Note: Descriptions are shown in the official language in which they were submitted.
WO 95114027 21 7 6 7 6 0 PCTICA94/00636
AN~II-VlRAL GUANlDlNO~Uh;~lllUl~ COMPOUNDS
Fi~l.l of th,. J
This invention relates to novel, O " ' cu T ' and to their anti-
viral use .~Lil~uLly for the treatment of h~ vi-~... infection.
r2 ~,U.. .1 tû rh.o I
The '~.~ YiLI...~,~ constitute a family of human pathogens related '~ ly by
life cycle, host range, i;UlylJ~IJ~i~il, '''"'1"'` ~;'~1' and genomic structure. The
k~ u~vi-~ family includes the ll~,.,U~i.Vi.i~i~ thatare divided into three sub-families;
p.~DViLilL4~ which includes herpes simplex virus (HSV) type 1, which manifests
as cold sores and type 2 which causes genitai lwions, . ' ~ virus, equine
abortion virus, and infectious bovine ~ virus; 1~ .U~.~ViLill~l., which
includwhumanandmurine~, ~ '~virus(CMV);and" '.~ vi. which
includes the r. B~u~ virus (EBV) which is r~,~ull~il,lc for infectious
Also included in the ll~,UWViLh. family is Varicella Zoster virus
(VZV) which is the cauSative agent of chicken pox, and recently discovered humanviruses HHV-6 and -7.
The current typw of herpw treatments are reviewed in "Antiviral Drugs:
rl ~ Adverse Effects and Therapeutic Use:, M.C. Nahata, 1. Pharm.
Technol., 1987, 3:100. A variety of drugs have been developed to treat l~p~ vi~
infection, including natural occurring proteins and sy-ntiletic nucleoside anaiogs. For
example, the natural antivirai protein, interferon, has been used in the treatment of
l..,.~,.,i,vi...~ infections, as have the nucleoside analogs, cytosine ~ ' ', adenine
_.,.1.:...,.: 1., iU iU~. " and acyciovir, which is presently the treatment of choice
for herpes sirnplex type I infection. Despite these significant advances in the
treatment of l,~."u~v i.. infections, the need for effective, safe Ih.,.. ~ iu agents for
treating ll.,.,U~/vi-u~ infections continues to exist.
It is a general object of the present invention to provide novel O ' substituted ' having anti-virai activity, and . ' containing these u~ u -
SUBSrITUTE SHE~
WO 95/14027 21~ 6 7 6 ~ PCT/CA94100636 --
It is a further object of the present invention to provide processes and i~useful for the production of such ~ ' ' ' , '
It is another object of the present invention to provide a method of treating
}~pWVil~ infection in a mammal.
SUMMA.RY OF T~ INVEi~TlON
It has now been discovered that certain guanidino-substituted ~ . _ ' are effective
inhibitors of h~ ~v;lua replication. The present invention provides, in one of its
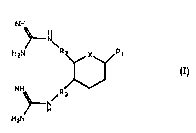
aspects, a compound of formula (1):
NH
~N
H2N R2 X~
~ H
H2N
or a salt, hydrate or solvate thereof, wherein
X is selected from C, O, N and S;
Rl is -(W)/-(R~)b-Z~ wherein
a and b are ' ' '~, 0 or 1;
W is NH, O or S;
R4 is Cl 8ailylene in which one or two of the carbon atoms thereof is
optionally replaced with nitrogen or oxygen; and
Z is H, OH or -NH-C(NH)-NH2;
R2 and R3 are, ' r ~ Cl galkylene in which one of the carbon atoms thereof
is optionally replaced with nitrogen or oxygen.
In another aspect, the present invention provides a L ' ~ useful
to treat l~ ,avilllJ infection in a mammai, comprising a compound as defined by
formula (1), or a salt, solvate or hydrate thereof, and a ~ ' lly acceptable
SUBSTITUTE SHE~T
Wo g5114027 21 7 6 7 6 Q PCT/CA94100636
carrier.
In a further aspect, the present invention provides a method for treating ll~ vilu~
infection in a mammal which comprises the step of ~ v to the infected
mammal a l ' . . . v a Ih ~ ly effective amount
of a compound of formula (1).
In yet another aspect of the present invention, there is provided a process of preparing
' of formula I which comprises the step of obtaining a precursor of a
compound of formula I having protecting groups coupled to the terminal N-groups of
the guanidino function, and then treating the precursor to remove the protectinggroups. r~u~iuul~uly for . ' of the invention in which W constitutes NH, tl'le
inveMion further provides an " of the formula:
Pr1
Pr20 ~,o ~N- Y
Pr
wherein:
Prl is an amino protecting group;
Pr~ is a hydroxyl protecting group; and
Y is -R~-Z wherein R and Z are as defined in formula (I).
Also provided is a process for preparing such an i.. ~ illg the
step of reacting a reagent of the formula noted below with a compound Y-NH-Prl in
an aprotic solvent in the presence of a Lewis acid:
pr20 o
p~ok~
OPrZ
F --' ' of the present invention are described in greater detail with reference
SUBSTITUTE SHEFT
WO9S114027 ~ 6~ 60 ~ PC11CA94100636
to the 2 . ,V;ll2; drawings in which:
RriPf Reference to thP ~
FIGURr 1 illustratw thé chemical process for preparing a ,, '
compound of the present invention.
FIGURES 2 to 7 illustrate Lhe chemiwal processes for preparing ",
' of examples 5 to 10, I~,~Li~,ly.
r~Pt'.ilP~ ~Pcrription of rhP I and Its Preferr~d r
The present invention provides novel ~ ' ' . . ' having anti-
},~ ,~Vil~ activity. The term "anti ~ DViUu," as used herein refers to Lhose
which inhibit repliwation of at least one member of the ~ wviu h.. family
as determined by ~. ' cell culture assay, such as the well established plaque
reduction assay. In the context of the plaque reduction assay, for example, the anti-
II~.llJ~,..Vilh.. activity of a compound is revealed by a reduction in plaque count, or a
reduction in plaque size, when virally infecLed cells are treated with the givencompound, relative to a virally infected, untreated control.
The term rl.~ ,.,vilu, is meant Lo encompass viruses which are ,' ' ~ 'ly
- similar to herpes simplex viruses and ~ir~lly . . ~ those viruses which
are enveloped, double-stranded DNA viruses. Included within this family of virusw
are herpw simplex virus type 1 (HSV-I), herpes simplex virus type 2 (HSV-2),
varicella-zoster virus (VZV), r~i.~;.. Ba~ virus (EBV), .,~ " ' viuu~ (CMV),
equine herpw virus (I~HV) and 1. -. 1~.., l. - ~ virus (PRV). Moreover, as one of skill
in Lhe art will appreciate, also included within the l..,.lJ~Vilh,~ family are the different
isolates and strains of each of the member virusw.
In one of its aspects, the present invention provides a compound of formula (1):
SUBSrITUTE SHE~
WO 95114027 2 ~ ~ G ~ 6 0 PCT~CA94100636
* 5
NH
~ N
H2N R ~ X~l/ R
~ H
HzN
or a sait, hydrate or solvate thereof, wherein
X is selected from C, O, N and S;
Rl is~ -(R~)b-Z, wherein
a and b are i l. ~. ..l. ly O or l;
W is NH, O or S;
R4 is C,.3aikylene in which one or two of the carbon atoms thereof is
optionally replaced with nitrogen or oxygen; and
Z is H, OH or -NH-C(NH)-NH2;
R2 and R3 are, i ' r ~ Cl gaikylene in which one of the caroon atoms thereof
is optionally replaced with nitrogen or oxygen.
With respect to substituent Rl as defined by the formula -(W),-(R~)b-Z, 'b' is suitably
1 and R4 is most suitably selected from a Cl ,alkylene group and a Cl aalkylene group
in which at least one carbon is replaced by oxygen. In specific ~ I ' , R4 is
selected from methylene, ethylene, n-propylene and iso-propylene, as well as ethers
such as
-Cl~aikylene-O-CIJalkylene- and -Cl *lkylene-O-C~ 2aikylene-O-C~ 2alkylene, e.g.,
-(CH2)2-O-(CH2)r and -(CH2)rO-(CH2)2-O-(CH2)r. In the case where 'b' is 1, 'a'
is also desirably 1, and W is selected most suitably from O and N. In this context,
specific ' '~ of the invention defined by tbe R1 formula include those in
which Z is H, e.g., methoxy, ethoxy, n-propoxy, i-propo~y, ethoxy-ethoxy and
ethoxy-ethoxy-ethoxy and analogues thereof in which W is N rather than O, e.g.,
~IU,U,~I~UllillO, cLi~y~. ..illU, ethoxy-eLi-~ -illù, etc. Also embraced by the Rl formuia
are ' in which Z is -OH, to yield the l~y~ilu~y' ~ uivak~ of the
SU5STITUTE SHE~
~095114027 ~ 61 6 PCT/CA94100636 --
just mentioned, sueh as hydroxy-ethoxy, hydroxy-methoxy and hydroxy-
ethoxy-ethoxy-ethoxy. In alternative . L ' , both 'a' and 'b' in the Rl formula
are zero, and Z is seleeted from -H, -OH and the guanidino function, -NH-C(NH)-
NH2
With respect to ,..1.~ R2 and R3, these are most suitably seleeted,' . ' 1~" from a Cl ,aikylene group and, desirably, a Cl halkylene group in
whieh at least one earbon is repiaced by oxygen. In c 1. " , R2 is C, ,alicylene-
O-CI 2aikylene, speeific examples of which include -(CH2)3-O-CH2-, ~(CH2)s~O-cHr;
and R3 is -Cl5alkylenoxy-, speeific examples of which include -(CH2)3-O- and -
(CHl)5-O-.
Compounds in which W is NH, may be ay ' ' from a protected glueal such as
tri-O-acetyl-glucal. As shown in the diagram below and described in detail in
example 11, a proteeted glueai is reaeted with an N-proteeted alkyl-amine to give the
l,U~ Ul~ o N-o y' ' compound wherein Y is aikyl, guanidino substituted
aikylene, hydroxy-aikylene or aikylenoxy-alkylene; Pr' is a suitable amino proteeting
group sueh as Boe or Cbz and pr2 is a suitable hydroxyi protecting group such asaeetyl. The reaetion is earried out in an aprotic solvent such as benzene, toluene or
dichlu.u.l.~,~h~.., in the presence of a Lewis acid sueh as SnCI~, AICI3 or BF3 in a
~ range of ap~ 'y 40 C and +20 C. The amino proteeting group
Pr~ is removed with a suitable reagent ailowing the N-~ lu~,ua~' ' eompound thento be substituted at the 4 and 6 positions, as previously deseribed, ie. alkylation
followed by a series of reaetions whieh introduce the desired ,,
groups at these positions.
P~Ok~ Y-NH-P- 1 Pr2Ok~ Y
OPr 2 pr2O
SUBSrlTUTE SHE~
woss/14027 676Q~ , PCTICAs4/00636
As set out in formula 1, X may be Qrbon, oxygen, sulfur or nitrogen atom. In themost preferred . ~ , X is an oxygen atom, and the Rl, R2 and R3
represent groups that, combined with the oxygen-containing ring, form a substituted
sugar backbone, i.e. a substituted pyranose such as a pyranoside. In this regard,
particular . ' in à~ll' with formula (I) include:
4,6-di-0-ylu~ l;d;l.~,-2,3-dideoxy h_AU~;dC,
methyl4-O e.}~'g ' -6-0-1JlU~ " ' -2,3-dideoxy-hexoside;
methyl-4-O-yluy ,1g ' 6-0 ell.~',g " ~-2,3-dideoxy-hexoside;
methyl-4,6-di-O ~'g " -2,3-dideoxy h_Ao~i~,
methyl4,6-di-O-ylu~!" ' -2,3-dideoxy ~._Au,ide,
ethyl-4,6-di-O e~h.~'g ' -2,3-dideoxy-hexoside;
ethyl-4,6-di-O-l,luyy'g " -2,3-dideoxy h_Au~id~,
ethyl-4,6-di-O-' .~,~" ' -2,3-dideoxy ~_AU:~;d~,
propyl-4,6-di-O e;ll.~'g " 2,3-dideoxy '- ",
propyl-4,6-di-O-yluy)!" ' ~-2,3-dideoxy ~_AU:~id~.,
propyl-4,6-di-O ~ , ' 2,3-dideoxy h_Ao~;.l~,
~ hyl ~lidill~4,6-di-O-I~luy ~ ~, ' -2,3-dideoxy-hexoside; and
ylu~ , " ~,6-di-0-el~ ' -2,3-dideox~ ' '
Such ~ , ' may be made using suitable sugar-based starting materials, such as,
for example, a protected gluQI. To provide an ill~. ' which is substituted at
position 1, the gluQI is reacted with the ayyLl r ' ' alcohol to yield an ~,~IV~ ~lallu~id~
substituted at position 1 by the d~ ' alcohol. The ~,I.UyJl. Iu,id~ is then
prepared for ' at positions 4 and 6 by alkylation at these positions, followed
by a seri~s of reactions which introduce the desired g ' substituted groups at
these positions. Included within these process steps is the ll.ydll" of the
double-bond between C2 and C3 of the ~,lluy.ylallu~id~, to yield a fully saturated
SUBSmUTE SHE~
WO 95114027 2 ~.~ 67 6 PCTICA94100636 ~
hexoside. Figure 1 illustrates the process steps for the synthesis of the compound,
methyl-4,6-di-0-~u.uyy'g ~ 2,3-dideoxy-hexoside, whieh is described in detail
in the specific examples. One of skill in the art will appreciate that similar process
steps can be applied to a~plu~ starting materials to yieid other sugar-based
~, ~ , ' such as propyl-4,6-di-0-,uluL,~'g - -2,3-
dideoxy-heAoside. the process for making which is described herein in Example 2.
Moreover, in accordance with further ~ of the present invention,
~IJIULJ I.~.~.u~,y~ starting materials can be used to prepare l;UIl~"lJUll~
substituted piperidine-based I , ' in which X is nitrogen, ~y~,lull~.A.~ -based
in which X is carbon and L~LI- ~y ilu~iliu~ ul-based . , ' in which
X is sulfur. For example, when X is sulfur, 5-tilio-D-glucose (Aldrich catalogue,
item no. 85,986-9) may be used as a starting materiai and when X is nitrogen, 1-~i~VAyllUji~ y~.;ll may be used, the synthesis of which is described in Shankar et ai,
Tetrahedron Letters, 1993, 34(45):7171. TT ' ~ of the oxygen-containing
Iisted above are prepared from these starting . ' by the steps:
I) protection at the 4 and 6 positions; 2) dcvAy~ followed by reduction at
positions 2 and 3; 3) oAidation followed by alkylation at position 1 to yield the
desireti Rl ' , 4) d~lu~,Iiull at positions 4 and 6 followed by alkylation
with N-Boc-aminoali~lblu.llide, hydrolysis of the N-Boc group to obtain free amino,
I,, r ,, ,, ~ ;,, of the free amino to bis-Boc-guanidino and hydrolysis to yield free
guanidino. Steps 1) to 4) may aiso be uscd to obtain l.... ,nlu~, ~ wherein X iscarbon from a suitable starting compound such as ~ l (Aldrich catalogue7
item no. 36,û6û-û) or inositol. In this case7 a carbon-carbon double bond is
introduced at position 5 prior to the protection step 1) by established techniques such
as the classical Wittig reaction or ulL llic chemistry.
As will be ~.1 7 by one of skill in the art7 17 of , ' of the
present invention may be separated using techniques well-established in the ar~, for
eAample, silica gel column ' ~ ~ A 7 Y-
Equivaient forms of , 7 according to formula (1), such as saits, solvates and
SUBSTITUTE SHE~T
= ~
WO95/14027 7676D PCT/CA94100636
hydrates, are also included within the scope of the present invention, Acid additionsalts are suitable and are formed with inorganic acids such as llydlu~hluric acid,
h~d~oblullli~i acid, sulfuric acid, nitric acid, I ' , ' i~, acid and the like, and organic
acids such as acetic acid, propionic acid, glycolic acid, pyruvic acid, oxalic acid,
malic acid, malonic acid, succinic acid, maleic acid, fumaric acid, tartaric acid, citric
acid, benzoic acid, cim]amic acid, mandelic acid, ~ acid,
; ~ ~r ' acid, p r ' acid, salicylic acid and the like. The
of an acid addition salt comprises admixing a compound of formula (l)
with the organic or inorganic acid. To convert an acid addition salt of a compound
to an alternative salt, an aqueous solution of the salt is treated with base, e.g. sodium
carbonate or potassium hydroxide, to liberate the free base which is then extracted
using an a~lJIUIJ ' ' solvent such as ether. The free base is then separated from the
aqueous portion, dried and treated with the ~/t)IUlJI' ' acid to yield the desired
alternative salt compound.
Once a compound according to the present invention has been ~ ' d, it is
desirable to analyze the compoumd furtber to ensure its chemical ' ~ and
purity. Typically, methods of a~ u~w~ are used to confirm the identity of a
compound including, for example, infra-red, ultra-violet, nuclear magnetic resonance
(NMR), electron spin resonance (~SR), and mass *~ .tlU~W~Jy (MS).
Having confirmed the identity of the , i, prior to its use as an anti-viral agent,
is purified to F ' grade~ to meet standards set by the various national
bodies which regulate quality of I ' I products. In this regard, it will be
,, ' that strict standards of purity may not be required for use of the present
' in vitro or in ~ , for use in the veterinary field. Any one of a
number of .. ' ~ procedures may be used to attain the required
purity. Reverse phase high-pressure liquid .,h., ,, ,' ~ (RP-HPLC), for
example, is one method that is commonly used to purify end products. r :-
of; , in the product mi~cture is generally , " ' ' by running a lineargradient, e.g. a mobile phase , ~ an increasing percentage of organic solvent
such as ', in an aqueous buffer usually containing a small amount of an ion-
SUBi3TlTUTE SHE~
WO 95/14027 ~ 2 ~ 7 ~ 60 PCTICA94100636 ¦~
10pairing agent such as 0.1% j .r. u~.i., acid (TFA), through alkylated silica
columns, e.g. C~-, C8-, or Cl8- silica. A ~ .,ly, metbods such as ion-exchange
clu~ may be used to purify the product.
Purified, I' I grade O ~ of formula (1), or
salts, solvates or hydrates thereof, are used to prepare anti ~ v i~ --r
according to an aspect of the invention. These ~ comprise a suitable
carrier and an effective amount ûf a ~ " . substituted . . 1, both of which
are of 1 ' ' grade as defined above The expression "an effective amount"
is meant to encompass amounts of the anti-viral compound sufficient to prevent or
cause a reduction in h~ vi~u~ ll . ' Such ~ have use in
preventing growth of ll.,li, vuu~, i.e by inhibiting viral replication, both in vitro as
well as in vivo. In an e~ample of an in vitro srrlirs~inn the anti l~ ~viuu~
may be added to cell culturing medium to prevent undesired 11~ vi~
Orowth during culturing.
Anti-ll~,lpwvil~ . for in vivo ' i.e. for treating infected
mammals, are also provided . i~ o at least one ~ u liy acceptable
carrier and a i' _r lly effective amount of a compound in accordance with the
invention. In this context, the term "I ' ~ly acceptable" means acceptable
for use in the pllal ' and veterinary arts, i.e. non-toxic and not adversely
effecting the anti-viral activity of the ~ . ' of the present invention. The term
effective amount" means an amount of the compound sufficient to
cause a reduction in the replication of the h~li, Vill... target in the infected mammal
without eliciting toxic or side effects intolerable by the recipient. Such reduction is
most properly revealed by assaying virus titer in serum or urine samples obtained
from the recipient before and after treatrnent. Further, as used herein, the term
rmammal" is meant to encompass humans, domestic animais such as cats, dogs and
horses, livestock such as cattle, pigs, goats, and sheep, and non-
~
mammals that may be in need of anti h_ll.~vu~ treatment.
rl,- ., - . .~ y acceptable carriers for inclusion into the present ~u~
SUBSTI~UTE SHE~
~ WO 95114027 76 760 PCT/CA94100636
include W.~ ull~d carriers generally selected fûr . ' with sugar-based
drugs such as diluents, e~cipients and the like. Reference may be made to
~Remington's r~ ' Sciencesr, 17th Ed., Mack Publishing Company,
Easton, Penn., 1985, for guidance on drug ~u. ' generally. As will be
i, the I ' ' carriers used to prepare r ' ' in accordance
with the present invention will depend on the _ ' ' ' form required to treat a
given }.~.~,.,,viuu~ infection. For example, herpes simplex virus manifests as
e~tternally-occurring lesions which may be treated topiQlly; however, some conditions
caused by l~ iU h,~ cause internal conditions which are best treated by
fi.. . ~ which are ' ' 'o, for example orally or by injection.
According to one ' ' of the invention, the, . ' are formulated for
r' by injection, either sub- ~/ or illLI~ Ju~ly, and are
~uldil~;ly provided as aqueous solutions in soerile or pyrogen-free form and
optionally buffered or made isotonic. Thus, the . ' may be ~ ' ' in
distilled water or, more desirably, in saline or 5 % dextrose solution. The .~, l v . I~
herein designated as preferred, . ' are ' 'Iy water-soluble. Water
solubility of these and other ~ A ' of the invention may be enhanced, if desired,
by i.l~.ul~ul~Li..t~ a solubility enhancer, such as cc~ ' bromide or
chloride. LYUt~ such as mannitol, sucrose or lactose and buffer systems,
such as acetate, citrate and phosphate may also be included in the ' ' as may
bulking agents such as serum albumin.
Al~.l~ii~.,ly, the ~ of the present invention may be formulated for
by routes other than injection. Oral dosage forms, such as tablets,
capsules and the like, formulated in ' with standard I ' ' practise,
may be employed. C~ . for topical 3~. ' such as eye drops, creams,
lotions or ointments can also be used, as may aerosol inhalable ' ' Cream,
lotion and ointment ' ' will be useful ~uLiuul~ly for zrplir~irn to virally-
induced skin lesions. Appropriate Ll i~ ly~. i~i~, bases and gels can be used to prepare
creams and ointments, which may: '' "!r include surfactants and ~ '
agents as is W.A~. '' I
SU13STITUTE SHE~
WO 95/14027 PCTICA94/00636 ~
2l7676~
'~he present invention provides, in another of its aspects, a method for treating a
L~vil~ infection in a mammal which comprises the step of a ~ to the
mammal a 1 ' ' ' ~ , in a form suitable to treat the infection, which
comprises a ' r '' 'Iy effective amount of a compound of the present invention
in r.."~h~ ;.... with a ~ y acceptable carrier. According to one
I ' ' of the invention, tne method is applied for the purpose of treating a
human patient diagnosed as having a l~ ,~vi l.~ infection. Suitable treatment
regimens are those which maintain at the desired site, e.g. in the infected tissue or
at a localized skin surface, an amount of the compound sufficient to control
~ J~vil~ rPrlir~tinn The precise dosage sizes a~ JI for treatment can
readily be established in a~ Jpl' ~ controlled trials, and will correspond to anamount of anti-viral compound that affords effective results against l~ vilh~
replication without causing any harmful or deleterious side effects to the host being
treated. It is anticipated that an effective treatment regimen for patients infected with
a l~ ~v il b~, will involve the systemic - ' of dosage sizes in the range of
from 1 ~Lg to about 10 mg per kg, e.g., between about 0.01 mg/kg to about 5 mg/kg.
It will be a~ , however, that effective dosage sizes will vary according to the
route of: ' ' ' the frequency of: ' and, of course, with the
particular host to be treated. For example, topical ' ' , which remain
localized at the infected site, may be ' ' less frequently than '
which are - ' c~ by injection.
Specific ' " of the present invention are described in more detail in the
following examples which are not to be coristrued as limiting.
F~ nlr 1 - S ' of Mrth~yl~.~di-O-r~~ h ' 2.3-dideoxy-r~ n~D
erythro-hi~ynr~
'l'he compound, methyl4,6-di-0-p-.~ '' -2,3-dideoxy-~,B-D-erythro-hexoside
was prepared as indicated in Figure 1, and as set out in detail in the following:
A: In a furst step, methyl 1,6-di-0-acetyl-2,3-dideoxy-hex-2~..~,~,y ' ' was
SUBST~TUTE SHE~
WO 95114027 ~ ~ 6 a PCTICA94100636
13
prepared as follows. To a stirred solution of tri-O-aoetyl-D-glucal (obtained from
Aldrich) (6 g, æ.o38 mmol) in dry benzene (20 ml) was added 2.2 mL of methanol
and 1.5 ml of boron 1- '' i~ cther. The reaction mixture was kept under nitrogenfor 45 min., and was then washed twice with saturated aqueous sodium bi~u~
and water, and dried with MgSO~. Removai of the solvent yielded methyl-4,6-di-O-acetyl-2,3-dideoxy-~,B-D-erythro-hex-2 e..vl,y.d.lu~iJc in the form of a colorless oil.
The product was purified by flash ' I ~ ' y with an ethyl r
(70:30) eluant.
B: Themethyl-2,3-dideoxy-~Y,B-D~rythro-hex-2 e.lv~ -u~ , r~ was
prepared next. The methyl-4,6-di-O-aoetyl-2,3-dideoxy-~Y,B-D-erythro-hex-2-
c..v,u, ' product of step A (2 g, 8.926 mmol) was stirred in 45 ml of dry
methanol. A catalytic amount of sodium methylate (240.9 mg, 4.463 mmol) was
added to the stirred solution and stirring at room . . was continued for I
hour. The solvent was evaporated and the remaining residue was diluted with ethyl
aoetate. This mixture was washed twioe with brine, dried over MgSO~ and the
remaining solvent was e~ Jl ' The resulting syrup was purified on silica gel
using an ethyl aoetate/60% hexane eluant to yield as a colorless oil the methyl-2,3-
dideoxy-~,B-D-erythro-hex-2~..v,uy-~wsidc ;-IL.I '' '
C: Methyl-4,6-di-O-[I'-N-Boc-~lup.~' ]-2,3-dideoxy-~,B-D-erythro-hex-2-ellv~ allu~ " the third ' in the pathway, was prepared by stirring
methyl-2,3-dideoxy-c~,B-D-erythro-hex-2-e..v~,,.~,v~ from B) (Ig, 6.248 mmol)
in 31 ml of dry i ' ,~ and then adding thereto freshly powdered potassium
hydroxide (1.4 g, 24.992 mmol), 18-Crown-6 cataiyst (660 mg, 2.499 mmol) and 1-
N-Boc-amino ~u~liJIl ' (3.7 g, 13.745 mmol). The reaction mixture was stirred
at room i , ~ until ail starting material was consumed as indicated by TLC
(approx. 2 hrs.). The mixture was then diluted with ethyl aoetate and washed several
times with water. The desired dialkylated : ' (crude) was obtained
following drying of the organic phase over MgSO~, and e~ vl.lLiu.. of remaining
solvent. rl ~ of the product was conducted on silica gel using ethyl
h.~ (50:50) as the eluant.
SUBST-iTUTE SHE~T
W095114~27 2 ~ 6~ 6 ~ `~ PCTIC~94100636--
. ~ l
14
D: T.'~.e next i.., 1; t~ in ti.e pathway, meti~.yl4,6-di-O-[1'-N-Boc-
~u~ lc]-2,3-dideoxy-~,B-D-erythro-hexoside, was prepared as follows. Ti-.e
diaiicylated product from C) (750 mg, 1.581 mmol) was dissolved in 10 mL of dry
eti.yl acetate. To ti-.e solution was added 50 mg of 10% Pd/c suspended in 5 ml of
ethyl acetate. Tl.e reaction mixture was stirred at room i , c under a bailoon
of hydrogen for 2 hours, and then filtered through celite. The filtrate was
' and passed through a short column (approx. 2-6 cm in lengti.) of silica
gel using an ethyl ~ ' (50:50) eluant. Th.e desired lly~ , ' product
was obtained as a coloriess oil.
E: Meti-.yl-4,6-di-0-,.1u,,,~ 2,3-dideo,cy-~ -D-erythro-hexoside was
prepared from the hy~ " ' product of D) which was dissolved (170 mg, 0.357
mmol) in 0.5 mL of 3N HCI in eti.yl acetate and stirred at room t~ iUlC for 2
hours. The solution was removed under vacuum and the resulting oil was triturated
with dry ether followed by C~ The crude sait was converted to the
Wl r ' ~, free base by passage through an ion exchange resin column (Amberlite
li~A400 OH) using pure metl.anol as the solvent. Ti~.e f.-ee base was obtained as a
white foam.
F: Methyl4,6-di-0-[di-Boc-,~lu,!rlg ' ]-2,3-dideoxy~,B-O-erytb.ro-hexoside
was then prepared from ti~.e product of E) as follows. To a stirred solution of methyl-
4,6-di-0-~1u~yl u~ ,-2,3-dideoxy-c~,~B-D-erythro-hexoside (100 mg), 0.359 mmol)
in dry DMF (1.5 ml) was adde~i 1.5 ml of dry ~ y' Following 15 minutes
of stir.-ing at room i r ' C~ a solution of di-Boc-thiourea (198.5 mg, 0.719
mmol) in 0.5 mL of dry DMF was added dropwise. The reaction mixture was stirred
at room . under argon ~ llu.~h_lc for 20 hr. T.'.e mixture was then diluted
witi~. ethyl acetate, quenched with water and washed twice with brine. The organic
phase was dried over MgSO~ and the solvent was c~ The crude di-guanidino
product was purifled on silica gel using eti~.yl I (40:60) as the eluant to
render a white foam.
SUBSTITUTE SHE~
WO 95/14027 ~ 7 6 o PCT/CA94/00636
G: Methyl-4,6-di-O-propylguanidine-2,3-dideoxy-~,B-D-erythro-hexoside
1' -' ,Y~IU~ IVI i~, the desired product, was made as follows. The methyl-4,6-di-O-[di-
Boc-yluy~" ' ]-2,3-dideoxy-~,B-O~I,Y' ~,_Aosid~ product of F (200 mg,
0.263 mmol) was dissolved in 6 ml of 3N-HCI in ethyl acetate, and the reaction
mixture was stirred at room h --r ' C; for 2 hours. The solution was removed
under vacuum and the resulting oil was triturated with dry ether. The solvent was
evaporated from the product to yield methyl-4,6-di-O-yluyy'& ' -2,3-dideoxy-
cY,B-D-erythro-hexoside in the form of a white solid. The melting point of the
product was ~' ' to be 135-139qC.
F-q-T~71P 2 - Sy~pcic of 1.4.6-tri-O- -2.3-~ lPnYy-hPYnoi~ip
Synthesis of the title , 1, 1,4,6-tri-O-yluyJI,, -2,3-dideoxy-hexoside
was essentially the same as the synthesis outlined in E~ample I for the compound of
example I with tbe following exception:
A: To a stirred solution of tri-O-acet,vl-D-glucal (6 g, 22.038 mmol) in dry
benzene (20 ml) was added 2.2 mL of l-N-Boc-propanol and 1.5 ml of boron
trifluoride-ether. The reaction mixture was kept under nitrogen for 45 min., and was
then washed twice with saturated aqueous sodium 1,;~1 and water, a nd dried
with MgSO~. Removal of the solvent yielded N-boc-O-propyl-4,6-di-O-acetyl-2,3-
dideoxy-~,B-D-erythro-hex-2-~.1vy~..l.lu~i~e in the form of a colorless oil.
Tbe remaining steps B through G were conducted as described in Example 1 to yield
the product 1 ,4,6-tri-O-ylu~,J 'g " -2,3-dideoxy-hexoside.
F~q~,nlP 3 - J ' ' of Tr u~ ~Yil~ - I ' " bv the ~ I of
The compound of example 1 was formulated as a 10 mM stock in water for in vitro
and cell culture procedures. The stock was then diluted into buffers used for specific
assays. or into cell culture media.
The following procedures were then used to determine inhibitory effects of the
compound of example I on the replication of HSV-1, strain F. First, confluent
SUBSTITUTE SHE~
WO 95/14027 2 ~ ~ 6 7 6 ~ PCT/CA94/00636 ~\
16
of the African green monicey kidney cell line, Vero (ATCC 733-VR CCL
81) in 24 well cell culture plates were pretreated with specifed ~ (0 ~lM,
0.5 ~M, 1 ~M, 5 ~M, 10 ~M, 20 IlM and 50 ~M) of the compound at 37C, 5%
CO1 for 24 hours. This was ~' ' ' by diluting the stock solution of the test
compound in growth medium (10% fetal bovine serum, lOOlU/ml peniciilin,
100,ug/ml . ~, and 0.02mg/ml gentamicin in Dulbecco's MEM (DMEM))
useci to overlay the IllUIIUl~
After l~IL.lti ' ' with the tcst compound, the ' ~...D were overlaid with 0. Imlof diluted virus stock at about 20 pfu/well. Virus was then allowed to adsorb for I
hour at 37C. The virus inoculum was removed and the ' ~ were overlaid
with DMEM containing 2 % FBS and the specified ~ of the test
compound. Virus was next allowed to replicate for 48 hours and then the l~wllol~were fixed and stained with a solution of 1% crystai violet in 70% eti]anol: formaiin
: acetic acid ao 2 l)- Finaily, plaques (each lq~ , a single infectious virus
particule or plaque forming unit (PFU)) were counted and checked lll;Llu~l,u~;~ily.
The results were as follows:
CONCENTRATION OF PI~QUE
COMPOUND ~M) NIIMBER
16
0.5 18
O 19
The IC,o was caiculated to be 2.6 ~M.
The results show that incubation of the cells with the O ' , ' ' compound
of example I in a . of more than I ~M induces significant reduction in
SUBSTITUTE SHE~T
wo 95/14027 ~ 7~o PCT/CA9410~636
17
the replication of herpes simplex virus. The vh~la~ , index of this rn~rol~nA, i.e.
the ratio of its CJtvtuAi~ y to its virai toxicity, was ~IPtPrminP~ to be 12.29
4 - I of IT~ vi., - l~P.i~lir~rinn by thP ~ ~ of P~
A procedure similar to that described in Example 3 was conducted to determine the
anti-virai activity of the compound of example 2. In this case, the virai host was
human embryonic lung line MRC-5 (ATCC CCL 171) and the ~ of the
test compound used to pretreat the MRC-5 cells were 0 ~uM, 1 ~M, 10 yM, 100 f.M
and 500 ~M, l~d,u~Li~,ly. The results showed that incubation of the cells with the
Example 2 compound in a of 1 ~M or more induces significant
reduction in the replication of herpes simplex virus (ED50 = 10.51 ~M). The
therapeutic index of this compound was ~PtPrrninPd to be > 50.
P 5 - Syr~hPcic of Pthyl-4.6-di-O-v~uv~ .3-riiAPn~y_hP~nci~ip
The compound ethyl4,6-di-0-1,.u~lg '~ 2,3-dideoxy-hexoside was prepared as
outiined in Figure 2.
A: i?~ alaliùll of ' (ethyl-2,3-dideoxy-D-erythro-hex-2-
CIIUIJ ~ . u.u,id~) _
To a stirred solution of ethyl-4,6-di-O-acetyl dideoxy-erythro-2~.,~ ...u~;dc 1 (2g,
7.744mmol) in dry metilanol (40ml), was added a catalytic amount of sodium
methylate (168mg, 3.097mmol). The reaction miAture was stirred at room
~IIItJ~l~l~UlC for 2hrs. The solvent was removed and the remaining residue was
diluted with ethyl acetate (200ml). This mixture was washed twice with brine. The
organic layer dried over MgSO4 and the remaining solvent was c~. ' The
resulting syrup was purified on silica gel column using an ethyl acetate/hexane (60%)
as eluant to yield as a white solid (mp 65-67'C) vhe ethyl-2,3-dideoxy-D-erythro-hex-
2-~ U~ (82%)-
B: F~Jal~lliUII of ' ethyl-4,6-di-0-[1'-N-Boc-l,lu,u~ llill.,]-2,3-
dideoxy-D-erythro-hex-2-~..u,u~ ' 3
To a stirred solution of ethyl-2,3-dideoxy-D-erythro-D-hex-2-~,1lu~
SUBSTlTUTE SHE~
~1l4027 ?,~ 6~ 6 ; . PCT/CA94/00636
18
(770mg, 47370mmol) in dry THF (22ml), were added au~ / freshly powderedpotaDsium hydroxide (980.8mg, 17.480mmol), 18-crown-6 catalyst tS77.5mg,
2.185mmol) and 1-N-Boc . ul~lvlu... Iu (2.63g, 9.614mmol). The reaction
mixture was stirred at room; , ~ until all starting material was consumed aD
indicated by TLC (2hrs). The mixture was then diluted with ethyl acetate and washed
several times with water. The organic layer was dried over MgSO4 after l,~ Uld~iU
of the solvent, the crude syrup product was purified on silica gel column using 50%
ethyl: t~_~.dll~, as eluant to give as a colorless syrup the ethyl~,6-di-0-[1'-N-
Boc-,ulut,~' ]-2,3-dideoxy-D-erythro-hex-2 UIIU~ IIVD;~L 3 (65%).
C: P~q~udLiull of ' ethyl~,6-di-0-[1'-N-Boc-u,u~,~ ]-2,3-
dideoxy-D-erythro-hexoside
Ethyl-4,6-di-0-[1'-N-Boc-,ulu~ ]-2,3-dideoxy-D-erythro-hex-2-~llu~ uluDid~
(lg, 2.046mmol) waD dissolved in 15ml of ethyl acetate. To the solution was added
8ûmg of 10% Pd/C suspended in Sml of ethyl acetate. The reaction mixture was
stirred at room i , .i under a balloon of hydrogen for 2hrs, and then filtered
through celite. The filtrate was: ' and passed through a short silica gel
column using an ethyl acetate hexane (50:50) eluant. The desired ll.vd
compound was obtained as a colorless syrup (approx. 100%).
D: rl~U~i~liull of " ethyl-4,6-di-0-,ulu~ ullil~-2,3-dideoxy-D-erythro-hexoside
Ethyl ~,6-di-O-[l'-N-Boc-P~u~ ]-2,3-dideoxy-D-erythro-hexoside _ (9SOmg,
1.936mmol) was dissolved in 9.6ml of 3N HCI in ethyl acetate and stirred at roomL~ for 2hrs. The solution was removed under vacuum and the resulting oil
was triturated with dry ether followed by ~UI..Liu... The crude salt obtained was
converted to the UUII~ ,, free base by passage through an ion exchange resin
column (Amberlite IRA~OO OH) using pure methanol as solvent to leave the free
base as a cûlorless syrup (83.8%).
SUBSTITUTE SHE~T
wo 95114027 6 7 ~ o PCTICA94100636
E: r., of " ethyl4,6-di-0-[bis-Boc-~u~y~,, " ]-2,3-
dideoxy-D-erythro-hexoside 6
To stirred solution of ethyl-4,6-di-O-I,Iu~ 2,3-dideoxy-D-erythro-hexoside _
(460mg, 1.504 mmol) in dry DMF (7ml) was added 2.8ml of dry Lli~,il.~' .
After 15 minutes of stirring at room i , , a solution of bis-Boc-thiourea
(962.2mg, 3.484mmol) in 2ml of dry DMF was added dropwise. The reaction
mixture was then stirred at rcom i ~ c under argon a~lllua~ for 20 hrs.
The mixture was then diluted with ethyl acetate quenched with water and washed
twice with brine. The organic layer was dried over MgSO4 and the solvent was
the crude product was purified on silica gel using ethyl ~ h~ nP
(40:60) as the eluant to yield a white foam (75%).
F: rltr of final product ethyl-4,6-di-0-1,.u~"'g " 2,3-dideoxy-
hexoside
The ethyl-4,6-di-0-[bis-Boc-p.up.~l,. ' ]-2,3-dideoxy-D erythro-hexoside 6
(llOmg, 0.141mmol) wa_ dissolved in 1.5ml of 3N.HCI in ethyl acetate, and the
reaction mixture was stirred at room t~ under argon ailllu~ for 2 hrs.
The solution was then removed under vacuum and the resulting oil was triturated with
dry ether. The solvent was evaporated from the product to yield the final product
ethyl~,6-di-0-~.u~ " ' -2,3-dideoxy-hexoside as a white foam (98%).
r ~,nle 6 - Syn~h~cic of ~ yl-4.6-di-O-v~ , " -2.3-di~ v-hexoside
The compound ethyl-4,6-di-0-~.,1lL~,, " 2,3-dideoxy-hexoside was prepared as
outlined in Figure 3. The synthesis of which was essentially the same as the synthesis
outlined for the compound of example 1.
A: F~aiiull of ' 5-bis-Boc-guanidine-1-pentanûl 5
To a stirred solution of ~i-amino-1-propanol _ (934ml, 9.053 mmol) in dry DMF
(23ml), was added 6.3 ml of dry l-;~ ' After 15 minutes of stirring at room
h~ i, a solution of bis-Boc-thiourea (2.5g, 9.053 mmol) in 5 ml of dry DMF.
The reaction mixture was then stirred at room i ~ c under argon ~ , ' ci
for 4 hrs. The mixture was then diiuted with cl~y'; , quenched with water and
SUBST~TUTE SHE~
WO95114027 ~17 6 7 6 0 PCT/CA94/00636 ~
washed twice with brine. The organic layer was dried over MgSO4 and the solvent
was removed. The crude product was purified on silica gel column using
cLh~'; ' , (40:60), as the eluaut, to yield 83% of bis-Boc guanidine
compound as a white foam.
B: r~cip~ualiull of 5-bis-Boc-guanidino-l-~
To a chilled (0C) solution of Lliph.,..~l~lluat/ll;l.~, (2.27g, 8.683 mmol) in dry CH2CI7
(60 ml) was added dropwise a solution of bromine (4~L7~1, 8.683 mmol) in CH2C12
(20 ml). A solution of 5-bis-Boc-guanidino-~ lv~ull~iJe _ (2g, 5.789 mmol) in
CH ,CI2 (20ml) was added dropwise and stirred 4 hrs. at room ~~ ,l dLulc. Saturated
K2CO3 (3ml) was added and the mixture was pertitioned between water (50ml) and
ether (SOml). The organic layer was separated, and the aqueous layer was extracted
with ether. The combined organic layers were dried (MgSO4), diluted with an equal
volume of hexane, and passed through a short column of silica gel. Removal of the
solvent gave 84.7% of bromide _ as a white powder.
C: r~c~udLiu.. of ' ethyl4,6-di-0-bis-Boc-!,~,...y~" ' 2,3-
dideoxy-O-hex-2-~1lvp.,.~ s;Je_
To a stirred solution of ethyl-2,3-dideoxy-D-erythro-hex-2 el.v~,J.dll~sid~ 1(143 mg,
o ~? I) indryTHFwereadded au~ ai~ powderedpotassiumhydroxide(185
mg, 3.294 mmol), 18-crown-6 catalyst (87mg, 0.32mmol) and bis-Boc-
. ~ lb~u~ilc _(672mg 1.6~L7mmol). Thereaction mixture was stirred at
room ~I.p~,l..~.C until . of total starting material as indicated by TLC
(~2hrs). The mixture was then diluted with cLII.~'; and washed several times
with water. The organic layer was dried over MgSO4. After ev~ . the crude
product was purified on silica gel column using cLh~'; ' , (30:70) as the
eluaunt, to afford as a white foam the ethyl 1,6-di-0-bis-Boc-~ 'g ' -2,3-
dideoxy-O-hex-2~l.vpy ' _ with a 25 % yield.
D: rlcpal~liiu.. of " ethyl4,6-di-0-bis-Boc-~ ulid;ll~,-2,3-
dideoxy-O-hexoside _
Ethyl4,6-di-0-bis-Boc-~ 'g ' -2,3-dideoxy-0-hex-2 e.lU~JldllUa;J~ 2 was
SUBSrITUTE SHE~T
~ WO95/14027 6760 21 PCrlCA94100636
dissolved in lSmi of ethyl acetate. To the solution was added 80mg of 10% Pd/C
suspended in 5ml of ethyl acetaoe. The reaction mi~ture was stirred at room
~ under a balloon of hydrogen for 2ilrs, and then filtered through celite.
The filtrate was ' and passed through a short silica gel column using an
ethyl acetate hexane (50:50) eluant. The desired llyJIl~ ' compound was
obtained as a colorle s syrup (78.5%).
E: rlc~alaliull of final product ethyl4,6-di-O~ 2,3-dideoxy-
hexoside 4
Ethyl4,6-di-0-bis-Boc-~ 'g " 2,3-dideoxy-O-hexoside3wasdissovedin3N
HCI ethyl acetate and stirred at room ~.I.p.,l~h~; for 2hrs. The solution was
removed under vacuum and triturated with dry ether followed by ,va~Ju-aliul- to give
the final product (86.9%).
r ~ - 7 - Syn~hf~cic of i~u~u~yl ~ .6-di-O-l~ , -2.3~ nYy-D-erythro-
h~
The titie compound was prepared as outlined in Figure 4. The synthesis was
essentially the same as the synthesis described for the compound of example 6, except
at the following steps:
: Preparation of " isopropyl4,6-di-O-acetyl-2,3-dideoxy-D-erythro-2-
allv~idci 2
To a stirreci solution of tri-O-acetyl-D-glucal (6g, 22.038)mmol) in dry benzene(20ml) was added (3.3ml, 44.076mmol) of 2-propanol. To the solution obtained wasa~ided boron trifluoride ether (l.Sml) The reaction mixture was stirred under argon
aL~Ilu~ ; at room i ,; .; for 45 minutes and then diluted with ethyl-acetate
and washed twice with saturated aqueous sodium L.;~L and water. The organic
layer was dried (MgSO~) and evaporated to afford the crude isopropyl-4,6-di-O-
acetyl-2,3-dideoxy-D-erythro-2 e~u~la~lui~;Ju as a colorless oil, which was purified
by flash ', ~ using ethyl - ' '?0:80) as eluant (88.3%)
SUBSrITUTE SHEiET
~1 ~ 6 ~ 6 0
WO95/14027 ,~ PCI~ 94/00636--
22
B: r . of ' isopropyl-2,3-dideoxy-D-erythro-2-c.. v~ u~idc~
The isv~ i,6-di-O-acetyl-2,3-dideoxy-D-erythro-2 cl~v~Jy~ v~icl~, 2 (5.2g,
lg.097mmol) was stirred in (lOOml) of dry methanol. To this solution a catalyticamount of sodium metilylate (515.8mg, 9.54mmol) was added and the stirring was
continued at room i , for overnight. The solvent was evaporated and the
remaining residue was diluted with ethyl acetate. This mixture was washed twice
with dried (MgSO") and ev_r ' The resulting syrup was purifed on silica gel
using ethyl _ h~-~n-~ (60:40) as the eluant to yield isopropyl-2,3-dideoxy-D-
erytilro-2-e..~.yylallv~ 3 as a white powder (76%).
py~ nl~8-syr~hr~cicof(~lhrly~y~thrlyy-ethyl)-4~6-di-o-~lu~y~ ;- -2.3~iiriPn~;~V-
d-erythro-h~ ~nQi,iP
The title compound was prepared as outlined in Figure 5. The synthesis was
essentially the same as the synthesis described for the compound of example I with
the exception that in the first step A, 2-(2-ethoxy ~ ' y).,~lalwl was used in place
of MeOH to give the " (ethoxy-ethoYy-ethyl)4 ,6-di-O-acetyl-2 ,3-dideoxy-
~-D-ery~ro-hex-2~ lu~Jylallvsid
r 1- 9 - S ~ of 4.6-di-0-vlu~y;~ ~ - 1,... 3-deox~y dihy~iro-D-~'
The titie compound was prepared as outlined Figure 6:
A: P~c. of ' dihydro-D-glucal_
A mi~cture of D-glucai 1 (Sg, 34.241mmol) was stirred in dry ethanol (170mL) with
catalytic amount of Pd/C (10%) (400mg) at room i~ ' c, under hydrogen
, ' c for 2 hrs. The catalyst was removed by filtration on celite. The solvent
was evaporated off and the residue was passed through a short column of silica gel,
using EtOAc-MeOH, 95:5 as eluant to give the title compound 2 as a colourless syrup
with r~ iialivc yields.
SUBSTITUTE SHE~
WO95/14027 1 76760 PC~/CA94100636
B: P,, of ' 4,6-0-bcl~y;id~ dihydro-D-glucal 3
To a miAture of dihydro-D-glucal 2 (3g, 20.267mmol), N,N~li..._llylrul '
(50ml), and benzaldehyde ." ' ~daL~ al (lSml, 101 .334mmol), was added a caulytic
amount of p-~..l. - ~l~....;- acid (771mg, 4.0S3 mmol). The reaction mixture wasstirred at room i . and under argon , ' c for 24 hrs. The solution was
then diluted with ethyl acetate (200ml), and quenched with water (lOOml). The
aqueous layer was eAtracted several times with ethyl acetate. The combined organic
layers were washed ~u~~ ly with saturated aqueous NaHCO3, and brine, and
dried (MgSO~) The solvent was removed, and the remaining grade product was
,, , ' ' on silica gel column using ethyl h_A~ulc~ (50:50), to give
4,6-0- ~l~ylid~ dihydro-D-glucal as a colorless syrup (75% yield).
C r~c~JauaLioll of 4.6-0 ~ ~ylidl,llc 3- benzyl dihydro-D-glucal _
To a stirred solution of 4,6-0-b.,~yli~ dihydro-D-glucal (lg, 4.462mmol) in dry
THF (22ml), were added ~u~i,i,i~,ly freshly powdered poussium hydroxide
(500.7mg, 8.924mmol), 18-crown-6 catalyst (589.7~1, 2.231mmol), and
lbl~ ' (636.8~1, 5.354mmol). The reaction mixture was stirred at room
c under argon ,' c until all starting material was consumed, as
indicated by TLC (~ lhr.). The reaction mixture was then diluted with ethyl acetate
and quenched with water. The organic layer was dried over MgSO~ and ~
to give the crude product which was purified on silica gel column using CLhy' ' '
heAane, (10:90) as the eluent, giving 82% yield of, 4,6-0-b~.~ylidu.,-3-benzyl
dihydro-D-glucal as a white solid (mp=75.76C).
D: r~ ualiu.. of ill~.l '' 3-benzyl dihydro-D-glucal _
To a solution of ' _ (Ig, 3.603mmol) in THF (154 ml), was added
dropwise lN aqueous HCI solution (ISml). The mixture reaction was then stirred at
room i , c overnight. The mixture was diluted with ethyl aceute and solid
sodium chloride was added with stirring. The aqueous layer was extracted twice with
ethyl aceute. The combined organic eAtracts were washed with saturated NaHCO~
solution and brine, dried (MgSO4) and L.~a~ r, ~ of the crude product
by flash " O , y using ethyl aceute and hexane, (SO:SO) provided 80 % of the
SUBSTITUTE SHE~
WO 95114027 ' PCTICA94100636
217~76`~ ~
24
expected compound as colorless oil.
E: T ' 4.6-di-0-[1'-N-Boc-~.u~y' ]-3-benzyl dihydro-D-glucal 6
The pll~ of 6 (step O was essentially the same as described for step C in
example I
F: F, of ' 4~6-di-o-[l~-N-Boc-~ulu~.~lallli~u~]-dihydro-D-gluca
To a solution of 4,6-di-0-[1'-N-Boc-l,.uL,~.. _]-dihydro-D-glucal 6 (850mg,1.537mmol) in dry ethanol (ISml) was added lOOmg of Pd/C (10%) as catalyst with
stirring. The mixture was stirred overnight at room L.,..I,U~ ILL.C under hydrogen
,' c, then filtered through celite. The filtrate was ~ ' and passed
through a short silica gel column using pure ethyl acetate as eluant to lead to desired
llydl~ ~ ~ compound as a colourless syrup, (98% yield).
G: P~cpcu~Liu~ of ' 4,6-di-0-[1'-N-Boc-lJluAuyl<ul.i-.~]-S-methyl
dilll;u~; -3-dihydro-D-glucal 8
To a stirred solution of the ' 6 (700nng, 1.513mmol) in dry THF (8ml) at -
78C, was added dropwise a solution of sodium I ' .~I.li~;l~ll~ (3ml, 3.000
mmol). The reaction mixture was stirred at -78C under argon for one hour. At the
same i . c (182 ml, 3.000 mmol of carbon disulfide was added dropwise, and
the stirring was coMinued another hour. Methyl iodide (lOOml, 3.000mmol) was then
added dropwise, and the reaction was stirred for another hour at room i
Ethyl acetate was added and the reaction was quenched with water. The aqueous
layer was e~tracted twice with ethyl acetate. The combined extracts were washed
several times with brine, dried and c~ r ~ ru.irl~Liull of the crude compound
by flash ~,L~ " ~' y (ethyl ~ , (40:60)), gave the title S-methyl
~' ' ' as a colourless oil, with 71% of yield.
H: rlc, of ' 4,6-di-0-[1-N-Boc-lnul~y' ]-3-deoxy dihydro-
D-glucal ~
To a solution of S-methyl lillliu~ ' (550~g, O.995mmol) in dry
SUBSTITUTE SHE~
wo ssA4027 1 7 ~ 76 ~ PCTICA94100636
dioxane (6 ml) was added '.Y'i' ,' (456.3~1, 4.975mmol) under argon
aLI..~ .c. The reaction mixture was then heated to reflux, and treated at 30 minute
intervais with 375,u1 portions of a solution of 967.5mg of b~.~V,~Ip~..UAi~il, in 6 ml of
dioxane. The reaction was monitored by TLC. When the reaction was complete, the
solvent was remove in vacuum and the product was isolated by column
,, ,',~ (CL~ ' , 40:60), to give 65.3% of the d~
d, as a coloriess syrup.
1: nc~JaldLiu.. of ~ - ' ethyl-4,6-di-O-~,Iup~ 2,3-dideoxy-D-
erythro-hexoside 10
The l. ~ " ' product of D, 4, 6-di-O-[ l -N-BOc-~Jl u~ r ' ] -3 -deoxy dihydro-D-
glucal ~, was dissolved 3N HCI in ethyl acetate and stirred at room ~.I.l)~,-aLll-c for
2 hours. The solution was removed under vacuum and the resulting oil was triturated
with dry ether followed by C~a~ulaiiU... The crude salt was converted to the
wll.-r ' ~ free base by passage through an ion exchange resin column (Amberlitc
Ii~A-400 OH) using pure methanol as the solvent to give 4,6-di-0-,ulu,u~la~ lc-3-
~i~V~yl"' y~ilu-D-glucal 10 (84.5%)
J: rl~, of ' 4,6-di-0-i-bis-Boc-~,lu~ ]-3-
d~,y.' ' ~ilu-D-glucal 11
To a stirred solution of 4,6-di-O-,ulupy' -3-~ A~ i;lly ilu-D-glucai 10 in dry
DMF (1.5 ml) was added 1.5 ml of dry Lli.,ii J' Following 15 minutes of
stirring at room i . c, a solution of di-Boc-thiourea in 0.5 mL of dry DMF
was added dropwise. The reaction mixture was stirred at room i . c under
argon - ,' c for 20 hr. The mixture was then diluted with ethyl acetate,
quenched with water and washed twice with brine. The organic phase was dried over
MgSO4 and the solvent was ~ ~~r ' The crude di 1 " product 11 was
purified on silica gel using ethyl ho~no (40:60) as the eluant to give a white
foam (46.3%)
SUBSTITUTE SHE~
W095/14027 217676!0 PCTICA94/0111636
26
K: r~ au..Liull of finai product 4.6-di-O-iJIu~J~l,, ' -3-deoxy dihydro-D- glucal 12
4,6-di-O-[bis-Boc-~lu~ ..a.l;.1;..~,]-3-~vAy~ ~ J~ilu-D-glucal 11 was dissolved in
1.5ml 3N.HCI in ethyl acetate, and the reaction mixture was stirred at room
C under argon ~ . ' c for 2 hrs. Tbe solution waD then removed under
vacuum and the resulting oil was triturated with dry ether. The solvent was
evaporated from the product to yield the finai product 4.6-di-O-~,.u~.~l" ' 3-
deoxy dihydro-D-glucai 12 (94.4%).
F ~71~ 10 - Synthesis of l.~ilu~ ,,1l11-4.6-di-O-vlu~ , ' -2.3-dideoxv-
h~Q~
The title compound was prepared as outlined in Figure 7. The synthesis was
essentially the same aD the syntheDis described for the compound of example 1 with
the exception that in the first step A, benzyloxy ethanol was used in place of MeOH
to give the ' ~ benzyloxy-4,6-di-O-acetyl-2,3-dideoxy-D-hex-2-
.,.lV~ .al~UDi~
E ' 11- ~ of N~
Tri-O-acetyl-D-glucal (200mg, 0.735mmol) and CBz-~,luLJyldll.;..~ (284mg,
1.469mmol) were dissoived in ~" ' ' , ' (3.7ml) at -10C. To this solution
was added SnCi~ (367~L of a 1.0M solution in diul.lulu...~ al~c, 367mmol~. The
reaction mixture was stirred and alloweci to come to 10C over a period of one hour.
After stirring for an additionai four hours at 10'C, the reac~ion mixture was diluted
with ii~ lu-~ ' (20ml) and Du~DD;~ washed with water (Sml), saturated
aqueous NaHCO~ (2XSml), and brine (Sml). The organic layer was dried over
MgsO~, filtered and ' Flash ~ ' , " . ' ~ using 35 % ethyl acetate and
65 % hexane as the elutant, afforded the protecteci N-~' ~ ' ' ' 1-(N-
CBz-iDu~.u~laullill~)-4,6-di-O-acetyl-1,2,3-trideox~ v~ allùDi~i~ as a colourless
syrup (106.2mg, 36% yield).
urtiler ~ . ' were prepared according to the procedure above with the
of the reactant CBz-l,.u~,~' with:
SUBSrITUTE SHE~
Wo95114027 1 7~ 7 6 o PCT/CA94100636
30c~ u~lyla~ to give 1-(N-Boc-propylamine)-4,6-di-0-acetyl-1,2,3-trideox~-
Jyldllu~i~e 115.31ng, 6% yield);
('Bz isopropylamille to give l-tN-CBz-iso~JIu~Jyldllline)-4~6-di-O-acetyl-1~2~3- trideoxy cl.~".,yldl~u~ide (54.6mg 18% );
C'3~-benzyL.mine to give 1-(N-CBz-benzylamine)-4,6-di-0-acetyl-1,2,3-trideoxy-
..vl~yld~oside (176.7mg 53%); and
30c-ben~ylamine to give 1-(N-Boc-i,~.lLyld",il";)-4,6-di-O-acetyl-1,2,3-trideoxy-
~,iul,y,a.,u~;de (96.01ng 31%).
~lie tollowing tdble illustrates results obtdined in inllibition and cylu~u~i~iLy studies
of l~erpesvirus replication by tlle . u~ .J~ exemplified.
COMPOUND ED~o(/lM) Tllerapeutic
Index
tle~lyl-~,6-di-O-propylguanidille-2,3-dideoxy-hexoside 2.6 12.29
1,~L,6-tri-0-propylguanidine-2,3-dideoxy-hexoside 10.51 >50
etllyl-4,6-di-0-propylguanidine-2,3-dideoxy-hexoside 2.5 9.96
etl~yl-4,6-di-0-pentylguanidine-2,3-dideoxy-hexoside; 9.3 17.^J~
isopropyl-4,6-di-0-p~,,Lyl~ud,,idine-2~3-dideoxy-hexoside 6.45 5.17
4.6-di-0-propylguanidine-3-deoxy dihydro-D-glucal ~50 ~350
hydroxyethyl-4.6-di-0-propylguanidine-2,3-dideoxy 10.78 31.
llexoside
ethoxyethoxy-ethyl-4,6-di-0-u,u~ ;ual~idi.,e-2,3-dideoxy- 10 > 20
hcxoside
~UBSTITUTE SHE~T