Note: Descriptions are shown in the official language in which they were submitted.
2 1 86 979
-2-
METHODS OF TESTING ANTAGONISTS FOR
l H~;l~ ABILITIES TO AFFECT THE ACTIVl~Y OF
G PROTEIN-COUPLED RECEPTORS
FIELD OF THE INVENTION
The present invention relates to methods of screening test substances for their abilities to
interact with and modulate the functional properties of G protein-coupled receptors. More
specifically, the present invention describes methods of testing and ranking substances for
their abilities to increase spontaneous G protein-coupled receptor activity and to sensitize G
protein-coupled receptors to agonists.
BACKGROUND OF THE INVENTION
G protein-coupled receptors (GPCRs) are proteins located in the plasma membrane of cells.
They function as one part of a multi-component complex involved in signal transmission.
GPCRs share a common ~ n~lin~ mech~ni~m, whereby signal transduction across the
membrane involves intracell~ r tr~n~d~1cer elements known as G proteins (named for their
ability to bind and hydrolyze the nucleotide GTP). When a chemical messenger binds to a
specific site on the extr~c~lhll~r surface of the receptor, the conformation of the receptor
changes so that it can interact with and activate an intr~cell~ r G protein. This causes
~l~nos;r,~ diphosphate (GDP), which is bound to the surface of the G protein, to be replaced
by guanosine triphosphate (GTP), triggering another conformational change in the G protein.
Once GTP is bound to its surface, the G protein re~ tes the activity of an effector. These
effectors include enzymes such as adenylyl cyclase and phospholipase C, ion channels that are
specific for calcium ions (Ca2+), potassium ions (K+), or sodium ions (Na+), and certain
transport proteins.
In general, activation of GPCRs by l,an~",;llers will induce one of the following effector
responses: activation of adenylyl cyclase, inhibition of adenylyl cyclase, or stimlll~tion of
phospholipase C activity. When the effector adenylyl cyclase is either activated or inhibited,
it produces changes in the concentration of the molecule cyclic adenosine monophosphate
(cAMP). Another effector, phospholipase C, causes one molecule of phosphatidylinositol-
bisphosphate ~PIP2) to be cleaved into one molecule each of inositol triphosphate (IP3) and
3 0 diacylglycerol (DAG); IP3 then causes calcium ions (Ca2+) to be released into the cytoplasm.
cAMP and Ca2+ are termed second messengers. Alterations in cellular levels of second
messengers act to alter the behavior of other target proteins in the cell.
GPCRs activate a number of di~ele.-l types of ~i~n~lin~ pathways in cells. This activation
occurs at the level of the G proteins, which detect and direct signals from diverse receptors
3 5 to the app~ul~iale effector-response pathway. Although there are many forms of G proteins,
the three main forms are as follows: Gs-like, which mediate activation of adenylyl cyclase;
Gi-like, which mediate inhibition of adenylyl cyclase; and Gq-like, which mediate activation
2 1 86979
-
-3 -
of phosphoplipase C. Since one receptor can activate many G proteins, the signal can be
greatly amplified through this signal tr~n~duction pathway.
A wide variety of chemical mess~ng~rs, involved in re~ *n~ key functions in the body, act through GPCRs.
These include ne~ol~ e, ~ such as dopamine, acetylcholine, and serotonin, hormones of the endocrine
system such as som~tost~tin~ glucagon, and adrenocorticotropin, lipid mediators such as prostaglandins and
leukotrienes, and immunomodulatory proteins such as interleukin-8 and monocyte-chemoattractant
polypeptide. The family of GPCRs also includes the receptors for light (rhodopsin), for odors (olfactory
receptors) and for taste (gustatory receptors). Over one hundred di~rel enl GPCRs have been identified in
hllm~ni, and many more are expected to be discovered. Most of these receptors are believed to utilize one
ofthe three principal G protein-effector ~ign~ling pathways (stimlll~tion or inhibition of adenylyl cyclase (AC)
or activation of phospholipase C (PLC)). Exa~lp!~s of neuro~l~nsllliller GPCRs and their respective effector
~ign~ling pathways are shown in Table 1.
Table 1. Examples of Neurotransmitter GPCRs
Effector Signaling Pathway
Neurotransmitter StimulatesAC Inhibits AC Stimulates PLC
15Acetylcholine m2, m4 m1, m3, m5
Adenosine A1, A2
Corticoll opin-Rele~ cin~ CRF-R
Factor
Cannabinoids Rc
20Dopamine D1, D5 D2
~i~t~mine H2
Neuropeptide Y Y1, Y2, Y3
Norepinephrine, 2-AR 1-AR 2-AR
epinephrine
25 Opioids '~1
Serotonin 5-HT4 5-HT1A, 5-HTlB~ 5-HT2
5-HTlD
A number of compounds can bind to GPCRs. These compounds are called ligands. Analysis
of the effects of ligands on the ability of GPCRs to activate effector ~ign~ling pathways
suggests that the receptors exist in two di~lelll conrollnalions: an inactive, silent
col~ollllation and an active col~llll~lion that triggers G protein activation and effector
sign~ling (Gilman, A.G., 1987, Annu. Rev, Biochem. 56: 615-649; Levitski, A., 1988,
Science 241 :800-806). An increasing amount of evidence suggests that GPCRs isomerize
rapidly between the active and inactive forms.
21 86979
-
-4 -
Ligands that bind to the ~ ",;l Içr recognition site on the receptor may be classified into two
types: 1) agonists, which mimic the action of n~tural transmitters and activate GPCRs; and
2) antagonists, which bind to the receptors but do not activate the CPR. Agonists cause the
receptor to assume the active conformation. The amplitude of activation of GPCRs by
agonists is termed efficacy and is an important parameter in ~sçssing the therapeutic potential
of a drug: a full agonist elicits a maximal response in a given tissue or system while a partial
agonist elicits only a fraction of the maximal response at full occupancy of the GPCR.
Antagonists prevent the activation of receptors. Until recently, it was generally believed that
~nt~t)ni~t.~ act by binding to the receptor and blocking agonist binding, without having any
effect on receptor activity. It has now become appare-ll that antagonists can also act by
turning off spontaneously-active GPCRs (GPCRs that are active even in the absence of
agonist) (Schutz and Frçii~ml~th (1992) Trends Pharmacol. Sci. 13:376-380.). These
negative regulators are called inverse agonists. Antagonists also exhibit efficacy: full
antagonists completely turn offreceptor activity while partial antagonists only partially inhibit
this activity. Thus, antagonists are now thought of as negative regulators, i.e. They turn
eceplol~ offin two ways: by blocking agonist action and by inhibiting spontaneous receptor
activity.
This evidence has led to a model in which active and inactive receptors co-exist in the cell in
equilibrium, with agonists pushing the equilibrium to the active form, inverse agonists pushing
it to the inactive form, and neutral antagonists blocking the agonist action without favoring
either collrollllaLion.
Effects of agonists and antagonists described above are acute effects, i. e. represent effects of
drug on receptor activity when the drug (agonist or antagonist) is bound to the receptor.
However, the effects of both agonists and antagonists on GPCR activity can change following
chronic exposure to these drugs.
Awell-characterized CA~IIPI~ of chronic effect is agonist-ind~lced receptor desen.~iti7~tion of
the ~2-adrenergic receptor: a decrease in the efficacy of an agonist is noted following an
extended exposure of receptor to agonist, removal of agonist followed by immediate
measuring of m~xim~l receptor response to the agonist. . The observed decrease in maximal
response upon second agonist challenge is referred to as desensitization and is believed to
occur through a co~ inalion of di~elenl mech~ni~m~, including removal of the receptor from
the cell surface (sequestration) and enzymatic phosphorylation of the activated receptor such
that it is unable to interact with G protein transducers. This phenomenon has a negative
impact on thel~p es, since the efficacy ofthe agonist drug decreases with time. As tolerance
to the drug increases, the dose of drug must be increased to m~int~in the therapeutic effect.
In some cases, chronic treatment with antagonist drugs has been reported to produce an
increase in the number of GPCRs (e.g. dopamine antagonists in animal models). In other
cases, however, chronic treatment with antagonists have been shown to promote decreases
2 1 86979
--5 -
in the number of GPCRs (e.g. 5HT2 receptors and serotonergic antagonists; see Meltzer and
Nash (l991)Pharmacol. Rev. 43:588-600.). In particular, it has been shown that antagonists
can decrease ligand binding capacity of the rat 5-hydroxytlyp~an~ille Type 2C (5HT2C)
receptor (Labrecque et al., (1995) Mol. Pharm. 48:150-159.). This decrease in GPCR
binding sites following antagonist treatment has been termed "atypical down-regulation". It
is thought that the down-regulation of GPCRs is a distinct action from inverse agonist
activity.
These effects of chronic antagonist treatment have not been associated previously with
changes in receptor response to subsequent agonist treatment. However, acute antagonist
effects have been described which suggest that antagonists may have effects on receptor
re~ons;~eness. A property termed "augmentative antagonism" has been described in tissue
studies, on a very few occasions whereby the ~imlllt~neous addition of an antagonist and an
agonist to the receptor causes an increase in the maximal response to the agonist. This
obseîvation has been interpreted as evidence that certain antagonists may increase the ability
of the receptor to activate G proteins in response to agonists.
In view of the diverse effects of GPCRs in the human body, it is not surprising that the
ph~rm~ceutical sector has great interest in the development of new drugs to target GPCRs.
These drugs have potential as therapeutic agents in a wide range of human pathologies,
inrl~lrling psychiatric disorders (depression, psychoses, bipolar disorder), metabolic disorders
(diabetes, obesity, anorexia nervosa), cancer, autoimmune disorders, cardiovascular disorders,
neurodegenerative disorders (e.g. Alzheimer's disease), and pain disorders.
There are no methods oftesting antagonists as drug candidates on the basis of their ability to
effect changes in activities of GPCRs. Specifically, while there is a limited body of evidence
suggesting that antagonists may promote an increase in receptor response to agonists in
certain tissue p~ ions, there exists no method to systematically rank antagonists for their
relative abilities to exert this effect. Moreover, there is no known method of comparing
antagonists for their relative abilities to increase spontaneous receptor activity, since this is
a property of antagonists which has not been previously described.
Thus, in determining whether particular antagonists are candidates for suitable drug
lre~ s) there is a need for methods of testing these substances for their ability to affect
the activities of GPCRs. It is therefore an object of the present invention to provide a method
of analyzing test substances for two manners of affecting receptor activities: 1) for their
abilities to increase the response of GPCRs to ~p;oni~t~, and 2) to increase spontaneous GPCR
activity.
This background information is provided for the purpose of making known information
believed by the applicant to be of possible relevance to the present invention. No admission
is necessarily intended, nor should be construed, that any of the preceding information
constitutes prior art against the present invention. Publications referred to throughout the
21 86979
,
-6-
specification are hereby incorporated by reference in their entireties in this application.
SUMMARY OF THE INVENTION
To meet the needs noted above, the present invention describes methods for testing and
comparing antagonists for their abilities to affect GPCR activities, that exploits two new
newly recognized abilities of antagonists: 1) to increase the response of GPCRs to agonists,
and 2) to increase spontaneous GPCR activity. These methods will have direct application
to the screening of antagonist drugs for potential therapeutic utility, and will be particularly
useful since the sçn~iti7ing effect of antagonist drugs is ligand selective.
These methods are applicable to any recombinant GPCRs that can be expressed in
heterologous host cells.
In one embodiment, the method ofthe present invention involves the following: 1) expressing
cloned cDNA encoding a GPCR in a heterologous host cell; 2) treating these cells with
di~e~ concentrations of test substance; 3) washing the cells to remove the test substance;
and 4) measuring the ability of the receptors to activate cellular response pathways in the
absence of added agonist (spontaneous receptor activity).
In another embodiment, the method of the present invention involves the following: 1)
les~ing cloned cDNA encoding a GPCR in a heterologous host cell; 2) treating these cells
with di~erenl concentrations of test substance; 3) washing the cells to remove the test
s~ ce, and 4) me~ in~ the ability ofthe receptors to activate cellular response pathways
in the presence of increasing concentrations of agonist drug (agonist response).
In yet another embodiment, this invention in~tolves a test kit including whole cells which can
express cloned GPCRs, appropliate reagents, and supporting documentation enabling the
working of this invention.
Various other objects and advantages ofthe present invention will become appar~ from the
detailed description of the invention.
BRIEF DESCR~PTION OF THE DRAWINGS
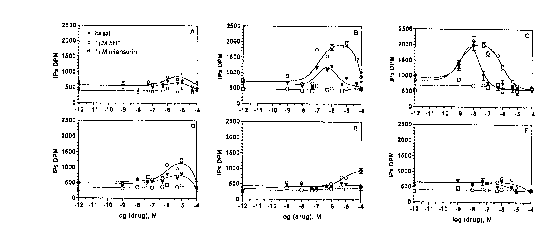
Figure 1. Effects of serotonergic antagonist pr~lea~lllent on IP release in SHT2c-expressing
Sf9 cells. Sf9 cells labeled with [3H]myo-inositol (1 IlCi/ml) were infected 48 hours with a
baculovirus encoding the SHT2C I eceplor and treated for 60 min. with various concentrations
of methysergide (A), mianserin (B), metergoline (C), clozapine (D), loxapine (E), and
chlorpromazine (F) as indicated on the abscissa. Treated and control cells were washed
extensively in parallel, then inc~1b~ted in the presence of 1 ',IM SHT (white circle), 1 IlM
mi~n~.rin (white square), or vehicle (black triangle). IP levels were measured in whole cells
in this representative experiment where the point shown represents averaged untransformed
21 86979
data from a triplicate assay. The half m~im~l estimates of the increase in receptor function
from 3 independent drug ~ are as follows: metergoline 3.8 + 0.7 nM; mianserin 1.9
~ 0.7 ~lM; and clozapine 4.9 ~ 1.7 ~M.
Figure 2. 5HT and mianserin dose response curves after di~lenl concentrations ofmetergoline p[~llealnlent on 5HT2C receptors expressed in Sf9 cells. These results
demonstrate effects of agonist on IP release in 5HT2c-e~pressillg Sf9 cells after various
pl~ll~llllents with metergoline. Sf9 cells labeled with [3H]myo-inositol were infected with
the 5HT2C baculovirus. At 48 hours after infection, cells were pre-treated for 60 min. with
either vehicle (control black square and dotted curve) or various concentrations of
metergoline (5M - white square; 6M - black circle; 7M - black lozenge; 8M - inverted black
triangle; 9M - upright black triangle) before the washout protocol was performed. IP levels
were then measured in pre-treated whole cells in the presence of various concentrations of
5HT (panel A) or mianserin in panel B as indicated on the abscissa. Data are from a
representative experiment of triplicate assays; averaged results from three independent
experiments (ECso) are reported in Table 2.
Figure 3 . 5HT2C receptors expressed in Sf9 cells. Receptor activity after di~el elll antagonist
pretreatments on Sf9 cells expressing the rat 5HT2C receptor demonstrating effects of
serotonergic antagonists on apparenl loss of [3H]mesulergine binding sites in 5HT2C-
cont~inin~ Sf9 membranes following antagonist plelleallllent. Cells were prepared at 48
hours post-infection from Sf~ cells infected with the 5HT2C baculovirus, then treated with the
indicated 5HT2C antagonists for 1 hour. Treated and control membranes were washed
extensively in parallel, then in~ b~ted with [3Hlmesulergine (10 nM) to determine the residual
binding after drug ll~l "~."I For each ligand, data from three independent experiments were
analyzed ~imlllt~neously ~qs lming a common value for EC50 (see Table 3), with Y(x-o) and
Y(x-)) unconstrained. The curves and averaged points (n=3) shown were derived from a
representative experiment obtained on the same day with all drugs tested in parallel on the
same membranes. Values from a representative set of data were scaled taking the fitted
values of Y(x-o) as 100%, and the points shown represent (the averages of) the scaled data
(+ SEM). Each curve was genel~led using the fitted values of EC50, with Y(x~o) set to 100%
and Y(x-)) set to the average of [((Y(X_)) /Y(~ 0)))*100%]. For clarity, points corresponding
to conce~ ion < (EC5J10000) were omitted from the figure. Values for EC50 are reported
in Table 2.
<IMG>
2 1 86979
DETAILED DESCRIPTION OF THE INVENTION
The following terms and abbreviations are used throughout the specification and in the claims:
A "chemical-mPq~P.ngP.r" is defined as any messenger, in the absolute broadest sense, natural
or unnatural, that induces an effect or blocks an effect on a proteinaceous receptor, including
cl~eln~-~l s l,~ ce ~ n~ Prs (e.g. neuro~lan~ P,rs, hormones, and lipid mediators such
as prostagl~n(linc~ and leukotrienes; usually act on chemoreceptors), light (e.g. sign~ling via
a rhodopsin receptor; usually acts on electromagnetic (photo) receptors), and stimuli such as
temperature and mechanical signals (e.g. physical or chemical damage, vibration, touch,
pressure, movement) which act on mechanoreceptors, thermoreceptors, and nociceptors.
An "effector protein" is defined as any protein that is activated or inactivated by a G protein.
Some examples include adenylyl cyclase and phospholipase C.
A "G protein" is defined as any member of the family of signal tr~n.cd~lc.ing guanine nucleotide
binding proteins.
A "G protein-coupled receptor" is defined to be any cell surface transmembrane protein that,
when activated by a chemical, mP~i~tes signal transduction by coupling with a heterotrimeric
guanine nucleotide-binding protein (G protein).
A "ligand" is intPn-led to include any substance that interacts with a receptor. It may
sinn~ P~ inhibit, or cause some effect for the activity of the receptor. An "agonist" is defined
as a ligand increasing the functional activity of a receptor (IE. Signal transduction through the
receptor). A "neutral antagonist" is defined as a ligand that can bind to the transmitter
recognition site on the receptor and thereby block receptor activation by agonists. An
"inverse agonist" is defined as a ligand that can decrease the spontaneous activity of the
receptor.
A "receptor" is intended to include any molecule present inside or on the surface of a cell,
which can affect cellular physiology when either stiml11~ted or inhibited by a ligand.
A"second messenger" is defined as an intermediate compound whose concentration, either
intercellularly or within the surrounding cell Ill~llll~l~le, is raised or lowered as a consequence
of the activity of an effector protein. Some examples of second messengers include cyclic
adenosine monophosphate (cAMP), phophatidylinositol (PI), calcium ions (Ca+2), and
arachidonic acid derivatives.
A "test substance" is intçn-led to include any drug, compound, or molecule with potential
biological activity.
2 ~ 86979
-10-
The present invention relates to methods of testing substances for their abilities to increase
the response of GPCRs to agonists and to increase spontaneous GPCR activity.
These methods are based on the discovery that certain antagonists can dirrerenlially increase
the efficacy of agonists or the spontaneous activity of GPCRs. These sensitizing effects
represent novel activities of antagonists which may involve improving receptor coupling
efflciency and reducing receptor density.
Antagonist induced sçn.~iti7Ation of GPCRs is likely to be of significance in the treatment of
disease since it could be used to revert a lack of agonist responsiveness to a normal
responsive level. It could also be used if poor coupling of agonist to the GPCR is implicated
in the etiology of disease. Specific therapeutic applications of drugs that re-sensitize GPCRs
could be important novel paradigms for drug development. These alternative methods of
antagonist action should be considered when exploring therapeutic processes involving
antagonist drugs.
Creation of Recombinant Vector and Infection of Cell Lines:
The p~ aly step ofthe assay method ofthe present invention is the expression of GPCRs
in heterologous host cells. For clarity, the 5HT2C receptor is used as an example; however,
the descriptions apply equally well to any GPCR whose cDNA has been cloned, such as
lLylùllupin, lutropin-choriogonadotropin, dopamine, and l~ e receptors. Likewise, once
cDNAs become available for other GPCRs, they can also be used in the present invention as
described below. This method is applicable to any GPCR once a cDNA clone for that
receptor has been generated.
The cDNA coding for functional GPCR can be inserted into any suitable vector, cloned, and
expressed in an appropliate cell line. An appropliate cell line is one that will be able to
express and process the receptor, in addition to possessing the necessary biochemical
mA-~hin~ry to respond to signal tr~n~duction through a given receptor. Transfection may be
pt;lrolllled according to known methods. In general, a cDNA sequence encoding a receptor
may conveniently be subjected to recolllbinalll DNA procedures.
The vector may be an autonomously replicating vector, such as a plasmid, or it may be one
which, when introduced into a host cell, is integrated into the host cell genome and replicated
together with the chromosome(s) into which it has been integrated. Using procedures that
are well known to those skilled in the art, DNA expression vectors incorporating coding
regions for the leceplor will be suitable for replication in the applupliate host cell. Eukaryotic
viral vectors such as insect, and "~A""~AliAn viral vectors can be constructed using vectors
such as the Bacculovirus, the Sindbus virus, the Semliki Forest virus or the vaccinia virus.
Retroviral and adenoviral vectors can also be used. The DNA sequence encoding the
receptor should be operably connected to a suitable promoter sequence and a suitable
terminator sequence, and may further comprise a DNA sequence enabling the vector to
replicate in the host cell in question. The procedures used to ligate the DNA sequences
2 1 869~9
1 1
coding for the receptor, the promoter, and the terminator, and to insert them into suitable
vectors COIIL~ lg the il~llll~ion ll.o,cess~.y for replication are well known to persons skilled
inthe art (Sambrooketal., (1989)Molecular Cloning: A LaboratoryManual, Cold Spring
Harbor Laboratory, Cold Spring Harbor, New York.).
Cells that may be used in the present method are cells that are able to express the cloned
receptor at a sufficiently high level, in addition to possessing the ability to activate signal
transduction in cell signal pathways. Such cells are typically eukaryotic cells, such as
m~mm~ n, or insect cells. Methods oftransfecting "~"""~ n cells and expressing DNA
sequences introduced in the cells are well known to persons skilled in the art (~ fm~n and
Sharp (1982) J. Mol. Biol. 159:601-621; Southern and Berg (1982) J. Mol. Appl. Genet.
1:327-341; Loyter et al., (1982) Proc. Natl. Aca~ Sci. USA 79:422-426; Wigler et al.,
(1978) Cell 14:725; Corsaro and Pearson (1981) Somatic Cell Genetics 7:603; Graham and
van der Eb 1973) Virology 52:456; Neumann et al., (1982) EMBO J. 1 :841-845; and Wigler
etal., (1977) Cell 11:223-232.).
One such vector and .,~res~;on system that works particularly well with this invention entails
constructing a recollll)inanl baculovirus expression vector, capable of expressing a GPCR in
a host insect cell line (e.g. Sf9 cells). Examples of how to construct suitable recombinant
baculovirus vectors are described in U.S. Patents No. 4,745,051 and 4,879,236. The general
procedures of reconlbillalll DNA technology pertaining to creation and manipulation of the
baculovirus system are well known to those skilled in the art (O'Reilly et al., (1992)
Baculovirus Expression Vectors, a Laboratory Manual (New York: W.H. Freeman and
Company); Davies (1994) Bio/Technology 12:47-50.).
One embodiment of this invention describes and demonstrates the expression of GPCRs in
a baculovirus-insect cell system. The Autographa californica nuclear polyhedrosis
baculovirus (AcNPV) has been shown to be suitable as a viral expression vector for the
efficient production in cultured insect Sf9 cells of m~mm~ n Ill~lllbl~ne proteins from foreign
genes (Luckow and Summers (1988) Bio/Technology 6:47-55; Miller (1988) Annu Rev.Microbiol. 42: 177-199.). A number of GPCRs have been expressed in the baculovirus-Sf9
cell expression system, and were found to m~int~in binding properties characteristic of the
"natural" receptors in tissues or expressed from cloned cDNAs in m~mm~ n cell lines
(Mouillac et al., (1992) J. Biol. Chem. 267:21733-21737; Parker (1991) . Biol. Chem.
266:519-527; Wong (1990) J. Biol. Chem. 265 :6219-6224.).
One particular embodiment of the invention involves 5HT2C receptors that are expressed
within a baculovirus ~A~les~ion system in Sf9 cells. It has been shown that the infection of
Sf9 cells with recombillall~ baculovirus encoding the 5HT2C receptor leads to the expression
of functional receptors capable of reg~ tin~ intracellular levels of IPs (Labrecque et al.,
(1995) supra.). The levels of 5HT2C receptor expression in the Sf9/baculovirus system are
2 1 86979
-12-
roughly 20-fold higher than those of the native receptor in rat choroid plexus and >5-fold
higher than the levels reported for NIH13T3 cells expressing the recombinant receptor. As
observed with m~mm~ n systems expressing either native or recombinant 5HT2C receptors,
the baculovirus-expressed 5HT2C receptor activates polyphosphoinositide hydrolysis in
response to serotonergic agonists via a pertussis toxin-in.cçn~itive pathway. The expressed
receptor exhibits spoll~ eo~ls activation of inositol phosphate production, which is inhibited
in a dose-dependent manner by serotonergic antagonists, consistent with inverse agonist
activity.
Screening of Test Substances:
Once a functional GPCR expression system is obtained, test substances can be screened to
determine whether they increase spontaneous GPCR activity or increase GPCR response to
agonists. This involves treating the recombinant cells with di~e. elll concentrations of test
substance, then washing the cells to remove the lest substance. The ability of the GPCRs to
activate cellular response pathways in the absence of added agonist (spontaneous receptor
activity) or in the presence of increasing concentrations of agonist drug (agonist response)
is then measured. Activation of cellular response pathways can be measured by any suitable
q~l~ntifi~ble parameter using biochemical or other assay procedures that indicate the activity
ofthe cloned receptor in the e?~ples~ion system. Many app-opliate biochemical and other
assay procedures using ~ d~d le~ les are well known to persons skilled in the art. One
example of such an assay involves detelll.il~ing the activity of effectors such as adenylyl
cyclase or phospholipase C. The activity of the effector is determined by measuring levels of
second mess~nger such as cAMP (for example, Gilman (1970) Proc. Natl. Acad. Sci. ~JSA
67:305-312.) or inositol phosphate ~Fargin etal., (1989) J. Biol. Chem. 254: 14818-14852.).
In a pre~lled embodiment ofthis invention, Sf9 cells infected with the 5HT2c/baculovirus
vector are pre-treated with varying concentrations of di~erenl test substances. A~er
extensive ~l ing of residual test substance, assays are performed on whole cell preparations
to determine the effect of these substances on the spontaneous activity of GPCRs and on the
response of receptors to agonists. This is achieved by measuring inositol phosphate levels.
The present invention is described in further detail in the following non-limiting examples.
It is to be understood that the examples described below are not meant to limit the scope of
the present invention. It is expected that numerous variants will be obvious to the person
skilled in the art to which the present invention pertains, without any departure from the spirit
ofthe present invention. The appended claims, properly construed, form the only limitation
upon the scope of the present invention.
F,XAMPLE I
Reagents:
Buffer chemicals and protease inhibitors were purchased from Sigma, and cell culture media
from Gibco/BRL. Unlabeled ligands were supplied by Research Biochemicals International,
2 1 86979
-13-
with the exception of Loxapine, which was a gift from Dr. B. Roth. [3H]mesulergine (78-82
Ci/mM) was purchased from Amersham and [3H]myo-inositol (10-20 Ci/mM) was purchased
from NEN-Dupont. The AG 1 X 8 ion exchange resin was supplied by Bio-Rad.
Construction of Recombinant Baculovirus:
The reco"~ baculovirus used for expression of the rat 5HT2C receptor was provided by
the Biotechnology Research Institute of Montreal. The virus was constructed using a
synthetic DNA fragment encoding the rat 5HT2C receptor, based on the published sequence
ofthe cloned cDNA from choroid plexus (Julius et al., (1988) Science 241 :558-564.). The
synthetic cDNA was prepared and its sequence verified by Allelix Biopharm~ce~lticals
(l~ie.~ie.e~l~, ON). The cDNA was inserted into the IpDC-126 baculovirus transfer vector
and a leco,..binal-l baculovirus was produced and purified as previously described (O'Reilly
etal., (1992)BaculovirusEi~pression Vectors: ALaboratoryManual).
Cell Culture and Receptor Expression:
Sf9 cells were cultured in 50 ml batches in 250 ml shaker flasks at 27OC in Sf-900 II serum
free medium co~ ining 50 ,ug/ml gentamicin sulfate. Cells were grown to a density of 3 X
106 cells/ml and infected with the 5HT2C recombinant baculovirus or with wild-type
Autographia californica nuclear polyhedrosis baculovirus, at a multiplicity of infection of 2.
Viral stocks for infections were in Grace's insect medium COI~ g 5% fetal bovine serum
(Hyclone) and were added to cultures at a dilution of applox;..~ ely 1/20 upon infection. The
infected cells were " ,~ ed in culture for various periods and used for analysis of ligand
binding and measurement of IPS production as described below.
Radioligand Binding Assays:
For the ~,etim~tion oftotal ~ecep~or numbers in intact Sf9 cells, the cells were pelleted by low-
speed centrifugation (3 min. at 800 rpm in Sorvall H6000A rotor), followed by resuspension
in PBS and re-centrifugation, and resuspended in either PBS or binding buffer (50 nM
TrislHCI, pH 7.4, 15 mM MgC12, 2 mM EDTA, 0.1% ascorbic acid, 5 mg/ml leupeptine, 10
mg/ml aprol~in, 20 mg/ml benzamidine, 50 mg/ml TPCK, and 50 mg/ml trypsin inhibitor).
Cell viability after washing was estim~ted at 60-70% by trypsan blue exclusion. Aliquots of
10,000 cells were in~lb~ted for 1 hour at 27OC in a final volume of 540 ml cont~ining 20 nM
of [3H]mesulergine. Incubations were termin~ted by vacuum filtration over GF/C filters and
washing with binding buffer at 40C. Bound radioactivity was measured on filters impregn~te~l
with MeltiLexTM melt-on sc.intill~nt using a Wallac MicroBeta counter. Nonspecific binding
was estim~ted in parallel incubations cont~ining 10mM mianserin or metergoline.
For analysis of ligand binding to membrane plepa~ ~ions, cultures were harvested at 48 hr
post-infection, Iysed, and a me~ e pellet was plepaled as previously described (Labrecque
et al., (1992) FEBS letters 304:157-162.). These were stored at -800C. Protein
concentrations were determined by the nitrocellulose amido black method (Schaffner and
WPic~m~nn (1973) Analytic. Biochem. 56:502-551.). Membranes were thawed on ice and
le~u~ended in binding buffer by homogenization in a Potter homogenizer. The membranes
21 86979
-14-
(5 mg) were in~lb~ted with [3H]mesulergine for 1 hour at 27OC in a final volume of 540 ml,
and the assays termin~ted as described above for intact cells. Saturation binding assays
yielded a Kd for [3H]mesulergine of 2 nM, and competition binding assays with unlabeled
drugs were carried out with 3 nM [3H]mesulergine (Table 1).
Inositol Phosphate Production:
Growing cell cultures (1 X 106 cells/ml) were prelabeled with 2 mCi/ml [3H]myo-inositol for
24 hours prior to infection, and the labeled cells were then transferred to 50 ml shaker flasks
for infection. At 48 hours after infection, Sf9 zell cultures were treated with antagonists by
adding drugs directly to the culture medium 1 hour before cell harvest. Cells from treated
cultures were extensively washed in culture m~ m (4 X lh) to remove unbound ligands and
resuspended for polyinositolphosphate production as described in Labrecque et al., (1995)
Mol. Pharm. 48:150-159.
Effect of Drug I ~ ~tl ~tment on [3H]r~es~ tine Binding:
Growing cell cultures (1 X 106 cells/ml) were prelabeled for 24 hours with 2 mCi/ml [3H]myo-
inositol prior to infection, and the labeled cells were then ~l~n~relled to 50 ml shaker flasks
for infection. At 48 hours after infection, Sf9 cell cultures were treated with antagonists by
adding drugs directly to the culture medium 1 hour before cell harvest. Cells from treated
cultures were extensively washed in culture medi~lm (4 X lh) to remove unbound ligands. and
resuspended for polyinositolphosphate production and for parallel binding with 10 nM
[3H]mesulergine as described (Labrecque etal., (1995)Mol. Pharm. 48:150-159. Further
details are described in Table and Figure legends.
Analysis of Data:
The binding of [3H]mesulergine (saturation expt;l~ s) and the inhibition of [3H]mesulergine
binding by unlabeled serotonergic ligands were analyzed in terms of a single class of binding
site using the computer program LIGAND (Munson and Rodbard (1980) Analytic. Biochem.
107:220-239.). Data from three independent binding experiments were fitted individually and
the affinities presented for [3H]mesulergine (Kd) and other ligands (Ki) represent as the
average values (+ SEM~ from 3 sets of data. Dose-response data for Ips and antagonist-
in~luced decreases in [3H]mesulergine binding capacity were scaled taking values measured
in the absence of added ligand as 100%. The scaled data were analyzed according to a four
p~ll~;lel logistic equation analogous to the Hill equation (ALL~Ll: A. DeLean, Department
of Pharmacology, Université de Montréal, or INPLOT: Prizm So~ware, San Diego, CA).
For the decrease in [3H]m~slll.o.rgine binding with each ligand, three to four sets of data were
fitted ~imlllt~neously using ALLFIT with the slope factor set equal to 1. Further details are
described in Tables and Figure legends. Maximal inverse agonist activities measured for each
drug tested were compared statistically by a two tailed t test (a=0.05).
Expression of functional 5HT2C r~ceptors in Sfg Cells:
A baculovirus encoding the rat 5HT2C receptor was used to express the recombinant
receptor in cultures of Sf9 insect cells. Receptor levels, as measured in whole cells by the
~ 1 86979
-
binding of [3H]mesulergine (lOnM), increased with time after infection to reach
approximately 1 X 106 sites/cell at 72 hours after infection (data not shown).
Analysis of Antagonist Pretreatment on Inositol Phosphate Activib:
Six serotonergic antagonists were selected to be screened for their abilities to affect GPCR
activities: metergoline, mianserin, methysergide, clozapine, loxapine, and chlorpromazine.
All six are known to demonstrate good affinities for the rat SHT2C receptor (Roth (1992)
JPET??). Cells were plellealed with varying concentrations ofthe antagonists, then washed
extensively. Cells were then incub~ted7 either unqtimul~ted to measure spontaneous activity,
or induced by agonist. The cells were then assayed for production of inositol phosphate (IP).
Results are shown in Figure 1.
Inere~re~l Spontaneous Activib of GPCRs:
Of the six prelle~l..,entq performed, metergoline, mianserin, and the atypical neuroleptic
clozapine had significant effects on the level of spontaneous receptor activity as compared to
control vehicle ple~lea~ed cells. These spectacular and unexpected increases in spontaneous
activity were concen~ ion dependent. These same three drugs inhibit spontaneous receptor
activity and display inverse agonist effect in acute assays (Labrecque et al., (1995) supra).
In contrast, pr~le~l~..ent with methysergide, loxapine, and chlorpromazine were unable to
change the spontaneous activity of the 5HT2C receptor.
~nt~oniqt pl~lea~ re~mP.ntq were performed across a range of concentrations varying
from vehicle to 100 ',lM. The IP activity demonstrates a byphasic response curve for the
sen.q1ti~in~ effect of metergoline, mianserin, and clozapine pl~ lllent on the spontaneous
ligand-independent activity of the receptor. The unusual bell shape curve suggests that
spontaneous receptor activation increased up to a peak level with increasing concentration
of antagonist treatment, but past a threshold of antagonist concentration, levels of
spontaneous receptor activation returned to a lower basal level.
These observations suggest that specific antagonist ple~leallllents have direct effects on the
basal level of ~oll~ eous activity of GPCRs and a concomitant effect on the level of GPCR
binding sites. The variation in receptor density observed would seem to be implicated in this
complex alteration of spontaneous levels of receptor activation.
Inc~ease d GPCR Response to Agonists:
Sen)~ol~in efficacy is greatly increased following pl e~l e~ s with metergoline, mianserin,
or clozapine (Fig. 1). This il~crease is concentration dependent. The ple~lea~lllent regiments
show a byphasic response curve for agonist response, similar to the bell shaped curve
corresponding to elevated spontaneous activity. Both curves display superimposable
~qcPn~ling components resulting in equivalent rank order potency (EC50) for the spnqiti7:ing
phenolllenon induced by increasing concentration of antagonist ple~leallllent. The descending
component of the basal lmqtimlll~ted curves drop at a concentration inferior to the same
component of the agonist response curves.
2 1 8~979
-
-16-
The resulting elevation in 5HT efficacy represents a phenomenal increase in agonist response
determined by the absolute level of sensitization and by the level of spontaneous activity
present in the system. The results indicate that lower concentrations of pr~l- eal---ent have
greater impact on spontaneous levels of activation than on the actual agonist efficacy.
The m~im~l agonist response observed after antagonist sçn~iti7~tion treatment indicates that
the rank order for the m~gnit~lde ofthis effect was metergoline (350% ~ 125) > mianserin
(300% + 125) > clozapine (60% + 20) 2 loxapine () 2 chlol~lro-llazine (30% ~ 35) 2
methysergide (20% ~t 20). This ranking of m~xim~l sçn~ g efficacy of antagonist
prell~l,nent is di~elel,l than their corresponding rank order potency for the increased 5HT
response described above. Although e~tim~ted m~xim~l percent IP stim~ tion with 1 ,uM
5HT after antagonist p~ edl~-lent varied between experiments, the extent of the stimlll~tion
observed was always within 100% to 350% over basal level for metergoline and mianserin
prt;l.eaL",ent. These stim~ tion levels correspond accurately to levels of maximal 5HT
stim~ tion observed in choroid plexus (400%) (Sanders-Bush and Breeding (1989) J.
Pharmacol.Exp. Ther. 252:984-988; Sanders-Bush and Breeding (1988) J. Pharmacol.Exp.
Ther. 247:169-173.), or in NIH/3T3 cells (200%) (Barker et al., (1994) J. Biol. Chem.
269: 11887-11890.).
Figure 2 shows a series of dose response curves with 5HT following antagonist (metergoline)
pl~LI~llllelll at various concentrations. Figure 2A reports the effect of metergoline treatment
on 5HT dose-response production of IPs. The results indicate that metergoline pretreatment
at concentrations inferior to 10 nM produce 5HT dose responses identical to vehicle treated
cells. In contrast, higher concentrations of metergoline have significant effects on 5HT dose
response.
Cell p-el~e~ .nt with 10 nM metergoline displayed a large increase in basal IP levels
s~l~e~ g an inclease in ~o"l~eous receptor activation. A ten-fold increase in metergoline
concentration to 100 nM significantly reduced the basal IP levels without significant effect
on m~im~l level. Further increases in metergoline to concentrations superior to 100 nM
progressively and significantly decreased 5HT potency (EC50) and reduced the maximal
response of IPs produced in the presence of 5HT (Fig. 3A, Table 2).
Table 2. Pharmacolo,5~ of 5HT2C receptors expressed in Sfg cells after metergoline
treatment.
(Results are expressed as a mean ~ SE for at least 3 experiments. IP responses and
[3H]mesulergine binding are measured in whole cells (48 hours post-infection) following
plel~eal~"ent with metergoline at various concentrations.)
Density of Maximum
[3H]mesulergine 5HT Dose Response Activity
Binding Sites
21 86979
-
-17-
Metergoline %Residual -pEC50~ SE pvalues2 % Magnitude
Plelre~ nt Sites Stimulation3
s
vehicle 100~ 10 7.69~0.10 12~2 0.03
-9M 57~ 14 7.33~0.06 >0.05 13 ~2 0.00
-8M 29~ 12 7.06~0.02 >0.05 39~5 0.94
-7M 11~4 7.07~0.08 0.03 112~4 1.00
-6M 6~4 6.42~0.12 0.01 129~3 0.87
-5M 5 + 4 5.40 ~ 0.09 0.01 40 ~ 4 0.22
Maximal IP levels from Figure 2 were n~ nn~li7ed to d~ the maximal efficacity following l~ -e--l of the cells
with metergoline.
2Student t test was p~. r.,. . ~d between vehicle treated and the various metergoline lleal~ (a=0.05).
3Values ~li. "ed from Figure 2.
From the foregoing description, one skilled in the art can easily ascertain the essential
characteristics ofthis invention, and without departing from the spirit and scope thereof, can
make various ch~nges and modifications to the invention to adapt it to various usages and
conditions. Such changes and modifications are properly, equitably, and intended to be within
the full range of equivalence of the following claims.