Note: Descriptions are shown in the official language in which they were submitted.
w0 95/21161 ~' 8 210 5 pCT~GB95/00225
-- -1-
Chloronvrimidine Intermediates
The present invention relates to certain novel pyrimidine intermediates,
processes for their
preparation and their conversion to 9-substituted-2-aminopurines, such as
certain
carbocyclic, heterocyclic and acyclic purine nucleoside analogues, and salts,
esters and
pharmaceutically acceptable derivatives thereof.
A number of 2-aminopurine nucleoside analogues have been shown to be useful in
the
treatment or prophylaxis of viral infections, for example the compound of
formula (A)
N~ '
N / N CA)
N NH2
HO
is described as having potent activity against human immunodeficiency virus
(HIS and
hepatitis B virus (IdB~ (EP 0434450).
Processes have been proposed for the preparation of 9-substituted-2-
aminopurines,
generally starting from a pyrimidine compound, coupling with a sugar analogue
residue,
and cyclisation to form the imidazole ring and introduction of any suitable 6-
substituent.
Pyrimidine compounds which have been identified as being usefi~l in the
preparation of 9-
substituted-2-aminopurines include 2,5-diamino-4,6-dichloropyrimidine, N,N-
(4,6-
dichloro-2,5-pyrimidinediyl)bis formamide and also N-2-acylated pyrimidine
derivatives
such as the 2-acetamido and 2-isobutryamide derivatives (US Patent 5087697).
Processes for the synthesis of these intermediates generally involve a number
of steps of
which some are difficult to perfoirn and produce poor yields, preventing any
practical
scale up of these processes above a few grams, and are thus di~cult and
uneconomical.
218 210 5 PCTIGB95/00225
WO 95121161
,~ , s . y
_2_
Processes For the synthesis of the intermediate 2,S-diamino-4,6-
dichloropyrimidine include
the direct chlorination of readily available 2,5-diamino-4,6-
dihydroxypyrimidine using
phosphorus oxychloride. The original examination of this reaction was carried
out by
Temple et aI. (J. Org. Chem. 1975, 40: 3141-3142). These workers concluded
that the
reaction was unsuccessful, apparently because of degradation of the pyrimidine
sing
system. Hanson (SmithKline Beecham, WO 91/01310, US Patent 5216161)
subsequently
described a process for the direct chlorination of 2,5-diamino-4,6-
dihydroxypyrimidine by
reffuxing with phosphorus oxychloride in the presence of large molar excesses
of
quaternary ammonium chlorides or amine hydrochlorides. We have examined this
process
and have obtained, repeatedly, much lower yields (<10%) of crude 2,5-diamino-
4,6-
dichloropyrimidine than those specified in the SmithKline Beecham patent
specification.
The extensive decomposition of the 2,5-diamino-4,6-dihydroxypyrimidine to tars
which
coat the equipment, combined with the problems of dealing with the copious
solids due to
the insoluble amine salts, constitute significant drawbacks and make scale-up
of such a
process impractical. The modifications of Legraverend et al. (Synthesis 1990:
587-589),
namely using acetonitrile as a solvent and adding phosphorus pentachloride to
the
phosphorous oxychloride and quaternary ammonium chloride, result, in our
experience in
the isolation of approximately 30% (after chromatographic purification) of 2,5-
diamino-
4,6-dichloropyrimidine on a 2-5 gram scale. Again, scale-up beyond a few grams
is
impractical due to the formation of tarry precipitates.
A recent Lonza AG patent specification (EP 0 552 758) suggests that higher
yields (35-
65%) may be obtained with phosphorus oxychloride chlorination when the 5-amino
group
of 2,5-diamino-4,6-dihydroxypyrimidine is protected with an alkoxycarbonyl
protecting
group. This modification is claimed to simplify the chlorination step in that
the amines and
phosphorus pentachloride, employed in the prior processes discussed above are
not
required. This creates a new problem, namely the need to remove the
alkoxycarbonyl
protecting groups in order to be able to convert the pyrimidine intermediates
to purines.
Indeed, the Lonza AG specification does not show that such 5-protected 2,5-
diamino-4,6-
dichloropyrimidines may be converted to purines in an advantageous manner.
WO 95/21161 218 210 5 pCT~GB95100225
- -3-
A process for the synthesis of N,N~-(4,6-dichloro-2,5-pyrimidinediyl)bis
formamide is the
reaction of 2,5-diamino-4,6-dichloropyrimidine with formic acid and acetic
anhydride
' (Harnden et al., J. Med. Chem. 1990, 33:187-196 and US Patent 5,159,076).
' The 5-step route to the N-2-acylated derivatives, and also to 2,5-diamino-
4,6-
dichloropyrimidine required for the synthesis of N,N-(4,6-dichloro-2,5-
pyrimidinediyl)bis-
formamide (Temple et al., J. Org. Chem. 1975, 40: 3141-3142), starts from 2-
amino-6-
chloropyiimidin-4-one and contains steps, which include the introduction of
the 5-nitro
group and the subsequent handling and reduction of very reactive S-nitro-4,6-
dichloropyrimidine intermediates, which make scale-up impractical. The yields
on a
number of the steps to these intermediates are poor (Legraverend et al.,
Synthesis 1990:
~ 87-589).
We have now discovered certain new pyrimidine intermediates which are useful
in a new
synthetic route for the preparation of the above 9-substituted-2-aminopurines
and in
addition which can be used in the synthesis of the known intermediates
described above.
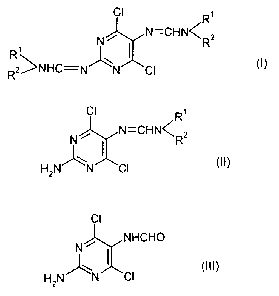
In one aspect of this invention we provide the following novel intermediates
which may be
utilised in the synthesis of 2-aminopurines, namely compounds of formulae (I),
(1T) and
ci
C1 R1
R1 C
N / N CHN C ~ N / R2
(II)
R1 (I)
~N C1
~NHC N N Cl H2
R2
C1
N / HCHO
(III)
r H N~N C1
2
PCTIGB95/00225
w0 95121161 y ' .-, .
-4- -
wherein R1 and R2, which be the same or different, are selected from C1_g
straight-chain
alkyl, C1_g branched alkyl, C3_g cycloalkyl, and aryl groups (such as phenyl
or naphthyl),
which may be optionally substituted, for example by C 1 ~ alkyl or halogen
(e.g. Cl). In a
preferred embodiment of the invention Rl and R2 are both methyl.
These novel intermediates can be readily prepared in good yields and are
useful for the
preparation of a wide variety of different types of 2-aminopurines including
the nucleoside
analogue of formula (A), famciclovir (EP 0182024), penciclovir (EP 0141927), I-
i2G
(EP 0343133), (1'S,3'S,4'S)-2-amino-1,9-dihydro-9-[3,4-dihydroxy-3-
hydroxymethyl-1-
cyclopentyl)-6H-purin-6-one (EP 0420518), and other 9-substituted-2-
aminopurines
provided that the 9-substituent is not attached by a glycosidic bond.
In a further aspect of this invention we provide processes for the synthesis
of the novel
intermediates of formulae (>7, ()I) and (III], and the known intermediate 2,5-
diamino-4,6-
dichloropyrimidine(I~. These processes are illustrated in the simplified
diagram below
which is designed for illustration only of the possible ways of synthesising
these
intermediates;
2,5-diamino-4,6-dihydroxypyrimidine.
m
1 s. ~ )l.
WO 95/21161 . ~ 2 1 0 ~ PCT/GB95100225
'- -5-
The present invention also provides a process for the preparation of compounds
of
fornmla (I) which comprises chlorination of 2,5-diamino-4,6-
dihydroxypyrimidine with a
' halomethylenimminium salt (V'~lsmeier reagent) of formula (~.
R1 + _
>N CHC1 C1
R2
(v~
wherein R1 and R2 are as defined above
Compounds of formula ('~, may be prepared from a variety of formamides of
secondary
amines by reaction with a variety of acid halides, such as phosphorus
oxychloride,
phosphorus pentachloride, thionyl chloride, phosgene, and oxalyl chloride, for
example as
detailed in a review by C. M. Marson, Tetrahedron 1992, 48: 3660-3720 and
references
therein.
The advantage of protecting the diaminopyrimidine from extensive decomposition
during
chlorination is achieved by the in situ protection of the amino groups with
two molar
equivalents of Vilsmeier reagent (~ to give a bis-formamidine intermediate
(detected by
thin-layer chromatography), which is subsequently chlorinated to a compound of
formula
(I~ as the reaction with additional equivalents of V'ilsmeier reagent
proceeds. The
improved solubility of such bis-formamidine derivatives is an added advantage
of this
process, facilitating the subsequent chlorination to compounds of formula (I)
and their
isolation and simple purification.
The disadvantage of the use of 5-alkoxycarbonyl protecting groups, as
described in the
Lonza specification (EP 0552758) is avoided since the formamidine groups in
compounds
of formula (1~ are readily hydrolysed under mild conditions in a step-wise
manner to form
the intermediates (II) and (III); or alternatively compounds of formula (I)
can be directly
hydrolysed to compounds of formula (III).
The compound 2,5-diamino-4,6-dichloropyrimidine (I~ can be prepared by:-
WO 95/21161 ~ v ~1 218 210 5 PCT/GB95100225
-6- --
A) the hydrolysis of a compound of formula (I);
B) the hydrolysis of a compound of formula (II); or '
C) the hydrolysis of a compound of formula (11T). '
The hydrolysis of (1], (In, or (II>7 to 2,5-diamino-4,6-dichloropyrimidine is
conveniently
carried out at pH 3 +/- 0. S by adding a water-miscible cosolvent, such as
ethanol. The
hydrolysis is more ei$cient at pH 1-2, with shorter reaction times required
than at a higher
pH. It is advisable at pH I-2, however, to protect 2,5-diamino-4,6-
dichloropyrimidine
from hydrolysis to hydroxypyrimidines by extraction, as it is formed, into an
organic layer
which is not miscible with the aqueous acid. When the pH of the aqueous layer
is below
1, extraction of the product into the organic layer is ine~cient (the pKa of
(I~ was found
to be ca. 0.5 and the pyrimidine ring is thus significantly protonated below
pH 1).
Preferably, the acid used for this hydrolysis should be one which is not
appreciably soluble
in the organic layer, e.g. phosphoric or sulfuric acid. The organic solvent
should be one
which is stable to aqueous acid and in which (I~ is soluble. Satisfactory
solvents for the
organic layer include toluene and halocarbon solvents such as methylene
chloride,
chloroform, and 1,2-dichloroethane. At completion, the organic layer is simply
washed,
e.g. with saturated aqueous bicarbonate, dried and concentrated to provide (I~
with no
purification required.
Compounds of formula (111) can be prepared by:-
A) selective hydrolysis of a compound of formula (I); or
B) selective hydrolysis of a compound of formula (11).
The hydrolysis of compounds of formula (I) or (1T) to (III) is most
efficiently carried out in
dilute aqueous acid, preferably in dilute aqueous mineral acid such as
sulfuric acid,
hydrochloric acid, or phosphoric acid. Prolonged exposure to pH below 1 should
be
avoided as the chloropyrimidine ring is protonated significantly below pH 1
and may
WO 95/21161
pCT~GB95/00225
_7_
therefore undergo attack by water, generating undesired hydroxypyrimidine by-
products.
Preferably, the pH is maintained above 2 and optimally at 3 +/- 0.5 for the
e$cient
' formation of (III). In this optimal pH range, the formamidine groups of (I)
and (11) are
selectively hydrolysed to give (11T) in approximately 70% yield. As the
hydrolysis of the
' formamidine groups of (17 and (11) proceed, the secondary amine from which
the V'~lsmeier
reagent (~ was formed (HNR1R2) is liberated and causes the pH of the solution
to rise,
thus slowing the hydrolyses. In addition, with certain reactive aliphatic
amines HNR1R2,
such as N,N-dimethylamine, it is necessary to maintain a pH sui~ciently low to
prevent the
chloro groups of the pyrimidine ring from displacement by the secondary amine.
We have
found that maintaining the pH of the reaction mixtures below 4 avoids
significant
displacement of the chloro groups by the secondary amine, even with amines as
reactive as
N,N-dimethylamine. It was thus found optimal to buffer the hydrolyses of (17
and (11) to
(11T) at pH 3 +/_ 0.5 or to add increments of acid throughout the hydrolyses
in order to
maintain the pH in this range.
Optimally, the hydrolysis of compounds of formula (I) or (1T) to (III) is
carzied out in a
minimum of water with the pH controlled as described above. Under these
conditions,
(III) precipitates as formed and is simply filtered off and washed with water.
The
hydrolysis is carried out at gentle reffux for 4 hours, or at lower
temperatures for longer
periods of time.
The compounds of formula (1TJ can be prepared by the selective hydrolysis of
the
compounds of formula (I~. Preferably the selective hydrolysis is carried out
with slightly
more than two molar equivalents of mineral acid in water or ethanol and warmed
for 15-
30 minutes.
The compounds of formula ()] can be prepared by reacting 2,5-diamino-4,6-
dihydroxypyrimidine with a V'~lsmeier reagent of formula (~.
The compound 2,5-diamino-4,6-dihydroxypyrimidine is commercially available
(Sigma,
' Maybridge BTB, Pfaltz and Bauer, Polyorganix).
WO 95121161 ~ * '~ ~ ~ ~ ~ PCT~GB95/00225
-8_ -
The novel bis-formamidines of formula (I) are formed and isolated conveniently
in high
yield when the 2,S-diamino-4,6-dihydroxypytZmidine (or a salt thereof, such as
the
hydrochloride or the hemisulfate) is treated with at least 4 molar equivalents
of a Vilsmeier
reagent (V). These chloiination reactions proceed under extremely mild
conditions
without the formation of copious tarry precipitates which characterises direct
chlorinations, as previously described with phosphorus oxychloride and
phosphorus
oxychloride / quaternary ammonium halides. The V'ilsmeier chlorination of 2,5-
diamino-
4,6-dihydroxypyrimidine may be carried out in an inert solvent, such as
toluene,
chloroallcenes, or chloroalkanes (such as methylene chloride, chloroform or
1,2-
dichloroethane). Preferably the solvent is 1,2-dichloroethane, chloroform, or
methylene
chloride. The chlorination may be carried out at 0 to 110°C, preferably
at 40-100°C,
conveniently at reflux for the solvent used. Reaction times are typically 12
to 48 hours.
Isolation of compounds of formula (I~ is simple and can be readily scaled-up,
involving
simply washing the reaction solution with an aqueous solution containing
sufficient base,
such as sodium bicarbonate, to neutralize any hydrogen chloride formed and
then
concentrating the dried organic layer to obtain the novel chlorinated
pyrimidines of
formula (I). The compounds of formula (I) are generally stable and may be
precipitated
from a variety of solvents, such as ethyl acetate, and stored or used without
further
purification.
Particularly preferred examples of the compounds of formulae (I), (1T) and
(III) are:
a) 4,6-Dichloro-2,5-bis-[(dimethylamino)methyleneamino]pyrimidine
b) 2-Amino-4,6-dichloro-S_[(dimethylamino)methyleneamino]pyiimidine
c) N-(2-Amino-4,6-dichloro-S-pyrimidinyl)formamide
According to a further aspect of this invention the novel intermediate of
formula (111) can
be used in the synthesis of 2-amino-6-chloropurines. In addition compounds of
formula
(I) or (1T) may also be used in the synthesis of 2-amino-6-chloropurine
nucleosides,
provided that the amine HNR1R2 (where Rl and R2 are defined earlier)
liberated, during
the conversion of the pyrimidine intermediate to the purine, is su~ciently
unreactive
towards the displacement of the chloro group of the 2-amino-6-chloropurines
generated. ,
:. G" _ ; r .-
WO 95/21161 ' ' ~ ~ ~ ~ PCT/GB95I00225
_9_
The compounds of formula (II17 share with the previously described N-2-
acylated
' derivatives the property of greater reactivity than 2,5-diamino-4,6-
dichloropyrimidine
toward displacement of a chloro group by an appropriate primary amine or
protected
' hydroxylamine. However, such condensations with (II>] (described in more
detail below)
may be carried out under milder conditions at lower temperatures and with
shorter
reaction times than with compound (I~, thus resulting in less decomposition of
the
amines. The condensation products (Vn are isolated in greater yield and purity
than the
corresponding products (V~ formed in condensations with 2,5-diamino-4,6-
dichloropyrimidine (I~. Another advantage of the use of the intermediate (III
over the
previously described N-2-acylated derivatives, in addition to greater ease of
synthesis, is
that the purines generated from (~ do not require deprotection, i. e.
hydrolysis of the N-
2-acyl group (these longer processes are described in US Patents 5,087,697 and
5,159, 076).
CI CI
NHCHO HCI ~N From
CH(~ NO ~ VIII N
H N N NHR 3 H N ~ ~N ~ ~ ~N
z N I HZN N I
R3 R3
Ma) - M~ (vua) - (vnt) pxd)
From Vlla (Famddovir)
From Vla ~-NH z
~-NH s EtOH
n-BuOH
NH
N
Z '>
H ZN N N
. i
R3
Wherein R3 is hereinafter defined. (Ixa)
2182105
WO 95/21161 " I~ . ~, ~ ' ~ ~, PCT/GB95/00225
-10-
The compound of formula (III) can be used to prepare the novel intermediates
of formula
(VI) which represent a further feature of the invention:-
NHCxo
N
(VI)
H2N N NHR3
wherein R3 may be hydrogen or any group which is not attached by a glycosidic
bond.
Preferably R3 is a hydroxyl or a protected hydroxyl; or a carbocyclic group
(e.g. C3_~
carbocyclic), an acyclic group (e.g. C2_g hydrocarbyl) wherein carbon atoms
may be
substituted by one or more heteroatoms such as N, O or S, or a heterocyclic
group (e.g.
C4_~ heterocyclic) in which at least one carbon atom is replaced by a N, 0, or
S atom, or
a substituted analogue of any thereof (e.g. such substituents are
independently selected
from C 1 ~allcyl, C 1 ~ alkoxy, hydroxyl or protected hydroxyl, azido,
phosphonyl, or
halogen), provided that such groups are not attached by a glycosidic bond .
Preferred groups for R3 are hydroxyl or protected hydroxyl.
Further preferred groups for R3 are
HO
a.
b. H;
0
HO
HO
C.
c~
i
r! n.
w0 95/21161 ° ' ' 218 210 5 PCT~GB95100225
- -11-
d. (AcOCH2)2CHCH2CH2- ;
e. HOCH2CH2~HCH2' ;
HZOH
HO
f.
HO
HO
g~ ; and
HO
H 0 ~~~~~ '
.,
h. H°\
A further preferred group for R3 is ;
HO
Suitable groups for R3 are selected from a; b; c; d; e; and ~ as defined
above.
By "hydrocarbyl" it is meant a group containing only hydrogen and carbon
atoms, which
may contain double and/or triple bonds and which may be straight, branched,
cyclic or
aromatic.
PCT/GB95/00225
W095/21161 '~ ,, ~ t~ , '
_ 12_
According to a further feature of the invention we provide a process for the
preparation of
compounds of formula (Vn which comprises reacting a compound of formula (1TT)
with
an amine of formula R3NH2, where R3 is defined above. Such condensations are
preferably carried out at reflux in a solvent such as ethanol, butanol, water
or acetonitrile
in the presence of at least one equivalent of a base, such as trialkylamine or
potassium or
sodium carbonate.
Subsequent references to compounds of formula (VIa, b, c, d, e, ~ g, or h)
denote a
compound of formula (V>] in which R3 is a group of a, b, c, d, e, i; g, or h
as defined
above.
A particularly preferred compound of formula(V~ is ( 1 S,4R)-4-[(2-amino-6-
chloro-S-
formamido-4-pyrimidinyl)amino]-2-cyclopentene-1-methanol (VIa)
The novel intermediates (V)7 can be converted by ring closure to the
corresponding
compounds of formula (VIn:-
c~
(VII)
N / N
H2H
R3
wherein R3 is defined above.
Ring closure of (Vn to (VIn is conveniently carried out in
trialkylorthoformates (e.g.
triethylorthoformate or trimethylorthoformate) with concentrated aqueous acid
(e.g. 2-4
molar equivalents of hydrochloric, sulfuric acid or methane sulfonic). For
example, the
hydrochloride salt of (Vaa) i.e. wherein R3 represents group a, begins to
precipitate from
such orthoformate solutions of (VIa) within minutes and yields above 90% may
be
achieved by filtration of the precipitate, optimally after several hours at
ambient
temperature.
. ~ .j . . . ~-
w0 95/21161 " ~ ' ' 218 210 5 p~'/GB95/00225
r-- - 13 -
The synthesis of 9-substituted-2-amino-6-chloropurines, such as compounds of
formula
(VIn, in this manner represents a significant improvement over previously
published
syntheses utilizing triaminopyrimidine intermediates such as (VIIl7:
ci
N / ~2
~N~N 3
(VIII)
as described US Patent 4,916,224. The previously-described routes to
intermediates such
as (V~ are longer and, more importantly, the number of steps to the purine
targets after
incorporation of the group R3 is greater. Also, triaminopyrimidine
intermediates such as
(VIII are air- and light-sensitive and extremely difficult to purify due to
their polarity and
metal-chelating abilities (the isolation from the zinc reduction of diazo
intermediates is
especially problematic). The novel 5-formamido intermediates of formula (Vn
are easily
and directly attainable from compounds of formula (IIn in one step and are
generally
solids which are stable and easily-purified by precipitation from a suitable
solvent.
(1'S,3'S,4'S)-2-Amino-1,9-dihydro-9-[3,4-dihydroxy-3-hydroxymethyl-1-
cyclopentyl]-6H-
purin-6-one (IXtI) (EP0420,518) may be prepared by condensation of the
compound of
formula(II~ with 4-amino-3-cyclopentene-1-methanol (US patent 5,049,671) to
form the
compound of formula (VIg) followed by ring closure of the compound of formula
(VIg)
to prepare the compound of formula (VBg), which may be hydroxylated, with
osmium
tetroxide/N-methyl-morpholine N-oxide to provide the compound of formula
(V>Zh). The
compound of formula (VIEa) is hydrolysed to form the compound of formula
(IXh).
218210
WO 95/21161 ' ~ ~~ PCT/GB95/00225
- 14- -
2-Amino-6-chloropurine (VIIb) may be prepared by ring closure of novel 2,4-
diamino-6-
chloro-5-formamidopyrimidine (VIb), conveniently synthesized by condensation
of the
compound of formula (III) with ammonia. The compound of formula (VIIb) is an
intermediate suitable for the synthesis of acyclic antiviral nucleosides, such
as famciclovir
wherein the 2-amino-6-chloropurine intermediate (Vtld) is conveniently
subjected to
hydrogenolysis to the 2-aminopurine nucleoside.
Carbocyclic nucleosides may also be synthesized from the compound of formula
(Vlib),
for example by (Pd-catalyzed coupling with an appropriate carbocyclic
intermediate as
described in Mac Keith et al., J.Chem.Soc.Perkin Trans 1. 1993: 313-314 and
references
therein.
The compounds of formula (VITa), (Vllc), (VITe), (VIIf), (Vltg) and (VIEa) are
conveniently hydrolyzed to the corresponding guanine compound by reffuxing
with
aqueous base or acid.
As a further feature of this invention we have found an alternative process
for the
synthesis of 2,6-diaminopurines (wherein the 6-amino group is substituted by
R4 and R5,
which may be the same or different, and are selected from H, C 1 _galkyl, C3-
6cycloalkyl,
aryl (such as phenyl), in particular R4 is H and RS is cyclopropyl) directly
from (VI) which
advantageously eliminates a step in the process. Such 2-aminopurine compounds
can be
synthesised directly from the intermediates (VI) by refluxing the compound of
formula
(VI) with an excess of the amine (HNR4R5) in a reiluxing solvent, such as
ethanol,
isopropanol, n-propanol, t-butanol or n-butanol.
In particular cases, it may be more useful to utilize 2,5-diamino-4,6-
dichloropyrimidine(I~ to prepare compounds of formula (VIII), useful in the
synthesis of
8-modified 2-aminopurine nucleoside analogues, such as 8-aza-2-aminopurines
(which
have broad-spectrum anti-herpes activities described in Storer et al., Spec.
Publ. Roy. Soc.
Chem (Rec. Adv. Chem. Anti-Infect. Agents) 1993, 119: 251-265); in such cases
the
intermediates (1), (II) and (111) can be used to provide (I~.
Zoa2io5
w0 95/21161 ~ P . t , ' PCT/GB95/00225
- -15-
Pharmaceutically acceptable esters of certain compounds of the invention may
be prepared
by esterification using conventional methods known in the art. Such methods
include , for
" example, the use of an appropriate acid halide or anhydride.
The compounds of the invention, including esters thereof, may be converted
into
pharmaceutically acceptable salts in a conventional manner by treatment with
an
appropriate acid or base. An ester or salt of a compound of the invention may
be
converted into the parent compound, for example, .by hydrolysis.
The following examples are intended for illustration only and are not intended
to limit the
scope of the invention in any way.
Example 1
4.6-Dichloro-2.5-bis-1 f ~dimethylamino)methylene)amino }pyrimidine
2,5-Diamino-4,6-dihydroxypyrimidine hemisulfate (Sigma, 25.0 g, 0.131 mole)
was stirred
in chloroform (AR Mallinckrodt, 400 mL) in a 2 L- 3-necked round bottom flask
equipped
with a reflux condenser (with source of nitrogen connected to the top of the
condenser)
and an exit for HCl gas connecting another neck of the flask to a NaOH trap.
(Chloromethylene)dimethyl ammonium chloride (V'~lsmeier reagent, Aldrich, 88.0
g, 0.651
mole as 95%) was washed into the flask with additional chloroform (400 mL).
The
reaction mixture was brought cautiously to reflux with nitrogen sweeping the
HCl
evolved into the trap. When the evolution of HCl slowed after about 1 hour of
reflux, the
sweep was stopped and the reaction kept under a gentle positive pressure of
nitrogen from
that point. Additional V'~lsmeier reagent (50.0 g, 0.370 mole) was added after
24 hours
and reflux continued for an additional 20 hours. The stirred reaction mixture
(yellow
solution with dark yellow solid) was cooled (ice bath) and diluted with water
(sufficient to
dissolve the solid, ca. 300 mL). The aqueous layer was adjusted to pH 7 with
sodium
hydroxide or solid sodium carbonate. The chloroform layer was separated,
washed with
water (3 x 400 mL), dried (sodium sulfate), and concentrated in vacuo to a
dark yellow
solid (36 g). This solid was dissolved in ethyl acetate (300 mL), stirred with
charcoal (1
g), and filtered with a silica gel pad (3x3 in., packed in ethyl acetate). The
pad was
PCT/GB95/00225
WO95121161 .~' "~
- 16- -
washed with additional ethyl acetate and eluents concentrated in vacuo to
leave the title
compound as a light tan solid (30.75 g, 81% ); m.p. 116-119°C; 1H-NMR
identical to that
of recrystallized samples.
Anal. Calcd. for C1pH14N6CI2Ø10 EtOAc: C, 41.92; H, 5.01; N, 28.20; Cl,
23.80.
Found: C, 42.23; H, 4.95; N, 28.46; CI, 24.11.
Recrystallization of such a sample from ethyl acetate gave the title compound
as white
granules; m.p. 123-125 °C; mass spectrum (CI/CH4): 291, 289 (M+1); 1H-
NMR
(DMSO-d6) 8: 8.49 and 8.69 (both s, 1 each, 2CH), 3.16 (s, 3, CH3), 3.03 (s,
6, 2CH3),
2.97 (s, 3, CH3); W (pH 7 phosphate buffer) ~,max 296 nM (E33,300), ,min 248
(5200).
Anal. Calcd. for C10H14N6CI2: C, 41.54; H, 4.88; N, 29.06; Cl, 24.52. Found:
C, 41.59;
H, 4.91; N, 29.01; CI, 24.47.
Example 2
2-Amino-4.6-dichloro-5-~ [tdimethylamino)methylene~] amino ~pyimidine
4,6-Dichloro-2,5-bis-{[(dimethylamino)methylene)amino}pyrimidine (Example 1,
5.87g,
20.3 mmol) was dissolved in 95% ethanol (200 mL) and 6 N aqueous hydrochloric
acid
(13.5 mL) added. The solution was heated in an oil bath at 55 °C under
nitrogen for 30
minutes, at which point TLC (silica gel, 5% methanol-chloroform) showed that
starting
material had been cleanly converted to a lower-Rf product. The cooled (ice
bath) solution
was adjusted to pH ~8 with concentrated ammonium hydroxide and the resulting
mixture
(white precipitate formed) concentrated on a rotary evaporator to ~5 mL to
remove
ethanol. Additional water (20 mL) was added and the cooled mixture was
filtered. The
white precipitate was washed with additional water (2 x 20 mL) and dried to
give the title
compound as a white powder (4.50 g, 95%), m.p. >dec 250 °C ; mass
spectrum
(CI/CH4): 236, 234 (M+1); 1H-NMR (DMSO-d6)8: 7.59 (s, 1, CH), 6.90 (s, 2,
NH2),
3.00 and 2.94 (both s, 3 each, 2CH3); LTV (pH 7 phosphate buffer) ~.max: 328
nM (s
4500), 255 (15,800).
PCT/GB95100225
w0 95/21161 '' "' ~ '~
- 17-
Anal. Calcd. for C7H9NSC12: C, 35.92; H, 3.88: N, 29.92; CI, 30.29. Found: C,
35.66; H,
3.86; N, 29.74; Cl, 30.54.
In another experiment, 2,5-diamino-4,6-dihydroxypyrimidine hemisulfate (Sigma,
48.0 g,
0.250 mole) was reacted as in Example 1 with less V'~lsmeier reagent (7.2
molar
equivalents) and the resulting 4,6-dichloro-2,S-bis-
{[(dimethylamino)methylene]amino}
pyrimidine (92%), without recrystallization, was hydrolyzed in 95% ethanol (1
L) and 6 N
aqueous hydrochloric acid (110 mL) to provide the title compound of the same
purity
(elemental analysis and 1H-NMR) as the characterized sample described above
(44.2 g.
76% overall from 2,5-diamino-4,6-dihydroxypyrimidine hemisulfate).
Example 3
N-(2-Amino-4.6-dichloro-S-pyiimidinyl)formamide lIIl7
A slurry of 2-amino-4,6-dichloro-5-{[(dimethylamino)methylene]amino}pyrimidine
(Example 2, I.50 g, 6.41 mmol) and 1.5 M aqueous potassium phosphate buffer
(35 mL,
prepared by adjusting the pH of a 1.5 M solution of KHZP04 to 3.2 by addition
of 85%
phosphoric acid) was gently refluxed (in an oil bath at 125 °C). After
4 hours of reflux,
the pH of the mixture was adjusted from 4 to 3 by addition of 4 drops of 85%
phosphoric
acid. After a total of 6 hours of reffux, TLC(silica gel plaxes developed in
5% methanol-
chloroform) showed that the starting material had been largely converted to a
lower-Rf
product. The solid was filtered and washed with water (5 mL), methanol (5 mL),
and
dried to give the title compound as a white solid (0.900 g, 68%), m.p.
>250°C dec.; mass
spectrum (CI/CH4): 209, 207 (M+I); 1H-NMR (DMSO-d6)8: 9.81 and 9.46 (s and d,
J =
11 Hz, total 1, NH), 8.25 and 8.00 (s and d, J = 11 Hz, total 1, CHO), 7.69
and 7.63 (both
s, total 2, NH2).
Anal. Calcd for CSH4N40C12: C, 29.01; H, 1.95; N, 27.07; Cl, 34.25. Found: C,
29.12;
H, 1.96; N, 27.13; Cl, 34.34.
WO95121161 ~ , : ~". ' a ,' 21821 O5
_; PCT/GB95l00225
- 18 - _.
In another experiment, a slurry of 2-amino-4,6-dichloro-S-{[(dimethyl-
amino)methylene]amino}-pyrimidine (Example 2, 25.0 g, 0.107 mol) in 1.5 M
aqueous
potassium phosphate buffer (300 mL, prepared as above) was gently refluxed for
4 hours.
pH was maintained at 3.2 by addition of 85% phosphoric acid, as required,
throughout
this period. The precipitate was filtered, washed with water (3 x 10 mL),
methanol (2 x
mL), and dried (50°C, 25 mm Hg) to give the title compound as an off
white powder
(16.0 g, 72%) with purity identical to that of the characterized sample
described above
(elemental analysis and 1H-NMR).
EXamDle 4
2.5-Diamino-4.6-dichloropyrimidine fIVI
4,6-Dichloro-2,5-bis-{[(dimethylamino)methylene]amino}pyrimidine (Example l,
1.00 g,
3.36 mmol) in ethanol (25 mL) and pH 3.2 aqueous potassium phosphate buffer
(1.5 M,
10 mL, prepared as described in Example 3) was refluxed for 24 hours. During
reflux, the
pH was maintained at ca. 3 by addition of 85% phosphoric acid, as required.
The ethanol
was evaporated in vacuo and waer added (10 mL). This solution was extracted
with
chloroform (3 x 25 mL). The comnined chloroform layers were dried (sodium
sulfate) and
chloroform evaporated to leave a solid (0.40 g). Crystallization of this solid
from ethanol-
water/ 4:1 gave the title compound (I~ as off white needles (0.324 g, 52%);
darkens and
shrinks to black solid above 185°C, does not become fluid below
300°C ; .[Lit. 198°C
(Legraverend et al., Synthesis 1990:587-589) and 188-191°C (Temple et
al., J. Org.
Chem. 1975, 40:3141-3142)]; mass spectrum (CI/CH4): 181, 179 (M+1); 1H-NMR
(DMSO-d6)8: 6.50 (br s, 2, NH2), 4.73 (br s, 2, NH2).
Anal. Calcd. for C4H4N4C12Ø12 EtOH: C, 27.60; H, 2.58; N, 30.36; Cl, 38.42.
Found:
C, 27.99; H, 2.39; N, 30.42; Cl, 38.74.
WO 95/21161 ' ~ ' ' ~ 18 210 5 p~'/GB95/00225
-- - 19 -
Example 5
2.5-Dia.mino-4.6-dichloropvrimidine (IVl
A mixture of 2-amino-4,6-dichloro-5-[(dimethylamino)methyleneJamino}pyrimidine
(Example 2, 500 mg, 2.14 mmol), pH 3.2 aqueous potassium phosphate buffer (1.5
M, 6
mL, prepared as described in Example 3), water (1 mL), and ethanol (5 mL) was
refluxed
gently for 28 hours. During the reflux period, pH was maintained at ca. 3 by
addition of
85% phosphoric acid. Volatiles were evaporated in vacuo and the residual
solids
partitioned between water (30 mL, adjusted to ph 8 with dilute ammonium
hydroxide) and
chloroform (75 mL). The chloroform layer was dried (sodium sulfate) and the
chloroform
evaporated to leave off white solid (0.30 g). Crystallization of this solid
from
ethanol:water/ 4:1 gave the title compound (IV) as light pink needles (332 mg,
61%);
darkens and shrinks to black solid above 185°C, does not become fluid
below 300°C;
1H-NMR (DMSO-d6) and mass spectra, identical to those described in Example 4.
Anal. Calcd. for C4H4N4C12: C, 26.83; I~ 2.25; N, 31.30; Cl, 39.61. Found: C,
26.93;
H, 2.25; N, 31.24; Cl, 39.52.
Example 6
2.5-Diamino-4.6-dichloropnimidine (T~
N-(2-Amino-4,6-dichloro-5-pyrimidinyl)formamide (Example 3, 500 mg, 2.42 mmol)
was
dissolved in 0.1 N hydrochioric acid (5 mL, 2.5 mequiv) and ethanol (7 mL) at
reflux.
The solution was refluxed for 5 hours. Volatiles were removed in vacuo. The
residue was
partitioned between water (30 mL) adjusted to pH 8 with dilute ammonium
hydroxide and
ethyl acetate (75 mL). The ethyl acetate layer was dried (sodium sulfate).
Evaporation of
the ethyl acetate left pink solid (0.40 g). Recrystallization of the solid
from 95% ethanol
gave the title compound (IV) as light pink needles (280 mg, 65%); darkens and
shrinks to
black solid above 185°C, does not become fluid below 300°C ; 1H-
NMR (DMSO-d6)
and mass spectra identical to those described in Example 4.
-A
WO 95/21161 . ' ' ~ ~ ~ PCTIGB95/00225
-20- ~-
Anal. Calcd. for C4H4N4C12: C.26.83; H.2.25; N.31.30; C1.39.61. Found C.26.95;
H.2.24; N. 31.19; Cl. 39.53.
Example 7
( 1 S.4R)-4-f (2-Amino-6-chloro-5-formamido-4-pvrimidinyl)amino] 2
cvclopentene 1
methanol (VIa~
N-(2-Amino-4,6-dichloro-5-pyiimidinyl)formamide (Example 3, 2.07 g, 10.0 mmol)
was
stirred in refluxing absolute ethanol (40 mL) under nitrogen to achieve
partial dissolution.
To this stirred mixture was added a solution of freshly prepared (1S,4R)-4-
amino-2-
cyclopentene-1-methanol (PCT Application 9204015.3, 1.57 g, 12.5 mmol as 90%)
in
ethanol ( 15 mL) followed by triethylamine (3 .5 mL, 25 mmol, freshly
distilled from
calcium hydride). After 14 hours of reflux, the resulting dark solution was
cooled and 1 N
sodium hydroxide ( 10 mL) was added. The volatiles were evaporated in vacuo.
The
residual tan solid foam was dissolved in 5% methanol-ethyl acectate, and the
solution was
washed through a silica gel pad to give the title compound as an off white
solid (2.50 g,
88%), after evaporation of solvents. Recrystallization of the solid from ethyl
acetate-
methanol (20:1) gave the title compound (VIa) as fine white crystals (2.29 g,
81%), m.p.
177-178°C; mass spectrum (CI/CH4): 286, 284 (M+1), 190, 188 (B+I~; 1H-
NMR
(DMSO-d6)8: 8.99 and 8.58 (s and d, J = 11.1 Hz, total 1, amide NH), 8.11 and
7.80 (s
and d, J = 11.1 Hz, total 1, amide CH), 6.77 and 6.61 (two d, J = 8.0 Hz)
overlapping
6.60 and 6.48 (two br s, total 3, NH and NH2), 5.85 and 5.70 (two m, 1 each,
CH=CH),
5.15-5.00 (m, 1, NCH), 4.71 (t, J = 5.1, 1, OH), 3.45-3.30 (m overlapping H20,
OCH2),
2.80-2.65 (m, 1, CH), 2.45-2.25 and 1.45-1.30 (both m, 1 each, CH2); [a]20 Sg9
+21.2°,
[a]20 578 + 22.2°, [a]20 546 + 25.2°, [ac]20 436 + 41.4°,
[oc]20 365 + 48.3° (c 0.50,
methanol).
Anal. Calcd. for C11H14N502C1: C, 46.57; H, 4.97; N, 24.69; Cl, 12.50. Found:
C,
46.63; H, 4.99; N, 24.58; Cl, 12.59.
.. . , ,,. Z' O 5 PCT/GB95100225
WO 95J21161
-21-
Example 8
(1 S.4R)-4.-(2-Amino-6-chloro-9-H-purin-9 y~-2-cvclopentene 1 methanol
Hydrochloride (V>Za)
A mixture of ( 1 S,4R)-4-[(2-amino-6-chloro-5-forrnamido-4-pyrimidinyl)amino]-
2-
cyclopentene-1-methanol (Example 7, 1.00 g, 3.50 mmol) and
triethylorthoformate
(Aldrich, Sure Seal, 18 mL) was stirred while concentrated hydrochloric acid
(37%, 1.25
mL) was added in one portion. The resulting clear, colorless solution was
stirred under
nitrogen. A white precipitate began to form after 15 minutes. After 4 hours,
TLC of a
drop of the reaction mixture dissolved in methanol and neutralized with sodium
hydroxide
(silica gel plates developed in 10% methanol-chloroform, visualized in tTV
light) showed
almost complete conversion of VIa to a higher-Rf material. The precipitate was
filtered,
washed with t-butyl methyl ether (15 mL) and dried at 0.2 mm Hg/ 25°C
for 18 hours to
give the title compound as a white powder (975 mg, 92%), m.p. >300°C
dec.; mass
spectrum (CI/CH4): 266(M+1); 1H-NMR (DMSO-d6)8: 8.18 (s, 1, purine CH), 7.2-
6.7
(br s, NH2, OH overlapped by water), 6.20 and 5.90 (both m, 1 each, CH=CH),
5.48 (m,
1, NCH), 3.47 (d, J = 5.7 Hz, 2, CH20), 2.90 (m, 1, CH), 2.75-2.55 and 1.75-
1.60 (both
m, 1 each, CH2).
Anal. Calcd. for C11HI2NSOCl.HCI: C, 43.73; H, 4.34; N, 23.18; Cl, 23.48.
Found: C,
43.62; H, 4.34; N, 23.07; Cl, 23.53.
Example 9
(IS.4R)-4-f2-Amino-6-fcvcloprop~o)-9H-purin-9 yIl 2 ~clopentene I methanol
A solution of (1S,4R)-4-chloro-5-formamido-6-{[(4-hydroxymethyl)-2-cyclopenten-
1-
ylJamino}pyrimidine (Example 8, 250 mg, 0.883 mmole) was refluxed gently (in
an oil
bath maintained at 130°C) in n-butanol (dried over 4 A molecular
sieves, 5 mL) under
nitrogen with cyclopropylamine (Aldrich, 0.30 mL, 4.4 mmol) for 16 hours. A
second
portion of cyclopropylamine (0.15 mL) was added and reflex continued for an
additional 5
hours. The volatiles were removed and the residual oil redissolved in ethanol-
water ( 1:1 )
218 ~ 10 5 PCT/GB95/00225
WO 95/21161
-22-
with 1 N sodium hydroxide (0.5 mL). Volatiles were again removed and the
residue
chromatographed on a silica gel flash column (1x10"). (1S, 4R)-[(2,5-Diamino-6-
chloro-
4-pyrimidinyl)amino]-2-cyclopentene-1-methanol (VI~a, 35 mg, 16%) eluted with
5%
methanol-ethyl acetate. Continued elution with 10% methanol-ethyl acetate gave
( 1 S,
4R)-4-[2-amino-6-(cyclopropylamino)-9H-pmin-9-yl]-2-cyclopentene-1-
methanol(IXa) as
a light tan solid foam (160 mg, 60%); H-NMR (DMSO-d6)8: 7.58 (s, 1, purine
CH),
7.25 (d, J = 4.5 Hz, 1, NH), 6.10 (m, 1, =CH), 5.80-5.75 (m, 3, =CH and NH2),
5.40 (m,
1, NCH), 4.72 (m, l, OH), 3.45 (m, 2, OCH2), 3.0 ( br m, 1, CH of
cyclopropyl), 2.80 (br
m, 1, CH), 2.70-2.50 (m overlapping solvent, CH), 1.50-1.05 (m,l, CH), 0.70-
0.50 (m, 4,
2 CH2 of cyclopropyl).
Anal. Calcd. for C14H18N60Ø20 H20Ø40 CH30H: C, 57.32; H, 6.35; N, 27.85.
Found: C, 57.59; H, 6.48; N, 27.70.
Example 10
11S.4R1-4-f2-Amino-6-( cloprop~)-9H-nurin-9-~l-2-~clopentene 1 methanol
(1S,4R)-4-(2-Amino-6-chloro-9-H-purin-9-yl)-2-cyclopentene-1-methanol (US
Patent
5,206,435) or the hydrochloride salt thereof (Example 8) was reffuxed in
ethanol with 10
molar equivalents of cyclopropylamine for 2 hours. The resulting solution was
cooled to
ambient temperature and 1 N sodium hydroxide (1 or 2 molar equivalerns,
depending on
whether the starting material was VIIa or the hydrochloride salt of VIIa) was
added. The
volatiles were evaporated in vacuo. (1S,4R)-4-[2-Amino-6-(cyclopropylamino)-9H-
purin-
9-yl]-2-cyclopentene -1-methanol (IXa) was washed from a silica gel pad eluted
with 5%
methanol-chloroform or 10% methanol-ethyl acetate and isolated as a white
solid foam
(80%); spectra identical to those of the product of Example 9.
WO 95/21161 3 ~ ~. ' ~~ 218 210 5 pCT~GB95100225
- 23 -
Example 11
(1'S.3'S.4'S)-2-Amino-1 9-dihydro-9(3 4-dihydroxy-3-hvdroxymethvl 1
cvclopentyl) 6H
purin-6-one
a) (4Rl-4-f (2-Amino-6-chloro-5-formamido-4-p '~~d'lnyl)aminol 1 cyclopentene
1
methanol
By the method of Example 7, N-(2-Amino-4,6-dichloro-5-pyrimidinyl)forrnamide
(Example 3, 2.56g, 52.4mmo1) was reacted with (4R)-4-amino-1-cyclopentene-1-
methanol ( 1.4g, 52.4mmo1), available from (-)-2-azabicyclo[2.2. I ]hept-5-en-
3-one
(Chiroscience) by methods described in Examples 1-4 and 42 of U.S. Patent
5,049,671. Crystallization from ethyl acetate - methanol gave title compound
as
white crystals, m.p. 148-150oC; mass spectrum (CI/CH4): 286, 284 (M+1), 190,
188 (B+I~; IH-NMR (DMSO-d6)S: 8.97 and 8.55 (s and d with J = 11.3 Hz, total
1, NHCHO), 8.12 and 7.80 (s and d with J = 11.5 Hz, total 1, CHO), 7.00 and
7.78 (both d, J = 7.4 Hz, total 1, NH), 6.60 and 6.40 (both 8, total 2, NH2),
5.48
(s, 1, = CH), 4.74 (t, J = 5.5 Hz, 1, OH), 4.74-4.60 (m, 1, NCH), 4.0-3.90 (m,
2,
CH20), 2.75-2.55 and 2.40-2.15 (both m, 2 each, 2CH~; [a]58920-4.40,
[a]57820-5.20, [a]54620-4.go, [a]43620-20.Oo, [a]36520-60.40 (c 0.25,
methanol).
Anal. Calcd. for CI1H14N502C1: C, 46.57; H, 4.97; N, 24.69; Cl, 12.50. Found:
C, 46.64; H, 5.01; N, 24.60; Cl, 12.45.
b) (4Rl-4-(2-Amino-6-chloro-9H-purin-9-yl -~cyclonentene 1 methanol
A mixture of (4R)-4-[(2-amino-6-chloro-5-foimamido-4-pyrimidinyl)amino]-1-
cyclopentene-1-methanol (Part a, 1.60g, 5.65mmo1) and triethylorthoformate
(29mL) was stirred while concentrated hydrochloric acid (37%, 2.OmL) was added
in one portion. The resulting clear, colourless solution was stirred under
nitrogen.
After 5 hours the resulting precipitate was filtered and washed with t-butyl
methyl
ether (3 x lOmLO and dried to provide white powder (1.25g). This powder was
PCT/GB95/00225
W0 95/21161
-24-
dissolved in water and the pH adjusted to 3 by addition of 1N hydrochloric
acid.
The solution was heated at 60oC for 4 hours, cooled, neutralized, and
evaporated
to a solid which was chromatographed on silica gel. Title compound was eluted
with S% methanol chloroform and crystallized from ethanol-ethyl acetate to
white
crystals, m.p. 145-147oC; mass spectrum (Cl/CH4): 268, 266 (M+1), 172, 170
(B+I~; 1H-NMR (DMSO-d6)8: 8.09 (s, 1, purine CH), 6.9 (br s, 2, NH2), 5.64
(m, 1, = CH), 5.2-5.0 (m, 1, NCH), 4.87 (t, J = 5.5 Hz, 1, OH), 4.05 (m, 2,
CH20), 3.0-2.5 (m, 4, 2 CH2).
Anal. Calcd. for C11H12N50C1: C, 49.06; H, 4.64; N, 26.01; Cl, 13.16. Found:
C, 49.18; H, 4.63; N, 26.11; Cl, 13.19.
c) (1S.2S.4R1-4-(2-Amino-6-chloro-9H-purin-9-vl)-2-fhydroJCymethyh' 1 2
cyclo_pentanediol
(4R)-4-(2-Amino-6-chloro-9H-purin-9-yl)-1-cyclopentene-1-methanol (Part b,
501mg, 1.89mmol), N-methyhnorpholine N-oxide (60% aqueous solution, Aldrich,
0.33mL, 1.89mmol), osmium tetroxide (2.5% in t-butyl alcohol, Aldrich,
0.47mL),
and t-butyl alcohol (l2mL) were heated at 60oC for 1.5 hours. Volatiles were
evaporated and the residual solids were chromatographed on silica gel. Title
compound was eluted with 10% methanol-chloroform as tan solid (210mg) and
resolidified from absolute ethanol to give white powder, m.p. 217-219oC; mass
spectrum (Cl/CH4): 302, 300 (M+1), 172, 170 (B+H); 1H-NMR (DMSO-d6)8:
8.29 (s, 1, purine CH), 6.9 (br s, 2, NHS, 5.15-4.90 (m, 1, NCH), 4:80 (d, J =
3.9
Hz) overlapping 4.78 (t, J = 3.5 Hz, total 2, 2 OH), 4.30 (s) overlapping 4.3-
4.2
(m, total 2, OH and OCH), 3.45-3.35 (m, overlapping water, CH20H), 2.25-2.05
(m, 4, 2 CHI.
Anal. Calcd. for C11H14N503C1: C, 44.08; H, 4.71; N, 23.37; Cl, 11.83. Found:
C, 43.89; H, 4.80; N, 23.16; Cl, 11.73.
WO 95/21161 PCT/GB95I00225
2182105
- 25 -
d) (1'S.3'S.4'Sl-2-Amino-1.9-dihvdro-9-(3 4-dihydroxy-3-hvdroxymethyl-1-
cyclopentyl)-6H~urin-6-one
(1 S,2S,4R)-4-(2-Amino-6-chloro-9H-purin-9-yl)-2-(hydroxymethyl)-1,2-cyclopen-
tanediol (Part c, 90mg, 0.27mmole) was refluxed in 1N hydrochloric acid
(2.7mL)
for 45 minutes. Volatiles were evaporated in vacuo. Portions of water were
evaporated and the residue was redissolved in water. The pH was adjusted to 5
with hydrochloric acid and the resulting mixture cooled, filtered, and the
precipitate dried to provide the title compound as an off white powder (5lmg,
68%), m.p. >300odec.; mass spectrum (CI/CH4): 283 (M+1); 1H-NMR(DMSO-
d6) identical with that described in U.S. Patent 5,233,041.
Example 12
N-(2.4-Diamino-6-chloro-5-pvrimidinyllformanvde
N-(2-Amino-4,6-dichloro-5-pyrimidinyl)formamide (Example 3, SOOmg, 2.14mmo1)
and
ammonia (150mL) was stirred in a Purr bomb at SOoC for 18 hours. The ammonia
was
evaporated and the residual solid triturated with water (lOmL). The solid was
dried to
give the title compound as red powder (400mg, 89%), m.p.>300oC; mass spectrum
(CI/CH4): 190, 188 (M+1); 1H-NMR(DMSO-d6)8: 9.05 and 8.60 (both br s, total 1,
NHCH_O), 8.1 and 7.8 (both br s, total 1, NH_CHO), 6.80-6.20 (4 br s, total 4,
2 NH2).
Anal. Calcd. for CSH6NSOC1: C, 32.01; H, 3.22; N, 37.34; Cl, 18.90. Found: C,
31.97;
H, 3.23; N, 37.26; Cl, 19.00.