvent (e.g. ether such as
tetrahydrofuran, dioxane, diglyme) under an inert atmosphere (nitrogen or argon).
10 Subsequently, the alkylating agent R9L is added wherein L represents a good
leaving group, e.g. L can be chloride, bromide, iodide, mesylate, or tosylate.
Preferred temperature range is 70~C to 160~C.
Step 9: This step is preferably carried out with any suitable
15 reducing agent (e.g. Iithium aluminum hydride, diborane, or aluminum hydride) in
an inert solvent (e.g. ether such as diethyl ether, tetrahydrofuran, or dioxane) under
an inert atmosphere (nitrogen or argon). Preferred temperatures range between
25~C and 65~C.
- -
-17- 2175313
Scheme 3
~y; step10 ~ step1
step 13 U~ ~ t' P U/~ ~
~--T/~ R1 ~T~ R1
u~ )~~ step 14 U/~ ~ step 15
~ ~R~ T~ R1
~ 1-
R7 R~ R7
~ N~R7
(Ra j5 H or alky~l) Ra
-
-18- 21 7531 3
Step 10: This step is preferably carried out by adding the
Grignard reagent of a suitably N-substituted 4-chloro-piperidine in an inert solvent
such as ether (e.g. diethyl ether, tetrahydrofuran, or dioxane) under an inert
atmosphere (nitrogen or argon). Preferred temperatures range between 0~C and
5 60~C.
Step 11: This step is preferably carried out with strong acid (e.g.
hydrochloric acid, sulfuric acid, or triflic acid) in water at temperatures between
25~C and 100~C.
Step 12: This step is preferably carried out by first adding a strong
base (e.g. n-butyl lithium, sec-butyl lithium, or lithium diisopropylamide) in an inert
solvent such as ether (e.g. diethyl ether, tetrahydrofuran, or dioxane) under an inert
atmosphere (nitrogen or argon) at temperatures between -78~C and 0~C.
15 Subse~uently, a proton source (e.g. methanol, ethanol or acetic acid) is ~dde~
Step 13: This step is preferably carried out by hydrogenation with
a catalyst (e.g. palladium on carbon or platinum oxide) in an inert solvent (e.g.
methanol, ethanol, ethyl acetate, or acetic acid) at 25~C.
Step 14: This step is preferably carried out by adding a
methylating reagent (e.g. methyl chloride, methyl bromide, methyl iodide, methyltosylate, or dimethyl sulfate) in an inert solvent such as a protic solvent (e.g.
methanol, ethanol, isopropanol, or butanol) or a silylating agent (e.g. trimethylsilyl
25 chloride, trimethylsilyl bromide, or trimethylsilyl iodide) in an inert solvent such as
an ether (e.g. tetrahydrofuran). Preferred temperature is between 25~C and
1 00~C.
Step 15: This step is preferably carried out by heating at high
30 temperatures between 1 50~C and 220~C.
- - .
-19- 21 7 531 3
GENERAL PROCESSES
Preparation of a compound of formula I wherein Z N
represents a double bond
U ~ +H2NOR9 U~
- R4 \ H 4 (CR5R6~o or
CHO \~N~
OR9
The process is preferably carried out by treating the aldehyde with a
10 hydroxyl amine derivative in an inert solvent such as chlorinated hydrocarbons
(e.g. dichloromethane, 1,2-dichloroethane, or chloroform) at ambient temperature.
If the hydroxyl amine derivative exists as a salt, the acid can be neutralized by the
~dd~tion of an amine base such as pyridine, collidine, or triethylamine.
Preparation of a compound of formula I wherein Z N
represe"ls a single bond
U~ + R8R7NH U~ ~
H (CR5R6)o 1 H 4 (CR5R6)o or 1
CHO ~N--R7
R8
The reductive amination process is preferably carried out by treating
the aldehyde with an amine (usually as a salt) in the presence of a reducing agent
-20- 21 7531 3
such as sodium cyanoborohydride and molecular sieves in a suitable solvent
mixture of ether (e.g. diethyl ether, tetrahydrofuran, or dioxane) and protic solvent
(e.g. methanol or ethanol) at ambient temperature.
C.
--~, + R8~ UQ~/~
R4 N~ - R7 R4 'N--R7
H R8
The process is preferably carried out by first adding an amine base
(e.g. pyridine, collidine, or triethylamine) in an inert solvent such as chlorinated
10 hydrocarbons (e.g. dichloromethane, 1,2-dichloroethane, or chloroform) or a strong
base (n-butyl lithium, sodium hydride, potassium hydride, lithium diisopropyl
amide, or potassium bis(trimethylsilyl)amide) in an inert solvent such as ether (e.g.
diethyl ether, tetrahydrofuran, or dioxane) or polar aprotic solvent (e.g. N,N-
dimethylfommamide or N,N-dimethylacetamide) to the tricyclic amine under an inert
15 atmosphere (nitrogen or argon). Subsequently, the alkylating or acylating agent
R8L is added wherein L represents a good leaving group, e.g. L can be chloride,
bromide, iodide, mesylate, or tosylate. Any suitable temperature can be used
between -78~C and 80~C.
D.
V--W R2 V--W R2
U/~( ~ reduction ~( ~/~
Q_T/~I R1 ~T~s
H ~--Z'N R7 R~ N~R7
O R10 R10
The process is preferably carried out with any suitable reducing agent
(e.g. Iithium aluminum hydride, alane, borane, or trichlorosilane) in an inert solvent
-21- 21 ~531 ;3
such as ether (e.g. diethyl ether, tetrahydrofuran, or dioxane) at temperatures
between 0~C and 60~C.
E.
hydrolysis ~ ~2Rt
R4 N - R7 R4 N - R7
~R10 H
o
The process is preferably carried out by treating the amide or
carbamate compound under basic (e.g. sodium hydroxide, potassium hydroxide, or
sodium peroxide in water with ethylene glycol, methanol, ethanol, tetrahydrofuran,
dioxane, or diglyme) or acidic (e.g. hydrochloric acid, sulfuric acid, or tosic acid in
10 water with tetrahydrofuran, dioxane, or diglyme) conditions. Any suitable
temperature can be used with preferable temperatures between 60~C and 150~C.
V~ R2 V~ R2
U~ hydrolysis ~R
~R7 ~--R7
~0 OR~~ ~0 OH
The process is preferably carried out by treating the tricyclic ester
compound with a base (e.g. sodium hydroxide or potassium hydroxide) in water
with tetrahydrofuran, dioxane, or diglyme. Any suitable temperature can be used
20 with preferable temperatures between 2~~C and 100~C.
2l l 53 ~ 3
SPEC~FIC PREPARATIVE EXAMPLES
COOMe
s
To synthesize an interrnediate (step 1 of Scheme 1):
Washed sodium hydride (5.71 9 of 60 wt%, 0.143 mol) two times with
hexane under a nitrogen atmosphere. Added 170 mL of dry DMF, and cooled to
0~C. Added trimethyl phosphonoacetate (25.99 9, 0.143 mol) dropwise via
10 ~ddition funnel. Hydrogen evolution was observed. Stirred at 0~C for 15 mins.then at room temperature for 15 mins. Added 6,11-dihydro-dibenz[b,eJoxepin~
one (15.00 9, 0.0714 mol) dissolved in 70 mL of dry DMF, and heated reaction
mix;ture in a 80~C oil bath for 45 hours. Cooled to room temperature, and added
250 mL of half saturated I~H4CI. Extracted with ethyl acetate. Washed combined
15 organic extracts with saturated NaHCO3, saturated NaCI, dried with MgSO4,
filtered, and evaporated. Purified crude product by flash chromatography on silica
gel eluting with a gradient of 5% ethyl acetate-hexane, 7% ethyl acetate-hexane,then 20% ethyl acetate-hexane. Combined appropriate fractions and evaporated
to give 3.16 9 (21% yield) of starting ketone and 13.16 9 (69% yield) of methyl 6,11-
20 dihydro-dibenz[b,e]oxepin-1 1 -ylidene acetate.
mass spectrum: (Cl, isobutane) m/e 267 (M+1)
OO Me
To synthesize an intermediate (ste~ 2 of Scheme 1):
Dissolved methyl 6,11-dihydro-dibenz[b,eJoxepin-11-ylidene ~cet~te
(12.35 9, 0.0464 mol) in 160 mL of dry DMF and 40 mL of DMPU. Added tetra-n-
butylammonium fluoride (2.00 9), 4 A molecular sieves, and then allyl
trimethylsilane (15.90 9, 0.139 mol) dropwise via addition funnel. Stirred at room
21 7531 3
-23 -
temperature for 90 mins. then added additionai allyi trimethylsilane (5.30 g, 0.0464
mol). Stirred at room temperature for 16 hours. Added 50 mL of 9:1 by volume
MeOH:1 N HCI, 400 mL of water, and 200 mL of ethyl acetate. Filtered through
celite, and separated layers. Extracted with ethyl acetate. Washed combined
5 organic extracts with water, saturated NaCI, dried with MgSO4, filtered, and
evaporated. Purified crude product by flash chromatography on silica gel elutingwith 5% ethyl ~cet~te-hexane then 10% ethyl acetate-hexane. Combined
appropriate fractions and evaporated to give 8.75 9 (61% yield) of methyl 2-[6,11-
dihydro-dibenzo[b,e]oxepin-1 1-yl]-pent-4-enoate.
mass spectrum: (Cl, CH4) m/e 309 (M+1)
OH
To synthesize an interrnediate (step 3 of Scheme 1):
Dissolved methyl 2-[6,1 1-dihydro-dibenzo[b,e]oxepin-1 1-yl]-pent-4-
enoate (9.00 g, 0.0292 mol) in 100 mL of dry THF, and cooled to 0~C under a
nitrogen atmosphere. Added 1.0 M lithium aluminum hydride in THF (29.2 mL,
0.0292 mol) via addition funnel. Stirred at room temperature for 45 mins. Added 1
mL of water, 1 mL of 1 N NaOH, then 3 mL of water. Stirred at room temperature for
20 30 mins. then filtered through celite. Washed celite cake with ethyl acetate.Washed filtrate with saturated NaCI, dried with MgSO4, filtered, and evaporated.Purified crude product by flash chromatography on silica gel eluting with 10% ethyl
~cet~te-hexane then 25% ethyl acetate-hexane. Combined appropriate fractions
and evaporated to give 6.78 9 (83% yield) of 2-[6,11-dihydro-dibenzo[b,e]oxepin-~5 1 1-yll-pent-4-en-1-ol.
mass spectrum: (Cl, CH4) m/e 281 (M+1 )
-
-24- 21753~3
HO
To synthesize an intemmediate (step 4 of Scheme 1):
Dissolved oxalyl chloride (3.83 9, 0.0302 mol) in 60 mL of dry
5 dichloromethane, and cooled to -78~C under a nitrogen atmosphere. Added
DMSO (4.3 mL, 4.72 9, 0.0604 mol) dissolved in 15 mL of dry dichloromethane
dropwise via addition funnel. CO and CO2 evolution observed. Stirred at -78~C for
10 mins. Added 2-[6,11-dihydro-dibenzo[b,eloxepin-11 -yl]-pent-4-en-1 -ol (6.77 9,
0.0241 mol) dissolved in 50 mL of dry dichloromethane via addition funnel. Stirred
10 at -78~C for 15 mins. Added triethylamine (10.1 mL, 7.33 9, 0.0724 mol) via
~ddition funnel, and warmed reaction mixture slowly to room temperature. Added
200 mL of water, and separated layers. Extracted aqueous solution with
dichloromethane. Washed combined organic extracts with 0.5 N HCI, saturated
NaCI, dried with MgSO4, filtered, and evaporated. Purified crude product by flash
15 chromatography on silica gel eluting with 10% ethyl acetate-hexane then 15%
ethyl acetate-hexane. Combined appropriate fractions and evaporated to give 6.449 (96% yield) of 2-16,11-dihydro-dibenzo[b,e]oxepin~ yl]-pent-4-en-1-al.
mass spectrum: (Cl/CH4) m/e 278 (M+)
NHMe
IA
For Com~ound IA:
Dissolved 2-16,11-dihydro-dibenzo~b,e]oxepin-11-yl]-pent-4-en-1-al
(5.0 9, 18.0 mmol) in 20 mL of dry THF and 60 mL of dry MeOH. Added 3 A
25 molecular sieves, methylamine hydrochloride (6.1 9, 89.8 mmol), and then sodium
cyanoborohydride (1.13 9, 18.0 mmol). Stirred at room temperature for 23 hours.
Evaporated reaction mixture. Added 80 mL of saturated NaHCO3 and 80 mL of
dichloromethane. Fllteredthrough celite. Separated layers. Extracted aqueous
- -
-25- 21 7531 3
solution with dichloromethane. Dried combined organic extracts with MgSO4,
filtered, and evaporated. Purified crude product by flash chromatography on silica
gel eluting with ~% MeOH-CH2CI2. Combined appropriate fractions and
evaporated to give 3.8 9 (73% yield) of N-methyl-2-[6,11-dihydro-
5 dibenzo[b,e]oxepin~ yl]-pent-4-en-1-amine. Dissolved free base in ethyl ~cet~te,
and added one equivalent of maleic acid dissolved in ethanol. Evaporated to givemaleate salt as a glass.
mass spectrum: (Cl, CH4) m/e 294 (M+1 for free base)
NHMe
K3
For ComDound K3:
Dissolved N-methyl-2-[6,1 1-dihydro-dibenzo[b,e]oxepin-1 1-yl]-pent-4-
en-1-amine (0.50 9, 2.08 mmol) in 1~ mL of absolute ethanol. Added 10 mg of
1~ 10% p~ diurn on carbon catalyst, and stirred under atmospheric hydrogen
balloon for 16 hours. Filtered through Celite, and washed with ethanol.
Evaporated filtrate. Purified crude product by flash chromatography on silica gel
eluting with 5% MeOH-CH2CI2. Combined appropriate fractions, and evaporated
to give 0.43 9 (86% yield) of N-methyl-2-[6,11 -dihydro-dibenzo[b,e]oxepin-1 1-yl]-
20 pentanamine. Dissolved free base in ethyl acetate, and added one equivalent ofmaleic acid dissolved in absolute ethanol. Evaporated to foamy glass.
mass spectrum: (Cl, CH4) m/e 296 (M+1 for free base)
The following compounds were obtained according to a similiar
2~ manner:
~Y
R NR'R"
-
-26- 21 7531 3
X Y R NR'R" salt Mass Spectrum
O H allyl NMe2 maleate (Cl, CH4) m/e 308 (M+1)
O H propyl NMe2 maleate (Cl, CH4) m/e 310 (M+1)
C Cl allyl NHMe maleate (Cl, CH4) m/e 326 (M+1)
C Cl allyl NMe2 maleate (Cl, CH4) m/e 340 (M+1)
C Cl propyl NHMe maleate (Cl, CH4) m/e 328 (M+1)
C Cl propyl NMe2 maleate (Cl, CH4) m/e 342 (M+1)
COO Et K4
For Compound K4:
Dissolved N-methyl-2-[6,11-dihydro-dibenzo[b,e]oxepin-11-yl]-
15 pentanamine (0.50 9, 1.70 mmol) in 20 mL of dry THF. Added triethylamine (0.28
mL, 0.21 9, 2.02 mmol) and ethyl chloroformate (0.18 mL, 0.20 9,1.86 mmol).
Stirred at room temperature for 16 hours. Added water, and extracted with ether.Dried combined organic extracts with MgSO4, filtered, and evaporated. Purified
crude product by flash chromatography on silica gel eluting with dichloromethane.
20 Combined appropriate fractions, and evaporated to give 0.45 9 (73% yield) of ethyl
[2-(6,11-dihydro-dibenzo[b,e]oxepin-1-yl)-pentyll-methylcarbamate as a colorlessoil.
mass spectrum: (Cl, CH4) m/e 368 (M+1)
-- i
-27- 21 7531 3
The following compounds were obtained according to a similar
manner
~Y
R'
X Y R R' mp Mass SDectrum
O H propyl acetyl oil (Cl, CH4) rn/e 338 (M+1)
O H allyl acetyl oil (FAB) m/e 336 (M~1)
O H allyl COOEt oil (FAB) m/e 366 (M+1)
C Cl allyl acetyl oil (Cl, CH4) Tle368 (M+1)
Me2N O
t 5 To synthesize an intermediate (step 6 of Scheme 2):
Washed sodium hydride (2.6 9, 64.6 mmol, 60 weight % in oil) two
times with hexane under a nitrogen atmosphere. Added 50 mL of dry ether, and
then added c~iber,~osuberol (13.6 9, 64.6 mmol) dissolved in 30 mL of dry ether
followed by 2-chloro-N,N-dimethylacetamide (7.8 9, 64.6 mmol) dissolved in 20 mLof dry ether. Stirred at room temperature for 16 hours. Added water, and
separated layers. Evaporated the organic solution, and triturated the residue with
carbon tetrachloride and hexane. Filtered the white solid to give 13.79 9 (72%
yield) of 10,11-dihydro-5H-dibenzo[a,d~cyclohepten-5-yl-oxy-N,N-
dimethylacetamide.
mp=73-75~C
mass spectrum: (FAB) m/e 193 (M-Me2NCOCH2O)
2 1 753 1 3
- 28 -
Ht~O
NMe2
To Synthesize an intemmediate (step 7 of Scheme ~):
Washed sodium hydride (0.9 9, 22 mmol, 60 weight % in oil) two
times with hexane under a nitrogen atmosphere. Added 100 mL of dry benzene
and 10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl-oxy-N,N-dimethylacetamide
(6.0 9, 20.3 mmol). Refluxed for 3 hours, and then cooled to room temperature.
Carefully added water, and filtered to give the first crop of product. Separated the
filtrate. Dried the organic solution with MgSO4, filtered, and evaporated to an oil
which was crystallized with ether. Filtered the solid, and combined with the first
crop. Recryst~ ed product from isopropanol to give 3.59 9 (60% yield) of 10,11-
dihydro-N,N-dimethyl-5H-dibenzo[a,d]cycloheptene-5-glycolamide as a white
solid.
mp=210-21 4~C
mass spectrum: (FAB) m/e 296 (M+1)
EtO~p
NMe2 IE
For Com~ound IE:
Washed sodium hydride (2.4 9, 60 mmol, 60 weight % in oil) two
times with hexane under a nitrogen atmosphere. Added 20 mL of dry dioxane and
10,11-dihydro-N,N-dimethyl-5H-dibenzo[a,d]cycloheptene-5-glycolamide (5.0 9,
16.9 mmol) dissolved in 350 mL of dry dioxane and 150 mL of dry DMF dropwise
via addition funnel. Added ethyl iodide (9.54 g, 4.5 mL, 60 mmol), and refluxed the
reaction mixture for 5 hours. Added minimal water (10 mL), and evaporated.
Added dichloromethane, and separated layers. Dried the organic solution with
MgSO4, filtered, and evaporated. Triturated the crude produc~ from petroleum
2 1 7 53 1 3
- 29 -
ether to give 5.25 9 (96% yield) of 2-ethoxy-2-~10,11 -dihydro-5H-
dibenzo~a,d]cyclohepten-5-yl]-N,N-dimethyl-acetamide as a white solid.
mp=86-90~C
mass spectrum: (FAB) m/e 324 (M+1)
EtO
NMe2 IB
For Compound IB:
Dissolved 2-ethoxy-2-~10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-
10 yl]-N,N-dimethyl-acetamide (4.45 9, 13.8 mmol) in 100 mL of dry tetrahydrofuran,
and added lithium aluminum hydride (0.6 9, 15.8 mmol) portionwise under a
nitrogen atmosphere. Heated the reaction mixture at 60~C for 2 hours. Cooled to
0~C, and carefully added 0.6 mL of water, 0.6 mL of 15 weight % NaOH, and then
1.8 mL of water in order to precipitate aluminum salts. Filtered precipitate, and
15 washed with tetrahydrofuran. Evaporated filtrate, and purified the crude product by
flash chromatography on silica gel eluting with 1:1 ethyl acetate:hexane.
Combined appropriate fractions, and evaporated to give 3.44 9 (81% yield) of ~-
ethoxy-10,11-dihydro-N,N-dimethyl-5H-dibenzo[a,d]cycloheptene-~-ethanamine
as a colorless oil. Dissolved free base in ethyl acetate, and added one equivalent
20 of maleic acid dissolved in methanol. Evaporated, and added ether to precipitate
maleate salt.
mp=138-141~C
mass spectrum: (FAB) m/e 310 tM+1 for free base)
- -
-30- 2175313
To synthesize an intermediate (steP 12 of Scheme 3~:
Dissolved 4-(10,1 1-dihydro-5H-dibenzo[a,d]cyclohepten-5-ylidene)-
1-methylpiperidine (12 9, 0.042 mol) in 250 mL of dry THF. Cooled to -78~C undera nitrogen atmosphere. Added n-butyl lithium (18 mL of 2.5 M in hexane) dropwisevia ~d~ition funnel. Maintain temperature at 0~C for 1 hour then recooled to -78~C.
Added 21 mL of dry methanol, and let warm to room temperature. Added saturated
NH4CI, and extracted with THF. Dried combined organic extracts with MgSO4,
filtered, and evaporated to give 11.3 9 (94% yield) of 4-(10,11-dihydro-5H-
dibenzo[a,dlcyclohepten-5-yl)-1-methyl-1,2,5,6-tetrahydropyridine as a white solid.
mp=96-98~C
mass spectrum: (FAB) m/e 290 (M+1 )
To synthesize an intermediate (steD 13 of Scheme 3~:
Dissolved 4-(10,1 1-dihydro-5H-dibenzo[a,d]cyclohepten-~-yl)-1-
methyl-1,2,5,6-tetrahydropyridine (10.5 9, 0.036 mol) in 100 mL of glacial acetic
acid and 60 mL of absolute ethanol. Added platinum oxide catalyst (1.5 9). Shakeon Paar shaker at 60 psi of hydrogen pressure for 24 hours. Filtered, and washedcatalyst with ethanol. Evaporated filtrate. Purified crude product by flash
chromatography on silica gel eluting with 17% MeOH-EtOAc. Combined
appropriate fractions, and evaporated to give 5.7 9 (54% yield) of as a white solid.
mp=88-89~C
-31- 21 753 1 3
mass spectrum: (FAB) m/e 292 (M+1)
~1-
To synthesize an intermediate (step 14 of Scheme 3):
Dissolved 4-(10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-1-
methyl-piperidine (1.0 9, 3.4 mmol) in 10 mL of methanol. Added iodomethane
(0.71 9, 5.0 mmoi), and stirred at room temperature for 18 hours. Filtered the
precipitate to give 1.22 9 (83% yield) of 4-(10,11-dihydro-5H-
dibenzo[a,d]cyclohepten-5-yl)-1,1-dimethyl-piperidinium iodide as a white solid. mp~300~C
mass spectrum: (FAB) m/e 306 (M-iodide for salt)
~q~
OH-
To synthesize an intermediate:
Mix 4-(10,11 -dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-1,1 -dimethyl-
piperidinium iodide (1.14 9, 2.6 mmol) and silver oxide (3.48 9, 15.0 mmol) in 150
mL of methanol and 15 mL of water. Stirred at room temperature for 24 hours.
Filtered, and evaporated filtrate to give 0.85 (100% yield) of 4-(10,11-dihydro-5H-
dibenzo[a,d~cyclohepten-5-yl)-1,1-dimethyl-piperidinium hydroxide as a white
solid.
mp=168-171~C
mass spectrum: (FAB) m/e 306 (M-hydroxide for salt)
-
-32- 21 7531 3
lc
For Compound IC:
Heated 4-(10,11~ihydro-5H-dibenzo[a,d]cyclohepten-5-yl)~
dimethyl-piperidinium hydroxide (0.84 9, 2.6 mmol) in a 180-185~C oil bath for 3hours. Cooled to room temperature, and purified the crude product by flash
chromatography on silica gel eluting with 10% MeOH-CH2Cl2. Combined
appropriate fractions, and evaporated to ~ive 0.3 9 (37~~O yield) of 3-(10,11-dihydro-
5H-dibenzo[a,d]cyclohepten-5-yl)-N,N-dimethyl-4-pentenamine as an oil.
Dissolved free base in absolute ethanol, and added 28 weight% HCl-EtOH until
acidic. Evaporated, and added 5:1 ether:ethyl acetate. Let stand to preci~ilate
hydrochloride salt.
mp= 173-175~C
mass spectrum: (Cl, CH4) m/e 306 (M+ 1)
As mentioned above, the compounds of formula I exhibit
good anti-TNF-oc activity. The compounds of the invention are,
therefore, useful when TNF-a activity is a factor in a given disease or
disorder such as in the case of septic shock and various allergic diseases
and inflammatory conditions.
The anti-TNF-a properties of the compounds of the present
invention may be demonstrated by use of a standard in vitro
pharmacological testing procedure as described below. This test
procedure is a standard test used to determine anti-TNF-a activity and
to evaluate the usefulness of said compounds for counteracting the
biological effects of TNF-a.
1. In Vitro Study: Inhibition of LPS-Induced TNF-a
Production From the Murine Cell Line WEHI-265
-33- 21 7531 3
1) Cells (obtained from cell cultures containing <106 cells/ml) are
suspended at 0.2x106 cells/ml in complete medium (RPMI1640,
with 10% FCS, 10-5 M 2-ME, 2 mM glutarnine and 10 mM HEPES
buffer) and plated in CoStar 24 well plates (1.0 mVwell).
2) Compounds are dissolved in the appropriate vehicle at 400 times
the concentration to be tested, and 5 ~l of compound is added to
the wells.
3) LPS (from E. coli Olll:B4) is diluted to 6 llg/ml and 1.0 ml is
added to wells.
4) Plates are incubated 20-24 hours in 37~ C02 incubator.
5) Supernatant fluids are collected and analyzed for TNF content as
described in J. Immunol., 142:3884.
The Results of this procedure are shown in TABLE 1 below.
J
- - -
34 21 7531 3
TABI F 1
COMPOUNI ) % INHIBITION AT 1
IA 54
IB 35
IC 46
IE 49
IF 4
IG 35
IH 23
IJ 34
IK 31
IL 49
IM 50
IN 28
K1 15
K2
K3 73
K4 48
K5 46
K6 64
K7 39
In addition to the in vitro test described above, the
5 following in vivo test was also performed on several of the compounds
of the present invention. Although the individual reported values may be
subject to a wide margin of error, collectively the in vivo data demonstrates that the
compounds of the invention are inhibitors of TNF-a in a mammalian species.
L
2 1 753 1 3
- 35 -
2. ~n Vivo Study- Inhibition of LPS-rnduced Serum TNF
1) Mice (C57BI/6J males, 6 - 8 weeks of age) are dosed with the
indicated compound (dissolved in CMC suspension vehicle;
compounds are given oraIly or i.p. one hour before LPS chalIenge).
2) Mice are challenged with LPS (from E. coli 0111:B4; 50 ~lg i.p.).
3) Mice are bled 90 min after LPS challenge.
4) Sera are analyzed for TNF content by ELISA as described in J.
Immunol. 142:3884.
Results are shown in TABLE 2 below.
TAB~.F. 2
COMPOUND % INH~BITION AT 25 MG/KG
IA 44
IB 25
IE 37
IF 24
IG 5 1
IH 30
IL 42
K1 71
K2 40
K3 27
K7 58
The effect of the compounds of the present in- ention against
septic shock may be demonstrated by use of a standard pharmacological
testing procedure as described below. This test procedure is a standard
20 test used to determine activity against septic shock.
2 1 753 1 3
- 36 -
3. In Vivo Study: Inhibition of
LPS/Galactosamine-Induced Lethalitv
1) Mice (C57BV6J males, 6 - 8 weeks of age) are dosed with the
indicated compound (dissolved in CMC suspension vehicle;
compounds are given orally or i.p. one hour before challenge with
LPS and d-galactos~mine).
10 2) Mice are challenged i.p. with a mixture of LPS (from E. coli
0111:B4; 100 ng) and d-galactosamine (8 mg).
3 ) Survival is determined 24 hours after challenge. See procedure
published in J. Exp. Med. 165:657 (1987)
Results are shown in TABLE 3 below.
TABLE 3
COM~O~JND ~ DEAD/TOTAL AT 25 MG/KG
IB 8/10
E 10/10
IF 8/1 0
IG
K1 1/10
K2 7/10
K5 10/10
For preparing pharmaceutical compositions from the
compounds described by this invention, inert, pharmaceutically
acceptable carriers can be either solid or liquid. Solid form preparations
include powders, tablets, dispersible granules, capsules, cachets and
suppositories. The powders and tablets may be comprised of from
about 5 to about 70 percent active ingredient. Suitable solid carriers are
known in the art, e.g. magnesium carbonate, magnesium stearate, talc,
sugar, lactose. Tablets, powders, cachets and capsules can be used as
solid dosage forms suitable for oral administration.
37 21 7531 3
For preparing suppositories, a low melting wax such as a
~IU1e of fatty acid glycerides or cocoa butter is first melted, and the
active ingredient is dispersed homogeneously therein as by stirring.
5 The molten homogeneous mixture is then poured into convenient sized
molds, allowed to cool and thereby solidify.
Liquid form preparations include solutions, suspensions and
emulsions. As an example may be mentioned water or water-propylene
10 glycol solutions for parenteral injection.
Liquid form preparations may also include solutions for
intranasal administration.
Aerosol preparations suitable for inhalation may include
soludons and solids in powder form, which may be in combination with
a pharmaceutically acceptable carrier, such as an inert compressed gas.
Also included are solid form preparations which are
20 intended to be converted, shortly before use, to liquid form
preparations for either oral or parenteral ~dministration. Such liquid
forms include solutions, suspensions and emulsions.
The compounds of the invention may also be deliverable
25 t~ansdermally. The ~ansdermal compositions can take the form of
creams, lotions, aerosols and/or emulsions and can be included in a
transdermal patch of the matrix or reservoir type as are conventional in
t~e art for this purpose.
Preferably the compound is administered orally.
Preferably, the pharmaceutical preparation is in unit dosage
form. In such form, the preparation is subdivided into unit doses
containing appropriate quantities of the active component, e.g., an
35 effective amount to achieve the desired purpose.
~175313
- 38 -
The quantity of active compound in a unit dose of
preparation may be varied or adjusted from about 0.1 mg to 1000 mg,
more pleferably from about 1 mg. to 300 mg, according to the
particular application.
The actual dosage employed may be varied depending upon
the requirements of the patient and the severity of the condition being
treated. Dete1mination of the proper dosage for a particular situation is
within the s~ill of the art. Generally, treatment is initiated with sm~ller
10 dosages which are less than the optimum dose of the compound.
Thereafter, the dosage is increased by small increments until the
optimum effect under the circulllstances is reached. For convenience,
the total daily dosage may be divided and ~dministered in portions
during the day if desired.
1!;
The amount and frequency of allministration of the
compounds of the invention and the pharmaceutically acceptable salts
thereof will be regulated according to the judgment of the attending
clinician considering such factors as age, condition and size of the
20 patient as well as severity of the symptoms being treated. A typical
recommended dosage regimen is oral a-lministration of from 10 mg to
2000 mg/day preferably 10 to 1000 mg/day, in two to four divided
doses to achieve relief of the symptoms.
DOSAGE FORMS
The following are e~amples of pharmaceutical dosage forms
which contain a compound of the invention. As used therein, the term
"active compound" is used to designate the compound
c$,~
I~ (IA).
~ NK:~ 3
,
,3g_ 21 7531 3
The scope of the invention in its pharmaceutical composition
aspect is not to be limited by the examples provided, since any other
compound of Formula I can be substituted into the pharmaceutical
composition examples.
Pharmaceutical Dosage Form Examples
EXA.~P~ F. A
Tablets
No. Ingredients mg/tablet mg/table
1 . Active compound 10 0 5 0 0
2. Lactose USP 122 113
3. Corn Starch, Food Grade, 30 40
as a 10% paste in
Purified Water
4. Corn Starch, Food Grade 45 40
5. Magnesium Stearate 3 7
Total 300 700
Method of Manufacture
Mix Item Nos. 1 and 2 in a suitable rnixer for 1~15
minnteS. Granulate the mixture with Item No. 3. Mill the damp
granules through a coarse screen (e.g., 1/4", 0.63 cm) if necessary. Dry
the damp granules. Screen the dried granules if necessary and mi~
15 with Item No. 4 and mix for 1~15 minutes. Add Item No. S and mix
for 1-3 minutes. Compress the mixture to appropriate size and weigh
on a suitable tablet machine.
FXAMp~ F. B
Cap~ules
No. In~redient m /car~sule m~/car)sule
1 . Active compound 1 0 0 5 0 0
- -
2175313
~ 40 -
2. Lactose USP 106 123
3. Corn Starch, Food Grade 40 70
4. Magnesium Stearate NF 7 7
Total 2 5 3 7 0 0
Method of Manufacture
Mix Item Nos. 1, 2 and 3 in a suitable blender for 10-15
minutes. Add Item No. 4 and mix for 1-3 minutes. Fill the mixture into
5 suitable two-piece hard gelatin capsules on a suitable encapsulating
machlne.
While the present invention has been described in
conjunction with the speciffc embodiments set forth above, many
t 0 alternatives, modifications and variations thereof will be apparent to
those of ordinary skill in the art. All such alternatives, modifications
and variations are intended to fall within the spirit and scope of the
present invention.