Note: Descriptions are shown in the official language in which they were submitted.
WO 96/14317 PCT/EP95/03487
1.
Novel piperidine derivatives with PAF antagonist activity.
Field of the invention.
The present invention relates to new piperidine derivatives which are
potent platelet activating factor (PAF) antagonists. The invention also
relates
S to a process for their preparation, to pharmaceutical compositions
containing
them and to their use in the treatment of diseases in which PAF is involved.
Description of the prior art.
Platelet activating factor (PAF) or (I-O-alkyl-2-acetyl-sn-glyceryl-3
phosphorylcholine), also called acetyl glyceryI ether phosphoryIcholine
(AGEPC) or PAF-acether, is a natural phospholipid synthesized by different
cells (basophiles, macrophages, neutrophiles, platelets) and tissues (heart,
lung
and kidney) of the organism.
PAF was described for the first time as a potent platelet aggregating
agent. Later on it was demonstrated to have other biological activities in
vivo,
such as peripheral vasodilatation, increase of the vascular permeability,
induction of bronchoconstriction and hyperreactivity of the respiratory tract.
PAF also produces immediate hypotension followed by pulmonary and renal
hypertension in rats, guinea pigs, rabbits and dogs, and it has been rated as
the
most potent ulcerogenic agent described until now.
2 0 Consequently, PAF is a mediator that is implicated in a large set of
pathological processes such as asthma, septic shock, transplant rejection,
thrombosis, ulceration, inflammation and renal diseases.
The closest prior art from the structural point of view is believed to be
EP 4-~I226, which discloses pyridylcyanomethylpiperazines and piperidines
2 5 having PAF antagonistic activity, different from the compounds of the
present
invention.
Description of the invention.
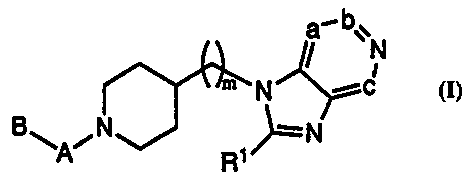
The present invention relates to new piperidine derivatives of general
formula I
a_ bvN
/ i
. .m~ N
B ~Ai N ~ ~=N
30 R
~ I
wherein:
m represents 0, 1 or Z;
a, b and c represent CR, wherein each R independently represents hydrogen or
3 5 C1~ alkyl;
SUBSTITUTE SHEET (RULE 26)
CA 02180660 2006-O1-06
2223 7-402
2
Rl represents C1...4 alkyl or C~~ cycloalkyl; .
A represents -CO-, -S02-, NHCO- or -0CO-;
B represents a group of formula (i), and when A represents -CO- or -S02-, then
B can also represent a group of formula (ii) or (iii)
R2 Y
Rs ~/N~
R2 Rs R
(i) (ii) (iii)
n represents 0, I, 2 or 3;
one of R2 or R3 represents C1~ alkyl, C3-~ cycloalkyl or aryl, and the other
represents hydrogen, Cl~ alkyl, Cl~ haloalkyl, C3_~ cycloalkyl, C1..4 alkoxy-
Cl~
alkyl aryl or aryl-C~_4alkyl;
Z represents hydrogen, Cl~ alkyl, -CH2-OR4, -COOR4 or -CONR4R$, and when
A represents -CO- or -SOz-, then Z can also be hydroxy, -NR4R5,
-NR6C(=O)OR4, -NR6C(=O)R4, -NR6C(=O)NR4R5, -N(OH)C(=O)NR4R5 or
-NR6S02R4;
or Z and R3 together form a C2_g polymethylene chain in which case R2
represents C1~ alkyl, C~~ cycloalkyl or aryl;
R4 represents hydrogen, C1.,4 alkyl; aryl or aryl-Cl~ alkyl;
2 0 R$ and R6 independently represent hydrogen or Ct~ alkyl;
R~ represents Cl~ alkyl, C~~ cycioalkyl, aryl, aryl-C1...4 allcyl or bisaryl-
C1.~ alkyl;
Y represents hydrogen, Cl~ alkyl, aryl, aryl-CIA alkyl, -C(=O)OR4, -C(=O)R4,
-C(=O)NR4R5, or -S02R4;
aryl, whenever appearing in the above definitions, represents phenyl or
2 5 phenyl substituted with 1, 2, 3 or 4 groups independently selected from
halogen, C1_4 alkyl, C~_4 alkoxy, hydroxy, C1~ haloalkyl, C1.4 haloalkoxy,
cyano, nitro, amino, Cl_4 alkylamino, Cl~ dialkylamino, C1..4 alkylcarbonyl,
C1_
4 alkylcarbonyloxy, Cl~ alkoxycarbonyl, Cl~ alkylsulfonyl, C1..4
alkylsulfinyl,
Cl~ alkylthio, Cl~ alkylcarbonylamino or Cl~ alkoxycarbonylamino;
3 0 and their salts and solvates.
The invention also provides a pharmaceutical composition which
comprises an effective amount of a compound of formula I or a
pharmaceutically acceptable salt or solvate thereof and a pharmaceutically
acceptable excipient.
3 5 The invention further provides the use of a compound of formula I.or a
pharmaceutically acceptable salt or solvate thereof, or a composition thereof
for the manufacture of a medicament for the treatment or prevention of
diseases mediated by PAF.
CA 02180660 2006-O1-06
22237-402
2a
The invention further provides a compound of
formula I or a pharmaceutically acceptable salt or solvate
thereof, or a composition thereof for the treatment or
prevention of diseases mediated by PAF.
The invention further provides a commercial
package comprising a compound of formula I or a
pharmaceutically acceptable salt or solvate thereof, or a
composition thereof, together with instructions for its use
for the treatment or prevention of diseases mediated by PAF.
CA 02180660 2006-O1-06
2223 7-402
3
Preferred is the use for the manufacture of a medicament for the treatment or
prevention of ischemia and shock states such as septic shock, anaphylactic
shock, hemorrhagic shock and myocardial ischemia; pancreatitis; and diseases
related with allergy and inflammation such as asthma, dermatitis, urticaria,
arthritis and psoriasis.
The invention also provides the use of a compound of formula I or a
pharmaceutically acceptable salt or solvate thereof , or a composition
thereof for the treatment or prevention of diseases mediated by PAF. Preferred
is
the use for the treatment or prevention of ischemia and shock states such as.
1 D septic shock, anaphylactic shock, hemorrhagic shock and myocardial
ischemia;
pancreatitis; and diseases related with allergy and inflammation such as
asthma,
dermatitis, urticaria, arthritis and psoriasis.
The invention further provides a method of treating or preventing
diseases mediated by PAF in a mammal, which comprises administering to
1 5 said mammal an effective amount of a compound of formula I or a
pharmaceutically acceptable salt or solvate thereof. Preferred is a method of
treating or preventing ischemia and shock states such as septic shack,
anaphylactic shock, hemorrhagic shock and myocardial ischemia; pancreatitis;
and diseases related with allergy and inflammation such as asthma, dermatitis,
2 0 urticaria, arthritis and psoriasis in a mammal in need thereof, the method
comprising administering to the mammal an effective amount of a compound
of formula I or a pharmaceutically acceptable salt or solvate thereof.
The invention still further provides a process for preparing a compound
of formula I which comprises:
2 5 (A) reacting a compound of formula II,
a.~b~
I
~.N ~c
HN ""'N
R~
It
3 0 wherein a, b, c, m and Rl have the previously described meaning, with an
arid
of formula BCOOH (III) or a suitable derivative thereof such as the and
chloride or the anhydride, a sulfonyl chloride of formula BS02CI (IV), a
compound of formula BOC(=O)G (V), a compound of formula BNHC(=O)G
(VI) or a compound of formula BN=C=O (VII), wherein B has the previously
WO 96/14317 ~~ ; '. PCT/F,P95/03487
~ , ;l . . ...
2~.~~6~d 4
described meaning and G represents a good leaving group such as chloro or
-0Ph; or
(B) converting in one or a plurality of steps a compound of formula I into
another compound of formula I; and
(C) if desired, after steps A or B, reacting a compound of formula I with an
acid
to give the corresponding and addition salt.
The invention yet further provides novel intermediates of formula II
a/b'N
l _,
~N ~c
HN -'N
R~
li
wherein a, b, c, m and Rl have the meaning described above for the
compounds of formula I. The compounds of formula II are valuable
intermediates in the preparation of the compounds of the present invention.
Compounds of formula I may have one or more aymmetric centers,
which can give rise to stereoisomers. The present invention covers each of the
individual stereoisomers as well as their mixtures. Moreover, some
compounds of the present invention may show cis/trans isomery. The present
2 0 invention covers each of the geometric isomers and the mixtures thereof.
In the above definitions, the term C1~ alkyl, as a group or part of a
group, means a linear or branched alkyl group that contains from one to four
carbon atoms. Therefore, it includes methyl, ethyl, propyl, isopropyl, butyl,
isobutyl, sec-butyl, and tent-butyl.
2 5 The term C1~ alkoxy, as a group or part of a group, means a group
derived from the union of a Cl_4 alkyl group like the above mentioned to an
oxygen atom of an ether functional group. Examples include methoxy, ethoxy,
propoxy, isopropoxy, butoxy, isobutoxy, sec-butoxy, and tert-butoxy.
A halogen group or its abbreviation halo represents fluoro, chloro,
3 0 bromo or iodo.
A group C3_~ cycloalkyl represents cyclopropyl, cyclobutyl, cyclopentyl,
cyclohexyl or cycloheptyl.
A group C2_5 polymethylene represents ethylene, propylene, butylene
and pentylene.
SUBSTITUTE SHEET (RULE 26)
WO 96!14317 PCT/EP95/03487
S
t.
A Cl.~ haloalkyl group means a group resulting from the substitution of
one or more hydrogen atoms of a C1~ alkyl group by one or more halogen
atoms (i.e. fluorine, chlorine, bromine or iodine), which can be the same or
different. Examples include trifluoromethyl, fluoromethyl, chloroethyl,
fluoroethyl, iodoethyl, 2,2,2-trifluoroethyl, pentafluoroethyl, fluoropropyl,
chloropropyl, 2,2,3,3,3-pentafluoropropyl, heptafluoropropyl, fluorobutyl, and
nonafluorobutyl.
A Clue haloalkoxy group means a group resulting from the substitution
of one or mare hydrogen atoms of a Clue alkoxy group by one or more halogen
1 0 atoms, which can be the same or different. Examples include
trifluoromethoxy,
fluoromethoxy, chloroethoxy, fluoroethoxy, iodoethoxy, 2,2,2-trifluoroethoxy
pentafluoroethoxy, fluoropropoxy, chloropropoxy, 2,2,3,3,3-
pentafluoropropoxy, heptafluoropropoxy, fluorobutoxy, and
nonafluorobutoxy.
1 5 A Cl_4 alkylamino or C1_4 dialkylamino group represents a group
resulting from the substitution of one or two hydrogen atoms, respectively, of
an amino group by one or two C1~ alkyl groups, which can be the same or
different. Examples include methylamino, dimethylamino, ethylamino,
diethylamino, ethylmethylamino, propylamino, dipropylamino,
2 0 isopropylamino and diisopropylamino.
A Cl.ø alkylcarbonyl group represents a group resulting from the union
of a C1~ alkyl group to a carbonyl group. Examples include acetyl, propionyl,
isopropionyl, and butanoyl.
A C1~ alkylcarbonyloxy group represents a group resulting from the
2 5 union of a Clue alkylcarbonyl group to an oxygen atom of an ether
functional
group. Examples include acetyloxy, propionyloxy, isopropionyloxy, and
butanoyloxy.
A C1_4 alkoxycarbonyl group represents a group resulting from the
union of a C1_4 alkoxy group to a carbonyl group. Examples include
3 0 methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, isopropoxycarbonyl,
butoxycarbonyl, isobutoxycarbonyl, sec-butoxycarbonyl and tent-butoxycarbonyl.
A Clue alkylsulfonyl group represents a group resulting from the union
of a Cl.~ alkyl group to a sulfonyl group. Examples include methylsulfonyl,
ethylsulfonyl, propylsulfonyl, isopropylsulfonyl, butylsulfonyl,
3 5 isobutylsulfonyl, sec-butylsulfonyl, and tent-butylsulfonyl.
A Clue alkylsulfinyl group represents a group resulting from the union
of a C1~ alkyl group to a sulfinyl group. Examples include methylsulfinyl,
ethylsulfinyl, ~ropylsulfinyl, isopropylsulfinyl, butylsulfinyl,
isobutylsulfinyl,
sec-butylsulfinyl, and tent-butylsulfinyl.
SUBSTITUTE SHEET (RULE 26)
WO 96/14317 \ pC'lyEP95/03487 ~ ,
6
A Cl~ alkylthio group represents a group resulting from the union of a
C1.~ alkyl group to a sulphur atom of a thioether funtional group. Examples
include methylthio, ethylthio, propylthio, isopropylthio, butylthio,
isobutylthio, sec-butylthio, and tent-butylthio.
A Cl~ alkylcarbonylamino group represents a group resulting from the
substitution of a hydrogen atom of an amino group by a Cl~ alkylcarbonyl
group. Examples include acetamido, propanamido and isopropanamido.
A Cl~ alkoxycarbonylamino group represents a group resulting from
the substitution of a hydrogen atom of an amino group by a C1-4
alkoxycarbonyl group. Examples include methoxycarbonylamino,
ethoxycarbonylamino, propoxycarbonylamino, isopropoxycarbonylamino,
butoxycarbonylamino, isobutoxycarbonylamino, sec-butoxycarbonylamino and
tent-butoxycarbonylamino.
A C~~ alkoxyCl~ alkyl group represents a group resulting from the
1 5 substitution of a hydrogen atom of a C1~ alkyl group by a C1~ alkoxy
group.
Examples include among others methoxymethyl, ethoxymethyl,
propoxymethyl, isopropoxymethyl, butoxymethyl, isobutoxymethyl, sec
butoxymethyl, tert-butoxymethyl, 2-methoxyethyl, 2-ethoxyethyl, 2
propoxyethyl, 2-isopropoxyethyl, 2-butoxyethyl, 2-isobutoxyethyl, 2-sec
2 0 butoxyethyl, 2-tent-butoxyethyl, I-methoxyethyl, I-ethoxyethyl, I-
propoxyethyl,
1-isopropoxyethyl, I-butoxyethyl, I-isobutoxyethyl, I-sec-butoxyethyl, and 1-
terf-butoxyethyl.
An aryl-C1_4 alkyl group represents a group resulting from the
substitution of one hydrogen atom of a C1_4 alkyl group by an aryl group as
2 5 defined above. Examples include among others, benzyl, 1-phenylethyl, 3
phenylpropyl, 2-phenylpropyl, 1-phenylpropyl, 4-phenylbutyl, 3-phenylbutyl,
2-phenylbutyl and I-phenyibutyl, wherein the phenyl groups can be
substituted as described above in the definition of an aryl group.
A bisaryl-C1_4 alkyl group represents a group resulting from the
3 0 substitution of two hydrogen atoms of a Cl~ alkyl group by two aryl groups
as
defined above, which can be the same or different. Examples include among
others, diphenylmethyl, 2,2-diphenylethyl, 1,1-diphenylethyl, I,2
diphenylethyl, 3,3-diphenylpropyl, 2,2-diphenylpropyl, I,I-diphenylpropyl, 3,2
diphenylpropyl, 1,3-diphenylpropyl, and 1,2-diphenyipropyl, wherein the
3 S phenyl groups can be substituted as described above in the definition of
an aryl
group.
Preferred compounds include those in which, independently or in any
compatible combination:
1
m represents 1 or 2;
SUBSTITUTE SHEET (RULE ?~';~
WO 96114317 PCT/EP95/03487
~I8~U6~0
7
n represents 0, 1 or 2;
A represents -CO- or -S02-;
A represents -NHCO- or -OCO-;
B represents a group of formula (i);
B represents a group of formula (ii);
B represents a group of formula (iii)
Z represents hydrogen, Cl.~ alkyl, -CH2-OR4, -COOR4; or -CONR4R5, and
when A represents -CO- or -S02-, Z can also represent hydroxy,
-NR6C(=O)OR4, -NR6C(=O)R4 or -NR6S02R4;
I 0 Z and R3 together form a C2_5 polymethylene chain;
R~ represents Cl~ alkyl, C~~ cycloalkyl or aryl;
aryl represents phenyl or phenyl substituted with I, 2, 3 or 4 groups
independently selected from halogen, C1_4 alkyl, CI_4 alkoxy, hydroxy, Cl_4
haloalkyl, Cl~ haloalkoxy or amino.
1 5 Accordingly, a preferred class of compounds of formula I is that
wherein:
m represents 1 or 2;
a, b and c represent CR, wherein each R independently represents hydrogen or
Cl~ alkyl;
2 0 Rl represents Cl.~ alkyl or C3_~ cycloalkyl;
A represents -CO- or -S02-;
B represents a group of formula (i), (ii) or (iii)
Z R2
n
RWN
2 5 (i) (ii) (iii)
n represents I or 2;
one of R2 or R3 represents C1_4 alkyl, C3_~ cycloalkyl or aryl, and the other
represents hydrogen, C1~ alkyl, C1~ haloalkyl, C3_~ cycloalkyl, C1..4 alkoxy-
C1..4
3 0 alkyl , aryl or aryl-C~-4alkyl;
Z represents hydrogen, C1~ alkyl, -CH2-OR4, -COOR4, -CONR4R5, hydroxy,
-NR6C(=O)OR4, -NR6C(=O)R4 or -NR6SOZR4;
or Z and R3 together form a C2_5 polymethylene chain in which case R2
represents C1.~ alkyl, C~~ cycloalkyl or aryl;
3 5 R4 represents hydrogen, Cl~ alkyl, aryl or aryl-C1~ alkyl;
RS and R6 independently represent hydrogen or C1..4 alkyl;
R~ represents Cl~ alkyl, C~~ cycloalkyl or aryl;
SU6STI T UTE SHEE T (RULE 26)
WO 96/14317 ' PCT/EP95/03487 ~ '
z~sosso~
8
Y represents hydrogen, Cl~ alkyl, aryl, aryl-C1~ alkyl, -C(=O)OR4, -C(=O)R4,
-C(=O)NR4R5, or -S02R4; and
aryl in the above definitions represents phenyl or phenyl substituted with I,
2,
3 or 4 groups independently selected from halogen, Cl_4 alkyl, C1~ alkoxy,
hydroxy, Cl~ haloalkyl, C1~ haloalkoxy or amino.
A preferred group of compounds within this class is that wherein:
B represents a group of formula (i) or (iii);
one of R2 or R3 represents C1_4 alkyl, C3_~ cycloalkyl or aryl, and the other
represents hydrogen, C1~ alkyl, Cl~ haloalkyl, C3_~ cycloalkyl, Cl~ alkoxy-C1~
1 0 alkyl , aryl or aryl-C ~ -4 alkyl ;
Z represents hydrogen, Cl~ alkyl, -CH2-0R4, -COOR4 or hydroxy;
or Z and R3 together form a C2_g polymethylene chain in which case R2
represents C1..4 alkyl, C~~ cycloalkyl or aryl;
R4 represents hydrogen, C1~ alkyl, aryl or aryl-C1~ alkyl;
1 5 R~ represents C~.~ alkyl, C~~ cydoalkyl or aryl; and
Y represents Cl.~ alkyl, aryl, or -S02R4.
Another preferred group of compounds within this class is that
wherein:
B represents a group of formula (ii); and
2 0 one of R2 or R3 represents C~.ø alkyl or aryl, and the other represents
hydrogen,
Cl~ alkyl, C1..4 alkoxy-C1~ alkyl , aryl or aryl C~-4 alkyl.
Another preferred class of compounds of formula I is that wherein:
m represents 1 or 2;
a, b and c represent CR, wherein each R independently represents hydrogen or
2 5 C1~ alkyl;
Rl represents Cl.ø alkyl or C~7 cycloalkyl;
A represents -NHCO- or -OCO-;
B represents a group of formula (i);
Z
R3
3 0 R2
(i)
n represents 0 or I;
one of R2 or R3 represents C1~ alkyl, C3_~ cycloalkyl or aryl, and the other
3 5 represents hydrogen, Cl~ alkyl, C1~ haloalkyl, C3_~ cycloalkyl, Cl~ alkoxy-
Cl.~
alkyl ~ aryl or aryl-C~-4 alkyl;
Z represents hydrogen, Clue alkyl, -CH2-OR4, -COOR4, or -CONR4R5;
SUBSTITUTE SHEI= T (RULE 261
WO 96/14317 ~ ~ I PCT/EP95/03487
.~
9
or Z and R3 together form a C2_S polymethylene gain in which case R2
represents C1~ alkyl, C~~ cycloalkyl or aryl;
R4 represents hydrogen, C1~ alkyl, aryl or aryl-Cl.~ alkyl;
RS represents hydrogen or C1~ alkyl; and
aryl in the above definitions represents phenyl or phenyl substituted with 1,
2,
3 or 4 groups independently selected from halogen, C1~ alkyl, Cl~ aIkoxy,
hydroxy, Cl~ haloalkyl, C1~ haloalkoxy or amino.
The formulae of some specific compounds are represented below,
together with the number corresponding to the example in which their
1 0 preparation is described:
SUBSTITUTE SHE/: T (RULE 261
WO 96/14317
PCT/EP95/03487
~N I N
I N N
N~~ N 1 I / N~~ N
O HO O
I/ I
N
I/ I N
I N 2 HN N
N~ '~~ z N N
I o i/ o
I ~N I ~N
Me0 N N~~ N 3 AcNH Nt J N N 10
r\
O O I / O
I
~N
N ~N
H ~ iN 4 H N
Me0 N N~ N N~ '-N 11
n
o / I o I / / o
I
~N I N
H NI~~
Bu'O H N
N N~~ N 5 N N~ ~ N 1..
O / O N02 I / / O
I I
\
NH ~N / N
z / H N ~
\ I N N~~ N 6 N N' J N N 13
O / O NHz I / / O
\I \I
I 'N / N
N ~ ~N
\ N N~~ N 7 N' J N 14
I H~
/ 0 0
SUBSTITUTE SHEET (RULE 26)
WO 96/14317
'. ,'" ,.,, ~ .,r, ,. ~ PGT/EP95/03487
,z...~ ~k.~m
11
N ~N / ~N
N~~N ~ 15 N' J LN 21
O HO /~a
/ ~ O
N~ / ~ N ~N
/ N ~ N
N~~ N 16 N~~ N 2 2
O O
NOz
N_~N N_~N
N~~N 17a ~~N 23
O O
NHz NHz
/ ~N / ~N
HzN \ ~ ' N~~N 17b ' N'J LN 24
fl r ~ 11
/ O N~ / O
~N ~N
N I 18 N 25
N~~ N ~ N~~ N
j[ il
HO O NH ~ / O
z
~N / ~N
/ N I~ N
N~)-N 19 EtOOC N~~N 26
EtOOC ~ O
NOz
~~ O I ~ N // N
NH H N ~ N
N~~=N 20 Et00C N~~N 27
R
O / O
NHz
SUBSTITUTE SHEET RULE 26)
WO 96/14317
,. , PCT/EP95/03487
12
~N / ~N
EeDOC N N 2 8 O N ~ 3 5
~~N EtO~ N~ ~N
N
O O
I I
~_N I N
I N O H N
\ N~~ N 19 E~ N N~ N 3 6
I O O
NOZ
I N N
N~ H N I
\ N~~ N EtOOC N N~ -N
30 ~ ~ 37
i O \ O
\I I~
NH2 OZN
~_N I N
H N N
EtO~ N N~~= N 31 EIOOC N N~ N 3 8
O / I O O
H2N I
NHZ
I 'N I N
N H N
N~,' N 3 2 EtOOC N N~ -N 3 9
O / O
\ I
N~N 33 I'N
N~ 40
HO N~~ N N~ -N
O
O
H2N ~N / N
I H '~ N
~S. N N~~ N 3 4 N~ N 41
Os O
O O
I I
~/
SU6STITUTE SNEFT (RULE 261
WO 96/14317
PCT/EP95/03487
13
N ~N N ~N
~N N~~N 4Z \ I N N~~N 49
H
I
~N I N
~N ~ N
N~~ N H
4 3 N N~~N 5 0
N
/ O O
\I I
NOZ
~N I
N ~l~'~,~
H N
N~~-~ N y.. N N~ -N
/ p 44 0 ~ 51
\I \i
NHy
~N I N
I \ H N ~ O N
N N~~-=N 4 5 \ N~ N S 2
N'
o i / / o
I/
~I
I N I
O N ~ N
N~ J
O O
I \ i ~'N N 46 \ ~S~N N~~N 53
HZN / ~O( I /
02N
I vN I N
N ~ 47 O N~ 54
L N N
FCC N N Et0 N
HO ~~ / O
I \ O I
~N NOZ N
HZN / I N ~~(~~, O ~N I
\ N~~N 48 E~~N N' J LN 55
/-O
/ O
\I
NHZ
SUBSTITUTE SNE.ET (R~1LE ~R1
WO 96/14317 ~" ~ ; PCT/EP95/03487
14
~N ~ N
~I J
N 56 H N~ 63
N~~=. N N N~ -N
O
O
I
~N N~ N
N ~ H N
N N~~-N 5 ~ N N -N 6 4
Y
O .I O . o
\ \I
NH2
/ N
N~ H~ ~ /'N
EtOOC N N -N ~ N
\ N '~ N
58 / ~~ 65
O
I O
\
N
H N I ~ I N
N
N N~~N 5 9 ~ N -N 6 6
~ N ~~-
p nneo
\I \i
~N H
N' ~~J p N ~N
Et0 N N~~-N 6 0 ~ O / ~ H N
~N N~~N 67
O O
\ I
~N HZN // N
S. ~N ~ 61 i ~ H ~N~~ 68
N~ J ~N \ N~N' J ~-N
O O O
\ I
I N / N
N ~ N
N~~=N 6 2 N~~N 6 9
0 0
I I
\
SUBSTITUTE SHEET (RULE 26)
WO 96/14317 ' PGT/EP95/03487
_~N ~N
O N~~ N 7 O N N~~=N 7 4
I v O I ~ O
N~N N / ~N
I ~ N~~ N I 71 ~ SOZ N N~~ N 7 S
~o 0
~N ~, JN .
N ~ N T
~ SOz N N~~=N 7 Z ~ SOZ N~~ N 7 6
N
NOZ I ~ ~ O HZN
I
// N
N ~7~''
~ SOz N N~~= N 7 3
H2N I ~ / O
I
The compounds of formula I contain basic nitrogen atoms and,
5 consequently, they can form salts with acids, which are also included in the
present invention. There is no limitation on the nature of these salts,
provided that, when used for therapeutic purposes, they are pharmaceutically
acceptable, which, as is well-known in the art, means that thev do not have
reduced activity (or unacceptable reduced activity) or increased toxicity (or
1 0 unacceptable increased toxicity) compared with the free compounds.
Examples
of these salts include: salts with an inorganic acid such as hydrochloric
acid,
hydrobromic aad, hydriodic acid, nitric acid, perchloric acid, sulfuric acid
or
phosphoric acid; and salts with an organic acid, such as methanesulfonic acid,
trifluoromethanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid, p-
15 toluenesulfonic aad, fumaric acid, oxalic acid, malefic acid, citric acid,
succinic
acid, tartaric acid; and other mineral and carboxylic acids well known to
those
skilled in the art. The salts are prepared by reacting the free base with a
sufficient amount of the desired acid to produce a salt in the conventional
manner. Free bases and their salts differ in certain physical properties, such
as
2 0 solubility, but they are equivalent for the purposes of the invention.
The compounds of the present invention can exist in unsolvated as well
as solvated forms, including hydrated forms. In general, the solvated forms,
SUBSTITUTE SNEFT SRI il F ~~~
~Z 8p6~~
WO 96/14317 PCT/EP95/03487
16
with pharmaceutically acceptable solvents such as water, ethanol and the like,
are equivalent to the unsolvated forms for the purposes of the invention.
Some compounds of the present invention can exist as different
diastereoisomers and/or optical isomers. Diastereoisomers can be separated by
conventional techniques such as chromatography or fractional crystallization.
The optical isomers can be resolved using any of the conventional techniques
of optical resolution to give optically pure isomers. Such a resolution can be
performed in any chiral synthetic intermediate as well as in the products of
general formula I. The optical resolution techniques include separation by
chromatography on a chiral phase or formation of a diastereoisomeric pair,
resolution and subsequent recovery of the two enantiomers. The optically pure
isomers can also be individually obtained using enantiospeczfic synthesis. The
present invention covers both the individual isomers and their mixtures (e.g.
racemic mixtures), whether as obtained by synthesis or by physically mixing
them up.
Furthermore, some of the compounds of the present invention may
present cis/trans isomery. The geometric isomers can be separated by
conventional techniques such as chromatography or recrystallization. Such a
separation can be performed either upon the products of formula I or upon
2 0 any synthetic intermediate thereof. The individual isomers can also be
obtained using stereospecific synthesis. The present invention covers each of
the geometric isomers and the mixtures thereof.
The compounds of formula I may be prepared using the methods
described below. It will be apparent to those skilled in the art that the
precise
2 5 method used for the preparation of a given compound may vary depending on
its chemical structure. The reactions are performed in a solvent appropriate
to
the reagents and materials employed and suitable for the transformation being
effected. Moreover, in some of the processes described below it will be
desirable
or necessary to protect reactive or labile groups using conventional
protecting
3 0 groups, for example the groups described below. Both the nature of these
protecting groups and the procedures for their introduction and removal are
well known in the art (see, e. g. Greene T. W., "Protective Groups in Organic
Synthesis", John Wiley & Sons, New York, 198/).
The compounds of formula I can be obtained in general from amines of
3 5 formula II by reaction with an acid chloride of formula BCOCl (VIII), a
sulfonyl
chloride of formula BS02C1 (IV), a compound of formula BOC(=O)G (V), a
compound of formula BNHC(=O)G (VI) or a compound of formula BN=C=O
(VII):
SUBSTITUTE SHEET (RULE 26)
WO 96/14317 PCT/EP95/03487
17
scoci (van
aso2ci (><~
N B~(~x M
t BNHC(-0~ (~'I)
N ~ c BN=C--o (~
HN~ ,=N I
R'
(In
wherein B, a, b, c, m and R1 have the previously described meaning and G
represents a good leaving group such as chloro or -OPh. This reaction is
carried
out in the presence of a proton scavenger amine such as triethylamine or
pyridine in a suitable solvent, or using the base as solvent. Examples of
suitable solvents include halogenated hydrocarbons, such as dichloromethane
and chloroform; ethers, such as diethyl ether, tetrahydrofuran and dioxane;
and aromatic hydrocarbons, such as benzene and toluene. The reaction is
1 0 carried out at a temperature preferably between 0 °C and the
boiling point of
the solvent. As an alternative to the acid chloride, the anhydride can be
employed. Isocyanate derivatives of formula VII may have been previously
prepared or may be generated in situ from the corresponding acid derivative of
formula BCOZH (III) by known procedures such as for example by treatment
1 5 with diphenylphosphorylazide in the presence of triethylamine.
As it will be apparent to those skilled in the art, a compound of formula
I wherein A represents -NHCO- may also be prepared by inverting the
functionality of the reactive groups involved, i.e. by reacting an amine of
formula BNHZ (IX) with a reactive carbamate derivative of amine II, for
2 0 example its phenylcarbamate derivative, which derivative can be prepared
from amine II by conventional procedures such as treatment with phenyl
chloroformate under standard conditions.
Alternatively, compounds of formula I wherein A represents -CO- can
also be prepared by a dehydration procedure between amines of formula II and
2 5 a carboxylic and of formula BCOOH (III). This process can be carried out
using
any conventional reaction of amide bond formation, such as reacting an amine
with an acid in the presence of an appropriate condensing agent such as a
diimide, e.g. dicyclohexylcarbodiimide, alone or in combination with I-
hydroxybenzotriazole. This reaction is carried out in an inert solvent such as
a
3 0 halogenated hydrocarbon, for example dichloromethane or chloroform; an
ether, for example tetrahydrofuran or dioxane; acetonitrile, or
dimethylformamide. The reaction is carried out at a temperature preferably
~I IIRSTITI1T~ CN~GT !AI II ~ ~R~
WO 96/14317
PCT/EP95/03487
18
comprised between 0 and 60°C during a reaction time preferably from 6
to 24
hours.
Moreover, a compound of formula I may also be obtained by
interconversion from another compound of formula I.
Thus, for example, the compounds of formula I in which B represents a
group of formula (i) wherein Z represents -NR6C(=O)OR4, -NR6C(=O)R4,
-NR6C(=O)NR4R5 or -NR6S02R4 or a group of formula (iii) wherein Y is
different from hydrogen can be prepared from the corresponding compounds
of formula I in which B represents a group of formula (i) wherein Z represents
1 0 -NHR6 or a group of formula (iii) wherein Y= H, respectively, by
conventional
reactions, which are well known to those skilled in the art, as shown in
Scheme I. Examples thereof include alkylations, acyiations, preparation of
sulfonamides, carbamates and ureas. These reactions are widely described in
the literature and are carried out in accordance with the usual conditions
1 5 employed in organic chemistry for such transformations.
These compounds of formula I in which B represents a group of
formula (i) wherein Z represents -NHR6 or a group of formula (iii) wherein Y=
H can be prepared by the general procedures described above for the
preparation of compounds I but starting from an acid of formula III (or the
2 0 corresponding acid chloride or anhydride) or a sulfonyl chloride of
formula IV
wherein the amino group is blocked with an amino protecting group (P), as
shown in Scheme 1. As amino protecting groups it is possible to use any
amino protecting group known in the art, such as for example a tert-
butoxycarbonyl group. In this case, it will be necessary a subsequent step for
2 5 removing the protecting group in order to obtain a compound of formula I.
Deprotection is carried out using conventional procedures, such as the
procedures described below.
SU$STITUTE SHEET (RULE 261
WO 96/14317
PG"T/EP95/03487
19
P
I P
Fis R3 Q or Rr~N~Q
R2
wherein Q= COzH (ITI)
S4iC1 (IV)
a. b~~N
_ I
_N / c
HN~ ,.... ~N
R~
11
a. b~~N a. b~~N
I
P N ~~ N / ~c
RsiN AiNJ ~~N RvN~AiN~ ~=N
Rs ~ R I R~
R2 P
~ ~b~N a~byN
i
H 'rtr'N ~c N ~c
Rs~N ANN ~=N Rv ~ iNJ ~=N
R3 Rv N A R~
R2 H
I I
I I
Scheme 1
wherein a, b, c, Rl, R2, R3, R6, R~, m and n have the previously described
meaning; A represents -CO- or -S02-; P represents an amino protecting group,
such as a tent-butoxycarbonyl group and Q represents a group C02H (thus
giving rise to acids of formula III) or a group S02C1 (thus giving rise to
sulfonyl
chlorides of formula IV).
Another example of interconversions between compounds of formula I
1 0 is the reduction of a nitro group in a compound of formula I to an amino
group. This reduction can be carried out by using any known reducing agent
for aromatic vitro groups which is compatible with the other functional
groups present in the molecule. Examples of suitable reducing agents include:
Zn under a wide range of pH conditions in a suitable solvent such as ethanol-
1 5 water mixtures at a temperature preferably between room temperature and
that of the boiling point of the solvent, more preferably between 50 and
60°C;
SUBSTITUTE SNFFT rpt n ~ ~~~
WO 96/14317 PCT/EP95/03487
NaZS204 in a suitable solvent such as mixtures of water and an organic
solvent, for example tetrahydrofuran, ethanol or pyridine; SnCl2 under a wide
range of pH conditions in a suitable organic solvent such as ethanol; Sn or Fe
under a wide range of pH conditions; NaBH4 in the presence of a suitable
5 catalyst such as a Sn, Co or Pd salt in a suitable organic solvent such as
ethanol;
and formic acid or ammonium formate in the presence of Pd/C. Alternatively,
the reduction can be carried out by hydrogenation in the presence of a
catalyst
such as palladium on carbon in a suitable solvent such as an alcohol at a
temperature preferably between room temperature and that of the boiling
1 0 point of the solvent, at a pressure preferably between atmospheric
pressure and
10 atmospheres and during a reaction time preferably between 1 and 48 h.
As it will be appreciated by those skilled in the art, the interconversion
of the substituents as described above can be effected either upon the final
compounds of formula I or upon any synthetic intermediate thereof.
15 The salts of the compounds of formula I can be prepared by
conventional procedures by treatment for example with an acid such as
hydrochloric acid, sulfuric aad, nitric aad, oxalic acid or fumaric aad.
Amines of formula II can be prepared following the process described in
Scheme 2, which is shown below:
SUfsSTIT(JTF SNFrT I~t It F ~Rv
WO 96/14317 , PCT/EP95/03487
21
vm-NH2 »~NH g
A ~b
~N .N I
P b.a~ L P N
(X) ~~ (XII) N02
N,
c N02
. (XI)
B
a, b~~N
1 l
N ~c
_ _ C ..,.. -N H
PiN ~N PAN I b
R~ ~ N
(XIV) NH2 c'
(XIII)
D
a, b.~N
l
N \/c
HN ~_- ~N
R'
Scheme Z ~ (n)
wherein a, b, c, m and Rl have the previously described meaning; P represents
an amino protecting group such as a tent-butoxycarbonyl group and L
represents halogen or C1_6 alkoxy.
In a first step (step A), a compound of formula X is allowed to react with
a compound of formula XI in the presence of a proton scavenger amine such
as triethylamine in a suitable solvent such as a halogenated hydrocarbon, for
example chloroform, at a suitable temperature, preferably between room
1 0 temperature and that of the boiling point of the solvent, to give a
compound
of formula XII.
The reduction of a compound of formula XII (step B) leads to a
compound of formula XIII. This reduction can be carried out by hydrogenation
in the presence of a catalyst such as palladium on carbon in a suitable
solvent
1 5 such as an alcohol at a temperature preferably between room temperature
and
SUBSTITUTE SMEFT (RULE 261
WO 96/14317 ~ ~ PCT/EP95/03487
22
that of the boiling point of the solvent, at a pressure preferably between
atmospheric pressure and IO atmospheres and during a reaction time
preferably between I and 48 h. Alternatively, this reduction may be carried
out
using a suitable reducing agent such as Na2S204 in a suitable solvent such as
mixtures of water and an organic solvent, for example tetrahydrofuran,
ethanol or pyridine.
In step C, a compound of formula XIII is allowed to react with an imino
ether salt of formula R1C(=NH)OR8.HX (XV, wherein Rl has the previously
described meaning, Rg represents C1_6 alkyl and X represents halogen) in a
1 0 suitable solvent such as an alcohol, for example ethanol, to give a
compound
of formula XIV. This reaction is carried out at a temperature preferably
between room temperature and that of the boiling point of the solvent, during
a reaction time preferably between 6 and 48 h. Alternatively, instead of an
imino ether it is possible to use a carboxylic acid of formula R1COOH (XVI),
an
1 5 and halide of formula R1COX (XVII), an anydride of formula (R1C0)20
(XVIII)
or a trialkylorthoester of formula R1C(OR8)3 (XIX), wherein Rl, X and R8 have
the previously described meaning.
Finally, deprotection of the piperidinic nitrogen atom of a compound of
formula XIV (step D) leads to a compound of formula II. The agent used for
2 0 this deprotection and the reaction conditions employed will depend upon
the
nature of the protecting group present. Thus, if the protecting group is a
tert-
butoxycarbonyl group, deprotection can be carried out by treatment with an
acid (for example an inorganic acid such as hydrochloric acid, phosphoric
acid,
sulfuric acid or the like or an organic acid such as toluenesulfonic acid,
2 5 methanesulfonic acid, acetic and, or trifluoroacetic and) in a suitable
solvent
such as water, an alcohol (e.g. methanol), an ether (e.g. tetrahydrofuran or
dioxane) or a halogenated hydrocarbon (e.g. dichloromethane), at a
temperature preferably between 0°C and room temperature.
The above reactions are all per se known ones and are carried out in
3 0 accordance with the described procedures.
Acids of formula II I and sulfonyl chlorides of formula I V are
commercially available, widely described in the literature or can be prepared
by
procedures known to those skilled in the art, starting from commercially
available products or products which have already been reported in the
3 5 literature. Examples of these reactions include alkylations, acylations,
conjugated additions to double bonds, Wittig reaction for the preparation of
double bonds, preparation of sulfonamides, reductive aminations, and the
like. All these reactions are known per se and are carried out in accordance
with the reported conditions.
SI IRRTITI ITF RNFFT fRl II F 2f1
z~s~ssa
WO 96/14317 '.s ~ .: j ~t ~ PC"T/EP95/03487
23
Compounds of formulae BOC(=O)G (V) and BNHC(=O)G (VI) can be
readily prepared from the corresponding alcohols and amines of formulae
BOH and BNH2, respectively, by conventional procedures, for example by
treatment with phenyl chloroforrna~e.
Isocyanates of formula BN=C=O (VII) can be prepared from acids of
formula BCOOH (III) using a sequence which comprises the following steps:
conversion of the acid to an acylazide by treatment for example with
diphenylphosphorylazide; and subsequent Curtius rearrangement of said
acylazide to give an isocyanate. This sequence for preparing isocyanates from
1 0 carboxylic acids is widely described in the literature and can be carried
~ out in
accordance with the reported conditions.
The compounds of formulae X, XI, XV, XVI, XVII, XVIII and XIX, as well
as alcohols of formula BOH and amines of formula BNH2 are commercially
available, widely described in the literature or can be prepared by methods
1 5 analogous to those known in the art starting from commercially available
products.
Compounds of general formula I, being potent PAF antagonists, are
useful as a preventive and therapeutic drugs for the treatment of circulatory
diseases where PAF is involved, such as thrombosis, cerebral apoplexy ( e.g.
2 0 cerebral hemorrhage, cerebral thrombosis), angina pectoris, thrombotic
phlebitis, thrombocitopenic purple, nephritis (e.g. glomerular nephritis),
diabetic nephrosis, pancreatiris; ischemia and shock states (e.g. septic shock
observed after severe infection or postoperatively, intravascular
agglutination
syndrome caused by endotoxin, anaphylactic shock, hemorrhagic shock,
2 5 myocardial infarction); digestive tract diseases wherein PAF is involved
(e.g.
gastric ulcer, inflamatory bowel disease); diseases related to allergy and
inflammation (e.g. asthma, dermatitis, urticaria, arthritis, psoriasis);
pneumonia; rejection due to inczeased PAF production after implantations of
organs; postoperative organodysfunction (e.g. in heart, liver and kidney) and
3 0 any other condition in which PAF is implicated. They may also be used for
contraception of female mammals by suppressing cell division and/or
ovoimplantation on the uterus, in the treatment of endometriosis and in the
prevention or treatment of hyperendothelinemia induced by excess secretion
of endothelin.
3 S According to the activity of the compounds disclosed, the present
invention further provides compositions that comprise a compound of the
invention together with an excipient and optionally other auxiliary agents, if
necessary. The compounds of the present invention can be administered in
different pharmaceutical preparations, the precise nature of which will
SUBSTITUTE SHEET (RULE 26)
WO 96/14317
PCT/EP95/03487
24
depend, as it is well known, upon the chosen route of administration and the
nature of the pathology to be treated.
Thus, solid compositions according to the present invention for oral
administration include compressed tablets, dispersible powders, granules and
capsules. In tablets, the active component is admixed with at least one inert
diluent such as lactose, starch, mannitol, miQOCrystalline cellulose or
calcium
phosphate; granulating and disintegrating agents for example corn starch,
gelatine, microcrystalIine cellulose or polyvinylpyrrolidone; and lubricating
agents for example magnesium stearate, stearic acid or talc. The tablets may
be
1 0 coated by known techniques to delay disintegration and absorption in the
gastrointestinal tract and, thereby, provide a sustained action over a longer
period. Gastric film-coated or enteric film-coated tablets can be made with
sugar, gelatin, hydroxypropylcelIulose, or acrylic resins. Tablets with a
sustained action may also be obtained using an excipient which provides
1 5 regressive osmosis, such as the galacturonic acid polymers. Formulations
for
oral use may also be presented as hard capsules of absorbable material, such
as
gelatin, wherein the active ingredient is mixed with an inert solid diluent
and
lubricating agents, or pasty materials, such as ethoxylated saturated
glycerides.
Soft gelatin capsules are also possible, wherein the active ingredient is
mixed
2 0 with water or an oily medium, for example peanut oil, Iiquid paraffin or
olive
oil.
Dispersible powders and granules suitable for the preparation of a
suspension by the addition of water provide the active ingredient in
admixture with dispersing or wetting agents, suspending agents, such as
2 5 sodium carboxymethylcellulose, methylcellulose, hydroxypropylmethyl-
cellulose, sodium alginate, polyvinylpyrrolidone, gum tragacanth, xantham
gum, gum acacia, and one or more preservatives, such as methyl or n-propyl-
p-hydroxybenzoate. Additional excipients, for example sweetening, flavouring
and colouring agents may also be present.
3 0 Liquid compositions for oral administration include emulsions,
solutions, suspensions, syrups and elixirs containing commonly used inert
diluents, such as distilled water, ethanol, sorbitol, glycerol, or propylene
glycol. Such compositions may also comprise adjuvants such as wetting
agents, suspending agents, sweetening, flavouring, perfuming, preserving
3 5 agents and buffers.
Other compositions for oral administration include spray or aerosol
compositions, which may be prepared by known methods. These
compositions, which can disperse the active ingredient in the form of drops of
SU~BS~TUTE Si~T (RI~.E 2s~
WO 96/14317
~~ ~~~ "'~"~~ . PCT/EP95/03487
a solution or suspension or in the form of a powder, will contain a suitable
propellent.
Preparations for injection, according to the present invention, for
parenteral administration by bolus injection or continuous infusion include
. 5 sterile aqueous or non-aqueous solutions, suspensions or emulsions, in a
non-toxic parentally-acceptable diluent or solvent. Examples of aqueous
solvents or suspending media are distilled water for injection, Ringer's
solution, and isotonic sodium chloride solution. Examples of non-aqueous
solvents or suspending media are propylene glycol, polyethylene glycol,
10 vegetable oils such as olive oil, or alcohols such as ethanol.. These
compositions may also include adjuvants such as wetting, preserving,
emulsifying and dispersing agents. They may be sterilized by any known
method or manufactured in the form of sterile solid compositions which can
be dissolved in sterile water or some other sterile injectable medium
15 immediately before use. When all of the components are sterile, the
injectables will maintain sterility if they are manufactured under a sterile
environment.
A compound of the invention may also administered in the form of
suppositories or enemas (which include aqueous or oily solutions as well as
2 0 suspensions and emulsions) for rectal administration of the drug. Such
compositions are prepared following conventional procedures; for example,
suppositories can be prepared by mixing the active ingredient with a
conventional suppository base such as cocoa butter or other glycerides.
Compositions for topical administration of a compound of the present
2 5 invention include creams, ointments, pastes, lotions, gels, sprays, foams,
aerosols, solutions, suspensions or powders. Such compositions are
conventional formulations and can be prepared by procedures known in the
art.
When the compounds of the present invention are to be administered
3 0 to the eye, they can be formulated into solutions or suspensions in
suitable
sterile aqueous or non-aqueous solvents. These compositions may also
include buffers and preservatives.
The dosage and frequency of dose may vary depending upon symptoms,
age and body weight of the patient, as well as upon the route of
administration,
3 5 but, in general, the compounds of the invention may be administered orally
in
a daily dose of from I-1000 mg for an adult, preferably a dosage from 5-250
mg,
which may be administered either as a single dose or as divided doses. A
preferred dosage for human patients is from 0.005 to 20 mg/kg of body weight,
more preferably from 0.05 to 5 mg/kg of body weight.
~I IRSTITI ITF SHFFT 1171 tl F ~R1
WO 96/14317 ~ i ; .
PCT/EP95/03487
26
The compositions for topical administration will typically contain 0.5-
IO% by weight of a compound of formula I.
Following are some representative preparations for tablets, capsules, -
syrups, aerosols, injectables and creams. They can be prepared following
standard procedures and they are useful in the treatment of PAF-mediated
conditions.
Tablets
Compound of formula I 100 mg
1 0 Dibasic calaum phosphateI25 mg
Sodium starch glycolate 10 mg
Talc IZ.S mg
Magnesium stearate 2.5 mg
1 5 250.0 mg
Hard gelatin capstles
Compound of formula I 100 mg
Lactose 197 mg
2 0 Magnesium stearate 3 mg
300 mg
2 5 Compound of formula I 0.4 g
Sucrose 45 g
Flavouring agent 0.2 g
Sweetening agent O.I g
Water to 100 m
L
30
A
erosol
Compound of formula I 4 g
Flavouring agent 0.2 g
Propylene glycol to I 00 m
1
3 5 Suitable propellent to 1 unit
Injectable ~repa_ration
Compound of formula I . 100 mg
Benrylic alcohol 0.05 m
I
SUBSTITUTE SHEET (RULE 26)
_ w0 96/14317 ~'i ~~ ~~6'~g' ~. ,
PCT/EP95/03487
27
Propylene glycol I m
1
Water to 5 m
1
Cream
Compound of formula I 2 g
Dimethyl acetamide 2 g
White paraffin 2,5 g
Stearic alcohol ~ g
Propylene glycol 12 g
1 0 Sodium lauryl sulfate 1.5 g
Methylparabene 0,3 g
Purified water 31.6 g
The following pharmacological tests explain the activity of the
1 5 compounds of the present invention in more detail.
PHARMACOLOGICAL TEST 1
Inhibition of platelet aQaregation induced by PAF
Platelet aggregation studies were done by the method of Born (J.
Physiol.,1962, I62. 67). Blood was collected in 3.16% sodium citrate (1 volume
2 0 per 9 volumes of blood) by cardiac puncture from male New Zealand rabbits
(2-2.5 Kg body weight). Platelet rich plasma (PRP) was prepared by
centrifuging
the blood at 250xg for IO min. at 4°C. The PRP was diluted with
platelet-poor
plasma (PPP) obtained by further centrifuging at 3000xg for 10 min. The
platelet count was adjusted to 3x105 cells/mm3. Platelet aggregation was
2 5 induced by Clg-PAF (15 nM) and measured with a dual-channel aggregometer
Chrono-log 560. Activity was expressed as the ICSp value, that is to say the
concentration of drug required to inhibit platelet aggregatory response by
50%.
The results are shown in table I below.
TABLIr II
3 0 Compound ICS (~M) .
No.
1 0.0076
16 0.0092
3 5 I7a 0.018
I~ 0.0050
2I 0.0069
O.OI I
SUBSTITUTE SHEET (RULE 26)
WO 96/14317 ~ ~ ~ : _ PCT/EP95/03487
28
32 0.011
39 O.OI6
40 0.012
41 0.0067
O.OI9
47 0.036
50 0.013
5~ O.OI7
60 O.OI7
1 0 6I 0.0062
70 0.034
PHARMACOLOGICAL TEST 2
1 S Inhibition of PAF-induced hwotension in normotensive rats
Male Sprague-Dawley rats, weighing I80-220 g, were anesthetized with
sodium pentobarbital (50 mg/Kg i.p.). Blood pressure was recorded from the
left carotid artery using a Statham pressure transducer coupled to a Beckman
R6I1 recorder. Right and left femoral veins were catheterized to inject test
2 0 compounds and PAF (0.5 ~tg/Kg). Test compounds were administered by
intravenous injection (1 mL/Kg, dissolved in saline) 3 min. before PAF. Blood
pressure was monitored and percent inhibition of PAF-induced hypotension
with respect to controls was calculated. The results were expressed as IDSo
values, that is to say the dose of test compound required to inhibit
hypotension
2 5 by 50%. Results are shown in Table II.
TABLE II
Compound IDSO (mg/Kg)
No.
3 0 1 0.0086
17a 0.029
1~ O.OI-0.025
21 0.043
30 O.OI5
3 5 32 0.0079
0.033
4I O.OI2
47 O.OI3
SUBSTITUTE S;iEFT IRULF ?Rv
WO 96/14317
y, PCT/EP95/03487
29
50 O.OI7
59 0.021
60 O.OI9
70 0.014
The following examples illustrate, but do not limit, the scope of the
present invention:
REFERENCE EXAMPLE 1
1-tent-Butoxycarbonyl-4-(aminomethyl)piperidine
1 0 To a cooled (0 °C) solution of 4-(aminomethyl)piperidine (40 g,
0.35 mol)
in CHCI3 (300 mL) was added a solution of di-tert-butyl dicarbonate (39.2 g,
O.I7
mol) in CHC13 (300 mL) and the reaction mixture was stirred at room
temperature for I8 h. The resulting solution was washed with H20 and the
aqueous phase was reextracted with CHC13. The combined organic phases were
dried and concentrated to a crude product (54.I g), which was directly used in
the next step as obtained.
IH NMR (SOMHz, CDC13) 8 (TMS): 4.1I (broad d, J= 13.4 Hz, 2H), 2.69 (m, 4H),
1.45 (s, 9H), I.8-0.8 (complex signal, 7I~.
REFERENCE EXAMPLE 2
2 0 4-[[1-(tent-Butoxycarbonyl)-4-piperidyl]methylamino]-3-nitropyridine
To a cooled (0 °C) solution of 4-chloro-3-nitropyridine (83.7 g,
0.53 mol)
in CHC13 (700 mL) a solution of the product obtained in reference example I
(140 g, 0.65 mol) and Et3N (110 mL) in CHCI3 (500 mL) was added and the
mixture heated at reflux for 18 h. It was then evaporated, and the residue was
2 5 partitioned between IN NaOH and EtOAc. The aqueous phase was reextracted
twice with EtOAc, and the combined organic extracts were dried and
concentrated to a total volume of 400 mL. After cooling (-20 oC) overnight, a
yellow solid was collected and dried (II5 g, 64%).
mp I3I-I38 °C;
3 0 1H NMR (80MHz, CDCl3) 8 (TMS): 9.20 (s, IH), 8.30 (d, J= 5.5Hz, IH), 8.19
(m,
1H), 6.72 (d, J= 5.5Hz, 1H), 4.18 (broad d, J= 13.4 Hz, 2H), 3.26 (t, J=
5.9Hz, 2H),
2.72 (broad t, J= I2.7Hz, 2H), I.46 (s, 9H), I.8-0.8 (complex signal, SH).
REFERENCE EXAMPLE 3
3-Amino-4-([1-(tent-butoxycarbonyl)-4-piperidyl]methylamino)pyridine
3 5 A mixture of the product obtained in reference example 2 (26.2 g, 0.077
mol) and Pd/C IO% (3.83 g) in MeOH (500 mL) was hydrogenated at
SUBSTITUTE SHEET (RULE 26)
WO 96/14317 PC"T/EP95/03487
~~846=~~.
atmospheric pressure for 18 h. The catalyst was filtered off and the filtrate
concentrated to a crude product (22.9 g, 96%), which was directly used in the
next step as obtained.
1H NMR (80MHz, CDCI3) 8 ('TMS): 7.65 (d, J= 5.5Hz, IH), 7.64 (s, IH), 6.59 (d,
J=
5 5.5Hz, IH), 4.I0 (broad d, J= 13.4 Hz, 2H), 3.9 (s, 3H), 3.25 (d, J= 6.5Hz,
2H), 2.74
(broad t, J= IZ.OHz, ZH), I.46 (s, 9H), I.8-0.8 (complex signal, 5H).
Alternatively, the title compound was prepared as follows: To a solution
of the product obtained in reference example 2 (82.2 g, 0.244 mol) in pyridine
(400 mL) was added a solution of Na2S204 (I70 g, 0.976 mol) in H20 (500 mL).
1 0 The mixture was stirred at room temperature for 24 h, then partitioned
between EtOAc (800 mL) and 5N NaOH (450 mL). The organic phase was
separated, dried and evaporated to give a yellow solid (101 g), which still
contained pyridine.
REFERENCE EXAMPLE 4
1 5 1-[[1-(tent-Butoxycarbonyl)-4-piperidyl)methyl]-1H-2-methylimidazo[4,5-
clpyridine
To a solution of the product obtained in reference example 3 (22.9 g, 0.07
mol) in EtOH (350 mL) was added ethyl acetimidate hydrochloride (9.2 g, 0.074
mot) and the mixture was refluxed for 4 h. A second equivalent of ethyl
2 0 acetimidate hydrochloride was added (9.2 g, 0.074 mol) and the mixture was
further refluxed for 18 h. Finally, a third equivalent of ethyl acetimidate
hydrochloride was added (9.2 g, 0.074 mol) and the mixture was heated for 4 h
more. The solvent was removed in vacuo and the residue partitioned between
CHC13 and 0.5N NaOH. The organic phase was dried and concentrated to a
2 5 residue (30 g), which was purified by chromatography on silica gel
(CHCI3:MeOH, IO%) to afford the title compound as a yellow solid (23.4 g,
95%).
1H NMR (80MHz, CDCl3) 8 (TMS): 8.98 (s, IH), 8.38 (d, J= 5.5Hz, IH), 7.22 (d,
J=
5.5Hz, IH), 4.I0 (broad d, J= I3.4 Hz, 2H), 3.96 (d, J= 7.3Hz, 2H), 2.64
(broad t, J=
12.7Hz, 2H), 2.63 (s, 3H), 1.46 (s, 9H), 2.2-1.0 (complex signal, 5H).
3 0 REFERENCE EXAMPLE 5
1-((4-Piperidyl)methyl)-IH-2-methylimidazo[4,5-c]pyridine
To a cooled (0 °C) solution of the product obtained in reference
example
4 (23.1 g, 0.07 mol) in MeOH (200 mL) was added dropwise 6.5N HChg~/dioxane
solution (44 mL). The mixture was stirred at room temperature for 2 h and
3 5 evaporated to dryness. The residue was cooled (0 °C), IN NaOH was
added and
the resulting solution was extracted with CHC13 (3x). The combined organic
extracts were dried and concentrated to a yellow solid (I5.8 g, 98%).
SUBSTITUTE SHEET (R' !!..~ ?"~
WO 96/14317 , PCT/EP95/03487
31
1H NMR (80MHz, CDCI3) 8 (TMS): 8.96 (s, IH), 8.35 (d, J= 5.5Hz, IH), 7.20 (d,
j=
S.SHz,1H), 3.95 (d, J= 7.3Hz, ZH), 3.06 (broad d, J= I2.01-iz, 2H), 2.6I (s,
3H), 2.51
(broad t, J= I2.7Hz, 2H), 2.2-L0 (complex signal, 6H).
REFERENCE EXAMPLE 6
1-(4-Piperidyl)-iH-2-methylimidazo[4,5-c]pyridine, hydrochloride
Following the procedure described above in reference examples I-5, but
starting from 4-aminopiperidine instead of 4-(aminomethyl)piperidine, the
title compound was obtained (56%).
1H NMR (80MHz, CD30D) S (T'MS): 9.32 (s, 1H), 8.82 (d, j= 5.5 Hz, IH), 8.62
(d,
J= 5.5 Hz, IH), 5.I7 (s, IH), 3.9-2.2 (complex signal, 9H).
REFERENCE EXAMPLE 7
3,3-biphenyl-3-ethoxycarbonylpropionic acid
a) Ethyl diphenylacetate
To a solution of diphenylacetic acid (20 g, 0.094 mol) in EtOH (70 mL),
1 5 was added toluene (70 mL). Next, H2S04 (3 mL) was added dropwise and the
reaction mixture was refluxed for IS h. Then, H20 and EtOAc were added, the
layers were separated, and the organic phase was washed with saturated
NaHC03 solution (3x), dried and concentrated to a white solid (23.2 g), which
was directly used in the next step as obtained.
2 0 1H NMR (80MHz, CDCl3) 8 (TMS): 7.28 (m, IOH), 5.00 (s, IH), 4.19 (q, j=
7Hz,
ZH), L22 (t, J= 7Hz, 3H).
b) Tent butyl 3,3-diphenyl-3-ethoxycarbonylpropionate
To a solution of NaH (60% suspension in parafine, 4.25 g) in DMF (100
mL), was added a solution of the product obtained in reference example 7a
2 5 (23.2 g, 0.965 mol) in DMF (50 mL) and the reaction mixture was stirred at
room temperature for 1 h. Tert-butyl bromoacetate (I3.5 mL, 0.965 mol) was
added dropwise and the mixture was stirred at 60°C for 18 h. The
resulting
solution was treated with H20 (1 mL) and the solvents were removed. More
H20 was added and then it was extracted with EtOAc, at basic pH. The organic
3 0 phase was dried and concentrated to a dark oil.
1H NMR (80MHz, CDC13) S (TMS): 7.26 (s, IOH), 4.21 (q, J= 7.2Hz, 2H), 3.43 (s,
2H), L30 (s, 9H),1.16 (t, J= 7.ZHz, 3H).
c) Title compound
To a cooled (0 °C) solution of the product obtained in reference
example
3 5 7b (6 g, O.OI7 mol) in CH2C12 (20 mL) was added dropwise trifluoroacetic
acid
(2.6 mL) and the mixture was stirred at room temperature for I8 h. The title
compound was then obtained by evaporating the resulting solution to dryness
(85%).
SUaSTI ,TUTE S~iEFT lRlll F ~~~
WO 96/14317 ~ ~ ~g PCT/EP95/03487
32
1H NMR (80MHz, CDC13) 8 (TMS): 11.26 (broad s, 1H), 7.25 (s, IOH), 4.21 (q, J=
7.IHz, ZH), 3.54 (s, ZH),1.I7 (t, J= 7.IHz, 3H).
REFERENCE EXAMPLE 8
3-Phenyl-3-(phenylamino)propionic acid
To a solution of aniline hydrochloride (3.37 g, 26 mmol) and ethyl
benzoylacetate (5 mL, 26 mmol) in MeOH (70 mL) was added NaBH3CN (1.75
g) and the mixture was stirred at room temperature for I8 h. The solvent was
removed and the residue partitioned between 0.5N HCl and Et20. The
aqueous phase was made basic with IN NaOH and extracted with CHC13. The
1 0 organic phase was dried and concentrated to a residue. Purification by
chromatography on silica gel (hexane : EtOAc, 5%) afforded ethyl 3-phenyl-3-
(phenylamino)propionate (4.3 g , 62%). This compound was then hydrolized
under basic conditions to give the title compound as a white solid.
mp: 1 I 0-I 11 °C;
1 5 1H NMR (80MHz, CDC13) b (TMS) 7.I0 (m, 8H), 6.63 (m, 2H), 4.85 (t, J =
6.3Hz,
IH), 4.05 (m, 2H), 2.85 (d, J = 6.5 Hz, 2H).
REFERENCE EXAMPLE 9
3-(4-Nitrophenyl)amino-3-phenylpropionic acid
A mixture of traps-cinnamic acid (Z g, 13 mmol) and HBr (30% solution
2 0 in AcOH, 40 mL) was stirred at room temperature for 18 h and then
evaporated to dryness. The resulting solid was taken up in 2-butanone (I00
mL) and p-nitroaniline (5 g, 36 mmol) was added. The reaction mixture was
refluxed for IS h, allowed to cool and partitioned between CHCl3 and IN HCI.
The organic phase was dried and concentrated to a residue. This was purified
2 5 by chromatography on silica gel (hexane : EtOAc, 50%), to afford the title
compound as a yellow solid (0.78 g, 2I %).
1H NMR (80MHz, CDCl3) 8 (TMS) 7.99 (d, J= 9.2 Hz, 2H), 7.33 (m, 5H), 6.56 (d,
J= 9.2 Hz, 2H), 4.92 (t, J = 6.3Hz, IH), 3.44 (m, 2H), 2.82 (d, J = 6.5 Hz,
2H).
REFERENCE EXAMPLE l0a and IOb
3 0 cis- and traps-3-(4-Nitrophenyl)-3-phenylpropenoic acid
To a cooled (0°C) suspension of 50% NaH (24.66 g, 0.5I mol) in THE
(375
mL) was added dropwise triethyl phosphonoacetate (88.2 mL, 0.44 mol). The
mixture was stirred for 45 min and 4-nitrobenzophenone (I02 g, 0.45 mol) in
THE (525 mL) was added. The resulting mixture was refluxed for I8 h under an
3 5 argon atmosphere, and then allowed to cool and partitioned between H20 and
EtOAc. The organic phase was dried and concentrated to a residue (l I5 g).
This
crude material was dissolved in MeOH (600 mL), a solution of K2C03 (87.2 g) in
H20 (288 mL) was added and the mixture was refluxed for 4 h. MeOH was
removed, water was added and the solution extracted with hexane. The
SUaSTITUTE SHEET fR(1fF SRI
WO 96/14317
_ ~ ~ P~~p95/03487
33
aqueous solution was then brought up to acid pH with 5N HC1 and extracted
with CHC13. Evaporation of the solvent afforded a brown solid as a czs/trans
mixture.
Pure cis isomer (l0a) can be obtained by recrystallization from EtOAc (34
g, 30%).
1H NMR (80MHz, CDCI3) 8 (TMS): 8.23 (d, J= 8.0 Hz, 2H), 7.33 (m, 7H), 6.70
(m),
6.44 (s, IH).
Pure traps isomer (lOb) can be obtained as follows: A mixture of ethyl
traps-cinnamate (4.4g, 27 mmol), 4-bromonitrobenzene (6 g, 29.7 mol),
triphenylphosphine (0.26 g), tributylamine (8 mL), and palladium acetate (57
mg) in acetonitrile (20 mL) was heated under argon at refIux for two days. The
cooled mixture was partitioned between 0.5 N NaOH and CHC13, the organic
phase separated, dried and concentrated. The residue was purified by
chromatography on silica gel (Hexane:EtOAc, 20%) to afford a white solid (2.4
g,
3I %).
1H NMR (80MHz, CDCl3) 8 (TMS): 8.I8 (d, J= 8.0 Hz, 2H), 7.33 (m, SH), 6.39 (s,
IH).
REFERENCE EXAMPLE 11
3-Hydroxy-3-phenyl-3-trifluoromethylpropionic acid
2 0 To a cooled (0 °C) solution of n-butyl lithium (I.6M in hexanes, 40
mL)
in dry THF (90 mL), was added dropwise diisopropylamine (9.45 mL) and the
mixture was stirred for 5 min. Keeping the temperature at 0°C, AcOH
(I.92 mL,
0.0336 mol) was added dropwise and the reaction mixture was stirred for 10
min and then heated at 50°C for 30 min. The resulting solution was
allowed to
2 5 cool, a solution of 2,2,2-trifluoroacetophenone (4.76 mL (0.0336 mol) in
dry
THF (I5 mL) was added at 0 °C and the resulting mixture was stirred
at room
temperature overnight. Finally, Et20 (I50 mL) and H20 (50 mL) were added,
the aqueous phase was separated, acidified with HCl and extracted with EtOAc
(3x). The organic phase was dried and concentrated to afford the title
3 0 compound as an orange solid (3.88 g, 49%).
1H NMR (80MHz, CDC13) 8 (TMS): 9.0 (complex signal, 2H), 7.38 (m, 5H), 3.2 (s,
2H).
REFERENCE EXAMPLE 12
3,3-Diphenylpropenoic acid
3 5 Following a similar procedure to that described in reference example 10,
but using benzophenone instead of 4-nitrobenzophenone, the title compound
was obtained.
1H NMR (80MHz, CDC13) b (TMS): 7.31 (m, IOH), 6.33 (s, IH), 5.9 (m, 1H).
REFERENCE EXAMPLE 13
SUBSTITUTE SHEET (RULE ?G)
WO 96/14317 '~
PGT/EP95/03487
34
3-(4-Nitrophenyl)butanoic acid
To cooled (0 °C) concentrated H2S04 (30 mL) was added 3-
phenylbutyric
and (IS g, 9I mmoI). Then, a cooled solution of HN03 (5 mL) in HZS04 (10 mL)
was added dropwise and the mixture was stirred at 0 °C for 30 min and
then at
room temperature for 30 min more. The mixture was poured into ice and the
resulting solution allowed to stand in the refrigerator overnight. The
precipitate was filtered, washed with H20 and dried to give a crude product
(28.3 g). This was purified by chromatography on silica gel (CHCI3:MeOH, 10%)
to afford the title compound (4.I g, 21 %).
IH NMR (80MHz, CDCl3+CD30D) 8 (TMS): 8.14 (d, J= 8.7 Hz, ZH), 7.47 (d, J=8.7
Hz, 2H), 4.77 (s, IH), 3.36 (quint, J= 7.6 Hz, IH), 2.61 (d, j= 7.4 Hz,
2H),1.3I (d, J=
7.0 Hz, 3H).
REFERENCE EXAMPLE 14
3-Ethoxycarbonyl-3-(4-nitrophenyl)propionic acid
1 5 Following a similar procedure to that described in reference example 7b,
but starting from ethyl 4-nitrophenylacetate instead of ethyl diphenylacetate,
and hydrolizing then the tert-butyl ester using p-toluenesulfonic acid in
benzene at reflux instead of using trifluoroacetic acid, the title compound
was
obtained.
2 0 1H NMR (80MHz, CDCI3) S (TMS): 9.61 (s, IH), 8.19 (d, J= 8.4 Hz, 2H), 7.49
(d, J=
8.4 Hz, ZH), 4.16 (q, J= 7.3 Hz, 2H), 4.14 (m, IH), 3.30 (dd, J= I7.5 Hz, J=
9.0 Hz,
IH), 2.76 (dd, J= I7.3 Hz, J= 6.I Hz, IH), I.20 (t, J= 7.3 Hz, 3H).
REFERENCE EXAMPLE 15
3-Ethoxycarbonyl-3-phenylpropionic acid
2 5 Following a similar procedure to that described in reference example I4,
but starting from ethyl phenylacetate, the title compound was obtained.
1H NMR (80MHz, CDCI3) 8 (TMS): 9.49 (m, IH), 7.28 (s, 5H), 4.I3 (m, 3H), 3.25
(dd, J= I7.2 Hz, J= 9.8 Hz, IH), 2.67 (dd, J= I7.I Hz, j= 5.3 Hz, 1H), I.17
(t, J= 7.3
Hz, 3H).
3 0 REFERENCE EXAMPLE 16
ris and traps-3-(3-Nitrophenyl)-3-phenylpropenoic acid
Following the procedure described in reference example I0, but starting
from 3-nitrobenzophenone, the title compound was obtained as a yellow solid.
1H NMR (80MHz, CDCl3) 8 (TMS): 8.1I (m, 2H), 7.25 (m, 7H), 6.37 (s, 0.67H),
3 5 6.34 (s, 0.33H), 6.I2 (s, IH).
REFERENCE EXAMPLE I7
3-[N-(Ethoxycarbonyl)amino]-3-(4-nitrophenyl)propionic acid
SUBSTITUTE SHEET (RULE 26)
WO 96/14317 ~ PCT/EP95/03487
Following the procedure described in reference example 13, but starting
from 3-[N-(ethoxycarbo~yl)~r~ino]-3-phenylpropionic acid, the title compound
was obtained.
1H NMR (80MHz, CDC13) b (TMS): 9.66 (s, 1H), 8.I7 (d, J= 6.5 Hz, 2H), 7.50 (d,
J=
5 6.5 Hz, 2H), 6.2 (m, IH), 5.22 (q, J= 7.5 Hz, IH), 4.I1 (q, J= 7.1 Hz, 2H),
2.9I (d, J=
6.2 Hz, 2H),1.20 (t, J= 7.I Hz, 3H).
REFERENCE EXAMPLE 18
3-Hydroxy-3-(2-methylpropyl)-5-methylhexanoic acid
Following the procedure described in reference example lI, but using
1 0 2,6-dimethyl-4-heptanone instead of 2,2,2-trifluoroacetophenone, . the
title
compound was obtained (56%).
1H NMR (80MHz, CDC13) b (TMS): 6.8 (m, 2H), 2.25 (s, 2H), I.7 (m, 2H), 0.95
(m,
16H).
REFERENCE EXAMPLE 19
1 5 3-Methyl-3-phenylbutanoic acid.
a) 3-Methyl-3-phenylbutyronitrile
A mixture of I-chloro-2-methyl-2-phenylpropane (150 g, 0.889 mol) and
NaCN (54.46 g) in DMSO (250 mL) was heated at 100 °C for 3 weeks.
The
solution was concentrated to half the initial volume, H20 (400 mL) was added
2 0 and it was extracted with Et20 (3x). The combined organic extracts were
dried
and concentrated to a crude product (115.1 g), which was directly used in the
next step as obtained.
b) Title compound
To the product obtained in a) above was added slowly H20 (375 mL) and
2 5 H2S04 (300 mL), and the mixture was refluxed for 48 h. Then, H?O was added
and the resulting solution was extracted with CHCl3. The organic phase was
washed with 2N NaOH (3x), and the aqueous phase was acidified with 5N HCl
and extracted with CHCl3. The combined organic extracts were dried and
concentrated to afford the title compound.
3 0 1H NMR (80MHz, CDCl3) 8 (TMS): 10.8 (m, IH), 7.29 (s, 5H), 2.6I (s, 2H),
1.43 (s,
6H).
REFERENCE EXAMPLE 20
N-Methyl-N-phenylaminoacetic acid
To a solution of N-phenylglydne (5 g, 33 mmol) and formaldehyde (37%
3 5 aqueous solution, 20 mL) in acetonitrile (I00 mL) was added NaBH3CN (6.8
g)
and AcOH (2 mL) and the reaction mixture was stirred at room temperature
overnight. Volatiles were removed in vacuo, the residue was acidified to
pH=3-4 and extracted with CHC13 several times. The combined organic extracts
were dried and concentrated to afford a crude product (5.73 g), which was
SUBSTITUTE SHEET (RULE 26)
WO 96/14317
PCT/EP95/03487 4.
~l8O.fi60
36
purified by chromatography on silica gel (EtOAc) to afford the title compound
(3.96 g, 73%).
1 H NMR (SOMHz, CDC13) 8 (TMS): 8.82 (s, I H), 7.38 (m, 2H), 6.75 (m, 3H),
4.05
(s, ZH), 3.03 (s, 3H).
REFERENCE EXAMPLE 21
3-Methyl-3-(4-nitrophenyl)butanoic acid
Following the procedure described in reference example I3, but starting
from the compound obtained in reference example I9, the title compound was
obtained (47%).
1H NMR (SOMHz, CDC13) 8 (TMS): 8.16 (d, J= 6.5 Hz, 2H), 7.55 (d, J= 6.5 H~,
2H),
3.5 (m, IH), 2.70 (s, 2H), L50 (s, 6H).
REFERENCE EXAMPLE ZZ
cis and traps-3-(4-Nitrophenyl)-2-butenoic acid
Following the procedure described in reference example I0, but using 4
1 5 nitroacetophenone instead of 4-nitrobenzophenone, the title compound was
obtained.
1H NMR (80MHz, CDCl3) 8 (TMS): 8.8 (m, IH), 8.23 (d, j= 6.4 Hz, 2H), 7.64 (d,
J=
6.4 Hz, 2H), 6.21 (d, J= 1.3 Hz, 0.7H), 6.07 (d, J= I.3 Hz, 0.3H), 2.62 (d, J=
1.3 Hz,
3H).
2 0 REFERENCE EXAMPLE 23
N-Ethoxycarbonyl-N-(4-nitrophenyl)aminoacetic acid
To a cooled (0 °C) suspension of 4-nitroaniline (10 g, 0.072 mol)
and Et3N
(10 mL) in CHC13 (120 mL), was added dropwise ethyl chloroformate (6.9 mL)
and the mixture was stirred under an argon atmosphere at room temperature
2 5 overnight. CHC13 was added and the resulting solution was washed with 1N
HCl. The layers were separated, the aqueous phase was washed with CHC13 and
the combined organic extracts were dried and concentrated. The residue was
chromatographed on silica gel (hexane:EtOAc, 30%) to afford N-
(ethoxycarbonyl)-4-nitroaniline (I.7 g).
3 0 This product was dissolved in THF (5 mL) and was then added dropwise
to a cooled (0 °C) suspension of NaH (0.48 g, IO mmol) in dry THF (IO
mL). The
mixture was stirred at room temperature for 30 min and then ethyl
bromoacetate (0.89 mL, 8 mmol) was added. The reaction mixture was stirred at
room temperature for 48 h and then refluxed for 24 h. The residue was taken
3 5 up in CHC13 and phosphate buffer, and extracted with CHC13 (2x).
Evaporation
of the solvent gave the title compound as the ethyl ester (I.54 g).
This product was dissolved in MeOH (35 mL), a solution of K2C03 (I.33
g) in H20 (I8 mL) was added and the mixture was refluxed for 3 h. Volatiles
were removed in vacuo and the resulting solution was extracted with hexane.
SUBSTITUTE SfiEFT !RULE 261
WO 96/14317 PCT/EP95/03487
218 '~ s~6 0,
37
The aqueous phase was made acid and extracted with CHC13. The organic
extracts were dried and concentrated to afford the title compound.
1H NMR (80MHz, CDC13) 8 (TMS): 8.21 (d, J= 6.5 Hz, 2I-~, 7.50 (d, J= 6.5 Hz,
2H),
5.89 (broad s, IH), 4.45 (s, 2H), 4.16 (q, J= 7.I Hz, 2H), L26 (t, J= 7.I Hz,
3H).
REFERENCE EXAMPLE 24
traps -3-Phenyl-2-pentenoic acid
Following the procedure described in reference example I0, but using
propiophenone instead of 4-nitrobenzophenone, the title compound was
obtained.
1 0 IH NMR (80MHz, CDC13) 8 CTMS): 10.68 (m, IH), 7.42 (m, 5H), 6.05 (s, IH),
3.I2
(q, J= 7.I Hz, 2H7,1.09 (t, J= 7.I Hz, 3H).
REFERENCE EXAMPLE 25
2-Methyl-2-phenylpropylsulfonyl chloride
In a flask under argon were placed magnesium turnings (0.8 g, 0.036
15 mol), dry THF (IO mL) and a iodine crystal. Then, I-chloro-2-methyl-2
phenylpropane (5 mL, 0.031 moI) in THF (I5 mL) was added slowly and the
reaction mixture was refluxed for 30 min. It was then allowed to cool to room
temperature and finally it was cooled to -70 °C and sulfuryl chloride
(2.5 mL,
0.031 mol) in THF (10 mL) was added dropwise. The reaction was allowed to
2 0 warm up to room temperature and was stirred at this temperature overnight.
The resulting solution was poured into diluted phosphate buffer and extracted
with EtOAc (3x). Evaporation of the solvent afforded the title compound (4.6
g,
65%).
1H NMR (SOMHz, CDCl3) S (TMS): 7.30 (m, 5H), 3.59 (s, 2H),1.39 (s, 6H).
2 5 REFERENCE EXAMPLE 26
[N-(2-Methoxyphenyl)-N-methylamino]acetic acid.
a) N-(2-Methoxyphenyl)aminoacetic acid.
To a cooled (0 °C) solution of 2-methoxyaniline (I3.2 g, 0.108 mol)
in
CHC13 (I00 mL) was added ethyl bromoacetate (6 mL, 0.05 mol) and the
3 0 mixture was stirred at room temperature overnight. The resulting solution
was washed with 0.5N NaOH and the organic phase was concentrated to a
crude product. This was purified by chromatography on silica gel (hexane-
EtOAc, IO%) to afford ethyl N-(2-methoxyphenyl)aminoacetate (4.2 g, 40%).
This compound was dissolved in MeOH (80 mL), a solution of K2C03 (4.4 g) in
3 5 H20 (50 mL) was added and the mixture was refluxed overnight. MeOH was
removed and the resulting aqueous solution was extracted with hexane. The
aqueous phase was acidified with 5N HCl and extracted with CHC13 (3x).
Evaporation of the solvent afforded the title compound.
SUBSTITUTE SiiEET (RULE 26?
~~fi6~ _
WO 96!14317 PCT/EP95/03487
38
1H NMR (80MHz, CDC13+ CD30D) 8 (TMS): 6.83 (m, 3H), 6.56 (m, IH), 3.91 (s,
2H), 3.86 (s, 3H), 3.62 (s, 2H).
b)Title compound
Following the procedure described in reference example 20, but starting
from the compound obtained in reference example 26a, the title compound
was obtained (76%).
1H NMR (80MHz, CDC13) 8 (TMS): 8.78 (broad s, IH), 6.87 (m, 4H), 3.85 (s, 3H),
3.71 (s, 2H), 2.88 (s, IH).
REFERENCE EXAMPLE 27
traps-3-(Methoxymethyl)-3-phenylpropenoic acid.
Following the procedure described in reference example I0, but using 2-
methoxyacetophenone instead of 4-nitrobenzophenone, the title compound
was obtained (30%a).
1H NMR (80MHz, CDC13) 8 (TMS): I1.I3 (broad s, IH), 7.23 (s, 5H), 6.49 (s,
IH),
1 5 3.66 (s, 3H), 3.52 (s, ZH).
REFERENCE EXAMPLE 28
N-Isobutyl-N-(4-nitrophenylsulfonyl)aminoacetic acid
a) N-Isobutyl-N-(4-nitrophenylsulfonyl)amine
To a solution of isobutylamine (5 mL, 0.052 mol) in CHZCl2 (100 mL) was
2 0 added Et3N (5.07 mL) and 4-nitrobenzenesulfonyl chloride (1 L7 g, 0.052
mol)
and the mixture was stirred at room temperature overnight. The resulting
solution was washed with H20 (3x), dried and concentrated to afford the
desired product (9.41 g, 70%).
1H NMR (80MHz, CDCl3) 8 (TMS): 8.38 (d, J= 6.5 Hz, 2H), 8.08 (d, J= 6.5 Hz,
2H),
2 5 S.I2 (t, J= 6.4 Hz, IH), 2.83 (t, J= 6.5 Hz, 2H), I.75 (hept, J= 6.5 Hz,
1H), 0.89 (d, J=
6.5 Hz, 6H).
b) Title compound
Following a similar procedure to that described in reference example
26a, but starting from the compound obtained in section a) above, the title
3 0 compound was obtained (3.20 g, 28%).
1H NMR (80MHz, CDCl3) S (TMS): 8.36 (d, J= 6.5 Hz, 2I-0, 8.03 (d, J= 6.5 Hz,
2H),
4.09 (s, 2H), 3.80 (s, IH), 3.I0 (d, J= 7.4 Hz, 2H), 1.87 (kept, J= 6.5 Hz,
1H), 0.91 (d,
J= 6.5 Hz, 6H).
EXAMPLE 1
3 5 1-([1-(3,3-biphenylpropionyl)-4-piperidyl]methyl]-1H-2-methylimidazo[4,r
c]pyridine
To a cooled (0 °C) mixture of the product obtained in reference
example
5 (0.5 g, 2.I7 mmol), 3,3-diphenylpropionic acid (0.49 g, 2.I7 mmol) and 1-
hydroxybenzotriazole (0.26 g) in DMF (25 mL), was added under a nitrogen
SUBSTITUTE SHEET (RULE 26)
'- WO 96/14317 PCT/EP95/03487
X180660
39
atmosphere dicyclohexylcarbodiimide (0.4 g) and the reaction mixture was
stirred at room temperature for I8 h. The solvents were removed in vacuo, the
resulting residue was stirred with EtOAc and the insoluble material was
filtered off. The organic solution was washed with saturated NaHC03 solution,
H20 and brine, dried and concentrated. The residue (1.44 g) was purified by
chromatography on silica gel (CHCI3:MeOH, IO%) to afford the title compound
as a white solid (0.59 g, 62%).
mp 79-84°C (CZgH3pN40);
1H NMR (80MHz, CDCl3) S (TMS): 8.96 (s, IH), 8.35 (d, J= 5.5Hz, IH), 7.23 (m,
1 0 lIH), 4.66 (t, J= 7.3Hz, IH), 4.65 (m, IH), 3.83 (d, J= 7.2Hz, 2H), 3.81
(m, IH), 3.OI
(dd, J= 7.8Hz, J= 3.2Hz, 2H), 2.57 (s, 3H), 3-0.5 (complex signal, 7H);
13C NMR (20.I5MHz, CDCI3) 8 (TMS): 169.25, I53.I7, 143.78, 143.37, 140.85,
140.71, 139.84, 138.97, 127.93, 127.58, 127,19, 125,87, 104.74, 48.62, 47.11,
44.90,
40.88, 38.00, 36.32, 29.66, 29.03,13.37.
1 5 The trihydrochloride was prepared by treatment of a solution of the
product (0.28 g) in a 1: 1 mixture of EtOAc and CH2CI2 with a solution of
HCl(g)
in Et20. The mixture was cooled for 1 h at -20°C and the solid was
collected by
filtration to afford the desired salt (0.3 g, 85%a).
mp 128-134°C (C28H3aN40.3HC1).
2 0 The hemifumarate was prepared by treatment of a solution of the
product (0.87 g) in EtOH with a solution of fumaric acid (0.46g) in EtOH. The
mixture was cooled for 1 h at -20°C, the solid was collected by
filtration and
recrystallized again in EtOH to afford the desired salt (0.308 g, 30%).
mp 190-194°C (C2gH3pN40.I /2 C4H404. H20).
2 5 EXAMPLE 2
1-[1-(3,3-Diphenylpropionyl)-4-piperidyl]-IH-2-methylimidazo[4,5-c]pyridine
Following the procedure described in example 1, but starting from the
compound obtained in reference example 6, the title compound was obtained
as a white solid (45%).
3 0 mp: 95-100°C (CZ~H2gN40);
IH NMR (80MHz, CDC13) 8 (TMS): 8.97 (s, 1H), 8.33 (d, j= 5.3Hz, IH), 7.29 (m,
lOH), 7.08 (d, J= 5.3Hz, IH), 4.83 (m, IH), 4.74 (t, J= 7.5Hz, IH), 4.30 (m,
2H), 3.10
(m, 3H), 2.61 (s, 3H), 3-I.5 (complex signal, 5H).
EXAMPLE 3
3 5 1-((1-[3-[N-(Methoxycarbonyl)amino]-3-phenylpropionyl]-4-piperidyl]methyl]-
1H-2-methylimidazo[4,5-c]pyridine
Following the procedure described in example I, but using 3-[N-
(methoxycarbonyl)amino)-3-phenylpropionic acid instead of 3,3-
SUBSTITUTE SHEET (RULE 26)
WO 96/14317 ~' ; PCT/EP95/03487
diphenylpropionic acid, the title compound was obtained as a white solid
(5I %).
mp: 97 I00°C (C24H29N5~3.1 /2H20);
IH NMR (80MHz, CDC13) S (TMS): 8.95 (s, IH), 8.35 (d, J= 5.5Hz, IH), 7.30 (m,
5 6H), 6.50 (m, IH), 5.08 (m, IH), 4.60 (m, 1H), 3.87 (m, 3H), 3.63 (s, 3H),
2.58 (s,
3H), 3-0.5 (complex signal, 9H).
EXAMPLE 4
1-[1-[3-[N-(Methoxycarbonyl)amino]-3-phenylpropionyl]-4-piperidyl]-1H-2
methylimidazo[4,5-c]pyridine
10 Following the procedure described in example 1, but starting. from the
compound obtained in reference example 6 and 3-[N-
(methoxycarbonyl)amino]-3-phenylpropionic acid, the title compound was
obtained as a white solid (56%).
mp: 102-I05°C (C23H2~N503.H20);
1 5 IH NMR (80MHz, CDC13) S (TMS): 8.96 (s, IH), 8.32 (d, J= 5.4Hz, IH), 7.36
(m,
5H), 7.08 (d, J =5.4 Hz, 1H), 6.30 (m, IH), 5.I9 (m, 1H), 4.87 (m, IH), 4.30
(m, 2H),
3.66 (s, 3H), 2.63 (s, 3H), 3.3-1.6 (complex signal, 8H).
EXAMPLE 5
1-[[1-[3-Phenyl-3-[N-(tent butoxycarbonyl)amino]propionyl]-4-piperidyl)methyl]-
2 0 1H-2-methylimidazo[4,5-c]pyridine
Following the procedure described in example 1, but using 3-phenyl-3-
[N-(tert-butoxycarbonyl)amino]propionic acid instead of 3,3-diphenylpropionic
acid, the title compound was obtained as a white solid (40%).
IH NMR (SOMHz, CDC13) S (TMS): 8.97 (s, IH), 8.38 (d, J= 5.5Hz, IH), 7.30 (m,
2 5 6H), 6.30 (m, IH), 5.08 (m, 1H), 4.60 (m, IH), 3.87 (m, 3H), 2.60 (s, 3H),
3-0.5
(complex signal, 9H),1.40 (s, 9H).
EXAMPLE 6
I-[[1-[3-[N-(4-aminobenzoyl)aminol-3-phenyipropionyl]-4-piperidyl]methyl]
IH-2-methylimidazo[4,5-c]pyridine
3 0 a) 1-[[1-(3-Amino-3-phenylpropionyl)-4-piperidyl]methyl]-1H-2-
methylimidazo[4,5-c]pyridine
Following the procedure described in reference example 5, but starting
from the compound obtained in example 5, the desired product was obtained
as a colourless oil.
3 5 IH NMR (80MHz, CDC13) b (TMS): 8.97 (s, IH), 8.38 (d, J= SHz, 1H), 7.31
(m, 6H),
4.70 (m, 1H), 4.51 (t, J= 7.3Hz, 1H), 3.91 (m, 3H), 2.60 (s, 3H), 3.0-0.7
(complex
signal, I1H).
b) Title compound
SUBSTITUTE SHEET RULE 26)
WO 96/14317 ' ; ~ PCT/EP95/03487
~; ..~ ~r i ,.
41
Following the procedure described in example I, but starting from 4-
aminobenzoic acid and the compound obtained in example 6a, the title
compound was obtair<ed as a white solid (75%).
mp: I32-142°C (C29H32N602.H20);
I H NMR (80MHz, CDCl3) b (TMS): 8.97 (s, 1H), 8.45 (m, 1 H), 8.38 (d, J=
5.5Hz,
IH), 7.72 (d, J= 8.3 Hz, 2H), 7.34 (m, 6H), 6.65 (d, J= 8.3 Hz, 2H), 5.50 (m,
IH), 4.64
(m, 1H), 3.80 (m, 5H), 2.57 (s, 3H), 3-0.5 (complex signal, 9H).
EXAMPLE 7
1-[[1-[N-(Diphenylmethyl)aminoacetyl)-4-piperidyl]methyl]-1H-2-
1 0 methylimidazo[4,5-c]pyridine
Following the procedure described in example I, but using [N-
(diphenylmethyl)amino]acetic acid instead of 3,3-diphenylpropionic acid, the
title compound was obtained as a white solid (65%).
mp: 77-79°C (C2gH31N50.I /2H20);
1 5 1H NMR (80MHz, CDCl3) b (TMS): 8.97 (s, IH), 8.38 (d, J= 5.5Hz, 1H), 7.29
(m,
I I H), 4.84 (s, I H), 4.65 (m, I H), 3.94 (d, J= 7.2 Hz, 2H), 3.50 (m, I H),
3.36 (s, 2H),
2.60 (s, 3H), 3.0-1.0 (complex signal, 8H).
EXAMPLE 8
1-[[1-(3,3-biphenyl-3-hydroxypropionyl)-4-piperidyl]methyl]-1H-2-
2 0 methylimidazo[4,5-c]pyridine
Following 'the procedure described in example I, but using 3,3-diphenyl-
3-hydroxypropionic acid instead of 3,3-diphenylpropionic acid, the title
compound was obtained as a white solid (6I %).
mp: ZIO-Z1I°C (C2gH3pN402.1 /2H20);
2 5 1H NMR (80MHz, CDCl3) 8 (TMS): 9.20 (s, IH), 8.60 (d, j= 5.5Hz, IH), 7.56
(m,
lOH), 7.38 (d, J= 5.5Hz, IH), 6.79 (m, IH), 4.82 (m, 1H), 4.I2 (d, J= 7.2H,
2H), 4.05
(m, IH), 3.42 (s, 2H), 2.8I (s, 3H), 3.3-1.0 (complex signal, 7H).
EXAMPLE 9
1-[[1-(2-Amino-2,2-diphenylacetyl)-4-piperidyl]methyl]-1H-2-
3 0 methylimidazo[4,5-c)pyridine
Following the procedure described in example I, but using 2-amino-2,2-
diphenylacetic acid instead of 3,3-diphenylpropionic acid, the title compound
was obtained as a white solid (88%).
. IH NMR (80MHz, CDCl3) b (TMS): 8.93 (s, IH), 8.36 (d, J= 5.5Hz, IH), 7.34
(m,
3 5 lOH), 7.03 (d, J= 5.5Hz, IH), 4.29 (m, 2H), 3.75 (d, J= 7.2H, 2H), 2.49
(s, 3H), 2.8-1.0
(complex signal, 9H).
EXAMPLE 10
1-[[1-[2-(N-acetylamino)-2,2-diphenylacetyl]-4-piperidyl]methyl]-iH-2
methylimidazo[4,5-c]pyridine
SUBSTITUTE SHEET (RULE 26)
WO 96/14317 ~ PCT/EP95/03487~
42
A solution of the compound obtained in example 9 (0.3 g, 0.68 mmol) in
pyridine (3 mL) and Ac20 (1 mL) was heated at 65 ~C for 18 h. The solvents
were removed in vacuo and the residue partitioned between CHC13 and O.SN
NaOH. The organic phase was dried and concentrated to a residue (0.38 g),
which was purified by chromatography on silica gel (CHCI3:MeOH 5%) to
afford the title compound (0.3 g, 92% ).
mp: I38-148°C (C29H31NSO?~;
1H NMR (80MHz, CDC13) 8 (TMS): 8.93 (s, IH), 8.35 (d, J= 5.5Hz, 1H), 8.27 (s,
IH), 7.60 (m, 4H), 7.30 (m, 6H), 7.00 (d, J= 5.5Hz, IH), 4.45 (m, 2H), 3.72
(d, J=
1 0 7.ZHz, 2H), 2.48 (s, 3H), 2.7 0.4 (complex signal, 7H), I.69 (s, 3H).
EXAMPLE 11
1-[[1-[3-Phenyl-3-(phenylamino)propionyl]-4-piperidyl]methyl]-1H-2
methylimidazo[4,5-c]pyridine
Following the procedure described in example I, but using 3-phenyl-3
1 5 (phenylamino)propionic acid (obtained in reference example 8) instead of
3,3
diphenylpropionic acid, the title compound was obtained as a white solid
(55%).
mp: 82-9I°C (C2gH31N50.1/2H20);
IH NMR (80MHz, CDC13) 8 (TMS): 8.95 (s, IH), 8.35 (d, J= 5.5Hz, IH), 7.32 (m,
2 0 7H), 7.06 (t, J= 8.OHz, 2H), 6.57 (t, j= 8.OHz, 2H), 5.40 (m, IH), 4.77
(m, 2H), 3.77 (d,
J= 7.IH, 2H), 3.70 (m, IH), 2.53 (s, 3H), 3.0-0.3 (complex signal, 9H).
EXAMPLE 12
1-[[1-[3-[(4-Nitrophenyl)amino]-3-phenylpropionyl)-4-piperidyl]methyl]-1H-2
methylimidazo[4,5-c]pyridine
2 5 Following the procedure described in example I, but using 3-(4-
nitrophenyl)amino-3-phenylpropionic acid (obtained in reference example 9)
instead of 3,3-diphenylpropionic acid, the title compound was obtained as a
white solid (90%).
mp: 230-232°C (CZgH3pN603.I /2H20);
3 0 IH NMR (80MHz, CDC13) 8 ('TMS): 8.98 (s, IH), 8.38 (d, J= 5.5Hz, IH), 7.97
(d, J=
9Hz, 2H), 7.35 (m, 5H), 7.12 (d, j= 5.5Hz, IH), 6.95 (m, IH), 6.42 (d, J=
9.OHz, 2H),
4.76 (m, 2H), 3.75 (m, 3H), 2.58 (s, 3H), 3.0-0.3 (complex signal, 9H).
EXAMPLE 13
1-[[1-[3-[(4-Aminophenyl)amino]-3-phenyIpropionyl)-4-piperidyl]methyl]-1H-2-
3 5 methylimidazo[4,5-c]pyridine
To a solution of the product obtained in example 12 (226 mg, 0.4 mmol)
in EtOH (5 mL) and HZO (0.6 mL) was added a solution of CaCl2 (33.6 mg) in
H20 (0.26 mL) and powdered zinc (0.58 g). The resulting mixture was heated at
50°C for 45 min, filtered through celite and the filtrate was
concentrated. The
SUeSTiTUTE S~iFFT ~R~ n F ~Fl
- WO 96/14317
PCT/EP95/03487
43
residue was purified by chr,,~matography on silica gel (CHCI3:MeOH IO%), to
afford the title compound as a white solid (O.I7 g, 9I %).
1H NMR (80MHz, CDCl3) 8 (TMS): 8.93 (s, IH), 8.34 (d, J= 5.5Hz, IH), 7.3I (m,
6H), 6.44 (broad s, 4H), 4.62 (m, 2H), 3.80 (d, J= 7.OHz, 2H), 3.52 (m, 4H),
2.55 (s,
3H), 3.0-0.5 (complex signal, 9H).
A solution of the title compound in CHCl3 was treated with a solution
of HCl(g) in EtZO, to afford the hydrochloride of the title compound.
mp: 189-195°C (C2gH32N6O.4HC1.2H20).
EXAMPLE 14
1 0 1-((I-(2,2-Dicyclohexylacetyl)-4-piperidyl)methyl]-1H-2-methylimidazo(4,5-
c]pyridine
Following the procedure described in example I, but using 2,2-
dicyclohexylacetic acid instead of 3,3-diphenylpropionic acid, the title
compound was obtained as a white solid (23%).
1 5 mp: I6I-164°C (C2~H4pN40.3/4H20);
1H NMR (80MHz, CDC13) 8 (TMS): 8.97 (s, IH), 8.37 (d, J= 5.5Hz, 1H), 7.I9 (d,
J=
5.5Hz, IH), 4.82 (m, IH), 4.03 (m, IH), 3.98 (d, J= 7.2Hz, 2H), 2.62 (s, 3H),
3.0-0.5
(complex signal, 30H).
EXAMPLE 15
2 0 1-((1-(3,3-Diphenylpropenoyl)-4-piperidyl]methyl]-1H-2-methylimidazo(4,5-
c)pyridine
Following the procedure described in example 1, but using 3,3-
diphenylpropenoic acid (obtained in reference example I2) instead of 3,3
diphenylpropionic acid, the title compound was obtained as a white solid
2 5 (85%).
mp: 85-92°C (C2gH2gN40.H20);
IH NMR (SOMHz, CDCl3) S (TMS): 8.95 (s, 1H), 8.37 (d, J= 5.5Hz, 1H), 7.32 (m,
lOH), 7.09 (d, J=5.4Hz, IH), 6.27 (s, IH), 4.65 (m, IH), 3.82 (m, 1H), 3.77
(dd, J=
6.7Hz, J= 2.2Hz, 2H), 2.54 (s, 3H), 2.8-0.5 (complex signal, 7H).
3 0 EXAMPLE 16
cis and traps -I-((I-(3-(4-Nitrophenyl)-3-phenylpropenoyl]-4-piperidyl]methyl)-
1H-2-methylimidazo(4,5-c)pyridine
Following the procedure described in example I, but using a cis/trans
mixture of 3-(4-nitrophenyl)-3-phenylpropenoic acid (obtained in reference
3 5 example 10) instead of 3,3-diphenylpropionic acid, the title compound was
obtained as a white solid (85%). .
mp:106-l I2°C (C2gH2~N5O3.1 /2HZO);
SUBSTITUTE SHEET (RULE 26)
WO 96/14317
PCT/EP95/03487~
44
1H NMR (80MHz, CDC13) S (TMS): 8.95 (s, IH), 8.37 (d, J= 5.5Hz, IH), 8.18 (dd,
J=
8.6Hz, J= 3.2 Hz, 2H), 7.37 (m, 8H), 6.49 (s, 0.5H), 6.40 (s, 0.5H), 4.66 (m,
IH), 3.84
(m, 3H), 2.57 (s, 3H), 2.8-0.5 (complex signal, 7H).
EXAMPLE 17a and 17b
a) ris -1-[[1-[3-(4-Aminophenyl)-3-phenylpropenoyl]-4-piperidyl]methyl]-1H-2-
methylimidazo[4,5-c]pyridine
b) traps -1-[[1-[3-(4-Aminophenyl)-3-phenylpropenoyl]-4-piperidyl]methyl]-IH-
2-methylimidazo[4,5-c]pyridine
Following the procedure described in reference example 3, but starting
from the compound obtained in example I6, the title compound was obtained
as a cis/trans mixture of isomers, which were separated by chromatography on
silica gel (CHCI3:MeOH, IO%).
Slower eluting, isomer cis (example I7a)(54%):
mp: I2I-I35°C (C2gH29Ng0.3/2H20);
1H NMR (80MHz, CDCI3) 8 (TMS): 8.95 (s, IH), 8.36 (d, J= 5.5Hz, 1H), 7.29 (s,
5H), 7.07 (m, 3H), 6.65 (d, J= 6.5Hz, 2H), 6.07 (s, 1 H), 4.70 (m, I H), 3.82
(m, 3H),
2.57 (s, 3H), 2.8-0.5 (complex signal, 9H).
Faster eluting, isomer traps (example I7b) (22%):
mp: 223-224°C (C2gH29N5O.I /2H20);
2 0 IH NMR (SOMHz, CDC13) 8 (TMS): 8.96 (s, 1H), 8.38 (d, J= 5.5Hz, IH), 7.30
(s,
5H), 7.05 (m, 3H), 6.60 (d, J= 6.5Hz, 2H), 6.I7 (s, IH), 4.60 (m, IH), 3.8I
(m, 3H),
2.55 (s, 3H), 2.8-0.5 (complex signal, 9H).
Following the same procedure described in example I6 but using pure
cis or pure traps-3-(4-nitrophenyl)-3-phenylpropenoic acid (described in
2 5 reference example l0a and IOb respectively) instead of a cis/trans mixture
and
then reducing the resulting compounds as described above, the title compound
was obtained in pure cis or traps form.
Alternatively, the pure cis isomer was obtained as follows: To a
solution of SnC12.2H20 (2I g) in HCl (21 mL) was added a solution of cis 1-[[1
3 0 [3-(4-nitrophenyl)-3-phenylpropenoyl]-4-piperidyl]methyl]-IH-2-methylimida
zo[4,5-c]pyridine (IO g, 20.7 mmol) in AcOH (35 mL). The mixture was stirred
at
room temperature overnight, and was then made basic with cooled aqueous
NaOH and extracted with CHC13 (3x), whereupon a solid precipitated in the
organic phase. This solid was collected and the organic solution was dried and
3 5 concentrated to give 8 g of the desired product. The first precipitate and
the
aqueous phase were combined and then treated with more NaOH solution and
extracted with CHC13 (3x). Evaporation of the solvent afforded I.73 g more of
the title product.
EXAMPLE 18
SU6STITUTE S;iEFT (RULE 2R~
_218~~60
WO 96114317 ' PCT/EP95/03487
I-[[1-(3,3-Dicyclohexyl-3-hydroxypropionyl)-4-piperidyl]methyl]-IH-2
methylimidazo(4,5-c]pyridine
Following the procedure described in example I, but using 3,3
dicyclohexyl-3-hydroxypropionic aad instead of 3,3-diphenylpropionic acid, the
5 title compound was obtained as a white solid (27%).
mp: 75-8I°C (C2sH42N402.5/4H20);
IH NMR (80MHz, CDCl3) 8 (TMS): 8.95 (s, IH), 8.37 (d, J= 5.5Hz, IH), 7.24 (d,
J=
5.5Hz, IH), 6.09 (s, 1H), 4.69 (m, 1H), 4.04 (d, J= 7.2Hz, 2H), 3.96 (m, 1H),
2.64 (s,
3H), 3.0-0.5 (complex signal, 3IH).
10 EXAMPLE 19
1-[[1-[3,3-biphenyl-3-(ethoxycarbonyl)propionyl]-4-piperidyl]methyl]-1H-2-
methylimidazo[4,5-c]pyridine
Following the procedure described in example I, but using 3,3-diphenyI
3-ethoxycarbonylpropionic acid (obtained in reference example 7) instead of
1 5 3,3-diphenylpropionic acid, the title compound was obtained as a white
solid
(60%).
mp: 79-89°C (C31H34N403.H20);
1H NMR (80MHz, CDCl3) 8 (TMS): 8.98 (s, IH), 8.38 (d, J= 5.5Hz, IH), 7.28 (m,
I1H), 4.62 (m, IH), 4.22 (q, J= 7.3Hz, 2H), 3.85 (d, J= 7.ZHz, 2H), 3.8I (m,
IH), 3.48
2 0 (m, 2H), 2.59 (s, 3H), 3-0.5 (complex signal, 7H), 1.I7 (t, J= 7.2Hz, 3H).
EXAMPLE 20
(R)-I-[[I-[2-(Methoxycarbonylamino)-2-phenylacetyl~-4-piperidyl]methyl]-1H-2
methylimidazo[4,5-c]pyridine
Following the procedure described in example I, but using (R)-2
2 5 (methoxycarbonylamino)-2-phenylacetic acid instead of 3,3-
diphenylpropionic
aad, the title compound was obtained as a white solid (94%).
mp: 108-I13°C (C23HZ~Ng03.I /2H20);
IH NMR (80MHz, CDC13) 8 (TMS): 8.95 (s, IH), 8.35 (d, J= 5.5Hz, 1H), 7.37 (m,
5H), 7.01 (d, J= 5.5Hz, IH), 6.28 (m, IH), 5.57 (m, IH), 4.70 (m, IH), 3.80
(m, 3H),
3 0 3.63 (s, 3H), 3-1 (complex signal, lOH).
EXAMPLE 21
1-[(1-(3-Hydroxy-3-phenylbutanoyl)-4-piperidyl]methyl]-1H-2
methylimidazo[4,5-c]pyridine
Following the procedure described in example 1, but using 3-hydroxy-3
3 5 phenylbutanoic aad instead of 3,3-diphenylpropionic acid, the title
compound
was obtained as a white solid (4I %).
mp: I99-200°C (C23HZgN40Z.I /4H20);
CI 11CTITI ITC C:~CCT !DI il C ~~1
WO 96/14317 PCT/EP95/03487
46
IH NMR (80MHz, CDC13) 8 (TMS): 8.90 (s, IH), 8.38 (d, J= 5.5Hz, IH), 7.22 (m,
6H), 6.0 (m, IH), 4.60 (m, IH), 3.9I (m, 3H), 2.57 (s, 3H), 3-0.5 (complex
signal,
9H), I.59 (s, 3H).
EXAMPLE ZZ
I-([I-(3-(4-Nitrophenyl)butanoyl]-4-piperidyl]methyl]-1H-2-methylimidazo[4,5-
c]pyridine
Following the procedure described in example 1, but using 3-(4-
nitrophenyl)butanoic acid (obtained in reference example I3) instead of 3,3
diphenylpropionic acid, the title compound was obtained as a white solid
X53%).
mp: 75-77°C (C23HZ~NSO3.1 /2H20);
I H NMR (80MHz, CDCl3) 8 (TMS): 8.92 (s, I H), 8.36 (d, J= 5.5Hz, I H), 8.28
(d, j=
9.5Hz, 2H), 7.42 (d, J= 9.5Hz, ZH), 7.I2 (d, J= 5.5Hz, IH), 4.60 (m, IH), 3.93
(m,
3H), 3.50 (m, IH), 2.61 (s, 3H), 3-0.5 (complex signal, 9H), I.36 (d, J=
6.9Hz, 3H).
EXAMPLE 23
I-([1-(3-(4-Aminophenyl)butanoyl]-4-piperidyl]methyl]-IH-2-
methylimidazo[4,5-c]pyridine
Following the procedure described in reference example 3, but starting
from the compound obtained in example 22, the title compound was obtained
2 0 as a white solid (35%).
mp: lI6-I17°C (C23H2gNg0.3/2H20);
IH NMR (80MHz, CDC13+CD30D) 8 (TMS): 8.88 (s, IH), 8.33 (d, J= 5.5Hz, 1H),
7.30 (d, J= 5.5Hz, IH), 7.02 (d, j= 9.OHz, 2H), 6.67 (d, J= 9.OHz, 2H), 4.64
(m, IH),
3.93 (m, 3H), 3.80 (m, ZH), 3.20 (m, 1H), 2.63 (s, 3H), 3-0.5 (complex signal,
9H),
2 5 1.3I (d, J= 6.9Hz, 3H).
EXAMPLE 24
1-[(I-[2-(4-Nitrophenyl)propionyl]-4-piperidyl]methyl]-IH-2-methylimidazo(4,5
c]pyridine
Following the procedure described in example I, but using 2-(4-
3 0 nitrophenyl)propionic acid instead of 3,3-diphenylpropionic acid, the
title
compound was obtained as a white solid (I2%).
mp: 82-87°C (C~H25N503.1 /2H20);
IH NMR (80MHz, CDCl3) b (TMS): 8.94 (s, IH), 8.33 (d, J= 5.5Hz, IH), 8.I6 (d,
J=
9.SHz, 2H), 7.44 (d, J= 9.5Hz, 2H), 7.I2 (m, IH), 4.73 (m, IH), 4.0 (m, 4H),
2.55 (s,
3 5 3H), 3-0.5 (complex signal, 7H),1.46 (d, J= 6.9Hz, 3H).
EXAMPLE 25
1-[[I-(2-(4-Aminophenyl)propionyl]-4-piperidyl]methyl]-1H-2
methylimidazo(4,5-c]pyridine
SUBSTITUTE SHEET (RULE 26)
'- WO 96/14317
PCT/EP95/03487
47
Following the procedure described in reference example 3, but starting
from the compound obtained in example 24, the title compound was obtained
as a white solid (34%).
mp: 9I-95°C (C~HZ~NSO.H20);
1H NMR (80MHz, CDCl3) 8 (TMS): 8.96 (s, 1H), 8.38 (d, J= 5.5Hz, 1H), 7.05 (m,
1H), 6.99 (d, J= 9.5Hz, 2H), 6.6I (d, J= 9.5Hz, 2H), 4.74 (m,1H), 3.77 (m,
4H), 2.53
(s, 3H), 3-0.5 (complex signal, 9H), I.37 (d, J= 6.9Hz, 3H).
EXAMPLE 26
1-[[1-[3-Ethoxycarbonyl-3-(4-nitrophenyl)propionyl]-4-piperidyl]methyl]-IH-2-
methylimidazo(4,5-c]pyridine
Following the procedure described in example I, but using 3-
ethoxycarbonyl-3-(4-nitrophenyl)propionic acid (obtained in reference example
14) instead of 3,3-diphenyl-propionic acid, the title compound was obtained as
a white solid (18%).
1 5 mp: 81-84°C (CZ5H29N505.2H20);
1H NMR (80MHz, CDCl3) 8 (TMS): 8.95 (s, 1H), 8.35 (d, J= 5.5Hz, IH), 8.16 (d,
J=
9.5Hz, 2H), 7.48 (d, J= 9.5Hz, 2H), 7.22 (m, 1H), 4.63 (m, 1H), 4.03 (m, 6H),
2.62 (s,
3H), 3:3-1 (complex signal, 9H),1.19 (t, J= 6.5Hz, 3H).
EXAMPLE 27
2 0 1-[[1-[3-(4-Aminophenyl)-3-ethoxycarbonylpropionyl]-4-piperidyl]methyl]-1H-
2-
methylimidazo[4,5-c]pyridine
Following the procedure described in reference example 3, but starting
from the compound obtained in example 26, the title compound was obtained
as a white solid (90%).
? 5 mp: 204-205°C (Cz5H31N5~3~H20);
1H NMR (SOMHz, CDCl3) 8 (TMS): 8.98 (s, IH), 8.41 (d, J= 5.5Hz, IH), 7.26 (m,
1H), 7.07 (d, J= 9.5Hz, 2H), 6.62 (d, J= 9.5Hz, 2H), 4.65 (m, IH), 3.98 (m,
6H), 2.62
(s, 3H), 3.3-I (complex signal, I1H), I.19 (t, J= 6.5Hz, 3H).
EXAMPLE 28
3 0 1-[[1-(3-Ethoxycarbonyl-3-phenylpropionyl)-4-piperidyl]methyl]-1H-2-
methylimidazo[4,5-c]pyridine
Following the procedure described in example 1, but using 3-
ethoxycarbonyl-3-phenylpropionic acid (obtained in reference example I5)
instead of 3,3-diphenylpropionic acid, the title compound was obtained as a
3 5 white solid (38%).
mp: I73-I74°C (Cz5H3pN403~H20);
1H NMR (80MHz, CDCl3) 8 (TMS): 8.99 (s, IH), 8.38 (d, J= 5.5Hz, IH), 7.30 (m,
6H), 4.66 (m, I H), 4.13 (m, 6H), 2.62 (s, 3H), 3.5-1 (complex signal, 9H),
1.19 (t, J=
7.I2Hz, 3H).
SUBSTITUTE SHEET (RULE 26)
WO 96/14317
PCT/EP95/03487
48
EXAMPLE 29
cis and tracts -1-[[1-[3-(3-Nitrophenyl)-3-phenylpropenoyl]-4-
piperidyl]methyl]-
1H-2-methylimidazo[4,5-c]pyridine
Following the procedure described in example 1, but using cis and trans-
3-(3-nitrophenyl)-3-phenylpropenoic acid (obtained in reference example 16)
instead of 3,3-diphenylpropionic acid, the title compound was obtained as a
white solid (95%).
mp: 99-I03°C (CZgHZ~N503.1 /2H20);
IH NMR (80MHz, CDC13) 8 (TMS): 8.97 (s, IH), 8.39 (d, J= 5.5Hz, 1H), 8.27 (m,
1 0 2H), 7.30 (m, SH), 6.54 (s, 0.66H), 6.39 (s, 0.34H), 4.62 (m, IH), 3.98
(m, 3H), 2.63 (s,
3H), 3.1-0.7 (complex signal, 7I~.
EXAMPLE 30
cis and traps -1-[[1-[3-(3-Aminophenyl)-3-phenylpropenoyl]-4
piperidyl]methyl]-IH-2-methyiimidazo[4,5-c]pyridine
Following the procedure described in reference example 3, but starting
from the compound obtained in example 29, the title compound was obtained
as a white solid (I4%).
mp: I22-133°C (C2gH29N50.H20);
1H NMR (80MHz, CDCl3) a (TMS): 8.94 (s, IH), 8.33 (d, J= 5.5Hz, IH), 7.28 (m,
2 0 8H), 6.63 (m, 2H), 6.20 (s, 0.4H), 6.16 (s, 0.6H), 4.67 (m, IH), 3.78 (m,
3H), 2.54 (s,
3H), 2.8-O.I (complex signal, 9H).
EXAMPLE 31
1-[[i-[3-(4-Aminophenyl)-3-[N-(ethoxycarbonyl)amino]propionyl]-4
piperidyl]methyl]-IH-2-methylimidazo[4,5-c]pyridine
2 5 Following the procedure described in example I, but using 3-[N-
(ethoxycarbonyl)amino]-3-(4-nitrophenyl)propionic acid (obtained in reference
example I7), and hydrogenating the compound thus obtained according to the
procedure described in reference example 3, the title compound was obtained
as a white solid (15%).
3 0 mp: I I3-I16°C (C25H32N603.I /2H20);
1H NMR (80MHz, CDCl3) 8 (TMS): 8.94 (s, 1H), 8.35 (d, J= 5.5Hz, IH), 7.21 (d,
J=
5.5Hz, IH), 7.07 (d, J= 9.5Hz, 2H), 6.63 (m, 3H), 4.95 (m, IH), 4.60 (m, IH),
4.06 (q,
J = 7.2Hz, 2H), 3.88 (m, 3H), 2.59 (s, 3H), 3.6-0.5 (complex signal, IIH),
I.I9 (t, j=
7.2Hz, 3H).
3 5 EXAMPLE 32
I-[[1-(3-Phenylhexanoyl)-4-piperidyl]methyl]-1H-2-methylimidazo[4,5
c]pyridine
SUBSTITUTE S;aEFT (RULE 261
' WO 96/14317 ~ ; =
PCT/EP95/03487
~~soss~
49
Following the procedure described in example 1, but using 3
phenylhexanoic acid instead of 3,3-diphenylpropionic acid, the title compound
. was obtained as a white solid (SI~%a).
mp: 39-55°C (C25H32N40.1 /2H20);
IH NMR (80MHz, CDCl3) 8 (TMS): 8.96 (s, IH), 8.36 (d, J= 5.5Hz, IH), 7.24 (m,
6H), 4.60 (m, 1H), 3.84 (m, 3H), 3.18 (m, IH), 2.58 (s, 3H), 2.8-0.5 (complex
signal,
13H), 0.97 (t, J= 6.9Hz, 3H).
EXAMPLE 33
1-[[1-[3-Hydroxy-3-(2-methylpropyl)-5-methylhexanoyl]-4-piperidyl]methyl]-iH
1 0 2-methylimidazo[4,5-c]pyridine
Following the procedure described in example 1, but using 3-hydroxy-3-
(2-methylpropyI)-5-methyIhexanoic acid (obtained in reference example I8)
instead of 3,3-diphenylpropionic acid, the title compound was obtained as a
white solid (25%).
1 5 mp: 38-SI°C (C24H3gN402.1/4H2O);
1H NMR (SOMHz, CDC13) 8 (TMS): 8.98 (s, 1H), 8.38 (d, J= 5.5Hz, IH), 7.21 (d,
J=
5.5Hz, IH), 4.98 (s, 1H), 4.69 (m, IH), 4.01 (d, J= 7.2Hz, 2H), 3.85 (m, 1H),
2.63 (s,
3H), 3.I-I.1 (complex signal, I5H), I.00 (broad s, IZH).
EXAMPLE 34
2 0 1-[[1-[3-[N-(4-Aminobenzenesulfonyl)amino]-3-phenylpropionyl]-4-
piperidyl]methyl]-1H-2-methylimidazo[4,5-c]pyridine
Following the procedure described in example I, but using 3-[N-(4-
nitrobenzenesulfonyl)amino]propionic acid, and hydrogenating the
compound thus obtained according to the procedure described in reference
2 5 example 3, the title compound was obtained as a white solid (22%).
mp: 126-134°C (C2gH32N603S.H20);
1H NMR (SOMHz, CDCl3+CD30D) 8 (TMS): 8.86 (s, IH), 8.32 (d, J= 5.5Hz, 1H),
7.53 (d, J= 8.3 Hz, 2H), 7.16 (m, 7H), 6.86 (d, J= 8.3 Hz, 2H), 4.65 (m, I H),
4.55 (m,
IH), 4.00 (m, 5H), 2.61 (s, 3H), 3-0.5 (complex signal, 9H).
3 0 Alternatively, the compound obtained in example 6a was reacted with 4-
nitrobenzenesulfonyl chloride in the presence of triethylamine to give I-[[I-
[3-
[N-(4-nitrobenzenesulfonyl)amino)-3-phenylpropionyl)-4-piperidyl]methyl]-
IH-2-methylimidazo[4,5-c]pyridine, which was hydrogenated according to the
procedure described in reference example 3 to give the title compound.
3 5 EXAMPLE 35
1-[[1-[(N-Ethoxycarbonyl-N-phenylamino)acetyl]-4-piperidyl]methyl]-1H-2-
methylimidazo[4,5-c]pyridine
Following the procedure described in example 1, but using N-
ethoxycarbonyl-N-phenylglycine (prepared from N-phenylglycine and ethyl
SUBSTITUTE SHEET (RULE 26)
WO 96/14317
PCT/EP95/03487
chloroformate) instead of 3,3-diphenylpropionic and, the title compound was
obtained as a white solid (41 %).
mp: I30-I37°C (C24H2gN503.H2O);
IH NMR (80MHz, CDCl3) 8 (TMS): 8.96 (s, IH), 8.35 (d, J= 5.5Hz, IH), 7.31 (m,
5 6H), 4.43 (m, IH), 4.40 (m, 2H), 4.I5 (q, J= 7.ZHz, 2H), 3.96 (d, J=6.9Hz,
2H) 3.90
(m, IH), 2.6I (s, 3H), 3-I.2 (complex signal, 7H), I.I9 (t, J= 7.2Hz, 3H).
EXAMPLE 36
(S)-I-([I-((N-(1-Ethoxycarbonyl-3-methylbutyl)amino]carbonyl]-4
piperidyl]methyl]-IH-2-methylimidazo(4,5-c]pyridine
10 A solution of the compound obtained in reference example 5 (0.5 g, 2.I
mmol) and N-phenoxycarbonyl-L-Leucine ethyl ester (0.78 g, 2.7 mmol,
prepared from L-Leucine ethyl ester and phenyl chloroformate) in pyridine (I5
mL) was refluxed for 18 h. The solvent was removed in vacuo and the residue
partitioned between CHC13 and 0.5N NaOH. The organic phase was dried and
1 5 concentrated to a residue (I.33 g), which was purified by chromatography
on
silica gel (CHCI3:MeOH, 5%) to afford the title compound as a white solid
(0.22
g, 44%).
mp: 60-63°C (C22H33N503.3/4H20);
IH NMR (SOMHz, CDC13) S (TMS): 8.96 (s, 1H), 8.36 (m, 1H), 7.26 (d, J= 5.5Hz,
2 0 IH), 5.30 (d, J= 8.I7Hz, IH), 4.51 (q, J = 8.I2Hz, IH), 4.18 (q, J= 6.5Hz,
2H), 4.I5 (m,
IH), 4.00 (d, J= 7.3Hz, 2H), 2.70 (m, 3H), 2.62 (s, 3H), 2.I-1.4 (complex
signal, 8H),
1.27 (t, J= 6.5Hz, 3H), 0.93 (d, J= 5.5 Hz, 6H).
EXAMPLE 37
(S)-1-[[1-[[N-[1-Ethoxycarbonyl-2-(4-nitrophenyl)ethyl]amino]carbonyl]-4-
2 5 piperidyl]methyl]-1H-2-methylimidazo[4,5-c]pyridine
Following the procedure described in example 36, but using N-
phenoxycarbonyl-4-nitro-L-phenylalanine ethyl ester instead of N-
phenoxycarbonyl-L-Leucine ethyl ester, the title compound was obtained as a
white solid (54%).
3 0 mp: 85-89°C (C25H3pN6O5.I /2H20);
I H NMR (80MHz, CDC13) 8 (TMS): 8.94 (s, I H), 8.35 (d, J= 5.5Hz, 1 H), 8.11
(d, J=
9.2Hz, 2H), 7.32 (d, J= 9.ZHz, 2H), 7.20 (d, j= 5.5Hz, IH), 5.32 (d, J=
8.I7Hz, 1H),
4.81 (q, J = 8.I2Hz, IH), 4.18 (q, J= 6.5Hz, 2H), 4.00 (m, 1H), 3.98 (d, J=
7.3Hz, ZH),
3.23 (d, J= 5.8Hz, 2H), 2.70 (m, 3H), 2.63 (s, 3H), 2.1-I.4 (complex signal,
5H), 1.25
3 5 (t, J= 6.5Hz, 3H).
EXAMPLE 38
(S)-1-[[1-[[N-[2-(4-Aminophenyl)-1-ethoxycarbonylethyl]amino]carbonyl]-4
piperidyl]methyl]-1H-2-methylimidazo(4,5-c]pyridine
SUBSTITUTE SHEET (RULE 26)
WO 96/14317
'~ ~ $ 0 6 ~6 rc~r/~r9s/o34s7
p, .
51
Following the procedure described in reference example 3, but starting
from the compound obtained in example 37, the title compound was obtained
as a white solid (94%).
mp: 87 96°C (C~,5H32N6~3~H2~):
1H NMR (80MHz, CDCl3) 8 (TMS): 8.96 (s, IH), 8.36 (d, J= 5.5Hz, IH), 7.20 (d,
j=
5.5Hz, IH), 6.88 (d, J= 9.2Hz, ZH), 6.57 (d, J= 9.2Hz, 2H), 4.97 (d, J= 8.IHz,
IH), 4.65
(q, J = 8.I2Hz, 1H), 4.18 (q, J= 6.5Hz, 2H), 4.00 (m, IH), 3.98 (d, J= 7.3Hz,
2H), 2.98
(d, J= 5.8Hz, 2H), 2.80 (m, 5H), 2.61 (s, 3H), 2.I-I.4 (complex signal, 5H),
L25 (t, J=
6.5Hz, 3H).
1 0 EXAMPLE 39
(S)-1-[[1-[[N-(1-Ethoxycarb onyl-1-phenylmethyl)amino]carb onyl]-4
piperidyl]methyl]-1H-2-methylimidazo[4,5-c]pyridine
Following the procedure described in example 36, but using N
phenoxycarbonyl-L-phenylglycine ethyl ester (prepared from L-phenylglycine
I S ethyl ester and phenyl chloroformate) instead of N-phenoxycarbonyl-L
Leucine ethyl ester, the title compound was obtained as a white solid (61%).
mp: I53-154°C (C24H29N5~3~1/2H20);
1H NMR (SOMHz, CDCl3) 8 (TMS): 8.96 (s, IH), 8.37 (d, J= 5.5Hz, IH), 7.33 (m,
5H), 7.20 (d, J= 5.5Hz, 1H), 5.5I (m, 2H), 4.18 (q, J= 6.5Hz, 2H), 4.00 (m,
IH), 3.98
2 0 (d, J= 7.3Hz, 2H), 2.72 (m, 3H), 2.6I (s, 3H), 2.1-I.4 (complex signal,
5H), L20 (t, J=
6.5Hz, 3H).
EXAMPLE 40
1-[[1-(3-Phenylbutanoyl)-4-piperidyl]methyl]-1H-2-methylimidazo[4,5
c]pyridine
2 5 Following the procedure described in example 1, but using 3-
phenylbutanoic and instead of 3,3-diphenylpropionic acid, the title compound
was obtained as a white solid (38%).
mp: 38-41°C (C23H2sN40.H20);
1H NMR (SOMHz, CDCl3) 8 (TMS): 8.87 (s, IH), 8.28 (d, J= 5.5Hz, IH), 7.16 (m,
3 0 6H), 4.60 (m, IH), 3.76 (m, 3H), 3.30 (m, IH), 2.50 (s, 3H), 3-0.5
(complex signal,
9H), L25 (d, J= 6.9Hz, 3H).
EXAMPLE 41
1-[[1-(3-Methyl-3-phenylbutanoyl)-4-piperidyl]methyl]-1H-2-methylimidazo[4,5-
c]pyridine
3 5 Following the procedure described in example I, but using 3-methyl-3-
phenylbutanoic acid (obtained in reference example 19) instead of 3,3-
diphenylpropionic acid, the title compound was obtained as a white solid
(58%).
mp: 37-45°C (C24H3pN40.1 /2H20);
SUBSTITUTE Sf-,EET (MULE 261
wo ~n43i7
PC"T/EP95/03487
52
IH NMR (80MHz, CDC13) 8 (TMS): 8.95 (s, 1H), 8.38 (d, J= 5.5Hz, IH), 7.26 (m,
6H), 4.65 (m, IH), 3.86 (d, J= 7.IHz, 2H), 3.50 (m, IH), 2.58 (s, 3H), 3-0.5
(complex
signal, 9H),1.25 (s, 6H).
EXAMPLE 42
S 1-[[1-[(N-methyl-N-phenylamino)acetyl]-4-piperidyl]methyl]-1H-2-
methyiimidazo[4,5-c]pyridine
Following the procedure described in example l, but using N-methyl-N-
phenylaminoacetic acid (obtained in reference example 20) instead of 3,3
diphenylpropionic acid, the title compound was obtained as a white solid
(39%).
mp: 74-78°C (C~H2~NgO.H20);
IH NMR (80MHz, CDC13) 8 (TMS): 8.96 (s, IH), 8.36 (d, J= 5.5Hz, IH), 7.21 (m,
3H), 6.7I (m, 3H), 4.60 (m, I H), 4.05 (s, 2H), 3.92 (d, J= 7.1 Hz, 2H), 3.80
(m, 1 H),
2.99 (s, 3H), 2.59 (s, 3H), 3-1 (complex signal, 7H).
1 5 EXAMPLE 43
1-[[1-(3-Methyl-3-(4-nitrophenyl)butanoyl]-4-piperidyl]methyl]-IH-2-
methylimidazo[4,5-c]pyridine
Following the procedure described in example I, but using 3-methyl-3
(4-nitrophenyl)butanoic acid (obtained in reference example ZI) instead of 3,3
2 0 diphenylpropionic acid, the title compound was obtained as an oil (28%).
1H RMN (80MHz, CDCl3) S (TMS): 8.96 (s, IH), 8.37 (d, J= 5.5Hz, 1H), 8.I1 (d,
J=
8.4 Hz, 2H), 7.51 (d, J= 8.4 Hz, 2H), 7.I8 (d, J= 5.5Hz, 1H), 4.55 (m, IH),
3.95 (d, J=
7.IHz, 2H), 3.83 (m, IH), 2.59 (s, 3H), 3-0.5 (complex signal, 9H), I.50 (s,
6H).
EXAMPLE 44
2 5 1-[[1-[3-(4-Aminophenyl)-3-methylbutanoyI]-4-piperidyl]methyl]-1H-2-
methylimidazo[4,5-c]pyridine
Following the procedure described in reference example 3, but starting
from the compound obtained in example 43, the title compound was obtained
as a white solid (57%).
3 0 mp: I72-I73°C (C24H31 N50.3 /4H20);
IH NMR (80MHz, CDC13) S (TMS): 8.96 (s, 1H), 8.38 (d, J= 5.5Hz, IH), 7.I8 (m,
3H), 6.62 (d, J= 5.5Hz, 2H), 4.65 (m, 1H), 3.86 (d, J= 7.IHz, 2H), 3.63 (m,
2H), 3.50
(m, IH), 2.59 (s, 3H), 3-0.5 (complex signal, 9H), I.25 (s, 6H).
EXAMPLE 45
3 5 1-[[1-[[N-(Diphenylmethyl)amino]carbonyl]-4-piperidyl]methyl]-IH 2-
methylimidazo[4,5-c]pyridine
Following the procedure described in example 36, but using N-
phenoxycarbonyl-N-(diphenylmethyl)amine (prepared from
aminodiphenylmethane and phenyl chloroformate) instead of N-
SUBSTITUTE S~ ~T (RULE 26)
WO 96/14317
PCT/EP95/03487
53
phenoxycarbonyl-L-Leucine ethyl ester, ~~the title compound was obtained as a
white solid (26%).
mp: ZI2-2I8°C (C2~H29N50.H20);
1H NMR (80MHz, CDC13) 8 (TMS): 8.96 (s, 1H), 8.35 (d, J= 5.5Hz, 1H), 7.27 (m,
lIH), 6.14 (d, J= 7.OHz, IH), 5.12 (d, J= 7.OHz, IH), 4.00 (m, IH), 3.96 (d,
j= 7.3Hz,
ZH), 2.70 (m, 2H), 2.61 (s, 3H), 2.I-1.I (complex signal, 6H).
EXAMPLE 46
I-[[I-[[N-(4-Aminobenzoyl)-N-methylamino]acetyl]-4-piperidyl]methyl]-IH-2
methylimidazo[4,5-c]pyridine
1 0 a) 1-[[I-[(N-methyl-N-tent butoxycarbonylamino)acetyl]-4-piperidyl]methyl]-
1H-
2-methylimidazo[4,5-c]pyridine
Following the procedure described in example I, but using N-(tert-
butoxycarbonyl)-N-methylamino]acetic acid instead of 3,3-diphenylpropionic
aad, the desired product was obtained (63%).
b) 1-[[I-[N-methylaminoacetyl]-4-piperidyl]methyl]-1H-2-methylimidazo[4,5-
c]pyridine
Following the procedure described in reference example 5, but starting
from the compound obtained in section a), the desired product was prepared
(quantitative yield).
2 0 c) Title compound
Following the procedure described in example 6b, but using the
compound obtained in example 46b instead of the compound obtained in
example 6a, the title compound was obtained (32%).
mp: I20-124°C (C23H28N6D2.H20);
2 5 1 H NMR (80MHz, CDC13) S (TMS): 8.97 (s, I H), 8.38 (d, J= 5.5Hz, I H),
7.25 (m,
3H), 6.61 (d, J= 9.OHz, 2H), 4.64 (m, 1H), 4.01 (m, 2H), 3.98 (d, J= 7.IHz,
ZH), 3.10
(s, 3H), 2.62 (s, 3H), 3.3-1 (complex signal, IOH).
EXAMPLE 47
1-[[I-(3-Hydroxy-3-phenyl-3-trifluoromethylpropionyl)-4-piperidyl]methyl]-IH-
3 0 2-methylimidazo[4,5-c]pyridine
Following the procedure described in example I, but using 3-hydroxy-3-
phenyl-3-trifluoromethylpropionic acid (obtained in reference example 11)
instead of 3,3-diphenylpropionic acid, the title compound was obtained as a
white solid (I5%).
3 5 mp: 210-21I°C (C23H25F3N402.1 /2H20);
IH NMR (80MHz, CDCl3) 8 (TMS): 8.90 (s, IH), 8.36 (d, J= 5.5Hz, IH), 7.35 (m,
6H), 4.68 (m, 1H), 4.60 (m, IH), 3.94 (m, 3H), 2.60 (s, 3H), 3-0.5 (complex
signal,
9H).
EXAMPLE 48
SUBSTITUTE SHEET (RULE 26)
WO 96/14317 ",
PCT/EP95/03487
54
traps -1-[[1-[3-(4-Aminophenyl)-2-butenoyl]-4-piperidyl]methyl]-IH-2
methylimidazo[4,5-c]pyridine
Following the procedure described in example I, but using 3-(4
nitrophenyl)-2-butenoic acid (obtained in reference example 22), and
hydrogenating the compound thus obtained according to the procedure
described in reference example 3, the title compound was obtained as a white
solid (22%).
mp: 106-I10°C (C23HZ~N50.I /2H20);
IH NMR (80MHz, CDC13) S (TMS): 8.98 (s, IH), 8.38 (d, J= 5.5Hz, IH), 7.27 (m,
1 0 3H), 6.60 (d, J= 8.5Hz, ZH), 6.I7 (s, 1H), 4.67 (m, IH), 3.90 (d, J=
7.2Hz, 2H), 3.81
(m,1H), 2.63 (s, 3H), 2.23 (s, 3H), 3.1-0.5 (complex signal, 9H).
EXAMPLE 49
1-[[1-[(Phenylamino)acetyl]-4-piperidyl]methyl]-1H-2-methylimidazo[4,5
c]pyridine
1 5 Following the procedure described in example I, but using N-tert-
butoxycarbonyl-N-phenylglycine instead of 3,3-diphenylpropionic acid, and
then subjecting the resulting compound to the procedure described in
reference example 5, the title compound was obtained as a white solid (49%).
mp: 2I9-220 °C (CZ1H25NgO.H20);
2 0 IH NMR (80MHz, CDCl3) b (TMS): 9.00 (s, IH), 8.40 (d, J= 5.5Hz, 1H), 7.20
(m,
4H), 6.66 (m, 2H), 4.72 (m, 2H), 3.89 (m, 5H), 2.63 (s, 3H), 3.2-I (m, 7H).
EXAMPLE 50
(R)-1-[[1-[(1-Phenylethylamino)carbonyl]-4-piperidyl]methyl]-1H-2-
methylimidazo[4,5-c]pyridine
2 5 Following the procedure described in example 36, but using (R)-N-
phenoxycarbonyl-I-phenylethylamine (prepared from (R)-I-phenylethylamine
and phenyl chloroformate) instead of N-phenoxycarbonyl-L-Leucine ethyl
ester, the title compound was obtained as a white solid (37%).
mp: 80-84 °C (C~H2~NgO.I /2H20);
3 0 IH NMR (80MHz, CDCI3) 8 (TMS): 8.97 (s, 1H), 8.38 (d, J= 5.5Hz, IH), 7.30
(m,
5H), 7.20 (d, j= 5.5Hz, IH), S.OI (quint, J= 6.8 Hz, IH), 4.70 (broad d, J=
7.1 Hz,
IH), 4.02 (m, IH), 3.97 (d, J= 7.3Hz, ZH), 2.68 (broad t, J= I2.7Hz, 2H), 2.62
(s, 3H),
1.48 (d, J= 6.5 Hz, 3H), 2.1-1.3 (m, 6H).
EXAMPLE 51
3 5 (S)-1-[[1-[(1-Phenylethylamino)carbonyl]-4-piperidyl]methyl]-1H-2-
methylimidazo[4,5-c]pyridine
Following the procedure described in example 36, but using (S)-N-
phenoxycarbonyl-1-phenylethylamine (prepared from (S)-1-phenylethylamine
SUBSTITUTE SHEET (RULE 26)
WO 96/14317
PCT/EP95/03487
SS
and phenyl chloroformate) instead of N-phenoxycarbonyl-L-Leucine ethyl
ester, the title compound was obtained as a white solid (41 %).
mp: 79-83 °C (C~H2~NSO.I /2H20);
IH NMR (SOMHz, CDCl3) 8 (TMS): 8.98 (s, 1H), 8.38 (d, J= 5.5Hz, IH), 7.26 (m,
5H), 7.20 (d, J= 5.5Hz, IH), S.OI (quint, J= 6.8 Hz, IH), 4.68 (broad d, J=
7.I Hz,
IH), 4.02 (m, IH), 3.97 (d, J= 7.3Hz, 2H), 2.68 (broad t, J= I2.7Hz, ZH), 2.62
(s, 3H),
1.48 (d, J= 6.5 Hz, 3H), 2.I-L3 (m, 6H).
EXAMPLE 52
I-[(1-[(N-Benzoyl-N-phenylamino)acetyl]-4-piperidyl]methyl]-IFi-2
1 0 methylimidazo[4,5-c]pyridine
To a cooled (0 °C) solution of the compound obtained in example 49
(0.3
g, 0.824 mmol) and Et3N (O.II mL) in CH2Cl2 (6 mL), was added dropwise a
solution of benzoyl chloride (0.09 mL) in CH2C12 (0.2 mL) and the mixture was
stirred at room temperature overnight. The resulting solution was treated
1 5 with 0.5N NaOH and extracted with CH2C12 (3x). The organic phase was dried
and concentrated to a residue that was purified by chromatography on silica
gel
(CHCl3-MeOH, 8%) to afford the title compound (70%).
1H NMR (80MHz, CDCl3) 8 CTMS): 8.96 (m, IH), 8.38 (m, IH), 7.I6 (m, IIH), 4.68
(m, 3H), 3.92 (m, 3H), 2.60 (s, 3H), 3.2-I.2 (m, 7H).
2 0 The hydrochloride was prepared following the procedure described in
example I3.
mp: 145-I52 °C (CZgH29N502.3HC1.Hz0).
EXAMPLE 53
1-[[1-[[N-methyl-N-(4-nitrobenzenesulfonyl)amino]acetyl]-4-piperidyl]methyl]-
2 5 iH-2-methyiimidazo[4,5-c]pyridine
Following the procedure described in example 52, but starting from the
compound obtained in example 46b) and using 4-nitrobenzenesulfonyl
chloride instead of benzoyl chloride, the title compound was obtained as a
white solid (48%).
3 0 mp: 98-I04 °C (CuH261V605S.H20);
IH NMR (80MHz, CDC13) b (TMS): 8.99 (s, IH), 8.39 (d, J= 5.5Hz, IH), 8.36 (d,
J=
6.9 Hz, 2H), 7.99 (d, J= 6.9 Hz, 2H), 7.22 (d, J= 5.5Hz, I H), 4.52 (m, I H),
3.97 (m,
SH), 2.89 (s, 3H), 2.64 (s, 3H), 3.2-I.3 (m, 7H).
EXAMPLE 54
3 5 1-([1-((N-Ethoxycarbonyl-N-(4-nitrophenyl)amino]acetyl]-4-
piperidyl]methyl]-
1H-2-methylimidazo[4,5-c]pyridine
Following the procedure described in example 1, but using N-
ethoxycarbonvl-N-(4-nitrophenyl)aminoacetic acid (obtained in reference
SULSTITUTE Si-~EET (RULE 26)
WO 96/14317 " PCT/EP95/03487
56
example 23) instead of 3,3-diphenylpropionic acid, the title compound was
obtained as a white solid (64%).
I H NMR (80MHz, CDC13) 8 (TMS): 8.96 (s, IH), 8.37 (d, J= 5.5Hz,1 H), 8.13 (d,
J=
7.05Hz, 2H), 7.48 (d, J= 7.05Hz, 2H), 7.21 (d, J= 5.5Hz, IH), 4.65 (m, IH),
4.SI (d, J=
4.3 Hz, 2H), 4.22 (q, J= 7.I Hz, 2H), 4.02 (d, J= 7.2 Hz, 2H), 3.80 (m, IH),
2.63 (s,
3H), 3.I-I.3 (m, 7H),1.24 (t, J= 7.I Hz, 3H).
EXAMPLE 55
1-[[1-[[N-(4-Aminophenyl)-N-ethoxycarbonylamino]acetyl]-4-piperidyl]methyl]-
1H-2-methylimidazo[4,5-c]pyridine
Following the procedure described in reference example 3, but starting
from the compound obtained in example 54, the title compound was obtained
as a white solid (74%).
mp: I37 I38 °C (C24H3pN603.H20);
1H NMR (80MHz, CDCI3) 8 (TMS): 8.98 (s, IH), 8.40 (d, J= 5.5Hz, IH), 7.20 (d,
J=
1 5 5.5Hz, 1H), 7.I0 (d, J= 7.05Hz, 2H), 6.60 (d, J= 7.05Hz, 2H), 4.65 (m,
IH), 4.30 (m
2H), 4.I0 (m, 4H), 3.80 (m, 3H), 2.62 (s, 3H), 3.I-I:3 (m, 7H), I.I7 (t, J=
6.9 Hz, 3H).
EXAMPLE 56
traps -1-[[1-(3-Phenyl-2-pentenoyl)-4-piperidyl]methyl]-1H-2-
methylimidazo[4,5-c]pyridine
2 0 Following the procedure described in example I, but using traps-3-
phenyl-2-pentenoic acid (obtained in reference example 24) instead of 3,3-
diphenylpropionic acid, the title compound was obtained as a white solid
(74%).
mp: 59-62 ° C (C24H2gN40.I /2H20);
2 5 1H NMR (80MHz, CDC13) 8 (TMS): 8.99 (s, IH), 8.40 (d, J= 5.5Hz, IH), 7.36
(m,
SH), 7.20 (d, J= 5.5Hz, 1 H), 6.09 (s, 1 H), 4.75 (m, I H), 4.05 (m, I H),
4.01 (d, j= 7.1
Hz, 2H), 2.72 (q, J= 7.4 Hz, 2H), 2.64 (s, 3H), 3.I-I.3 (m, 7H),1.02 (t, j=
7.4 Hz, 3H).
EXAMPLE 57
1-[[1-[[N-(2-Methoxyb enzyl )amino] carbonyl]-4-piperidyl]methyl]-1H-2-
3 0 methylimidazo[4,5-c]pyridine
Following the procedure described in example 36, but using N
phenoxycarbonyl-N-(2-methoxybenzyl)amine (prepared from 2
methoxybenzylamine and phenyl chloroformate) instead of N
phenoxycarbonyl-L-Leucine ethyl ester, the title compound was obtained as a
3 5 white solid (I7%).
mp: 76-85 °C (CuH2~N50~;
1H NMR (80MHz, CDCI3) b (TMS): 8.96 (s, IH), 8.35 (d, J= 5.5Hz, IH), 7.25 (m,
3H), 7.89 (t, J= 7.2Hz, 2H), 5.I4 (t, J= 5.3 Hz, 1H), 4.40 (d, J= 5.6 Hz, ZH),
3.91 (m
4H), 3.82 (s, 3H), 2.60 (s, 3H), 2.8-I.3 (m, 71-0.
SUBSTITUTE SHEET (RULE 26)
WO 96/14317 0 PCT/EP95/03487
$7
EXAMPLE 58
(R)-1-[(I-(((1-Ethoxycarbonyl-1-phenyl)methyiamino]carbonyl]-4
piperidyl]methyl]-IH-2-methylimidazo(4,5-c]pyridine
Following the procedure described in example 36, but using (R)-N-
phenoxycarbonyl-2-phenylglycine ethyl ester (prepared from (R)-2-
phenylglycine ethyl ester and phenyl chloroformate) instead of N-
phenoxycarbonyl-L-Leucine ethyl ester, the title compound was obtained as a
white solid (27%).
mp: 78-80 °C (C24H29N503~ I /2H20);
1 0 1 H NMR (80MHz, CDCl3) b (TMS): 8.96 (s, I H), 8.35 (d, J= 5.5Hz, 1H),
7.33 (s,
SH), 7.I9 (d, J=5.5Hz, 1H), 5.52 (m, 2H), 4.I3 (m, 6H), 2.7I (m 2H), 2.60 (s,
3H),
2.I0 (m, IH),1.45 (m, 4H), LI9 (t, J= 7.1 Hz, 3H).
EXAMPLE 59
I-[[I-[(I-Phenyl-1-cyclopropylamino)carbonyl]-4-piperidyI]methyl]-1H-2
1 5 methylimidazo(4,5-clpyridine
To a solution of I-phenyl-1-cyclopropanecarboxylic acid (I.62 g, O.OI mol)
and Et3N (1.I4 mL) in benzene (40 mL) was added dropwise
diphenylphosphorylazide (2.14 mL). The mixture was heated at 90 °C for
Z h.
The compound obtained in reference example 5 (1.6 g, 6.8 mmol) was then
2 0 added and the mixture was heated at 90 °C overnight. After cooling,
1N NaOH
was added and it was extracted with EtOAc (3x). The organic phase was dried
and concentrated to a residue which was purified by chromatography on silica
gel (CHCI3-MeOH, IO%a) to afford the title compound as a white solid (1.24 g,
47%).
2 5 mp: 227-228 °C (C23H2~N50);
IH NMR (80MHz, CDCI3) 8 (TMS): 8.98 (s, IH), 8.38 (d, J= 5.5Hz, 1H), 7.23 (s,
6H), 5.44 (s, I H), 4.02 (m, 2H), 3.97 (d, J= 8.0 Hz, 2H), 2.64 (m 2H), 2.62
(s, 3H),
2.I0 (m, IH), I.55 (m, 4H), L22 (s, 4H).
EXAMPLE 60
3 0 (S)-1-((1-[(2-Ethoxy-I-phenylethylamino)carbonyl]-4-piperidyl]methyl]-IH-2-
methylimidazo(4,5-c]pyridine
Following the procedure described in example 36, but using (S)-N-
phenoxycarbonyl-2-phenylglycinol ethyl ether (prepared from 2-phenylglyanol
ethyl ether and phenyl chloroformate) instead of N-phenoxycarbonyl-L-
3 5 Leucine ethyl ester, the title compound was obtained as a white solid
(24%).
mp: 68-70 °C (C24H31N5~:
I H NMR (80MHz, CDCl3) b (TMS): 8.97 (s, I H), 8.38 (d, J= 5.5Hz, IH), 7.29
(s,
5H), 7.21 (d, J=5.5Hz, IH), 5.36 (d, J= 6.4 Hz, IH), 5.01 (q, J= 5.4 Hz, 1H),
4.05 (m,
SUBSTITUTE SHEET (RULE 26)
WO 96/14317 PCT/EP95/03487
S8
2H), 3.96 (d, J= 7.I Hz, 2H), 3.54 (m, 4H), 2.75 (m, 2H), 2.61 (s, 3H), 2.i0
(m, IH),
1.45 (m, 4H),1.I5 (t, J= 6.9 Hz, 3H).
EXAMPLE 61
1-[[1-((2-Methyl-2-phenylpropyl)sulfonyl]-4-piperidyl]methyl]-1H-2
methylimidazo[4,5-c]pyridine
To a solution of the compound obtained in reference example 5 (I g, 4
mmol) and Et3N (0.6 mL) in CHC13 (20 mL), was added 2-methyl-2-
phenylpropylsulfonyl chloride (2.32 g, IO mmol, obtained in reference example
25) and the mixture was stirred at room temperature overnight. The resulting
solution was diluted with CHCl3, washed with 0.5N NaOH, dried and
concentrated. The residue was purified by chromatography on silica gel
(CHCl3-MeOH, 5%) to afford a solid which was recrystallized from hot EtOAc.
The title compound was obtained as a white solid (0.4 g, 25%).
mp: I65-I66 °C (C23H3pN402S);
1 5 1H NMR (80MHz, CDC13) 8 (TMS): 8.98 (s, IH), 8.38 (d, J= 5.5Hz, IH), 7.32
(m,
5H), 7.I7 (d, J=5.5Hz, IH), 3.94 (d, J= 6.9 Hz, 2H), 3.65 (broad d, J= 12.0
Hz, 2H),
3.I6 (s, 2H), 2.60 (s, 3H), 2.I8 (broad t, J=12.0 Hz, 2H), I.58 (s, 6H), I.48
(m, 5H).
EXAMPLE 62
1-[[1-(3-Phenylpropionyl)-4-piperidyl)methyl]-1H-2-methylimidazo[4,5-
2 0 c]pyridine
Following the procedure described in example 1, but using 3-
phenylpropionic acid instead of 3,3-diphenylpropionic acid, the title
compound was obtained as a white solid.
mp: 52-58 °C (C??H261V40.3/4H20);
2 5 IH NMR (80MHz, CDC13) S (TMS): 8.96 (s, 1H), 8.38 (d, J= 5.5Hz, 1H), 7.23
(m,
6H), 4.71 (broad d, J= I3.6 Hz, IH), 3.94 (d, J= 7.2I Hz, 2H), 3.56 (broad d,
J= 13.6
Hz, 1H), 2.80 (m, 2H), 2.62 (m, 2H), 2.61 (s, 3H), 2.7-0.8 (m, 7H).
EXAMPLE 63
1-([1-[[1-(4-Nitrophenyl)ethylamino]carbonyl]-4-piperidyl]methyl)-1H-2-
3 0 methylimidazo[4,5-c)pyridine
Following the procedure described in example 59, but using 2-(4-
nitrophenyl)propionic acid instead of I-phenyl-I-cyclopropanecarboxylic acid,
the title compound was obtained as a white solid (57%).
IH NMR (80MHz, CDCl3) 8 (TMS): 8.95 (s, IH), 8.35 (d, J= 5.5Hz, IH), 8.22 (d,
J=
3 5 9.I Hz, 2H), 7.45 (d, J= 9.I Hz, 2H), 7.20 (d, J= 5.5Hz, 1H), 5.03 (m,
2H), 4.05 (m,
2H), 3.96 (d, J= 7.I Hz, 2H), 2.7I (m, 2H), 2.62 (s, 3H), I.48 (d, J=6.6 Hz,
3H), 2.2-1.0
(m, 5H).
EXAMPLE 64
SUBSTITUTE SHEET (RULE 26)
WO 96/14317 :.j ;~'~.i~'-~ ~y ~ pCT/Ep95/03487
59
1-[[1-[[1-(4-Aminophenyl)ethylamino]carbonyl]-4-piperidyl]methyl]-1H-2
methylimidazo[4,5-c]pyridine
A solution of the compound obtained in example 63 (0.8 g, I.89 mmol)
and SnC12.2H20 (2.128 g, 9.4 mmol) in EtOH (25 mL) was heated at 60 °C
and
y 5 then a solution of NaBH4 (0.035 g, 0.94 mmol) in EtOH (I5 mL) was added
dropwise. The reaction mixture was heated at 60 °C for I h and was then
cooled
to IO °C, made basic and extracted with CHC13, washing with water. The
organic phase was dried and concentrated to a residue which was
chromatographed on silica gel (CHCI3:MeOH:NH3, 60:20:0.2) to afford the title
1 0 compound (27 mg, 4%).
mp: I22-128 °C (CuH2gN60.3/2H20);
1H NMR (80MHz, CDCI3) S (TMS): 8.96 (s,1H), 8.36 (d, J= 5.5Hz, IH), 7.18 (d,
J=
5.5Hz, IH), 7.II (d, J= 8.3 Hz, 2H), 6.62 (d, J= 8.3 Hz, ZH), 4.90 (quint, J=
6.7 Hz,
1H), 3.96 (d, J= 6.4 Hz, IH), 3.96 (m, 4H), 2.64 (m, 2H), 2.6I (s, 3H), 2.I0
(m, IH),
1 5 I.43 (d, J=6.6 Hz, 3H),1.26 (m, 6H).
EXAMPLE 65
traps -1-[[1-[3-(4-Aminophenyl)propenoyl]-4-piperidyl]methyl]-1H-2-
methylimidazo[4,5-c]pyridine
Following the procedure described in example I, but using traps-4-
2 0 aminocinnamic acid hydrochloride instead of 3,3-diphenylpropionic acid,
the
title compound was obtained as a white solid (7I %a).
mp: I IS-I20 °C (C~Hz5N50.1 /2H20);
1H NMR (80MHz, CDC13) 8 (TMS): 8.99 (s, IH), 8.40 (d, J= 5.5Hz, IH), 7.62 (d,
J=
I5.3Hz, I H), 7.32 (d, J= 8.4 Hz, 2H), 7.20 (d, J= 5.5Hz, I H), 6.64 (d, j=
I5.3 Hz, I H),
2 5 6.63 (d, J= 8.3 Hz, 2H), 4.40 (m, 1 H), 4.00 (d, J= 7.3 Hz, 2H), 3.94 (m,
I H), 2.80 (m,
2H), 2.64 (s, 3H), 2.4-I.2 (m, 7H).
EXAMPLE 66
1-[[I-[[N-(2-Methoxyphenyl)-N-methylamino]acetyl)-4-piperidyl]methyl]-1H-2
methylimidazo[4,5-c]pyridine
3 0 Following the procedure described in example l, but using [N-(2-
methoxyphenyl)-N-methylamino)acetic acid (obtained in reference example
26) instead of 3,3-diphenylpropionic acid, the title compound was obtained as
a
white solid (7I %).
mp: 59-64 °C (C23H29N502.I /2H20);
3 S 1H NMR (80MHz, CDC13) 8 (TMS): 8.98 (s, IH), 8.38 (d, J= 5.5Hz, IH), 7.20
(d, J=
5.5Hz, 1H), 7.06 (m, 3H), 4.45 (m, IH), 4.42 (m, IH), 3.97 (m, 4H), 3.82 (s,
3H), 2.87
(s, 3H), 2.63 (m, 3H), 2.62 (s, 3H), 2.4-I.2 (m, 5H).
EXAMPLE 67
SUBSTITUTE SHEET (RULE 26)
WO 96/14317
PCT/EP95/03487
1-[[I-[[4-(tent Butoxycarbonylamino)phenylmethylamino)carbonyl]-4
piperidyl]methyl]-1H-2-methylimidazo[4,5-c]pyridine
Following the procedure described in example 59, but using 4-(N-fert
butoxycarbonylamino)phenylacetic acid instead of I-phenyl-1
5 cyclopropanecarboxylic acid, the title compound was obtained as a white
solid
(94%).
mp:125-I30 °C (C26H3~N603.I /2H20);
IH NMR (80MHz, CDC13) 8 (TMS): 8.96 (s, IH), 8.36 (d, J= 5.5Hz, IH), ?.28 (m,
5H), 6.90 (s, IH), 5.05 (m, IH), 4.3I (d, J= 5.2 Hz, 2H), 4.03 (m, 2H), 3.96
(d, J= 7.1
1 0 Hz, 2H), 2.62 (m, 2H), 2.60 (s, 3H), 2.I-I.2 (m, 5H),1.50 (s, 9H). .
EXAMPLE 68
1-[[1-[(4-Aminophenylmethylamino)carbonyl]-4-piperidyl]methyl]-1H-2
methylimidazo[4,5-c)pyridine
Following the procedure described in reference example 5, but starting
1 5 from the compound obtained in example 67, the title compound was obtained
as a white solid (70%).
mp: 104-109 °C (C21H26N60.H20);
IH NMR (80MHz, CDC13) 8 (TMS): 8.92 (s, 1H), 8.34 (d, J= 5.5Hz, IH), 7.I9 (d,
j=
5.5Hz, IH), 7.05 (d, J= 8.2 Hz, 2H), 6.58 (d, J= 8.2 Hz, 2H), 5.05 (m, IH),
4.26 (d, J=
2 0 5.2 Hz, 2H), 4.08 (m, 2H), 3.96 (d, J= 7.I Hz, 2H), 3.40 (m, 2H), 2.60 (m,
2H), 2.59
(s, 3H), 2.I-I.2 (m, SH).
EXAMPLE 69
1-[[1-[3-(2-Methoxyphenyl)propionyl)-4-pip eridyl]methyl]-1H-2
methylimidazo[4,5-c]pyridine
2 5 Following the procedure described in example I, but using 3-(2-
methoxyphenyl)propionic acid instead of 3,3-diphenylpropionic acid, the title
compound was obtained as a white solid (20%).
mp: 54-56 °C (C23HZgN402.I /2H20);
IH NMR (80MHz, CDCl3) 8 (TMS): 8.97 (s, 1H), 8.37 (d, J= 5.5Hz, IH), 7.15 (m,
3 0 3H), 6.86 (m, 2H), 4.7I (broad d, J= I3.6 Hz, IH), 3.94 (d, J= 7.2I Hz,
2H), 3.81 (m,
1H), 3.80 (s, 3H), 2.97 (m, ZH), 2.62 (s, 3H), 2.6I (m, 4H), 2.3-0.8 (m, SH).
EXAMPLE 70
1-[[1-[[(1-Phenyl-1-cyciopropyl)methoxy)carbonyl)-4-piperidyl]methyl]-1H-2
methyiimidazo[4,5-c]pyridine
3 5 Following the procedure described in example 36, but using phenyl (I-
phenyl-1-cyclopropyl)methyl carbonate (prepared from 1-phenyl-I-
cyclopropanemethanol and phenyl chloroformate) instead of N-
phenoxycarbonyl-L-Leucine ethyl ester, the title compound was obtained as a
white solid (23%).
SUBSTITUTE SHEET (RULE 26)
WO 96/14317 i J a PCT/EP95/03487
61
mp: I38-140 °C (C2qH2gN4O2.I /2H20);
IH NMR (80MHz, CDC13) 8 (TMS):~8.97 ~s, IH), 8.37 (d, j= 5.5Hz, IH), 7.27 (s,
5H), 7.I9 (d, J= 5.5Hz, 1H), 4.16 (s, 2H), 4.I5 (broad d, J= I3.6 Hz, 2H),
3.94 (d, J=
7.2I Hz, 2H), 2.60 (s, 3H), 2.60 (m, 2H), 2.3-0.8 (m, 5H), 0.92 (s, 4H).
EXAMPLE 71
traps-1-[[1-[3-(Methoxymethyl)-3-phenylpropenoyl)-4-piperidyl]methyl)-IH-2
methylimidazo[4,5-c)pyridine
Following the procedure described in example I, but using traps-3
(methoxymethyl)-3-phenylpropenoic acid (obtained in reference example 27)
instead of 3,3-diphenylpropionic acid, the title compound was obtained as a
white solid.
mp: 63-67 °C (C24HzgN402.H2O);
1H NMR (80MHz, CDC13) 8 (TMS): 8.98 (s, IH), 8.40 (d, J= 5.5Hz, 1H), 7.27 (m,
5H), 7.I8 (d, j= 5.5Hz, IH), 6.39 (s, IH), 4.60 (m, IH), 3.98 (m, IH), 3.96
(d, J= 7.2
1 5 Hz, ZH), 3.7I (s, 3H), 3.54 (d, J= 7.2 Hz, 2H), 2.60 (s, 3H), 3.1-1.3 (m,
7H).
EXAMPLE 72
1-[[1-[[N-(4-Nitrophenylsulfonyl)-N-phenylamino)acetyl)-4-piperidyl]methyl)
1H-2-methylimidazo[4,5-c)pyridine
To a solution of the compound obtained in example 49 (2.5 g, 6.9 mmol)
2 0 in pyridine was added 4-nitrobenzenesulfonyl chloride (I.54 g) and the
resulting mixture was heated at 60 °C for 18 h. The solvent was removed
and
the residue partitioned between 0.5 N NaOH and CHC13. The organic phase
was dried and concentrated to a residue which was chromatographed on silica
gel (CHCI3:MeOH, IO%) to afford the title compound as a yellow solid (2.93 g,
2 S 78%).
mp: 1 I 0-1 I5 °C (CZ~H2gN605S.1 / 2H20);
IH NMR (80MHz, CDC13) 8 (TMS): 9.00 (s, IH), 8.40 (d, J= 5.5Hz, IH), 8.26 (d,
J=
8.8Hz, 2H), 7.84 (d, J= 8.8 Hz, 2H), 7.27 (m, 5H), 7.I8 (d, J= 5.5Hz, 1 H),
4.70 (m,
IH), 4.52 (s, 2H), 4.00 (d, J= 7.2 Hz, 2H), 3.80 (m, IH), 2.64 (s, 3H), 3.I-
I.3 (m, 7H).
3 0 EXAMPLE 73
1-j[1-[[N-(4-Aminophenylsulfonyl)-N-phenylamino)acetyl)-4-piperidyl)methyl)-
iH-2-methylimidazo[4,5-c)pyridine
Following the procedure described in reference example 3, but starting
from the compound obtained in example 72, the title compound was obtained
3 5 as a white solid (78%).
mp: 147-I57°C (C2~H3pN603S.H2O);
1H NMR (80MHz, CDC13) 8 (TMS): 8.79 (s, 1H), 8.38 (d, J= 5.5Hz, 1H), 7.45 (d,
J=
8.8Hz, ZH), 7.26 (m, 6H), 6.94 (d, J= 8.8Hz, 2H), 4.36 (m, IH), 4.35 (s, 2H),
4.00 (d,
J= 7.2 Hz, 2H), 3.98 (m,1H), 2.63 (s, 3H), 3.3-1.0 (m, 9H).
CI !F~CTITI ITF ~IlCC'T flat tl C ~Rv
WO 96/14317 ~ PCT/EP95/03487
62
EXAMPLE 74
1-[[1-[(2-Methyl-2-phenylpropylamino)carbonyl]-4-piperidyl]methyl]-1H-2
methylimidazo[4,5-c]pyridine
Following the procedure described in example 59, but using 3-methyl-3-
phenylbutanoic acid (obtained in-reference example I9) instead of 1-phenyl-1-
cyclopropanecarboxylic acid, the title compound was obtained as a white solid
(36%).
mp: 66-69 °C (C24H31N50.1 /2H20);
IH NMR (80MHz, CDCl3) 8 (TMS): 8.98 (s, IH), 8.39 (d, J= 5.5Hz, IH), 7.25 (m,
5H), 7.I8 (d, J= 5.5Hz, IH), 4.08 (m, 1H), 3.94 (d, J= 7.3 Hz, 2H), 3.78
(broad d, J=
I6.0 Hz, 2H), 3.43 (d, J= 5.7 Hz, 2H), 2.61 (s, 3H), 2.60 (m, 2H), 2.I-0.8 (m,
5H), L33
(s, 6H).
EXAMPLE 75
1-[[1-[[N-Isobutyl-N-(4-nitrophenylsulfonyl) amino]acetyl]-4-piperidyl]methyl]
1H-2-methylimidazo[4,5-c]pyridine
Following the procedure described in example I, but using N-isobutyl-
N-(4-nitrophenylsulfonyl)aminoacetic acid (obtained in reference example 28)
instead of 3,3-diphenylpropionic acid, the title compound was obtained as a
white solid (71 %).
2 0 IH NMR (SOMHz, CDCl3) S (1'MS): 8.98 (s, IH), 8.40 (d, J= 5.5Hz, IH), 8.31
(d, J=
8.8 Hz, 2H), 8.00 (d, J= 8.8 Hz, 2H), 7.2I (d, J= 5.5Hz, I H), 4.50 (m, I H),
4. I6 (s,
ZH), 4.01 (d, J= 7.2 Hz, ZH), 3.85 (m, IH), 3.08 (d, J= 7.4 Hz, ZH), 2.63 (s,
3H), 3.1-
1.2 (m, 8H), 0.87 (d, J= 6.5 Hz, 6H).
EXAMPLE 76
2 5 1-[[1-[[N-(4-Aminophenylsulfonyl)-N-isobutylamino]acetyl]-4-
piperidyl]methyl)-1H-2-methylimidazo[4,5-c]pyridine
Following the procedure described in reference example 3, but starting
from the compound obtained in example 75, the title compound was obtained
as a white solid (67%).
3 0 mp: II2-I16 °C (C25H34N603S.3/2H20);
IH NMR (80MHz, CDCl3) S (TMS): 8.96 (s, IH), 8.39 (d, j= 5.5Hz, IH), 7.5I (d,
J=
8.8 Hz, 2H), 7.20 (d, J= 5.5Hz, IH), 6.63 (d, J= 8.8 Hz, 2H), 4.50 (m, 3H),
3.89 (m,
5H), 3.00 (m, 2H), 2.62 (s, 3H), 3.I-I.2 (m, 8H), 0.84 (d, J= 6.5 Hz, 6H).
~I IR~,T~ i 1 ITF CI-I~CT 1171 II G '7~1