Note: Descriptions are shown in the official language in which they were submitted.
V~ 95/00482 , PCTIUS94/06873
PIPERIDINE DERIVATIVES AND
PROCESS FOR THEIR PRODUCTION
FIELD OF THE INVENTION .
The present invention relates to piperidine derivatives and a process
for their production.
BACKGROUND OF TIC IIWENTION
Terfenadine, 1-(p-tent-butylphenyl~4-[4'-(a-hydroxydiphenylmethyl)-
1'-piperidinyl]-butanol is a non-sedating anti-histamine. It is reported to be
a
specific H=-receptor antagonist that is also devoid of any anticholingeric,
anti-
serotoninergic, and anti-adrenergic effects both in- and in vivo. ee
D. McTavish, K.L. Goa, M. Ferrill, ~ 1990, 39, 552; C.R. Kingsolving,
N.L. Monroe, A.A. Carr, Pharmacolo ist, 1973, I5, 221; J.K. Woodward, N.L.
Munro, Arzneim-Forsch, 1982, 32,1154; K.V. Mann, K.J. Tietze, Clin. Pharm.
1989,
6, 331. A great deal of effort has been anade investigating structure-activity
relationships of terfenadine analogs, and this is reflected in the large
number of
U.S. patents disclosing this compound and related strictures as follows:
U.S. Patent No. 3,687,956 to Zivkovic
U.S. Patent No. 3,806,526 to Carr, et. al.
U.S. Patent No. 3,829,433 to Carr, et. al.
U.S. Patent No. 3,862,173 to Carr, et. al.
U.S. Patent No. 3,878,217 to Carr, et. al.
U.S. Patent No. 3,922,276 to Duncan, et: al:
U.S. Patent No. 3,931,197 to Carr, et. al.
U.S. Patent No. 3,941,795 to Carr, et. al.
U.S. Patent No. 3,94b,022 to Carr, et. al.
U.S. Patent No. 3,956,296 to Duncan, et. al.
U.S. Patent No. 3,965,257 to Carr, et. al. '
U.S. Patent No. 4,742,175 to Fawcett, et. al.
WO 95/00482 PCT/US94/06873
2181089
-2-
Terfenadine has been linked to potentially fatal abnormal heart
rhythms in some patients with liver disease or who also take the antifungal
drug
ketoconazole or the antibiotic erythromycin. In animal and human metabolic
studies, terfenadine was shown to undergo high first-pass effect, which
results in
readily measurable plasma concentrations of the major metabolite 4-[4-[4-
(hydroxydiphenylmethyl)-1-piperidinyl]-1-hydroxybutyl]-a"a-
dimethylphenylacetic
and, also known as terfenadine carboxylic acid metabolite. The terfenadine
carboxylic acid metabolite also possesses anti-histaminic activity in animal
models
and may lack the cardiac side effects seen with terfenadine.
Piperidine derivatives related to the terfenadine carboxylic acid
metabolite are disclosed in the following U.S. patents:
U.S. Patent No. 4,254,129 to Carr, et. al.
U.S. Patent No. 4,254,130 to Carr, et. al.
U.S. Patent No. 4,285,957 to Carr, et. al.
U.S. Patent No. 4,285,958 to Carr, et. al.
In these patents, 4-[4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]-1-
hydroxybutyl]-
a,a-dimethylbenzeneacetic acid and related compounds are prepared by
alkylation
of a substituted piperidine derivative of the formula:
with an w-haloalkyl substituted phenyl ketone of the formula:
WO 95/00482 PCT/US94/06873
~_~g~oss
halo ~ 2)~ ~ C R6
CH ~ ~ ( H'
Z CHa
wherein the substituents halo, Rl, Rv n, z, and R6 are described in .column 6
of
U.S. Patent No. 4,254,130.
It is further described that the txrhaloalkyl substituted phenyl ketone
wherein Z is hydrogen are prepared by reacting an appropriate straight or
branched lower alkyl Cl.~ ester of a-a-dimethylphenylacetic acid with the
compound of the following formula:
0
halo (CH2)m C halo
under the general conditions of a Friedel-Crafts acylation, wherein halo and m
are
described in column 11 of U.S. Patent No. 4,254,129. The reaction is carried
out in
2 0 carbon disulfide as the preferred solvent.
Applicant has discovered that the preparation of ethyl 4-(4-chloro-l-
oxobutyl)-a,a-dimethylphenylacetate by reaction of 4-chlorobutyryl chloride,
aluminum chloride, and ethyl a,a-dimethylphenylacetate in carbon disulfide, as
described in Example 1 of U.S. Patent Nos. 4,254,130 and 4,285,958 provides an
inseparable mixture of monosubstituted aromatic regioisomers of the formula:
~H3
C~ C02CZH~
CI-(CHQ)'~C
CHy
e~ ~aeTiri irc euccr ioi ii ~ ~a~
WO 95!00482 PGT/US94106873
.~. 2181089
-4-
wherein the chlorobutyryl substituent is attached at either of the three
aromatic
carbons which are mete or pare to the dimethylacetate substituent. These
regioisomers are not separable by standard techniques of thin layer
chromatography, or column chromatography, and low field proton nuclear
magnetic resonance spectroxopy is inconclusive in identifying the product of
this
_ reaction as a mixture. When the mixture of monosubstituted aromatic
regioisomers of the preceding formula is reacted with a piperidine of the
formula:
H
a second mixture of aromatic regioisomers is obtained of the formula:
c- R,
R2
I H3
O
COOCZHS
(CHZ)~- C
CHI
wherein the monosubstituted mete, pare mixture,of regioisomers is obtained.
It is known in the art that a monoalkyl substituent on a benzene ring
is ortho, pare directing in electroplulic aromatic substitution reactions such
as a
Friedel-Crafts reaction. Thus, it would be expected that the Friedel-Crafts
reaction
WO 95/00482 PCT/US94106873
-5-
of a-chlorobutyryl chloride with ethyl a,a-dimethylphenylacetate would yield
predominantly the pare substituted product of the fortriula:
H3
CI (CHZ)~ C ~ C COOEt
CH3
because of the electron donating, pare-directing character of the
dimethylalkyl
substituent combined with the steric hindrance associated with reaction of the
ortho positions. In practice, the inductive electronic withdrawing effect of
the
carboxylic ester of ethyl a,a-dimethylphenylacetate counteracts the expected
alkyl
electron donating effect, resulting in no significant directing effect for the
aromatic
substitution reaction. For the described reaction, a statistical mixture of
mete to
pare regioisomers results, with the two mete positions predominating.
2 0 The above second mixture of regioisomers can be converted to a
third mixture of regioisomers of formula:
~ R~
Rz
CH'
NJ OH
C COOH
(CHz)~ CH
~ cH~
WO 95/00482 PCT/US94/06873
~islo~9
-6-
Although the second mixture of regioisomers and the third mixture of
regioisomers can be analyzed by HPLC experiments, a practical separation to
obtain gram quantities of substantially pure regioisomers has not been
achieved.
Each mixture (including the first), would be expected to contain 33% of the
para
isomer and 67%a of the meta isoir~er. Since these components are inseparable,
it
- has not been possible to obtain either of the regioisomers in each mixture
in
substantially pure form.
SUMMARY OF THE INVENTIpN
The present invention relates to substantially pure piperidine
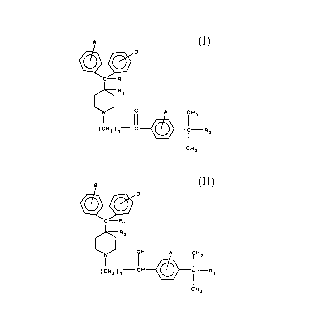
derivative compounds of the formulae:
s
D
C- R ~
R2
N/ A ~H~
(CHZja C ~ ~ - Ra
or cHa
D
IH A
~3
( CHI j 3' CH
Ra
CHI
Y WO 95/00482, PCT/US94106873
wherein
Rl is hydrogen or hydroxy;
Ri is hydrogen;
or R, and RZ taken together form a second bond between the carbon
atoms bearing Ri and R=;
R3 is -COOH or -COOK,;
R, is an alkyl with 1 to 6 carbon atoms;
A, B, and D are the subsHtuents of their rings, each of which
may be different or the same, and are selected from the group
consisting of hydrogen, halogens, alkyl, hydroxy, alkoxy, or other
substituents
or a salt thereof. These compounds are useful in pharmaceutical compositions,
particularly as antihistamines, antiallergy agents, and bronchodilators.
The piperidine derivative compound is prepared by a process which
is initiated by providing a substantially pure regioisomer of the following
formula:
A
~H~
C ~ ~ -R3
CHI
The substantially pure regioisomer is converted to the piperidine derivative
having a keto group with a piperidine compound of the formula:
30
H
WO 95100482 PCT/US94106873
.- 21~108g
_8_
A number of synthetic pathways for preparing the substantially pure
regioisomer
and for reacting it with the piperidine compound having a keto group are
disclosed. The piperidine derivative having a keto group can be converted to
the
above piperidine derivative having a hydroxyl group by reduction.
Although a wide variety of piperidine derivatives can be produced
by the process of the present invention, it is particularly useful in forming
a
hydroxylated piperidine derivative of the formula:
C-'_ OH
NJ H
CH3
( CHZ ) ~--. C I
C"'-COOH
H CHa
Alternatively, the process of the present invention can be used to produce a
piperidine derivative with a .keto group of the following formula:
30
WO 9,,100482 PCTIUS94I06873
_g_
i i I ~H~
( CHZ ) ~-C ~ ~-COOH
CH3
DETAILED DESCItII'TTON OF THE INVENTION
The present invention relates to substantially pure piperidine
derivative compounds of the formulae:
s
D
C-R ~
Rz
i ~ I I A ~H,
(CHZ ) ~ C ~ ~ R~
CHI
Or
WO 95/00482
218 ~ 0 8 9 ~T~~4/06873
-10-
B
D
-R~
a2
5- J
I H A ~H3
~CH2~a CH
CH3
wherein
R, is hydrogen or hydroxy;
RZ is hydrogen;
or R, and R= taken together form a second bond between the carbon
atoms bearing R, and Rz
R3 is --COOH or --COOR,;
R, is an alkyl with 1 to 6 carbon atoms;
A, B, and D are the substituents of their rings, each of which
may be different or the same, and are selected from the group
consisting of hydrogen, halogens, alkyl, hydroxy, alkoxy, or other
substituents
or a salt thereof.
These substantially pure piperidine derivative compounds may be in
the form of 4-diphenylmethylpiperidine derivatives represented by the
following
2 5 formulae:
WO 95/00482 Z PCT/US94/06873
--.,
-11-
B o
a ~J
CH
J o CH3
i II I
(CHz)a C C R
A CHa
B D
a a
CH
IH IH3
(CHZ)~ CH C R3
A I
CH3
where A, B, D, R3 are defined above. The substantially pure piperidine
derivative
compounds include 4-(hydroxydiphenylmethyl)piperidine derivatives according to
the following formulae:
B D
C OH
N p CH9
(CHZ)~ C C R~
A CHI
WO 95/00482 PCT/US94106873
-12-
s o
C OH
NJ OH CHa
(CH2)a CH ~ Ra
A CHa
where A, B, D, R3 are defined above. Another useful class of piperidine
derivative compounds are 4-diphenylmethylenepiperidine derivatives in
accordance with the following formulae:
B D
C
NJ p A CHa
(CHp)a C ~ ~ Ra
CHa
D
v
c
OH A CHa
(CHz)a CH ~ C Ra
CHa
WO 95/00482 PCT/US94I06873
msios~
-13-
where A, B, D, R, are defined above. Examples of R, are straight or branched
alkyl groups, including methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-
butyl, tert-
butyl, n-pentyl, neopentyl, and n-hexyl groups.
Illustrative examples of compounds of the present invention are as
follows:
4-[4-[4-(hydroxydiphenylmethyl~l-piperidinyl]-1-hydroxybutyl]-a,a-
dimethylbenzeneacetic add;
4-[4-[4-(diphenylmethyl)-1-piperidinyl]-1-hydroxybutyl ]-ay a-
dimethylbenzeneacetic add;
4-[4-[4-(diphenylmethylene)-1-piperidinyl]-1-hydroxybutyl]-a,a-
dimethylbenzeneacetic add;
4-[4-[4-(hydroxydiphenylmethyl)-I-piperidinyl]-1-hydroxybutylJ-a,a-
dimethyl-3-hydroxybenzeneacetic add;
4-[4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]-1-hydroxybutyl]-a,a-
dimethyl-2-hydroxybenzeneacetic acid;
4-[4-[4-(diphenylmethylene)-1-piperidinyl]-1-hydroxybutyl)-a,a-
dimethyl-3-hydroxybenzeneacetic add;
5-[4-[4-(diphenylmethylene)-I-piperidinyl]-I-hydroxybutyl]-a,a-
dimethylbenzeneacetic add;
2 0 ethyl 4-[4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]-1-
hydroxybutyl]-a,a-dimethylbenzeneacetic;
n-pentyl 4-[4-[4-(diphenylmethyl)-1-piperidinyl]-I-hydroxybutyl]-a,a-
dimethylbenzeneacetate;
ethyl 4-[4-[4-(diphenylmethylene)-I-piperidinyl]-I-hydroxybutyl]-a,a-
2 5 dimethylbenzeneacetate;
methyl 4-[4-[4-(hydroxydlphenylmethyl)-I-piperidinyl]-I-
hydroxybutyl]-a,a-dimethylbenzeneacetate;
ethyl 4-[4-[4-(hydroxydiphenylmethyl)-I-piperidinyl]-1-
hydroxybutyl]-a,a-dimethyl-(3-hydroxybenzene)acetate;
WO 95/00482 PGT/US94/06873
2181089
-I4-
n-propyl 4-[4-[4-(hydroxydiphenylmethyl)-I-piperidinyl]-1-
hydroxybutyl]-a,a-dimethyl-(2-hydmxybenzene)acetate;
n-hexyl 4-[4-[4-(diphenylmethylene)-I-piperidinyl]-1-hydroxybu tyl]-
a,a-d i methyl-(3-hyd roxybenzene)aceta te;
ethyl 5-[4-[4-(diphenylmethylene)-1-piperidinyl]-1-hydroxybutyl]-oc,a-
dimethylbenzeneacetate;
a,a~iphenyl-1-(4-(4-tert-butyl-2-hydroxy)phenyl)-hydroxybutyl-4-
piperidinemethanol;
a,a-Biphenyl-1-(4-(4-tert-butyl-3-hydroxy)phenyl)-4-hydroxybutyl-4-
piperidinemethanol;
a,a-Biphenyl-1-(3-(4-tert-butyl-2-hydroxy)phenyl)-3-hydroxypropyl-4-
piperidinemethanol;
a,a-Biphenyl-1-(5-(4-tert-butyl-2-acetyloxy)phenyl)-5-hydroxypentyl-
4-piperidinemethanol;
a,a-Biphenyl-1-(4-(4-hydroxy-tert-bu tyl-2-hydroxy)-phenyl)-4-
hydroxybutyl-4-piperidinemethanol;
a,a-Biphenyl-1-(4-(4-hydroxy-tert-butyl-3-hydroxy)-phenyl)-4-
hydroxybutyl-4-piperidinemethanol;
a,a-Biphenyl-I-(3-(4-hydroxy-tert-butyl-2-hydroxy)-phenyl)-3-
hydroxybutyl-4-piperidinemethanol;
a,a-Biphenyl-1-(4-(4-hydroxy-tert-butyl)phenyl)-4-hydroxybutyl-4-
piperidinemethanol;
I-(4-tert-butyl-2-hydroxyphenyl)-4-(4-diphenylmethylene)-1-
(piperidinyl)butanol;
2 5 1-(4-tert-bu tyl-3-hydroxyphenyl)-4-(4-diphenylmethylene)-1-
(piperidinyl)butanol;
1-(4-tert-butyl-3-hydroxyphenyl)-2-(4-diphenylmethylet~e)-1-
(piperidinyl)butanol;
I-(4-tert-butyl-2-butyryloxyphenyl)-6-(4-(diphenylmethyl)-I-
3 0 piperidinyl)hexanol;
WO 95/00482 PCT/US94/06873
2181089
-15-
1-(4-hydroxy-tert-butyl-2-hydroxyphenyl)-4-(4-(diphenylmethylene)-1-
(piperidinyl)butanol;
1-(4-hydroxy-tert-butyl-3-hydroxyphenyl)-4-(4-(diphenylmethylene)-1-
(piperidinyl)butanol;
1-(4-hydroxy-tert-butylphenyl)-4-(4-(diphenylmethylene)-1-
(piperidinyl)butanol;
Particularly preferred are compounds of the formulae:
C~ OH
NJ H Ha
(CHZ)3~C ~ C COOH
H CHa
and
C- OH
NJ O H,
i II
( CHz ) 3-C O C-COOH
CHI
PCT/US94/06873
w~.95/°°4~' 21810 8 9
-16-
Optionally, both diphenyl groups from the piperidine compound may be alkyl
(e.g., methyl) substituted at the position para to the methylene.
This invention also includes pharmaceutically acceptable salts in the
form of inorganic or organic acid or base addition salts of the above
compounds.
Suitable inorganic acids are, for example, hydrochloric, hydrobromic,
sulfuric, and
phosphoric acids. Suitable organic acids include carboxylic acids, such as,
acetic,
propionic, glycolic, lactic, pyruvic, malonic, succinic, fumaric, malic,
tartaric, citric,
cyclamic, ascorbic, malefic, hydroxymaleic, dihydroxymaleic, benzoic,
phenylacetic,
4-aminobenzoic, anthranillic, cinnamic, salicyclic, 4-aminosalicyclic, 2-
phenoxybenzoic, 2-acetoxybenzoic, and mandelic acid. Sulfonic acids, such as,
methanesulfonic, ethanesulfonic, and ~i-hydroxyethane-sulfonic acid are also
suitable acids. Non-toxic salts of the compounds of the above-identified
formulas
formed with inorganic and organic bases include, for example, those alkali
metals,
such as, sodium, potassium, and lithium, alkaline earth metals, for example,
calcium and magnesium, light metals of group IIIA, for example, aluminum,
organic amines, such as, primary, secondary, or tertiary amines, for example,
cyclohexylamine, ethylamine, pyridine, methylaminoethanol, and piperazine.
These salts are prepared by conventional means, for example, by treating the
piperidine derivative compounds of the formula: .
g
D
c-a,
az
N~ O A H~
I
(CH:)a. C U I R~
CHI
or
W"p..95/00482 PCT/US94/06873
-17-
B
D
C- R ~
R2
5- J
I H A ~H3
(CHZ)3 CH ~ ~ R3
CH3
where R,, R2, and R3 are defined above, with an appropriate acid or base.
The piperidine derivative compounds of the present invention can be
utilized as the biologically active components in pharmaceutical compositions.
The compounds of this invention are useful as antihistamines, antiallergy
agents,
and bronchodilators. They may be administered alone or with suitable
pharmaceutical carriers, and can be in solid or liquid form such as, tablets,
capsules, powders, solutions, suspensions or emulsions.
The compounds of this invention can be administered orally,
parenterally, for example, subcutaneously, intravenously, intramuscularly,
2 0 intraperitoneally, by intranasal instillation or by application to mucous
membranes, such as, that of the nose, throat and bronchial tubes. Such
application to mucous membranes can be achieved with an aerosol spray
containing small particles of a compound of this invention in a spray or dry
powder form.
The quantity of the compound of the present invention administered
will vary depending on the patient and the mode of administration and can be
any effective amount. The quantity of the compound administered may vary over
a wide range to provide in a unit dosage an effective amount of from about
0.01
to 20 mg/kg of body weight of the patient per day to achieve the desired
effect.
For example, the desired antihistamine, antiallergy, and bronchodilator
effects can
'~='~"' 95/00482 21810 8 9 ~T~S94/06873
-18-
be obtained by consumption of a unit dosage form such as a tablet containing 1
to
50 mg of the compound of the present invention taken 1 ~ to 4 times daily.
The solid unit dosage forms can be of the conventional type. This,
the solid form can be a capsule, such as an ordinary gelatin type containing
the
_ compound of the present invention and a carrier, for example, lubricants and
inert
fillers such as, lactose, sucrose, or cornstarch. In another embodiment, these
compounds are tableted with conventional tablet bases such as lactose,
sucrose, or
cornstarch in combination with binders like acacia, cornstarch, or gelatin,
disintegrating agents such as, cornstarch, potato starch, or alginic acid, and
a
lubricant like stearic acid or magnesium stearate.
The compounds of this invention may also be administered in
injectable dosages by solution or suspension of the compounds of the present
invention in a physiologically acceptable diluent with a pharmaceutical
carrier.
Such carriers include sterile liquids such as water and oils, with or without
the
addition of a surfactant and other pharmaceutically acceptable adjuvants.
Illustrative oils are those of petroleum, animal, vegetable, or synthetic
origin, for
example, peanut oil, soybean oil, or mineral oil. In general, water, saline,
aqueous
dextrose and related sugar solution, and glycols such as, propylene glycol or
polyethylene glycol, are preferred liquid carriers, particularly for
injectable
2 0 solutions.
For use as aerosols the compounds of this invention in solution or
suspension may be packaged in a pressurized aerosol container together with
suitable propellants, for example, hydrocarbon propellants like propane,
butane,
or isobutane with conventional adjuvants. The compounds of the present
invention also may be administered in a non-pressurized form such as in a
nebulizer or atomizer.
The compounds of the present invention can be used to treat warm
blooded animals, birds, and mammals. Examples of such beings include humans,
cats, dogs, horses, sheep, cows, pigs, lambs, rats, mice, and guinea pigs.
WO 95100482 PGT/US94106873
X181089
-19-
The piperidine derivative compounds of the present invention are
prepared by providing a substantially pure regioisomer of the following
formula:
Ha
C ~ C R
3
CH3
and then converting the substantially pure regioisomer to the piperidine
derivative compounds of the invention having a keto group with a piperidine
compound of the formula:
H
The resulting piperidine derivative compounds with a keto group can be
converted by reduction to the above-described piperidine compounds with a
hydroxyl group.
There are several techniques of providing these substantially pure
regioisomers.
WQ 95/00482 PCT/CTS94106873
~1810~9
-20-
Process One For Producing Substantially Pure Re~ioisomer
In one embodiment of the present invention, the substantially pure
regioisomer is formed by initially acylating a starting compound of the
formula:
A ~ H3
~CORS
CH3
wherein
Rs is -OR6, -N(R6)~, and -SRd and
R6 is an alkyl with 1 to 6 carbons,
with a compound of the formula:
ci cox
wherein
X is a halogen,
under conditions effective to produce a first mixture of regioisomers of the
formula:
A ~H~
-coRa
ci
CHI
O
WO 95/00482 PC'T/US94/06873
2181089
-21-
Such conditions include those rnnventionally utilized in a Friedel-Crafts
acylation
reaction catalyzed by, for example, A1C13. The reaction~is carried out in a
solvent
such as, carbon disulfide, tetrachloroethane, or nitrobenzene with carbon
disulfide
being the preferred solvent. The reaction is carried out for a time period of
1 /2 to
12 hours, preferably 3 to 5 hours, at a temperature of 0 to 25 C.
The first mixture of regioisomers can be hydrolyzed under
conditions effective to form a second mixture of regioisomers of the formula:
A øH
I3
l
'~ COOH
O CHI
Typically this reaction is carried out by base hydrolysis procedures which are
well
known in the art. For example, the first mixture of regioisomers can be
treated
with an inorganic base, such as, sodium hydroxide or potassium hydroxide, in
an
aqueous lower alcohol solvent. Suitable solvents include aqueous methanol,
ethanol, isopropanol, or n-butanol solutions. Hydrolysis is carried out at
reflux
temperatures of the solvent for 1 /2 to 12 hours..
2 0 Following such hydrolyzation, the substantially pure regioisomer of
the formula:
O~ A I Ha
i
C ~ C""~COOH
d I
CH3
WO 95/00482 P- i/US94/06873
zl~~os9
is recovered from the second mixture of regioisomers. Such recovery is carried
out by crystallizing the substantially pure regioisomer'salt of the formula:
O A ~ H3
C--COO' X~
U
CH3
wherein
X' is a Lewis Acid
Such crystallization is carried out by fractional crystallization techniques
known in
the art. Generally, such procedures involve dissolving the second mixture of
regioisomers in a solvent containing a salt at temperatures of 20 C to the
reflux
temperature of the solvent. The resulting solution is then slowly cooled to
temperatures of -20 to 25 C.
Suitable solvents for fractional crystallization include: alcohol
solvents, like methanol, ethanol, isopropyl alcohol, and n-butanol; ketone
solvents,
such as acetone or methyl ethyl ketone; ester-containing solvents, like ethyl
acetate
or isopropyl acetate; ethereal solvents such as tetrahydrofuran; acetonitrile;
and
dimethylformamide. Ethyl acetate is preferred.
Suitable salts for fractional crystallization are those where X~ is an
alkali metal salt, like sodium and potassium salts, or, more, preferably,
ammonium
salts of the form NR~RsR9, where R~, Ra, and R, is hydrogen or a straight or
branched alkyl of 1 to 6 carbon atoms which magi be substituted at any
position
with a phenyl ring or a substituted phenyl ring. The ammonium salt can also be
cinchonidine, quinine, quinidine, quinuclidine, b ,urine, thebaine, or
cinchonine.
Of these salt complexes, cinchonidine is preferred.
The substantially pure regioisomer salt is then isolated by filtration
and converted to the substantially pure regioisomer of the formula:
WO 95/00482 P(:'1'/US94/06873
2lsio$9
O A ~ H3
'C
C COOH
CHa
by procedures well known in the art. Typically, such conversion is
accomplished
by treatment with acid.
Process Two For Producing Substantially Pu~~iois
In another embodiment of the process of the present invention, the
substantially pure regioisomer is produced by acylating a starting compound of
the formula:
2 0 wherein
A ~ Ha
~Ra
CHI
R~ is -COOH, -COpalkyl, -CON(alkyl)=, -COSalkyl where the
alkyl moieties have 1 to 6 carbon atoms and are straight or branched
with a compound of the formula:
c I-C=o
wherein
X, is a halogen, trialkyl tin, trialkyl borate, triflate, or
organometallic reagents of lithium or magnesium derived from
WC~"~95/00482 . ~ 1810 8 e7 ~Trt1s94/06873
-24-
bromine or iodine, with any alkyl groups having 1 to 4 carbon atoms
and being straight or branched under conditions effective to produce
the substantially pure regioisomer of the formula: -
10
O~ A ~Ha
'C
. I .-Ra
CH3
This acylation reaction is carried out in a suitable solvent in the presence
of an
appropriate catalyst for about 1 to 120 hours and at temperatures of about 0 C
to
the reflux temperature of the solvent. Suitable solvents for acylation
include:
hydrocarbon solvents, such as benzene, toluene, xylene, or cyclohexane;
halogenated hydrocarbons, such as chlorobenzene, dichloroethane, methylene
chloride, chloroform, or carbon tetrachloride; carbon disulfide;
dimethylformamide; ethereal solvents, like tetrahydrofuran and diethylether;
or
dioxane.
A variety of catalysts may be utilized when A is hydrogen. Suitable
catalysts include palladium catalysts, like palladium chloride, palladium
acetate,
tetrakis(triphenylphosphine)palladium(0), dichlorobis(triphenylphosphine
palladium(II), or benzylchlorobis(triphenylphosphine)palladium(iI); or nickel-
phosphine catalysts. Acylation may also be carried out in the presence of
added
lithium chloride or triphenylphosphine. The latter acylation reaction is known
in
the art as organometallic cross coupling reactions, and are conducted by the
general procedures of D. Milstein, et al.,1. Org. Chem., 1979, 44, 1613; ].W.
Labadie, et al., I. Orb. Chem., 1983, 48, 4b34; C. Sahlberg, et al.,
Tetrahedron
Letters. 1983, 24, 5137; D. Milstein, et al., l.Am. Chem. Soc., 1978, 100,
3636; and K.
Tamao, et al., Tetrahedron, 1982, 38, 3347.
WO 95/00482 PCTIUS94/06873
=~ z~sio$g
Process Three For Producing Substantially Pure Reeioisomer
In another embodiment ~of the process of the present invention, the
substantially pure regioisomer is produced by acylating a starting compound of
the formula:
A ~ H3
_,_CORS
cH3
wherein
Rs is -ORd -N(R6)Z, and -SRd and
R6 is an alkyl with 1 to 6 carbon atoms
with a compound of the formula:
c i -c.= o
under conditions effective to produce a first mixture of regioisomers of the
formula:
A ~ H~
C-CORE
CHI
WO 95/00482 PCT/US94106873
~1810~9
-26-
Typically, such acylation is carried out by a Friedel-Crafts reaction, as
described
above in Process One for Producing Substantially Pure Regioisomers.
The substantially pure regioisomer salt is recovered by fractional
crystallization, isolation, and converting, as dexribed above with reference
to
Process One for Producing Substantially Pure Regioisomers.
Once the substantially pure regioisomer of the present invention is
produced by one of the above (or some other) process, there are a number of
procedures for using that compound to produce the piperidine derivatives of
the
present invention.
Process One Of Converting The Substantially Pure Regioisomer to The
Substantially Pure Piperidine Derivative Havine A Keto Group
According to one aspect of the present invention, the substantially
pure regioisomer can be halogenated under conditions effective to form a first
intermediate compound of the formula:
A ~H3
~ c-a3
x " ~ '-' I
CH3
O
wherein X is a halogen.
Suitable halogens include chlorine, bromine, and iodine. Suitable conditions
for
carrying out such halogenating include reacting the substantially pure
regioisomer
with a halogen nucleophile and a Lewis Acid. The ring opening reaction is
carried out in a suitable solvent, optionally in the presence of a catalytic
amount
of base for about 0.5 to 24 hours and a temperature of about -40 degrees C to
the
reflux temperature of the solvent. Suitable halogen nucleophiles include
sodium
iodide, sodium bromide, potassium iodide, potassium bromide, cesium iodide,
PCT/US94106873
wo 95~oo4sZ, ~ 1810 $
-27-
cesium bromide, trimethylsilyl iodide, manganese iodide, cerium iodide,
magnesium bromide, magnesium iodide, magnesium carbonate, calcium bromide,
and calcium iodide. Suitable Lewis Acids include silicon compounds such as
trimethylsilyl chloride and trimethylsilyl iodide; aluminum compounds such as
aluminum chloride, trimethyl aluminum, diethyl aluminum chloride, ethyl
aluminum dichloride, and diethyl aluminum cyanide; magnesium salts; and boron
salts. Suitable solvents for the ring opening reaction include hydrocarbon
solvents, such as, benzene, toluene, xylene, or cyclohexane; ethereal solvents
such
as ether, tetrahydrofuran, dioxane, or dimethoxyethane; or halogenated
hydrocarbons, such as, chlorobenzene, methylene chloride, carbon
tetrachloride,
chloroform, or dichloroethane.
After such halogenation, the first intermediate compound is reacteri
with a piperidine compound of the formula:
s
0
c-R,
R2
H
under conditions effective to form the piperidine derivative compound having a
keto group of the formula:
30
R~
CHI
WO 95/00482 PCT/US94/06873
-28-
Tlus alkylation reaction is carried out in a suitable solvent preferably in
the
presence of a base and, optionally, in the presence of a catalytic amount of
potassium iodide for about 4 to 120 hours at a temperature of about 70 C to
the
reflux temperature of the solvent. Suitable solvents for the alkylation
reaction
include alcohol solvents, such as, methanol, ethanol, isopropyl alcohol, or n-
butanol; ketone solvents, such as, methyl isobutyl ketone; hydrocarbon
solvents,
such as, benzene, toluene, or xylene; halogenated hydrocarbons, such as,
chlorobenzene or methylene chloride; or dimethylformamide. Suitable bases for
the alkylation reaction include inorganic bases, for example, sodium
bicarbonate,
potassium carbonate, or potassium bicarbonate or organic bases, such as a
trialkylamine, for example, triethylamine or pyridine, or an excess of the
piperidine compound can be used.
When R3 is -COOalkyl, the alkylation reaction is followed by base
hydrolysis to convert R, substituents that are -COOalkyl groups to -COOH
groups. Such base hydrolysis involves treatment of the substantially pure
piperidine derivative with an inorganic base, such as, sodium hydroxide in an
aqueous lower alcohol solvent, such as, aqueous methanol, ethanol, isopropyl
alcohol, or n-butanol at reflux temperature for about 1/2 hour to 12 hours.
2 0 Piperidine compounds where each of Rl and R2 is hydrogen or
wherein Rl is hydroxy and RZ is hydrogen are commercially available or may be
prepared according to procedures well known in the art (e.g. F.j. McCarty,
C.H.
Tilford, M.G. Van Campen, 1. Am. Chem. Soc.. 1961, 26, 4084). Piperidine
compounds wherein R, and RZ form a second bond between the carbon atoms
bearing R, and R= may be prepared by dehydration of the corresponding
compound wherein R, is hydroxy by procedures generally known in the art.
~'~'n 95~~Z PGT/US94/06873
~18i0~9
-29-
Second Process For Convertine Substantially Pure ReEioisomer To SubstanHallv
Pure Piperidine Derivative Havin~~ A Keto Grouy
In another embodiment of the present invention, the substantially
pure regioisomer of the formula:
O A
Ha
C
C _--R
a
CH3
is reacted directly with a piperidine compound of the formula:
H
under conditions effective to form the piperidine derivative compound having a
keto group of the formula:
s
0
C-R~
R2
O A ~Ha
,
( CHz j ~--C ~ ~ Ra
CHI
z 1 s i o ~ 9 ~T/US94,06873
-30-
This alkylation reaction is carried out in a suitable solvent preferably in
the
presence of a base and optionally in the presence of a L,ewis Acid such as
magnesium, cesium, or calcium salts or trimethylsilyl chloride or in -the
presence
of a catalytic amount of potassium iodide for about 4 to I20 hours at a
5_ temperature of about 70 C to the reflux temperahue of the solvent. Suitable
solvents for the alkylation reaction include alcohol solvents, such as,
methanol,
ethanol, isopropyl alcohol, or n-butanol; ketone solvents, such as, methyl
isobutyl
ketone; hydrocarbon solvents, such as, benzene, toluene, or xylene; and
halogenated hydrocarbons, such as, chlorobenzene or methylene chloride; or
dimethylformamide. Suitable bases of the alkylation reaction include inorganic
bases, for example, sodium bicarbonate, potassium carbonate, or potassium
bicarbonate or organic bases, such as, a trialkylamine, for example,
triethylamine
or pyridine, or an excess of a compound of the piperidine compound may be
used.
Processes for Reduction of Keto Group in Substantially Pure Piperidine
Derivative
As discussed above, the process of the present invention is useful in
producing substantially pure piperidine derivatives with either a keto group
or a
hydroxyl group. Derivatives with keto groups can be converted to similar
compounds with hydroxyl groups by reduction reactions which are well known in
the art.
Reduction can be carried out with sodium borohydride or potassium
borohydride in Ivwer alcohol solvents, such as, methanol, ethanol, isopropyl
alcohol, or n-butanol.
When lithium aluminum hydride or, diborane are used as reducing
agents, suitable solvents are ethers, for example, diethyl ether,
tetra~ydrofuran, or
dioxane. These reduction reactions are carried out at temperatures ranging
from
about 0 C tv the reflux temperature of the solvent, and -the reaction time
varies
from about 0.5 to 8 hours.
WO 95/00482 - '~':~ . FG"T/US94/06873
-3I-
Catalytic reduction may also be employed using, for example, Raney
nickel, palladium, platinum or rhodium catalysts in lower alcohol solvents,
such
as, methanol, ethanol, isopropyl alcohol, or n-butanol or acetic acid or their
aqueous mixtures, or by the use of aluminum isopropoxide in isopropyl alcohol.
Reduction using sodium borohydride is generally preferred over catalytic
reduction when forming carboxylic acids or esters. When the starting material
is
an ester, lithium aluminum hydride is the preferred reducing agent, while
diborane is preferred when starting with an acid.
When esters with hydroxyl groups have been formed, base
. hydrolysis can be used to produce a carboxylic acid. Such procedures are
well
known and generally involve treatment with an inorganic base, such as, sodium
hydroxide or potassium hydroxide, in an aqueous Lower alcoholic solvent, such
as
aqueous methanol, ethanol, isopropyl alcohol, or n-butanol. Base hydrolysis is
carried out at about the solvent reflux temperature for about I/2 hour to I2
hours.
EXAMPLES
Example 1 - Preyaration of Ethyl 3- and 4-(4-chloro 1-oxobutyl) a,a
2 0 dimethylphenylacetate
Aluminum chloride (44 80.33 mol) was added slowly in portions to
a solution of freshly distilled 4-chlorobutyryl chloride (17 mL; O.I5 mol) in
460 mL
of carbon disulfide at -IO C. under a nitrogen atmosphere. The mixture was
stirred for 15 minutes, then the cooling bath was removed and the mixture was
allowed to warm to ambient temperature. The mixture was stirred then for I5
minutes more, then cooled again to -10 C and ~ solution of ethyl
a,a-dimethylphenyl acetate (26.6 g; O.I4 mol) in 70 mL of carbon disulfide was
added dropwise. The mixture was maintained with stirring for 3 hr, then
stirred
overnight at room temperature.
"' Trademark
C
wc,~,~9siooas2
X181089
-32-
The reaction mixture was partitioned between H=O and CHC13. The
combined organic portions were washed with saturated aqueous NaHC03
solution, dried over MgSO,, filtered and concentrated in vacuo. The residue
was
dissolved in CHzCI= and filtered through a plug of SiOZ, eluting with 10%
EtOAc
in hexane. Concentration of the product-containing factions afforded 39.4 g of
ethyl 3- and 4-(4-chloro-1-oxobutyl)-a,a~iimethylphenylacetate as a mixture of
aromatic regioisomers.
Example 2 - Preparation of 4-(C3rclopropyl-oxo-methyl)-a.a-
dimethvlyhenvlacetic
acid
To a solution of 39.4 g of ethyl 3- and 4-(4-chloro-1-oxobutyl)-a,a-
dimethylphenylacetate obtained in Example 1 dissolved in 800 mL of CH30H and
200 mL of HIO was added 40 g of NaOH. The resulting mixture was refluxed for
one hour. The cooled mixture was then concentrated in vacuo to remove the
CH~OH. The rnncentrate was diluted with H20 and washed with two portions of
EtOAc. The aqueous layer was acidified with concentrated HCl and extracted
with two portions of EtOAc. The extracts were dried over MgSO,, filtered, and
concentrated in vacuo to afford 30.3 g of crude product.
The crude product was dissolved in 600 mL of EtOAc,.38 g of
cinchonidine was added, and the mixture was stirred overnight. The resulting
solids were filtered and washed with EtOAc and sucked dry under a rubber dam
to afford 2S g of a tan solid.
The solids were partitioned between EtOAc and ZN HCI. The
aqueous layer was extracted with EtOAc. The combined organics were dried over
MgSO,, filtered, and concentrated in vacuo to afford 10.6 g of an oil (33%
from
ethyl a,a-dimethyl-phenylacetate).
Example 3 - Preparation of 4-(4-Iodo-1-oxobutvl)-a.a-dimeth l~phenylacetic
acid
A solution of 10.5 g of 4-(cydopropyl-oxo-methyl)-aa-
dimethylphenylacetic acid, prepared in accordance with Example 2, in 250 mL of
WO 95/00482 PCT/US94/06873
X181089
-33-
CH=C1Z was cooled in an ice-MeOH bath and 25 g of trimethylsilyliodide was
then
added rapidly via pipette. The mixture was stirred in the ice bath for one
hour,
warmed to ambient temperature, and stirred for one hour. A solution of aqueous
sodium bisulfite was then added and the mixture was stirred well. The phases
were partitioned and the aqueous layer was extracted with CH=Cl=. The combined
_ organics were washed with saturated aqueous NaCI, dried over MgSO,,
filtered,
and concentrated in vacuo to afford 12.6 g (77%) of 4-(4-iodo-l-oxobutyl)-a"a-
dimethylphenylacetic acid.
Example 4 - Preparation of Methvl 4-(4-Iodo-1-oxobutyl)-~a
dimethylphen~rlacetate
To a solution of 12.6 g of 4-(4-iodo-I-oxobutyl)-a,a
dimethylphenylacetic acid, prepared in accordance with Example 3, in 100 mL of
EtzO cooled in an ice bath, was added 40 mL of ethereal CHiIV2. The mixture
was
stirred at 0 C for few minutes, then let stand for 2 hr. A few drops of AcOH
were added to decompose excess CHZN=, then the mixture was filtered and
stripped to afford 12.6 g (96%) of methyl 4-(4-iodo-l-oxobutyl)-aya-
dimethylphenylacetate.
2 0 Example 5 - Preparation of Methvl 4-(4-(4-(Hvixydyvhenvlmethvl) 1
piveridinyll-1-oxobut~yll-oc,a-dimethy~henylacetate
A solution of 12.6 g of methyl 4-(4-iodo-1-oxobutyl)-a,a-
dimethylphenylacetate, prepared in accordance with Example 4, in 500 mL of
toluene in a one liter three neck flask with mechanical stirring was added 8.8
g of
4-(a,a-diphenyl)piperidinemethanol and 23 g of IC=CO~ and the mixture was
refluxed for 7 hr. The cooled reaction mixture was then filtered and
concentrated
inin-, The residue was dissolved in EtxO and treated with excess ethereal
HCI. The mixture was then concentrated to a solid. The solid was treated with
EtOAc and collected by filtration. The product was then partitioned between
EtOAc and 2N NaZCO,. The organics were dried over MgSO,, filtered, and
WO 95/00482 PCTIUS94106873
X181089
-34-
concentrated in vacuo to afford 13.5 g (79%) of methyl 4-[4-[4-
(hydroxydiphenylmethyl)-1-piperidinyl]-1-oxobutyl]-aa-dimethylphenylaceta te.
Example 6 - Preparation of Methyl 4-(4-I4-(Hydroxyd~hen,~, ly )-1-
piperidin, ly lil-hYdtO -xybutvll-a.a-dimethylphenylacetate
A solution of 13.5 g of methyl 4-[4-[4-(hydroxydiphenylmethyl)-1-
piperidinyl)-1-oxobutyl]-a,a-dimethylphenylacetate, prepared in accordance
with
Example 5, in 250 mL of CH~OH was cooled in an ice- CHjOH bath and 1.8 g of
NaBH, was added in portions. After 1 hr, the mixture was concentrated to a
solid. The residue was partitioned between EtOAc and saturated aqueous
NaHCO,. The aqueous portion was extracted with EtOAc. The combined
organics were washed with saturated aqueous NaCI, dried over MgSO,, filtered,
and concentrated in varuo to afford 9.5 g (70%) of methyl 4-~4-[4-
(hydroxydiphenylmethyl)-1-piperidinyl]-1-hydroxybutyl]-aa-
dimethylphenylacetate as a foam.
Examvle 7 - Preparation of 4-I4-(4-Hydroxydiph~nylmethyl)-1=pi~eridin, 1
hydro ~x~butyll-a~a-dimethylphenylacetic Acid
To a solution of 9.5 g of methyl-4-(4-[4-(hydroxydiphertylinethyl)-1-
piperidinyl]-1-hydroxybutyl]-a,a-dimethylphenylacetate, prepared in accordance
with Example 6, in 300 mL of CH,OH and 150 mL of H20 was added 10 g of
NaOH. The mixture was refluxed for lhr, then cooled. The CH~OH was
removed in vacuo. The concentrate was diluted with HIO and CHC13 and the pH
adjusted to approximately 5.5 to 6Ø The phases were separated and the
aqueous
phase was extracted with CHCh. The combined organics were dried over MgSO,,
filtered, and stripped to afford 9.0 g of crude product.
The crude product was dissolved in CHZCI= and chromatographed
on Davisil Grade 633 Si0= eluting with a gradient of CHCIy to 10% CH~OH in
CHCI~, to 25% CH30H in CHCh. The product containing fractions were
concentrated to afford 5.2 g of white crystals. An analytical sample was
prepared
WO 95/00482 ~~ PCTlUS94/06873
-35-
by treatment of the product with EtOAc, mp 199-203 C. Calc. for C32H3gI~IQ~;
C,
76.62; H, 7.84; N, 2.79. Found: C, 76.24; H, 7.76; N, 2.75.
Examvle 8 - Preparation of Methvl 4-(4-I4-(Bis(4-methvlphenvl)hvdroxvmethvl)
~-oiyeridinvll-1-oxobutvll-a,a-dimethvlohenvlacetate
- To a solution of 6.4 g (0.017 mol) of methyl 4-(4-iodo-I-oxobutyl)-a,a-
dimethylphenylacetate, prepared in accordance with Example 4, in 500 mL of
toluene in a one liter round bottom flask equipped with a mechanical stirrer
was
added 5.1 g (0.017 mol) of 4-(a,a-bis(4-methylphenyl)-piperidinemethanol,
followed by 11.8 g (0.086 mol) of solid potassium carbonate. The solution was
heated to reflux for 24 hr. After cooling, the mixture was filtered and the
toluene
was removed in vacuo. The residue was partitioned between ethyl acetate and 2
N sodium bicarbonate solution. The aqueous layer was extracted twice with
ethyl
acetate, the combined organic layers were dried with sodium sulfate and the
ethyl
acetate was removed in vacuo to provide 6.8 g (73%) of methyl 4-[4-[4-(bis(4-
methylphenyl)hydroxymethyl)-1-piperidinyl]-1-oxobutyl]-a,a-
dimethylphenylacetate as a viscous, dark colored oil.
Examyle 9 - Preyaration of Methyl 4-f4-f4-(Bis(4-Methylyhenyl)hydroxymeth,~,
2 0 1-yiyeridinyll-1-hydrox~utyll-oca-dimeth~phenylacetate
To a -10 C solution of 6.8 g (0.013 mol) of methyl 4-[4-[4-(bis(4-
methylphenyl)hydroxymethyl)-1-piperidinyl]-1-oxobutyl]-a,a-
dimethylphenylacetate, prepared in accordance with Example 8, in 150 mL of
methanol in a 500 mL round bottom flask equipped with a mechanical stirrer was
slowly added 0.86 g (0.023 mol) of sodium borohydride, and the reaction was
stirred for 2 hr. The methanol was removed in vacuo and the residue was
partitioned between ethyl acetate and aqueous sodium bicarbonate solution. The
aqueous layer was extracted with ethyl acetate, the combined organic layers
were
dried with sodium sulfate, and the ethyl acetate was removed inin vacuo to
provide
6.9 g of a dark colored foam. The resultant material was purified by column
_. , .
1 . ~.' ~_.. PCT/US94/06873
,_~O 95/00482 . . .
-36-
chromatography (Davisil grade 633 silica gel, packed in methylene chloride,
material applied in chloroform, and eluted with a gradient of Z% methanol to
methylene chloride to 5% methanol to methylene chloride) to afford 5.3 g (77%)
of
methyl 4-[4-[4-(bis(4-methylphenyl)hydroxymethyl)-I-piperidinyl]-I-
hydroxybutyl]-a,a-dimethylphenylacetate.
Example 10 - Pzeyaration of 4-(4-f4-(Bis(4-methylphenyl)hydroxyntethyl) 1-
piperidinyl]'-1-hydzooybut~l]-a,a-dimethylphenylacetic Acid
To 350 mL of methanol in a I L round bottom flask equipped with a
mechanical stirrer was added 5.3 g (9.8 mmol) of methyl 4-[4-[4-(bis(4-
methylphenyl)hydroxymethyl)-I-piperidinyl]-1-hydroxybutyl]-a,a-
dimethylphenylacetate, prepared in accordance with Example 9, 5.I g (O.I3 mol)
of
solid sodium hydroxide, and 100 mL of water. The mixture was heated to reflux
for 3 hr. After cooling, the methanol was removed in vacuo, and 6 N
hydrochloric acid was added dropwise until the solution was no longer basic
(pH
= 7). The solution was extracted three times with ethyl acetate. The organic
layers were combined and a white crystalline solid precipitated out of
solution.
The solid was washed with ether to provide 1.8 g (34%) of 4-[4-[4-(bis(4-
methylphenyl)hydroxymethyl)-1-piperidinylJ-I-hydroxybutyl]-a,a-
2 0 dimethylphenylacetic acid, as the dehydrate, mp 208-2I5 C. Analysis.
Calcd. for
C34H~N04 2(HZO): C, 72.18; H, 8.37; N, 2.47. Found: C, 72.02; H, 8.36; N,
2.41.
Example 11 Preparation of 4-(1-H~droxy-4-iodobutyl)-a,a-dimethylphenylacetic
acid
To a solution of 50 mg of 4-(4-iodo-1-oxobutyl)-a,a-
dimethylphenylacetic acid, prepared in accordance with Example 3, in 3 mL of
methanol was added 50 mg of NaBH4. The mixture was stirred for 30 minutes,
acidified with 2N HCI, and the methanol removed in vacuo. The concentrate was
extracted with EtOAc. The organics were dried over NarSO" filtered, and
~' Trademark
v
WC? 9S/00482 ~ 1810 8 9 PCT/US94/06873
- 37 -
concentrated to afford 40 mg of 4-(1-hydroxy-4-iodobutyl)-a,a-
dimethylphenylacetic acid.
Examyle 12 - Preyaration of 4-(4-f4-(Hydmxydiphenylmeth" l~-1-piperidinyll-1-
~obutyll-a.a-dimeth~phenvlacetic acid
A mixture of 800 mg of 4-(4-iodo-1-oxobutyl)-a,a-
dimethylphenylacetic acid, prepared in accordance with Example 3, 80b mg of 4-
(a,a-diphenyl)piperidinemethanol, and 2.4 g of K=CO' in 25 mL of toluene was
stirred for 48 hours at room temperature. The mixture was concentrated in
vacuo.
The residue was treated with EtOAc, filtered, and concentrated to afford 4-[4-
[4-
(hydroxydiphenylmethyl)-1-piperidinyl]-1-oxobutyl]-ova-dimethylphenylacetic
acid.
Examflle 13 - Preparation of 4-L4-(4-Hpdmxydiphen~rlmethyl)-1-pperidinyll 1
hydrox~utyl]-a,a-dimethylphenylacetic Acid
A mixture of 4-[4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]-1-
oxobutyl]-a,,a-dimethylphenylacetic acid, prepared in accordance with Example
12,
and 300 mg of IvaBH, in 25 mL of CH30H was stirred overnight at room
temperature. The mixture was then concentrated in vacuo. The residue was
partitioned between EtOAc and H=O. The aqueous portion was treated with
concentrated HCl until pH 6, then extracted with EtOAc. The organics were
concentrated in vacuo: The residue was dissolved in EtOAc, filtered, and
concentrated in vacuo to an .oil. The oil was dissolved in CH~OH and
concentrated to a solid. The solid was slurried with EtOAc, filtered, and
rinsed
with EtOAc to afford 4-[4-[4-hydroxydiphenylmethyl)-1-piperidinyl]-I-
hydroxybutyl]-a,a-dimethylphenylacetic add.
Although the invention has been described in detail for the purpose
of illustration, it is understood that such detail is solely for that purpose,
and
W~ 95/00482 ~ 1810 8 9 PCT/US94/06873
-38-
variations can be made therein by those skilled in the art without departing
from
the spirit and scope of the invention which is defined by the following
claims.