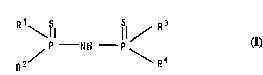Note: Descriptions are shown in the official language in which they were submitted.
~7~4
~ 095115329 PCT/GB94102485
AMIDES OF ACIDS OF PHOSPHORUS FOR THE EXTRACTION OF METALS
This invention relates to chemical compounds and more
particularly to certain aryloxy-substituted amidobis(thiophosphoryl)
compounds and to their use as extractants in a solvent extraction
process for extracting metal values from aqueous solutions of metal
salts.
The use of solvent extraction techniques for the
hydrometallurgical recovery of metal values from metal ores has been
practised commercially for a number of years. In general, the
technique involves contacting an aqueous solution of metal salt,
obtained for example by treating the crushed ore with acid, with a
solution in a water-immiscible organic solvent of an organic
extractant which complexes with the metal and extracts it into the
non-aqueous phase. The metal may then be recovered by a further
extraction step in which the organic solution cont~i n i ng the metal
complex is contacted with another aqueous phase containing an agent,
usually a strong acid, capable of decomposing the complex so that the
metal is extracted into the aqueous phase from which it can be
recovered by suitable procedures such as electrowinning.
Since metals are usually found in their ores in
association with other metals, it is essential that the organic
extractant extracts the desired metal selectively so as to achieve a
degree of separation from the other metals present. Selective
extractants are known for some metals, for example copper, and their
use is well-established. The search for a suitably selective
extractant for zinc has been less successful.
The use of extractants containing the phosphoric acid
group, especially di(2-ethylhexyl)phosphoric acid (D2EPHA), has been
proposed, see "Productivity and Technology in the Metallurgical
Industries", edited by M.Koch and J.C.Taylor, in an article by A.Selke
and D.de Juan Garcia, pages 695 to 703. However, as is apparent from
the Selke et al article, ferric iron is extracted together with the
zinc. To prevent build-up of the ferric iron in the organic solution,
it is necessary to remove the iron from the organic solution in a
stripping stage subsequent to that used to recover the zinc. In this
separate stripping stage, the organic solution is contacted with 5 to
6 molar hydrochloric acid to give ferric chloride in hydrochloric
acid. Free hydrochloric acid is recovered by a further step in which
the ferric chloride in hydrochloric acid is contacted with an organic
solution containing tributyl phosphate from which the ferric chloride
is stripped using water. The additional stages required to remove
iron add to the complexity and cost of the procedure and hence are
undesirable.
-
W 095/15329 2 ~ 7 7 0 6 ~ 2 PCT/GB94/02485
The behaviour of bis(2,4,4-trimethylpentyl)
monothiophosphinic acid as an extractant for zinc has been studied by
C.Caravaca and F.J.Alguacil (Hydrometallurgy, 27, l991, 327-338) who
found that at relatively acidic pH values (near l.0) the proportion of
zinc extracted was only slightly higher than the proportion of iron.
It has now been found that certain compounds containing
the aryloxy-substituted amidobis(thiophosphoryl) group are excellent
metal extractants, particularly for the separation of other metals
from solutions containing iron. In particular, it has been found that
these amidobis(thiophosphoryl) compounds are highly effective in
selectively extracting zinc from acidic aqueous solutions containing
both zinc(II) and iron(III) ions. It has also been found that certain
of the amido-bis(thiophosphoryl) compounds are strong extractants,
that is that they extract zinc from aqueous solutions at pH values
below 2 without requiring the addition of base to neutralise the
protons liberated by the complexation reaction.
Accordingly, the present invention provides a compound of
the formula:
R ~ / R 3
P N H p ~
R2/ \ R4
wherein Rl is an optionally substituted 2-alkylpheno~y group, each of
R2, R3 and R~ is a group selected from optionally substituted 2-
alkylphenoxy, optionally substituted phenyl, optionally substituted
alkyl and optionally substituted alkoxy and at least one optionally
substituted 2-alkylphenoxy group has a tertiary alkyl substituent
except the compound wherein each of Rl and R2 is 2-isopropyl-4-tert-
nonylphenoxy and each of R3 and R4 is phenyl and the compound wherein
Rl is 2-methyl-4-tçrt-nonylphenoxy, R2 is 2,4-dimethylphenQxy and each
of R3 and R4 is phenyl.
It will be appreciated that the structure of the compounds
of Formula I is such that they may exist in more than one tautomeric
form, another such form having the structure:
Rl~ll SIH~ R3
P N P l I
p2/ \ R4
0
wherein R1-R4 are as already defined. Whilst the invention is
described herein with reference to compounds of Formula I, it is to be
understood that it relates to said compounds in any of their possible
tautomeric forms.
~ 095/1532g 2 17 7 Q 6 4 PCTIGB94/02485
~ . j
As examples of optionally substituted 2-alkylphenoxy
groups which may be present in the compounds of Formula I, there may
be mentioned phenoxy groups wherein the alkyl substituent in the 2-
position relative to the oxygen atom contains from 1 to 20, for
example from 1 to 10, carbon atoms. Such alkyl groups may be primary
alkyl groups having one or more carbon atoms, secondary alkyl groups
having three or more carbon atoms or tertiary alkyl groups having four
or more carbon atoms. In addition to the alkyl substituent in the 2-
position, the phenoxy residue may optionally carry one or more
additional alkyl substituents, for example an alkyl substituent in the
4-position.
When the optionally substituted 2-alkylphenoxy group has a
tertiary alkyl substituent, the latter may be present in addition to
the alkyl substituent in the 2-position and/or the alkyl substituent
in the 2-position may itself be a tertiary alkyl group.
Examples of optionally substituted 2-alkylphenoxy groups
include 2-tert-butylphenoxy, 2-tert-butyl-4-methylphenoxy, 2-tert-
butyl-5-methylphenoxy, 2,4-di-tert-butylphenoxy, 2,4-di-tert-
pentylphenoxy, 2-methyl-4-tert-nonylphenoxy, 2-tert-butyl-4-tert-
nonylphenoxy, 4-octylphenoxy, 4-tert-dodecylphPnnxy, 4-tert-dodecyl-2-
methylphPno~y, 2-sec-butylphenoxy and the like.
In particularly useful compounds of Formula I, at least
one 2-alkylph~nnxy group is a 2-tert-alkylphenoxy group and preferably
at least two 2-alkylph~nnxy groups are 2-tert-alkylphenoxy groups.
Preferred 2-tert-alkyl groups include 2-tert-butyl groups.
Optionally substituted phenyl groups which may be
represented by R2 and/or R3 and/or R4 in the compounds of Formula I
include alkyl substituted phenyl groups, for example o-tolyl, m-tolyl,
p-tolyl and xylyl groups and mixtures of such groups. However,
because of the commercial availability of suitable intermediates, the
preferred optionally substituted phenyl group is the unsubstituted
phenyl group.
As examples of optionally substituted alkyl and optionally
substituted alkoxy groups which may be represented by R2 and/or R3
and/or R4, there may be mentioned alkyl and alkoxy groups contAin;ng
from 1 to 20, for example from 1 to 10, carbon atoms. The alkyl
groups and the alkyl moieties of alkoxy groups may be primary alkyls
having one or more carbon atoms, secondary alkyls containing three or
more carbon atoms or tertiary alkyls containing four or more carbon
atoms. As examples of substituents which may be present in
substituted alkyl or substituted alkoxy groups, there may be mentioned
halogen, nitro, cyano, hydrocarbyloxy, hydrocarbyloxycarbonyl, acyl
and acyloxy groups. More than one substituent may be present in which
case the substituents may be the same or different.
W O 9S/15329 PCTIGB94/02485 ~
2~7~0~4 4
One valuable class of compounds falling within the scope
of Formula I comprises the compounds wherein R1 is an optionally
substituted 2-alkylphenoxy group, each of R2, R3 and R4 is either an
optionally substituted 2-alkylphenoxy group or an optionally
substituted phenyl group and at least one optionally substituted 2-
alkylphenoxy group has a tertiary alkyl substituent except the
compound wherein each of R1 and R2 is 2-isopropyl-4-tert-nony]ph~nn~y
and each of R3 and R4 is phenyl and the compound wherein R1 is 2-
methyl-4-tert-nonylphenoxy, R2 is 2,4-dimethylphenoxy and each of R3
and R4 is phenyl.
When only R1 in this class of compounds is an optionally
substituted 2-alkylphenoxy group, R2, R3 and R4 being phenyl groups, it
is preferred that the phenoxy group is heavily substituted with
aliphatic groups as in, for example, the 2-tert-butyl-4-tert-
nonylph~nn~y group to provide the extractant compound with good
solubility in hydrocarbon solvents.
It is preferred, in this class of compounds, that at least
one of R2, R3 and R4 is an optionally substituted 2-alkylphenoxy group
so that the compound of Formula I contains at least two optionally
substituted 2-alkylphenoxy groups and it is further preferred that at
least two optionally substituted 2-alkylphenoxy groups have a tertiary
alkyl substituent.
Compounds of Formula I in which R1 and R2 are optionally
substituted 2-alkylphenoxy groups, R3 and R4 being optionally
substituted phenyl are strong metal extractants. In an example of
such a compound, each of R1 and R2 is 2-methyl-4-tert-nonylphenoxy and
each of R3 and R4 is phenyl.
Useful compounds in which R1 and R3 are optionally
substituted 2-alkylphenoxy groups, R2 and R~ being optionally
substituted phenyl, include compounds in which the alkyl substituents
in the 2-position are tertiary alkyl groups, for example the compound
in which each of Rl and R3 is 2-tert-butyl-4-methylphenn~y and each of
R2 and R4 i s phenyl.
In especially valuable compounds of Formula I, each of R1,
R2 and R3 is an optionally substituted 2-alkylphenoxy group and R4 is
optionally substituted phenyl. Preferably, at least one of the 2-
alkylphenoxy groups is a 2-tert-alkylphenoxy group, the others
preferably being 2-tert-alkyl and/or 2-sec-alkylphenoxy, preferred
sec-alkyl groups having at least four carbon atoms. Examples of
compounds containing one 2-tert-alkyl substituent and two 2-sec-alkyl
substituents include the compound wherein each of R1 and R2 is 2-sec-
butylphenoxy, R3 is 2,4-di-tert-pentylphenoxy and R4 is phenyl.
Examples of compounds containing two 2-tert-alkyl substituents and one
~ O 95/15329 2 ~ 7 7 0 6 '1 PCTIGB94/02485
5 ~ i
2-sec-alkyl substituent include the compound wherein each of Rl and R3
c is 2-tert-butylphenoxy, R2 is 2-sec-butylphenoxy and R4 i8 phenyl.In compounds of Formula I in which each of Rl, R2, R3 and
R4 is an optionally substituted 2-alkylphenoxy group, preferably at
least one is a 2-tert-alkylphenoxy group and more preferably two and
especially three of the optionally substituted 2-alkylphennxy groups
are 2-tert-alkylphenoxy groups. Useful structures include those
compounds in which Rl and R2 are 2-tert-alkylphenoxy groups especially
when R3 and R4 are 2-sec-alky]phennxy groups.
A second valuable class of compounds falling within the
scope of Formula I comprises the compounds wherein Rl is an optionally
substituted 2-alkylphenoxy group, at least one of R2, R3 and R4 is an
optionally substituted alkyl or optionally substituted alkoxy group,
any remaining group or groups from R2, R3 and R4 being selected from
optionally substituted 2-alkylphenoxy and optionally substituted
phenyl and at least one optionally substituted 2-alkylphenoxy group
has a tertiary alkyl substituent.
Within this second class of compounds, mention may be made
of compounds of Formula I in which Rl is an optionally substituted 2-
tert-alkylphenoxy group, R2 is an optionally substituted alkoxy group
and each of R3 and R4, independently, is an optionally substituted 2-
alkylph~noxy group or an optionally substituted alkoxy group.
Preferably, at least one of R3 and R4 is a 2-tert-alkylphenn~cy group.
Also within this second class of compounds, mention may be
made of compounds of Formula I in which Rl is an optionally
substituted 2-tert-alkylphenoxy group, R2 is an optionally substituted
alkoxy group or an optionally substituted phenyl group and R3 and R4
are optionally substituted alkoxy groups which may be the same or
different.
Also within this second class of compounds, mention may be
made of compounds of Formula I in which Rl is an optionally
substituted 2-tert-alkylphenoxy group, R2 is an optionally substituted
2-alkylphenoxy group or an optionally substituted phenyl group and R3
and R4 are optionally substituted alkyl groups which may be the same
or different.
Compounds of Formula I may be obtained from a
chlorophosphorus compound of formula A, B, C or D
R 1 \ R3 \ R 1 \iSI R3\iSI
~P Cl ~P Ci ~P Cl ~P Cl
R R~ i-i R2 C Rs D
W O95llS329 ~ PCT/GB94102485 -
2~ ~7~4 6
A chloro compound of formula C or D may be reacted with an
excess of ammonia to give a phosphoramide, E.
R ~ R l \ll
/ P-- Cl ~2NH3 ~ /P NH2~NH4C I
R2 R2 E
The amide E may be reacted with a chloro compound C or D
in the presence of a strong base.
Rl\ll R3\ll Rl\~l ll R3
/P ~H2~2N~H~ /P Cl ~ /P --N --P
R2/ R4 / R2 / Na t \R4
~H?~N~C I
Instructions for the preparation of amides (cf E) and
reaction with chloro compounds for oxygenated analogues are given by
L.Meznik and A.Maracek, Z.Chem. 21(8), 1981 at page 259, but more
satisfactory reaction conditions are provided herein.
General methods for preparation of the chloro compounds C-
D are well known to the art, for example when Rl-RZ are aryl groups,
Grignard reagents may be reacted with phosphorus trichloride (if
re~uired in two stages, to give a mixed product) as described by
W.Voskuil and J.F.Arens, Rec.Trav.Chim., 82, 302, (1963):
R1MgCI~PCl3 ~ R1PCl2 /P Cl
and the diarylchlorophosphine may be oxidised by reaction with
thiophosphoryl chloride to give C.
Alternatively, 3 e~uivalents of a Grignard reagent may be
reacted with a dialkyl phosphite to give a dialkyl phosphine oxide
which is converted into the acid chloride by reaction with phosphorus
trichloride as described by Robert H Williams and Lyle A Hamilton,
J.Am.Chem.Soc., 74, 5418, 1952.
~l77n6~
~ 095/lS329 ' PCTIGB94/02485
P 1 0
C4HgO\ 1I R \ 11 PC I 3
3RlMgBr ~ /PH ~ /PH
R
C4Hs
R1 \
/ P Cl
R
Useful available compounds of formula A or B include
chlorodiphenylphosphine.
To prepare compounds of formulae C and D in which Rl and
R2 are aryloxy, a dithiophosphoric acid may be reacted with chlorine
15or sulphuryl chloride:
R 1 \ 15l C I ~R 1 \ 11
/P SH ~ /P C I
R2 / R2 /
as described by John H.Fletcher et al, J.Am.Chem.Soc., 72, 2461, 1950.
Where Rl-R4 are aryloxy groups, a wide range of
dithiophosphoric acids for use in the reaction above may be prepared
by reaction of the appropriate phenol either with phosphorus
pentasulphide or with thiophosphoryl chloride in the presence of an
acid acceptor. When thiophosphoryl chloride is used, different
phenols may be reacted sequentially to provide compounds C and D in
which R' is different from R2 and R3 is different from R4.
Rl \ISI
4 R lH ~ P255 2 /P ~ SH ~H25
R
[ F I e t c h e r , I o c c i t )
R l\lSI
2 RlH ~ PSCI3 --2 /PCI i2HCI
R
See N.A.Meinhardt, S.Z.Cardon and P.W.Vogel, J.Org.Chem.,
25, 1991, (1960) and J.H.Fletcher et al, J.Am.Chem. Soc., 70, 3943,
(1948).
W 09S/lS329 ~ ~ 7 7 ~ ~ ~ PCT/GB94102485
Compounds of formula C in which Rl is phenyl and R2 is
aryloxy may be obtained by reacting C6H5PSCl2 with a phenol.
In a further aspect, the present invention provides a
process for extracting metal values from aqueous solutions of metal
salts with an organic phase comprising a compound of the invention as
hereinbefore defined.
The organic phase employed in the extraction process
typically contains a water-immiscible inert organic solvent, that is
to say a water-immiscible organic liquid that is inert under the
extraction conditions and is a good solvent for the extractant
compound and the metal complex thereof.
It will be appreciated that the extraction process may be
incorporated into a wide variety of different procedures for the
recovery of metals from their ores or from other metal-bearing
sources. Details of these procedures will vary depending on the metal
concerned and the nature and composition of the leach solution. An
integrated process which is especially suitable for sulphate leach
solutions can be carried out using operations well known to the
skilled person.
Typically, the extractive process comprises a sequence of
stages in which the metal is extracted into an organic solution,
stripped into an aqueous phase and recovered from the aqueous phase by
any suitable means, for example by electrowinning.
Thus, as a particular aspect of the invention, there is
provided a process for extracting metal values from aqueous solution
by a sequence of stages comprising:
(l) contacting the aqueous solution cont~;ning metal values
with a solution of an extractant compound as hereinbefore
defined in a water-immiscible organic solvent whereby to
extract metal values into the solvent in the form of a
complex of the metal with the extractant;
(2) separating the solvent phase containing metal complex from
the extracted aqueous phase;
(3) contacting the solvent phase containing metal complex with
an aqueous strip solution whereby the metal complex is
unstable and metal ions transfer into the a~ueous phase,
and
(4) separating the aqueous phase containing metal ions from
the stripped solvent phase.
The extraction process may be applied to the extraction
from aqueous solution of any metal capable of forming a stable complex
with a compound of the invention in the organic phase. The process is
especially suitable for the solvent extraction of zinc from aqueous
solutions of zinc salts, especially solutions obtained by the acid
a O 95/lS329 2 1 7 7 0 6 ~ PCT/GB94/02485
leaching of zinc ores. Examples, however, of other metals which can
be extracted from acidic solutions having pH values of pH 2 and below
are bismuth, cadmium, silver, mercury and copper but many other metals
may also be extracted at higher pH values.
In operating stage (1) of the aforementioned process, the
amount of extractant compound to be used will depend upon the
concentration of metal salt in the aqueous solution and also on the
plant design. It is preferred, however, to use from 5g to 400g of
compound of the invention per dm3 (litre) of organic solution. Higher
concentrations may be used but tend to afford organic phases of too
high viscosity for convenient handling. Lower concentrations can also
be used but involve the use of unnecessarily large volumes of solvent.
For use with aqueous solutions containing lg or more per
dm3 of a metal such as zinc, it is preferred to use from 50 to 400g of
compounds of the invention per dm3 of organic solution. If desired,
the extractant compound can be used together with an agent which
modifies the behaviour thereof in the extraction process, for example
an alkylphenol, alcohol or ester which may be used in an amount of
from 10~ to 200~, especially from 20~ to 100~ by weight of extractant
compound. Such compounds weaken the extractant but facilitate the
subsequent stripping of metal therefrom. In this way, a very strong
extractant may be adjusted in strength to the requirements of
different feed solutions and different stripping solutions.
Alkylphenols which may be used as modifiers in conjunction
with the extractant compounds of the invention include alkylphenols
containing from 3 to 15 alkyl carbon atoms, for example 4-tert-
butylphenol, 4-heptylphenol, 5-methyl-4-pentylphenol, 2-chloro-4-
nonylphenol, 2-cyano-4-nonylphenol, 4-dodecylphenol, 3-
pentadecylphenol and 4-nonylphenol and mixtures thereof. The
preferred phenols contain alkyl groups having from 4 to 12 carbon
atoms, especially the mixed 4-nonylphenols obtained by contl~n~ation of
phenol and propylene trimer.
Alcohols which may be used as modifiers in conjunction
with the extractant compounds of the invention include saturated and
unsaturated hydrocarbon alcohols and polyols containing 14 to 30,
preferably 15 to 25 carbon atoms. The alcohols are preferably highly
branched with the hydroxyl group located approximately midway along
the hydrocarbon backbone. Especially preferred are the br~n~-hP~l chain
alcohols that may be made by condensation of short chain alcohols by
the Guerbet process, such alcohols sometimes being referred to as
Guerbet alcohols. Optionally, the alcohols may contain an aromatic
group or other functional group, particularly an ester group.
Especially useful alcohols may be synthesised from highly
br~n~-he~ precursors leading to very highly branched Guerbet alcohols
W O9S/lS329 PCTIGB94/02485
~77~64 lo
containing a large number of terminal methyl groups. Examples of
particularly efficient alcohol modifiers include highly branched
isohexadecyl alcohol and iso-octadecyl alcohol, the latter being 2-
(1,3,3-trimethylbutyl)-5,7,7-trimethyloctanol.
Esters which may be used as modifiers in conjunction with
the extractant compounds of the invention include saturated and
unsaturated aliphatic and aromatic-aliphatic esters containing from 10
to 30 carbon atoms. The esters may be mono-esters or polyesters,
especially di-esters. The esters are preferably highly branched.
Optionally, the esters may contain other functional groups,
particularly a hydroxyl group. Especially useful esters include
2,2,4-trimethyl-1,3-pentanediol isobutyrate and the benzoic acid ester
thereof.
In the context of the present invention, 'highly branched'
as applied to the alcohols and esters means that the ratio of the
number of methyl carbon atoms to non-methyl carbon atoms is higher
than 1:5. Preferably, this ratio is higher than 1:3.
If desired, mixtures of alkylphenols and/or alcohols
and/or esters may be employed as modifiers.
The aforementioned modifiers may be used in the
preparation of extractant compositions containing one or more
extractant compound of the invention and one or more modifier.
It has been found that for some of the pure extractant
compounds of the invention the rate at which zinc is extracted is
rather slow, but that a wide range of compounds may be added to
increase this rate even in amounts of 1.0~ and below. Useful rate
increasing additives include compounds which are known to be
extractants for zinc and are soluble in the organic phase but with
fast rates of extraction. Compounds found to be effective in this
aspect are other known zinc extractants such as esters of phosphoric
acid, (eg D2EHPA), and particularly surface active agents capable of
transferring metal ions such as alkyl and aryl sulphonic acids having
solubility in the organic phase.
Stages (1) and (2) of the aforementioned process may
conveniently be carried out using well known conventional solvent
extraction techniques. Typically, the aqueous solution containing
metal values is intimately contacted, in a single stage or in multiple
stages but preferably continuously, with the organic phase (for
example by agitating the two phases together in a suitable vessel) for
a time sufficient to allow substantial extraction of the metal values
from the aqueous solution, the two phases then being separated in any
conventional manner. The extraction is usually carried out at ambient
temperature although somewhat higher temperatures, for example up to
~ O95/15329 2 7 7 7 0 6 4 PCTIGB94/02485
100C but preferably not more than 50C, may be used if operationally
convenient.
Organic solvents which may be used in the extraction
include any mobile organic solvent, or mixture of solvents, which is
immiscible with water and is inert under the extraction conditions to
the other materials present. ~xamples of suitable solvents include
aliphatic, alicyclic and aromatic hydrocarbons and mixtures of any of
these as well as chlorinated hydrocarbons such as trichloroethylene,
perchloroethylene, trichloroethane and chloroform. Preferred solvents
are hydrocarbon solvents including high flash point solvents with a
high aromatic content such as SOLVESSO 150 commercially available from
Exxon (SOLVESSO is a trade mark) and AROMASOL H which consists
essentially of a mixture of trimethylbenzenes and is commercially
available from Imperial Chemical Industries PLC (AROMASOL is a trade
mark~. Especially preferred, however, on grounds of low toxicity and
wide availability are hydrocarbon solvents of relatively low aromatic
content such as kerosene, for example ESCAID 100 which is a petroleum
distillate comprising 20~ aromatics, 56.6~ paraffins and 23.4~
naphthenes commercially available from ~xxon (ESCAID is a trade mark).
The conditions under which the solvent extraction is
performed are chosen to suit the metal or metals present in the
aqueous solution. It is generally desirable that conditions are
selected such that any other metals present do not form stable complex
compounds with the extractant compound in order that substantially
oRly the desired metal is extracted from the aqueous solution. Since
formation of the complex may involve the liberation of acid, it may be
necessary to add, for example, alkali during the process to maintain
the pH within a desired range but it is generally preferred to avoid
this, especially in a continuously-operated process. It is a
particular advantage of the process of the invention that zinc can be
extracted selectively even in the presence of iron.
Stages (3) and (4) of the process may conveniently be
carried out by intimately contacting the solution of metal complex in
the organic solvent obtained in stage (2) with an aqueous solution of
a mineral acid at a suitable temperature, the two phases then being
separated in conventional manner. The operations are usually
performed at ambient temperature although somewhat higher
temperatures, for example up to 100C but preferably not more than
50C, may be used if operationally convenient.
The aqueous strip solution used in stage (3) preferably
contains sulphuric acid, suitable strengths being from 100 to 250g of
acid per dm3 of solution. After removal of a convenient part of the
metal by, for example, electrolysis, the recovered aqueous acid,
containing residual metal salt, may be re-used in stage (3) of the
W 095/lS329 2 1~ 7 ~ ~ ~ PCTIGB94/02485 -
12
process. The extractant compound regenerated in stage (3) may be
recycled for use in stage (1).
Suitable relative volumes of organic to aqueous phases are
those conventionally used in metal extraction processes and in the
stripping stage will be typically not more than 10:1. The stripped
organic layer, containing regenerated extractant compound and some
residual metal, may be re-used in stage (1) of the process. The
aqueous layer from stage ~4~, containing metal salt, may be treated in
any conventional manner to obtain the metal.
The effectiveness of the compounds of the invention as
extractants for zinc may be determined by the procedure of Test 1
which is described below.
The extractant compounds of the invention are valuable in
that, when subjected to Test 1, they are generally capable of
providing an organic solution containing from 1500 to 4500 parts per
million, often 3000 to 4500 parts per million and preferably 3600 to
4500 parts per million of zinc, a range which provides the most
efficient combination of extraction and stripping. In the case of
extractant compounds of the invention which provide a figure much
above the 3600-4500 ppm range, it is advantageous to incorporate one
of the above mentioned modifiers to have a weakening effect on the
extractant.
The invention is illustrated but not limited by the
following Examples. Examples 1-5, relating to compounds in which no
2-alkylphenoxy group contains a tertiary alkyl substituent, are
included for the sake of comparison.
ExamDle 1
This Example describes the preparation of O,O'-bis(2-
isopropyl-5-methylphenyl)chlorothiophosphate which is the chloro
compound listed in Table 1 and the preparation of O,O'-bis(2,4-
dimethylphenyl)thiophosphoramide which is the amino compound listed in
Table 1, and reaction of the chloro compound with the amino compound
to give the compound of Formula 1 in which R1=R2- 2-isopropyl-5-
methylph~no~y, and R3=R4= 2,4-dimethylphenoxy, which is the product of
this Example. The General Method designated Test 1 of testing the
product for its strength as an extractant for zinc, with the result
which is ennnmerated in Table 1, is also described.
Sodium hydride (0.40M, 9.6g) was added in portions during
15 minutes to a stirred solution of 2-isopropyl-5-methylphenol (0.40M,
60g) in tetrahydrofuran (350 cm3) in an atmosphere of nitrogen.
During this addition the temperature rose to about 50C. The solution
was allowed to cool and was then added during 45 minutes under
nitrogen atmosphere to a stirred solution of thiophosphoryl chloride
VO 9S/15329 21 7 7 0 6 4 PCT/GB94/02485
13
(0.20M, 33.9g~ in tetrahydrofuran (50 cm3) which was maintained at -
40C by external cooling. The reaction mixture was allowed to warm to
ambient temperature when a sample analysed by HPLC indicated that
reaction was complete. The chloro compound was isolated by diluting
the mixture with diethyl ether (300cm3 ); the ether solution was
extracted with water (three 100 cm3 portions) and the organic layer was
separated, dried with magnesium sulphate, filtered and concentrated by
evaporation of the ether under reduced pressure yielding 0,O'-bis (2-
isopropyl-5-methylphenyl) chlorothiophosphate which was an oil
(73,1g): 31p NMR in CDCl3, singlet 56.7 ppm downfield of phosphoric
acid.
By the same procedure 2,4-dimethylphenol was reacted with
sodium hydride and then with thiophosphoryl chloride to give a
solution of O,O'-bis(2,4-dimethylphenyl)chlorothiophosphate in
tetrahydrofuran. This reaction intermediate was not isolated but,
instead, ammonia gas was bubbled through the solution for 2 hours.
Precipitated ammonium chloride was removed by filtration and the
solu~ion was concentrated by evaporation under reduced pressure
yielding an oil which was 0,O'-bis(2,4-
20 dimethylphenyl)thiophosphoramide: 31p NMR in CDCl3, singlet, 59.6 ppm
downfield of phosphoric acid.
The final stage of the reaction was carried out as
follows. The amino compound prepared as described above (0.05M,
16.0sg, assuming 100~ purity at M.W. 321) and the chloro compound also
25 prepared as described above (0.05M, 19.8g assuming 100~ purity at M.W.
396.5) were dissolved in a mixture of hexane (100 cm3) and
tetrahydrofuran (50 cm3) and the solution was stirred whilst a
suspension of sodium hydride (0.115M, 2.76g) in hexane (25 cm3) was
added during 30 minutes. The mixture was then stirred for 18 hours at
30 ambient temperature (Note: in later Examples it was found that the
reaction could be completed without detriment by boiling the mixture
under reflux, at about 65-70C, for 3 hours; the necessary reaction
time increased with increasing bulk of the substituents at position 2-
of the phenoxy groups) . The reaction mixture was filtered and
35 concentrated by evaporation of the solvents under reduced pressure
yielding an oil which was the sodium salt of the crude reaction
product. The crude reaction product was purified and isolated by the
following general method. The oil was dissolved in hexAn~ (100 cm3)
and the hPx~ne solution was twice extracted with 100 cm3 portions of a
40 mixed solvent prepared by adding 5 parts by volume of water to 95
parts by volume of methanol. The hexane solution was then discarded.
The methanolic solutions were combined and dilute sulphuric acid was
added to reduce the pH to about 2Ø This solution was then extracted
with hexane (two 100 cm3 portions) and the hexane solutions were
W O 9S/15329 PCTIGB9410248~ -
~ - - 14
2~7~4
combined and dried (magnesium sulphate) and concentrated by
evaporation of the hexane under reduced pressure yielding an oil
(19 5g) which was the compound of Formula 1 in which Rl=R2= 2-
isopropyl-5-methylphenoxy and R3=R~= 2,4-dimethylphenoxy.
The purity of this compound was estimated by
potentiometric titration of a sample (0.3576g) dissolved in 50~
aqueous tetrahydrofuran with 0.1 molar sodium hydroxide solution. The
acidic (~H) proton was neutralised between pH 4.0 and pH 8.6,
requiring 4.4 cm3.of alkali; hence it was calculated that the compound
was 84~ pure based on M.W. 681.
The compound was ~X~mi ned for its strength in extraction
of zinc by Test 1 described below with the result enumerated in Table
1 which indicates that although it is a good extractant for zinc, it
does not have the very high strength ideally required.
TEST 1
A 0.20 molar solution of the compound to be tested in
ESCAID 1~0 is shaken with an equal volume of a 0.1 molar aqueous
solution of zinc sulphate containing sufficient sulphuric acid to give
an initial pH value of 2Ø Samples of the dispersion are withdrawn
periodically and the aqueous layer is separated and analysed for zinc
by titration with EDTA according to the usual procedure, until
successive samples give the same zinc value denoting that equilibrium
has been reached. The amount of zinc which has passed into the
organic solution, expressed as a concentration in parts of zinc by
weight per million parts of solution by volume (ppm~ is calculated.
These results are listed in Table 1. If the compound to be tested was
not sufficiently soluble in ESCAID then SOLVESSO lS0 was used as
solvent, and this is noted in the Table.
EXAMPLES 2-5
The chloro compounds and amino compounds listed in Table 1
were prepared by the methods of Example 1, using as a~lopliate 2-se~-
butylphenol, 2,4-dimethylphenol, 2-isopropyl-5-methylphenol or 2,6-
dimethylphenol as starting materials. The chloro and amino compounds
were then reacted together, also by the method of Example 1 to give
the compounds of Formula 1 having the groups R1-R~ listed. The
strengths of these compounds as zinc extractants were examined by Test
1 with the results listed. The results show that the compounds of
these Examples have strengths similar to the compound of Example 1.
R~MPLES 6-9
These Examples demonstrate compounds of Formula 1 in whiçh
two phenoxy groups each bearing a 2-tert-alkyl substituent are
~ O 95/15329 PCT/GB94/02485
~1~7064
attached to the same phosphorus atom. The compounds, which are listed
in Table 1, were prepared by the methods of Example 1 using 2-tert-
butyl-4-methylphenol or 2-tert-butylphenol as starting materials where
required, with the following modifications:
(i) in preparing the chloro compounds listed in Table 1
it was found advantageous to carry out the reaction
without external cooling, and finally to boil the
reaction mixture under reflux for 18 hours.
(ii) in reacting the chloro compounds with the amino
compounds it was found necessary to boil the
reaction mixture under reflux for up to 72 hours to
complete the reaction.
The compounds of Formula 1 were assessed by Test 1 with
the results listed in Table 1 which show that they are all stronger
extractants for zinc than the products of Examples 1-5.
EXAMPLE 10
This Example demonstrates the preparation of a compound of
Formula 1 in which differently substituted phenoxy groups are attached
to the same phosphorus atom.
Preparation of the chloro compound for this reaction was
carried out as described in Example 9.
Sodium hydride (0.60M, 14.4g) was added during 10 minutes
to a stirred solution of 2-tert-butyl-5-methylphenol (0.60M, 98.4g) in
hexane (400 cm3) and tetrahydrofuran t50 cm3) whilst the temperature
was maintained below 30C by external cooling. This solution was then
added during 45 minutes to a stirred solution of thiophosphoryl
chloride (0.60M, 101.7g) in hexane (100 cm3) under nitrogen atmosphere
whilst the reaction temperature was maintained at -40C. The reaction
mixture was allowed to warm to a~bient temperature to complete the
reaction and after 30 minutes was cooled again to -40C. To this
solution was then added a solution of sodium 4-tert-nonylphennx;de
prepared by adding sodium hydride (0.60M, 14.4g) to commercial mixed-
isomer 4-tert-nonylphenol (0.60M, 132g) dissolved in ~X~n~ (400 cm3).
The addition lasted 30 minutes, and the temperature during addition
was maintained at -40C. The reaction mixture was allowed to warm to
ambient temperature when analysis by HPLC indicated that reaction was
complete. The solution was extracted with water (three 200cm3
portions), dried with magnesium sulphate, filtered and concentrated by
evaporation of the hexane under reduced pressure, yielding an oil
which was substantially 0-(2-~ert-butyl-5-methylphenyl)-0'-(4-tert-
nonylphenyl)chlorothiophosphate (31p NMR in CDCl3, multiplet 55.3 ppm
down field of phosphoric acid). This material (130g) was dissolved in
tetrahydrofuran (400 cm3) and ammonia gas was bubbled through the
W 095/15329 PCT/GB94/02485
~7~
solution for 45 minutes whilst the temperature was kept below 35C by
external cooling. Analysis by HPLC then showed complete conversion of
the chloro compound. The mixture was diluted with diethyl ether (400
cm3) and extracted with water (three 200 cm3 portions), dried with
magnesium sulphate and concentrated by evapoLation of the solvents
under reduced pressure yielding the amino compound listed in Table 1
(115.3g); 31p NMR in CDCl3 multiplet, 59.8 ppm downfield of phosphoric
acid.
The amino compound (0.22M, 101.4g) was reacted with O,O'-
bis(2-tert-butyl-5-methylphenoxy) chlorothiophosphate (0.22M, 93.4g)
and sodium hydride (0.55M, 13.2g) in tetrahydrofuran (200 cm3)
solution; it was found necessary to boil this reaction mixture under
reflux for 70 hours before HPL~ analysis showed that the starting
materials had been almost completely converted (see Example 9,(ii)).
After purification and isolation as described in Example 1 the
reaction product was obtained (103.1g), but found by titration to have
a purity of only 57~ (M.W. 833). Accordingly it was dissolved in
toluene (400 cm3i and the toluene solution was extracted twice with 2M
sodium carbonate solution (200 cm3 portions). The toluene solution
was then extracted twice with aqueous methanol (95ppv methanol mixed
with 5 ppv water, 200 cm3 portions) and the methanolic solutions were
combined and extracted with toluene (three 100 cm3 portions).
Concentrated hydrochloric acid (40 cm3) was then added to the
methanolic solution and this acidified solution was extracted with
hexane (400 cm3). The hexane solution was extracted three times with
fresh aqueous methanol solution (lO0 cm3 portions). The hexane
solution was then dried with magnesium sulphate, filtered and
concentrated by evaporation of solvents yielding the product of this
Example (49.6g) which was found by titration to be 87~ pure. The
performance of this compound in Test l (Table 1) showed it to be a
very strong extractant for zinc.
EXAMPLES 11-13
These compounds of Formula 1 were prepared by the methods
of Example 10, except that in the final reaction stage the second
purification step was omitted as unnecessary. The compounds together
with their performance in Test l are set out in Table 1. The results
show that the products of Examples 6-10 and Example 13, in which two
phenoxy groups having 2-tert-alkyl substituents are joined to the same
phosphorus atom are stronger extractants for zinc than the products of
Examples 11 and 12.
1770S4
09511S329 PCTIGB94/02485
17
EXAMPLES 14-24
The products of these Examples, which are set out in Table
1, each contain a substituted phenoxy group and a phenyl group
attached to the same phosphorus atom. They were prepared by reacting
the chloro compounds set out in Table 1 with the corresponding amino
compounds set out in Table 1 in the presence of sodium hydride. The
chloro compounds required for these reactions are prepared by the
methods set out in Examples 1-13. The amino compounds required for
these reactions are prepared by reacting phenylphosphonothioic
dichloride (Ph.PS.Cl2, obtainable from Jansen Chemical Company)
firstly with the ap~lopLiate substituted phenol and secondly with
ammonia by procedures detailed for Example 24 which follow.
Sodium hydride (0.60M, 14.4g) was added to a solution of
2-tert-t-butylphenol (0.6M, 90.0g) in tetrahydrofuran (250 cm3) during
10 min at -40C. This solution was then added during 30 min. to a
solution of thiophosphoryl chloride (0.60M, 101.7g) in hexane (250
cm3) keeping the temperature throughout at -40C. The solution was
stirred at this temperature for a further 30 min. A solution of the
sodium salt of 2-sec-butylphenol was then prepared from 90.0g of the
phenol, sodium hydride (14.4g) and tetrahydrofuran (250 cm3) and added
to the reaction mixture, again m~int~;n;ng the temperature at -40C.
The mixture was allowed to warm to room temperature. Hexane (250 cm3)
was added and the mixture was extracted with water (three 200cm3
portions). The hexane solution was dried with magnesium sulphate and
concentrated by evaporation of hexane under reduced pressure yielding
an oil (231g) which is the chloro compound of Example 24. By 31p NMR
the product prepared in this way contained < 9~ of the two isomers
having identical phenoxy groups.
Sodium hydride (0.6M, 14.4g) was added to a stirred
solution of 2-tert-butylphenol (0.60M, 90.0g) in tetrahydrofuran (250
cm3), in portions during 10 minutes so as to keep the reaction
temperature below -40C. This solution was then added during about 40
min. to a solution of phenylphosphonothioic dichloride (0.60M, 126.6g)
in hexane (250 cm3) held at -40C. The mixture was then allowed to
warm to ambient room temperature during about 1 hr. Analysis by HPLC
indicated that reaction was complete. Ammonia was bubbled through the
mixture for about 45 min. (raising the temperature from ambient to
40C) when HPLC showed conversion of all the chloro compound to amino
compound. The mixture was extracted with water (three 200cm3
portions), and the organic solution was dried with magnesium sulphate
and filtered and the hexane was evaporated under reduced pressure
leaving an oil (178g) which crystallised on st~n~;n~, which is the
amino compound of Example 24.
W 095/1S329 ~ PCT/GB94/02485
~177~6~ 18
The chloro compound described above (198.3g), and the
amino compound also described above (152.5g) were dissolved in hexane A
(2~0 cm3) and tetrahydrofuran (250 cm~). Sodium hydride (30.0g) was
added during 15 min. at room temperature. After addition was complete
the mixture was stirred and boiled under reflux at 60-70C for 18
hrs. The mixture was cooled to room temperature and isopropanol (80
cm3) was added cautiously (frothing occurred) to destroy excess sodium
hydride.
The product was purified and isolated as follows: the
reaction mixture was extracted twice with 300 cm3 portions of a
solution comprising methanol (540 cm3) and water (60 cm3). The
methanolic solutions were combined, and extracted with hexane (200
cm3), and the hexane extract was discarded. The methanol solution was
acidified with concentrated hydrochloric acid (lOOcm3) and water (100
cm 3) was added. The product was extracted into hexane (400 cm3) and
the hexane was washed with three 100 cm3 portions of methanol/water
made up as previously described. The hexane solution was dried with
magnesium sulphate and the hexane was evaporated under reduced
pressure leaving an oil (213g) which is the product o~ Example 24. The
purity was estimated by titration as 89.1~ of theoretical for M.W.
665. 31p NMR in CDCl3: a triplet of doublets centred at 45.lppm
(phosphorus 1) and a doublet of multiplets centred at 65.0 ppm
(phosphorus 2); measurements are downfield of phosphoric acid.
The performance of the products of these Examples in Test
1 which is set out in Table 1 shows them all to be very strong
extractants for zinc. The product of Example 24 was found to have
particularly high solubility in hydrocarbon solvents.
~XAMPLE 25
This Example describes the preparation of a compound o~
Formula 1 in which one phenyl group is attached to each phosphorus
atom.
The chloro compound listed in Table 1 was prepared by
reacting 2-tert-butyl-4-methylphenol (l.OM, 164.3g) with sodium
hydride (1.0 M, 24.0g) and then with phenylphosphonothioic dichloride
(1ØM, 211g) as described in Example 24. On this occasion, the
reaction solution was not treated with ammonia but, instead, was
concentrated by evaporation of the tetrahydrofuran under reduced
pressure, and the concentrate was redissolved in ethyl acetate (400
cm3) and the solution was extracted with water (three 100 cm3
portions). The ethyl acetate solution was dried with magnesium
sulphate, filtered , and again concentrated yielding the chloro
compound which was a crystalline solid (281g, m.p. 71-73C).
~ 095/15329 21 7 7 ~ 6 ~ PCT/GB94102485
lg '
Part of this chloro compound (169.3g) was reacted with
ammonia as described in Example 1 to give the amino compound listed in
Table 1 tl30g, m.p. 108.5-111C). This amino compound (0.2M, 63.8g)
was then reacted with a further part of the chloro compound (0.2M,
67.7g) and sodium hydride (0.5M, 12.0g) in tetrahydrofuran (400 cm3)
solution. It was necessary to heat the solution at 50-60C for two
hours to complete the reaction. The product was purified and isolated
by the procedure of Example 1 but using an aqueous methanol solution
of altered composition, i.e. 90 parts methanol to 10 parts of water,
yielding the compound of Formula 1 in which R1=R3= 2-tert-butyl-4-
methylphenoxy and R2=R~= phenyl (60.6g, purity 80~ of theoretical for
M.W. 621). The behaviour of this compound in Test 1, which is
enumerated in Table 1, shows it to be a stronger extractant than the
product of Examples 1-5.
Exam~le 26
Firstly this Example describes the preparation of 2-
methyl-4-tert-nonylphenol.
Secondly this Example describes the preparation of
diphenylphosphonothioic amide (Ph2PS.NH2) and its use in preparation
of a compound of Formula 1 in which two phenyl groups are attached to
the same phosphorus atom.
Propylene trimer (2.OM, 252g) and 2-methylphenol (2.OM,
216g) and an activated Fullers earth catalyst (FULLCAT 22B supplied by
Laporte Industries, 5.4g) and phosphoric acid (4 drops) were stirred
and heated at 80C for 48 hours. The mixture was then allowed to cool,
filtered and distilled, yielding 2-methyl-4-tert-nonylphenol (245g) as
the fraction of b.p. 114-132C under a pressure of 0.2-0.3mm of
mercury.
Thiophosphoryl chloride (0.45M, 76.9g) was added to
chloro~;ph~nylphosphine (0.45M, lOOg) in nitrogen atmosphere during
ten minutes so that the reaction temperature did not rise above 70C.
The solution was allowed to stand for 18 hours and then distilled
yielding diphenylphosphonothioic chloride (100.6g) as the fraction of
b.p. 178-180C at a pressure of 0.6mm of mercury. All this compound
was reacted with ammonia by the procedure of Example 1 yielding
diphenylphosphonothioic amide ( 84,3g) which is a white crystalline
solid: 31p NMR in CDCl3, singlet 53.8ppm downfield of phosphoric acid.
2-Methyl-4-tert-nonylphenol was reacted with
thiophosphoryl chloride by the procedure of Example 5, except that
hexane was used as the solvent instead of tetrahydrofuran, to give the
chloro compound listed in Table 1. This chloro compound (O.lM, 56.5g)
was reacted with diphenylphosphonothioic amide (O.lM, 23.3g) and
sodium hydride (0.2M, 8.0g) using the procedure of Example 1 to give
W 095115329 PCT/GB94102485
~177~4 20
the compound of Formula 1 in which Rl5R2= 2-methyl-4-tert-nonylphenoxy
and R3=R45 phenyl (66.lg, purity 88~ of theoretical for M.W. 761). The
result of Test 1 shows this compound to be a stronger extractant for
zinc than the products of Example 25, teaching that stronger
extractants are obtained when two phenyl groups are attached to the
same phosphorus atom than when each phenyl group is attached to a
different phosphorus atom.
Exam~le 27
The procedure of Example 26 was used to prepare 2-sec-
butyl-4-t~t-nonylphenol (b.p. 148-150C at a pressure of 1.5mm of
mercury) from 2-sec-butylphenol and propylene trimer. This compound
was further reacted with phenylphosphonothoic dichloride using the
procedure of Example 24 to give the chloro compound listed in Table 1
(31p NMR in CDCl3, multiplet 82.6ppm downfield of phosphoric acid).
This chloro compound was then reacted with diphenylphosphonothioic
amide using the procedure of Example 26 to give the compound of
Formula 1 in which Rl= 2-sec-butyl-4-tert-nonylphenoxy and R2=R3~R4=
phenyl. This compound was subjected to Test 1 with the result listed
in Table 1 which shows it to be a strong extractant for zinc.
217706~
~/0 95115329 PCT/GB94/0248S
21
4~ V
o ~3 C
.~ a
. ,¢~ ~
E~ ~ ~1 ~ o u~ o
f~ 0 o n ~ r u
n 0 0 ~ ~ n o
z E~ EQ '
U~ N H-- 0~n
a~
.c
~ 0 In ~ 0 ~ O ~ ~ In ~ ~ ~ ~ ~1
C ~ a~ 0 0 o~ 0 o r o 0 o 0 o 0 ,~ 0 In
~ ~ E E tJl~ E ~ E~ E E E E ~ F U1 E
C
u .~ E~
'' H o~ o~ o\ o\O o\O o\O o\O o\O o\O o\O o\O o~) m o ~ 0 t~ o
0 ~D 0 U~ ` 0 0 O~ 0 0
-
~ , , ,
C~ I I Ul ~Ln " Ul `~ I
a) -' ` ' ~ V . . v . O
Q - ~ Q ~ y ~
Z 1~ ~ I Ir I ~
~ ~ U ~ U U 1 5J U
a~ ~ X ~ u ~ a~ a) a~ ~ a~
. Z ~ ~ -, . -, . u . ' a~ ' ' . I ~ m
S ~DJ
U O -
-'' Z ~ ~ ~ I In ~
H ~ -Q ~ ~ Q ~ -- ~ _
'C 't7 ~ ~ Ul '~ ui Ui ~.)i Vl~ hl '.
. a,) . ~I) Q) ~I) ~ Il)
~, . ~ , . ~ m ., ~ m m m ~ ' i . .
~_~
I I
~ ~ ~, . ' V
u ~U¦ ~' ~ Ul q '-~ ~¦ h~ ~ h¦ ~ "I
~_ ~ ,. m ~,. ~m J~ v ~ . J- . m
5~ m ~0 ~
O . V ~n ' ~ ,, ~" ~, :~ >. >~ ~ .~ v
0~ 3 Q
- ' ~1 ~ 'Ul ~r~ ~ V
. m l l- l ~- l.l I 1. 1 1 1 1 a~ I I I I
~4 s
~3
0 ~ O ,,
~ z
E~
WO 9S/15329 2 ~ 7 7 0 6 ~ 22 PCIIGB94/0248!;
o o Ul o o o o o o o o
D u~ ~ o c
V ~ F
Z Q
W H--
~ a~o o~ ~ ,~ ` O~D ~ 0~ t-~ oa~
~ . . . . . . . . . . . . . . .
Z U~ t~ O cl:l N t` O Itl ~ I` ~ 1` 0 ~ a~ 0 'd'
F f F ~12 U~ ~ ~ F F ~ ~ E F ~ El E~ F
o
H o~ o\ ol~o o~ o\O o\O o\O o\O o\O o\O o
r c~ 0 c~
C~
~ >~
a
~:~ a) I :
Z un U
'~~ 'J '~ ~ S
O
z ~r ~. U') Lf)
~ I t I I I I ~ I I .,
>~ ~ . ~ ~ ~ V I . . I
_l , , Q R ' ` ~- ~ . . ,
~ u o s.~l ~ts~ Vl 1~
a~ ~ IS a) a~ ~ ~ a~ S 1~ I a) .'
U~ V ~ I~ JJ J- Ul V ~ ~ ~ LJ V .
,, ,, ,, ,, I o `E ' ~ '
E t~ ~ N E ~ t~ N
~t ~ ~ ~ ~t ~t ~ ~ ~t
~ u 5~ 'u ~ V~
G) a) a) s ~ a~ ~ 1. a) ~ ~
I v ~ v ~ . ~ . ~ v u~ , ~n v . ~ .
V
I `~ I " I `- I l l " I I '.
~ >- ~ ~ ~ >, >,
r~ ~ S l ~ ~ h ~ I S~ S~ ~ I Ul ~¦ 5~
a~ SI S o S I I a~ ~ . I l l ~1
J_l L~ V V V V V I ~ JJ . b~ U~l Vl ~1 .
~`i ~ E ~1~1 E ~ ~ E ~ . t~ N ~ ~ U'l
~3
~: m ~ ~ o ,~
~Z
~1~706~
¦~10 95/15329 PCT/GB94/02485
23
o o Ln o o
VE~ E
z Q
H Z
1~;1 H--
0 ~ ~1 a~ ~ ~ ~ u~
U~ ~D ~ ~0
3 r~
o ~ ~ tq F
E~
H o\O o\O o\O o\O o\O
o 0 a~
a~ co 0 0
a 'T~ '~
~Z
, cq ~ a~
o~
Z .,~ ~
H ~ I I I "
5' ~1 ~1 ~
Q Q
V ~ ~ ,,
~ a) a) ,
V JJ J .
tY ~ N ~
N ~^1 ` , ~ ~1 ,
. I O ~, , I
a) ,,, .
U~
a
Z N . N .
~ I
o ca s-, ~
c: 11 a) , . '
0~ 1 , ,, ~
_, ~, l 1_' ~ ~
V V ,~ o U~
~ a5 N ~ C (~
E~
W
N ~ C~l
c3æ
W O9S/lS329 , . - PCTIGB94102485
2~ 77~6~ 24
EXAMPLE 28
In order to exemplify further the utility of materials of
this invention for the extraction of metals, a distribution curve was
obtained for the extraction of zinc from an aqueous metal bearing
solution by an organic solution of the extractant. This was done by
equilibrating various volume ratios of extractant and metal bearing
feed solutions, separating and analysing for the metal contained in
each phase.
An organic solution was prepared containing 0.5 moles per
litre of the extractant of Example 24 in the hydrocarbon solvent
Escaid 100. A simulated high concentration zinc feed solution was
prepared that contained 20.1 g/l zinc, 12.5 g/l ferric iron, 0.47 g/l
calcium, and 2.6 g/l magnesium in an aqueous sulphate medium at pH
1.8.
In a series of experiments, various volume ratios of the
organic extractant solution and aqueous feed solution were
equilibrated by vigorous stirring at 25C for a period of 24 hours.
The phases were then allowed to disengage, separated, filtered and
analysed for zinc. The distribution of zinc between the organic and
aqueous phases after contacting at various volume ratios was found to
be as follows:
TABLE 2
Volume Organic Volume aqueous Zinc in Zinc in aqueous
contacted (ml) contacted (ml) organic (g/l) (g/l)
5.19 2.31
8.46 5.10
11.64 10.75
13.06 14.86
13.76 18.44
14.28 21.00
= 35 These data demonstrate the ability of the reagent to
attain both high loadings of zinc in the organic phase and high
recovery of zinc from the aqueous phase. In practice this would be
achieved by a number of equilibration stages with the organic and
aqueous flows running counter current wise.
In a solvent extraction process for the recovery of a
metal, it is essential that the extractant is not only capable of
extracting metal efficiently from the aqueous feed solution, but that
the metal can be recovered subsequently from the metal loaded organic
095/lS329 25 PCTIGB94/02485
phase by a stripping operation. Ideally, for use of the extractant in
a process for zinc recovery based on solvent extraction combined with
electrowinning, it is desirable that the stripping is carried out with
an acidic aqueous solution such as a spent electrowinning electrolyte.
In order to demonstrate this, a portion of the extractant solution of
the composition described in the first part of this Example was loaded
with zinc by contacting for at least 12 hours one part by
volume with four parts by volume of the aqueous feed solution also
described in the first part of this Example.
Portions of this zinc loaded organic solution were then
contacted at various volume ratios with an aqueous strip solution
containing 30g/1 zinc and 180 g/l sulphuric acid. Contacting was
carried out by vigorous stirring at 50C for 2 hours. The phases were
then separated and each analysed for zinc. The distribution of zinc
after stripping at various volume ratios was found to be as follows:
TABLE 3
Volume organic Volume aqueous Zinc in Zinc in aqueous
contacted (ml) contacted (ml) organic (g/l) (g/l)
5.63 31.90
7 6.98 47.95
8.58 56.05
EXAMPLE 29
In order to exemplify further the ability of compounds of
the invention to be able to extract zinc with high selectivity over
iron and for the zinc to be stripped subsequently with acidic zinc
electrolyte solution, a number of compounds were loaded with zinc, and
then stripped as described below.
One part of a 0.5 molar solution of extractant in Escaid
100 was contacted by vigorous stirring for 24 hours at 25C with four
parts of the aqueous zinc feed solution of composition given in
Example 28. The phases were allowed to disengage, separated, the
organic filtered and analysed for both zinc and iron.
A portion of the zinc loaded organic phase was then
contacted with an aqueous strip solution as used in Example 28 in the
ratio of 15 parts of organic to 7 parts of strip solution. Contacting
was carried out by vigorous stirring for 2 hours at 50C. After the
phases had disengaged, the phases were separated, filtered, and
analysed for zinc. The results for the compounds tested in this way
are as follows:
W 095/lS329 217 7 0 6 4 PCTIG~94102485
26
TABLE 4
Extraction into Stri~inq
Compound of orqanic ~hase Zinc in Zinc in
Example No.Zinc extracted Iron extracted aqueous (g/l) organic (g/l)
(g/l) mg/l
16 14.25 42 56.37 3.92
24 13.76 413 47.95 6.98
13.63 75
26 17.34 113 57.88 5.1
27 18.03 150 52.80 l.9
These results clearly show the ability of solutions o~ the
extractants to extract zinc, with high selectivity over iron, and to
transfer it satisfactorily to an acidic strip solution.
EXAMPLE 30
Solutions were prepared containing (A) 0.5 moles per litre
of the extractant of Example 24 in the hydrocarbon solvent Escaid 100.
In addition, one of the solutions (B) contained 50 g/litre (0.25
molar) of isotridecyl alcohol added as modifier. Yet a third solution
of the extractant (C) contained as modifier 72 g/l, being 0.25 moles
per litre, of the ester 2,2,4-trimethyl-1,3-pentanediol diisobutyrate
available commercially under the name KODAFLEX TXIB (Eastman Kodak).
Portions of each of these extractant solutions were
contacted by vigorous stirring for 24 hours at 25C with the aqueous
zinc feed solution described in Example 28 and in the ratio of 10
parts of extractant solution contacted with 20 parts of aqueous feed.
The phases were allowed to disengage, separated and portions set aside
for analysis.
Portions of each organic phase, pre-loaded with zinc feed
solution were then stripped by contact at an organic to aqueous phase
ratio of 10 parts organic to 20 parts aqueous using the conditions
described in Example 28. After 2 hours, the phases were allowed to
disengage, separated and analysed. The results, in terms of zinc in
the organic phase after extraction and again, after stripping, are as
shown in the table below.
O9S/lS329 ~ 1 7 ~ ~ 6 ~ PCT/GB94102485
27
TABLE 5
Modifier ~inc in loaded Zinc in stri~Ded Zinc
extractant (~/1) extractant (q/l) transferred (q/l)
A) None 13.06 5.63 7.43
B) isotridecylalcohol 12.39 3.94 8.45
C) Kodaflex TXIB (ester) 11.98 3.18 8.80
These results show that under the same strip conditions
for each extractant composition, more zinc is removed at the strip
stage in the presence of the modifier than when the modifier is
absent. There is also some reduction in the amount of zinc loaded at
the extraction stage, but this is outweighed by the improvement at
stripping, so that there is a net gain in the amount of zinc
transferred between the extraction and stripping stages.
EXAMPLE 31
This Example demonstrates the preparation of a compound of
Formula I in which two optionally substituted 2-tert-alkyl~hennxy
groups are attached to the same phosphorus atom and two optionally
substituted alkyl groups are attached to the other phosphorus atom.
The Example describes the preparation of bis-(2-
pentyl)chlorothiophosphate, which is the chloro compound listed in
Table 6, and the reaction of the chloro compound with O,O -(bis-2-
tert-butylphenoxy)thiophosphoramide, which is the amino compound
listed in Table 6 :
A solution of 2-pentylmagnesium bromide was prepared by
adding 2-bromopentane (2554.74g, 16.93M) to magnesium turnings (422g,
17.6M) in diethyl ether over a period of 2~2 hours. The temperature of
the reaction mixture was maintained at 38-40C by external cooling.
The diethyl ether solution of 2-pentylmagnesium bromide was then added
to a solution of phosphorus trichloride (946g, 6.88M) in diethyl ether
(946 cm3) during 3 hours. Throughout the addition, the temperature of
the reaction mixture was m~;nt~;n~d at -20C or lower by external
cooling. After the addition was complete, the reaction mixture was
stirred at 0C for 2 hours, over which time, a greyish solid resulted.
Water (3000cm3) was added to the reaction mixture over a period of 2
hours and the temperature was allowed to rise freely to 20C.
The diethyl ether solution was separated and washed with
brine (1 x 2000cm3), then with brine/Na2Co3 (15~ wt Vol NaCl, 2~
wt.vol. Na2CO3; 5 x 3000cm3) and again with brine (1 x 2000cm3). The
W 095tlS329 j PCT/GB94102485 ~
2177~64 28
diethyl ether solution was then concentrated by evaporation under
reduced pressure. Low boiling impurities were removed by heating the
residue under reduced pressure (25 mmHg) at a temperature of 80C for
30 minutes. The clear oil (541.2g) was taken as the product bis-(2-
pentyl)chlorophosphine 31p, n.m.r in CDC13, singlet at 130.7 ppm.
Thiophosphoryl chloride (90.Og) was added dropwise to the bis-(2-
pentyl)chlorophosphine, under a nitrogen atmosphere, over a period of
90 minutes. The temperature of the mixture rose to 45C over this
time. The reaction mixture was then cooled to ambient temperature and
stirred for a further 2 hours. The PCl3 by-product formed was
distilled from the reaction mixture under reduced pressure. The
remaining clear oil was taken as the product, bis(2-
pentyl)chlorothiophosphate, 31p n.m.r in CDCl3, singlet at 120.6 ppm.
The amino compound, O,O'-bis(2-tert-
butylphenoxy)thiophosphoramide was prepared from bis(2-tert-
butylphenoxy)chlorothiophosphate by the method of Example 1, and O,O'-
bis(2-tert-butylphenoxy)chlorothiophosphate was also prepare by the
method of Example 1.
The compound of Formula 1 was prepared by reacting bis(2-
pentyl)chlorothiophosphate and O,O'-bis(2-tert-
butylphenoxy)thiophosphoramide by the method o~ Example 1. The
compound isolated was a brown oil (44.04g). The purity of the
compound was estimated by titration as 89~ of theoretical for M.W. of
580.6. 3lp n.m.r. in CDCl3, having two sets of multiplets centred at
41s80 ppm and 90.85 ppm. The performance of the product of this
Example in Test l, which is set out on Table 6, shows it to be a very
strong extractant for zinc.
Exam~le 32-33
These compounds of Formula I were prepared by the methods
of Example 31. The compounds together with their performance in Test
1 are set out in Table 6. The results show that the compounds pf
Examples 32 & 33 are weaker extractants for zinc than the product of
Example 31.
ExamDle 34
This Example describes the preparation of compounds of
Formula 1, wherein R3 and R~ are alkoxy groups and Rl and R2 are 2-
alkylphenoxy groups. The Example describes the preparation of O,O'-
(bis-2-ethylhexyl)chlorothiophosphate, its conversion to phosphoramide
and the preparation of O,O'-(bis-2-~ -butylphenoxy)thiophosphoryl
chloride. The final stage is the reaction of chloro and amino
compound to produce the compound of Formula I where Rl~ RZ= 2-
ethylhexyloxy and R3=R~C 2-tert-butylphenoxy.
~ 09S/15329 2 17 7 0 ~ 4 PCT/GB94/02485
To 0,0'-(bis-2-ethylhexyl)phosphorodithioate (0.7M, 254g),
sulphuryl chloride (l.lM, 150g) was added during 2 hours. The
temperature rose to 40C over this time. The reaction mixture was
then stirred at ambient temperature for a further 10 hours, after
which time a sample analysed by GC indicated the reaction was
complete.
The excess sulphuryl chloride was removed by vacuum
evaporation (0.2 mmHg) of the crude product, at ambient temperature,
for 2 hours.
The oil residue was the product 0,0'-(bis-2-tert-
ethylhexyl)chlorothiophosphate (225.24g), 31p n.m.r. in CDCl3 singlet
at 67.28 ppm. The chloro compound (lOOg) was dissolved in
tetrahydrofuran (250 cm3) and ammonia gas was bubbled through the
solution over 2 hours. Analysis of a sample by GC showed complete
lS conversion of the chloro compound. Precipitated ammonium chloride was
filtered from the mixture and the tetrahydrofuran solution was
evaporated under reduced pressure to leave O,O'-(bis-2-
ethylhexyl)phosphoramide (85.45g) as an oil. 31p n.m.r. in CDCl3
singlet at 70.64 ppm.
The amino compound (0.2M, 67.4g) was reacted with 0,0'-
(bis-2-tert-butylphenn~y)chlorothiophosphate, prepared as described in
Example 10, (0.2M, 79.3g) and sodium hydride (0.46M, 11.06g) in
tetrahydrofuran (500cm3) and refluxed for 12 hrs. A~ter purification
and isolation as described in Example 1, the reaction product was
obtained (90.3g), which was found by titration to be 89~ pure. The
purity of the sample was further improved to 93~ by redissolving the
product in hexane and washing the solution with a methanol/H20 (90~
Vol : 10~ Vol) mixture 4-5 times. The purified product was isolated
by evaporation under reduced pressure. 31p n.m.r. in CDC13 : a doublet
of doublets centred at 41.75ppm & 59.23ppm. The performance of the
product in Test 1, which is listed in Table 6, showed it to be a very
strong extractant for zinc.
Exam~les 35-38
The compounds of Formula I, listed in Table 6, were
prepared by the methods of Example 34 using, where a~Lu~Liate~ 0,0'-
(bis-1,3-dimethylbutoxy)phosphorodithioate, O,O'-(bis-
ethoxy)phosphorodithioate, O,O'-(bis-n-propoxy)phosphorodithioate,
O,O'-(bis-isopropoxy)phosphorodithioate and 0,0'-(bis-2-tert-
butylphenoxy)chlorothioate as the starting materials. The chloro and
amino compounds were then reacted together, by the methods of Example
1 to give compounds of Formula 1. The compounds, together with their
performance in Test 1, are set out in Table 6. The results show that
W 09S/lS329 ' PCTIGB94/0248~ ~
21~7~4 30
the compounds of these Examples have strengths similar to the compound
of Example 34.
~xamDle 39
This Example demonstrates the preparation of a compound of
Formula I in which 2-tert-butylphenoxy and isopropoxy groups are
attached to the same phosphorus atom.
Preparation of a chloro compound for this reaction was
carried out as described as in Example 10.
A suspension of sodium hydride (l.OM, 24g) in
tetrahydrofuran (150cm3) was added portionwise to a stirred solution
of 2-te~t-butylphenol (150g) in tetrahydrofuran (250cm3) over a period
of 2 hours, whilst the temperature was maintained below 30C by
external cooling. This solution was then added during 2 hours to a
stirred solution of thiophosphoryl chloride (l.OM, 169.4g) in
tetrahydrofuran (350cm3) whilst the reaction temperature was
maintained at -50C. The reaction mixture was allowed to warm up to
ambient temperature to complete the reaction, and after 30 minutes,
was cooled to -30C. To this solution was then added a solution of
sodium isopropoxide prepared by adding a suspension of sodium hydride
(l.OM, 24g) in tetrahydrofuran (150cm3) to a solution of commercial
isopropanol (60g) in tetrahydrofuran (250cm3). The addition lasted 2
hours and the temperature was maintained below -30C. The reaction
mixture was allowed to warm to ambient temperature after which
analysis by GC indicated that reaction was complete. The
tetrahydrofuran was removed by vacuum evaporation and the residue was
diluted with diethyl ether (lOOOcm3). The diethyl ether solution was
washed with water (2 x 500cm3 portions), dried with magnesium
sulphate, filtered and concentrated by evaporation of the diethyl
ether under reduced pressure. The re~in-ng oil was substantially 0-
(2-tert-butylphenyl)-0'-(2-isopropyl)chlorothiophosphate. 31p n.m.r
in CDCl3 singlet at 57.63ppm. This compound (52.42g) was dissolved in
tetrahydrofuran (350cm3) and ammonia gas was bubbled through the
solution for 2 hours. Analysis of a sample by GC showed complete
conversion of the chloro compound. Precipitated ammonium, chloride
was removed by filtration, and the tetrahydrofuran solution was
evaporated under reduced pressure to give a brown oil (42.00g) which
was the expected amide.
The amide (O.lM, 28.7g) was reacted with the chloro
compound (O.lM, 30.65g) and sodium hydride (0.23M, 5.52g) in
tetrahydrofuran (150cm3). The reaction mixture was heated under
reflux for 12 hours until analysis by HPLC indicated that the reaction
was complete. The product was purified and isolated as described in
Example 1. The reaction product obtained (30.7g) was found by
095/15329 ~ 17 7 0 ~ 4 PCT/GB94102485
titration to have a purity of 70~ for a M.W. of 557. The product was
further purified by dissolving it in hexane (250cm3) and washing the
solution with a mixture of 90~ vol MeOH and 10~ vol H20 (4-5 x 100cm3
portions). The hexane solution was dried over magnesium sulphate,
filtered and the hexane was removed by evaporation under reduced
pressure to give a brown oil (15.25g), which was found by titration to
be 89.7~ pure : 31p n.m.r in CDCl3, a singlet (with fine splitting) at
50.05 ppm.
The performance of this compound in Test 1, which is
enumerated in Table 6, shows it to be as strong as the products of
Examples 1-5.
Exam~le 40
The chloro compound listed in Table 6 was prepared by the
method of Example 39. The amino compound listed in Table 6 was
prepared by the method of Example 34. The chloro and amino compounds
were reacted together by the methods of Example 1 to give the compound
of Formula I having the groups Rl - R4 listed. The purity of the
compound was estimated by titration as 86~ for MW of 557.
Exam~le 41
This Example demonstrates the preparation of a compound of
Formula I, wherein R~ is a 2-alkylphenoxy group, R2 is a phenyl group
and R3 and R4 are alkoxy groups.
The chloro compound listed in Table 6 was prepared by the
methods of Example 24. The amino compound listed in Table 6 was
prepared by the methods of Example 34. The chloro and amino compounds
were reacted together by the methods of Bxample 1 to give the compound
of Formula I listed in Table 6. The purity of the compound was
estimated by titration as 91~.
The performance of this compound in Test 1, which is
enumerated in Table 6 shows it to be a weak extractant for zinc.
Exam~le 42
This Example demonstrated the preparation of a compound of
Formula I by the methods described in Example 1. The chloro compound
listed in Table 6 was prepared by the methods of Example 31. The
amino compound was prepared by the methods of Example 24. The chloro
and amino compounds were reacted together by the methods of Example 1.
The purity of the compound having Rl - R4 listed in Table 6 was 84~
for a compound of MW 509. The behaviour of this compound in Test 1,
is enumerated in Table 6.
W 09S/lS329 PCT/GB94/02485
~1770~ 32
Exam~le 43
This Example demonstrates the preparation of a compound of
Formula I by the methods described in Example 1. The chloro compound
listed in Table 6 was prepared by the methods of Example 39. The
amino compound listed in Table 6 was prepared by the methods of
Example 10. The chloro and amino compounds were reacted together by
the methods of Example 1. The purity of the compound having R1 - R4
listed in Table 6 was 76~ for a compound of MW 647. The behaviour of
this compound in Test 1, is enumerated in Table 6.
~17~06~
~0 95/15329 PCT/GB94/02485
33
-~ o u~ o Ln o o o In o o
~ ~n Ln ~ ~ ~ o ~ a~ ~D ~1 0
Z Q
H Z ~
~D D LO
' ~ O ~D O ~ 1` ~1 a~ ~I t~ ~ Ln ~ In ~ ~ In o
a -- E E E E E E E E E E '0~ E E E E ~ ~
Ho\ o\ o\ o\ o\ o\ o\ 0\ 0\ \
~D ~7 a~ o o ~ o ~
3 0 ~ ~ 0
. o
V ~s . C
~S Q
C N ~ R ~ l ` ~ ~I X
~) Z h t~l h ~ O `,
~L) ~ O V ~
~ U~ L) ~ I Ll q) X - - ~ 3 , v
D~
,.7' U~ " X X
~~ , , , o o
P~ ~ ~ ~ ~ ~ ,,
E~ V . . X . `~
R , . L: , ,
a)¦ v h¦ r
v v. ~v a) . ~. v a)
I
v . . V .
S R , , R
hl hl vhl hl L
~.I ~ I ~ a)
~ L .LJ L V L _ _
Z ~ '. ~ ~ ~ . il
~ V .
O U~ . . . V
1V Ll V V V V V
h l I h h h h
va I a a~a~ a~ ~
1~ V L L V V V V
~3
tr:
~; m_, ~ ~ ~ u- ~ t~ a) a~ O
~,3æ
WO9S/lS329 21 770~ 34 PCTtGB94/02485
~ Ln U7
p U~ ~D~ O O
V E~
æ ~ ~0
H Z ~ -rl Ul
N H--
O ~D O ~`
E ~ 0 ~L
pV ~ ~ ~~ a~
o ~ ~E E ~ ~
C~
Cll E~
H o~Oo~O o\O
X~
>
a ~
~ æ c~ s
Il I V I r
æ . ~
~ o
`
,~, ~ "
V
a~ ~ v
N t'`l
~: ~ ~
- ~ - J '~
~ V ~ ~ ~
O U~ ~ ~ >t
O ~ _ _ I
V ~ hl hl h¦
~ I
~: ~ V V
~ C 3
~æ ~ ~ ~
~3 Z
~17706~
O95/15329 PCTIGB94/02485
Exam~le 44
An aqueous solution cont~;n;ng up to 350 parts per million
of each of 15 different metals or metalloids and sufficient nitric
acid to give a pH value of 2.0 was made up. The metals were taken as
their nitrate or acetate salts except for arsenic which was taken as
the trioxide. This solution was stirred rapidly with a 0.1 molar
solution of the product of Example 24 in ESCAID 100, for one hour at
20-25C. The a~ueous and organic phases were separated and each was
analysed for metals content with the results tabulated below :
Metal Concentration of metal found, in Darts ~er million
In the a~ueous Dhase In the orqanic Dhase
Zinc(II) 35 365
Bismuth(III) cl 40
Manganese(II) 305 cl
Cadmium(II) cl 375
Chromium(III) 250 2
Copper(II) cl 315
Calcium(II) 285
Mercury(II) cO.l 175
Arsenic(III) 20 c5
Silver (I) cl 335
Lead(II) 7 90
Iron(III) 195 19
Magnesium(II) 290 cl
Nickel(II) 305 cl
Cobalt(II) 330 cl
The results show that at an initial pH value as low as
2.0, zinc, bismuth, cadmium, silver and mercury are strongly extracted
and may be separated from the other metals listed.
ExamDle 45
In a separate test, a 0.1 molar solution of the product of
Example 26 in ESCAID 100 was stirred for one hour at 20-25C with the
mixed metal solution of Example 44. the aqueous and organic phases
were separated and the organic phase was analysed for metal content
with the results tabulated below :
W 095/lS329 ~17 7 0 ~ 4 36 PCT/GB94/02485 ~
Concentration of metal found in Parts ~er million
Mçtal In the oraanic ~hase
Iron~III) 60
Chromium(III) <l
Manganese~II) <1
Zinc(II) 315
Nickel(II) <1
Arsenic(III) <2
Calcium(II) <1
Magnesium(II) <l
Cadmium(II) 340
Copper(II) 305
Bismuth(III) 40
Mercury(II) 180
Silver(I) 290
Lead~II) 100