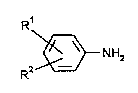Note: Descriptions are shown in the official language in which they were submitted.
2 1 78 700
LeA 31 091 -Foreign Countries
A PROCESS FOR PRODUCING AROMATIC
AMINES BY GAS PHASE HYDROGENATION
AND A CATALYST USEFUL THEREFOR
BACKGROUND OF THE INVENTION
The present invention relates to an improved process for
producing aromatic amines by catalytic h~dluu~lldliùll of ~ ludlullldliu
compounds in the gas phase and to a new catalyst which is useful in this
process.
Aromatic amines are known to those skilled in the art as important
intermediates for the production of dyes polyurethanes and plant
protection products.
Various procedures for the hyd~uytlndlioll of ~ u~ el1e and
other ~ UdlUllldli~ compounds are known to those skilled in the art. Due
to the high enthalpy of reaction which is released during these known
~, u~ sesl they are usually conducted in reactors equipped with
. integrated heat transfer systems. Examples of some of these known
procedures are hyd, U9t:1 Idliul~ in the liquid phase on suspended catalysts
such as Pd catalysts (described in EP 476,404) h~lug~l~dliullin the gas phase onfluidized solid catalysts (disclosed in U.S. 3136 818) h~ u~lldliul1in
the gas phase over fixed catalysts such as supported Pd catalysts
(described in DE-A 2,244 401 2 849 002 and 4,039 026).
DE-A 2 244 401 and 2 849 002 describe Pd catalysts on alumina
supports which are operated as fixed catalyst beds in heat e,~..l Idl ,g~,
tubes under normal pressure at loadings of less than 1 9 I ~ ub~ e
25 (NBz)/ml (cat)/hour at low hydrogen/nitrobenzene ratios. Between 6 and
11 moles of hydrogen are used for each mole of llillu~
DE-A 4 039 026 describes Pd catalysts on graphitic supports
which are operated under conditions similar to those used for Pd
catalysts on alumina. These ca alysts provide ill~ulll~ conversion at
Le A 31,091 -2- 2, 7~700
loadings cullsid~ldL,ly less than 1 9 (NBz) / ml (cat)/ hour and at a
hyd~ug~ ' u~e,l~"e ratio of from 14 to 26 moles to 1 mole. Between
1000 and 4000 ppm r, ' uue"~ e, with respect to the aniline formed, are
fûund in the colldt:, ISdl~.
Both an increase in the loading of lli~lUdlUllldLiC compound and an
increase in the ratio of hydrogen to nitroaromatic compound increase the
volume flow through the bed and thus reduce the dwell time on the
contact catalyst. It would therefore be expected that both of these
measures would lead to an increase in the breakthrough of the
llilludlullldlic compound (i.e., to incomplete conversion).
A general measure of this gas flow through the catalyst bed is the
gas hourly space velocity (GHSV), expressed in hour 1.
Even small amounts of lI 'ludlullldlk, compounds in aromatic
amines cause signlficant ~ ,cul~ldliul, of the aromatic amine which is
otherwise colorless. Such nitroaromatic compounds are therefore
u~de~ dule. Separation of l~i~ludlullldliu compounds by distillation is
costly in terms of both equipment and energy.
In each of these known processes, the high heat of reaction has to
be withdrawn from the reactor via an expensive heat transfer system.
H~lu~el~dliu" processes in the gas phase with simple adiabatic
catalyst packed beds are particularly economical, due to the simple
construction of the apparatus with reactors that do not include integrated
heat ~x-:l Idl ,g~l systems. In an adiabatic type of process, the highly
~xull ~e~ iu nature of the hy.ll u~l ldliul~ of nitro groups imposes particular
demands on the catalyst. Due to the col~sid~,dùl~ exothemm, there is a
lar~e temperature difference between the beginning and the end of the
catalyst. In order to control this temperature difference in an adiabatic
process, a heat transfer medium (generally hydrogen in h~dlu~u~lldliull
processes) is admixed with the mixture of starting materials resulting in
very short dwell times or high GHSVs. This means that the catalyst must
2 1 7870(~
L~: A 31 091 3
be both active and selective over a very large temperature range in order
to achieve complete conversion of "il,ube"~ne to aniline even at low
UU~ le loadings.
~iUMMARY OF THE INVENTION
It is an object of the present invention to provide a h~ ugelldli
catalyst which can be more highly loaded than the known catalysts.
It is also an object of the present invention to provide a
h~l ug~l Id~iUI I catalyst which exhibits a higher selectivity than the known
prior art catalysts.
It is another object of the present invention to provide a
hy~JI us ~l Id~iUI I catalyst which can be used in simple reactor
constructions.
It is a further object of the present invention to provide a
hyd,uut:l~d~iu, I catalyst having a long service life.
It is also an object of the present invention to provide a
hydlUy~lldliUI, process in which extraction of heat from the catalyst bed
with an additional heat transfer system is not necessary.
These and other objects which will be apparent to those skilled in
the art are accu",~ 71led by h~dlu~u~:lld~il,g ~, udlurlld~i~; compounds in
:20 the gas phase in the presence of a Pd catalyst on a graphite or a
graphitic carbon support and large excesses of hydrogen. This process
may be carried out in simple adiabatic reactors.
~ETAILED DESCRIPTION OF THE PRESENT INVENTION
The present invention relates to a process for producing aromatic
amines l~pl~ d by the formula
R1
2~3NH2 (1)
R
2 ~ 7~700
~e A 31,091 4_
in which
R' and R2, i~l~e~ d~ y of each other, represent hydrogen, a
methyl group or an ethyl group and R' may also represent
an amino group,
by the hydlu~ lldliull of ~ udlullldli~; compounds rt,u,~su,,~d by the
formula
R~No2 (Il)
in which
R2 and R3, il Id~,ut:l ,de, Illy of each other, represent hydrogen, a
methyl group or an ethyl group and R3 may ad.iiliu, I...ly
1 û represent a nitro group
with hydrogen over fixed catalysts in the gas phase. The catalyst used is
palladium on a graphite or a graphitic coke support. The graphite or
graphitic coke support has a BET specific surface of from û.2 to 1û m2/g.
The catalyst has a palladium content (based on the total weight of
catalyst) greater than 1.5% by weight and up to 7% by weight, preferably
from 1.6 to 6% by weight, most preferably from 1.9 to 5% by weight.
From 3û to 6ûûû, preferably from 5û to 3ûûû, more preferably from 8û to
1ûûû, most preferably from 1ûû to 3ûû equivalents of hydrogen for each
equivalent of nitro groups are fed to the catalyst.
2û The present invention also relates to a catalyst having a graphite
or a graphite-containing coke support. The graphite or graphite-
~.ullldillillg support has a BET specific surface of from û.2 to 1û m2/g.
The palladium is deposited on the support by illl~ ul ldLiol~ with a
suitable palladium-containing solution. The palladium content of the
2 1 7~700
L~! A 31,091
-5
catalyst is from greater than 1.5% (e.g., 1.5001%) by weight up to 7% by
weight (based on the total weight of catalyst). The preferred lower limit
of the Pd content is 1.6% by weight, most preferably 1.9% by weight.
The preferred upper limit of the Pd content is 6% by weight, most
5 preferably 5% by weight.
Graphite-containing materials are used as the support for the
catalysts of the present invention. Suitable support materials include
graphite itself (e.g., t~le-:tlUyld,UIlil~) and cokes such as needle coke 2nd
petroleum coke. These supports have BET specific surfaces of from 0.2
10 to 10 m2/g.
The catalyst is prepared by depositing palladium on the support in
from 1 to 50, preferably from 2 to 30, most preferably from 4 to 10
impregnation steps. ~etween each illl,Ult::~Jlld~iUIl step the catalyst
support is dried in a hot gas stream, preferably a stream of air or
15 nitrogen.
The catalysts of the present invention may be prepared by
depositing palladium in the form of suitable salts on the support material
in the form of tablets, spheres, lengths of granulated material, Raschig
rings, Pall rings, cart wheels or honeycomb structures of d~ameters from
20 1 to 30 mm. A plurality of illl,Ul~JlldliUI~ steps may be used with drying
after each d~posiLiu". Drying is effected in an air current at temperatures
of from 30 to 140C, preferably from 30 to 60CC and preferably under
nonmal pressure. Aqueous and organic solvents and mixtures thereof
may be used for the illlpl~ylld~iull of the support. Examples of solvents
25 which may be used include water, NH3, simple alcohols, amines,
ketones, esters, cyclic ethers, lldlug~lld~t:d hydro-carbons and nitriles.
Specific examples of organic solvents include methanol, ethanol,
propanol, isopropanol, ethylamine, isopropylamine, acetone, methyl ethyl
ketone, dioxane, methylene chloride, acetonitrile and cull~,udldble
ao solvents. Examples of suitable palladium salts include palladium chloride,
2 ~ 7~700
Le A 31,091 -6-
palladium nitrate, palladium acet~ldc~Lu,~d~, palladium acetate and
amine wlllpi~ S of palladium The catalyst pl~:pdldLiùll is preferably
carried out without the use of halogens in the solvent or the metal salt
used. After illlpl~ dliul~ and final drying, the catalyst of the present
5 invention is ready for use.
Before it is first placed in operation, the catalyst is generally
activated by treating it with a flow of hydrogen at 1 to 10 bar and
temperatures of from 250 to 450C (preferably from 300 to 400C) for
from 1 to 50 hours (preferably from 5 to 30 hours).
The hydlug~lld~iun process of the present invention is conducted
at a pressure of from 1 to 30 bar, preferably from 1 to 15 bar, most
preferably from 1 to 7 bar.
The gaseous mixture of starting materials wntaining the
nitroaromatic compound and hydrogen is at a temperature of from 20û to
400C, preferably from 230 to 370C, most preferably 250 to 350C,
upstream of the catalyst bed. The maximum catalyst temperature is
600C, preferably 55~C, more preferably 500C, most preferably 460C.
The catalyst of the present invention may be used in reactors
without a system for the removal of heat.
The short dwell times or high GHSVs of the process of the present
invention are particularly l~,,,dlhd~le. These short dwell times make it
possible to use catalyst loadings of from 0.5 to 40 kg, preferably from 1
to 30 kg, most preferably from 2 to 20 kg of l~i~lUdlUllld~iU compound per
liter of catalyst per hour.
The space-time yields which can be obtained are therefore an
order of magnitude higher than those of the known processes for the
h~,~,u~elld~iu" of nitroaromatic compounds. Such higher space-time
yields are particularly important for the economic production of large
quantities of aromatic amine because only small amounts of catalyst and
~0 .,ull~:,,uul~dillgly small reactors are required.
21 7g700
A 31,091 7
Another important advantage of the process of the present
invention is the quantitative conversion of the llilludlullldli~ compound
which is surprisingly and ~l ,e,x,oeule~ly obtained, even at the high
loadings used.
The process of the present invention is also distinguished by the
absence of a so-called catalyst induction phase and by its selectivities
which are higher than 99.4% from the outset.
In the process of the present invention, the conversion of
nltroaromatic compound is higher than 99.95%, preferably higher than
99.99%, more preferably higher than 99.995%, most preferably higher
than 99.999%. This should not, however, be ~l ld~ uod as being
: limiting because any desired lower degree of conversion can be achieved
by selection of the d,U,UlUplidl~ process conditions.
The catalysts of the present invention can be used in any reactor
having a fixed catalyst bed.
One industrial implementation of the process can be described as
follows:
The catalyst is fixed in an adiabatic reactor of known design (See
Ullmann, EnzykloPadie der l~-.l " li~ l Çhemie [Ullmann's Encyclopedia
:20 of Industrial Chemistry~, 4th Edition, Volume 3, pages 468-649; and Kirk-
Othmer, EncvcloPedia Qf Chemical Technoloav. Vol. 19 (1982), pages
880-914, for example.). However, the catalyst may also be distributed
between a plurality of reactors which are cu"lle~ d in series or in
parallel. These reactors may be any of those known in the art to be
:25 useful for the oxidation of methanol to roll"dld~l"/de, for example.
The packed catalyst beds are provided on or between gas-
permeable walls, as in the prior art. Satisfactory gas distribution must be
ensured.
Instead of being used as a bulk packing, the catalyst may also be
prepared and used on suitable packings as a support material.
2 1 78700
Le A 31,091
-8-
Fresh ~ UdlUllldli~ compound is metered into the circulating gas
stream which is ~""ùosed primarily of recycled and freshly added
hydrogen upstream of the catalyst packing. It is preferred however that
the nitroaromatic compound be completely volatilized in the fresh
hydrogen before being introduced in gaseous form into the circulating
gas stream. After passing through the catalyst bed the product gas is
cooled with the recovery of steam. The steam may be recovered by
means of any of the heat e~.l,d"~e,~ known to those skilled in the art.
Thereafter the product gas is cooled to remove the aromatic amine and
the water of reaction from the reaction mixture by, u,,de,,~dLiu,l. The
remaining circulating gas is recycled after removing a small amount of
gas for the outward transfer of gaseous constituents in the circulating
gas. Before recycling the circulating gas should generally be preheated
to its inlet temperature and admixed with fresh starting material.
The above d~s~i,uLiu" of one ~",bodi",~"l of the present invention
is of an elementary nature and should not have a limiting effect or be
judged as limiting.
The process of the present invention is particularly suitable for the
h~,d,u~elld~iù,l of l,il,uL~ e or nitrotoluene.
The process of the present invention makes it possible to use
catalyst loadings or GHSVs which are extremely high (i.e. higher by a
power of ten than those of the prior art). Despite these high catalyst
loadings however selectivities greater than 99.4% are obtained with
complete conversion even at the commencement of the reaction. The
catalysts of the present invention exhibit the greatest productivity
achieved without production l 'I .I,~aPc due to catalyst deactivation.
The invention is further illustrated but is not intended to be limited
by the following Examples in which all parts and p~l~llldU~:S are parts
by weight or p~, ,e"ld~es by weight unless otherwise specified.
2 1 78700
L~ ~ 31,091 9
E~AMPLES
The GHSV (gas hourly space velocity), u~ se"l~ the hourly
space velocity of the gas under normal conditions or at a given pressure,
wlth respect to the empty volume which the packed catalyst bed
5 occupies.
Catalvst ~ Udl~iull
EG 17 granulated graphite supplied by Ringsdorff which had a
BET specific surface of about 0.4-0.8 m2/g, was used as the support
material. The grain size was between 1 and 3 mm and the tap density
10 was from 650 to 1000 9/l.
Results similar to those reported below have also been obtained
with other graphites and graphite-containing materials having a low BET
speclfic surface.
The catalysts used in the Examples which follow were prepared in
15 the following manner:
EG 17 granulated graphite with an ~LJSOIIJ~IICY of 7 ml au~ il,ile
per 100 9 support was placed in a rotatable vessel and mixed, by
rotation, with a solution of palladium acetate in ~Le~u"il,ile. The mixture
was agitated until the solvent had been completely absorbed by the
20 support. Thereafter, the solid was dried for 5 minutes in a strongly
ascending current of hot air at 40C. The i",,~ ull~iull and drying steps
were repeated until the desired amount of palladium had been deposited.
The dried catalyst was subsequently activated in a stream of hot
hydrogen under normal pressure.
~ Le A 31,091 -10- 2 j1 78700
ExamPle 1
Catalyst 1 which had a palladium content of 1.6% by weight on
200 9 of EG17 as support was prepared by 7 il~, nt-ylldliol~s, each
with 0.95 9 PdAc2 in 14 9 dct lullillilt~ and then activated for 20
hours at 370C.
ExamPle 2
Catalyst 2 which had a palladium content of 2.4% by weight on
200 9 of EG17 as the support was prepared by 10 illlpl~:ylldliull:~, each
with 1 9 of PdAc2 in 14 9 aut lu"il,ilt and then activated for 20 hours at
0 370C.
ExamPle 3
Catalyst 3 which had a palladium content of 2% by wei3ht on 2000
9 of EG17 as support was prepared by 9 illl,Ul~lldliUIls, each with 9.25 9
of PdAc2 in 140 9 acetonitrile and then activated for 20 hours at 370C.
ExamPle 4
220 ml (219.0 9) of Catalyst 3, which contained 2.0% Pd, were
introduced into a very well insulated reactor to give a poured bed height
of 180 mm. The reactor was fitted at its upper end with a vaporizer and
su,ùe, I It3dlt'1. A well insulated tube was connected to the reactor outlet forthe continuous removal of the product gas. This tube conveyed the
product into a system made up of a tube bundle and a spiral UUl nit~ t~l
for the purpose of cu, Idt~l ISdliul ,. The temperature upstream, in and
downstream of the catalyst bed packing was measured by means of a
)le thermocouple. The catalyst was first treated in the reactor
:25 at 200C for 10 hours while passing in hydrogen via the vaporizer and
superheater at normal pressure. Thereafter, the hydrogen flow was
adjusted to 1620 I/hour. 110 g/hour of nitrobenzene were metered into
the hydrogen stream by means of a metering pump via the vaporizer-
superheater unit at initial temperatures of Tjnjt = 210C. This
~0 corresponded to a hydrogen/llilluuel,~t l,e m~lar ratio of 81 to 1. The
2~ 78700
Le A 31,091
resuitant temperature difference betweèn the starting material and
product gas streams was about 200CC for quantitative conversion under
adiabatic conditions. After a few hours, a temperature profile was
attained in the catalyst bed which co" eS,uul ,-;led to a heat loss of about
5 10% through the reactor walls. The remainder of the heat of reaction left
the catalyst bed with the product gas mixture. Analysis of the
cu"de":,dle by gas uillullld~o~u,ld,ully gave the contents listed in the Table
below. After 1000 hours, the catalyst showed no signs of deactivation.
GHSV = 7460 hour '
10Run time Cat. No. NBz* Selectivity Tjnjt Tm~x
(hours) (ppm) (%) (C) (C)
4 3 0 99.49 201 376
3 0 99.54 201 376
214 3 0 99.63 201 375
151004 3 0 99.73 201 377
*NBz = Nillube"~elle
Example 5 tComparatlve)
220 ml of a catalyst prepared analogously to Example 1 of DE
2,849,002 with 9 9 Pd and 9 9 V on alpha-alumina (SPH 512 supplied by
Rhone-Poulenc), were introduced into the same reactor as was used in
Example 4 (the procedure was anaiogous to that used in Example 4 aboYe).
The following results were obtained after activatlon and hydrogenation
under the same e~.~Jel i, Ldl conditions as were used in Example 4:
GHSV = 7460 hour 1
2 1 78700
Le A 3~,091 -12-
Run time NBz~ Selectivity Tjnjt Tm~x
(hours) (ppm) (%) (C) (C)
2 0 98.0 201 385
0 98.5 199 375
5 301 100 98.9 200 370
~NBz = Nil~ ube~ t-
ln an oil-heated reactor at the same " ube"~ e loadingas in Exa;ple 4 and
a.t a hydrogen/nitrobenzene ratio of 611 (GHSV = 637 hour~') the catalyst had
a life of about 1000 hours and a selectivity of about 98.0% as ~ lt"l"i"ed
10 over the conversion cycle.
The catalyst was unsuitable for a process in which a large excess
of hydrogen and a high loading of "illube, I~ ,e were used.
ExamPles 6 and 7
The following examples were carried out in the same reactor as
15 that which was used in Example 4 at an absolute pressure of about 5
atm.
Hydrogen and lli~, u~e" ~"e were ~,, upu, ~iu, ,ed in a molar ratio of
81 to 1. For a quantitative conversion under ideal adiabatic conditions
this would result in a temperature difference of about 200C between the
20 starting material and product streams. Catalyst 1 (Example 6) or
Catalyst 2 (Example 7) was loaded with 10 g ~it~ube~ e per ml
catalyst per hour.
GHSV (5 atm.) = 29860 hour~'
2~ 78700
; L,~ A 31,091
13
Run time Cat. No. NBz~ Selectivity Tjnjt Tm~x
(ppm) (~0) (C) (C)
4 1 0 99.5 250 449
40 1 0 99.7 250 448
400 1 0 99.7 250 447
51100 1 26 99.7 250 448
4 2 0 99.4 250 449
40 2 0 99.5 250 451
400 2 0 99.7 250 450
1600 2 0 99.7 250 449
lO2400 2 14 99.8 250 450
~NBz = ~ ' u~e"~t:"e
The very low "il~ ubd~ e breakthroughs indicate the
commencement of a slight deactivation of the contact catalyst.
- Due to the high loading (10 g/ml hour), the same aniline
production per unit volume of catalyst was not obtained in a prior art
process (loading < 1 glml hour) until after more than 11,000 hours or
24,000 hours, respectively.
Long run times or high productivities of this type without
intermediate regel-dldliu" have not been described previously.
.20 The t-~lldul dil Idl ily high selectivity of the process over the entire
production cycle is also It:llldlkdLI~.
Examr le 8
Hy.l, u~, IdLiul I of o-nitrotoluene
The experimental set-up was the same as that used in Example 4.
The loading was 2.5 ~ o-nitrotoluene per ml catalyst per hour. Hydrogen
2~ 7~7~0
Le A 31,091 -14-
and o-nitrotoluene were proportioned in a ratio of 81 to 1. The results
obtained were as follows:
Run time Cat. No. NTol Selectivity Tinie Tm~x
(ppm) (%) (C) (C)
24 2 0 99.8 250 447
5100 2 0 99.1 250 445
400 2 0 99.4 250 442
600 2 0 99.6 250 440
NTol = o-ni trotol uene
Examples 9 and 10 were carried out at normal pressure using
10 about 2 liters of Catalyst 3 in oil-heated heat e~.ul,d"~, tubes made of
V2A having a length of about 300 cm, which is customary for industrial
reactors, and an inside diameter of about 3 cm.
The loading was 0.65 g/ml hour, and the temperature of the heat
transfer medium was adjusted to 250C.
ExamPle 9
GHSV = 9707 houf'
H2/NBz = 81/1 Service life > 5000 hours (no llillu~e~ lle
breakthrough)
Examole 10 (Comparative)
GHSV = 2131 hour~1
H2/NBz = 17/1 Service life 95 hours ("il, u~"~"e breakthrough >
100 ppm)
A deactivation of the catalyst, which was clearly progressive,
occurred below a critical ratio of hydrogen to "i~,ub~"~ e.
Although the invention has been described in detail in the
foregoing for the purpose of illustration, it is to be ~" Id~l bluod that such
detail is solely for that purpose and that variations can be made therein
21 78700
Le A 31,091 -15-
by those skilled in the art without departing from the spirit and scope of
the inventlon extept as it may be limlted by th~ cl~ims