Note: Descriptions are shown in the official language in which they were submitted.
CA 02176183 2005-12-23
1 CT-2294A (BMMA-10A)
3-SUBSTITUTED OXINDOLE DERIVATIVES AS
POTASSIUM CHANNEL MODULATORS
10
The present invention is directed to novel substituted 3-phenyl
oxindole derivatives which are modulators of the large-conductance
calcium-activated potassium (Maxi-K) channels and, therefore, useful in the
protection of neuronal cells, especially in the treatment or prevention of
ischemic stroke. The present invention also provides a method of treatment
with the novel oxindole derivatives and to pharmaceutical compositions
thereof.
Potassium channels play a key role in regulation of cell membrane
potential and modulation of cell excitability. Potassium channels are largely
regulated by voltage, cell metabolism, calcium and receptor mediated
processes. [Cook, N.S.; Trends in Pharmacol. Sciences (1988), 21; and
Quast, U., et al, Trends in Pharmacol. Sciences (1989), 10, 431 ]. Calcium-
activated potassium (KCa) channels are a diverse group of ion channels that
share a dependence on intracellular calcium ions for activity. The activity of
KCa channels is regulated by intracellular [Ca2+], membrane potential and
phosphorylation. On the basis of their single-channel conductances in
symmetrical K+ solutions, KCa channels are divided into three subclasses:
large conductance (maxi-K) > 150 pS; intermediate conductance 50-150
pS; small conductance < 50 pS. Large-conductance calcium-activated
potassium (Maxi-K) channels are present in many excitable cells including
2176183
2 CT-2294A (BMMA-10A)
neurons, cardiac cells and various types of smooth muscle cells.
[Singer, J. et al., Pflugers Archiv. (1987) 408, 98; Baro, I., et al.,
Pflugers
Archiv. (1989) 414 (Suppl. 1), S168; and Ahmed, F. et al., Br. J. Pharmacol.
(1984) 83, 227].
Potassium ions play a dominant role in controlling the resting
membrane potential in most excitable cells and maintain the
transmembrane voltage near the K+ equilibrium potential (Ek) of about -90
mV. It has been shown that opening of potassium channels shift the cell
membrane potential towards the equilibrium potassium membrane potential
(Ek), resulting in hyperpolarization of the cell. [Cook, N.S., Trends in
Pharmacol. Sciences (1988), 9, 21]. Hyperpolarized cells show a reduced
response to potentially damaging depolarizing stimuli. Maxi-K channels
which are regulated by both voltage and intracellular Ca2+ act to limit
depolarization and calcium entry and may be particularly effective in
blocking damaging stimuli. Therefore cell hyperpolarization via opening of
Maxi-K channels may result in protection of neuronal cells under ischemic
conditions.
A range of synthetic and naturally occuring compounds with maxi-K
opening activity have been reported. The avena pyrone extracted from
avena sativa-common oats has been identified as a maxi-K channel opener
using lipid bi-layer technique [International Patent application WO
93/08800, published May 13, 1993]. 6-Bromo-8-(methylamino)imidazo[1,2-
a]pyrazine-2-carbonitrile (SCA-40) has been described as a maxi-K
channel opener with very limited electrophysiological experiments
[Laurent, F. et al., Br. J. Pharmacol. (1993) 108, 622-626]. The flavanoid,
Phloretin has been found to increase the open probability of Ca2+-activated
potassium channels in myelinated nerve fibers of Xenopus laevis using
outside-out patches [Koh, D-S., et al., Neuroscience Lett. (1994) 165,
167-170].
A number of substituted oxindoles have been disclosed as
neuroanabolic agents by H. Kuch et al in U.S. Patent Nos. 4,542,148,
issued September 17, 1985 and 4,614,739, issued September 30, 1986.
3 CT-2294A (BMMA-10A)
In European patent application EP-477,819 published
January 4, 1992 and corresponding U.S. Patent No. 5,200,422, issued
April 6, 1993 to Olesen, et al., a number of benzimidazole derivatives were
disclosed as openers of maxi-K channels by using single-channel and
whole-cell patch-clamp experiments in aortic smooth muscle cells. Further
work was reported by Olesen, et al in European J. Pharmacol., 251, 53-59
(1994).
Stroke is presently recognized as the third leading cause of adult
disability and death in the United States and Europe. In the past decade,
several therapeutic approaches for the minimization of stroke-related brain
damage have been pursued including inhibitors of AMPA/kainate, N-
methyl-D-aspartate (NMDA) and adenosine reuptake inhibitors. It is the
object of the present invention to provide novel compounds that will
modulate potassium channels, in particular, large-conductance calcium-
activated potassium (Maxi-K) channels which will be useful in reducing
neuronal damage during ischemic stroke.
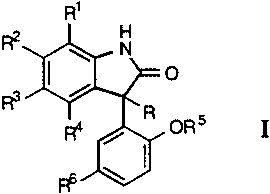
The present invention provides novel oxindole derivatives having the
general formula
R'
F? H
\ N
~ O
R3 ~
R
~ / ORS I
FO \
wherein R, Rl, R2, R3, R4, R5 and R6 are as defined below, or a non-toxic
pharmaceutically acceptable salt, solvate or hydrate thereof which are
openers of the large conductance calcium-activated K+ channels also
known as maxi-K or BK channels. The present invention also provides
pharmaceutical compositions comprising said oxindole derivatives and to
the method of treatment of disorders sensitive to potassium channel
. 4 CT-2294A (BMMA-10A)
opening activity such as ischemia, convulsions, asthma, urinary
incontinence and traumatic brain injury.
The present invention provides novel oxindole derivatives which are
potent openers of the high conductance, calcium-activated K+-channels
(BK channel) and which have the formula
R'
F? H
~ N
( 0
R3 ~
~ R OR5 I
Fe
wherein
R is hydrogen, hydroxy or fluoro;
Rl, R2, R3
and R4 each are independently hydrogen, C1-4 alkyl, halogen,
trifluoromethyl, phenyl, p-methylphenyl or p-
trifluoromethylphenyl; or Rl and R2, R2 and R3 or R3 and R4
are joined together to form a benzo fused ring;
R5 is hydrogen or C1-4 alkyl; and
R6 is chlorine or trifluoromethyl;
or a nontoxic pharmaceutically acceptable salt, solvate or hydrate thereof.
The present invention also provides a method for the treatment or
alleviation of disorders associated with BK channels, especially ischemia,
convulsions, asthma, urinary incontinence and traumatic brain injury, which
comprises administering together with a conventional adjuvant, carrier or
diluent a therapeutically effective amount of a compound of formula I or a
nontoxic pharmaceutically acceptable salt, solvate or hydrate thereof.
= 5 CT-2294A (BMMA-10A)
The term "C1_4 alkyl" as used herein and in the claims (unless the
context indicates otherwise) mean straight or branched chain alkyl groups
such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t-butyl.
Preferably,
these groups contain from 1 to 2 carbon atoms. Unless otherwise specified,
the term "halogen" as used herein and in the claims is intended to include
bromine, chlorine, iodine and fluorine while the term "halide" is intended to
include bromide, chloride and iodide anion.
As the compounds of the present invention may possess an
asymmetric carbon atom at the 3-position of the oxindole ring, the present
invention includes the racemate as well as the individual enantiometric
forms of the compounds of Formula I as described herein and in the claims.
The use of a single designation such as (3R) or (3S) is intended to include
mostly one stereoisomer. Mixtures of isomers can be separated into
individual isomers according to methods which are known per se, e.g.
fractional crystallization, adsorption chromatography or other suitable
separation processes. Resulting racemates can be separated into
antipodes in the usual manner after introduction of suitable salt-forming
groupings, e.g. by forming a mixture of diastereosiomeric salts with optically
active salt-forming agents, separating the mixture into diastereomeric salts
and converting the separated salts into the free compounds. The possible
enantiomeric forms may also be separated by fractionation through chiral
high pressure liquid chromatography columns. Altematively, the optically
active enantiomers of the compounds of Formula I may be prepared by
stereoselective synthetic procedures, some of which are described herein.
The use of optically active reagents in combination with the appropriate
intermediate described herein would produce the desired enantiomer of the
compound of Formula I.
The term "nontoxic pharmaceutically acceptable salt" as used herein
and in the claims is intended to include nontoxic base addition salts with
inorganic bases. Suitable inorganic bases such as alkali and alkaline earth
metal bases include metallic cations such as sodium, potassium,
magnesium, calcium and the like.
2176183
6 CT-2294A (BMMA-10A)
Certain of the compounds of the present invention can exist in
unsolvated forms as well as solvated forms including hydrated forms such
as monohydrate, dihydrate, hemihydrate, trihydrate, tetrahydrate and the
like. The products may be true solvates, while in other cases, the products
may merely retain adventitious solvent or be a mixture of solvate plus some
adventitious solvent. It should be appreciated by those skilled in the art
that
solvated forms are equivalent to unsolvated forms and are intended to be
encompassed within the scope of the present invention.
In the method of the present invention, the term "therapeutically
effective amount" means the total amount of each active component of the
method that is sufficient to show a meaningful patient benefit, i.e., healing
of
acute conditions characterized by openers of large conductance calcium-
activated K+ channels or increase in the rate of healing of such conditions.
When applied to an individual active ingredient, administered alone, the
term refers to that ingredient alone. When applied to a combination, the
term refers to combined amounts of the active ingredients that result in the
therapeutic effect, whether administered in combination, serially or
simultaneously. The terms "treat, treating, treatment" as used herein and in
the claims means preventing or ameliorating diseases, tissue damage
and/or symptoms associated with dysfunction of cellular membrane
polarization and conductance.
The compounds of Formula I may be prepared by various
procedures such as those illustrated herein in the examples, in the reaction
schemes and variations thereof which would be evident to those skilled in
the art. The various oxindole derivatives of Formula I may advantageously
be prepared from isatin intermediates which are generally well-known and
a general method of preparation is illustrated in Reaction Scheme 1 and a
specific example in Reaction Scheme 2.
In the process for the preparation of isatin intermediates of the
Formula VII, a number of commonly known and well-established
procedures may be employed such as those described by Sandmeyer, T.,
Helv. Chim. Acta, 2, 234 (1919); Stolle, R., J. Prakt. Chem., 105, 137 (1922);
and Gassman, P., et al., J. Ocr . Chem., 42, 1344 (1977). However, a more
preferred method for the preparation of isatins of Formula VII starting from
7 CT-2294A (BMMA-10A)
the appropriately substituted anilines of Formula V is generally described
by Hewawasam, P., et al., Tetrahedron Lett., 35, 7303 (1994) and is
illustrated in Reaction Scheme 1. This method appears to be insensitive to
the electronic nature of substituents bound to the aromatic ring and is
characterized by predictable regiochemical control.
REACTION SCHEME 1
R1 R1 Li
RZ NHCOR BuLi F:?I NCOR (CO2 Et) 2
Li
Ra Re
v
R'
R2 I NHCOR H3O
+ F? I~ N O
~2Q
R4 O R 4 0
Vi Vil
It will be appreciated by those skilled in the art that when the amino
group of an aniline compound of Formula V is suitably protected such as
with N-pivaloyl and N-(tert-butoxycarbonyl) protecting groups, it can direct
metalation to the ortho position. Once the dianions are formed, the reaction
with about 1.2 equivalents of diethyl oxalate at low tempeatures such as
-78 C may be used to introduce an a-ketoester moiety ortho to the
protected amino group of the aniline derivative to produce the compound of
Formula VI. Removal of the protecting group followed by spontaneous
cyclization will advantageously produce the isatin of Formula VII. To
elaborate further on the process of Reaction Scheme 1, the dianions of N-
2176183
8 CT-2294A (BMMA-10A)
pivaloylanilines or N-(tert-butoxycarbonyl) anilines are advantageously
generated using about 2.2 to 2.4 fold excess of a variety of butyllithium
reagants, such as n-butyl-, s-butyl- and t-butyl-lithium reagents in THF at
about 0 to -40 C for 2 to 7 hours.
In a typical procedure, neat dry diethyl oxalate (1.2 equivalents) was
added to a solution of the dianion stirred at -78 C under nitrogen. After
being stirred for 30-45 minutes, the reaction was quenched with 1 N HCI
and diluted with ether to afford the compound of Formula VI. Although the
intermediate a-ketoesters of Formula VI may be purified for purposes of
characterization, this step is not necessary and the crude product can be
advantageously deprotected directed to afford the isatins in excellent
overall yield. Deprotection of the N-(tert-butoxycarbonyl) or pivaloyl
moieties may be carried out using 3N HCI/THF or 12N HCI/DME,
respectively, at reflux temperature. Upon evaporation of the volatile
solvents, the isatins generally precipitated from the aqueous residue and
isolated by filtration.
An alternative method for the preparation of isatins using the method
of Gassman, et al is illustrated in Reaction Scheme 2 for the preparation of
4,6-bis(trifluoromethyl) isatin of the Formula Vlla.
REACTION SCHEME 2
F3C NH2 1.tBuOCI, CH2CI2, -65 C NC 2
Ihexanes2. MeSCH2CO2Et, -65 C CO2Et reflux
3. Et3 N, -65 C to r. t.
CF3 CF3 SMe
NC \ N NCS NC N NC N
C -' I O H90 ( o
/ CCI4, r.t BF3 O 2tE
CF3 SMe CF3 CI SMe THF-H20 CF3 0
VIII Vila
.~17il 1~? e~
9 CT-2294A (BMMA-10A)
As shown in Reaction Scheme 2, N-chlorination of 3,5-
bis(trifluoromethyl)aniline with freshly prepared tert-butyl hypochlorite
followed by consecutive addition of ethyl (methylthio)acetate and
triethylamine gave the aminoester which, upon heating in boiling hexanes,
underwent cyclization to afford the (methylthio)indolone of the Formula VIII.
Chlorination of the compound of Formula VIII with N-chlorosuccinimide
(NCS) gave the corresponding a-chloro(methylthio)indolone which upon
hydrolysis with HgO-BF3=OEt2 in THF-H20 gave the desired isatin of
Formula Vlla.
Isatins of Formula VII, prepared as described in the above Reaction
Schemes 1 and 2 or by well-known literature procedures, were converted to
the hydroxyindolones of Formulas Ila and IIb as shown in Reaction Scheme
3. Addition of a THF solution of either a Grignard or aryllithium reagent
derived in-situ from an anisole, to the sodium salt of the isatin of Formula
VII
conveniently prepared with NaH in THF, gave the desired
hydroxyindolones of Formula Ila. Most of the hydroxyindolones were
purified by either recrystallization or trituration with suitable organic
solvents. Demethylation of the methyl ether moiety of the compound of
Formula Ila with BBr3 in CH2CI2 under carefully controlled conditions from
-78 to 0 C afforded the desired phenols of Formula Ilb. It was found that
the reaction should advantageously not be warmed above 0 C and, after
completion of the demethylation, quenching of the reaction with saturated
NaHCO3 followed by acidification with dilute HCI is required before
extracting into an organic solvent to afford the hydroxyindolones of
Formula Ilb.
CT-2294A (BMMA-10A)
REACTION SCHEME 3
R~ 1. NaH, THF F? H H
Rz H 0OC DO- J=0 BBr3 R2 N
N O
2. -20 C to r.t.
O M OH ke'
OH
~ OMe OMe R4 OH
R4-
Ff 1 j 10!5~ Rs Z~~ Ff ~_ I
VII M- MgBr or Li Ila
Ilb
An altemative and more direct approach to the hydroxyindolones of
5 Formula Ilb was developed by the addition of magnesium phenolates to
isatins of Formula VII as illustrated in Reaction Scheme 4.
REACTION SCHEME 4
R'
H
H
R~ \ OMgBr ~ N
H
~ 'P
oH
R2 xQ=o
Fe CH2CI2 30 R OH
~- o \
Rs
VII lib
Unlike the process described in Reaction Scheme 3 involving
Grignard addition reaction followed by demethylation of the methyl ether
product, the free phenol of Formula IIb is directly obtained by this alternate
route. The process of Reaction Scheme 4 which involves the reaction of
magnesium phenolates prepared by mixing the desired phenol and ethyl
magnesium bromide with isatins of Formula VII in methylene chloride,
tolUene or DMF advantageously afforded the desired hydroxyindolone of
Formula Iib. Further, it was found that a variety of electron-deficient
11 CT-2294A (BMMA-10A)
magnesium phenolates could be added to the isatins of Formula VII in an
advantageously convenient one-pot reaction to directly produce the
hydroxyindolones of Formula Ilb.
When it is desired to prepare the indolones of Formulas Illa and Illb,
the corresponding hydroxyindolone of Formula Ila is selectively
dehydroxylated with triethylsilane and trifluoroacetic acid (TFA) as
illustrated in Reaction Scheme 5.
REACTION SCHEME 5
w R1 w
H
Ff N 03SiH, TFA F? N ~
N
O 110-120 C O BBr3 O
FO O sealed tube ~ ~ ~
H
OMe F OMe Ra OFi
Rs FP Ff
Ila Illa Illb
The deoxygenation was carried out in refluxing dichloroethane for
several days and additional amounts of TFA were added to replenish any
losses. More preferably, the deoxygenation was carried out in a sealed
tube with neat triethylsilane and TFA at about 110-120 C. As will be
appreciated by those skilled in the art, the rate of deoxygenation was
dependent upon the electronic nature of the substituents present in the
hydroxyindolone of Formula Ila and unsubstituted and electron-rich
compounds of Formula Ila deoxygenated much more readily compared to
electron-deficient compounds of Formula Ila. Demethylation of the methyl
ether moiety of the compound of Formula Illa with BBr3 in CH2CI2 under
carefully controlled conditions from -78 C to 0 C afforded the
corresponding phenol of Formula Illb.
12 CT-2294A (BMMA-10A)
In an altemate route to the indolone of Formula Illa which avoids the
use of triethylsilane and trifluoroacetic acid at temperatures of 110-120 C
for several days is illustrated in Reaction Scheme 6. The process depicted
in Reaction Scheme 6 illustrates the preparation of a specific indolone of
Formula Illc, wherein Rl, R3 and R4 are hydrogen, R2 is trifluoromethyl and
R6 is chloro. Thus, chlorination of commercially available 2-methoxy-
phenylacetic acid with S02CI2 in acetic acid gave 5-chloro-2-
methoxyphenylacetic acid which was converted to the methyl ester of
Formula X using dimethyl sulfate and anhydrous K2C03 in CH3CN.
Reaction of the potassium enolate of ester of Formula X with 4-fluoro-3-
nitrobenzotrifluoride in the presence of one equivalent of additional
potassium bis(trimethylsilyl)amide (KHMDS) in THF at -78 C resulted in
formation of a dark blue solution of potassium enolate of Formula XI and
acidic workup of the reaction provided the desired ester of Formula XI in
75% yield. Upon reduction of the nitro group in the compound of Formula
Xi with iron in acetic acid, the resultant anilino-ester spontaneously
cyclized
to provide the desired indolone of Formula IIIc in 84% recrystallized yield.
REACTION SCHEME 6
CH2CO2H 1. SO 2CI2, ACOH CH2C02Me 1. KHMDS (2.1 eqt), THF
OMe 2. Me2SO4, K2CO3 jjOMe 2.
F3 C NO
CH3CN 2
CI
X -78 to -40 C; 75%
FsC NO2 F3C N
~ ~
C02Me O
/
Fe, AcOH
OMe OMe
reflux, 1 hr
CI CI
XI IIIc
1176 18~
13 CT-2294A (BMMA-10A)
When it is desired to prepare the fluoroindolones of Formula IV, the
corresponding hydroxyindolone of Formula Ila is reacted with
diethylaminosulfur trifluoride (DAST) as illustrated in Reaction Scheme 7.
Preferably, the reaction with DAST is carried out in an organic solvent such
as methylene chloride at a temperature of about -78 to 0 C. It should be
appreciated by those skilled in the art that the hydroxyindolone of Formula
Ila will thereby produce the corresponding fluoroindolone of Formula IV.
The substantially pure enantiomeric forms of the fluoroindolone of
Formula IV may readily be obtained by the separation of the racemic
mixture using chiral high pressure liquid chromatography methods as
described herein and by other well-known methods.
REACTION SCHEME 7
RO
Re
H
F I \ N F? N
O
OH DAST, CH 2G 2 0
Fe F
R4 / OMe R3
-78 to 0 C Ra OMe
Rs ~ Ft
Ila IV
When it is desired to prepare the substantially enantiomeric pure
form of the hydroxyindolone of Formulas Ila and IIb, the corresponding
indolone of Formula Illa is selectively oxidized using the appropriate
commerically available chiral (+)-(2R, 8aS)- or (-)-(2S, 8aR)-
(camphorsulfonyl) oxaziridine of Formula lXa or IXb, respectively as
illustrated in Reaction Scheme 8.
14 CT-2294A (BMMA-10A)
REACTION SCHEME 8
Re RO
1. KHMDS, dry degased THF
Fe ' N O argon, -78 C to 0 oC F H
0
FF R3 OH
Fe OMe Ra OMe
I (+~Ixa I
Rs g\I~ Rs
IIia Op 0
/ `
lic
KHMDS
THF, Ar (-)-IXb BBr3, CH2CI2
-78 to 0 C N -78 to 0 C
o S\
O
RO
Re R2 N
~ BBr3, :':
aõ~ -78 to ~ Ra OH O
H
p4 OMe
Ff6
Ff \ I I
lid lie or Ilf
In the process of Reaction Scheme 8, the hydroxylation of the
indolone of Formula Illa is treated with potassium bis(trimethylsilyl)amide
(KHMDS) in THF and the resulting potassium enolate of the indolone of
Formula Illa is oxidized with the chiral oxaziridine IXa or IXb at about -78
C
followed by gradual warming to O C and then quenching with glacial acetic
acid gave the desired corresponding hydroxyindolone of Formula IIc or Ild,
respectively in high yield and high enantiomeric purity as determined by the
application of the NMR chiral-shift solvent (L)-trifluoromethylphenyl
carbinol.
In the hydroxylation process of Reaction Scheme 8, it is most preferred to
utilize degassed dry THF under an argon atmosphere to prevent
~1'7G18~
15 CT-2294A (BMMA-10A)
hydroxylation with molecular oxygen and retain a high degree of
asymmetric hydroxylation. Finally, the methyl ether moieties of
hydroxyindolones of Formula lic and lid may be demethylated with BBr3 in
methylene chloride to afford the corresponding phenois of Formula Ile and
Ilf, respectively. In a specific example described herein, the compound of
Example 12 and the compound of Example 13 were each prepared with
greater than 95% enantiomeric purity.
In a preferred embodiment of the invention the compounds of
Formula II have the formula
R1
R2 H
~ N
~ O
R3 ~
Rw' ~ OR5 II
F:P
wherein Rl, R2, R3 and R4 each are independently hydrogen, methyl,
halogen, or trifluoromethyl, and when Rl, R3 and R4 are hydrogen, R2 is
phenyl, p-methylphenyl or trifluoromethylphenyl; or Rl and R2, R2 and R3 or
R3 and R4 are joined together to form a benzo fused ring;
R5 is hydrogen or methyl; and R6 is chlorine or trifluoromethyl; or a nontoxic
pharmaceutically acceptable salt, solvate or hydrate thereof.
In another preferred embodiment of the invention the compounds of
Formula III have the formula
R'
4
F? H
N
O
R3
w ORS jII
FO
wherein Rl, R2, R3 and R4 each are independently hydrogen, methyl,
halogen, or trifluoromethyl, and when R1, R3 and R4 are hydrogen, R2 is
phenyl, p-methylphenyl or trifluoromethylphenyl; or Rl and R2, R2 and R3 or
~
~17 6 18
16 CT-2294A (BMMA-10A)
R3 and R4 are joined together to form a benzo fused ring; R5 is hydrogen
or methly; and R6 is chlorine or trifluoromethyl; or a nontoxic
pharmaceutically acceptable salt, solvate or hydrate thereof.
In still another preferred embodiment of the invention the compounds
of Formula IV have the formula
R1
F? H
~ N
~ 0
R3 ~
R4 F ~5 IV
F~
wherein Rl, R2, R3 and R4 each are independently hydrogen, methyl,
halogen, or trifluoromethyl, and when Rl, R3 and R4 are hydrogen, R2 is
phenyl, p-methylphenyl or trifluoromethyl; or Rl and R2, R2 and R3 or
R3 and R4 are joined together to form a benzo fused ring; R5 is C1-4 alkyl;
and R6 is chlorine or trifluoromethyl; or a nontoxic pharmaceutically
acceptable salt, solvate or hydrate thereof.
In another aspect, this invention provides a method for the treatment
of or protection from disorders which are mediated by opening of the large
conductance calcium-activated K+ channels (BK channels) in a mammal in
need thereof, which comprises administering to said mammal a
therapeutically effective amount of a compound of Formula I or a nontoxic
pharmaceutically acceptable salt, solvate or hydrate thereof. Preferably, the
compounds of Formula I are useful in the treatment of ischemia,
convulsions, asthma, urinary incontinence and traumatic brain injury and
other disorders sensitive to BK channel activating activity. Most preferably,
the compounds of Formula I are useful in the treatment of cerebral
ischemia.
In still another aspect, this invention provides pharmaceutical
compositions comprising at least one compound of Formula I in
combination with a pharmaceutical adjuvant, carrier or diluent.
17bI
17 CT-2294A (BMMA-10A)
Biological Activity
Potassium (K+) channels are structurally and functionally diverse
families of K+-selective channel proteins which are ubiquitous in cells,
indicating their central importance in regulating a number of key cell
functions [Rudy, B., Neuroscience, 25: 729-749 (1988)]. While widely
distributed as a class, K+ channels are differentially distributed as
individual
members of this class or as families. [Gehiert, D.R., et al., Neuroscience,
52:
191-205 (1993)]. In general, activation of K+ channels in cells, and
particularly in excitable cells such as neurons and muscle cells, leads to
hyperpolarization of the cell membrane, or in the case of depolarized cells,
to repolarization. In addition to acting as an endogenous membrane
voltage clamp, K+ channels can respond to important cellular events such
as changes in the intracellular concentration of ATP or the intracellular
concentration of calcium (Ca2+). The central role of K+ channels in
regulating numerous cell functions makes them particularly important
targets for therapeutic development. [Cook, N.S., Potassium channels:
Structure, classification, function and therapeutic potential. Ellis Horwood,
Chinchester (1990)]. One class of K+ channels, the large-conductance
Ca2+-activated K+ channels (maxi-K or BK channels), is regulated by
transmembrane voltage, intracellular Ca2+, and a variety of other factors
such as the phosphorylation state of the channel protein. [Latorre, R., et
al.,
Ann. Rev. Pysiol., 51: 385-399 (1989)]. The large, single channel-
conductance (generally > 150 pS) and high degree of specificity for K+ of
BK channels indicates that small numbers of channels could profoundly
affect membrane conductance and cell excitability. Additionally, the
increase in open probability with increasing intracellular Ca2+ indicates
involvement of BK channels in the modulation of Ca2+-dependent
phenomena such as secretion and muscular contraction. [Asano, M., et al.,
J. Pharmacol. E~x . Ther., 267: 1277-1285 (1993)].
Openers of BK exert their cellular effects by increasing the open
probability of these channels [McKay, M.C., et al., J. Neurophysiol., 71:
1873-1882 (1994); and Olesen, S.-P., Exp. Opin. Invest. Druas, 3: 1181-
1188 (1994)]. This increase in the opening of individual BK channels
collectively results in the hyperpolarization of cell membranes, particularly
- ~176 18 3
18 CT-2294A (BMMA-10A)
in depolarized cells, produced by significant increases in whole-cell BK-
mediated conductance.
The ability of compounds described in the present invention to open
BK channels and increase whole-cell outward (K+) BK-mediated currents
was assessed under voltage-clamp conditions by determining their ability to
increase cloned mammalian (mSlo or hSlo) BK - mediated outward current
heterologously expressed in Xenopus oocytes [Butler, A., et al., Science,
261: 221-224 (1993); and Dworetzky, S.I., et al., Mol. Brain Res., 27: 189-
193 (1994)]. The two BK constructs employed represent nearly structurally
identical homologous proteins, and have proven to be pharmacologically
identical in our tests. To isolate BK current from native (background, non-
BK) current, the specific and potent BK channel-blocking toxin iberiotoxin
(IBTX) [Gaivez, A., et al., J. Biol. Chem, 265: 1 1 083-1 1 090 (1990)] was
employed at a supramaximal concentration (50 nM). The relative
contribution of BK channels current to total outward current was determined
by subtraction of the current remaining in the presence of IBTX (non-BK
current) from the current profiles obtained in all other experimental
conditions (control, drug, and wash). It was determined that at the tested
concentration the compounds profiled did not effect non-BK native currents
in the oocytes. All compounds were tested in at least 5 oocytes and are
reported at the single concentration of 20uM; the effect of the selected
compounds of Formula I on BK current was expressed as the percent of
control IBTX-sensitive current and is listed in Table I. Recordings were
accomplished using standard two-electrode voltage clamp techniques
[Stuhmer, W., et al., Methods in Enzymology, Vol. 207: 319-339 (1992)];
voltage-clamp protocols consisted of 500-750 ms duration step
depolarizations from a-holding potential of -60 mV to +140 mV in 20 mV
steps. The experimental media (modified Barth's solution) consisted of (in
mM): NaCI (88), NaHCO3 (2.4), KCI (1.0), HEPES (10), MgSO4 (0.82),
Ca(N03)2 (0.33), CaCI2 (0.41); pH 7.5.
19 CT-2294A (BMMA-10A)
TABLE I
Effect of Selected Compounds on BK Channels
Example No. BK Current*
1 +
3 ++
4 +++
6 +
7 +
9 ++
13 ++
14 ++
15 +++
16 +++
22 +++
25 +
31 +
33 +++
* at 20 M expressed as percent of controls;
+ = 100-125%
++ = 125-150%
+++_> 150%
The compound of Example 3 was also examined with single-channel
inside-out excised patches from both COS cells stably transfected with
mSio and with patches from oocytes expressing hSlo and was found to
effectively increase the open probability in both systems at a concentration
of 1 M. Other compounds were examined using whole-cell and single-
channel patch-clamp techniques in HEK 293 cells transiently and stably
expressing hSlo BK channels at high levels, for example, the compounds of
Examples 14, 37 and 38 were found to be very potent and effective openers
of hSlo BK channels under whole-cell and (inside out and outside-out)
excised patch clamp recording conditions.
20 CT-2294A (BMMA-10A)
To determine the ability of these compounds to reduce cell loss
resulting from neuronal ischemia, a standard rodent model of permanent
focal ischemia, involving occlusion of the middle cerebral artery in the
spontaneously hypertensive rat (MCAO model) was employed [Tamura, A.,
et al., Journal of Cerebral Blood Flow and Metabolism, Volume 1, 53-60,
(1981)].
Selected compounds have been evaluated in the focal stroke model
involving permanent middle cerebral artery occlusion (MCAO) in the
spontaneously hypertensive rat. This procedure results in a reliably large
neocortical infarct volume that is measured by means of vital dye exclusion
in serial slices through the brain 24 hours after MCAO. In the present test,
compounds were administered using an i.v. or i.p. route of administration at
two hours after occlusion. For example, in this model, the compound of
Example 4 reduced the cortical infarct volume by about 18% when
administered intravenously (0.03 mg/kg) and about 26% when
administered intraparitoneally (10 mg/kg) as a single bolus 2 hours after
middle cerebral artery occlusion as compared to vehicle-treated (2%
DMSO, 98% PG) control. Also, in this model, the compound of Example 14
reduced the cortical infarct volume by about 18% when administered
intravenously (0.3 mg/kg) as a single bolus 2 hours after middle cerebral
artery occlusion as compared to vehicle-treated (2% DMSO, 98% PG)
control.
The results of the above in vitro and in vivo tests demonstrate that the
compounds of the instant invention are potent openers of the large-
conductance calcium-activated K+ channels (maxi-K or BK channels).
Thus, the compounds of the present invention are useful for the treatment of
human disorders arising from dysfunction of cellular membrane polarization
and conductance and, preferably, are indicated for the treatment of
ischemia, convulsions, asthma, urinary incontinence and traumatic brain
injury and other disorders sensitive to BK channel activating activity. Most
preferably, the compounds of Formula I are useful in the treatment of
cerebral ischemia.
21 CT-2294A (BMMA-10A)
Therefore, the compounds of Formula I or pharmaceutical
compositions thereof are useful in the treatment, alleviation or elimination
of
disorders or other disorders associated with the BK channels. Such
disorders include ischemia, convulsions, asthma, urinary incontinence and
traumatic brain injury, and other disorders sensitive to potassium channel
openers.
In another embodiment, this invention includes pharmaceutical
compositions comprising at least one compound of Formula I in
combination with a pharmaceutical adjuvant, carrier or diluent.
In still another embodiment, this invention relates to a method of
treatment or prevention of disorders responsive to opening of potassium
channels in a mammal in need thereof, which comprises administering to
said mammal a therapeutically effective amount of a compound of Formula I
or a nontoxic pharmaceutically acceptable salt, solvate or hydrate thereof.
In yet another embodiment, this invention relates to a method for
treating an ischemic condition in a mammal in need thereof, which
comprises administering to said mammal a therapeutically effective amount
of a compound of Formula I or a non-toxic pharmaceutically acceptable salt,
solvate or hydrate thereof.
For therapeutic use, the pharmacologically active compounds of
Formula I will normally be administered as a pharmaceutical composition
comprising as the (or an) essential active ingredient at least one such
compound in association with a solid or liquid pharmaceutically acceptable
carrier and, optionally, with pharmaceutically acceptable adjuvants and
excipients employing standard and conventional techniques.
The pharmaceutical compositions include suitable dosage forms for
oral, parenteral (including subcutaneous, intramuscular, intradermal and
intravenous) bronchial or nasal administration. Thus, if a solid carrier is
used, the preparation may be tableted, placed in a hard gelatin capsule in
powder or pellet form, or in the form of a troche or lozenge. The solid
carrier
may contain conventional excipients such as binding agents, fillers,
tableting lubricants, disintegrants, wetting agents and the like. The tablet
22 CT-2294A (BMMA-10A)
may, if desired, be film coated by conventional techniques. If a liquid
carrier
is employed, the preparation may be in the form of a syrup, emulsion, soft
gelatin capsule, sterile vehicle for injection, an aqueous or non-aqueous
liquid suspension, or may be a dry product for reconstitution with water or
other suitable vehicle before use. Liquid preparations may contain
conventional additives such as suspending agents, emulsifying agents,
wetting agents, non-aqueous vehicle (including edible oils), preservatives,
as well as flavoring and/or coloring agents. For parenteral administration, a
vehicle normally will comprise sterile water, at least in large part, although
saline solutions, glucose solutions and like may be utilized. Injectable
suspensions also may be used, in which case conventional suspending
agents may be employed. Conventional preservatives, buffering agents
and the like also may be added to the parenteral dosage forms. Particularly
useful is the administration of a compound of Formula I directly in parenteral
formulations. The pharmaceutical compositions are prepared by
conventional techniques appropriate to the desired preparation containing
appropriate amounts of the active ingredient, that is, the compound of
Formula I according to the invention. See, for example, Remington's
Pharmaceutical Sciences, Mack Publishing Company, Easton, PA, 17th
edition, 1985.
The dosage of the compounds of Formula I to achieve a therapeutic
effect will depend not only on such factors as the age, weight and sex of the
patient and mode of administration, but also on the degree of potassium
channel activating activity desired and the potency of the particular
compound being utilized for the particular disorder of disease concerned. It
is also contemplated that the treatment and dosage of the particular
compound may be administered in unit dosage form and that the unit
dosage form would be adjusted accordingly by one skilled in the art to
reflect the relative level of activity. The decision as to the particular
dosage
to be employed (and the number of times to be administered per day) is
within the discretion of the physician, and may be varied by titration of the
dosage to the particular circumstances of this invention to produce the
desired therapeutic effect.
A suitable dose of a compound of Formula I or pharmaceutical
composition thereof for a mammal, including man, suffering from, or likely to
2 17 6 18~
23 CT-2294A (BMMA-10A)
suffer from any condition as described herein is an amount of active
ingredient from about 0.1 g/kg to 100 mg/kg body weight. For parenteral
administration, the dose may be in the range of 1 g/kg to 10 mg/kg body
weight for intravenous administration The active ingredient will preferably
be administered in equal doses from one to four times a day. However,
usually a small dosage is administered, and the dosage is gradually
increased until the optimal dosage for the host under treatment is
determined.
However, it will be understood that the amount of the compound
actually administered will be determined by a physician, in the light of the
relevant circumstances including the condition to be treated, the choice of
compound of be administered, the chosen route of administration, the age,
weight, and response of the individual patient, and the severity of the
patient's symptoms.
The following examples are given by way of illustration and are not to
be construed as limiting the invention in any way inasmuch as many
variations of the invention are possible within the spirit of the invention.
DESCRIPTION OF SPECIFIC EMBODIMENTS
In the following examples, all temperatures are given in degrees
Centigrade. Melting points were recorded on a Gailenkamp capillary
melting point apparatus and boiling points were measured at specific
pressures (mm Hg) and both temperatures are uncorrected. Proton
magnetic resonance (1H NMR) and carbon magnetic resonance
(13C NMR) spectra were recorded on a Bruker AC 300 and fluorine
magnetic resonance (19F NMR) were recorded on a Bruker AM 300,
equipped with a QNP probe. All spectra were determined in the solvents
indicated and chemical shifts are reported in S units downfield from the
intemal standard tetramethylsilane (TMS) and interproton coupling
constants are reported in Hertz (Hz). Splitting pattems are designated as
follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br,
broad
peak; dd, doublet of doublet; bd, broad doublet; dt, doublet of triplet; bs,
broad singlet; dq, doublet of quartet. Infrared (IR) spectra using potassium
- ~176133
24 CT-2294A (BMMA-10A)
bromide (KBr) were determined on a Perkin Elmer 781 spectrometer from
4000 cm-1 to 400 cm-1 , calibrated to 1601 cm-1 absorption of a polystyrene
film and reported in reciprocal centimeters (cm-1 ). Optical rotations [a]p
were determined on a Perkin-Elmer 41 polarimeter in the solvents
indicated. Low resolution mass spectra (MS) and the apparent molecular
(MH+) or (M-H)- was determined on a Finnigen TSQ 7000. The element
analysis are reported as percent by weight.
The following preparations Nos.1-5 illustrate representative actual
procedures for the preparation of intermediates and methods for the
preparation of products according to this invention. It should also be
evident to those skilled in the art that appropriate substitution of both the
materials and methods disclosed herein will produce the examples
illustrated below and those encompassed by the scope of this invention.
Preparation No. 1
6-(Trifluoromethyl)-1 H-indol-2.3-dione
A stirred neat mixture of 3-aminobenzotrifluoride (82.4 g, 0.51 mol)
and (Boc)2O (123 g, 0.56 mol) was heated at 80 C for 2-3 hours until C02
evolution was ceased. The mixture was allowed to cool and the tBuOH was
rotary evaporated. The resultant white solid was recrystallized from
hexanes to provide white needles (119 g, 89%) of N-(tert-
butoxycarbonyl)aminobenzotrifluoride.
Sec-BuLi (338 mL, 0.44 mol, 1.3M in cyclohexane) was added over
20 minutes to a cold (-78 C) stirred solution of N-Boc-
aminobenzotrifluoride (52.25 g, 0.2 mol) in dry THF (200 mL) under
nitrogen. The resultant yellow partial solution was warmed to -45 -40 C
and maintained for 2 hrs. The resultant thick yellow slurry of the dianion
was cooled to -78 C and neat dry diethyl oxalate (35.1 g, 0.24 mol) was
added rapidly. The resultant orange-brown solution was stirred at -78 C
for 1 hour. The reaction was diluted with ether (200 mL) and quenched with
3N HCI (150 mL) and then allowed to warm to room temperature. The
25 CT-2294A (BMMA-10A)
organic layer was separated, washed with water, brine and then dried
(Na2SO4). Evaporation of solvents gave a golden-yellow oil (80.7 g) which
was flash chromatographed (silica gel /CH2CI2) to afford the pure keto-ester
(61.1 g, 85%): IR (film, cm-1) 3320, 1740, 1670, 1540, 1370, 1340, 1250,
1140; 1 H NMR (300 MHz, CDCI3) 8 1.40 (3 H, t, J = 7.1 Hz), 1.51 (9 H, s),
4.45 (2 H, q, J = 7.1 Hz), 7.25 (1 H, d, J = 8.3 Hz) 7.79 (1 H, d, J = 8.3
Hz),
8.86 (1 H, s), 10.40 (1 H, brd s); MS m/e 362 (MH+).
A stirred solution of the keto-ester (57 g, 0.158 mol) in THF (1 L) and
3N HCI (250 mL) was heated to reflux for 6 hours. The mixture was allowed
to cool and the THF was rotary evaporated. The resultant orange
suspension was allowed to cool. The solid was filtered, washed with water
and then air dried overnight to afford the desired 6-(trifluoromethyl)isatin
(29.6 g, 87%): mp 196-198 C; IR (KBr, cm-1) 3100, 1750, 1710, 1320,
1170, 1125; 1H NMR (300 MHz, DMSO-d6) S 7.09 (1 H, s), 7.39 (1 H, d, J
7.7 Hz), 7.68 (1 H, d, J = 7.7 Hz), 11.26 (1 H, brd s); MS m/e 216 (MH+).
Anal. calcd. for C9H4F3NO2: C, 50.28 H, 1.91; N, 6.47.
Found: C, 50.25; H, 1.87; N, 6.51.
The following isatin was prepared according to a similar procedure.
5-(Trifluoromethyl)-1 H-indol-2,3-dione: mp 188-190 C
Preparation No. 2
4 6-bis-(Trifluoromethyl)-1 H-indol-2.3-dione
Freshly prepared tBuOCI (2.2 g, 20 mmol) was added dropwise to a
stirred cold (-65 C) solution of 3,5-bis-(trifluoromethyl)aniline (4.58 g, 20
mmol) in anhydrous CH2CI2 (25 mL). After 10 minutes, neat ethyl
(methylthio)acetate (2.68 g, 20 mmol) was added and the mixture stirred at
-65 C for 1 hr. Triethylamine (2.68 g, 20 mmol) was added and then the
mixture was allowed to warm to room temperature. The reaction mixture
was quenched with water, the organic layer was separated and then rotary
evaporated. The oily residue (6.35 g) was dissolved in hexanes and boiled
26 CT-2294A (BMMA-10A)
for several hours and then allowed to cool. The precipitated beige solid
was filtered and washed with hexanes to afford 3.67 g of pure (methylthio)
indolone intermediate.
N-chlorosuccinimide (1.31 g, 9.8 mmol) was added to a stirred
solution of (methylthio) indolone (2.95 g, 9.4 mmol) in CC14 (150 mL). The
mixture was stirred at room temperature for 6.5 hours. The suspension was
filtered, washed with CCI4 and the filtrate was rotary evaporated at 25-30
C. The residual light-brown oil was dissolved in a minimum volume (-- 5-
10 mL) of CCI4 and kept in an ice bath. The precipitated last traces of
succinimide were filtered and washed with hexanes. Evaporation of the
filtrate gave a red-brown oil (3.56 g) of a-chlorooxindole which was used in
the next step without further purification.
Neat BF39OEt2 (1.16 mL, 9.4 mmol) was added to a stirred
suspension of HgO in 4:1 THF-H20 (100 mL). A solution of the a-
chlorooxindole (3.56 g) in THF (20 mL) was added and the mixture was
stirred for 2-3 days. The suspension was filtered through a celite pad and
the filtrate was washed with saturated brine and then dried (Na2SO4).
Evaporation of THF gave 3.2 g of crude isatin which was recrystallized from
ether to afford pure 4,6-bis-(trifluoromethyl)-1 H-indol-2,3-dione (1.27 g):
mp 200-203 C ; IR (KBr, cm-1) 1778, 1748, 1278, 1138; 1 H NMR (300
MHz, DMSO-d6) S 7.40 (1 H, s), 7.61 (1 H, s), 11.52 (1 H, s); MS m/e 284
(MH+).
The following isatins were prepared according to known literature
procedures, P.M. Maginnity, et al, J. Am. Chem. Soc., 73, 3579 (1951) and
C.S. Marvel, et al, Or anic Synthesis Coll. Vol. 1, 327-330.
4-(Trifluoromethyl)-1 H-indol-2,3-dione: mp 210-212 C
7-(Trifluoromethyl)-1 H-indol-2,3-dione: mp 190-192 C
4,6-Dichloro-1 H-indol-2,3-dione: mp 258-260 C
The following isatins can also be prepared by the methods in the cited
reference.
~17 61
27 CT-2294A (BMMA-10A)
1 H-Benz[g]indol-2,3-dione: mp 256-259 C (dec.) [H. Cassebaum, Chem.
Ber., 90, 2876 (1957)].
1 f I Benz[f]indol-2,3-dione: mp 250-255 C (dec.) [A. Etienne et al., Bull.
Soc. Chem. Fr., 6, 743-748 (1954)].
1 H-Benz[e]indol-2,3-dione: mp 252-254 C (dec.) [W. Wendelin, et al., J.
Het. Chem., 24, 1381 (1987)].
6-Phenyl-1 H-indol-2,3-dione: mp 230-235 C [P. W. Sadler, J. Ocr . Chem.,
21, 169 (1956)].
6-lodo-1 H-indol-2,3-dione: mp 196-198 C [Von. W. Langenbeck, et al., J.
Prakt. Chemie., 4, IV, 136-146 (1956)].
Preparation No. 3
General Procedure for preparation of 1.3-dihydro-3-hydroxX 3-(2-
hydroxyaryl)-2H-indol-2-ones:
R'
F? H
N
~ 0
R3 ~
OH
R4 OH
Method A: A solution of the 2-methoxyaryl Grignard reagent (1-2 eqt.) in
THF was added to a stirred cold (-20 C) solution of sodium salt of the isatin
in THF under nitrogen. The mixture was allowed to warm to room
temperature and maintained until (1-2 hours) the isatin was consumed. The
reaction mixture was diluted with ether, cooled in a ice-bath and then
quenched with 1 N HCI. The organic layer was separated, washed with
0.5'N NaOH, water, brine and then dried (Na2SO4). The crude solid
isolated after evaporation of the solvents was triturated with CH2CI2 to
~176183'
28 CT-2294A (BMMA-10A)
afford pure 1, 3-d ihyd ro-3-hyd roxy-3- (2-methoxyaryl) -2 H-i n dol -2 -ones
in
70-95% yield.
Demethylation of methyl ether moiety of the above product was
carried out with BBr3 in CH2CI2. To a cold (-78 C) stirred solution of 1,3-
dihydro-3-hydroxy-3-(2-methoxyaryl)-2H-indol-2-one in anhydrous CH2CI2,
BBr3 (3 eqt.; 1 M solution in CH2CI2) was added under nitrogen. The
mixture was warmed in an ice bath and maintained until starting material
disappeared by TLC (1-2 hours). The reaction was quenched with
saturated NaHCO3 and then acidified with 1 N HCI. When the product is
soluble in CH2CI2, the organic layer was separated, washed with brine and
then dried (MgSO4). If product is insoluble in CH2CI2, the organic layer
was rotary evaporated at room temperature and the aqueous residue was
extracted with EtOAc. The EtOAc extract was washed with brine and then
dried (Na2SO4). Evaporation of the organic solvent gave 1,3-dihydro-3-
hydroxy-3-(2-hydroxyaryl)-2H-indol-2-ones. The crude product was purified
by either trituration or recrystallization from a suitable solvent to afford
pure
product in 80-95% yield.
Method B: Bromomagnesium phenolate was prepared by reacting 1
equivalent of the phenol in ether with one equivalent of ethereal solution of
ethyl magnesium bromide at 0 C and then allowing to warm to room
temperature. The resultant suspension of the bromomagnesium phenolate
in ether was rotary evaporated at 25 C to dryness and then dissolved in
anhydrous CH2CI2. A solution of the isatin (1 eqt) in CH2CI2 was added to
the bromomagnesium phenolate solution and the mixture stirred at room
temperature until the isatin was fully consumed (1-24 hours). The reaction
was quenched with either saturated NH4CI solution or 1 N HCI and then
extracted with CH2CI2. The crude product was purified by either trituration
or recrystallization from a suitable solvent to afford pure 1,3-dihydro-3-
hydroxy-3-(2-hydroxyaryl)-2H-indol-2-ones.
~17fi1.83
29 CT-2294A (BMMA-10A)
Preparation No. 4
General Procedure for preparation of 1.3-dihydro-3-(2-hydroxyaryl)-2H-
indol-2-ones
w
FF H
N
0
R3 \
R' OH
\
R6
A neat stirred mixture of 1,3-dihydro-3-hydroxy-3-(2-methoxyaryl)-2H-indol-
2-one (1 eqt.), triethylsilane (3 eqt.) and trifluoroacetic acid (3 eqt.) was
heated at 110-120 C in a sealed tube for 1-3 days until deoxygenation was
complete by TLC analysis. Excess Et3SiH and TFA were rotary evaporated
and the residue was flash chromatographed (silica gel/3% MeOH in
CH2CI2) to afford the desired deoxygenated product 1,3-dihydro-3-(2-
methoxyaryl)-2H-indol-2-one (80-90%). Demethylation of the methyl ether
moiety was carried out with BBr3 (3 eqt.) at -78 C to 0 C followed by usual
work up as described in Preparation No. 3 to afford the title product.
Preparation No. 5
General Procedure for preparation of 1.3-dihydro-3-fluoro-3-(2-
methoxyark)-2H-indol-2-ones
R'
F? H
N
~ 0
R3 \
F
::i0c
176183
30 CT-2294A (BMMA-10A)
Neat diethylaminosulfur trifluoride (DAST) was added dropwise to a cold
(-78 C) stirred solution or suspension of the 3-aryl-1,3-dihydro-3-hydroxy-
214 indol-2-one under nitrogen. The resultant mixture was allowed to warm
to 0 C. The progress of the reaction was followed by TLC. The reaction
mixture was quenched with water at 0 C. When the product was soluble in
CH2CI2 , the organic layer was separated, washed with brine and then
dried (MgSO4). If the product is insoluble or partially soluble in CH2CI2, the
organic layer was rotary evaporated at room temperature and the aqueous
residue was extracted with EtOAc. The EtOAc extract was washed with
brine and then dried (Na2SO4). Evaporation of the organic solvent afforded
the 3-aryl-1,3-dihydro-3-fluoro-2H-indol-2-ones. The crude products were
purified by either trituration or recrystallization from suitable solvents to
afford the pure products in 90-95% yield.
Preparation No. 6
Methyl (5-chloro-2-methoxxphenyI)acetate
Neat SO2CI2 (30.5 g, 18 mL, 0.225 mol) was added dropwise over
minutes to a cold (5 C) partial solution of (2-methoxyphenyl)acetic acid
(25 g, 0.15 mol) in glacial AcOH (500 mL). The mixture was stirred at room
temperature for 16 hours and then poured into cold water (2.5 L) with
25 vigorous stirring. The resultant white precipitate was allowed to stand at
room temperature for 2-3 hours, then filtered, washed with water and then
air dried overnight to afford (5-chloro-2-methoxyphenyl)acetic acid (19.8 g,
66%).
30 A stirred suspension of (5-chloro-2-methoxyphenyl)acetic acid (10 g,
0.05 mol), anhydrous K2C03 (8.3 g, 0.06 mol) and dimethyl sulfate (7.6 g,
0.06 mol) in anhydrous CH3CN (60 mL) was heated to reflux under nitrogen
for 2 hours. The reaction mixture was allowed to cool and the excess
dimethyl sulfate was quenched with Et3N (1 mL) and then filtered. The
filtrate was rotary evaporated and the residue was suspended in water and
extracted with ether, washed with satd. NaHCO3, water, brine and then
dried (Na2SO4). Filtration and evaporation of the ether gave a colorless oil
7G1~ ~
31 CT-2294A (BMMA-10A)
which was distilled in vacuo to afford methyl (5-chloro-2-
methoxyphenyl)acetate (10.1 g, 94%): bp 96-98 C/0.5 torr; IR (film, cm-1)
1742,1250, 1150, 1028; 1H NMR (300 MHz, CDC13) S 3.56 (2 H, s), 3.66 (3
H, s), 3.76 (3 H, s), 8.75 (1 H, d, J = 8.6 Hz), 7.13 (1 H, d, J = 2.5 Hz),
7.17
(1 H, dd, J = 8.6 and 2.5 Hz); MS m/e 215 (MH+).
Example 1
( )-3-(5-Chloro-2-methoxyphenyl)-1.3-dihydro-3-hydroxy-6-
(trifluoromethyl)-2H- indol-2-one
mp 207-210 C; IR (KBr, cm-1) 3400, 1730, 1320, 1250, 1125, 1170;
1 H NMR (300 MHz, DMSO-d6) S 3.41 (3 H, s), 6.91 (1 H, d, J = 0.73 Hz),
6.94 (1 H, s), 7.05 (2 H, m), 7.19 (1 H, d, J = 8.2 Hz), 7.35 (1 H, dd, J =
8.6
and 2.7 Hz), 7.80 (1 H, d, J = 2.7 Hz), 10.67 (1 H, s); 13C NMR (75 MHz,
DMSO-d6) S 55.95, 74.25, 105.21. 113.52, 118.45, 122.28, 124.27, 124.46,
126.83, 128.70, 129.47 (q), 131.31, 136.60, 143.88, 154.30, 177.29; MS
m/e 358 (MH+).
Anal. calcd. for C16H11CIF3NO3: C, 53.72; H, 3.09; N, 3.83.
Found: C, 53.43; H, 2.99; N, 3.83.
Example 2
( )-3-(5-Chloro-2-methoxyphenyl)-1.3-dihydro-6-(trifluoromethyl)-2H-indol-
2-one
A stirred suspension of methyl (5-chloro-2-methoxyphenyl)[2-nitro-4-
(trifluoromethyl)phenyl]acetate (2.02 g, 5 mmol) and iron powder (1.18 g, 20
mmol) in gla. AcOH (25 mL) was heated to reflux for 1 hour. The
suspension was allowed to cool and then poured into cold water (100 mL)
with vigorous stirring. The product was extracted with ether (2x50 mL),
washed with 6N HCI (50 mL), water, brine and then dried (Na2SO4).
Evaporation of ether gave a beige solid (1.8 g) which was triturated with
f;176 18
32 CT-2294A (BMMA-10A)
ether to afford the desired product as an off-white solid (1.61 g, 95%): mp
210-213 C; IR (KBr, cm-1) 3200, 1710, 1320, 1250, 1170, 1120, 1050; 1 H
NMR (300 MHz, DMSO-dg) S 3.57 (3 H, s), 4.91 (1 H, s) 7.02 (1 H, d, J = 8.8
Hz), 7.06 (2 H, m), 7.21, (1 H, dd, J = 7.7 and 0.6 Hz), 7.35 (2 H, m), 10.77
(1 H, s); 13C NMR (75 MHz, DMSO-ds) S 48.25, 56.07, 105.05. 113.58,
118.26, 122.42, 124.18, 126.03, 127.38, 128.60 (q), 128.70, 130.65,
134.37, 143.58, 156.09, 176.73; MS m/e 342 (MH+).
Anal. calcd. for C16H1jCIF3NO2: C, 56.24; H, 3.24; N, 4.09.
Found: C, 56.37; H, 3.25; N, 4.07.
Example 3
( )-3-(5-Chloro-2-hydroxyphenyl)-1.3-dihydro-3-hydroxy-6-(trifluoromethyl)-
2H-indol-2-one
mp 210-213 C; IR (KBr, cm-1) 3300, 1725, 1320, 1250, 1170, 1140;
1 H NMR (300 MHz, DMSO-d6) S 6.61 (1 H, dd, J = 8.5 and 2.6 Hz), 6.81
(1 H, s), 7.02, (1 H, s), 7.05 (1 H, d, J = 7.7 Hz), 7.17 (2 H, m), 7.71 (1 H,
d,
J = 2.7 Hz), 9.72 (1 H, s), 10.60 (1 H, s); 13C NMR (75 MHz, DMSO-d6) S
74.37, 105.17, 116.48. 118.30, 122.34, 124.32, 125.92, 126.84, 128.33,
129.14, 129.26 (q), 136.87, 144.11, 152.37, 177.30; MS m/e 344 (MH+).
Anal. calcd. for C15H9CIF3NO3: C, 52.42; H, 2.64; N, 4.08.
Found: C, 52.19; H, 2.57; N, 3.97.
Example 4
( )-3-(5-Chloro-2-hydroxyphenyl)-1.3-dihydro-6-(trifluoromethxl)-2H-indol-
2-one
A solution of BBr3 (12 mL, 12 mmol; 1 M in CH2CI2) was added to a
stirred cold (-78 C) solution of ( )-3-(5-chloro-2-methoxyphenyl)-1,3-
dihydro-6-(trifluoromethyl)-2H-indol-2-one (1.37 g, 4 mmol) in anhydrous
CH2CI2 (20 mL). The mixture was allowed to warm to ambient temperature
Ewi7 6 1:J3
33 CT-2294A (BMMA-10A)
and maintained for 2 hours. The reaction was quenched with saturated
NaHCO3 and then acidified with 1 N HCI. The organic layer was separated,
washed with water, brine and then dried (MgSO4). Evaporation of CH2CI2
gave an off-white solid which was triturated with warm CH2CI2 to afford the
title compound (1.21 g, 93%): mp 266-268 C; IR (KBr, cm-1) 3320, 1690,
1310, 1250, 1160, 1125; 1 H NMR (300 MHz, DMSO-d6) S 4.85 (1 H, s), 6.75
(1 H, d, J = 8.6 Hz), 7.04 (1 H, s), 7.11, (1 H, d, J = 7.7 Hz), 7.17 (1 H,
dd, J
8.6 and 2.6 Hz), 7.22 (1 H, d, J = 8.0 Hz), 7.26 (1 H, d, J = 2.4 Hz), 9.82 (1
H, s), 10.73 (1 H, s); 13C NMR (75 MHz, DMSO-d6) S 48.47, 104.94, 116.84.
118.16, 122.27, 124.20, 125.61, 126.06, 128.05, 128.41, 130.76, 134.72,
143.69, 154.26, 176.92. MS m/e 328 (MH+).
Anal. calcd. for C15H9CIF3N02: C, 54.98; H, 2.77; N, 4.27.
Found: C, 54.84; H, 2.64; N, 4.16.
Example 5
( )T3-(5-Chloro-2-hydroxXphenyl)-l.3-dihydro-3-hydroxy-5-(trifluoromethyl)-
2H-indol-2-one
mp 156-158 C; IR (KBr, cm-1) 3350, 1740, 1325, 1260, 1160, 1120;
1 H NMR (300 MHz, DMSO-d6) S 6.62 (1 H, d, J = 8.5 Hz), 6.80 (1 H, s), 6.98
(1 H,d,J=8.1 Hz),7.09(1 H, d, J = 1.2 Hz), 7.17 (1 H, dd, J = 8.5 and 2.7
Hz), 7.56 (1 H, dd, J = 8.1 and 1.2 Hz), 7.73 (1 H, d, J = 2.7 Hz), 9.73 (1 H,
s), 10.72 (1 H, s); 13C NMR (75 MHz, DMSO-d6) 8 74.37, 109.44, 116.52,
120.16, 121.75 (q), 122.37, 126.42, 126.75, 126.93, 128.37, 129.28,
133.33, 146.94, 152.36, 177.56; MS m/e 344 (MH+).
Anal. calcd. for C15H9CIF3N03: C, 52.42; H, 2.64; N, 4.08.
Found: C, 52.19; H, 2.48; N, 4.13.
2176 18 3
34 CT-2294A (BMMA-10A)
Example 6
(+_)-3-(5-Chloro-2-hydroxyphenyl)-4.6-dichloro-1.3-dihydro-3-hydroxy-2-H-
indol-2-one
mp 232-235 C (dec.); IR (KBr, cm-1) 3400, 1730, 1275; 1 H NMR (300 MHz,
DMSO-d6) S 6.61 (1 H, d, J = 8.5 Hz), 6.79 (1 H, d, J = 1.7 Hz), 6.81 (1 H,
s),
6.94(1 H, d, J = 1.7 Hz), 7.14 (1 H, dd, J = 8.5 and 2.7 Hz), 7.71 (1 H, d, J
=
2.7 Hz), 9.71 (1 H, s), 10.71 (1 H, s); 13C NMR (75 MHz, DMSO-d6) 5 74.77,
108.31, 116.32, 121.22, 122.07, 127.55, 128.20, 128.39, 130.40, 134.05,
146.26, 152.27, 176.89; MS m/e 344 (MH+).
Anal. calcd. for C14H8C13N03: C, 48.80; H, 2.34; N, 4.06.
Found: C, 48.70; H, 2.35; N, 4.01.
Example 7
( )-3-(5-Chloro-2-hydroxyphenyl)-1,3-dihydro-3-hydroxy-7-(trifluoromethyl)-
2H-indol-2-one
mp 205-207 C; I R (KBr, cm-1) 3250, 1745, 1340, 1240, 1175, 1120;
1H NMR (300 MHz, DMSO-d6) 5 6.59 (1 H, d, J = 8.5 Hz), 6.99 (1 H, t,
J = 7.7 Hz), 7.10 (1 H, d, J = 7.1 Hz), 7.16 (1 H, dd, J = 8.5 and 2.7 Hz),
7.44 (1 H, d, J = 7.8 Hz), 7.72 (1 H, d, J = 2.7 Hz), 9.79 (1 H, s), 10.79 (1
H,
s); 13C NMR (75 MHz, DMSO-d6) 573.41, 110.10 (q), 116.37, 121.43,
122.27, 125.16, 125.61, 126.83, 127.53, 128.30, 129.37, 134.40, 140.72,
152.38, 178.02; MS m/e 344 (MH+);
Anal. calcd. for C15H9CIF3NO3-0.2H20: C, 51.88; H, 2.73; N, 4.03.
Found: C, 51.87; H, 2.75; N, 3.98.
2"176183
35 CT-2294A (BMMA-10A)
Example 8
( )-3-(5-Chloro-2-hydroxyphenyl)-1.3-dihydro-3-hydroxy-4-trifluoromethxl)-
2H-indol-2-one
mp 239-242 C; IR (KBr, cm-1) 3300, 1725, 1330, 1250, 1170, 1140;
1H NMR (300 MHz, DMSO-d6) S 6.56 (1 H, d, J = 8.5Hz), 6.76 (1 H, s),
7.07-7.13 (3 H, m), 7.39 (1 H, t, J = 7.9 Hz), 7.66 (1 H, s), 9.57 (1 H, s),
10.71
(1 H, s); 13C NMR (75 MHz, DMSO-d6) S 74.93, 113.23, 116.01, 118.26,
121.60, 121.84,125.50 (q), 127.77, 127.95, 128.76, 129.57, 129.73, 145.00,
152.40, 176.89; MS m/e 344 (MH+).
Anal. calcd. for C15H9CIF3N03: C, 52.42; H, 2.64; N, 4.08.
Found: C, 52.16; H, 2.87; N, 4.06.
Example 9
( )-1.3-Dihydro-3-hydroxy-3-[2-hvdroxy-5-(trifluoromethyl)phenkl-6-
(trifluoromethyl)-2H-indol-2-one
mp 175-177 C; 1H NMR (300 MHz, DMSO-d6) S 6.77 (1 H, d, J = 8.3 Hz),
7.02-7.09 (2 H, m), 7.19 (1 H, d, J = 7.7 Hz), 7.51 (1 H, d, J = 8.3 Hz), 8.08
(1
H, s), 10.40 (1 H, s), 10.67 (1 H, s); MS m/e 378 (MHi-).
Anal. calcd. for C16H9F6N03: C, 50.94; H, 2.40; N, 3.71.
Found: C, 50.85; H, 2.34; N, 3.76.
Example 10
( )-3-(5-Chloro-2-hydroxyphenyl)-1.3-dihydro-3-hydroxy-4.6-
bis(trifluoromethyl)-2H-indol-2-one
mp.191-193 C; I R (KBr, cm-1) 3700-2500, 1740, 1280, 1170, 1130;
1 H NMR (300 MHz, DMSO-d6) 8 6.59 (1 H, d, J = 8.5 Hz), 7.06 (1 H, s), 7.15
36 CT-2294A (BMMA-10A)
(1 H, dd, J = 8.5 and 2.6 Hz), 7.34 (1 H, s), 7.45 (1 H, s) 7.68 (1 H, d, J =
2.6
Hz), 9.74 (1 H, s), 11.07 (1 H, s); MS m/e 412 (MH+).
Anal. calcd. for C16H8CIF6NO3=0.2H20: C, 46.24; H, 2.05; N, 3.37.
Found: C, 46.24; H, 2.18; N, 3.27.
Example 11
(-)-3-(5-Chloro-2-methoxyphenyl)-1.3-dihvdro-3-hydroxy-6-(trifluoromethyl)-
2H-indol-2-one
Potassium bis(trimethylsilyl)amide solution (2.2 mL, 1.1 mmol, 0.5 M
in toluene) was added dropwise to a cold (-78 C) stirred solution of ( )-3-
(5-Chloro-2-methoxyphenyl)-1,3-dihydro-6-(trifluoromethyl)-2f / indol-2-one
342 mg, 1 mmol) in dry degased THF (3 mL) under argon. The resultant
light yellow solution of the potassium enolate was stirred at -78 C for 30
minutes. A solution of (1 S)-(+)-(10-camphorsulfonyl)oxaziridine (252 mg,
1.1 mmol) in dry degased THF (2 mL) was added dropwise over 5 minutes
to the enolate solution at -78 C. The mixture was stirred at -78 C for 1
hour and then allowed to warm in an ice-bath (0-5 C). The reaction was
quenched with glacial AcOH (0.1 mL), diluted with ether (25 mL) followed
by addition of a saturated NH4CI (10 mL) solution. Organic layer was
separated, washed with saturated NaHCO3, water, brine and then dried
(Na2SO4). Filtration and evaporation of solvents gave 0.54 g of crude
product which was triturated with ether to remove the insoluble,
(camphorsulfonyl)imine by-product through filtration. Evaporation of the
filtrate gave 0.39 g of product slightly contaminated with the by-product.
The crude product (0.39 g) was triturated with boiling CH2CI2 to afford 230
mg of pure desired hydroxyindalone. Concentration of the mother liquor
followed by re-trituration with CH2CI2 gave an additional 72 mg to afford
302 mg (84%) of combined product : mp 244-245 C; [a]p 25 -166.78
(CHCI3); IR (KBr, cm-1) 3300-3100, 1722, 1320, 1250, 1125; 1H NMR (300
MHz, DMSO-d6) 5 3.42 (3 H, s), 6.90 (1 H, s), 6.93 (1 H, d, J = 8.7 Hz), 7.04
(1 H, d, J = 7.7 Hz), 7.05 (1 H,s),7.19(1 H, d, J = 7.7 Hz), 7.35 (1 H,dd,J=
17 6 183
37 CT-2294A (BMMA-10A)
8.7 and 2.7 Hz), 7.79 (1 H, d, J = 2.7 Hz), 10.67 (1 H, brd s); MS m/e 358
(MH+).
Anal. calcd. for C16H1lCIF3N03: C, 53.72; H, 3.10; N, 3.92.
Found: C, 53.77; H, 2.95; N, 3.95.
Example 12
(+)T3-(5-Chloro-2-hydrox.yphenyl)-l.3-dihydro-3-hydroxy-6-(trifluoromethyl)-
2H-indol-2-one
A solution of BBr3 (1.4 mL, 1 M in CH2CI2) was added dropwise to a
cold (-78 C) stirred solution of (-)-3-(5-chloro-2-methoxyphenyl)-1,3-
dihydro-3-hydroxy-6-(trifluoromethyl)-2H-indol-2-one [prepared in
Example 11 ](170 mg, 0.475 mmol) in anhydrous CH2CI2 (10 mL). The
resultant mixture was warmed in an ice bath and maintained for 2 hours.
The reaction was quenched with saturated NaHCO3 and then acidified with
1 N HCI. The cloudy organic layer was separated, rotary evaporated, re-
dissolved in EtOAc (25 mL) and then combined with the aqueous layer.
The EtOAc layer was separated and washed with water, brine and then
dried (Na2SO4). Filtration and evaporation gave 198 mg of crude product
which was flash chromatographed (silica geV10% MeOH in CH2CI2) to
afford 164 mg (100%) of pure title compound as a white solid: mp
200-201 C; [a]p 25 + 29.90 (CHCI3); IR (KBr, cm-1) 3540, 3350, 1725,
1320, 1160, 1130; 1H NMR (300 MHz, DMSO-d6) S 6.60 (1 H, d, J = 8.5 Hz),
6.82 (1 H, brd s), 7.02, (1 H, s), 7.05 (1 H, d, J = 7.6 Hz), 7.17 (2 H, m),
7.71
(1 H, d, J = 2.7 Hz), 9.75 (1 H, brd s), 10.61 (1 H, s); MS m/e 344 (MH+).
Anal. calcd. for C15H9CIF3NO3: C, 52.42; H, 2.64; N, 4.08.
Found: C, 52.62; H, 2.48; N, 4.04.
~:lr6 1
38 CT-2294A (BMMA-10A)
Example 13
(-)-3-(5-Chloro-2-hydroxyphenyl)-1.3-dihydro-3-hydroxy-6-(trifluoromethKl)-
2H-indol-2-one
mp 198-200 C; [a]p 25 - 22.18 (CHCI3); IR (KBr, cm-1) 3540, 3300, 1725,
1320, 1125; 1H NMR (300 MHz, DMSO-d6) 5 6.60 (1 H, d, J = 8.6 Hz), 6.82
(1 H, brd s), 7.02, (1 H, s), 7.05 (1 H, d, J = 7.6 Hz), 7.17 (2 H, m), 7.72
(1 H,
d, J = 2.7 Hz), 9.75 (1 H, brd s), 10.61 (1 H, s); MS m/e 344 (MH+).
Anal. calcd. for C15H9CIF3NO3: C, 52.42; H, 2.64; N, 4.08.
Found: C, 52.40; H, 2.59; N, 4.01.
Example 14
( )-3-(5-Chloro-2-methoxyphenyl)-1.3-dihyd ro-3-fl uoro-6-(trifluoromethyl)-
2H-indol-2-one
mp 168-170 C; IR (KBr, cm-1) 3200, 1734, 1320, 1268, 1132; 1H NMR
(300 MHz, CDCI3) 5 3.53 (3 H, s), 6.76 (1 H, dd, J = 8.7 and 1.0 Hz), 7.14
(1 H, d, J = 2.0 Hz), 7.18 (1 H, dd, J = 7.8 and 2.0 Hz), 7.26 (1 H, d, J =
7.8
Hz), 7.33 (1 H, dd, J = 8.7 and 2.6 Hz), 7.78 (1 H, dd, J = 2.6 and 1.0 Hz),
9.00 (1 H, s). 19F NMR (282 MHz, CDCI3) 8 -63.10 (6-CF3), -159.87 (3-F);
MS m/e 360 (MH+).
Anal. calcd. for C16H10CIF4N02: C, 53.43; H, 2.80; N, 3.89.
Found: C, 53.44; H, 2.79; N, 3.84.
Example 15
( )-3-(5-Chloro-2-hydroxyphenyl)-1.3-dihydro-3-hydroxy-2 H-benz[-Qlindol-
2-one
mp 170-172 C; 1H NMR (300 MHz, DMSO-d6) S 6.58 (1 H, d, J = 8.5 Hz),
6.66 (1 H, s), 7.02 (1 H, d, J = 8.2 Hz), 7.14 (1 H, dd, J = 8.5 and 2.6 Hz),
39 CT-2294A (BMMA-10A)
7.42 (1 H, d, J = 8.2 Hz), 7.45-7.53 (2 H, m), 7.76 (1 H, d, J = 2.6 Hz), 7.85
(1 H, dd, J = 7.2 and 2.1 Hz), 8.10 (1 H, d, J = 7.2 Hz), 9.57 (1 H, brd s),
11.03 (1 H, s); MS m/e 324 [M-H)-].
Anal. calcd. for C18H12CINO3=0.25H20: C, 65.46; H, 3.82; N, 4.24.
Found: C, 65.48; H, 3.60; N, 3.89.
Example 16
( )-3-(5-Chloro-2-hydroxyphenyl)-1.3-dihydro-6-phenyl-2H-indol-2-one
mp 221-225 C (dec.); 1 H NMR (300 MHz, DMSO-d6) 8 6.78 (1 H, J = 9.1
Hz), 6.99 (1 H, d, J = 7.6 Hz), 7.05 (1 H, d, J = 6.6 Hz), 7.12-7.16 (3 H, m),
7.34 (1 H, t, J = 7.3 Hz), 7.44 (2 H, t, J = 7.2 Hz), 7.58 (2 H, d , J = 7.6
Hz),
9.83 (1 H, s), 10.59 (1 H, s); MS m/e 336 (MH+).
Anal. calcd. for C20H14CINO2=H20: C, 68.29; H, 4.01; N, 3.98.
Found: C, 68.47; H, 3.81; N, 3.89.
Example 17
( )-3-(5-Chloro-2-hydroxyphenyl)-1.3-dihydro-2H-benz[g]indol-2-one
mp 211-215 C (dec.); 1H NMR (300 MHz, DMSO-d6) 5 4.97 (1 H, s), 6.80 (1
H, d, J = 8.9 Hz), 7.10-7.17 (3 H, s), 7.46-7.53 (3 H, m), 7.87 (1 H, d, J =
7.5
Hz), 8.10 (1 H, d, J = 8:0 Hz), 9.83 (1 H, s), 11.25 (1 H, s); MS m/e 308
[M-H)-].
Anal. calcd. for C18H12CIN02=H20: C, 65.96; H, 4.31; N, 4.27.
Found: C, 65.87; H, 3.99; N, 3.88.
2176183
40 CT-2294A (BMMA-10A)
Example 18
( )-3-(5-Chloro-2-methoxyphenyl)-1.3-dihydro-3-fluoro-6-phenyl-2H-indol-
2-one
IR (KBr, cm-1) 3200, 1738, 1338, 1266, 1120, 766, 693; 1H NMR (300 MHz,
CDCI3) 5 3.56 (3 H, s), 6.76 (1 H, d, J = 8.7 Hz), 7.11 (1 H, s), 7.14 (1 H,
d,
J = 2.3 Hz), 7.20 (1 H, d, J = 7.9 Hz), 7.31 (1 H, dd, J = 8.6 and 2.3 Hz),
7.37
(1 H, d, J = 7.0 Hz), 7.43 (2 H, m), 7.54 (2 H, d, J = 6.9 Hz), 7.80 (2 H, d,
J = 2.5 Hz); 19F NMR (282 MHz, CDCI3) 8 -156.62 (3-F); MS m/e 366
[M-H)-].
Example 19
( )-3-(5-Chloro-2-methoxyphenyl)-1.3-dihydro-3-fluoro-6-iodo-2 H-indol-2-
one
mp 205-210 C; IR (KBr, cm-1) 3600-3200, 1736, 1266; 1H NMR (300 MHz,
CDCI3) 5 3.55 (3 H, s), 6.74 (1 H, d, J = 9.8 Hz), 6.80 (1 H, dd, J = 7.8 and
2.5 Hz), 7.26 (1 H, s), 7.28-7.35 (2 H, m), 7.66 (1 H, brd s), 7.74 (1 H, d,
J=
2.1 Hz); 19F NMR (282 MHz, CDCI3) 5-28.75(?) (3-F); MS m/e 416 [M-H)-].
Example 20
( )-3-(5-Chloro-2-hydroxyphenyl)-1.3-dihydro-6-(4-methkphenyl)-2H-indol-
2-one
mp 277-279 C (dec.); IR (KBr, cm-1) 3200, 1686; 1H NMR (300 MHz,
DMSO-d6) 5 2.32 (3 H, s), 4.78 (1 H, s), 6.81 (1 H, d, J = 9.1 Hz), 6.97 (1 H,
d, J = 7.7 Hz), 7.03 (1 H, d, J = 9.1 Hz)),7.09(1 H, d, J = 1.4 Hz), 7.12-7.16
(2 H, m), 7.24 (2 H, d, J = 8.5 Hz), 7.47 (2 H, d, J = 8.1 Hz), 9.87 (1 H, s),
10.58 (1 H, s); MS m/e 348 [(M-H)-].
2~ 17
41 CT-2294A (BMMA-10A)
Example 21
( )-3-(5-Chloro-2-methoxyphenyl)-1.3-dihydro-3-fluoro-7-(trifluoromethyl)-
2H-indol-2-one
mp 230-233 C; IR (KBr, cm-1) 3230, 1754, 1322, 1212, 1128; 1H NMR
(300 MHz, CDCI3) 5 3.52 (3 H, s), 6.73 (1 H, d, J = 8.5 Hz), 7.08 (1 H, t, 8.1
Hz), 7.24 (1 H, s), 7.31 (1 H, dd, J = 8.5 and 2.1 Hz), 7.51 (1 H, d, J = 7.4
Hz), 7.62 (1 H, brd s), 7.76 (1 H, d, J = 2.1 Hz); 19F NMR (282 MHz, CDCI3)
8 -61.03 (7-CF3), -159.54 (3-F); MS m/e 358 [M-H)-].
Anal. calcd. for C16HioCIF4NO2: C, 53.43; H, 2.80; N, 3.89.
Found: C, 53.09; H, 2.88; N, 3.78.
Example 22
( )T3-(5-Chloro-2-hydroxyphenyl)-1,3-dihydro-2H-benz[e]indol-2-one
mp 158-160 C; 1 H NMR (300 MHz, DMSO-d6) 5 5.17 (1 H, brd s), 6.79
(1 H, brd s), 7.13 (1 H, d, J = 6.5 Hz), 7.21-7.24 (3 H, m), 7.29-7.31 (2 H,
m),
7.83 (3 H, m), 10.63 (1 H, s); MS m/e 308 [M-H)-].
Anal. caicd. for C18H13CIN02=1.25H20: C, 65.07; H, 4.40; N, 4.22.
Found: C, 64.70; H, 4.38; N, 4.08.
Example 23
( )T3-(5-Chloro-2-methoxyphenyl)-1.3-dihydro-3-fluoro-5-methyl-2H-indol-
2-one
mp 193-195 C; IR (KBr, cm-1) 3200, 1732, 1270, 1216; 1H NMR (300 MHz,
CDCI3) 8 2.22 (3 H, s), 3.53 (3 H, s), 6.74 (1 H, d, J= 8.4 Hz), 6.77 (1 H, d,
J
= 7.5 Hz), 6.87 (1 H, s), 7.07 (1 H, d, J = 8.0 Hz), 7.29 (1 H, dd, J = 8.8
and
2.5 Hz), 7.77 (1 H, d, J = 2.5 Hz), 8.16 (1 H, brd s); 19F NMR (282 MHz,
CDCI3) 8-157.38 (3-F); MS m/e 304 [M-H)-].
42 CT-2294A (BMMA-10A)
Anal. calcd. for C16H13CIFN02: C, 62.86; H, 4.29; N, 4.58.
Found: C, 62.67; H, 4.29; N, 4.49.
Example 24
( )T3-(5-Chloro-2-methoxyphenXl)-1.3-dihydro-3-fluoro-4.6-bis-
(trifluoromethyl)-2H-indol-2-one
mp 262-264 C; IR (KBr, cm-1) 3200, 1750, 1316, 1280, 1202, 1140;
1H NMR (300 MHz, CDCI3) 5 3.38 (3 H, s), 6.63 (1 H, dd, J = 8.7 and 1.1
Hz), 7.21 (1 H, dd, J = 8.7 and 2.5 Hz), 7.29 (1 H, s), 7.33 (1 H, s), 7.63 (1
H,
d, J = 2.1 Hz), 10.91 (1 H, brd s); 19F NMR (282 MHz, CDCI3) 6 -60.00
(CF3), -63.40 (CF3), -163.42 (3-F); MS m/e 426 [M-H)-].
Anal. calcd. for C17H9CIF7NO2: C, 47.74; H, 2.12; N, 3.27.
Found: C, 47.58; H, 2.18; N, 3.19.
Example 25
( )-3-(5-Chloro-2-methoxyphenyl)-1.3-dihydro-3.5-difluoro-2H-indol-2-one
mp 205-207 C; IR (KBr, cm-1) 3200, 1732, 1272, 1218, 1144; 1 H NMR
(300 MHz, CDCI3) 5 3.53 (3 H, s), 6.75 (1 H, dd, J = 8.8 and 1.1 Hz), 6.78-
6.84 (2 H, m), 6.95- 7.02 (1 H, m), 7.31 (1 H, dd, J = 8.7 and 2.6 Hz), 7.75
(1 H, d, J = 2.1 Hz), 8.48 (1 H, brd s); 19F NMR (282 MHz, CDCI3) 8 -119.53
(5-F), -158.81 (3-F); MS m/e 308 [M-H)-].
Anal. calcd. for C15H10CIF2NO2: C, 58.17; H, 3.25; N, 4.52.
Found: C, 58.02; H, 3.45; N, 4.41.
2176183
43 CT-2294A (BMMA-10A)
Example 26
( )-5-Bromo-3-(5-chloro-2-methoxyphenyl)-1,3-dihydro-3-fluoro-2H-indol-2-
one
mp 206-208 C; IR (KBr, cm-1) 3200, 1738, 1300, 1262, 1216, 1126, 820;
1 H NMR (300 MHz, CDCI3) S 3.53 (3 H, s), 6.75 (1 H, d, J = 8.2 Hz), 6.78
(1 H, d, J = 7.4 Hz), 7.17 (1 H, s), 7.32 (1 H, dd, J = 8.5 and 2.3 Hz), 7.41
(1 H, d, J = 8.2 Hz), 7.74 (1 H, d, J = 2.4 Hz), 8.37 (1 H, brd s); 19F NMR
(282 MHz, CDCI3) 8 -158.55 (3-F); MS m/e 368 [M-H)-].
Anal. calcd. for C15H10BrCIFNO2: C, 48.61; H, 2.72; N, 3.78.
Found: C, 48.71; H, 2.36; N, 3.58.
Example 27
( )-3-(5-Chloro-2-hydroxyphenyl)-1.3-dihydro-6-[4-(trifluoromethyl)phenkl-
2H-indol-2-one
mp 221-225 C; IR (KBr, cm-1) 3278, 1686, 1326, 1276; 1H NMR (300 MHz,
DMSO-d6) S 4.81 (1 H, s), 6.77 (1 H, d, J = 8.4 Hz), 7.04 (1 H, d, J = 7.6
Hz),
7.10(1 H, d, J = 1.3 Hz), 7.14 (1 H, d, J = 2.6 Hz), 7.18-7.23 (3 H, m), 7.77-
7.84 (3 H, m), 9.83 (1 H, s), 10.64 (1 H, s); MS m/e 402 [M-H)-].
Anal. calcd. for C21 H13CIF3N02=0.25H20: C, 61.78; H, 3.33; N, 3.43.
Found: C, 61.97; H, 3.63; N, 3.62.
Example 28
( )-3-(5-Chloro-2-hydroxyphenyl)-1.3-dihydro-2H-indol-2-one
mp.256-258 C; IR (KBr, cm-1) 3300, 3200, 1680, 820, 750; 1H NMR (300
MHz, DMSO-d6) S 4.75 (1 H, s), 6.78 (1 H, d, J = 8.6 Hz), 6.85 (2 H, t, J =
8.1
Hz), 6.92 (1 H, d, J = 7.2 Hz), 7.08 (1 H, d, J = 2.5 Hz), 7.11-7.17 (2 H, m),
2176 18~
44 CT-2294A (BMMA-10A)
8.78 (1 H, s), 10.46 (1 H, s); 13C NMR (75 MHz, DMSO-d6) S 48.04, 109.09,
116.87, 121.31, 122.20, 123.72, 126.61,127.64, 127.99, 130.01, 130.05,
142.78, 154.40, 177.17; MS m/e 260 (MH+).
Anal. calcd. for C14H10CIN02: C, 64.75; H, 3.88; N, 5.39.
Found: C, 64.63; H, 3.93; N, 5.23.
Example 29
( )-3 -(5-Chloro-2-methoxyphenxl)-1.3-dihydro-3-hydroxk 5-
(trifluoromethyl)-2H-indol-2-one
mp 218-220 C; I R (KBr, cm-1) 3350, 1730, 1325, 1260, 1150, 1120; 1 H
NMR (300 MHz, DMSO-d6) S 3.41 (3 H, s), 6.88 (1 H, s), 6.93 (1 H, d, J 8.8
Hz), 7.01 (1 H, d, J = 8.1 Hz), 7.09 (1 H, d, J = 1.6 Hz), 7.35 (1 H, dd, J
8.7 and 2.7 Hz), 7.56 (1 H, dd, J = 8.1 and 1.1 Hz), 7.80 (1 H, d, J = 2.7
Hz),
10.77 (1 H, s); 13C NMR (75 MHz, DMSO-d6) S 55.97, 74.26, 109.51,
113.61, 120.06, 120.11, 121.8 (m, CF3), 124.50, 126.37, 126.94, 128.72,
131.38, 133.10, 146.70, 154.31, 177.55; MS m/e 358 (MH+).
Anal. calcd. for C16Hl1 CIF3NO3: C, 53.72; H, 3.10; N, 3.92.
Found: C, 53.51; H, 3.00; N, 3.91.
Example 30
( )-5-Bromo-3-(5-chloro-2-methoxyphenyl)-1.3-dihydro-3-hydroxy-2 H-
indol-2-one
mp 245-247 C; IR (KBr, cm-1) 3450-3200, 1712, 1246; 1H NMR (300 MHz,
DMSO-d6) 5 3.42 (3 H, s), 6.79 (2 H, d, J = 8.3 Hz), 6.91 (1 H, d, J = 7.1
Hz),
6.93 (1 H, s), 7.31-7.36 (2 H, m), 7.76 (1 H, d, J = 2.7 Hz), 10.50 (1 H, s);
13C
NMR (75 MHz, DMSO-d6) S 55.95, 74.60, 111.25, 112.82, 113.49, 124.41,
126.25, 126.85, 128.59, 131.58, 131.68, 134.62, 142.34, 154.32, 177.12;
MS m/e 370 (MH+).
2176183
45 CT-2294A (BMMA-10A)
Anal. calcd. for Cl 5H1 1 BrCINO3: C, 48.88; H, 3.01; N, 3.80.
Found: C, 49.52; H, 3.03; N, 3.58.
Example 31
( )-3-(5-Chloro-2-hydroxyphenyl)-4.6-dichloro-1.3-dihydro-2H-indol-2-one
mp 238-240 C; IR (KBr, cm-1) 3400, 1694, 1318; 1H NMR (300 MHz,
DMSO-d6) S 4.83 (1 H, brd s), 6.70 (1 H, brd s), 6.82 (1 H, s), 7.01 (1 H, s),
7.14 (1 H, dd, J = 8.7 and 2.4 Hz), 7.4 (1 H, brd s), 9.70 (1 H, brd s), 10.82
(1
H, s); MS m/e 328 (MH+).
Example 32
( )-3-(5-Chloro-2-methoxyphenyl)-1.3-dihydro-3-hyd roxy-6-iodo-2 H-indol-
2-one
mp 209-211 C; 1H NMR (300 MHz, DMSO-d6) 5 3.42 (3 H, s), 6.62 (1 H, d,
J = 7.7 Hz), 6.73 (1 H, s), 6.91 (1 H, d, J = 8.8 Hz), 7.13 (1 H, d, J = 1.4
Hz),
7.18 (1 H, dd, J = 7.7 and 1.4 Hz), 7.32 (1 H, dd, J =8.8 and 2.7 Hz), 7.75
(1 H, d, J = 2.7 Hz), 10.46 (1 H, s); 13C NMR (75 MHz, DMSO-d6) S 55.99,
74.34, 94.43, 113.48, 117.55, 124.36, 125.60, 126.76, 128.48, 129.99,
131.75, 132.06, 144.60, 154.33, 177.28; MS m/e 416 (MH+).
Anal. calcd. for C15H11CIINO3Ø25CH2CI2: C, 41.93; H, 2.65; N, 3.21.
Found: C, 41.98; H, 2.73; N, 3.19.
Example 33
( )-3-(5-Chlo ro-2-hydroxyphenyl)-1.3-dihyd ro-6-iodo-2H-indol-2-one
mp 199-203 C (dec.); MS m/e 386 (MH+).
Anal. calcd. for C14H9CIINO2.H20: C, 39.04; H, 2.71; N, 3.14.
Found: C, 38.82; H, 2.40; N, 3.04.
2176183
46 CT-2294A (BMMA-10A)
Example 34
( )-3-(5-Chloro-2-methoxyphenyl)-1.3-dihydro-3-hydroxy-2H-benz[f indol-2-
one
mp 305-307 C (dec.); IR (KBr, cm-1) 3356, 1728, 1248; 1H NMR (300 MHz,
DMSO-d6) S 3.32 (3 H, s), 6.79 (1 H, s), 6.89 (1 H, d, J = 8.7 Hz), 7.14 (1 H,
s), 7.24 (1 H, t, J = 7.0 Hz), 7.32 (1 H, d, J = 2.7 Hz), 7.35-7.41 (2 H, m),
7.75
(2 H, t, J = 8.5 Hz), 7.85 (1 H, d, J = 2.7 Hz), 10.71 (1 H, s); MS m/e 357
(M+N H4)+.
Example 35
( )-3-(5-Chloro-2-hydroxyphenyl)-1.3-dihydro-3-hvdroxy-2H-benz~flindol-2-
one
mp 160-165 C (dec.); IR (KBr, cm-1) 3400, 1706; 1H NMR (300 MHz,
CDCI3-CD3OD) S 6.71 (1 H, dd, J = 7.4 and 1.4 Hz), 7.03 (1 H, s), 7.05 (1 H,
d, J = 2.6 Hz), 7.12 (1 H, s), 7.22 (2 H, m), 7.29 (1 H, d, J = 8.1 Hz), 7.38
(2
H, t, J = 8.2 Hz), 7.64-7.69 (3 H, m); MS m/e 324 (M-H)-.
Example 36
( )-3-(5-Ch loro-2-hydroxyphenyl)-1.3-dihydro-2H-benz[fJindol-2-one
mp 254-256 C (dec.); IR (KBr, cm-1) 3300, 1690, 1250, 740; 1 H NMR (300
MHz, DMSO-d6) S 4.88 (1 H, s), 6.75 (1 H, d, J = 8.6 Hz), 7.15 (1 H, s), 7.18
(1 H, d, J = 2.7 Hz), 7.22-7.27 (2 H, m), 7.37 (1 H, t, J = 8.1 Hz), 7.42 (1
H,
s), 7.55 (2 H, t, J = 9.4 Hz), 9.75 (1 H, s), 10.77 (1 H, s); MS m/e 308 (M-H)-
.
~~'~~:-~~'~
47 CT-2294A (BMMA-10A)
Example 37
(3S)-(+)-(5-Chloro-2-methoxyphenyl)-1.3-dihydro-3-fluoro-6-
(trifluoromethyl)-2H-indol-2-one
The racemic compound of Example 14 was separated into its
enantiomers using a Chiracel-OD analytical HPLC column (250 x 4.6 mm)
using 9:1 hexanes/isopropyl alcohol as the eluting solvent at a flow rate of
0.75 mUmin. The detection method employed a HP 1090 UV detector with
diode array at a wavelength of 220 nm. The first enantiomer which eluted
from the column had a retention time of about 8.22 minutes and was
determined to be the (+)-enantiomer of the title compound. On a
preparative scale, up to one gram of the racemate may be resolved with a
single injection on a 5X50 cm Chiracel-OD preparative HPLC column using
9:1 hexanes/isopropyl alcohol at a flow rate of 85 mUmin with baseline
separation. Recrystallization of the enantiomer from methylene chloride/
hexanes provided crystals suitable for single crystal X-ray analysis. Using
the anomalous scattering from the chlorine atom, the absolute configuration
at the asymmetric carbon atom was established as S for the (+)-enantiomer.
The enantiomer was identical to the racemate with respect to NMR, mass
spectra, TLC and I R. The title compound was found to have a mp = 198-
200 C and [a]p 25 + 149.84 (MeOH).
Example 38
(3R)-(-)-(5-Chloro-2-methoxyphenyl)-1.3-dihydro-3-fluoro-6-
(trifluoromethyl)-2H-indol-2-one
The racemic compound of Example 14 was separated into its
enantiomers using a Chiracel-OD analytical HPLC column (250 x 4.6 mm)
using 9:1 hexanes/isopropyl alchohol as the eluting solvent at a flow rate of
0.75 mUmin. The detection method employed a HP 1090 UV detector with
diode array at a wavelength of 220 nm.
2176183
48 CT-2294A (BMMA-10A)
From the experimental process described in Example 37, the second
enantiomer eluted from the same column at a retention time of about 11.58
minutes and was determined to be the (-)-enantiomer of the title compound.
On a preparative scale, up to one gram of the racemate may be resolved
with a single injection on a 5X50 cm Chiracel-OD preparative HPLC column
using 9:1 hexanes/isopropyl alcohol at a flow rate of 85 mUmin with
baseline separation. Recrystallization of the individual enantiomer from
methylene chloride/hexanes provided crystals suitable for single crystal
X-ray analysis. Using the anomalous scattering from the chlorine atom, the
absolute configuration at the asymmetric carbon atom was established as R
for the (-)-enantiomer. The enantiomer was identical to the racemate with
respect to NMR, mass spectra, TLC and IR. The title compound was found
to have a mp = 199-201 C and [a]p 25 - 149.43 (MeOH).