Note: Descriptions are shown in the official language in which they were submitted.
WO 95/13379 PCTIU594J12976
2175703
1
DESCRIPTION
Alteration of Seauence of a Target Molecule
This invention relates to therapy of diseases using
ribozymea.
The following is a brief history of the discovery and
activity of enzymatic RNA molecules or ribozymes. This
history is not meant to be complete but is provided only
for understanding of the invention that follows. This
summary is not an admission that all of the work described
below is prior art to the claimed invention.
Prior to the 1970s it was thought that all genes were
direct linear representations of the proteins that they
encoded. This simplistic view implied that all genes were
like ticker tape messages, with each triplet of DNA
"letters" representing one protein "word" in the transla
tion. Protein synthesis occurred by first transcribing a
gene from DNA into RNA (letter for letter) and then trans-
lating the RNA into protein (three letters at a time). In
the mid 1970s it was discovered that some genes were not
exact, linear representations of the proteins that they
encode. These genes were found to contain interruptions
in the coding- sequence which were removed from, or
"spliced out" of, the RNA before it became translated into
protein. These interruptions in the coding sequence were
given the name of intervening sequences (or introns) and
the process of removing them from the RNA was termed
splicing.--A general reference for spliceosomes and how
they are- related to self-splicing introns is Guthrie, C.,
253 Scier~ce 157, 1991. After the discovery of introns,
two questions immediately arose: (i) why are introns
present in genes in the first place, and (ii) how do they
get removed from the RNA prior to protein synthesis? The
first question is still being debated, with no clear
answer yet available. The second question, how introns
get removed from the RNA, is much better understood after
WO 95113379 PCT/US94112976
X175703
2
a decade and a half of intense research on this question.
At least three different mechanisms have been discovered
for removing-introns from RNA. Two of these splicing
mechanisms involve the binding of multiple protein factors
which then act to correctly cut and join the RNA. A third ' F
mechanism involves cutting-and joining of the RNA by the
intron itself, in what was the first discovery of-catalyt-
ic RNA molecules.
Cech and colleagues were trying to understand how RNA
splicing was accomplished in a single-celled pond organism
called _T~trahvmena thermoDhs~a They had chosen
Tetrahvmena thermobhila as a matter of convenience, since
each individual cell contains over 10,000 copies of one
intron-containing gene (the gene for ribosomal RNA). They
reasoned that such a large number of intron-containing RNA
molecules would require a large amount of (protein) splic-
ing factors to get the introns removed quickly. Their
goal was to purify these hypothesized splicing factors and
to demonstrate that the purified factors could splice the
intron-containing RNA,i~ vi r . Cech rapidly succeeded in
getting RNA splicing to work in vitro, but something funny
was going on.- -As expected, splicing occurred when the
intron-containing RNA was mixed with protein-containing
extracts from Tetrahymena, but splicing also occurred when
the protein extracts were left out. Cech.proved that the
intervening sequence RNA was acting as its own splicing
factor to snip itself out of the surrounding RNA. They
published this startling discovery in 1982. Continuing
studies in the early 1980's served to elucidate the
complicated structure of the Tetrahvmena intron and to
decipher the mechanism by which self-splicing occurs. -
Many research groups helped to demonstrate that the
specific folding of the Tetrahymena_intron is critical for
bringing-together the parts of the RNA that will be cut
and spliced. Even after- splicing is complete, the
released intron maintains its catalytic structure. As a
consequence, the released intron is capable of carrying
W O 95113379 PCTIUS94112976
217~7~3
i
3
out additional cleavage and splicing reactions on itself
(to form intron circles). By 1986, Cech was able to show
that a shortened form of the Tetrahvmena intron could
carry out a variety of cutting and joining reactions on
other pieces of RNA. The demonstration proved that the
Tetrahvmena intron canact as_a true enzyme,: (i) each
intron molecule was able to cut many substrate molecules
while the intron molecule remained unchanged, and (ii)
reactions were specific for RNA molecules that contained
a unique sequence (CUCU) which allowed the intron to
recognize and bind the RNA. Zaug and Cech coined the term
"ribozyme" to describe any ribonucleic acid molecule that
hae enzyme-like properties. Also in 1986, Cech showed
that theRNA substrate sequence recognized by the
Tetrahymena ribozyme could be changed by altering a
sequence within the ribozyme itself. This property has
led to the development of a number of site-specific
ribozymes that have been individually designed to cleave
at other RNA sequences. The Tetrahvmeria intron is the
most well-studied of what is now recognized as a large
class of introns, Group I -introns. The overall folded
structure, including several sequence elements, is
conserved among the Group I_ introns, as is the general
mechanism of splicing. Like the Tetrahvmena intron, some
members of this class are catalytic, i.e.. the intron
itself is capable of the self-splicing reaction. Other
Group I introns require additional (protein) factors,
presumably to help the intron fold into and/or maintain
its active structure. While the Tetrahvmena intron is
relatively large, (413 nucleotides) a shortened form of
at
least one other catalytic intron (SunY intron of phage T4,
180 nucleotides) may prove advantageous not only because
. of its smaller-size but because it undergoes self-splicing
at an even faster rate than the Tetrahvmena intron.
Ribonuclease P (RNAseP) is an enzyme comprised of both
RNA and protein components which are responsible for con-
verting precursor tRNA molecules into their final form by
WO 95/13379 PCTIUS94112976
2~ 7 5~ ~3
4
trimming extra RNA off one of their ends. RNAseP activity
has been found in all organisms tested, but the bacterial
enzymes have been the most studied. The function of
RNAseP has been studied since the mid-1970s by many labs.
In the late 1970x, Sidney Altman and his colleagues showed
that the RNA component of _RNAaeP is essential for its
processing activity; however, they also showed that the
protein component also was required for processing under
their experimental conditions. After Cech's discovery of
self-splicing by the Tetrahvmena intron, the requirement
for both protein and RNA components in RNAseP was reex-
amined. In 1983, Altman and Pace showed that the RNA was
the enzymatic component of- the RNAaeP complex. This
demonstrated that an RNA molecule was capable of acting as
a true enzyme, processing numerous tRNA molecules without
itself undergoing any change. The folded structure of
RNAseP RNA has been determined, and while the sequence is
not strictly conserved between RNAs from different organ-
isms, this higher order structure is. It is thought that
the protein component of the BNAseP complex may serve to
stabilize the folded RNA in 'vo. At.least one RNA posi-
tion important both to substrate recognition arid to
determination of the cleavage site has been identified,
however little else is known about the active site.
Because tRNA sequence recognition is minimal, it is clear
that some aspects) of the tRNA structure must also be
involved in substrate recognition and cleavage activity.
The size of RNAseP RNA (>350 nucleotides), and the com-
plexity of the substrate recognition, may limit the
potential for the use of an RNAseP-like RNA in thera-
peutics. However, the size--of RNAaeP is being trimmed '
down (a molecule of only 290 nucleotides functions
reasonably well). In addition, substrate recognition has
been simplified by the recent discovery that RNAseP RNA
can cleave small RNAs lacking the natural tRNA secondary
structure if an additional RNA (containing a "guide's
PCTIUS94I12976
WO 95113379
sequence and a sequence element naturally present at the
end of all tRNAs) is present-as well.
Symons and colleagues identified two examples of a
self-cleaving RNA that differed from other forms of
5 catalytic RNA already reported- Symons was studying the
propagation of the avocado sunblotch viroid (ASV), an RNA
virus that infects avocado plants. Symons demonstrated
that as little- as 55 nucleotides of the ASV RNA was
capable of folding in such a way as to cut itself into two
pieces. It is thought that j,~ vivo self-cleavage of these
RNAs is responsible- for cutting the RNA into single
a=nome-length pieces during viral propagation. Symons
discovered- that variations on the minimal catalytic
sequence from ASV could be found in a number of other
plant pathogenic RNAa as well. Comparison of these
sequences revealed a common structural design consisting
of three. stems and loops connected by central loop con-
taining many conserved (invariant from one RNA to the
next) nucleotides. The predict-~d secondary structure for
this catalytic RNA reminded the researchers of the head of-
a hammer; thus it was named as such. Uhl: peck was
successful in separating the catalytic region of the
ribozyme from that of the substrate. Thus, it became
possible to assemble a hammerhead ribozyme from 2 (or 3)
small synthetic RNAs. A-19-nucleotide catalytic region
and a 24-nucleotide -substrate were sufficient to support
specific cleavage. The catalytic domain of numerous
hammerhead ribozymes have now been studied by both the
Uhlenbeck and Symons groups with regard to defining the
nucleoides required for specific assembly and catalytic
activity and determining the rates of cleavage under
various conditions.
Haseloff and Gerlach showed it was possible to divide
the domains of the hammerhead ribozyme in a different
manner. By doing so, they placed most of the required
sequences in the strand that didn't get cut (the ribozyme)
and only a required UH where H = C, A, or B in the strand
WO 95/13379 PCT/US94/12976
21 ~ 5103
that did get cut (the substrate). This resulted in a
catalytic ribozyme that could be designed to cleave any UH
RNA sequence embedded within a longer "substrate recogni-
tion" sequence. The specific cleavage of a long mRNA, in
a predictable manner using several such hammerhead
ribozymes, was reported in 1988.
One plant pathogen RNA (from the negative strand of
the tobacco ringspot virus) undergoes self-cleavage but
cannot be folded into the consensus hammerhead structure
described above. Bruening and colleagues have indepen-
dently identified a 50-nucleotide catalytic domain for
this RNA. In 1990, Hampel and Tritz succeeded in dividing
the catalytic domain into--two partsthat could act as
substrate and ribozyme in a-multiple-turnover, cutting
reaction. As with the hammerhead ribozyme, the hairpin
catalytic portion contains most of the sequences required
for catalytic activity while only a short.sequence (GUC in
this case) is required in the target. Hampel and Tritz
described the folded structure of this RNA as consisting
of a single hairpin and coined the term "hairpin" ribozyme
(Bruening and colleagues use the term "paper clip" for
this ribozyme motif). -Continuing experiments suggest an
increasing number of similarities between the hairpin and
hammerhead ribozymes in respect to both binding of target.
RNA and mechanism of cleavage. At the same time, the
minimal size of the hairpin ribozyme is still 50-60%
larger than the minimal hammerhead ribozyme.
Hepatitis Delta Virus (HDV) is a virus whose genome
consists of single-stranded RNA. A small region (-.80
nucleotides) in both the genomic RNA, and in the comple
mentary anti-genomic RNA, is sufficient to support
self-cleavage. As the most recently discovered ribozyme,
HDV's ability to self-cleave has only been studied for a
few years, but is interesting because of its connection to
a human disease. In 1991, Been and Perrotta proposed a
secondary structure for the I3DV RNAs that is conserved
betweenthe genomic and anti-genomic RNAa and is necessary
W O 95113379 ~ , ~ J ~ ~ ~ PCTIUS94112976
7
for catalytic activity. Separation of the HDV RNA into
"ribozyme" and "substrate" portions has recently been
achieved by Been, but the rules for targeting different
substrate RNAs have not yet been determined fully. Been
has also succeeded in reducing the size of the HDV
ribozyme to -.60 nucleotides.
The table below lists some of the characteristics of
the ribozymes discussed above:
TABLE 1
rh-,-acteristics of ribozvmes
Group I Introns
Size: -300 to >1000 nucleotides.
Requires a U in the target sequence immediately 5' of the
cleavage site.
Binds 4-6 nucleotides at 5' side of cleavage site.
Over 75 known members of this class. Found in Tetrahvmena
thermobhila rRNA, fungal mitochondria, chloroplasts, phage
T4, blue-green algae, and others.
RNAseP RNA (M1 RNA)
Size: -290 to 400 nucleotides.
RNA portion of a ribonucleoprotein enzyme. Cleaves tRNA
precursors to form mature tRNA.
Roughly 10 known members of this group all are bacterial
in origin.
Hammerhead R~bozvme
~ Size: ~30 to 40 nucleotides.
Requires the target sequence UH immediately 5' of the
~ cleavage site.
Binds a variable number nucleotides on both sides of the
cleavage site.
14 known members of this class. Found in a number of
plant pathogens (virusoids) that use RNA as the infectious
agent.
R'O 95/13379
PCTIUS94/12976
1151 ~3
8
Fia~ Yp~ n R~ bozvm
Size: -50 nucleotides.
Requires the target sequence GUC immediately 3' of the
cleavage site.
Binds 4 nucleotides at 5' side of the cleavage site and a
variable number to the 3' side of the cleavage site.
Only 1 known member of this class. Found in one plant
pathogen (satellite RNA of- thetobacco ringspot virus)
which uses RNA as the infectious agent.
Hepatit;a Dei a Vim
(HDTT1 17; hnv _
Size: -60 nucleotides (at present).
Cleavage of target RNAs recently demonstrated.
Sequence requirements not fully determined.
Binding sites and structural requirements not -fully
determined, although no sequences 5' of cleavage site are
required.
Only 1 known member of this class. Found in human HDV.
As the term is used in this application, ribozymes are
RNA molecules having an enzymatic activity which is able
to cleave and splice other separate RNA mol-ecules in a
nucleotide base sequence specific manner. Such enzymatic
RNA molecules can be targeted- to virtually any RNA
transcript, and efficient cleavage and splicing achieved
.~ vitro. Kim et al., 84 Proc. Nat Pcad of ~r.; rrer
8788, 1987, Hazeloff et al., 234 Na ,r 585, 1988, Cech,
260 AJ N~ 3030, 1988, and Jefferies et al., I7 Nu ~ ; p ;d
Research 1371, 1989.
Ribozymes act by first binding to a target RNA. Such
binding occurs through the target RNA binding portion of
a ribozyme which is held in close proximity to an
enzymatic portion of the RNA which acts to cleave the
target RNA. Thus, the ribozyme first recognizes and then
binds a target RNA through complementary base-pairing, and
once bound to the correct site, acts to cut and splice the
target RNA. Strategic cleavage and splicing of such a
CA 02175703 2002-09-16
76909-169
9
target RNA will destroy its ability to direct synthesis of
an encoded protein. After a ribozyme has bound, cleaved
and spliced its RNA target it is released from that RNA.
By the phrase "catalytic" or "enzymatic RNA molecule"
is meant an RNA molecule which has complementarity in a
substrate binding region to a specified gene target, and
also has an enzymatic activity wh:~~h is active to
.specifically cleave and splice RNA in shat target. That
is, ._~e. $.nzymaGic-RNA-mo~.ecule-- is-- able to iritermolecularly
cleave and splice RNA and thereby alter a target RNA
molecule. This complementarity functions to allow
sufficient hybridization of the enzymatic RNA molecule to
the target RNA to allow the cleavage to occur. 100%
complementarity is preferred, but complementarity as low
as 50-75% may also be useful in this invention..
In preferred embodiments of this invention, the
enzymatic RNA molecule is formed in a hammerhead motif,
but may also be formed in the motif of a hairpin,
hepatitis delta virus, group I intron or RNAsE~P RNA (in
association with an RNA guide s,equence). Examples of such
hammerhead motifs are described by Rossi et al., 8 AIDS
RESEARCH AND HUMAN RETROVIRUSES 183, 1992,
Hampel and Tritz, 28 Biochemistrv
4929, 1989 and Hampel et 'al. , 18 Nucl,e~c Acids Research
299, 1990, and an example of the hepatitis delta virus
motif is described by Perrotta and Been, 31 Biochemistry
16, 1992, of the RNAseP motif by Guerrier-Takada et al.,
Cell 849, 1983, and of the group I intron by Cech et
al., U.S. Patent 4,987,071. These specific motifs are not
limiting in the invention and those skilled in the art
will recognize that all that is important in an enzymatic
35 RNA molecule of this invention is that it has a specific
substrate binding site which is complementary to one or
more of the target gene RNA regions, and that it have
WO 95/13379 PCT/US94/12976
a C ~ ~ '~ Q3
1~
nucleotide sequences within or-surrounding that substrate
binding site which impart an RNA cleaving activity to the
molecule.
The invention provides a method for designing a class _
of enzymatic cleaving and splicing agents which exhibit a
high degree of specificity for the RNA of a desired
target. The ribozyme molecule is preferably targeted to
a highly conserved sequence region of a-target such that
specific treatment of a disease or condition can be
provided with a single ribozyme. Such enzymatic RNA
molecules can be delivered exogenously to specific cells
as required.
Synthesis of ribozymes greater than 100 nucleotides in
length is very difficult using automated-methods, and the
therapeutic cost of - such molecules is prohibitive.
However, delivery of such ribozymes by expression vectors
is primarily feasible using-~x v'vo treatments.
moue et al., 43 Cell -431, 1985, state that short
oligonucleotides of 2-6 nucleotides can undergo inter
molecular exon ligation or splicing in traps, It indi
cates that "long 5' exons should be reactive provided that
three conditions are met: the exonmust have-a 3'
hydroxyl group, it must terminate in a sequence similar to
that of the 3' -end of the 5' exon, and the 3' terminal
sequence must be available as opposed to being tied up in
some secondary structure. Thus, it appears that exon
switching is possible in this system, though limited by
the availability of alternative 5' exons that meet the
above criteria.- These could-include transcripts that are
not 5' exons from other precursors, since RNA polymerases
always leave 3' hydroxyl ends". .
~ummarv of the Invention
This invention features a method in which natural
transcripts are altered by use of a splicing reaction ~
v'vo or ~ v' ro. It involves the manipulation of genetic
CA 02175703 2002-09-16
76909-169
11
information to ensure that a useful transcript is provided
within a cellular system or extract.
In a first aspect, the invention features a method for
splicing a target nucleic acid molecule with a separate
nucleic acid molecule. Such splicing-generally causes
production of a chimerie protein with advantageous
features over that protein naturally produced from the
target nucleic acid prior 'to splicing. The method
includes contacting the target nu._-cleic. acid mole~u~.e -w-ith
a catalytic nucleic acid molecule including the separate
nucleic acid molecule. Such contacting is performed under
conditions in which at least a portion of the separate
nucleic acid molecule is spliced with at least a portion
of the target nucleic acid molecule to form a chimeric
nucleic acid molecule. In this method, the catalytic
nucleic acid molecule is chosen so that it is not
naturally associated with the separate nucleic acid
molecule.
The target nucleic acid molecule can be~any desired
molecule with which a splicing reaction can occur.
Generally, this will be an RNA molecule, preferably a
messenger RNA molecule, but it may also include molecules
that have one or more non-ribonucleotides substituents,
such as deoxyribonucleotides or other analogs as described
by Eckstein et al. EP90j01731.
Generally, the target nucleic acid molecule is present
within a cell and is chosen or targeted because it encodes
a defective protein or is deleterious to that cell.
Splicing of the separate nucleic acid molecule with such
a target nucleic acid molecule is designed to alter the
protein product of that nucleic acid molecule. Such
alteration causes production of a useful protein which
will allow that cell to either survive or die, as desired.
Thus, for example, in a gene therapy setting, the target
nucleic acid molecule may encode a non-functional protein
necessary for normal life. This molecule can be spliced
WO 95/13379 PCT/U594/12976
2~~~~03
i
12
with a separate nucleic acid molecule to allow appropriate
expression of a functional protein. Alternatively, the
splicing may cause production of a more stable protein,
or of a protein which acts as an agonist or antagonist of
a function, e-g., a viral ,or bacterial replication
function.
The separate nucleic acid molecule is generally chosen
such that it encodes a 3' exon which it is desirable to
express within a cell. This exon will generally not
include control sequences such as promoter regions, but
may include poly(A) tails and other stabilizing or
enhancing functions well known in the art. As with the
target nucleic acid molecule, the separate nucleic acid
molecule generally is a ribonucleic acid molecule but may
be substituted as described above.
By "enzymatic" or "catalytic nucleic acid molecule" is
meant a molecule having a motif generally as described
above in the Background of the Invention, which and is
preferably selected from the moti~ of a group I or group
II intron having a cleavage and splicing activity.
Alternatively, the splicing or cleavage activity may be
provided by a different nucleic-acid molecule, or may
supplement the catalytic nucleic acid molecule. Those of
ordinary skill in the art will recognize that other motifs
than those of the group I and group II introns may also be
manipulated to provide useful splicing activity.
The conditions chosen for the contacting step may be
those naturally occurring within a cell, or may be manipu-
lated ~n v'troto ensure that the splicing reaction will
occur. These conditions are well known to those in the
art, for example, as described by Inoue et al., sur~ra. ,
By at least a portion of the respective nucleic acid
molecules is meant that the 5' end of the target nucleic.
acid molecule will be spliced with the 3' end of the
separate nucleic acid molecule. Such a portion may be
only a few nucleotides (10-500 nucleotides) or may be
significantly greater and may represent almost all of a
CA 02175703 2002-09-16-
76909-169
1. 3
molecule encoding a gene product (i.e., at least 1 to 5
kbases) .
Tre chimeric nucleic acid molecule is one which may
occur naturally in nature but is not present prior to the
splicing reaction. Alternatively, it may be a completely
novel structure which does not occur in nature, but which
is useful in gene therapeutic treatment of an organism.
The catalytic nucleic acid molecule is not naturally
assoc3at-ed-w3t~ the separate nucleic acid tttoie~ule s~.nce
it is not generally desired to splice the 3' end of a
naturally occurring catalytic nucleic acid molecule with
a target nucleic acid molecule. Rather, the separate
nucleic acid molecule is chosen or selected to have a
beneficial function once spliced with the target nucleic
molecule.
In a related aspect, the invention features a method
f or splicing a target nucleic acid molecule with a separ-
ate nucleic acid molecule by contacting those molecules in
the presence of one or more splicing factors or
20. spliceosomes under splicing conditions. Such molecules
are not naturally spliced together in nature, although the
final splice product may be a natural product.
The various splicing factors and spliceosomes are well
known in the art, and this activity is generally described
by Bruzik and Maniatis in 360 Nature 692, 1:992.
The invention concerns
splicing of target nucleic acid molecules and separate
nucleic acid molecules which are not normally spliced
together within a cell as described by Bruzik and
Maniatis, supra. Rather, as described above, a separate
nucleic acid molecule is selected such that a useful
function can be achieved in a gene therapeutic fashion.
In preferred embodiments, the catalytic nucleic acid
is able to cleave and splice, e~ct. , it has a group I or
group II intron motif; the method is performed sn vitro or
in vivo with an RNA target; and the method can be used to
treat g?netic disease in a gene therapy type manner, for
WO 95113379 PCT/US94/12976
2175?03
14
example, by correcting an abnormal transcript, or by
providing antiviral activity such as a dominant negative
allele to a viral RNA.
In other aspects, the invention features catalytic
nucleic acid molecules having a selected separate nucleic
acid molecule as a 3' exon encoding at least a portion of -
a useful gene which can be..used in gene therapy. Such a
molecule can be spliced with and thereby correct or modify
the expression of- other target RNA molecules. The
invention also features vectors encoding such catalytic
nucleic acid molecules.
The observation that ribozymes can specifically cleave
targeted RNAs in vitro hassled to much speculation about
their potential usefulness as gene inhibitors. By
cleaving targeted mRNAs ~ ivo, ribozymes can be used to
atop the flow of genetic information. Here we describe a
different application of ribozymes. For example, a group
I intron ribozyme can be used to manipulate the flow of
genetic information by targeted trans-splicing. Defective
cellular transcripts may be-repaired, or pathogen-derived
transcripts may be altered=to encode antagonists to the
pathogen using such technology.
In nature the group I intron ribozyme from Tetrahvmena
thermophila self-splices itself from precursor ribosomal
RNAs (7.R. Cech, A.J. Zaug, P.J. Grabowski, Cell 27 487
(1981); K. Kruger et al., Cell 31, 147 (1982)). This
process is accomplished in two successive steps. First
the phosphodieater bond at the 5' exon-intron border is
cleaved. Then the 3' hydroxyl group on the 5' exon is
covalently attached to the3' exon, and the intron is -
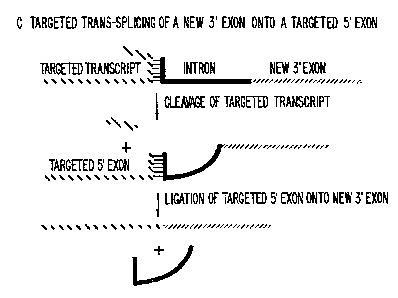
removed (Fig. 1A). It has been previously demonstrated _in '
vitro that the 5' exon in this reaction-can be mimicked by
RNA molecules supplied ~ traps; the minimum active unit
is the dinucleotide substrate rCU (Fig. 1B) (T. moue,
F.X. Sullivan, T.R. Cech, Cell 43, 431 (1985)). We
propose use of-traps-splicing reactions to ligate foreign
sequences onto targeted transcripts after cleavage (Fig.
R'O 95143379 PCTfUS94112976
2175703
I5
1C). In this manner, ribozymes can be employed to
manipulate the flow of genetic information inside cells by
changing what a-targeted RNA encodes.
Other features and advantages of the invention will be
apparent from the following description of the preferred
embodiments thereof, and from the claims.
nPacrip ion of the Preferred Embodiments
The drawings will briefly be described
Drawings
FIGS. 1A, 1B, and 1C are diagrammatic representations
showing reactions of the group I intron from Tetrahymena
for targeted trans-splicing.
FIGS. 2A and 2B are a comparison of cis- and trans-
splicing reactions for LacZ transcripts.
FIG. 3 is a copy of an autoradiogram showing targeted
trans-splicing to correct truncated transcripts from the
alpha complement of LacZ 39 nucleotides long. L-21 (or L-
21 4e1) ribozyme-3' exon chimeric RNAs (see Fig. 2B) (;2P-
body-labeled) (200nM) were preheated in reaction buffer
[50mM Hepes (pH 7.0), 150mM NaCl, and 5mM MgCl,] at 50C
for 5 minutes and then equilibrated at 37C for 2 minutes.
The 13 (5'-A5: GGCCCUCUAS) or 39 (5'L-A2: see Fig. 2B)
nucleotide substrate RNAs (1~M) and GTP (100~M) were
preheated to 37C and added to the ribozymes to start the
reactions which proceeded at 37C. Portions containing
one fifteenth of the reactions were removed at 0, 2, 10,
60; and 180 minutes and added to an equal volume of lOmM
EDTA to stop the reactions. Reaction products were
analyzed upon a 4g polyacrylamide gel with 8M urea. The
inactive L-21 del ribozyme was generated by deleting 93
nucleotide of--the ribozyme (nucleotides 237-330 comprising
L6b to P9).
FIG. 4 is a graphical representation of the targeted
traps-splicing rate for- correcting the 39 nucleotide
truncated LacZ transcript. The products from the trans-
CA 02175703 2002-09-16
76909-169
16
splicing reaction time course containing the action L-21
ribozyme and the 39 nucleotide substrate shown in figure
2 were quantified with an AMBIS* Image Acquisition and
Analysis System (AMBTS, Tnc., San Diego, CA). The
percentage of the ribozyme-3' exon RNA remaining is
plotted versus time.
FTG. 5 is a copy of an autoradiogram showing
hydrolysis of the 3' LaeZ exon attached to the L-21
ribozyme .. L-21 _ (.0r_.. L-_21.. del ) ribQZ~rme_ 3 ~_ _ exon. .chimeric
RNAs ('2P-body-labeled) (100nM) wee incubated at 37°C in
reaction buffer (50mM Hepes (pH 7.0), 150mM NaCl, 5mM
MgCl2, and 100~.M GTPl . A portion of the reaction was
removed after 0, 2, 10, and 60 minutes and added to an
equal, volume of lOmM EDTA to stop the reaction. Products
were analyzed upon a.4% polyacrylamide gel containing 8M
urea.
FIGS. 6A and 6B are representations of trans-splicing
to recreate an entire 3074 nucleotide LacZ messenger RNA
from a 1106 nucleotide truncated trans,cri.pt. A. Scheme
for correcting transcript. 8. Trans-splicing reaction.
L-21 (or L-21 del) ribozyme-3' exon chimeric RNAs (20nM)
were preheated in reactian buffer (50mM Hepes (pH 7.0) ,
150mM NaCl, and 5mM MgClz] at 50°C for 5 minutes and then
equilibrated at 37°C for 2 minutes. The 1106 nucleotide
substrate RNA ('2P-end labeled) (200nM) and GTP (100uM)
were preheated at 37°C and added to the ribozymes to start
the reactions which proceeded at 37°C. One sixth of each
reaction was removed at 0, 2, 10, 60; and 1B0 minutes and
added to an equal volume of lOmM EDTA to stop the
reaction. Reaction products were analyzed upon a 1.2%
agarose gel containing 1.1% formaldehyde. rRNAs from
mouse NIH 3T3 cells were used to as 5100 and 1900 nt
molecular weight markers: The remaining sixth of the
reactions, which had proceeded for 120 minutes, were in
vitro translated.
FIG. 7 is a scheme for correcting genetic mutations
using targeted trans-splicing.
*Trade-mark
W095/13379 2 i ~ 5 7 0 3 pCT~s94112976
17
FIG. S is a scheme for mutating HIV transcripts using
targeted trans-splicing.
Taraeted Trans-splicina
The general scheme for a targeted trana-splicing is
shown in Fig. 1 using ,the group I intron of Tetrahvmena
thermophila assn example. Those in theart will recog
nize that this example is not limiting in the invention
and that other enzymatic RNA molecules having the appro
priate splicing activity can be used in the invention.
Alternatively, as discussed above, these molecules can be
supplemented by other molecules having a suitable splicing
activity, or by spliceosomes or splicing factors.
Generally, the reaction involves base pairing of the
catalytic nucleic acid molecule with the targeted
transcript, cleavage of the targeted transcript, and then
ligation of the 3' exon (separate nucleic acid molecule)
with this targeted 5' exon. The catalytic nucleic acid is
removed in the reaction. As will be noted, the
specificity of-the reaction can be changed by alteration
of the substrate binding site in the catalytic nucleic
acid molecule by methods well known in the art.
The following is an example of various constructs used
to show the operability of the claimed invention. Those
in the art will recognize that this example indicates the
utility of the invention for both in vitro and in vivo
splicing reactions. While significant utility will be
attained in vivo by use of the present invention, those in
the art will also recognize that in vitro utility is
important and can be used to create chimeric transcripts
for use in laboratory situations or in a clinical setting.
Example l LaCZ Fusion _
To assess the feasibility of- the targeted trans-
splicing approach, we tested the ability of the
Tetrahvmena ribozyme to correct truncated LacZ transcripts
with targeted trans-splicing. It has previously been
shown that in ~. coli the Tetrahvmena self-splicing group
WO 95/13379 PCTlU594/12976
2~ ~ ~ 03
18
I intron can efficiently splice itself from transcripts
encoding the alpha-complement of,Q-galactosidase ((3-gal) ,
(Fig. 2A) (J. V. Price, T.R. Cech, Science 228, 719 (1985);
blaring et al., Cell 40, 371 (1985)). Since this reaction
proceeded very efficiently in cis, we decided to determine
if the ribozyme could perform a similar reaction in traps.
This system consists oft RNA molecules (Fig. 2B): a
ribozyme-3' exon RNA and a 5' exon RNA. The group I ribo-
zyme used in this study lacks the first 21 nucleotides
present in the full length intron from which it is derived
(A.J. Zaug, T.R. Cech, Science 231, 470 (1986)). The
first 23 nucleotides of the=3' exon arederived from the
pre-rRNA 3' exon sequence from Tetrahymena (M. D. Been,
T.R. Cech, Cell 47, 207 (1986)). This 23 nucleotide
sequence is fused in-frame to 200 nucleotides of the
alpha-complement of the LacZ gene (Been and Cech, supra).
The 39 nucleotide 5' exon =contains a ribosome binding
site, the first 21 coding nucleotides of an alpha-
complement LacZ transcript, the ribozyme recognition
sequence CCCUCU, and two adenosines. These adenosines
must be removed if traps-splicing is to correct these LacZ
transcripts. (Fig. 2B). (Previous studies have shown
that the sequence and length of the RNA following the
CCCUCU is not critical for Tetrahvmena ribozyme action
(A.J. Zaug, M.D. Been, T.C. Cech, Nature 324, 429
(1986))1.
In vitro, the ribozyme can quickly and accurately
traps-splice this LacZ 3' exon onto the truncated 39
nucleotide LacZ 5' exon to generate an RNA product which
encodes the alpha-complement of f~-galactosidase (Fig. 3).
The reaction proceeds with speed and efficiency similar to '
those seen in a reaction with a short 13 nucleotide
substrate. The t1~2 for the arans-splicing reaction with
the 39 nucleotide substrate was determined to be 13
minutes under conditions of substrate excess (Fig. 4).
In these experiments, traps-splicing (production of
5'-3' or 5'L-3') occurred faster than hydrolysis (produc-
W O 95/13379 PCTIUS94112976
2175703
19
tion of free 3' exon;-see Fig. 2). The rate of hydrolysis
of the 3' exon from the ribozyme was determined to be tl/z -
60 minutes in a separate experiment (Fig. 5). An inactive
version of the L-21 ribozyme (L-21 del) was not able to
perform either- she traps-splicing or the hydrolysis
reaction (Fig. 3 and 5}. Sequencing of the traps-splicing
product confirmed that the ultimate and penultimate 3'
adenosine nucleotide were correctly removed from the 5'
exon-substrate RNA, and this cleaved 5' exon was
accurately spliced onto the 3' exon (data not shown). The
splice junction gave the proper reading frame for ,Q-gal
expression.
Example 2: mRNA Solicins
To determine if targeted traps-splicing could be
1.. employed to correct mRNA-size RNA fragments, a transcript
which contained the first 1106 nucleotides of the LacZ
coding sequence as well as signals for in vitro
translation was created and targeted for alteration by
traps-splicing. The L-21 ribozyme was directed to cleave
2D the truncated LacZ transcript 19 nucleotides from its 3'
end and traps-splice a 3' exon brought in by the ribozyme
onto the cleaved LacZ target RNA (Fig. 6A). The 3' exon
sequence attached to the ribozyme encoded the last 1987
nucleotides of the LacZ coding sequence and no sequences
25 from the Tetrahymena pre rRNA. Accurate traps-splicing of
the 3' exon sequences onto the truncated transcript
resulted in a 3074 nucleotide product which encoded the
entire LacZ coding sequence (Fig. 6B).
Once again the inactive version of the ribozyme (L-21
30 del) was unable to perform this reaction, confirming its
expected dependence of the catalytic--activity of the RNA
itself. The traps-splicing products from the 120 minute
time points of the reactions shown in figure 6 were
vitro translated in wheat germ extract, and the in vitro
35 translated proteins were assayed for-;Q-gal activity using
R'O 95/13379 PC."f/US94/12976
2~~~~03
a standard ONPG assay (C. Smith et al., Leukemia 7, 310
(1993)).
Proteins from traps-splicing reactions containing
active ribozymes were shown.to Contain 1500 units [1000 x
5 OD920/(ml-min)] of (3-gal activity, while no activity was
found in proteins translated from reactions containing the
inactive ribozyme. Therefore, traps-splicing can be
employed to correct the coding sequence of large defective
transcripts.
10 In the reaction shown in -figure- 6, the labeled
substrate RNA is in a 10 fold excess to the ribozyme-3'
exon RNA. Therefore, only 10% of the labeled substrate
RNAa could at best be converted to traps-spliced products.
In this reaction however, we roughly estimate (by
15 comparing different X-ray film exposures of the gel) that
at most 1% of the truncated RNAs are corrected: This lack
of efficiency is probably a result of the targeted RNAs
adopting conformations which inhibit the ribozyme from
correctly interacting with them. To improve the
20 efficiency of this traps-splicing reaction, alternative
sites for cleavage and splicing which are more accessible
to the ribosome can be targeted by standard manipulation
of this experiment. In vivo, cellular proteins may
improve the efficiency of formation of the-correct RNA
interaction (Z. Tsuchihashi-, M. Khosla, D. Herschlag,
Science 262, 99 (1993)).
Uses
Gene mapping and human genome sequencing provides the
genetic basis for an increasing number .of inherited
diseases. With each discovery or identification of a new
disease-related gene there is an opportunity to develop
gene therapy based treatments. Conventional gene therapy
approaches attempt to correct a genetic deficiency by
transferring a wild-type cDNA copy of a gene under the
control of a heterologous promoter to cells harboring a
defective copy of the gene. One obstacle for implementing
WO 95113379 PCTIUS94I12976
2175703
21
such treatments is an inability to Faithfully recapitulate
the normal expression pattern of endogenous genes after
gene transfer (R. A. Morgan, W.F. Anderson, Ann. Rev. Bio-
chem. 62, 191 (1993); E.A. Dzierzak, T. Papayannopoulou,
R.C. Mulligan, Nature 331, 35 (1989)). This may limit the
number of genetic diseases treatable by gene therapy.
Targeted trans-splicing offers a solution to this problem.
Ribozymes can be -used to correct the defective
transcripts issuing from mutant genes: This approach will
be valuable for the treatment of the many genetic diseases
caused by a common set of specific mutations which do not
affect the expression of the mutant gene. For example,
the genetic basis of- many globin diseases is well
understood. However, gene therapy based treatments for
such diseases have been slow in coming, perhaps, because
the expression -patterns of the globin genes cannot be
recapitulated after gene transfer: Targeted trans-
aplicing can potentially repair or correct globin
transcripts that are either truncated or contain pout
mutations. In the process, the cellular expression
pattern of these genes is maintained (Fig. 7). Therefore,
targeted trans-splicing represents an important, novel
strategy for the treatment of many genetic diseases.
Trans-splicing ribozymes based on any of the self
splicing group I introns can be designed to cleave a
targeted transcript upstream of a specific mutation or
upstream of a premature 3' end at essentially any uridine
residue -(F. L. Murphy, T.R. Cech, Proc. Natl. Acad. Sci,
USA 86, 9218 (1989)). One simply changes the sequence of
the internal -guide sequence within the ribozyme (5'-
GNNNNN) to match the sequence preceding the site of target
RNA cleavage (5'-N'N'N'N'N'U), where N-N' represent any
allowable base pair. -The 3' exons attached to the ends of
these ribozymes are comprised of a sequence designed to
correct the mutant transcripts being targeted. The ribo-
zyme will both cleave the mutant transcript and replace
the mutant 3' region by a functional sequence. There is
WO 95/13379 PCT/US94/12976
21 Z 5103
22
very little sequence requirement for a 3' exon in these
reactions, so virtually any sequence can serve (J. V.
Price, T.R. Cech, Genes and-Development 2, 1439 (1988)).
Thus, traps-splicing ribozymea.can be made to correct es-
sentially any mutant transcript because sequence require-
ments for 5' cleavage sites and 3' exons are minimal.
Traps-splicing -ribozymes are also be effective
antiviral agents. Several groups have employed trana
cleaving ribozymes to inhibit viral replication. Use of
such ribozymes results in the destruction ofthe targeted
viral RNA inside cells (N. Sarver et al., Science 247,
1222 (1990)). Thus, the effectiveness of these trans-
cleavage ribozymes rests upon their ability to destroy the
vast majority of the targeted viral RNAS. We propose
employing traps-splicing ribozymes not to destroy viral
RNAs, but to change the sequence of the viral RNAs to give
them antiviral activity. For example, the HIV transcripts
that encode the aaa protein can be changed to encode a
dominant negative version of this protein via targeted
traps-splicing (Fig. 8) (M.H. Malim, E. Bohniein, J.
Hauber, B.R. Culien, Cell 58, 205 (1989); D. Trono, M.B.
Feinberg, D. Baltimore, Cell 59, 1I3 (1989)) or to contain
a large number of TAR or RRE decoy RNAs (B. A. Sullenger,
H.F. Gallardo, G.E. Ungers, E. Gilboa, Cell 63, 601
(1990)).
In contrast to traps-cleaving ribozymes, such
antiviral traps-splicing ribozymes would have to affect
only a small percentage of the targeted HIV transcripts to
be effective at inhibiting viral replication. In general,
the ability to change the information encoded by targeted
transcripts by traps-splicing represents a broad new
approach to gene inhibition-because now transcripts can be
altered to encode proteins or RNAs which can inhibit the
function of the targeted gene. In other words, with
targeted traps-splicing, deleterious transcripts can be
turned against themselves.-
WO 95113379 ~ ~ 7 J 7 ~ ~ PCTfUS94112976
23
As noted above, trans-splicing may also be
accomplished without the use of ribozymes. It has been
demonstrated that spliced leader sequences from lower
eucaryotes can be trana-spliced onto mammalian 3' splice
sites in tissue culture cells (J. P. Bruzik, T. Maniatis,
Nature 360, 692 (1992)). Trans-splicing in this case is
mediated by the spliceosome or splicing factors. Thus, it
is possible to employ spliceosomes to alter the sequence
of targeted transcripts for some desired end via targeted
trans-splicing.
Thus, this invention provides a means for performing
molecular reconstructive surgery. A defective part of a
useful RNA molecule can be cut away from the rest of the -
molecule and subsequently replaced by a functional part.
Alternatively, a functional portion of a disease-causing
or -deleterious RNA can be replaced by an inhibitory
portion.
Administration
The above trans-splicing factors or agents can be
administered by standard techniques, some of which are
discussed below. They may be administered as RNA or
expressed from expression vectors. Selected agents, e-a.,
oligonucleotides or ribozymes can be administered
prophylactically, or to patients suffering from a target
disease, e-a., by exogenous delivery of the agent to an
infected -tissue by means of an appropriate delivery
vehicle, e--a., a liposome, a controlled release vehicle,
by use of iontophoreais, electroporation or ion paired
molecules, or covalently attached adducts, and other
pharmacologically approved methods of delivery. Routes of
administration include intramuscular, aerosol, oral
(tablet or pill form), topical, systemic, ocular, intra
peritoneal and/or intrathecal. Expression vectors for
immunization with ribozymes and/or delivery of oligo
nucleotides are also suitable.
W O 95/13379 PCTIUS94112976
2~~ ~~~3
24
The specific delivery route of any selected agent will
depend on the use of the agent. Generally, a specific
delivery program for each agent will focus on naked agent
uptake with regard to intracellular localization, followed
by demonstration of efficacy. Alternatively, delivery to
these same cells in an organor tissue of an animal can be
pursued. Uptake studies will include uptake assays to
evaluate, e-g., cellular oligonucleotide uptake, regard-
less of the delivery vehicle or strategy. Such assays
will also determine the intracellular localization of the
agent following uptake, ultimately establishing the
requirements for maintenance of steady-state concentra-
tions within the cellular compartment containing the
target sequence (nucleus and/or cytoplasm). Efficacy and
cytotoxicity can then be tested. Toxicity will not only
include cell viability but also cell function.
Some methods of delivery-that may be used include:
a. encapsulation in liposomes,
b. traneduction by retroviral vectors,
c. conjugation with cholesterol,
d. localization to nuclear compartment utilizing
antigen binding site found on most snRNAs,
e. neutralization of charge of ribozyme by using
nucleotide derivatives, and
f. use of blood stem cells to distribute ribozymea
throughout the body.
At least three types of delivery strategies are useful
in the present invention, including: ribozyme
modifications,--particle carrier drug delivery vehicles,
and retroviral expression vectors. Unmodified ribozymes
and antisenae oligonucleotides, like most small molecules,
are taken up by cells, albeit slowly. To enhance cellular
uptake, the ribozyme may be modified essentially at
random, in ways which reduce its charge but maintain
specific functional groups required for RNA cleavage and
splicing activity. This results in a molecule which is
WO 95/d3379 PCTIU5941d2976
1 2?75703
able to diffuse across the cell membrane, thus removing
the permeability barrier.
Modification of ribozymes to reduce charge is just one
approach to enhance the cellular uptake of these larger -
5 molecules. The random approach, however, is not advisable
since- ribozymes are structurally and functionally more
complex than small drug molecules. The structural
requirements necessary to maintain- ribozyme catalytic
activity are well understood by those in the art. (See,
10 Cech, Curr. Op. Structural Biol., 1992) These
requirements are taken into consideration when designing
modifications to enhance- cellular delivery. The
modifications are also designed to reduce susceptibility
to nuclease degradation. Both of these characteristics
15 -should greatly improve the efficacy of the ribozyme.
Cellular -uptake can be increased by several orders of
magnitude without .having to alter the. phosphodiester
linkages necessary for ribozyme cleavage activity.
Chemical modifications of the phosphate backbone will
20 reduce the negative charge thereby facilitating diffusion
across the membrane. This principle has been successfully
demonstrated for antisenae DNA technology. The
similarities in chemical composition between DNA and RNA
make this a feasible approach. In the body, maintenance
25 of an external concentration will be necessary to drive
the diffusion of the modified ribozyme into the cells of
the tissue. Administration routes which allow the
diseased tissue to be exposed to a transient high
concentration of the drug, which is slowly dissipated by
systemic adsorption are preferred. Intravenous adminis-
tration with a drug carrier designed to increase the
circulation half-life of the ribozyme can be used. The
size and composition of the drug carrier restricts rapid
clearance from the blood stream. The carrier, made to
accumulate at -the site of infection, can protect the
ribozyme from degradative processes.
WO 95113379 PCT/US94112976
2~-~ 5~ 03
26
Drug delivery vehicles are effective for both systemic
and topical administration. They can be designed to serve
as a slow release reservoir, onto deliver their contents
directly to the. target cell. ,Aw advantage o~-using direct
delivery drug vehicles is..that multiple molecules are
delivered per uptake. Such vehicles have been shown to
increase thecirculation half-life of drugs which would
otherwise be rapidly cleared from the blood stream. Some
examples ofsuch specialized drug delivery vehicles which
fall into this category are liposomes, hydrogels, cyclo-
dextrins, biodegradable nanocapsules, and bioadhesive
microspheres.
From this category of delivery systems, liposomes are
preferred. Liposomes -increase .intracellular stability,
increase uptake efficiency and improve biological
activity. Liposomes are hollow spherical vesicles
composed of lipids arranged in a, similar fashion as those
lipids which make up the cell membrane. They have an
internal aqueous space for entrapping water soluble
compounds and range in size from 0.05 to several microns
in diameter. Several studies have shown that liposomes
can deliver RNA to cells and that the RNA remains
biologically active.
For example, a liposome delivery vehicle originally
designed-as a research tool, Lipofectin, has been shown to
deliver intact mRNA molecules to cells yielding production
of the corresponding protein.
Liposomes offer several advantages: They are non- -
toxic and biodegradable in composition; they display long
circulation half-lives; and-recognition molecules can be
readily attached to theirsurface. for targeting to
tissues. Finally, cost effective manufacture of lipoaome-
based pharmaceuticals, either in a liquid suspension or
lyophilized product, has demonstrated the viability of
this technology as an acceptable drug delivery system.
Other controlled release drug delivery systems, such
as nonoparticles and hydrogels may be potential delivery
W O 95113379 PCTIfi594112976
~1~3703
27
vehicles for a ribozyme. These carriers have been
developed for chemotherapeutic .agents and protein-based
pharmaceuticals, and consequently, can be adapted for
ribozyme delivery.
Topical administration of traps-splicing ribozymes is
advantageous since it allows localized concentration at
the site of administration with minimal systemic
adsorption. This simplifies the delivery strategy of the
ribozyme to the disease site and reduces the extent of
toxicological characterization. Furthermore, the amount
of material to be applied is far less than that required
for other administration routes. Effective delivery
requires the-ribozyme to diffuse into the infected cells.
Chemical modification of the ribozyme to neutralize
negative charge may be all that is required for
penetration. However, in the event that charge
neutralization is insufficient, the modified ribozyme can
be co-formulated with permeability enhancexs, such as
Azone or oleic acid, in a liposome. The liposomes can
either represent a slow release presentation vehicle in
which the modified ribozyme and permeability enhancer
transfer from the liposome into the infected cell, or the
liposome phospholipids can participate directly with the
modified ribozyme and permeability enhancer in
facilitating cellular delivery. In some cases, both the
ribozyme and permeability enhancer can be formulated into
a suppository formulation for slow release.
Such ribozymes may also be systemically administered.
Systemic absorption refers to the accumulation of drugs in
the blood stream followed by distribution throughout the
entire body. Administration routes which lead to systemic
absorption include: intravenous, subcutaneous=, intra-
peritoneal, intranasal, intrathecal and ophthalmic. Each
of these administration routes expose the-ribozyme to an
accessible diseased tissue. Subcutaneous administration
drains into a localized lymph node which proceeds through
the lymphatic network into-the circulation. The rate of
R'O 95/13379 PCT/US94112976
1
21?~~03
28
entry into the circulation has been shown to be a function
of-molecular weight or size. The use of a liposome or
other drug carrier localizes the ribozyme at the lymph
node. The ribozyme can bemodified to diffuse into the
cell, or the liposome can- directly participate in the
delivery of either the unmodified or modified ribozyme to
the cell.
A liposome formulation which can associate ribozymes
with the surface of lymphocytes and macrophages is also
useful. This will provide enhanced delivery to HIV
infected cells by taking advantage of the specificity of
macrophage and lymphocyte immune recognition of infected
cells. Whole blood studies show that the formulation is
taken up by 90% of the lymphocytes after 8 hours at 37°C.
Preliminary biodistribution and pharmacokinetic studies
yielded 70% of the injected dose/gm of tissue in the
spleen after one hour - following intravenous
administration.
Intraperitoneal administration also leads to entry
into the circulation with the molecular weight or size of
the ribozyme-delivery vehicle complex controlling the rate
of entry.
Liposomes injected intravenously show accumulation in
the liver, lung and spleeri.- The composition and size can
be adjusted so that this accumulation represents 30% to
40% of the injected dose. The rest is left to circulate
in the blood-stream for up to 24 hours.
The chosen method of delivery will result in
cytoplasmic accumulation in the afflicted cells and
molecules should have some nuclease-resistance for pptimal
dosing. Nuclear delivery may be used but is less prefer-
able. Most preferred delivery methods include liposomes
(10-400 nm?, hydrogels, controlled-release polymers,
microinjection or electroporation-(for ex vivo treatments)
and other pharmaceutically applicable vehicles. The
dosage will depend upon the disease indication and the
route of administration but should be between 100-200
WO 95/fi3379 PCTlUS94112976
2 J 7~~03
29
mg/kg of- body weight/day. The duration of treatment will
extend through the course of the disease symptoms, usually
at least 14-16 days and possibly continuously. Multiple
daily doses are anticipated for topical applications,
ocular applications and vaginal applications. The number
of doses will depend upon disease delivery vehicle and
efficacy data from clinical trials.
Establishment of therapeutic levels of ribozyme within
the cell is dependent upon the rate of uptake and degrada
tion. Decreasing the degree of degradation will prolong
the intracellular half-life of the ribozyme. Thus,
chemically modified ribozymes, e.a., with modification of
the phosphate backbone, or capping of the 5' and 3' ends
of the ribozyme with nucleotide analogs may require
different dosaging. Descriptions of useful systems are
provided in the art cited above, all of which is hereby
incorporated by reference herein.
Particular diseases that may be treated in this manner
include any disease which can be treated by such RNAs, for
example, HSV, HBV, EBV, and HIV infection; as well as
various carriers (where-the target molecule is located in
a known cellular compartment).
Any disease -caused by a specific set of mutations in
a given genes RNA is potentially treatable by using target
trans-splicing to correct such defective RNAs. Such
diseases would include:
A. ,Q-globin diseases (such as sickle cell anemia),
cystic fibrosis, as well as any other genetic diseases
caused by a point mutations or deletions in RNA.
B. Cancers caused by specific mutant oncogene
encoding RNAs (e-a. bcr-abl mRNAs, mutant p53 mRNAs).
C. Genetic diseases caused by unstable trinucleotide
repeats in RNAs (e.a. Huntington's disease, fragile X
syndrome).
Other embodiments are within the following claims.
'"'r,.. ~_,_. t ; ;~;:~t:=,'-a