Note: Descriptions are shown in the official language in which they were submitted.
WO 95/14007 PCT/US94/12717
_ , _ 217b551
Phenyl-Alhyl imidazoles as H3-receptor antagonists
FIELD OF THE INVENTION
The present invention relates to phenyl-alkyl-imidazoles having valuable
pharmacological properties, especially CNS activities and activity against
inflammatory disease. Compounds of this invention are antagonists of the H3
receptor.
BACKGROUND OF THE INVENTION
European Patent Application No. 0 420 396 A2 (Smith Kline & French
Laboratories Limited) and Howson et al., Bioorg. & Med Chem. Letters, Vol. 2
No. 1 (1992), pp. 77-78 describe imidazole derivatives having an amidine group
as H3 agonists. Van der Groot et al. (Eur. J. Med Chem. (1992) Vol. 27, pp.
511-517) describe isothiourea analogs of histamine as potent agonists or
antagonists of the histamine H3 receptor, and these isothiourea analogs of
histamine overlap in part with those of the two references cited above.
Clapham
et al. ["Ability of Histamine H3 Receptor Antagonists to improve Cognition and
to
increase Acetylcholine Release in vivo in the Rat", British Assn. for
Psychopharmacology, July 25-28 1993, reported in J. Psychopharmacol. (Abstr.
Book), A17] describe the ability of histamine H3 receptor antagonists to
improve
cognition and to increase release of acetylcholine in vivo in the rat. Clapham
et
al. ["Ability of the selective Histamine H3 Receptor Antagonist Thioperamide
to
improve Short-term Memory and Reversal Learning in the Rat", Brit. J. Pharm.
Suppl., 1993, 110, Abstract 65P] present results showing that thioperamide can
improve short-term memory and reversal learning in the rat and implicate the
involvement of H3 receptors in the modulation of cognitive function. Yokoyama
et al. ["Effect of thioperamide, a histamine Ha receptor antagonist, on
electrically
induced convulsions in mice", Eur. J. Pharmacol., vol. 234 (1993), pp. 129-
133]
report how thioperamide decreased the duration of each phase of convulsion
and raised the electroconvulsive threshold, and go on to suggest that these
and
other findings support the hypothesis that the central histaminergic system is
involved in the inhibition of seizures. International Patent Publication No.
W09301812-A1 (SmithKline Beecham PLC) describes the use of S-[3-(4(5)-
imidazolyl)propyl]isothiourea as a histamine H3 antagonist, especially for
r f I t f f f
1 f
. ~ r 1 ~ f f f
~1~~~~~ . f.. . . f ... fff
.. .
f ff .. .f ..
-2-
treating cognitive disorders, e.g. Alzheimer's disease and age-related memory
impairment. Schlicker et al. ["Novel histamine H3 receptor antagonists:
affinities
in an H3 receptor binding assay and potencies in two functional H3 receptor
models"] describe a number of imidazolylalkyl compounds wherein the
imidazolylalkyl group is bonded to a guanidine group, an ester group or an
amide group (including thioamide and urea), and compare these to
thioperamide. Leurs et al. ["The histamine H3-receptor: A target for
developing
new drugs", Progr. Drug Res. (1992) vol. 39, pp. 127-165] and Lipp et al.
["Pharmacochemistry of H3-receptors" in The Histamine Receptor, eds.:
Schwartz and Haas, Wiley-Liss, New York (1992), pp. 57-72] review a variety of
synthetic H3 receptor antagonists, and Lipp et al. (ibid. ) have defined the
necessary structural requirements for an H3 receptor antagonist. WO 93/14070
discloses imidazole derivatives described as antagonists of the H3 receptors.
and therefore useful as antihistamines.
SUMMARY OF THE INVENTION
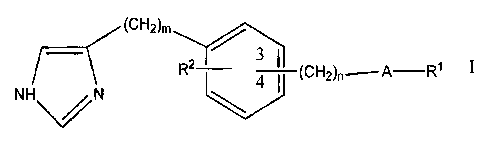
The present invention provides a compound of the formula I or IC
(CH2)m
R2 3~1 (CH2)n A-R' I
HN~N
wherein:
A is selected from -O-CO-NR~-, -O-CO-, -NR~-CO-NR~-, -NR~-CO-,
-CO-NR~-, -CH2-NR1- and -C(:NR1)-NR1-;
the groups R~, which may be the same or different when there are two or three
such groups in the molecule of formula I, are selected from hydrogen, and
lower
alkyl, aryl, cycloalkyl, heterocyclic and heterocyclyl-alkyl groups, and
groups of
the formula -(CH2)y-G, where G is selected from C02R3, COR3, CONR3R4, OR3,
SR3, NR3R4, heteroaryl and phenyl, which phenyl is optionally substituted by
halogen, lower alkoxy or polyhaloloweralkyl, and y is an integer from 1 to 3;
R2 is selected from hydrogen and halogen atoms, and alkyl, alkenyl, alkynyl
and
trifluoromethyl groups, and groups of the formula OR3, SR3 and NR3R4;
ANiElVCED SHEET
IPEAIEP
er r~~'
v c ~~n
v r , v , r
r ", , a r ",
21'~ ~ ~ ~'~ ~ .. ~.r rr ...
-3-
R3 and R4 are independently selected from hydrogen, and lower alkyl and
cycloalkyl groups, or R3 and R4 together with the intervening nitrogen atom
can
form a saturated ring containing 4 to 6 carbon atoms that can be substituted
with
one or two lower alkyl groups;
with the proviso that, when y is 1 and G is OR3, SR3 or NR3R4, then neither R3
nor R4 is hydrogen;
the group -(CH2)~-A-R~ is at the 4-position, and the group RZ is at any free
position;
m is an integer from 1 to 3;
and n is 0 or an integer from 1 to 3;
or a pharmaceutically acceptable acid addition salt thereof;
or a pharmaceutically acceptable salt thereof with a base when G is C02H;
including a tautomeric form thereof;
or a compound of the formula J
(CI-12)m
(CHZ)n NR12
HN~N '~ 4 IC
X
wherein:
X is HZ or NH;
the groups R1, which may be the same or different when there are two or three
such groups in the molecule of formula I, are selected from hydrogen, and
lower
alkyl, aryl, cycloalkyi, and heterocyclic groups, and groups of the formula
-(CH2)y-G, where G is selected from C02R3, COR3, CONR3R4, OR3, SR3,
NR3R4, heteroaryl and phenyl, which phenyl is optionally substituted by
halogen, lower alkoxy or polyhaloloweralkyl, and y is an integer from 1 to 3;
m, n, R3 and R4 are as defined above;
the group -(CH2)n-CX-NR~ R2 is at the 3- or 4-position;
AIIiENDED SHEET'
IPEA/EP
- rr
r r
r r o n r .
rrr v r r rrr
r r
r rr rr rr
217!657
-4 -
or a pharmaceutically acceptable acid addition salt thereof;
or a pharmaceutically acceptable salt thereof with a base when G is C02H;
including a tautomeric form thereof;
wherein the term "lower" as applied to alkyl and alkoxy groups in a compound
of
formula I or IC indicates groups having from 1 to 6 carbon atoms.
The present invention also provides a method for treating inflammation,
allergy, diseases of the GI-tract, cardiovascular disease, or disturbances of
the
central nervous system, which comprises administering to a patient suffering
from such a disease an effective amount of a compound of the formula I or IC
or
salt thereof as defined above; with the proviso that, when the method is for
treating disturbances of the central nervous system, then A in the compound
of.
formula I can also represent -CO-O-.
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS
Compounds of the formula I can exist in tautomeric forms by virtue of the
imidazole ring: the N-hydrogen atom can tautomerize from one nitrogen atom to
the other of that ring. Furthermore, compounds wherein A is a group of the
formula -C(:NH)-NR~-, so that the side chain is -(CH2)n-C(:NH)-NR~2, where
only one group R1 is hydrogen, can exist in tautomeric forms. For example, if
just one group R~ is hydrogen, then one tautomeric form can be represented by
the formula
(CH2)m ~ N R'
~CH2)n IA
N~NH \ NHZ
wherein m, n and R~ are as defined above, except that R~ is not hydrogen. The
interconversion of the tautomers is catalyzed by acids. All such tautomeric
forms
are covered by the invention; in particular, where a compound of formula I is
referred to or a compound is named according to formula I, then all such
tautomeric forms of the compound are covered.
The compounds of the invention are basic and form pharmaceutically
acceptable salts with organic and inorganic acids. Examples of suitable acids
for such salt formation are hydrochloric, sulfuric, phosphoric, acetic,
citric, oxalic,
malonic, salicylic, mafic, fumaric, succinic, ascorbic, malefic,
methanesulfonic
AMENDED SHEET
lrE.~,%~P
r.
. . r
..
.. .
~1'~~557 ,
.. ..
-5-
and other mineral and carboxylic acids well known to those skilled in the art.
The salts are prepared by contacting the free base form with a sufficient
amount
of the desired acid to produce a salt in the conventional manner. The free
base
forms may be regenerated by treating the salt with a suitable dilute aqueous
base solution such as dilute aqueous sodium hydroxide, potassium carbonate,
ammonia and sodium bicarbonate. The free base forms differ from their
corresponding salt forms somewhat in certain physical properties, such as
solubility in polar solvents, but the salts are otherwise equivalent to their
corresponding free base forms for purposes of this invention.
Certain compounds of the invention are zwitterionic in nature, in particular
the compounds that possess a carboxyl group in G. These compounds can form
pharmaceutically acceptable salts with bases also. Examples of such salts are
the sodium, potassium, calcium, aluminum, gold and silver salts, and also
silts.
formed with pharmaceutically acceptable amines such as ammonia,
alkylamines, hydroxyalkylamines, N-methylglucamine and the like.
When used herein, the following terms have the given meanings:
lower alkyl (including the ail~yl portions of lower alkoxy) - represents a
straight or branched, saturated hydrocarbon chain having from 1 to 6 carbon
atoms, preferably from 1 to 4;
lower alkenyl (in R2) - represents a straight or branched aliphatic hydro-
carbon radical having at least one carbon-to-carbon double bond (preferably in
conjugation with the benzene ring that the group RZ substitutes) and having
from
2 to 6 carbon atoms;
lower alkynyl (in R2) - represents a straight or branched aliphatic hydro-
carbon radical having at least one carbon-to-carbon triple bond (preferably in
conjugation with the benzene ring that the group R2 substitutes) and having
from
2 to 6 carbon atoms;
aryl - represents a carbocyclic group having from 6 to 14 carbon atoms and
having at least one benzenoid ring, with all available substitutable aromatic
carbon atoms of the carbocyclic group being intended as possible points of
attachment, said carbocyclic group being optionally substituted with 1 to 3 Y
groups, each independently selected from halo, alkyl, hydroxy, loweralkoxy,
phenoxy, amino, loweralkylamino, diloweralkylamino, and polyhaloloweralkyl.
Preferred aryl groups include 1-naphthyl, 2-naphthyl and indanyl, and
especially
phenyl and substituted phenyl;
cycloalkyl - represents a saturated carbocyclic ring having from 3 to 8 carbon
atoms, preferably 5 or 6;
AMENDED SHEET
1 P EaIE~
r « . r . rrfr
r r s r r r
. . ~ r . , r t r r
. .r~ - r ~ err r r
f ~ t
~~,~~~5,~ r:.. .r rr rr fr fr
-6-
halogen - represents fluorine, chlorine, bromine and iodine;
heterocyclic - represents, in addition to the heteroaryl groups defined
below, saturated and unsaturated cyclic organic groups having at least one O,
S and/or N atom interrupting a carbocyclic ring structure that consists of one
ring or two fused rings, wherein each ring is 5-, 6- or 7-membered and may or
may not have double bonds that lack delocalized pi electrons, which ring
structure has from 2 to 8, preferably from 3 to 6 carbon atoms; e.g., 2- or
3-piperidinyl, 2- or 3-piperazinyl, 2- or 3-morpholinyl, or 2- or 3-
thiomorpholinyl;
heteroaryl - represents a cyclic organic group having at least one O, S
and/or N atom interrupting a carbocyclic ring structure and having a
sufficient
number of delocalized pi electrons to provide aromatic character, with the
aromatic heterocyclic group having from 2 to 14, preferably 4 or 5 carbon
atoms,
e.g., 2-, 3- or 4-pyridyl, 2- or 3-furyl, 2- or 3-thienyl, 2-, 4- or 5-
thiazolyl, 2- or: -
4-imidazolyl, 2-, 4- or 5-pyrimidinyl, 2-pyrazinyl, or 3- or 4-pyridazinyl,
etc.
Preferred heteroaryl groups are 2-, 3- and 4-pyridyl;
heterocyclyl-alkyl - represents a heterocyclic group defined above
substituting an alkyl group; e.g., 2-(3-piperidinyl)-ethyl, (2-piperazinyl)-
methyl,
3-(2-morpholinyl)-propyl, (3-thfomorpholinyl)-methyl, 2-(4-pyridyl)-ethyl,
(3-pyridyl)-methyl, or (2-thienyl)-methyl.
Prefered compounds of formula IC include those wherein m is 1 or 2, and
n is 0, 1 or 2, more especially those of the formula
(Cf"(2)m ~ NR'2
(CH2)n
H N / N ~.. 4 ~ N H
In these compounds, the groups R1 are as defined above, and the side
chain [-(CH2)n-C(=X)-NR~2 or-(CH2)n-C(=NH)-NR~2 ] is preferably at the
4-position.
In formula I, A is preferably -CH2-NR~- or especially -C(:NH)-NR~-.
Another particularly preferred value of A is -O-CO-NR~-.
In compounds of formula I and especially in compounds of formulae IC
and IB, the groups R~, which may be the same or different, are as defined
above, and are preferably selected from hydrogen, and aryl groups, and groups
of the formula -(CH2)y-G, where G is selected from pyridyl and phenyl, which
phenyl is optionally substituted by halogen, lower alkoxy or
polyhaloloweralkyl,
and y is 1 or 2. One group R~ is preferably selected from hydrogen, 2-phenyl-
ethyl, 4-chlorophenylmethyl, 4-methoxyphenylmethyl, 4-trifluoromethylphenyl-
AMENDED SHEET
~ ~H_!E- ~
. '..".
. . .,.
..: . .. '.
~1'~G55'~ .:.. .. .. .. .
-7-
methyl and 4-pyridylmethyl, but is especially 4-chlorophenylmethyl; any other
group R~ that is present is preferably a hydrogen atom or a methyl group.
Preferred compounds of the formula I or IC include those selected from
the following formulae, where the compounds bear the same numbering as in
the Examples (except that the compounds in the Examples are salts, e.g., the
dihydrochlorides):
NH2 NH
HN~N 17 \ / N ,\. NH2
HN,,~N /
NH
HN~N I / NH2 HN ~ ,
_ ~,/ ~ ~ N H 2
21 ~N 16
HN,~N I / NH2 I ~ NH / I
HN,~%N /
NH 6 NH C
HN~N I / NH ~
-CI
NH
HN,~N ~ / NH ~ )
\ 'CF3
N H 6b
HN~N ~ / NH ~ I
~OCH3
NH
/ I
HN~N ~ / NH
I
N H 6d
AMENDED SHEET
r, ~.~,:
~ I I r r f
r r s r r r r s
s r r s rst~
r ~ r r r . . r r r r r r
srr rrr -~ rrr rsrr
~17fi55'~
- 7a -
NH / I
HN~N / NH ' CI
29
NH
HN~N /
N H 6c
CI
H
HN~N ( ~. N
33 ~ CI
O
HN~N I / O
i
CI O 43
HN~N I / O
36 ,~ CI
i
HN~N I / O \ I
O N , 40 p
H N N ( O ''~ (
CI
38 / ~ CI
H
HN~N .~ I N ~ I
and
30 O
The following compounds of this invention are of special interest:
N-[(4-chlorophenyl)methyl]-4-(1b,-imidazol-4-ylmethyl)benzamide;
N-[2-(4-chlorophenyl)ethyl]-4-[2-(1 jj-imidazol-4-yl)ethyl]benzamide;
N-phenylmethyl-4-(1b,-imidazol-4-ylmethyl)benzamide;
N-[(4-chlorophenyl)methyl]-3-(1,~-imidazol-4-ylmethyl)benzamide;
N-[(4-chlorophenyl)methyl]-4-[2-(1 ~-imidazol-4-yl)ethyl]benzamide;
N-[2-(4-chlorophenyl)ethyl]-4-(1 ~-imidazol-4-ylmethyl)benzamide;
(4-chlorophenyl)methyl 4-(1 jj-imidazol-4-ylmethyl)benzoate;
2-(4-chlorophenyl)ethyl4-[2-(1~-imidazol-4-yl)ethyl]benzoate;
AMENDED SHEET
If L~r,_lC
WO 95/14007
PCf/IJS94I12717
-8 -
phenylmethyl 4-(1 H-imidazol-4-ylmethyl)benzoate;
(4-chlorophenyl)methyl 3-( 1~- -imidazol-4-ylmethyl)benzoate;
(4-chlorophenyl)methyl 4-[2-{1 H-imidazol-4-yl)ethylJbenzoate;
2-{4-chlorophenyl)ethyl 4-(1~1-imidazol-4-ylmethyl)benzoate;
4-[[4-[[(4-chlorophenyl)methoxy]methyl]phenyl]methylJ-1 H-imidazole;
4-[2-[4-[2-[(4-chlorophenyl)methoxy]ethyl]phenyl]ethyl]-1~i-imidazole;
4-[[4-[(phenylmethoxy)methyl]phenyl]methyl]-1 ~-im idazole;
4-[[3-[[(4-chlorophenyl)methoxy]methyl]phenylJmethylJ-1~-imidazole;
4-[2-[4-[[(4-chlorophenyl)methoxy]methyl]phenyl]ethyl]-1 H-imidazole;
4-[[4-[2-[(4-chlorophenyl)methoxy]ethyl]phenyl]methyl]-1 H-imidazole;
[4-(1 I-~-imidazol-4-ylmethyl)phenyl]methyl 4-chlorobenzoate;
2-[4-[2-(1 -,~i-imidazol-4-yl)ethyl]phenyl]ethyl 4-chlorobenzoate;
[4-(1 H-imidazol-4-ylmethyl)phenyl]methyl benzoate;
[3-(1~-L-imidazol-4-ylmethyl)phenyl]methyl 4-chlorobenzoate;
2-[4-{1~-I-imidazol-4-ylmethyl)phenyl]ethyl4-chlorobenzoate;
[4-[2-(1~-imidazol-4-yl)ethyl]phenyl)methyl 4-chlorobenzoate;
[4-(1 H-imidazol-4-ylmethyl)phenyl]methyl N-(4-chlorophenyl)carbamate;
2-[4-[2-(1L-II-imidazol-4-yl)ethyl)phenylJethyl N-{4-chlorophenyl)carbamate;
[4-(1 H-imidazol-4-ylmethyl)phenyl]methyl N-phenylcarbamate;
[3-(1 H-imidazol-4-ylmethyl)phenylJmethyl N-(4-chlorophenyl)carbamate;
2-[4-(1L-II-imidazol-4-ylmethyl)phenyl]ethyl N-(4-chlorophenyl)carbamate;
[4-[2-(1H-imidazol-4-yl)ethyl]phenyl]methyl N-(4-chlorophenyl)carbamate;
N-[(4-ch lorophenyl)methyl]-4-( 1 ~-im idazol-4-ylm ethyl)benzene-
carboximidam ide;
N-[(4-chlorophenyl)methyl]-4-[2-(1J~-imidazol-4-yl)ethylJbenzene-
ethanimidamide;
N-phenylmethyl-4-(1 -~I-imidazol-4-ylmethyl)benzenecarboximidamide;
N-[(4-chlorophenyl)methylJ-3-( 1 H_-im idazol-4-ylm ethyl)benzene-
carboximidam ide;
N-[(4-chlorophenyl)methyl]-4-(1,~-I-imidazol-4-ylmethyl)benzeneethanimidamide;
N-[(4-chlorophenyl)methyl]-4-[2-(1,~1-imidazol-4-yl)ethyl]benzene-
carboximidamide;
4-chloro-N-[[4-(1 H-imidazol-4-ylmethyl)phenyl]methylJbenzamide;
4-chloro-N-[2-[4-[2-{1 f~- -imidazol-4-yl)ethylJphenyl]ethyl]benzamide;
N-[[4-(1~i-imidazol-4-ylmethyl)phenylJmethyl]benzamide;
4-chloro-N-[[3-(1 H-imidazol-4-ylmethyl)phenyl]methylJbenzamide;
4-chloro-N-[2-[4-(1 H-imidazol-4-ylmethyl)phenyl]ethylJbenzamide;
4-chloro-N-[[4-[2-(1 H-imidazol-4-yl)ethyl]phenyl]methylJbenzamide;
' WO 95/14007 PGT/US94I12717
-9 -
4-(1,~,-imidazol-4-ylmethyl)-N-[(4-methoxyphenyl)methyl]benzene-
carboximidamide;
4-(1~-imidazol-4-ylmethyl)-N-[[(4-(trifluoromethyl)phenyljmethylJbenzene-
carboximidamide;
4-(1b,-imidazol-4-ylmethyl)-N-(4-pyridinylmethyl)benzenecarboximidamide;
4-(1b,-imidazol-4-ylmethyl)-N-(2-phenylethyl)benzenecarboximidamide;
2-[4-(1~-imidazol-4-yl)ethyl]-N-(2-phenylethyl)benzeneethanimidamide;
3-(1,~-imidazol-4-ylmethyl)-N-(2-phenylethyl)benzenecarboximidamide;
4-(1~[-imidazol-4-ylmethyl)-N-(2-phenylethyl)benzeneethanimidamide;
2-[4-{1~-imidazol-4-yl)ethyl]-N-(2-phenylethyl)benzenecarboximidamide;
4-( 1 ~[-im idazol-4-ylmethyl)benzenecarboxim idamide;
3-(1b,-imidazol-4-ylmethyl)benzenecarboximidamide;
3-[2-(1~-imidazol-4-yl)ethylJbenzenecarboximidamide;
4-[2-(1~-imidazol-4-yl)ethyl]benzenecarboximidamide;
4-(1rj-imidazol-4-ylmethyl)benzenemethanamine;
2-[4-(1~-imidazol-4-ylmethyi)phenyljethyl N-[(4-chlorophenyl)methylj-N-
methylcarbamate;
2-[4-[2-(1~-imidazol-4-yl)]ethyl]phenyl]ethyl N-[(4-chlorophenyl)methyl]-N-
methylcarbamate;
2-[4-{1Jj-imidazol-4-ylmethyl)phenyl]ethyl N-(phenylmethyl)-N-
methylcarbamate;
2-[3-(1,~-I-imidazol-4-ylmethyl)phenyl]ethyl N-[(4-chlorophenyl)methyl]-N-
methylcarbamate;
2-[4-[2-(1~-imidazol-4-yl)Jethyl]phenyljethyl N-[(4-chlorophenyl)methyl]-
carbamate;
2-[4-{1 -~i-imidazol-4-ylmethyl)phenyljethyl N-[(4-chlorophenyl)methylj-
carbamate;
2-[4-(1~-imidazol-4-ylmethyl)phenyl)ethyl 4-chlorobenzeneacetate;
2-[4-[2-(1,~-imidazol-4-yl)ethyl]phenyl]ethyl 4-chlorobenzeneacetate;
2-[4-(1~1-imidazol-4-ylmethyl)phenyl]ethyl benzeneacetate;
2-[3-{1 I~-,-imidazol-4-ylmethyl)phenyl]ethyl 4-chlorobenzeneacetate;
N'-[(4-chlorophenyl)methyl]-N-[[4-(1b,-imidazol-4-ylmethyl)phenyl]methyl]-N,N'-
dimethylurea;
N'-[(4-chlorophenyl)methyl]-N-[2-[4-[2-(1~-I-imidazol-4-yl)ethyl]phenyljethyl]-
N,N'-
dimethylurea;
N'-(phenylmethyl)-N-[[4-(1~i-imidazol-4-ylmethyl)phenyl]methyl]-N,N'-
dimethylurea;
WO 95114007 PCTIUS94112717
- 10 -
N'-[(4-chlorophenyl)methyl]-N-[[3-(1 H-imidazol-4-ylmethyl)phenyl]methyl]-N,N'-
dimethylurea;
N'-[(4-chlorophenyl)methyl]-N-[2-[4-(1~-I-imidazol-4-ylmethyl)phenyl]ethyl]-
N,N'-
dimethylurea;
N'-[(4-chlorophenyl)methyl]-N-[[4-[2-(1L-II-imidazol-4-yl)ethyl]phenyl]methyl]-
N,N'-
dimethylurea;
4-chloro-N-[[4-(1 H-imidazol-4-ylmethyl)phenyl]methyl]-N-methylbenzene-
acetamide;
4-chloro-N-[2-[4-[2-(1 L-II-imidazol-4-yl)ethyl]phenyl]ethyl]-N-methylbenzene-
acetamide;
N-[[4-(1~1-imidazol-4-ylmethyl)phenyl]methyl]-N-methylbenzeneacetamide;
4-chloro-N-[[3-(1~-imidazol-4-ylmethyl)phenyl]methyl]-N-methylbenzene-
acetamide;
4-chloro-N-[[4-(1 ~-imidazol-4-ylmethyl)phenyl]ethyl]-N-methylbenzene-
acetamide;
4-chloro-N-[[4-[2-(1 I!- -imidazol-4-yl)ethyl]phenyl]methyl]-N-methylbenzene-
acetamide;
(4-chlorophenyl)methyl 4-(1~-imidazol-4-ylmethyl)benzeneethanoate;
(4-chlorophenyl)methyl 4-[2-(1 ~-imidazol-4-yl)ethyl]benzenepropanoate;
phenylmethyl4-(11~--imidazol-4-ylmethyl)benzeneethanoate;
(4-chlorophenyl)methyl 3-(1L-II-imidazol-4-ylmethyl)benzeneethanoate;
(4-chlorophenyl)methyl 4-(1 ~-imidazol-4-ylmethyl)benzenepropanoate;
(4-chlorophenyl)methyl 4-[2-(1~i-imidazol-4-yl)ethyl]benzeneethanoate;
4-[[4-[[(3-chlorophenyl)methoxy]methyl]phenyl]methyl]-1 ~-imidazole;
N-[(4-chlorophenyl)methyl]-[4-(11~-imidazol-4-ylmethyl)-N-methyl-benzene-
ethanimidamide;
N-[(4-chlorophenyl)methyl]-4-[2-( 1 ~-I-im idazol-4-yl)ethyl]-N-m ethyl-
benzene-
propanimidamide;
N-(phenylmethyl)-[4-(1 I~-imidazol-4-ylmethyl)-N-methyl-benzene-
ethanimidamide;
N-[(4-chlorophenyl)methyl]-[3-( 1 ~.-im idazol-4-ylmethyl)-N-methyl-benzene-
ethanimidamide;
N-[(4-chlorophenyl)methyl]-[4-(1~-imidazol-4-ylmethyl)-N-methyl-benzene-
propanimidamide;
N-[(4-chlorophenyl)methyl]-4-[2-(1~-I-imidazol-4-yl)ethyl]-N-methyl-benzene-
ethanim idam ide;
N-[(4-chlorophenyl)methyl]-4-[2-(1 H-imidazol-4-yl)ethyl]benzene-
propanimidamide;
2176557
WO 95/14007 PCT/US94/12717
_11
N-(phenylmethyl)-4-(1~,-imidazol-4-ylmethyl)benzeneethanimidamide;
N-[(4-chlorophenyl)methyl]-3-(1~-imidazol-4-ylmethyl)benzeneethanimidamide;
and
N-[(4-chlorophenyl)methyl]-4-(l,d,-imidazol-4-ylmethyl)benzene-
propanimidamide.
Compounds of this invention are antagonists.of the H3 receptor. As such,
they may be useful for the treatment of various allergic, inflammatory, GI-
tract, or
cardiovascular diseases. In addition, they possess CNS activity; they may be
useful as sleep regulators, -anticonvulsants, cognition enhancers, anti-
depressants, regulators of hypothalamo-hypophyseal secretions, and the like.
A further feature of the invention therefore is pharmaceutical compositions
containing as active ingredient a compound of the formula I defined above (or
salt or tautomer), especially a compound of the formula IC, together with a
pharmaceutical carrier or excipient.
Further features of the invention are methods for treating inflammation,
allergy, diseases of the GI-tract, cardiovascular disease, or disturbances of
the
central nervous system, which comprise administering to a patient suffering
from
the corresponding disease an effective amount of a compound of the formula I
defined above (or salt or tautomer).
Preparation of Final Products
Compounds of the formula I can be prepared by standard methods.
Typical methods appropriate for the preparation of the compounds of the
formula
I are illustrated below, wherein the radicals, m and n are as defined above
(unless otherwise stated) and ~1 represents a phenyl group; for convenience,
the group R2 has been omitted from the reaction schemes, but its absence does
not affect the operability of the chemical reactions. The particular process
chosen should not cause significant decomposition elsewhere in the molecule;
for example, removal of a protecting group by hydrogenolysis should not cause
the loss of an essential phenylmethyl group. The first processes A through E
illustrate methods for the preparation of compounds of the formula IC.
A. For the preparation of a compound of formula IC wherein X is NH,
reduction of an amidoxime of the formula:
WO 95/14007 PCTIUS94/12717
-12-
Z'
2 NR~z
I
HN / N \\
NOH
(or an acid addition salt thereof) wherein Z~ is a group (CH2)m, or a dehydro
derivative thereof when m is 2 or 3, and Z2 is a group (CH2)n, or a dehydro
derivative thereof when n is 2 or 3. The reduction can be effected for example
by means of catalytic hydrogenation in an inert solvent, e.g., over Rh/AI203
or
Pd/C, but especially over Raney nickel; the solvent is preferably a lower
alkanol.
(A dehydro derivative of a compound of formula II wherein m and/or n is 2 or 3
will have a double bond in the carbon chain between the imidazole ring and the
phenyl ring or between the phenyl ring and the group C(:NOH)NR~2; this
double bond will be reduced in the same step as the reduction of the group
C(:NOH)NR~2.)
B. Removal of a protecting group from a compound of the formula:
(CH2)m
(CE"'~~n NR~2
PgN~N
X
wherein Pg is a protecting group. The protecting group is preferably one that
can be removed by hydrolysis or hydrogenolysis; it can for example be a trityl
group (C6H5)3C-, which is preferably removed by hydrolysis in an aqueous
organic solvent. The hydrolysis can for example be effected by means of
mineral acid in an aqueous water-miscible organic solvent such as a lower
alkanol, especially methanol or ethanol. Other protecting groups that can be
used (and their method of removal) include t-Bu-O.CO- [often abbreviated to
t-BOC] (which can be removed with acid, or with hydrazine, ammonia and a
lower alkanol, e.g., methanol or ethanol), (2-triloweralkylsilyl)ethoxymethyl
groups, especially Me3Si.(CH2)2.O.CH2- [often abbreviated to SEM] (which can
be removed with acid or fluoride ion), and disubstituted aminosulfonyl,
especially Me2N.S02 (which can be removed with acid or base).
C. For the preparation of a compound of formula IC wherein X is NH,
reaction of an imidate salt containing a cation of the formula
2176557
WO 95/14007 PGTlUS94/12717
-13-
N H2 2+
~CH2)m ,/r
l (CHI aIA
n
Pg"N~,,;_., ~NH ~ OR
wherein m and n are as defined above, Pg' is a hydrogen atom or preferably a
protecting group (such as Me2N.S0~), and R is a lower alkyl group, especially
a
methyl or ethyl group, with an amin~ of the formula NHR~2, wherein the groups
R~, which may be the same or different, are as defined above, according to the
general method disclosed by Pinner in "Die Imidp~ther and ihre Derivate", R.
Oppenheim, Berlin, 1892 (when Pg' is preferably a hydrogen atom), or the
adaptation disclosed by Dox, Org. Synth., Coll. Vol. 1, 5 (1941). The anion
associated with the cation of the formula IIIA may for example be methosulfate
or fluoroborate {as disclosed by Weintraub et al., J. Org. Chem., Vol. 33 no.
4
(April 1968), pp. 1679-1681), but is most preferably a halide (e.g.,
chloride).
D. Reaction of a nitrite of the formula IV:
Z' ~
~ II
ZZ-CN
HN~N
with an ammonium salt, to yield a compound of the formula I wherein X is NH,
or
reduction of the nitrite of the formula IV to yield a compound of the formula
IC
wherein X is H2. These processes provide a compound of the formula IC
wherein both groups R~ are hydrogen. Preferred reducing agents include
lithium aluminum hydride {when Z~ and Z2 do not contain double bonds), or
catalytic hydrogenation, e.g., with Raney nickel and chloroplatinic acid
{especially when Z~ and Z2 do contain double bonds).
E. For the preparation of a compound of formula IC wherein X is H2,
reaction of an aldehyde of the formula
(CH?~m
(CH~n-CHO
~g"N~ N .\ IVA
wherein Pg' is a hydrogen atom or a protecting group, with an amine of the
formula NHR~2, in the presence of a reducing agent and of an inert organic
WO 95/14007 PCTlU594/12717
~~~s~~~
- 14 -
solvent. The reducing agent may for example be Raney nickel or sodium
cyanoborohydride. Any protecting group Pg" that is present after the reduction
can be removed, for example as described above under Process B.
In all of these processes, reactive or functional groups that might become
modified during the process (or might even cause large-scale decomposition of
the compounds) should be protected with protecting groups that can be readily
removed when the process has been carried out. (Such groups may include,
but are not restricted to, for example, a hydroxy group OH in G.) Details of
such
groups and of their removal are well known in the art and are given in
standard
textbooks, for example "Protective Groups in Organic Synthesis", by Greene and
Wuts (2nd Edition, John Wiley & Sons, Inc., 1991 ).
Preparation of Starting Materials and Intermediates
Starting materials for processes A, B, C, D and E can be prepared by the
methods discussed below, wherein: Y represents a group convertible into
-(CH2)".CX.NR~, e.g., CN or a group convertible into Z2-CN; M and Yi are as
defined in Table 1 below; and Z~, Z2, Pg and Pg" are as hereinabove defined;
except that, when Y~ represents a divalent group, then Z~, to compensate,
lacks
a hydrogen atom from the carbon atom bonded to Y~; and, when Y~ contains a
carbon atom, then Z~ contains one carbon atom fewer.
Main preliminary reaction stage:
M ~ Z~ Z~
Y ~ ~ / ~ Y
rgN~%N ~ rgN~%N
V VI VII
Reaction conditions (for both steps, where there is more than one) are
given in the following Table, wherein OTs stands for toluene-4-sulfonyloxy,
TCDI
stands for thiocarbonyldiimidazole, and AIBN stands for azoisobutylniorile.
,.~ WO 95/14007 ~ ~ PGTIUS94/12717
-15-
TABLE 1
M Y~ Solvent Temper-Comments
ature
MgBr, I, Br, CH2C12 0C to
OTs
CuI or THF reflux
MgBr =O CH2C12 0C to Reduction of hydroxy group in
second
or THF reflux step can be effected with TCDI,
Bu3SnH and AIBN in benzene/toluene
from 0 to reflux
MgBr, COCI, CH2C12 0C to Reduction of carbonyl group in
second
CuI C02alkylor THF reflux step can b~ effected under Wolff-
Kishner conditions, or with an
alkane-
dithiol (to form a thioketal)
and Raney
nickel under reflux
Values of Y (and Z2) in Intermediates
The group Y representing CN or a group convertible into Z2--CN can also
be represented by ZZY2 wherein Z2 is as hereinabove defined and Y2 is a group
convertible into CN; except that, when Y2 represents a divalent group, then
22,
to compensate, lacks a hydrogen atom from the carbon atom bonded to Y2.
When n in the compound of formula IC is to be 0 (i.e., when Z2 is simply a
bond),
Y is preferably CN. When n is to be 1, 2 or 3, the group Y can be provided by
the
following reaction schemes, wherein the radicals are as defined above, and f~
represents a phenyl group:
I) For n = 1, Y is preferably -CH(OR)2, wherein R is lower alkyl,
preferably Me or Et, or the two groups R together form an ethylene or a
trimethylene group:
Zi Acid, Z~
Acetone/water /='~ ~ 1 CHO
PgN i N .,~~ -". PgN N
or THF/water
VIIA
This yields an aldehyde of the formula VIIB having Y = CHO; it can be
converted into a compound of the formula VII wherein Y = CN by reaction with
tosylmethylisocyanide and t-BuOK and then methanol, or with KCN and
~I'~6557
WO 95/14007 PCTlUS94/12717
-~s-
i-Pr
i-Pr ~_~ S02NHNH2
i-Pr (2,4,6-triisopropylbenzenesulfonylhydrazide;
II) For n = 2, Y again is preferably -CH(OR)2, which can be converted
as above and then through -CH=CH-CN into -CH2CH2-CN:
Z'
(Et0)2PO.CH2CN,
CHO Z' CN
PgN~N ~ t-BuOK, THF ~ /
VIIB pgN~N
Z' CN ~~H / ridine
/ 4 PY
PgN~N ~ ' 0°-reflux
Vlm
III) For n = 3, Y is preferably -CH(OR)2, which can be converted
through -CHO, -CH2-CHO and -CH2-CH=CH-CN into -CH2CH2CH2-CN,
analogously to methods given above:
Z' / ~3P=CHOR and THF, Z~ CHO
;t CHO then acid
pgN~N ~ pgN~N
Q
VIIB
(Et0)2PO.CH2CN, t-BuOK, THF
Z' / ' NaBH4/pyridine, ~ Z' /
PgN~N ~ ~ 0°-reflux PgN~N
VIIS N C VIIR ~ N
Compounds of the formula V can be prepared from the corresponding
iodide by the methods of the references given in J. Org. Chem. 1991, 56,
5739-5740.
Aldehydes for use in process E above are for example compounds of the
formulae VII and VIIQ in paragraph III) above wherein Z~ is a group (CH2)m, or
can be prepared therefrom by lengthening of the aldehyde side-chain.
WO 95/14007 PCT/US94/12717
_17_
Values of Z~ in Intermediates
Compounds of the formula VII can be prepared by the following schemes,
wherein the radicals are as defined above, ~ represents a phenyl group, and Q
represents a hydrocarbyl group, preferably a lower alkyl group, especially
methyl or ethyl:
i) For m = 1, a metal derivative of an N-protected imidazole of the
formula V (wherein M is e.g. MgBr) can be reacted with a Y-substituted-benz-
aldehyde of the formula VIA, and the resulting substituted benzyl alcohol of
the
formula VIIE can be reduced, for example as indicated in the following scheme:
THF Or CH2C12, O H
M O H C / 0° to ambient
n Y - ,l
PgN~N + .,," ' temperature PgN~N
V ' VIA VILE
r i1 Y TCDI, inert organic solvent, 0° to reflux;
PgN,,~N \ ' then Bu3SnH and AIBN in an inert
organic solvent
ii) For m = 2, one of the following schemes can be used:
a)
~CHO BI / Base, e.g., t-BuOK or ~G
+ + ;~ Y
P9N~N ~aP ~"~ NaH, in THF or ether ~ ~ Y
\
PgN~N
Y
PgN~N \ ' Reduce; e.g., Pd/C, alcohol,
0° to ambient temperature
WO 95/14007 PGT/US94/12717
~1?65~'~
b)
M UH
V /
PgN~N + ~ Y VnJ
\ /~
Y
PgN~ N \
'r Reduce, e. ., with TCDI, and
_ /~ g
P N N \ ' Y then with Bu3SnH and AIBN
g ~ VIgi
c)
COCI q3Sn Pd°,THF, dioxane;
/ VIIK
Y 0° to reflux
PgN~N \
/~
~ Y
PgN~N \
/ 1 Y Wolff-Kishner, etc.
PgN~N \ ~ (as in Table 1)
VBH
iii) For m = 3, one of the following schemes can be used:
a)
Organic solvent, e.g.
M OHC / ~,
/ 1 Y ether, dioxane, OH
PgN~N \ ~ THF, CH2C12
1
i Y
PgNVN \
_ a /~ Reduce; e.g.
PgN ~ N \ ~ Y (i) by catalytic hydrogenation;
vmvt
(ii) with TCDI and then with
Bu3SnH and AIBN.
~~..
WO 95/14007 PCT/US94/12717
_19_
b)
~~COCI Q3Sn , Pd°, THF, dio~ane; VuT
+ '~ Y 0° to reflex O
PgN,~ N
v /
P9N~N 'w. ~ Y
~olff Kishner, etc.
y (as in Table 1 )
pgN~N
c)
OH Mild ~'CHO
PgN~N oxidation pgN N Br / ,
y, base,
TIC
Reduce, e.g. by /
n y .",~. __ - - o y
Pg N~ N \ ~ catalytic Pg N ~ N ~,, '
\/
hydrogenation
d)
OH Br
- 1 ) CBr4/~ P+~3
pgN~N 2) ~3P pgN~N t-BuOH, THF,
OHC
/ 1
Y
Reduce, e.g. by
1
- I y .,~.-__- - Y
PgN~N '~,, catalytic p9N~N
hydrogenation
Conversion of Nitrite
The compound of the formula VII (designated VIIA through VIIS above,
but VII' or VII" in the following schemes) wherein Y represents CN or a group
WO 95/14007 PCTIUS94I12717
~~~s5~~r
- 20 -
convertible into Z2-CN can then be converted into the starting material for
Process A, B, C or D by the following processes I, II, or III, respectively:
I. ~ Conversion of a compound of the formula VB' into an amidoxime of
the formula IIA:
/~CZ1 ~ ; Z2-CN H2NOH, etc. ~Z~ ~ ~ 2 NHOH
PgN ~ N w ----~ ~ l Z ~ N H
VIr Pg NON ~ IIA
This reaction can be carried out with a hydroxylamine salt, e.g., the
hydrochloride, in the presence of a base such as potassium hydroxide in an
inert organic solvent such as ethanol at elevated temperature, e.g., under
reflux.
The group Pg' represents a protecting group (if that group is stable to the
reaction conditions) or a hydrogen atom (if the protecting group is displaced
under the reaction conditions, as for example a trityl group is).
II. Conversion of a compound of the formula VII' into an amidine of
the formula IIIB:
Z2-CN R12N-A1R2 ~ Z' / ~ 2 NR' 2
PgN~N w
PgNvN w ' ~NH
VII 1118
This reaction can be carried out in the presence of an inert organic
solvent, preferably an aromatic hydrocarbon such a benzene or toluene, at
elevated temperature, e.g., 50° to reflux, and under an inert
atmosphere. The
dialkylaluminoamine of the formula R~2N-AIR can be generated by reaction of a
trialkylaluminum R3AI with an amine R~2NH (or its hydrochloride) in the
solvent
in which the main reaction will be carried out, under an inert atmosphere.
III. Conversion of a compound of the formula VII" into an imidate salt
containing a cation of the formula IIIA:
~ {CH~m
;' (CH~~-CN
PgNvN
VIr' pier method
NH2 2+
{CH2~m
II (CH~n
I
Pg~~N,,,,__.; NH OR
WO 95/14007 ~ PGT/US94/11717
- 2y -
This reaction is carried out by passing a stream of dry HCI through a
solution of the nitrite in a lower alkanol, preferably ethanol or methanol.
For process A, C, D or E, any remaining protecting group (designated Pg,
Pg' Or Pg' ) can be removed, for example as set out under Process B above.
PREPARATION OF FURTHER COMPOUNDS
Compounds of the formula I wherein A is selected from -O-CO-NR~-,
-O-CO-, -NR~-CO-NR~-, -NR~-CO-, -O-, -CO-NR~-, and -CO-O-, can be
prepared by a process in which the left-hand part of the molecule represented
by
(CH~m
(CH ~n
rgN,~N
is coupled to a compound providing the remainder of the molecule, including
the
group R~. Specific examples of processes for the preparation of such
compounds follow:
1. For the preparation of a compound of the formula I wherein A is
-O-CO-NR~-, reaction of a hydroxy compound with an isocyanate:
(CHI"., / ~ CH _ R1NC0,
PgN~N ~,, '' ( ~n ~H Solvent
(CH ~,r, / ~
PgN~N .,,1 y (Ch'~~n~0-CO-NHR
The resulting compound can then if desired be reacted with an alkylating
agent to introduce another group R~ on to the nitrogen atom in the side chain.
The hydroxy compound can be prepared by reaction of a cyanide of the
formula VII' with an alkanol and an alkoxide (e.g., sodium or potassium
methoxide or ethoxide) and then with the alkanol and an acid to form an ester,
which can then be reduced with a hydride reducing agent such as DiBALH and
lithium aluminum hydride. However, it should be noted that the number of
carbon atoms in the group Z2 in the compound of the formula VII' will increase
by 1 in this process.
WO 95/14007 PCT/US94I12717
21'7 6 5 5'~
- 22 -
2. For the preparation of a compound of the formula I wherein A is
-NR1-CO-NR~-, reaction of a amino compound with an isocyanate:
(CH2)m / CH -NHR R1NC0,
PgN ~ N ~ 'I ( ~n 1 Solvent
(CH 2) m
(CHI"-NR~-CO-NHR~
rgN~N ~
The resulting compound can then if desired be reacted with an alkylating
agent to introduce another group R~ on to the N-(H) atom in the side chain.
Any
resulting mixture of products can then be resolved by standard methods such as
chromatography.
The amino compound can be prepared for example by reduction of a
cyanide of the formula VII' with a hydride reducing agent such as DiBALH or
lithium aluminum hydride, or by catalytic hydrogenation with e.g. Raney nickel
or
palladium on carbon.
3. For the preparation of a compound of the formula I wherein A is
-0-CO- or -NR~-CO-, reaction of a hydroxy or amino compound (as given in
process 1 or 2 above) with a reactive derivative of an acid, especially the
acid
chloride:
(CH~rt, / I CH _ H R1COC1,
PgN ~ N ~ ' ( ~~ ~ Solvent
(CHI / CH
PgN ~ N ~ ~~ ( 2.)n-O-CO-R ~
or
(CH~~., / i CH -NH R1COC1,
i
PgN ~ N ~ '' ( ~~ 2 Solvent
(CH2)m
(CH~~-NH-CO-R~
~gN~N
4. For the preparation of a compound of the formula I wherein A is
-O-, reaction of a hydroxy compound with a halide (according to the Williamson
2176557
WO 95114007 PGT/US94112~17
- 23 -
synthesis of ethers):
(CHI / ~ CH ~;~Hal, NaH
PgN~N ~ y ( 2~n-OH
Solvent
~ (CHI
P N N .. ~ (CH~n O
\%
5. For the preparation of a compound of the formula I wherein A is
-CO-NR~-, reaction of an ester with an dialkylaluminoamine, preferably one of
the formula Me2AINR~2:
(CH ~
PgN ~ N .~ ~l (CH2~n-OCO.R
a v
Me 2A1NR12 ~ (CH ~ m ,
PgN N i (CH~n-CO-NR 2
6. For the preparation of a compound of the formula I wherein A is
-CO-O-, reaction of a reactive derivative of an acid, preferably a chloride,
with
an alcohol R~ OH in the presence of a base, preferably an organic base such as
a tertiary amine (e.g., pyridine or triethylamine):
(CH ~
PgN / N ~ .~- (CH~Jn-(: O.OCI
R10H, base ~ (CH ~ m
PgN~N ~~ (CH~n-CO.OR
The acid chloride can be prepared by reaction of the corresponding acid
with SOC12, and this acid can be prepared by hydrolysis of a cyanide of the
formula VII' with an alkanol and an alkoxide (e.g., sodium or potassium
methoxide or ethoxide).
All these reactions can be carried out by methods that are well known
and/or disclosed in the literature. Further details are given in the Examples.
EXAMPLES
The following Examples illustrate but do not in any way limit the present
invention:
WO 95/14007 PCT/US94/12717
~,1'~ 6 5 5'~
- 24 -
Example 1 ~ N-[(4-Chloro~vl)methyll-4-[(1 H-imidazol-4-yrl)methyl]benzene
methanimidamide (as di ,ydrochloride)
Fart A.
I OH
1. EtMgBr, CH2C12
TrN ~N
NC~-CHO TrN~N I ~ CN
2 3
A solution of ethyl magnesium bromide in ether (8.4 mL of a 3 M solution)
was added to a solution of iodide 1 (synthesized according to the references
given in J. Organic Chem. 1991, 56, 5739-5740; 10 g, 22.9 mmol) in CH2C12
(90 mL) at room temperature under a nitrogen atmosphere. The reaction was
stirred at room temperature for 30 minutes and a solution of aldehyde 2 (3.0
g,
22.9 mmol) in CH2C12 (15 mL) was then added. The reaction was stirred
overnight (about 18 hours) and then quenched by the addition of half-saturated
NH4C1 (100 mL). The organic layer was separated and the aqueous layer was
extracted with additional CH2C12 (100 mL). The combined organic layers were
dried (MgS04), filtered, and concentrated to give a white solid (11 g), which
was
triturated with CH2C12 (100 mL) to yield 5.5 g of the desired material.
Repetition
of this procedure gave an additional 1.55 g of product. The filtrate was
concentrated and the residue subjected to column chromatography (85:15
ether:acetone) which yielded a further 1.77 g of the desired material. Total
yield
8.82 g (87%).
Part B.
OH
I. TCDI> THF
~ TrNVN I ~ CN
TrNvN / CN 2. Bu3SnH, AIBN
3
A suspension of 3 (6.5 g, 14.7 mmol) and thiocarbonyldiimidazole (TCDI,
3.94 g, 22.1 mmol) was heated to reflux in THF (150 mL) under a nitrogen
atmosphere. After 2 hours, additional thiocarbonyldiimidazole (1 g) was added
and the reaction stirred overnight at room temperature. The reaction mixture
was concentrated and the dark residue was dissolved in CH2C12 and washed
with half-saturated NH4C1. The organic layer was separated, and the aqueous
layer was extracted with additional CH2CI2 (2x 75 mL). The combined organic
layers were washed successively with water and brine, and dried with MgS04.
m,~,., w0 95/14007 PCT/US94/12717
- 25 -
Filtration and concentration gave a dark solid that was purified on a flash
column
(70:30 hexane:ethyl acetate) to give the thioimidazolide (7 g, 86%). A
solution of
the thioimidazolide (10 g, 18.15 mmol) and AIBN (azoisobutylnitrile) (0.45 g)
in
dry toluene (200 mL) was slowly added to a solution of n-Bu3SnH (11.1 g, 38.1
mmol) in refluxing dry toluene (200 mL) over about 2 hours under a nitrogen
atmosphere. Additional AIBN (0.2 g) and n-Bu3SnH (3 g) were added, and the
reaction was stirred overnight at reflux. It was then cooled to room
temperature
and washed with 0.1 N HCI, saturated aqueous sodium bicarbonate and water,
and dried (MgS04). Concentration gave a crude solid which was purified on a
flash column (85:15 hexane:isopropanol) to give 6.5 g (85%) of 4 as a white
solid.
NHAIMe2
CI I .,~ H , I CI
TrN vN ~/ .C N TrN,~N ~ N
NH
A solution of 4-chlorobenzyl amine (0.16 g, 1.1 mmol) in toluene (3 mL)
was added dropwise to a solution of Me3Al (0.55 mL of a 2M solution in
toluene,
1.1 mmol) in toluene (5 mL) at room temperature and under a nitrogen
atmosphere. After 45 minutes at room temperature, 4 (0.213 g, 0.5 mmol) in
toluene (3 mL) was added and the reaction was heated to 80°C overnight.
An
additional two equivalents of the aluminum reagent (from Me3Al and 4-chloro-
benzyl amine) was added and the reaction stirred for 2 hours at 100°C.
The
reaction was then cooled to room temperature and quenched by the addition of
saturated aqueous sodium sulfate. When gas evolution ceased, solid sodium
sulfate was added. The mixture was filtered, concentrated, and purified on a
flash column (100 g Si02, 92:8 CH2C12:MeOH/NH3). A white solid was obtained
(230 mg, 81%).
CI
_ ~. H ~ CI w H i
TrN ~ N ~ i N ~. ~ 1N HCI, EtOH HN~N ~ i N
v x 60° C
5 N H 6 N H 2HC1
1 N HCI (20 mL) was added to a solution of 5 (0.51 g, 0.9 mmol) in
ethanol (25 mL) and the reaction heated to 60°C for 2 hours. After
cooling to
room temperature, water (50 mL) was added and the solid that precipitated was
WO 95/14007 PCT/US94112717
- 26 -
removed by filtration. The aqueous layer was washed with ether and
concentrated in vacuo. A white glassy solid, 6, was obtained (0.24 g, 67%).
Example 2
In a similar manner to that of Example 1, the following compounds were
obtained:
OMe
_ H
3
HN ~N I / N \ I ~ H / CF
NH 2HC1 HN ~N I / N \ I
6 b N H 2HC1
\ H / 'N
HN ~N I / N ~ I ~ ~ H
NH HN ,N ( / N /
3HCl v N H \
6 d 2HC1
6a: N-[(4-Methoxyphenyl)methyl]-4-[(1 H-imidazol-4-yl)methyl]benzene
methanimidamide (as dihydrochloride);
6b: N-[(4-trifluoromethylphenyl)methyl]-4-[(1 H-imidazol-4-yl)methyljbenzene
methanimidamide (as dihydrochloride);
6c: N-[(4-Pyridyl)methyl]-4-[(1 H-imidazol-4-yl)methyljbenzene
methanimidamide (as dihydrochloride);
6d: N-(2-Phenylethyl)-4-[(1 H-imidazol-4-yl)methyl]benzene
methanimidamide (as dihydrochloride):
Example 3: 4-[(1 H-Imidazol-4-yl)methyljbenzene methanimidamide (as
ihvdrochloride~
Part A.
_ \ 1. H2NOH-HCl \
TrN ~ N I / KOH, EtOH HNVN I / '~1HOH
a CN fl
4 2. O.SN HCl g N H
Hydroxylamine hydrochloride (0.90 g, 13 mmol) and potassium hydroxide
(0.73 g, 13 mmol) were stirred for 5 minutes in absolute ethanol at room
temperature. Compound 4 (0.55 g, 1.3 mmol) in absolute ethanol (25 mL) was
added and the reaction heated to reflux for three hours. It was then cooled to
room temperature, filtered, and concentrated to give a white solid. This solid
W095/14007 ; .~ PC1YU594/12717
- 27 -
was dissolved in 0.5N HCI and heated to 50°C for one hour. The mixture
was
cooled, filtered, and washed with ether. The aqueous layer was concentrated
and the residue applied to a flash column (85:15 CH2C12:MeOH/NH3).
Compound 8 was obtained as a white solid (0.125 g, 44%).
Part B.
H2, Ra-Ni
HNVN / NHOH EtOH ~ HN~N / NH2
N H 9 N H 2HC1
A solution of 8 (0.12 g, 0.56 mmol) and Raney-Nickel (about 0.1 g wet)
were hydrogenated on a Parr shaker under 4.4 kg.cm.-2 H2 pressure (63 psi H2)
at room temperature overnight. The heterogeneous mixture was filtered through
Celite, and the filter cake washed with additional ethanol. The ethanol was
removed on the rotary evaporator, and the residue purified by HPLC (RCM
25x10 silica gel column eluted with acetonitrile:water:conc. HCI 1600 mL:400
mL:0.5 mL at 3 mUmin.). Compound 9 (0.123 g, 81 %) was obtained as a glass.
NH
In the same manner as that used
to prepare compound 9, compound 10 - ~ \ 1 NH2
was prepared. ~ HN~N l 0 2HC1
Example 5: 3-[(1 H-Imidazol-4-yrl)eth~~)benzene ,~gtl~,nimidamide ~~,
~yrdrochlorir,~
Br ~ ~ 1 1 P~3, Toluene B~ P / I 1 2
CN 3 ~ CN
The nitrite 11 (9.8 g, 0.05 mole) and triphenylphosphine (14.4 g, 0.055
mole) were combined in toluene (100 mL) and heated to reflux under a nitrogen
atmosphere for 8 hours. A white precipitate formed. The reaction was cooled,
and the solid was collected by filtration and washed with toluene (150 mL),
and
dried under vacuum. A white solid was obtained (19.7 g, 86%) and used without
further purification in the next step.
CA 02176557 2005-04-20
- 28 -
1. t-BuOK, THF i
CHO
Br~3P ~ ~ 2. ~ ~ CN
12 CN TrN~N 1 3 TrNvN 14
A solution of potassium t-butoxide in THF ( 16.3 mL of a 1 M
solution) was added dropwise to a suspension of the phosphonium salt 12
(7.45 g, 16.3 mmol) in dry THF (45 mL) under a nitrogen atmosphere at
room temperature. The orange suspension was stirred for three minutes
and a solution of the aldehyde 13 (5 g, 14.8 mmol) [prepared according to
Bernabe and Burger, J. Med. Chem., 14 (1971) 883-885] in dry THF (45
mL) was added. After 3.5 hours at room temperature, the reaction was
diluted with ether and filtered through Celite.~ The Celite~ was washed
~~ additional ether. The organic layer was dried (MgS04) and
concentrated to give a solid that was purified on a flash column (Si02, 1:1
hexane:ethyl acetate) to give 5.03 g (78%) of 14 as a white solid.
H2NOH-HCI
C N KOH, EtOH ~ ~ N H
TrN~N 14 HN~N 15 NHOH
Hydroxylamine hydrochloride (8 g, 115 mmol) and potassium hydroxide
(6.8 g, 121 mmol) were combined in ethanol (100 mL) and heated to 50°C
for 10
minutes. A solution of 14 (5.03 g, 1 i.5 mmol) in ethanol (100 mL) was added
and the reaction heated to reflux for 2 hours. It was cooled, filtered and
concentrated. The solid that was obtained was dissolved in 1 N HCl (80 mL) and
heated to 60°C. After 1.5 hours the reaction mixture was filtered and
the
aqueous layer was washed with ether and concentrated. The residue was
dissolved in methanoUNH3 and stirred for twenty minutes. The solvent was
removed and the residue dissolved in ethyl acetate and methylene chloride
(80:20) and washed with water. The aqueous layer was extracted again and the
combined organic layers were dried (MgSOa), filtered, and concentrated to give
2.5 g (94 % ) Of 15 as a white Solid.
"~ wo 9snaoo7 ~ 1 ~ rcr~s9anz~m
- 29 -
I N H Ra-Ni, H2, EtOH ~~,~ I N H2
HN,~N 15 NHOH HN~N 16 2HC1 NH
Raney nickel (about 0.5 g wet) and compound 15 (0.46 g, 2 mmol) in
ethanol (50 mL) were combined in a Parr bottle and hydrogenated under 4.2
kg.cm.-2 H2 pressure (60 psi H2) 60 psi H2. After 20 hours, the mixture was
filtered and the residue was washed with additional ethanol. Upon
concentration, an amber gum was obtained which was purified by HPLC (RCM
25x10, Si02, acetonitrile:water:conc. HCI 1600 mL:400:0.5, 3 mUmin, 254 nm).
Compound 16 was obtained as a glass (0.31 g, 54%).
Exam,;[(1 Hllmi~~ oZ~.l=4-yrl)ethyllbenzene methanimidamide (as
dihydrochloride)
In a manner similar to that used to
prepare compound 16, compound 17 was
HN~N I / NH2
prepared. 17 2HC1
NH
Example 7: 4-j(1H-Imidazol-4-~rlymethyr~)benze~Qemethanamine (~
dihydrochloride)
Part A.
Ra-Ni, H2 "~ /
1 1
TrNVN .,~ I CN HzptCl6 TrNVN ,,~ I NH2
MeOH/NH3 2 0
Compound 4 (1.53 g, 3.6 mmol) was combined with Raney-Nickel (about
1 g), methanol saturated with ammonia (50 mL), and chloroplatinic acid (0.8 mL
of a solution of 1.0 g of the acid in 10 mL of water) in a Parr bottle and
shaken
under 4.2 kg.cm.-2 H2 pressure (60 psi H2) for 24 hours. The reaction was
filtered through Celite and concentrated on the rotary evaporator. The crude
material was purified on a flash column (200 g Si02; 95:5 CH2C12:MeOH/NH3)
to give 1.31 g (85%) of 20 as a white solid.
~ ~.'~ 6 5 5'~ PCTJUS94/1271 i
WO 95/14007
- 30 -
TrN ~/~ ~~ 1 N HCI, E~ H N ''
uN ~NH2 ~N ~ NH2
2 0 21 2HCl
To a solution of 20 (1.31 g, 3.1 mmol) in absolute ethanol (30 mL) was
added 1 N HCI (20 mL) and the heterogeneous mixture heated to 70°C for
2
hours. The reaction was cooled, filtered, and concentrated. Water (50 mL) was
added and the solution washed with ether. The aqueous layer was
concentrated to give a white solid (0.8 g, 99%).
Fxam~ N-j(4-Chlorophenyrl)methv!]-4-((1 H-imidazol-4-yl)methyrllbenzene
~ooanimidamide (as dihydrochloridel
Part A.
I OH
1. EtMgBr, CHzCl2
TrN~N OHC
~ TrN i N i OEt
oEt ~
OEt 22 OEt
In an analogous manner to that described in Example 1 Part A,
compound 22 was obtained in 81% yield by the reaction of the Grignard reagent
derived from 1 and terephthalaldehyde mono-(diethylacetal).
Part B.
1. TCDI
~ TrN~N I / OEt
T 2. n-Bu3SnH,
AIBN 2 3 OEt
In a manner similar to that described in Example 1 Part B above,
compound 23 was derived from compound 22 in 36% overall yield.
Part C.
TrN ~ N I ~ OEt '~m~rlyst-1~ TrN i N i
U AcetonelH 20 ~ CHO
23 OEt 24
WO 95/14007 PCT/US94/12717
- 3r -
To a solution of 23 (1.37 g, 2.73 mmol) in acetone (15 mL) was added
Amberlyst-15 resin (0.15 g) and water (0.2 mL). The reaction was stirred
overnight at room temperature and filtered, and the resin washed with
additional
acetone (25 mL). After drying (MgS04) and concentration, a white solid was
obtained (1.03 g, 88%) that was used without further purification.
Et0 ~ C N
( )2~
TrN~N ~CHO .~~. TrN ~ N
2 4 and NaH ~ 2 5 C N
Neat diethyl cyanomethylphosphonate {2.6 mMol, 0.46 g) was added
dropwise over 10 min to a pentane-washed suspension of NaH (0.104 g of a
60% suspension in mineral oil, 2.6 mmol) in THF (30 mL) under argon at
0°C .
After 45 min., aldehyde 24 (0.86 g, 2 mmol) in THF (30 mL) was added and the
reaction stirred for 4 hours. The reaction was poured into water and extracted
with chloroform (3 x 75 mL). The combined organic layers were washed with
12% NaOH, dried (MgS04), and concentrated. The crude olefins 25 were
purified on a flash column (150 g Si02; 90:10 ether:hexane) to give 0.63 g of
a
7.5:1 trans:cis mixture of olefins (70%).
TrN ~ N I ~ N~H4--~ TrN,~ N I ~ C N
~CN ~~H/p~dine 26
To a solution of 25 (0.43 g, 0.95 mmol) in methanol (0.5 mL) and pyridine
20 (1.5 mL) was added NaBH4 (0.04 g, 1.05 mmol) portionwise. The reaction was
heated to 120°C for 36 hours, cooled and poured into saturated aqueous
NH4C1. The aqueous layer was extracted with ethyl acetate, and the combined
organic layers were dried with MgS04. Filtration and concentration on the
rotary
evaporator gave a thick oil which was purified on a flash column (75 g Si02;
25 ether) to give 0.3 g (70%) of 26.
WO 95/14007 ~ PCT1US94/12717
- 32 -
H
w CN CI I ~ N I w H N / I
I i ~ ' HN N i w
TrNvN ~ AIMeZ v ~/~/ CI
-~' 2HCl
2 6 2 . 1 N HCl/MeOH 2 ~
In an analogous manner to that described in Example 1 parts C and D
above, compound 26 was transformed into compound 27 (36% overall yield).
ExamRle 9N-[(4-Chlorophenyrl)methyrl~-4-[~1 H-imidazol-4-yl)methyllbenzene
Qthanimidamide las dihydrochloride)
Part A.
1. t-BuOK, TosMic
TrN~ N I ~ CHO 2. MeOH TrN~ N I~CN
24 28
To a solution of t-BuOK in THF (6.5 mL of a 1 M solution) at -40°C
was
added a solution of TosMic (tosylmethylisocyanide) (0.66 g, 3.4 mmol) in THF
(5 mL) followed by the aldehyde 24 (1.31 g, 3.1 mmol) in THF (5 mL). After
1 hour at this temperature, MeOH (10 mL) was added and the reaction heated to
reflux for 20 min. It was then cooled to room temperature and the solvent
removed under a stream of nitrogen. The residue was dissolved in CH2C12 and
washed with water/acetic acid (10 mU0.4 mL). The aqueous layer was
extracted with CH2C12, and the combined organic layers were washed with
aqueous NaHC03 and dried (MgS04). The residue obtained upon filtration and
evaporation was purified on a flash column (85:15 hexane:isopropanol) to give
0.55 g (40%) of 28.
Part B.
CI~H
CI
1. ' N'AIMeZ I ~ NH (
TrNvN ~CN ~ HN~N l~N
2 g 2. 1 N HCI/EtOH 2 9 H 2HC1
In a manner analogous to that described in Example 1 Parts C and D
above, compound 28 was converted into compound 29.
'WO 95/14007 217 6 5 5 7 pCT/US94/12717
- 33 -
Part A.
CI
CI / CI
I O T
TrN,~ N I~~ N H 2 .~. TrN N ~~~s~ N
Et3N, CH2C12 ~
20 30 O
Distilled Et3N (0.25 g, 2.5 mmol) was added to a solution of 20 (0.43 g,
1.0 mmol) in dry methylene chloride (10 mL). The solution was cooled in an ice
water bath, and 4-chlorobenzoyl chloride (0.19 g, 1.1 mmol) was added slowly
(25 min.). After 1 hour, the reaction was poured into ice-water and extracted
with
methylene chloride (2 x 50 mL). The combined organic layers were washed
with brine, dried (MgS04), and concentrated to give a white solid that was
purified by flash column chromatography (95:5 methylene chloride:
methanol/NH3). Compound 30 was obtained as a white solid (0.55 g, 97%).
Part B.
CI
TrNvN I / N ~ ~ ~ H / CI
H N,~,N I / N
O '"
30 H~ O
1N HCl / MeOH 31
In a manner analogous to that described in Example 1, Part D,
Compound 30 was transformed into compound 31.
1. 2N NaoHIEto~-I
TrN ~ N / C N 2. H~S04/EtOH T~.,"i, N / OEt
3. TrCI, Et3N
4 32
Compound 4 (3 g, 7.1 mmol), 2N NaOH (7.5 mL), and ethanol (35 mL)
were heated to reflux for 20 hours. The reaction mixture was cooled to room
temperature and concentrated to a paste. Ethanol (50 mL) was added and the
mixture concentrated again. This procedure was repeated with toluene.
Sulfuric acid (3 mL) and ethanol (30 mL) were added to the residue, and the
WO 95/14007 PCT/US94/12717
~ 1'~ 6'~ ~ ~
- 34 -
mixture was heated to reflux for 20 hours. The reaction mixture was cooled to
room temperature and adjusted to pH 8 with 2N NaOH. Water was added, and
the aqueous mixture extracted with methylene chloride (4 x 35 mL). The
combined organic layers were dried and concentrated. The crude product was
redissolved in dry methylene chloride (60 mL) and triethylamine (1.96 mL), and
trityl chloride (2.34 g) were added. After 4 hours, additional methylene
chloride
(100 mL) was added and the reaction mixture was washed with water and brine.
The crude material obtained upon drying and concentration was purified on a
flash column (ether) to give 32 as a white solid (1.9 g, 57%).
Part B
NHAIMe2 ~ / CI
TrN ~ N I / OECI HN~N ( / N
2. 1 N HCI, EtOH O HCl
32 33
In a manner analogous to that described in Example 1 Parts C and D
above, compound 32 was converted into compound 33.
~~ple 12: 4~[4-~(4-Chloroohenyl)~methox)r)methyljohenyl]methyrlJ-1 H-
imidazole
Part A
DiBAL-H, THF'
TrN ~N I ~ OEt TrN~N / OH
a
32 O 34
DiBAL-H was added dropwise over 3 min. to a solution of 32 (1 g, 2.1
mmol) in dry THF (14 mL) at 0°C. After 30 min., the reaction was
quenched by
the slow addition of 2N NaOH. The reaction mixture was poured into ether (60
mL) and additional 2N NaOH (1.5 mL) and water (1.5 mL) were added. After
stirring for 10 min., the turbid mixture was washed with water. The water
layer
was back-extracted with additional ether (25 mL), and the combined ether
layers
were dried (MgS04). The crude material was purified on a flash column (ether)
to give 0.84 g (93%) of 34 as a white solid.
WO 95/14007 PGT/US94I12717
F,
1. NaH, THF
TrN N I / O H 2~ I B r -. I / I ~C I
TrN N
CI .,r ~~O \
NaH (0.032 g of a 60% dispersion in mineral oil, 0.8 mmol) was added to
a solution of 34 (0.26 g, 0.6 mmol) in dry THF (5 mL) at 0°C. The
reaction was
5 allowed to warm to room temperature and then stirred for 20 min.; it was
then
recooled to 0°C, and 4-chlorobenzyl bromide (0.12 g, 0.6 mmol) was
added.
The reaction was slowly warmed to room temperature and stirred overnight.
Additional NaH (0.008 g) and 4-chlorobenzyl bromide (0.041 g) were added and
the reaction mixture was stirred an additional 6 hours. The reaction was
diluted
10 with ether and washed with water and brine. After drying (MgS04), the crude
material was purified on a flash column (90:10 ether:hexane) to give 35 as a
white SOlld (0.15 g, 46%).
/ ( CI
TrN ~ N / O '''~ '\ / C I
35 1 N HCI, MeOH H N ~ N I~,,~ O
v
36 HCl
15 In a manner analogous to that described in Example 1, Part D, compound
35 was transformed into compound 36.
Examr~le 13' [4-(1 H-Imida7ol-4-ylmethyrly hen~tl]methyrl N-(4-chloro-
~yl)~carbamate
Part A
NCO H
'\ I ~ ~. p N ',.
TrN~N ( / OH C~ -~..T~~N I~~O I /
P~~nc C I
20 34 37
4-Chlorophenyl isocyanate (0.1 g, 0.66 mmol) was slowly added to a
solution of 34 (0.26 g, 0.6 mmol) in dry pyridine (4 mL) at 0°C. When
TLC
(ether) indicated complete reaction, the pyridine was removed under reduced
pressure. The residue was dissolved in methylene chloride (50 mL) and
WO 95/14007 PGTJUS94/12717
~ 1'~ ~ ~'~-'~ - 3s -
washed with saturated aqueous NaHC03 and water and dried (MgS04). The
residue obtained upon concentrating was purified on a flash column (80:20
ether: hexane) to give 0.15 g (43%) of 37 as a white solid.
Part B
H
\ Oy N \
TrN ~ N ( / 'O I / O N
CI
1 N HC1, MeOH I ~ I
HN~N / O / CI
38 HCl
In a manner analogous to that described in Example 1, Part D, compound
37 was transformed into compound 38.
ExamQle 14: [4-(1 H-Imidazol-4-ylmethyl)~yllmethyrl 4-chlorobenzoate
Part A
CI
\ ~ ~ CI \ \ CI
I O
TrN ~ N I~~ O H TrN ~ N ( / O ( /
Et3N, CH2Cl2 O
34 39
4-Chlorobenzoyl chloride (0.12 g, 0.66 mmol) was slowly added over 20
min. to a solution of 34 (0.26 g, 0.6 mmol) and triethylamine (0.15 g, 1.5
mmol)
in dry methylene chloride (10 mL) at 0°C. After 30 min., the reaction
was diluted
with additional methylene chloride (30 mL) and poured into half-saturated
NaHC03 (20 mL). The organic layer was separated, and the aqueous layer was
further extracted with methylene chloride (25 mL). The combined organic layers
were dried (MgS04) and concentrated to give a foam that was purified on a
flash
column (80:20 ether:hexane). Compound 39 (0.22 g, 64%) was obtained as a
white solid.
,Part B
\ I \ C CI
I \ \
TrN ~ N I~~ O / I I
HN ~ N / O /
39 O 1 N HCI, MeOH~ HCl
40 O
In a manner analogous to that described in Example 1, Part D, compound
39 was transformed into compound 40.
WO 95114007 PGT/US94112717
21765~'~
- 37 -
',~, 1. 2N NaOH/EtOH
TrN ~ N / C N 2. HCI, EtOH H N ~;/ N H~ / O H
4 41 O
A suspension of 4 (1 g, 2.4 mmol) in ethanol (5 mL) and 2N NaOH (5 mL)
was heated to reflux for 20 hours. After cooling, the solvent was removed
under
reduced pressure and the residue was suspended in 1 N HCI (25 mL) and
heated to 60°C for 2 hours. The remaining solid was removed by
filtration after
cooling, and the aqueous layer was concentrated to give a solid. This was used
in the next step without purification.
Part B
'~ socl2
HN ~ N / OH '' HN ~ N / CI
H~ ~ HCI~
41 O 42 O
The residue from Part A was suspended in SOC12 (20 mL) and stirred for
hours at room temperature. The excess SOC12 was removed under reduced
pressure and the residue dried by azeotropic removal of toluene. The resulting
15 yellow solid was used directly in the next step without purification.
OH ~,,~ / CI
CI _
HNvN I / CI Et CH 1 HN~N I / O
C
j-~C] 3N 2 2
42 O 2. HCI, ether 43 O HCl
4-Chlorobenzyl alcohol (0.71 g, 5 mmol) and triethylamine (1.01 g, 10
mmol) were added to a suspension of the acid chloride from Part B in dry
20 methylene chloride (15 mL) at 0°C. The reaction mixture was warmed
to room
temperature and stirred for 24 hours. Additional methylene chloride (50 mL)
was added and the organic layer was washed with saturated aqueous NaHC03.
The organic layer was separated and dried (MgS04). Concentration gave an
amber oil that was purified on a flash column (97:3 CH2C12:MeOH/NH3). A
white solid was obtained (0.36 g, 46% from nitrite 4). This material was
dissolved in methylene chloride (10 mL) and 1 N HCI in ether (5 mL) was added.
WO 95/14007 PCT/US94/1Z717
- 3B -
The solvent was evaporated under a stream of dry argon to give 43 as a white
solid (0.4 g, 100%).
Other compounds named herein can be prepared analogously, together
with the following compounds:
N-[(4-chlorophenyl)methyl]-2-fluoro-4-[(1 H-imidazol-4-yl)methyl]benzene
methanim idam ide;
N-[(4-chlorophenyl)methyl]-2-chloro-4-[(1 H-imidazol-4-yl)methyl]benzene
ethanim idam ide;
N-[(4-chlorophenyl)methyl]-3-methyl-4-[(1 H-imidazol-4-yl)methyl]benzene
methanimidamide;
N-[(4-chlorophenyl)methyl]-2-(1-propenyl)-4-[(1 H-imidazol-4-
yl)methyl]benzene ethanimidamide;
N-[(4-chlorophenyl)methyl]-3-trifluoromethyl-4-[(1 H-imidazol-4-
yl)methyl]benzene methanimidamide;
N-[(4-chlorophenyl)methyl]-2-(1-propynyl)-4-[(1H-imidazol-4-
yl)methyl]benzene ethanimidamide;
N-[(4-chlorophenyl)methyl]-3-methoxy-4-[(1 H-imidazol-4-
yl)methyl]benzene methanimidamide;
N-[(4-chlorophenyl)methyl]-2-dimethylamino-4-[(1 H-imidazol-4-
yl)methyl]benzene ethanimidamide; and
N-[(4-chlorophenyl)methyl]-3-methylthio-4-[(1 H-imidazol-4-
yl)methyl]benzene methanimidamide.
CA 02176557 2005-04-20
- 39 -
TABLE 2: MASS SPECTRAL DATA FOR COMPOUNDS OF THE EXAMPLES:
Compound Mass Compound Mass
Number S ectrum Number S ectrum
6 Calc: 325.1220 21 Calc: 187.1109
Found:325.1231 Found:187.1122
6 8 Calc: 320.1637 2 7 Calc: 352.1455
Found:320.1620 Found:352..1476
6 b Caic: 358.1404 2 9 Calc: 338.1298
Found:358.1411 Found:338.1314
6c Caic: 291.1484 31 Calc: 326.1060
Found:291.1500 Found:326.1059
6d Calc: 304.1688 3 3 Calc: 326.1060
Found:304.1702 Found:326.1059
9 Calc: 200.1062 3 6 Calc: 313.1108
Found:200.1074 Found:313.1108
Calc: 201.1140 3 8 Caic: 342.1009
Found:201.1152 Found:342.0998
16 Calc: 215.1297 4 0 Calc: 327.0900
Found:215.1305 Found:327.0891
1 7 Calc: 215.1297 4 3 Calc: 327.0900
Found:215.1292 Found:327.0897
H3 Receptor Binding:
Ha Receptor Binding Assey
The source of the H3 receptors in this experiment was guinea pig
5 brain. The animals weighed 400-600 g. The brain tissue was homogenized
using a Polytron~ in a solution of 50 mM Tris, pH 7.5. The final
concentration of tissue in the homogenization buffer was 10% w/v. The
homogenates were centrifuged at 1,000 x g for 10 min. in order to remove
clumps of tissue and debris. The resulting supernatants were then
10 centrifuged at 50,000 x g for 20 min. in order to sediment the membranes,
which were next washed three times in homogenization buffer (50,000
x g for 20 min. each). The membranes were frozen and stored at -70°C
until needed.
All compounds to be tested were dissolved in DMSO and
then diluted into the binding buffer (50 mM Tris, pH 7.5) such that
the final concentration was 2 ~,g/mL with 0.1 % DMSO. Membranes
were then added (400 dug of protein) to the reaction tubes. The reaction
was started by the addition of 3 nM [3H]R-a-methylhistamine (8.8
Ci/mmol) or 3 nM [3H]N"-methylhistamine (80 Ci/mmol)
WO 95/140(17 217 6 5 5 l pCTlUS94112717
- 40 -
and continued under incubation at 30°C for 30 min. Bound ligand was
separated from unbound ligand by filtration, and the amount of radioactive
ligand bound to the membranes was quantitated by liquid scintillation
spectrometry. All incubations were performed in duplicate and the standard
error was always less than 10%. Compounds that inhibited more than 70% of
the specific binding of radioactive ligand to the receptor were serially
diluted to
determine a K; (~.M). The results are given in Table 3 and Table 4.
Table 3: K;(~.M) values
Compound K; Compound Ki Compound K;
Number (p.M) Number (~.M) Number (~,M)
6 0.0140 9 0.038 21 0.078
6a 0.56 10 0.31 27 0.18
6 b 0.14 1 6 0.22 2 9 0.0072
6 d 0.45 1 7 0.17 3 8 0.024
Table 4: inhibition of binding of radioactive ligand
Compound Inhibition (%) Compound Inhibition (%)
Number at 2 p.g/ml Number at 2 ~g/ml
6c 48 36 6-87
30 61 40 10
33 61 43 16-76
From these test results and the background knowledge about the
compounds described in the references in the section "Background of the
Invention", it is to be expected that the compounds of the invention would be
useful in treating inflammation, allergy, diseases of the GI-tract,
cardiovascular
disease, or disturbances of the central nervous system.
Pharmaceutically acceptable inert carriers used for preparing
pharmaceutical compositions from the compounds of Formula I and their salts
can be either solid or liquid. Solid form preparations include powders,
tablets,
dispersible granules, capsules, cachets and suppositories. The powders and
tablets may comprise from about 5 to about 70 percent active ingredient.
2176551
WO 95/14007 PGT/US94/12717
-a~ -
Suitable solid carriers are known in the art; e.g. magnesium carbonate,
magnesium stearate, talc, sugar, lactose. Tat~lets, powders, cachets and
capsules can be used as solid dosage forms suitable for oral administration.
Liquid form preparations include solutions, suspensions and emulsions,
for example water or water-propylene glycol solutions for parenteral
injection.
Liquid form preparations may also include solutions for intranasal
administration.
Also included are solid form preparations which are intended for
conversion, shortly before use, into liquid form preparations for either oral
or
parenteral administration. Such liquid forms include solutions, suspensions
and
emulsions.
Aerosol preparations suitable for inhalaticm may include solutions and
solids in powder form, which may be in combination with a pharmaceutically
acceptable carrier, such as an inert compressed gas.
For preparing suppositories, a iow melting wax such as a mixture of fatty
acid glycerides or cocoa butter is first melted, and the active ingredient is
dispersed homogeneously therein as by stirring. The molten homogeneous
mixture is then poured into conveniently sized molds, and allowed to cool and
thereby solidify.
Preferably the compound is administered orally.
Preferably, the pharmaceutical preparation is in unit dosage form. In such
form, the preparation is subdivided into unit doses containing appropriate
quantities of the active component, e.g., an effective amount to achieve the
desired purpose. The quantity of active compound in a unit dose of preparation
may be varied or adjusted from about 0.1 mg to 1000 mg, more preferably from
about 1 mg to 500 mg, according to the particular application.
The actual dosage employed may be varied depending upon the
requirements of the patient and the severity of the condition being treated.
The
determination of the proper dosage for a particular condition is within the
skill of
the art. Generally, treatment is initiated with smaller dosages which are less
than the optimum dose of the compound. Thereafter, the dosage is increased by
small amounts until the optimum effect under the circumstances is reached. For
convenience, the total daily dosage may be divided and administered in
portions during the day if desired.
The amount and frequency of administration of the compounds of the
invention and the pharmaceutically acceptable salts thereof will be regulated
according to the judgement of the attending clinician considering such factors
as
age, condition and size of the patient as well as severity of the symptoms
being
WO 95/14007 PCT/US94112717
- 42 -
treated. A typical recommended dosage regimen is oral administration of from
1 mg to 2000 mg/day, preferably 10 to 1000 mg/day, in one to four divided
doses
to achieve relief of the symptoms. The compounds are non-toxic when
administered at therapeutic doses.
The following are examples of pharmaceutical dosage forms which
contain a compound of the invention. As used therein, the term 'active
compound' is used to designate one of the compounds of the formula I or salt
thereof, especially compounds 6 and 29 herein (as free base), namely N-[(4-
chlorophenyl)methyl]-4-[(1 H-imidazol-4-yl)methyl]benzene methanimidamide
and N-[(4-chlorophenyl)methyl]-4-[(1 H-imidazol-4-yl)methyl]benzene
ethanimidamide, or the dihydrochloride thereof, but any other compound of the
formula I or salt thereof can be substituted therefor:
,fy~. Ingredients Lnq,/tabletmg"(tablet
1. Active compound 100 500
2. Lactose USP 122 113
3. Corn Starch, Food Grade, 30 40
as a 10% paste in
Purified Water
4. Corn Starch, Food Grade 45 40
5. Magnesium Stearate 3 7
Total 300 700
Method of Manufacture
Mix Items No. 1 and 2 in a suitable mixer for 10 to 15 minutes. Granulate
the mixture with Item No. 3. Mill the damp granules through a coarse screen
(e.g., 1/4', 0.63 cm) if necessary. Dry the damp granules. Screen the dried
granules if necessary and mix with Item No. 4 and mix for 10-15 minutes. Add
Item No. 5 and mix for 1 to 3 minutes. Compress the mixture to appropriate
size
and weigh on a suitable tablet machine.
WO 95/14007 PGT/US94/12717
-43 - 2176557
I~~redi,~ _m_a/caosulema/cac~sule
1. Active compound 100 500
2. Lactose USP 106 123
3. Corn Starch, Food Grade 40 70
4. Magnesium Stearate NF 4 7
Total 250 700
Method of Manufacture
Mix Items No. 1, 2 and 3 in a suitable blender for 10 to 15 minutes. Add
Item No. 4 and mix for 1 to 3 minutes. Fill the mixture into suitable two-
piece
hard gelatin capsules on a suitable encapsulating machine.
While a number of embodiments of this invention are described herein, it
is apparent that the embodiments can be altered to provide other embodiments
that utilize the compositions and processes of this invention. Therefore, it
will be
appreciated that the scope of this invention includes alternative embodiments
and variations which are defined in the foregoing Specification and by the
Claims appended hereto; and the invention is not to be limited to the specific
embodiments that have been presented herein by way of example.