Note: Descriptions are shown in the official language in which they were submitted.
2187515
SPECIFICATION
4,6-DI-t-BUTYL-2,3-DIHYDROHENZOTHIOPHENE DERIVATIVES
Technical Field
This invention relates to compounds which prevent
the oxidative modification of LDL, more particularly, to
compounds useful as therapeutics of arteriosclerosis,
myocardial infarction, etc.
Background Art
Atherosclerosis is one of the principal causes
of ischemic diseases such as angina pectoris, myocardial
infarction and cerebral apoplexy. The mechanism of initia-
tion and progression of atherosclerosis is closely related
to the oxidative modification of LDL. The modified LDLs
are not recognized by the LDL receptor but by the scavenger
receptor, to induce the foam cell formation which is
characterized by cholesterol accumulation.
The modification of LDL is caused by endothelial
cells, smooth muscle cells, macrophages, etc. and the
modified LDLs are eventually taken by macrophages via the
scavenger or other pathways. It is additionally known that
the modification of LDL by these cells is similar to the
oxidative modification of LDL by Cu2'.
LDL is chiefly composed of cholesterol esters,
phospholipids and apo-B-100. The oxidative modification of
LDL is shown from various aspects, for example fragmentation
of apo-B-100 by the generated radicals, the reaction between
the lipid peroxidation products and the free amino groups
in apo-B-100 lysine residues, and the transformation of
- 1 -
2187315
phosphatidyl choline to a lyso-form. One of the most
established phenomena in LDL oxidation is an increase of
thiobarbituric acid reactive substances (TBARS) as a result
of the lipid peroxidation. Oxidized LDL, or LDL that has
undergone such oxidative modification, causes the foam cell
formation and the cholesterol accumulation by the scavenger
and other pathways.
Under these circumstances, it is expected that
compounds having the inhibitory action on lipid peroxidation
can inhibit the initiation and progression of atherogenic
lesions by preventing the oxidative modification of LDL
and, hence, have the potential to work as therapeutics of
arteriosclerosis.
In ischemic diseases such as cerebral apoplexy
and myocardial infarction, various active oxygen species
are generated during blood reperfusion at ischemic sites
and tissue disorders can be exacerbated by the disruption
of cell membranes and other effects caused by the lipid
peroxidation. It is expected that compounds having the
anti-oxidative activity can prevent the tissue disorders
in ischemic lesions by removing the various active oxygen
species and lipid peroxidation and, hence, have the
potential to work as therapeutics of ischemic diseases.
Vitamin E is known as a natural antioxidant and
studies have been made to develop synthetic antioxidants
using vitamin E as the basic skeleton but no completely
satisfactory products have yet been synthesized.
- 2 -
- 2187315
Some of the compounds of the present invention which
are represented by formula (I) shown below are described,
by their generic concept, in British Patent Publication GB
2224028, but the publication has no mention of antioxidant
activity and applicability as therapeutics of
arteriosclerosis.
Disclosure of the Invention
An object of the present invention is to provide
antioxidants useful for the treatment of arteriosclerosis
and other ischemic diseases such as myocardial infarction
and cerebral apoplexy, as well as intermediates useful for
preparing said compounds.
The inventors of the present invention thought that
the reason for the insufficient efficacy of conventional
antioxidants, such as the compounds disclosed in Japanese
Patent Publication Laid-Open No. 121975/90, is that their
activity is lost before they reach the target site. Because
they easily react with various free radical species besides
lipid peroxidation related radicals. Based on this assump-
tion, the inventors have conducted extensive investigations
for the purpose of developing efficient antioxidants having
higher reaction specificity. It has been found as a result
that the above object is accomplished by the compounds
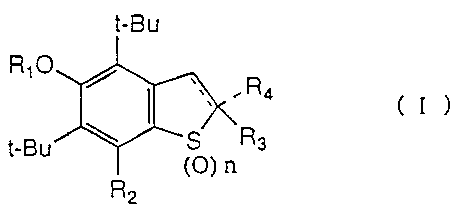
represented by formula (I):
t-BU
RIO
'~ R
4
' ( I )
t-B a
(O) n
R2
- 3 -
CA 02187315 2004-03-25
wherein R, represents a hydrogen atom, a lower alkyl group or an acyl group;
RZ
and R3, which may be the same or different, each represents a hydrogen atom,
an
optionally substituted alkyl group, or an optionally substituted alkenyl
group; R4
represents a hydrogen atom, an optionally substituted alkyl group, or an
optionally
substituted alkenyl group, or R4 forms a double bond between the carbon atom
to
which R3 is bonded and the adjacent carbon atom to form a benzothiophene
skeleton, or R3 and R4 are taken together to form a 5- to 8-membered spiro
ring
which may contain a hetero atom, e.g., oxygen, sulfur or nitrogen; and n
represents
an integer of 0 to 2.
It also has been found that the above object is accomplished by the
compounds represented by formula (I')
t-Bu
Rt0
( I~ )
t-Bu ~
(O) n
R2
wherein R, represents a hydrogen atom, a lower alkyl group or an acyl group;
R2
and R3 are the same or different, each represents a hydrogen atom, an
optionally
substituted alkyl group, or an optionally substituted alkenyl group; R4
represents a
hydrogen atom, an optionally substituted alkyl group, or an optionally
substituted
alkenyl group; and n represents an integer of 0 to 2, or a pharmaceutically
acceptable salt thereof.
-4-
CA 02187315 2005-02-O1
According to another aspect, there is also provided a pharmaceutical
composition comprising a compound represented by formula (I'):
t-Bu
R10
a ( I' )
t-Bu ~ S R3
(O) n
R2
wherein RI represents a hydrogen atom, a lower alkyl group or an acyl group;
R2
and R3 are the same or different, each represents a hydrogen atom, an
optionally
substituted alkyl group, or an optionally substituted alkenyl group; R4
represents a
hydrogen atom, an optionally substituted alkyl group, or an optionally
substituted
alkenyl group; and n represents an integer of 0 to 2,
or a pharmaceutically acceptable salt thereof; and
a pharmaceutical carrier.
Preferably, n represents 0.
According to another aspect, there is also provided a composition
comprising a compound dissolved in a lipid, the compound being represented by
formula (I):
t-Bu
RIO
'~ R
( I )
t-Bu ~ S R3
(0) n
R2
-4a-
CA 02187315 2005-02-O1
wherein R~, represents a hydrogen atom, a lower alkyl group or an acyl group;
R2
and R3 are the same or different, each represents a hydrogen atom, an
optionally
substituted alkyl group, or an optionally substituted alkenyl group; R4
represents a
hydrogen atom, an optionally substituted alkyl group, or an optionally
substituted
alkenyl group, or R4 forms a double bond between the carbon atom to which R3
is
bonded and the adjacent carbon atom to form a benzothiophene skeleton; and
n represents an integer of 0 to 2,
or a pharmaceutically acceptable salt thereof.
Preferably, R4 represents a hydrogen atom, an optionally substituted alkyl
group, an optionally substituted alkenyl group, or R4 forms a double bond
between the carbon atom to which R3 is bonded and the adjacent carbon atom to
form a benzothiophene skeleton; and n represents 0.
It has also been found that compounds represented by formula (II):
t-Bu
RIO / RS
(II)
t-Bu ~ S
R2 O~N.Rs
R~
wherein R1 and R2 are as defined above; RS represents a group of formula
(III):
R ( III )
-CH =C~Re
9
-4b-
CA 02187315 2005-02-O1
wherein R8 and R9, which may be the same or different, each represents a
hydrogen atom, an optionally substituted alkyl group or an optionally
substituted
alkenyl group,
-4c-
_ 2187315
a group of formula (IV):
-CH2C=C~ R9 ( N)
Rto
B
wherein R8, R9, and. Rlo, which may be the same or different,
each represents a hydrogen atom, an optionally substituted
alkyl group or an optionally substituted alkenyl group,
or a group of formula (V):
-H-C-Re ( V )
wherein R8 is as defined above;
and R6 and R~, which may be the same or different, each
represents a lower alkyl group,
are novel compounds that have not been reported in the
10 literature and intermediates useful for synthesizing the
compounds represented by formula (I).
It should be mentioned that the compounds of the
invention which are represented by the general formula (I)
have the following three characteristic features:
15 (1) They are lipid-soluble antioxidants which
inhibit lipid peroxidation efficiently;
(2) While there are many species of free radicals
that are involved in oxidation, the compounds react
efficiently with those radical species which are responsible
20 for the chain reaction of lipid peroxidation, therefore they
inhibit lipid peroxidation intensely.
- 5 -
- 2187315
(3) In order to develop the specific lipid
peroxidation inhibiting action to be specific in lipids, the
compounds have low reactivity for so-called "active oxygen"
species (e. g. superoxides and singlet oxygen) in aqueous
solution.
The Best Mode for Carrying out the Invention
The compounds represented by formula (I) are novel
compounds having two t-butyl groups on both ortho-positions
of the phenolic hydroxyl group, not being found in the
literature. British Patent Publication GB 224028 discloses
the generic concept including some of the compounds of the
present invention, but no specific mention of the compounds
is given.
The present invention is based on the fact that the
compounds represented by formula (I) having two t-butyl
groups on both ortho-positions of the phenolic hydroxyl
group exert markedly excellent effects as demonstrated in
Test Examples hereinafter given. The present invention
provides the compounds represented by formula (I) and
pharmaceutically acceptable salts thereof. The invention
also includes optically active isomers of the compounds.
In the formulae used herein, the term "lower alkyl
group" is intended to mean a straight-chain or branched
alkyl group having 1 to 6 carbon atoms and includes methyl,
ethyl, n-propyl, isopropyl, n-butyl, s-butyl, and t-butyl
groups. The acyl group includes acetyl, formyl, propionyl,
benzoyl, and benzyloxycarbonyl group, with an acetyl group
being preferred.
- 6 -
2187315
The term "optionally substituted alkyl group" is
intended to mean a straight-chain or branched optionally
substituted alkyl group having 1 to 20 carbon atoms, such as
methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, t-
butyl, pentyl, hexyl, heptyl, octyl, nonyl and decyl groups.
The term "optionally substituted alkenyl group" means
a straight-chain or branched optionally substituted alkenyl
group having 2 to 20 carbon atoms, such as vinyl, allyl,
butenyl, pentenyl, geranyl and farnesyl groups.
The substituent of the substituted alkyl or alkenyl
group includes a halogen atom, a hydroxyl group, an amino
group which may be substituted with a straight-chain or
branched alkyl group having 1 to 6 carbon atoms, an alkoxy
group, and an aryloxy group.
Specific examples of the compound of formula (I)
according to the invention are shown below.
4,6-Di-t-butyl-5-hydroxy-2,2-di-n-pentyl-2,3-
dihydrobenzothiophene,
4,6-Di-t-butyl-5-hydroxy-2-methyl-2,3-
dihydrobenzothiophene,
4,6-Di-t-butyl-5-hydroxy-2,2-dimethyl-2,3-
dihydrobenzothiophene,
4,6-Di-t-butyl-5-hydroxybenzo[b]thiophene,
4,6-Di-t-butyl-5-hydroxy-2,3-dihydrobenzothiophene,
4,6-Di-t-butyl-5-hydroxy-2,2-diethyl-2,3-
dihydrobenzothiophene,
4,6-Di-t-butyl-5-hydroxy-2,2-di-n-propyl-2,3-
dihydrobenzothiophene,
-
2181315
4,6-Di-t-butyl-5-hydroxy-2,2-diisopropyl-2,3-
dihydrobenzothiophene,
4,6-Di-t-butyl-5-hydroxy-2,2-di-n-butyl-2,3-
dihydrobenzothiophene,
4,6-Di-t-butyl-5-hydroxy-2,2-diisoamyl-2,3-
dihydrobenzothiophene,
4,6-Di-t-butyl-5-hydroxy-2,2-di-n-hexyl-2,3-
dihydrobenzothiophene,
4,6-Di-t-butyl-5-hydroxy-2,2-di-n-heptyl-2,3-
dihydrobenzothiophene,
4,6-Di-t-butyl-5-hydroxy-2,2-di-n-octyl-2,3-
dihydrobenzothiophene,
4,6-Di-t-butyl-5-hydroxy-2,2-diphenyl-2,3-
dihydrobenzothiophene,
4,6-Di-t-butyl-5-hydroxy-2,2-dibenzyl-2,3-
dihydrobenzothiophene,
4,6-Di-t-butyl-5-hydroxy-2-methyl-2-(4,8,12-
trimethyltrideca-3(E),7(E),11-trienyl)-2,3-
dihydrobenzothiophene,
4,6-Di-t-butyl-5-hydroxy-2-methyl-2-(4,8,12-
trimethyltridecyl)-2,3-dihydrobenzothiophene,
4,6-Di-t-butyl-5-hydroxy-2-n-octyl-2,3-
dihydrobenzothiophene,
2,4,6-Tri-t-butyl-5-hydroxy-2,3-
dihydrobenzothiophene,
4,6-Di-t-butyl-5-hydroxy-2,2-dimethyl-7-n-propyl-2,3-
dihydrobenzothiophene,
_ g -
218731
4,6-Di-t-butyl-5-hydroxy-2,3-dihydrobenozothiophene-
2-spiro-1'-cyclopentane,
4,6-Di-t-butyl-5-hydroxy-2,3-dihydrobenzothiophene-2-
spiro-1'-cyclohexane,
4,6-Di-t-butyl-5-hydroxy-2,3-dihydrobenzothiophene-2-
spiro-1'-cycloheptane,
4,6-Di-t-butyl-5-hydroxy-2,3-dihydrobenzothiophene-2-
spiro-1'-cyclooctane,
4,6-Di-t-butyl-2-methyl-5-hydroxybenzo[b]thiophene,
2,4,6-Tri-t-butyl-5-hydroxybenzo[b)thiophene,
4,6-Di-t-butyl-2-octyl-5-hydroxybenzo[b]thiophene_,
4,6-Di-t-butyl-5-hydroxy-2-(N,N-dimethylaminomethyl)-
2-methyl-2,3-dihydrobenzothiophene,
4,6-Di-t-butyl-5-hydroxy-2-hydroxymethyl-2-methyl-
2,3-dihydrobenzothiophene,
4,6-Di-t-butyl-5-hydroxy-2-methyl-2-(4,8-
dimethylnona-3(E),7-dienyl)-2,3-dihydrobenzothiophene, and
4,6-Di-t-butyl-5-hydroxy-2-methyl-2-(4,8-
dimethylnonyl)-2,3-dihydrobenzothiophene.
The compounds according to the invention can be
synthesized, for example, as follows.
_ g _
2181315
Method A:
t-Bu t-Bu t-Bu
HO ~ RIO ~ RIO
t-Bu I / OCH t-Bu I / OCH3 t-Bu I / OH
3
R2 R2 R2
(~) (2) (3)
t-Bu O t-Bu
R1p \ NCI R10 \ NCI
H
t-Bu / off t-Bu OJ
R2 R2
(4) (S).
t-Bu t-Bu
RIO I ~ NH2 RIO I ~ CHO
t-Bu / OH t-Bu / OH
R2 R2
(6) (7)
t-Bu OH t-Bu t-Bu
RIO ~ R8 RIO ~ \ Rs R10 IW \ Re
~ ~ I ~ '
t-Bu / OHR9 t-Bu / OHR9 t-Bu / O Rg
R2 R2 R S~N. Rs
) (9) (10) R
t-Bu t-Bu t-Bu
RIO ~ \ Ra RIO ~ Re HO ~ Re
~ t-Bu I ~ S R9 ~ t-Bu 1 / S R9 ~ t-Bu / S R9
R2 ~ ~ Rs R2 R2
O N
(12) (13)
( 1 1 ) R~
- 10 -
2187515
wherein R1 represents a hydrogen atom, a lower alkyl group or
an acyl group; Rz represents a hydrogen atom, an optionally
substituted alkyl group, or an optionally substituted
alkenyl group; R6 and R~, which may be the same or different,
each represents a lower alkyl group; and R8 and R9, which may
be the same or different, each represents a hydrogen atom,
an optionally substituted alkyl group, or an optionally
substituted alkenyl group; or R8 and R9 are taken together
to form a 5- to 8-membered spiro ring which may contain
a hetero atom, such as an oxygen atom, a sulfur atom or
a nitrogen atom.
- 11 -
2187315
Method H:
t-Bu t-Bu
RIO I ~ RIO I ~ R R
9 t0
t-Bu ~ OH t-Bu ~ O
R2 R2 Ra
(3) (14)
t-Bu Rio t-Bu R5o
RIO I ~ / R ~ RIO ~ ~ / Rs
t-Bu ~ OH Ra t-Bu ~ O Ra
R2 R S~N~ Rs
i
R~
(1 S) (1 6)
t-Bu Rio t-Bu
Rs0 ~ / Rs HO ~ Ra
-~ ~ ~
t-Bu ~ S Ra t_gu I ~ S Rio
R~~N~R6 R2 Rs
R
(1 ~) ' (18)
wherein Rl, Rz, R6, and R~ are as defined above; R8, R9,
and Rlo, which may be the same or different, each represents
a hydrogen atom, an optionally substituted alkyl group, or
an optionally substituted alkenyl group.
- 12 -
2187315
Method C:
t-Bu R'° t-Bu
Ri0 ~ / RIO ~ Ra
Re -~ ~ , ~O -~-
t-Bu ~ 's a t-BU ~ 'S
R O~N. Rs R O~N~ Rs
i i
R~ R~
(17) (19)
t-Bu t-Bu
R~ O ~ HO
Ra ~- ~ ~ S~ Ra
t-Bu ~ t-Bu
R2 R2
(20) (21 )
t-Bu t-Bu t-Bu
RIO ~ RIO ~ HO
~,--Ra ~- ~ ~Re ~ ~ ~Ra
t-Bu ~ ~ t-Bu ~ ~ t-Bu ~ S
2
R2 R2 R2
(22) (23) (24)
wherein Rl, Rz, R6, R~, R8, R9, and Rlo are as defined above.
- 13 -
218731
Method D:
~'Bu Rio
Ri0 / / R
9
t'Bu ~ S Re
RzO~N.Rs
(17) R~
t'Bu '-gu ''Bu
RIO / Rg RIO / RB R» HO / RB . R»
_ ~ %~N'R~2~ ~ %~N'R~z
~'Bu S R9 Rio ~ Bu ' S R9 Rio ~.gu \ S Rs Rio
R2 Rz Rz
(25) (26) (27)
''Bu ''Bu
RB OR~3 ~ O / ~ R8 ORia
t'Bu S R9 Rio t Bu \ S R~R~o
Rz Rz
(28) (29)
t'Bu ~'Bu
RIO / ~ R HO / Re
a O~R~a~ I ~~OH
i
~'Bu \ S RXR~o O Bu S R9 Rio
R2 Rz
(3d) (31 )
r_
~'Bu R~5 Bu
HO / R8 SOz HO / RB
t'Bu ~ ~ g~R~ R~s agu ~ S~~ R~s
9 10 9 10
R2 Rz
(32) (33)
- 14 -
21$7315
wherein R1, Rz, R6, R~, R8, R9, and Rlo are as defined
above; Rll and Rlz, which may be the same or different,
each represents a straight-chain or branched alkyl group
having 1 to 6 carbon atoms; R13 represents a straight-chain
or branched alkyl group having 1 to 20 carbon atoms; R14
represents a lower alkyl group; R15 represents a phenyl group
which may be substituted with a lower alkyl group; and Rls
represents a hydrogen atom, an optionally substituted alkyl
group, or an optionally substituted alkenyl group.
In method A, the phenolic hydroxyl group of compound
(1) is protected to give compound (2). Compound (2) is
demethylated by reaction with, e.g., iodotrimethylsilane
to obtain compound (3). Compound (3) is reacted with N-
hydroxymethyl-2-chloroacetamide in an acetic acid/sulfuric
acid mixture at room temperature to obtain a mixture of
compound (4) and compound (5). The mixture of compound
(4) and compound (5) is heated under reflux in an
ethanol/concentrated hydrochloric acid mixture to form
compound (6). Compound (6) is dissolved in an acid
aqueous solution, and hexamethylenetetramine is added to
the solution, followed by heating to obtain compound (7).
This reaction is preferably carried out by dissolving
compound (6) in an acetic acid aqueous solution, adding
hexamethylenetetramine, heating under reflux, and adding
thereto a hydrochloric acid aqueous solution, followed by
heating under reflux. Compound (7) is subjected to Grignard
reaction to obtain compound (8). Compound (9) is obtained
from compound (8) by dehydration reaction, for example,
- 15 -
21~731~
reaction with thionyl chloride in pyridine at room
temperature. Compound (9) is thiocarbamoylated with
N,N-dialkylthiocarbamoyl chloride to give compound (10).
Compound (10) is heated under reflux in a solvent, e.g.,
diphenyl ether, to give compound (11). Compound (11) is
reacted with a Lewis acid, e.g., boron trifluoride etherate
in a solvent, e.g., chloroform, dichloromethane or diethyl
ether, at room temperature to afford compound (12). The
protective group of compound (12) is removed to yield
compound (13).
In method B, compound (3) is reacted with an alkenyl
halide, such as 3-chloro-2-methyl-1-propene, in a solvent,
e.g., tetrahydrofuran, N,N-dimethylformamide, N,N-
dimethylacetamide or acetone, in the presence of a base,
e.g., sodium hydride, potassium carbonate, sodium carbonate,
sodium hydroxide or potassium hydroxide, to obtain compound
(14), which is then subjected to rearrangement by heating in
a solvent, e.g., N,N-dimethylaniline or N,N-diethylaniline,
to give compound (15). Compound (15) is thiocarbamoylated
with an N,N-dialkylthiocarbamoyl chloride to give compound
(16). Compound (16) is heated under reflux in a solvent,
e.g., diphenyl ether to afford compound (17). Compound (17)
is subjected to protective group removal and cyclization
simultaneously to obtain compound (18).
In method C, compound (17) obtained in method B
is reacted with sodium periodate etc. in the presence of
a catalytic amount of osmium tetroxide in a mixed solvent
of water and tetrahydrofuran, dioxane, methanol, ethanol
- 16 -
2187313
etc. at room temperature to give compound (19). Compound
(19) is cyclized by heating under reflux in a solvent, e.g.,
benzene or toluene, in the presence of a catalytic amount of
p-toluenesulfonic acid, or in a solvent, e.g., chloroform,
dichloromethane or diethyl ether, in the presence of a Lewis
acid, e.g., boron trifluoride etherate, to give compound
(20). Removal of the protective group of compound (20)
affords compound (21). Separately, compound (20) is
oxidized by reacting with, e.g., hydrogen peroxide, in
a solvent, such as acetic acid, to give compound (22),
which is then catalytically reduced in a solvent, e.g.,
ethyl acetate, methanol or ethanol, in the presence of
palladium-on-carbon or a like catalyst to give compound
(23). Compound (23) is reacted with, for example, lithium
aluminum hydride in a solvent, e.g., tetrahydrofuran, to
remove the protective group and conduct reduction
simultaneously to yield compound (24).
In method D, compound (25) is derived from compound
(17) obtained in method B by reacting compound (17) with
iodine in a mixed solvent of water and diethyl ether, etc.
in the presence of a base, e.g., sodium hydrogencarbonate,
at room temperature. From compound (25) are obtained
various derivatives according to the following four routes.
1. Compound (25) is reacted with ammonia or an
alkylamine (a primary amine, a secondary amine, etc.) in
a solvent, e.g., N,N-dimethylformamide, in the presence of
a base, e.g., potassium carbonate, to obtain compound (26).
Removal of the protective group of compound (26) gives
- 17 -
2187315
compound (27).
2. Compound (25) is reacted with an alkyl alcohol, etc.
in a solvent, e.g., N,N-dimethylformamide, in the presence
of a base, e.g., sodium hydride, to obtain compound (28).
Removal of the protective group of compound (28) yields
compound (29).
3. Compound (25) is reacted with a carboxylic acid
alkali metal salt etc. e.g., sodium acetate, in a solvent,
e.g., N,N-dimethylformamide or hexamethylphosphoric
triamide, to afford compound (30). Removal of the
protective group of compound (30) yields compound (31).
4. Compound (25) is reacted with a 1-(p-
toluenesulfonyl)alkyl, a 1-benzenesulfonylalkyl, etc.
in a solvent, e.g., tetrahydrofuran, in the presence of
a base, e.g., n-butyl lithium, simultaneously with removal
of the protective group to give compound (32). Compound
(32) is reacted with lithium triethylborohydride, etc.
in a solvent, e.g., tetrahydrofuran, in the presence of a
catalyst, e.g., [1,4-bis(diphenylphosphono)butane]palladium
chloride, or reacted with sodium amalgam, etc. in a solvent,
e.g., methanol, to obtain compound (33).
Examples
The present invention will now be illustrated in
greater detail with reference to Examples, but it should be
understood that the present invention is not deemed to be
limited thereto.
The chemical formulae of the compounds prepared in
Examples are shown below.
- 18 -
2187315
Example No. Chemical Formula
t-BU
HO
\
t-Bu S
t-Bu
HO
>--Me
t-Bu \
t-B a
HO / Me
t-Bu ~ S Me
t-Bu
HO
t-Bu
t-Bu
HO j
5
t-Bu \ S
- 19 -
218731
Example No. Chemical Formula
~~Bu
HO
~'Bu ~ S
''Bu
HO
j'Bu
''Bu
HO
~'Bu ~ I S~OH
''Bu
HO
w
t'Bu
HO
10
S~ v ~ v
~'Bu
'-Bu
HO
11 t_Bu \ ~ S w
''Bu
HO
12
v a
t'Bu
- 20 -
2187315
EXAMPLE 1: Synthesis of 4.6-Di-t-butyl-5-hydroxy-2 2-di-n-
pentyl-2.3-dihydrobenzothiophene
1) Synthesis of 4-Acetoxy-3,5-di-t-butylanisole:
In 150 ml of acetic anhydride was dissolved 23.6 g of
4-hydroxy-3,5-di-t-butylanisole, and 0.5 ml of concentrated
sulfuric acid was added thereto, followed by stirring at
70°C for 2 hours. The reaction mixture was concentrated
under reduced pressure, and to the residue was added
a saturated aqueous solution of sodium hydrogencarbonate.
The solution was extracted with ethyl acetate, and the
extract was dried over anhydrous magnesium sulfate and
concentrated. The precipitated solid was recrystallized
from methanol/water (2/1) to give 24.5 g (yield: 88%) of
4-acetoxy-3,5-di-t-butylanisole as a white solid.
1H-NMR (60 Mhz, CDC13)
Sppm: 1.06 (s,l8H), 2.02 (s,3H), 3.47 (s,3H), 6.53 (s,2H)
Mass : 278 ( M' )
m.p.. 96.6°C
2) Synthesis of 4-Acetoxy-3,5-di-t-butylphenol:
In 2 ml of dichloromethane was dissolved 0.50 g of
4-acetoxy-3,5-di-t-butylanisole, and the solution was cooled
with ice. To the solution was added dropwise 0.31 ml of
iodotrimethylsilane. The temperature was slowly raised
to room temperature, at which the reaction mixture was
stirred for 2 days. To the reaction mixture was added
a saturated aqueous solution of sodium hydrogencarbonate,
and the mixture was extracted with diethyl ether. The
organic layer was washed with a saturated sodium chloride
- 21 -
- 2187315
aqueous solution and dried over anhydrous magnesium sulfate.
After concentration, the residue was purified by silica
gel chromatography (15~ ethyl acetate in n-hexane) to give
0.38 g (80~) of 4-acetoxy-3,5-di-t-butylphenol as a white
solid.
1H NMR (60 Mhz, CDC13)
Sppm: 1.27(s,l8H), 2.27(s,3H), 5.22(bs,lH), 6.67(s,2H)
Mass: 222(M')
m.p.. 156.9°C
3) Synthesis of 4-Acetoxy-3,5-di-t-butyl-2-
(chloroacetylaminomethyl)phenol and 6-Acetoxy-5,7-di-t-
butyl-3-(2-chloroacetyl)-2,3-dihydro-1,3,4H-benzoxazine:
In 200 ml of a 9:1 mixture of acetic acid and
sulfuric acid was dissolved 29 g of 4-acetoxy-3,5-di-t-
butylphenol, and 34 g of N-hydroxymethyl-2-chloroacetamide
was added thereto, followed by stirring at room temperature
for 48 hours. The reaction mixture was poured into water,
neutralized with a 1N sodium hydroxide aqueous solution,
and extracted with ethyl acetate. The organic layer was
dried over anhydrous magnesium sulfate and concentrated.
The resulting concentrate was used as such in the subsequent
reaction. Silica gel chromatography (20% ethyl acetate
in n-hexane) of part of the concentrate revealed that
the product consisted of 4-acetoxy-3,5-di-t-butyl-2-
(chloroacetylaminomethyl)phenol and 6-acetoxy-5,7-di-t-
butyl-3-(2-chloroacetyl)-2,3-dihydro-1,3,4H-benzoxazine.
4-Acetoxy-3.5-di-t-butyl-2-(chloroacetylaminomethyl)phenol
(colorless oil):
- 22 -
2187315
1H NMR (60 MHz, CDC13)
Sppm: 1.30(s,9H), 1.43(s,9H), 2.28(s,3H), 4.00(s,2H),
4.73(d,2H,J=6.OHz), 6.88(s,lH), 7.54(t,lH,J=6.OHz)
Mass: 369(M+)
6-Acetoxy-5,7-di-t-butyl-3-(2-chloroacetyl)-2 3-dihydro-
1.3.4H-benzoxazine (colorless oil):
1H NMR (60 MHz, CDC13)
8ppm: 1.30(s,9H), 1.47(s,9H), 2.30(s,3H), 4.17(s,2H),
5.00(s,2H), 5.33(s,2H), 6.83(s,lH)
Mass: 381(M+)
4) Synthesis of 4-Acetoxy-2-aminomethyl-3,5-di-t-
butylphenol:
The concentrate obtained in Example 1-3) was
dissolved in 550 ml of a 10:3 mixture of ethanol and
concentrated hydrochloric acid, and the solution was
heated under reflux for 2 hours. After cooling, the
reaction mixture was poured into water, neutralized with
a 1N sodium hydroxide aqueous solution, and extracted with
ethyl acetate. The organic layer was dried over anhydrous
magnesium sulfate and concentrated. The resulting concen
trate was used as such in the subsequent reaction. Silica
gel chromatography (20o ethyl acetate in n-hexane) of part
of the concentrate revealed that the product consisted
mainly of 4-acetoxy-2-aminomethyl-3,5-di-t-butylphenol.
1H NMR (60 MHz, CDC13)
8ppm: 1.27(s,9H), 1.37(s,9H), 2.25(s,3H), 4.22(s,2H),
5.18(bs,3H), 6.85(s,lH)
Mass: 293(M+)
- 23 -
2187315
5) Synthesis of 5-Acetoxy-4,6-di-t-butyl-2-
hydroxybenzaldehyde:
The concentrate obtained in Example 1-4) was
dissolved in 636 ml of a 11:3 mixture of acetic acid and
water, and 19.3 g of hexamethylenetetramine was added
thereto, followed by heating under reflux for 4 hours. To
the reaction mixture was added 85 ml of 4.5N hydrochloric
acid, followed by refluxing for additional 20 minutes.
After cooling, the reaction mixture was poured into water,
neutralized with a 1N sodium hydroxide aqueous solution, and
extracted with ethyl acetate. The organic layer was dried
over anhydrous magnesium sulfate and concentrated. The
concentrate was purified by silica gel column chromatography
(chloroform) to afford 19.0 g of 5-acetoxy-4,6-di-t-butyl-2-
hydroxybenzaldehyde as a pale yellow solid.
1H-NMR (60 MHz, CDC13)
8ppm: 1.35(s,9H), 1.54(s,9H), 2.35(s,3H), 6.92(s,lH),
10.67(s,lH), 12.32(s,lH)
IR(cm-1): 2976, 1758
Mass: 292(M')
m.p.. 79.0°C
6) Synthesis of 4-Acetoxy-3,5-di-t-butyl-2-(1-hydroxy-2-n-
pentylheptyl)phenol:
A solution of 96.4 g of 6-bromoundecane prepared
in a usual manner in 300 ml of tetrahydrofuran was added
to 10 g of magnesium under a nitrogen atmosphere to prepare
a Grignard reagent. To the resulting Grignard reagent was
added dropwise a solution of 40 g of 5-acetoxy-4,6-di-t-
- 24 -
- 217315
butyl-2-hydroxybenzaldehyde in 200 ml of tetrahydrofuran.
After stirring the mixture at room temperature for 2 hours,
a saturated aqueous solution of ammonium chloride was added
to the reaction mixture, followed by extraction with ethyl
acetate. The organic layer was dried over anhydrous
magnesium sulfate and concentrated. The concentrate was
purified by silica gel chromatography (10% ethyl acetate
in n-hexane) to yield 24.4 g (39%) of 4-acetoxy-3,5-di-t-
butyl-2-(1-hydroxy-2-n-pentylheptyl)phenol as a white solid.
1H NMR ( 270 MHz, CDC13 )
sppm: 0.91(m,6H), 1.29(s,9H), 1.33(br,l6H), 1.40(s,9H),
2.17(m,lH), 2.28(s,3H), 5.22(m,lH), 6.77(s,lH),
7.89(s,lH).
Mass : 448 ( M+ )
7) Synthesis of 4-Acetoxy-3,5-di-t-butyl-2-(2-n-pentyl-1-
heptenyl)phenol:
To 23.0 g of 4-acetoxy-3,5-di-t-butyl-2-(1-hydroxy-2-
n-pentylheptyl)phenol was added 100 g of pyridine, and
4.6 ml of thionyl chloride was added thereto dropwise under
cooling with ice. The mixture was stirred at room tempera-
ture for 1 hour, and pyridine was removed by evaporation
under reduced pressure. Water was added to the residue,
and the mixture was extracted with ethyl acetate. The
organic layer was dried over anhydrous magnesium sulfate
and concentrated. Purification of the concentrate by silica
gel chromatography (10% ethyl acetate in n-hexane) gave
19.1 g (87%) of 4-acetoxy-3,5-di-t-butyl-2-(2-n-pentyl-1-
heptenyl)phenol as a colorless oil.
- 25 -
- 218731'_
1H NMR ( 60 MHz, CDC13 )
8ppm: 0.72-0.99(m,6H), 1.12-1.97(m,l4H), 1.30(s,9H),
1.33(s,9H), 2.25(m,2H), 2.27(s,3H), 5.35(d,lH),
6.14(s,lH), 6.85(s,lH)
Mass : 430 ( M' )
8) Synthesis of 0-{4-Acetoxy-3,5-di-t-butyl-2-(2-n-pentyl-
1-heptenyl)phenyl) N,N-Dimethylthiocarbamate:
In 10 ml of N,N-dimethylformamide was suspended
0.14 g of 60% oily sodium hydride under a nitrogen
atmosphere, and a solution of 1.25 g of 4-acetoxy-3,5-di-
t-butyl-2-(2-n-pentyl-1-heptenyl)phenol in 10 ml of N,N-.
dimethylformamide was added dropwise to the suspension under
cooling with ice, followed by stirring at room temperature
for 1 hour. The reaction mixture was cooled with ice, and a
solution of 0.43 g of N,N-dimethylthiocarbamoyl chloride in
10 ml of N,N-dimethylformamide was added thereto dropwise.
After stirring at room temperature for 1 hour, a saturated
aqueous solution of ammonium chloride was added to the
reaction mixture, and the mixture was extracted with ethyl
acetate. The organic layer was washed successively with
water and a saturated aqueous sodium chloride solution,
dried over anhydrous magnesium sulfate, and concentrated.
The concentrate was purified by silica gel chromatography
(10% ethyl acetate in n-hexane) to afford 0.79 g (53%) of 0-
{4-acetoxy-3,5-di-t-butyl-2-(2-n-pentyl-1-heptenyl)phenyl}
N,N-dimethylthiocarbamate as a colorless oil.
1H NMR ( 270 MHz, CDC13 )
6ppm: 0.75(t,3H,J=6.6Hz), 0.91(t,3H,J=6.8Hz),
- 26 -
2187315
1.11-1.82(m,l4H), 1.33(s,9H), 1.35(s,9H),
2.08(t,2H,J=7.8Hz), 2.32(s,3H), 3.21(s,3H),
3.43(s,3H), 6.14(s,lH), 6.89(s,lH)
Mass: 517(M')
9) Synthesis of S-{4-Acetoxy-3,5-di-t-butyl-2-(2-n-pentyl-
1-heptenyl)phenyl} N,N-Dimethylthiocarbamate:
In 10 ml of diphenyl ether was dissolved 0.7 g of O-
{4-acetoxy-3,5-di-t-butyl-2-(2-n-pentyl-1-heptenyl)phenyl}
N,N-dimethylthiocarbamate under a nitrogen atmosphere,
followed by heating under reflux for 16 hours. After
cooling, the reaction mixture was purified by silica gel.
chromatography (20$ ethyl acetate in n-hexane) to give 0.2 g
(29$) of S-{4-acetoxy-3,5-di-t-butyl-2-(2-n-pentyl-1-
heptenyl)phenyl~ N,N-dimethylthiocarbamate as a colorless
oil.
1H NMR (270 MHz, CDC13)
Sppm: 0.74(t,3H,J=6.8Hz), 0.91(t,3H,J=7.OHz),
1.08-1.76(m,l4H), 1.33(s,9H), 1.35(s,9H),
2.12(t,2H,J=7.4Hz), 2.31(s,3H), 3.04(s,6H),
6.31(s,lH), 7.41(s,lH)
Mass: 517(M')
10) Synthesis of 5-Acetoxy-4,6-di-t-butyl-2,2-di-n-pentyl-
2,3-dihydrobenzothiophene:
To 0.2 g of S-{4-acetoxy-3,5-di-t-butyl-2-(2-n-
pentyl-1-heptenyl)phenyl} N,N-dimethylthiocarbamate was
added 10 ml of boron trifluoride etherate under a nitrogen
atmosphere, followed by stirring at room temperature for
3 hours. The reaction mixture was poured into a saturated
- 27 -
217315
aqueous solution of sodium hydrogencarbonate and extracted
with chloroform. The organic layer was dried over anhydrous
magnesium sulfate and concentrated. The concentrate was
purified by silica gel chromatography (10$ ethyl acetate
in n-hexane) to provide 0.1 g (57$) of 5-acetoxy-4,6-di-
t-butyl-2,2-di-n-pentyl-2,3-dihydrobenzothiophene as
a colorless oil.
1H NMR (270 MHz, CDC13)
8ppm: 0.88(m,6H), 1.29(s,9H), 1.30(br,l2H), 1.38(s,9H),
1.76(m,4H), 2.28(s,3H), 3.26(d,lH,J=15.2Hz),
3.33(d,lH,J=15.2Hz), 7.07(s,lH)
Mass: 446(M')
11) Synthesis of 4,6-Di-t-butyl-5-hydroxy-2,2-di-n-pentyl-
2,3-dihydrobenzothiophene:
In 10 ml of tetrahydrofuran was suspended 0.07 g of
lithium aluminum hydride under a nitrogen atmosphere, and
a solution of 0.85 g of 5-acetoxy-4,6-di-t-butyl-2,2-di-n-
pentyl-2,3-dihydrobenzothiophene in 10 ml of tetrahydrofuran
was added dropwise to the suspension. The mixture was
heated under reflux for 3 hours and cooled to room tempera-
ture. A 10$ hydrochloric acid aqueous solution was added
to the reaction mixture, followed by extraction with ethyl
acetate. The organic layer was washed with a saturated
sodium chloride aqueous solution, dried over anhydrous
magnesium sulfate, and concentrated. The concentrate was
purified by silica gel chromatography (n-hexane) to give
0.55 g (72$) of 4,6-di-t-butyl-5-hydroxy-2,2-di-n-pentyl-
2,3-dihydrobenzothiophene as a colorless oil.
- 28 -
2187315
1H NMR (270 MHz, CDC13)
8ppm: 0.88(t,6H,J=6.8Hz), 1.29(br,l2H), 1.39(s,9H),
1.52(s,9H), 1.73(m,4H), 3.33(s,2H), 5.08(s,lH),
6.95(s,lH)
IR( cm'1 ) : 3648, 2952
Mass : 404 ( M' )
EXAMPLE 2: Synthesis of 4 6-Di-t-butyl-5-hydroxy-2-methyl
2,3-dihydrobenzothiophene
1) Synthesis of 4-Acetoxy-3,5-di-t-butyl-1-(2-
propenyloxy)benzene:
In 300 ml of acetone was dissolved 10 g of 4-acetoxy-
3,5-di-t-butylphenol obtained in Example 1-2) and 15.6 g
of potassium carbonate, and 0.39 ml of 3-bromo-1-propene
was added thereto, followed by refluxing for 24 hours.
The reaction mixture was concentrated under reduced pres-
sure, water was added to the residue, and the mixture was
extracted with diethyl ether. The organic layer was washed
successively with water and a saturated sodium chloride
aqueous solution, dried over anhydrous magnesium sulfate,
and concentrated. The concentrate was purified by silica
gel chromatography (10% ethyl acetate in n-hexane) to
quantitatively provide 11.0 g of 4-acetoxy-3,5-di-t-butyl-
1-(2-propenyloxy)benzene as a colorless oil.
1H NMR ( 60 MHz, CDC13 )
sppm: 1.30(s,l8H), 2.27(s,3H), 4.47(d,2H,J=5.OHz),
5.05-5.57(m,2H), 5.68-6.37(m,lH), 6.81(s,2H)
Mass: 304(M')
- 29 -
2187315
2) Synthesis of 4-Acetoxy-3,5-di-t-butyl-2-(2-
propenyl)phenol:
In 50 ml of N,N-dimethylaniline was dissolved 11.0 g
of 4-acetoxy-3,5-di-t-butyl-1-(2-propenyloxy)benzene, and
the solution was heated under reflux for 18 hours under
a nitrogen atmosphere. After cooling to room temperature,
the reaction mixture was concentrated under reduced pres-
sure, and the concentrate was purified by silica gel
chromatography (15% ethyl acetate in n-hexane) to give
8.84 g (77%) of 4-acetoxy-3,5-di-t-butyl-2-(2-
propenyl)phenol as a white solid.
1H NMR (60 MHz, CDC13)
8ppm: 1.30(s,9H), 1.42(s,9H), 2.28(s,3H), 3.52-3.84(m,2H),
4.88-5.42(m,3H), 5.68-6.45(m,lH), 6.79(s,lH)
Mass : 304 ( M' )
m.p.: 103.6°C
3) Synthesis of 0-~4-Acetoxy-3,5-di-t-butyl-2-(2-
propenyl)phenyl} N,N-Dimethylthiocarbamate:
In 10 ml of N,N-dimethylformamide was suspended
0.32 g of 60% oily sodium hydride under a nitrogen
atmosphere. A solution of 2.0 g of 4-acetoxy-3,5-di-t-
butyl-2-(2-propenyl)phenol in 10 ml of N,N-dimethylformamide
was added dropwise to the suspension while cooling with
ice, followed by stirring at room temperature for 1 hour.
The reaction mixture was cooled with ice, and a solution
of 0.99 g of N,N-dimethylthiocarbamoyl chloride in 10 ml
of N,N-dimethylformamide was added thereto dropwise. After
stirring at room temperature for 1 hour, a saturated aqueous
- 30 -
2187315
solution of ammonium chloride was added to the reaction
mixture, and the mixture was extracted with ethyl acetate.
The organic layer was washed successively with water and
a saturated aqueous solution of sodium chloride, dried
over anhydrous magnesium sulfate, and concentrated. The
concentrate was purified by silica gel chromatography (10%
ethyl acetate in n-hexane) to afford 2.05 g (79$) of O-{4-
acetoxy-3,5-di-t-butyl-2-(2-propenyl)phenyl} N,N-
dimethylthiocarbamate as a white solid.
1H NMR (60 MHz, CDC13)
sppm: 1.33(s,9H), 1.43(s,9H), 2.30(s,3H), 3.27(s,3H),
3.42(s,3H), 3.62(m,2H), 4.72-5.05(m,2H),
5.63-6.18(m,lH), 6.95(s,lH)
Mass: 391(M')
m.p.: 134.3°C
4) Synthesis of S-{4-Acetoxy-3,5-di-t-butyl-2-(2-
propenyl)phenyl} N,N-Dimethylthiocarbamate:
In 10 ml of diphenyl ether was dissolved 1.0 g
of O-{4-acetoxy-3,5-di-t-butyl-2-(2-propenyl)phenyl~ N,N-
dimethylthiocarbamate under a nitrogen atmosphere, and
the solution was heated under reflux for 16 hours. After
cooling, the reaction mixture was purified by silica
gel chromatography (20a ethyl acetate in n-hexane) to
give 0.74 g (74%) of S-{4-acetoxy-3,5-di-t-butyl-2-(2-
propenyl)phenyl} N,N-dimethylthiocarbamate as a white solid.
1H NMR (270 MHz, CDC13)
8ppm: 1.33(s,9H), 1.43(s,9H), 2.31(s,3H), 3.05(bs,6H),
3.88(d,2H,J=S.OHz), 4.71(d,lH,J=17.2Hz),
- 31 -
2187315
5.00(d,lH,J=10.2Hz), 5.83-6.00(m,lH}, 7.42(s,lH)
Mass : 391 ( M' )
m.p.. 133.6°C
5) Synthesis of 4,6-Di-t-butyl-5-hydroxy-2-methyl-2,3-
dihydrobenzothiophene:
In 10 ml of tetrahydrofuran was suspended 0.14 g of
lithium aluminum hydride under a nitrogen atmosphere, and
a solution of 0.7 g of S-{4-acetoxy-3,5-di-t-butyl-2-(2
propenyl)phenyl~ N,N-dimethylthiocarbamate in 10 ml of
tetrahydrofuran was added dropwise to the suspension.
The mixture was heated under reflux for 3 hours and
cooled to room temperature. To the reaction mixture was
added carefully 10 ml of acetic acid, and the mixture was
refluxed for additional 30 minutes. After cooling, a 10%
hydrochloric acid aqueous solution was added to the reaction
mixture, and the mixture was extracted with ethyl acetate.
The organic layer was washed with a saturated sodium
chloride aqueous solution, dried over anhydrous magnesium
sulfate, and concentrated. The concentrate was purified
by silica gel chromatography (5$ ethyl acetate in n-hexane)
to yield 0.35 g (70%) of 4,6-di-t-butyl-5-hydroxy-2-methyl-
2,3-dihydrobenzothiophene as a white solid.
1H NMR (270 MHz, CDC13)
Sppm: 1.39(s,9H), 1.42(d,3H,J=6.6Hz), 1.52(s,9H),
3.17(m,lH), 3.64(m,lH), 3.80(m,lH), 5.11(s,lH),
7.01(s,lH)
IR( cm-1 ) : 3620, 2956
Mass : 278 ( M' )
- 32 -
218731
m.p.. 96.7°C
EXAMPLE 3: Synthesis of 4 6-Di-t-butyl-5-hydroxy 2 2
dimethyl-2 3-dihydrobenzothiophene
1) Synthesis of 4-Acetoxy-3,5-di-t-butyl-1-(2-methyl-2-
propenyloxy)benzene:
In 10 ml of N,N-dimethylformamide was suspended
0.18 g of 60% oily sodium hydride under a nitrogen
atmosphere, and a solution of 1.0 g of 4-acetoxy-3,5-di-
t-butylphenol obtained in Example 1-2) in 5 ml of N,N-
dimethylformamide was added thereto dropwise under cooling
with ice, followed by stirring for 30 minutes. The
temperature was raised to room temperature, 0.45 ml of
3-chloro-2-methyl-1-propene was added thereto dropwise,
and the reaction mixture was stirred at room temperature
for 2 hours. To the reaction mixture was added 15 ml of
a saturated aqueous solution of ammonium chloride, followed
by extraction with diethyl ether. The organic layer was
washed successively with water and a saturated sodium
chloride aqueous solution, dried over anhydrous magnesium
sulfate, and concentrated. The concentrate was purified
by silica gel chromatography (10% ethyl acetate in n-hexane)
to give 1.08 g (90%) of 4-acetoxy-3,5-di-t-butyl-1-(2-
methyl-2-propenyloxy)benzene as a colorless oil.
1H NMR (60 MHz, CDC13)
8ppm: 1.30(s,l8H), 1.83(s,3H), 2.30(s,3H), 4.37(s,2H),
5.02(br,2H), 6.83(s,2H)
Mass: 318(M+)
- 33 -
2187315
2) Synthesis of 4-Acetoxy-3,5-di-t-butyl-2-(2-methyl-2-
propenyl)phenol:
In 100 ml of N,N-dimethylaniline was dissolved
24.0 g of 4-acetoxy-3,5-di-t-butyl-1-(2-methyl-2-propenyl-
oxy)benzene, and the solution was heated under reflux for
18 hours under a nitrogen atmosphere. After cooling to room
temperature, the reaction mixture was concentrated under
reduced pressure, and the concentrate was purified by silica
gel chromatography (10°s ethyl acetate in n-hexane) to give
6.66 g (28%) of 4-acetoxy-3,5-di-t-butyl-2-(2-methyl-2-
propenyl)phenol as a white solid.
1H NMR (60 MHz, CDC13)
8ppm: 1.30(s,9H), 1.37(s,9H), 1.88(s,3H), 2.28(s,3H),
3.34(br,2H), 4.60(bs,lH), 4.88(bs,lH), 5.02(bs,lH),
6.79(s,lH)
Mass: 318(M+)
m.p.. 102.0°C
3) Synthesis of 0-{4-Acetoxy-3,5-di-t-butyl-2-(2-methyl-2-
propenyl)phenyl~ N,N-Dimethylthiocarbamate:
In 20 ml of N,N-dimethylformamide was suspended
0.75 g of 60% oily sodium hydride under a nitrogen
atmosphere. A solution of 4.57 g of 4-acetoxy-3,5-di-t-
butyl-2-(2-methyl-2-propenyl)phenol in 20 ml of N,N-
dimethylformamide was added dropwise to the suspension under
ice-cooling, followed by stirring at room temperature for
1 hour. The reaction mixture was cooled with ice, and a
solution of 1.82 g of N,N-dimethylthiocarbamoyl chloride in
20 ml of N,N-dimethylformamide was added thereto dropwise.
- 34 -
2187315
After stirring at room temperature for 1 hour, a saturated
ammonium chloride aqueous solution was added to the reaction
mixture, and the mixture was extracted with ethyl acetate.
The organic layer was washed successively with water and
a saturated sodium chloride aqueous solution, dried over
anhydrous magnesium sulfate, and concentrated. The concen-
trate was purified by silica gel chromatography (10% ethyl
acetate in n-hexane) to afford 3.04 g (52%) of 0-{4-acetoxy-
3,5-di-t-butyl-2-(2-methyl-2-propenyl)phenyl~ N,N-
dimethylthiocarbamate as a white solid.
1H NMR (270 MHz, CDC13)
8ppm: 1.33(s,9H), 1.40(s,9H), 1.77(s,3H), 2.31(s,3H),
3.25(s,3H), 3.29-3.60(m,2H), 3.45(s,3H), 4.29(bs,lH),
4.76(bs,lH), 6.96(s,lH)
Mass: 405(M+)
m.p.. 152.1°C
4) Synthesis of S-{4-Acetoxy-3,5-di-t-butyl-2-(2-methyl-2-
propenyl)phenyl} N,N-Dimethylthiocarbamate:
In 10 ml of diphenyl ether was dissolved 1.0 g of
0-{4-acetoxy-3,5-di-t-butyl-2-(2-methyl-2-propenyl)phenyl}
N,N-dimethylthiocarbamate under a nitrogen atmosphere, and
the solution was heated under reflux for 16 hours. After
cooling, the reaction mixture was purified by silica gel
chromatography (20% ethyl acetate in n-hexane) to afford
0.57 g (57%) of S-{4-acetoxy-3,5-di-t-butyl-2-(2-methyl-2-
propenyl)phenyl) N,N-dimethylthiocarbamate as a white solid.
1H NMR (270 MHz, CDC13)
Sppm: 1.33(s,9H), 1.40(s,9H), 1.83(s,3H), 2.31(s,3H),
- 35 -
2187315
3.06(bs,6H), 3.70(m,2H), 4.00(bs,lH), 4.74(bs,lH),
7.41(s,lH)
Mass : 405 ( M' )
m.p.. 132.1°C
5) Synthesis of 4,6-Di-t-butyl-5-hydroxy-2,2-dimethyl-2,3-
dihydrobenzothiophene:
In 10 ml of tetrahydrofuran was suspended 0.1 g of
lithium aluminum hydride under a nitrogen atmosphere, and
a solution of 0.5 g of S-{4-acetoxy-3,5-di-t-butyl-2-(2
methyl-2-propenyl)phenyl} N,N-dimethylthiocarbamate in 10 ml
of tetrahydrofuran was added thereto dropwise, followed by
heating under reflux for 3 hours. After cooling to room
temperature, 10 ml of acetic acid was carefully added to
the reaction mixture, followed by refluxing for 30 minutes.
After cooling, a 10% hydrochloric acid. aqueous solution
was added thereto, and the mixture was extracted with ethyl
acetate. The organic layer was washed with a saturated
sodium chloride aqueous solution, dried over anhydrous
magnesium sulfate, and concentrated. The concentrate was
purified by silica gel chromatography (5% ethyl acetate in
n-hexane) to give 0.25 g (70%) of 4,6-di-t-butyl-5-hydroxy-
2,2-dimethyl-2,3-dihydrobenzothiophene as a white solid.
1H NMR (270 MHz, CDC13)
8ppm: 1.39(s,9H), 1.51(s,6H), 1.52(s,9H), 3.34(s,2H),
5.11(s,lH), 6.98(s,lH)
IR( cm-1 ) : 3644, 2956
Mass : 292 ( M' )
m.p.. 79.0°C
- 36 -
2187315
EXAMPLE 4: Synthesis of 4 6-Di-t-butyl-5-
hydroxybenzofblthiophene
1) Synthesis of S-(4-Acetoxy-3,5-di-t-butyl-2-
formylmethylphenyl) N,N-Dimethylthiocarbamate:
In 20 ml of a 3:1 mixture of tetrahydrofuran and
water was dissolved 1.0 g of S-{4-acetoxy-3,5-di-t-butyl-2-
(2-propenyl)phenyl} N,N-dimethylthiocarbamate prepared in
Example 2-4), and 50 mg of osmium tetroxide and 1.1 g of
sodium periodate were added to the solution, followed by
stirring at room temperature for 24 hours. A saturated
aqueous solution of sodium thiosulfate was added to the
reaction mixture, and the mixture was extracted with ethyl
acetate. The organic layer was washed with a saturated
aqueous solution of sodium chloride, dried over anhydrous
magnesium sulfate, and concentrated. The concentrate was
purified by silica gel chromatography (20o ethyl acetate in
n-hexane) to give 0.52 g (52%) of S-(4-acetoxy-3,5-di-t-
butyl-2-formylmethylphenyl) N,N-dimethylthiocarbamate as
a white solid.
1H NMR (60 MHz, CDC13)
8ppm: 1.30(s,9H), 1.43(s,9H), 2.33(s,3H), 3.01(s,6H),
4.10(bs,2H), 7.47(s,lH), 9.62(bs,lH)
Mass: 393(M')
2) Synthesis of 5-Acetoxy-4,6-di-t-butylbenzo[b]thiophene:
In 15 ml of benzene was dissolved 0.5 g of S-(4-
acetoxy-3,5-di-t-butyl-2-formylmethylphenyl) N,N-dimethyl-
thiocarbamate, and a catalytic amount of p-toluenesulfonic
acid was added thereto, followed by heating under reflux
- 37 -
_ 2187315
for 1 hour. After cooling, a saturated aqueous solution of
sodium hydrogencarbonate was added to the reaction mixture,
and the mixture was extracted with ethyl acetate. The
organic layer was dried over anhydrous magnesium sulfate
and concentrated. The concentrate was purified by silica
gel chromatography (lOs ethyl acetate in n-hexane) to give
0.3 g (780) of 5-acetoxy-4,6-di-t-butylbenzo[b]thiophene as
a colorless oil.
1H NMR (60 MHz, CDC13)
Sppm: 1.37(s,9H), 1.54(s,9H), 2.31(s,3H),
7.28(d,lH,J=6.OHz), 7.62(d,lH,J=6.OHz), 7.72(s,lH.)
Mass: 304(M')
3) Synthesis of 4,6-Di-t-butyl-5-hydroxybenzo[b]thiophene:
In 10 ml of tetrahydrofuran was suspended 0.11 g
of lithium aluminum hydride under a nitrogen atmosphere,
and a solution of 0.9 g of 5-acetoxy-4,6-di-t-butyl-
benzo[b]thiophene in 10 ml of tetrahydrofuran was added
thereto dropwise. The mixture was heated under reflux for
3 hours, followed by cooling to room temperature. A 10$
hydrochloric acid aqueous solution was added thereto, and
the mixture was extracted with ethyl acetate. The organic
layer was washed with a saturated sodium chloride aqueous
solution, dried over anhydrous magnesium sulfate, and
concentrated. The concentrate was purified by silica gel
chromatography (n-hexane) to yield 0.7 g (90~) of 4,6-di-t
butyl-5-hydroxybenzo[b]thiophene as a pale yellow solid.
1H NMR (270 MHz, CDC13)
8ppm: 1.48(s,9H), 1.71(s,9H), 5.64(s,lH),
- 38 -
2i8731~
7.31(d,lH,J=5.9Hz), 7.66(s,lH), 7.72(d,lH,J=5.9Hz)
IR(cm-1): 3644, 2952
Mass: 262(M+)
m.p.: 107.4°C
EXAMPLE 5: Synthesis of 4 6-Di-t-butyl-5-hydroxy-2 3-
dihydrobenzothiophene
1) Synthesis of 5-Acetoxy-4,6-di-t-
butyldioxobenzo[b]thiophene 1,1-Dioxide:
In 2 ml of acetic acid was dissolved 0.3 g of
5-acetoxy-4,6-di-t-butylbenzo[b]thiophene obtained in
Example 4-2), and 2.2 ml of a 35% hydrogen peroxide aqueous
solution was added thereto, followed by heating under reflux
for 1 hour. After cooling, water was added thereto, and
the mixture was extracted with ethyl acetate. The organic
layer was washed with a saturated aqueous solution of sodium
chloride, dried over anhydrous magnesium sulfate, and con-
centrated. The concentrate was purified by silica gel
chromatography (50% ethyl acetate in n-hexane) to give 0.3 g
(89$) of 5-acetoxy-4,6-di-t-butylbenzo[b]thiophene 1,1-
dioxide as a white solid.
1H NMR (60 MHz, CDC13)
Sppm: 1.35(s,9H), 1.43(s,9H), 2.33(s,3H),
6.63(d,lH,J=7.OHz), 7.56(s,lH), 7.68(d,lH,J=7.OHz)
Mass: 336(M+)
m.p.: 195.0°C
2) Synthesis of 5-Acetoxy-4,6-di-t-butyl-2,3-
dihydrobenzothiophene 1,1-Dioxide:
- 39 -
2187315
To a solution of 0.3 g of 5-acetoxy-4,6-di-t-
butylbenzo[b]thiophene 1,1-dioxide in 10 ml of ethyl acetate
was added 0.03 g of 10% palladium-on-carbon, and the mixture
was stirred under a hydrogen atmosphere for 24 hours.
After palladium-on-carbon was separated by filtration,
the filtrate was concentrated, and the concentrate was
purified by silica gel chromatography (50% ethyl acetate in
n-hexane) to give 0.27 g (90~) of 5-acetoxy-4,6-di-t-butyl-
2,3-dihydrobenzothiophene 1,1-dioxide as a white solid.
1H NMR (270 MHz, CDC13)
Sppm: 1.35(s,9H), 1.44(s,9H), 2.36(s,3H), 3.33-3.69(m,4H),
7.65(s,lH)
Mass : 338 ( M' )
m.p.. 182.0°C
3) Synthesis of 4,6-Di-t-butyl-5-hydroxy-2,3-
dihydrobenzothiophene:
In 10 ml of tetrahydrofuran was suspended 0.15 g
of lithium aluminum hydride under a nitrogen atmosphere.
A solution of 0.27 g of 5-acetoxy-4,6-di-t-butyl-2,3-
dihydrobenzothiophene 1,1-dioxide in 10 ml of tetrahydro-
furan was added to the suspension dropwise, followed by
heating under reflux for 3 hours. After cooling to room
temperature, a 10% hydrochloric acid aqueous solution was
added to the reaction mixture, and the mixture was extracted
with ethyl acetate. The organic layer was washed with
a saturated sodium chloride aqueous solution, dried over
anhydrous magnesium sulfate, and concentrated. The
concentrate was purified by silica gel chromatography
- 40 -
- 2187315
(n-hexane) to give 10 mg of 4,6-di-t-butyl-5-hydroxy-
2,3-dihydrobenzothiophene as a pale yellow solid.
1H NMR ( 270 MHz, CDC13 )
8ppm: 1.41(s,9H), 1.54(s,9H), 3.22(t,2H,J=7.6Hz),
3.53(t,2H,J=7.6Hz), 5.13(s,lH), 7.08(s,lH)
IR( cm-1 ) : 3640, 2956
Mass: 264(M')
m.p.. 82.0°C
EXAMPLE 6: Synthesis of 4 6-Di-t-butyl-5-hydroxy-2 3
dihvdrobenzothiophene-2-spiro-1'-cvclohexane
The title compound was obtained in the same manner as
in Example 1.
1H NMR (270 MHz, CDC13)
8ppm: 1.39(s,9H), 1.45-1.65(m,lOH), 1.53(s,9H), 3.34(s,2H),
5.10(s,lH), 6.96(s,lH)
IR(cm'1): 3644, 3620, 2924
Mass : 332 ( M+ )
m.p.: 128.5°C
EXAMPLE 7: Synthesis of 4 6-Di-t-butyl-5-hydroxy 2 (N N
dimethylaminomethyl)-2-methyl-2 3-dihydrobenzothiophene
1) Synthesis of 5-Acetoxy-4,6-di-t-butyl-2-iodomethyl-2-
methyl-2,3-dihydrobenzothiophene:
In 400 ml of a 3:1 mixture of diethyl ether and water
was dissolved 40 g of S-(4-acetoxy-3,5-di-t-butyl-2-(2-
methyl-2-propenyl)phenyl} N,N-dimethylthiocarbamate obtained
in Example 3-4), and 16.6 g of sodium hydrogencarbonate and
37.7 g of iodine were added to the solution, followed by
stirring at room temperature for 30 minutes. A saturated
- 41 -
2181315
aqueous solution of sodium thiosulfate was added to the
reaction mixture, and the mixture was extracted with ethyl
acetate. The organic layer was washed with a saturated
sodium chloride aqueous solution, dried over anhydrous
magnesium sulfate, and concentrated to give 45.3 g (990)
of 5-acetoxy-4,6-di-t-butyl-2-iodomethyl-2-methyl-2,3-
dihydrobenzothiophene as a pale yellow oil.
1H NMR (270 MHz, CDC13)
8ppm: 1.29(s,9H), 1.42(d,9H,J=0.7Hz), 1,69(d,3H,J=6.9Hz),
2.30(d,3H,J=2.OHz), 3.17(dd,lH,J=15.2Hz,J=l.3Hz),
3.52-3.72(m,2H), 3.79(d,lH,J=15.2Hz),
7.07(d,lH,J=4.3Hz)
Mass: 460(M+)
2) Synthesis of 5-Acetoxy-4,6-di-t-butyl-2-(N,N-
dimethylaminomethyl)-2-methyl-2,3-dihydrobenzothiophene:
In 40 ml of a 3:1 mixture of N,N-dimethylformamide
and water was dissolved 2.0 g of 5-acetoxy-4,6-di-t-butyl-
2-iodomethyl-2-methyl-2,3-dihydrobenzothiophene. To
the solution were added 2.47 g of N,N-dimethylamine
hydrochloride and 4.2 g of potassium carbonate, and the
mixture was stirred at room temperature for 24 hours.
Water was added to the reaction mixture, and the mixture
was extracted with n-hexane. The organic layer was washed
with a saturated sodium chloride aqueous solution, dried
over anhydrous magnesium sulfate, and concentrated. The
concentrate was purified by silica gel chromatography (33%
ethyl acetate in n-hexane) to give 1.6 g (98$) of 5-acetoxy-
4,6-di-t-butyl-2-(N,N-dimethylaminomethyl)-2-methyl-2,3-
- 42 -
2i873i5
dihydrobenzothiophene as a colorless oil.
1H NMR (270 MHz, CDC13)
8ppm: 1.29(s,9H), 1.39(s,9H), 1.54(d,3H,J=18.5Hz),
2.29(s,3H), 2.34(s,3H), 2.37(s,3H),
2.56(d,lH,J=5.9Hz), 2.66(d,lH,J=4.9Hz),
3.21(dd,lH,J=15.2Hz,J=5.9Hz),
3.44(dd,lH,J=17.5Hz,J=15.2Hz), 7.08(d,lH,J=3.3Hz)
Mass : 377 ( M' )
3) Synthesis of 4,6-Di-t-butyl-5-hydroxy-2-(N,N-
dimethylaminomethyl)-2-methyl-2,3-dihydrobenzothiophene:
In 10 ml of tetrahydrofuran was suspended 0.16 g
of lithium aluminum hydride under a nitrogen atmosphere,
and a solution of 1.6 g of 5-acetoxy-4,6-di-t-butyl-2-(N,N-
dimethylaminomethyl)-2-methyl-2,3-dihydrobenzothiophene in
30 ml of tetrahydrofuran was added dropwise thereto. The
mixture was heated under reflux for 3 hours and cooled to
room temperature. A saturated ammonium chloride aqueous
solution was added to the reaction mixture, and the mixture
was extracted with ethyl acetate. The organic layer was
washed with a saturated sodium chloride aqueous solution,
dried over anhydrous magnesium sulfate, and concentrated.
The concentrate was purified by silica gel chromatography
(20~ ethyl acetate in n-hexane) to give 1.29 g (91~) of
4,6-di-t-butyl-5-hydroxy-2-(N,N-dimethylaminomethyl)-2-
methyl-2,3-dihydrobenzothiophene as a colorless oil.
1H NMR (270 MHz, CDC13)
8ppm: 1.39(s,9H), 1.52(s,3H), 1.53(s,9H), 2.35(s,6H),
2.52(d,lH,J=13.5Hz), 2.58(d,lH,J=13.5Hz),
- 43 -
2187315
3.19(d,lH,J=15.2Hz), 3.55(d,lH,J=15.2Hz), 5.09(s,lH),
6.96(s,lH)
IR(cm'1): 3640, 2960
Mass: 335(M')
EXAMPLE 8: Synthesis of 4 6-Di-t-butyl-5-hydroxy-2
hydroxymethyl-2-methyl-2 3-dihydrobenzothiophene
1) Synthesis of 5-Acetoxy-2-acetoxymethyl-4,6-di-t-butyl-2-
methyl-2,3-dihydrobenzothiophene:
In 30 ml of hexamethylphosphoric triamide was
dissolved 2.0 g of 5-acetoxy-4,6-di-t-butyl-2-iodomethyl-2-
methyl-2,3-dihydrobenzothiophene prepared in Example 7-1),
and 0.71 g of sodium acetate was added thereto, followed
by stirring at room temperature for 24 hours. Water was
added to the reaction mixture, and the mixture was extracted
with ethyl acetate. The organic layer was washed with
a saturated sodium chloride aqueous solution, dried over
anhydrous magnesium sulfate, and concentrated. The
concentrate was purified by silica gel chromatography
(20% ethyl acetate in n-hexane) to afford 1.0 g (59%) of
5-acetoxy-2-acetoxymethyl-4,6-di-t-butyl-2-methyl-2,3-
dihydrobenzothiophene as a colorless oil.
1H NMR ( 270 MHz, CDC13 )
8ppm: 1.29(s,9H), 1.38(d,9H,J=l.OHz), 1.56(d,3H,J=3.3Hz),
2.05(d,3H,J=15.2Hz), 2.29(s,3H),
3.24(dd,lH,J=25.4Hz,J=15.2Hz),
3.57(dd,lH,J=18.1Hz,J=15.2Hz),
4.16(dd,lH,J=37.3Hz,J=11.2Hz), 4.18(s,lH),
7.08(d,lH,J=l.7Hz)
- 44 -
2181.315
Mass : 392 ( M' )
2) Synthesis of 4,6-Di-t-butyl-5-hydroxy-2-hydroxymethyl-2-
methyl-2,3-dihydrobenzothiophene:
In 10 ml of tetrahydrofuran was suspended 0.14 g of
lithium aluminum hydride under a nitrogen atmosphere, and
a solution of 0.6 g of 5-acetoxy-2-acetoxymethyl-4,6-di-
t-butyl-2-methyl-2,3-dihydrobenzothiophene in 20 ml of
tetrahydrofuran was added dropwise to the suspension. The
mixture was heated under reflux for 3 hours and cooled to
room temperature. A loo aqueous solution of hydrochloric
acid was added thereto, and the mixture was extracted
with ethyl acetate. The organic layer was washed with
a saturated aqueous solution of sodium chloride, dried
over anhydrous magnesium sulfate, and concentrated. The
concentrate was purified by silica gel chromatography
(20% ethyl acetate in n-hexane) to give 0.39 g (84%) of
4,6-di-t-butyl-5-hydroxy-2-hydroxymethyl-2-methyl-2,3-
dihydrobenzothiophene as a colorless oil.
1H NMR (270 MHz, CDC13)
8ppm: 1.39(s,9H), 1.52(s,9H), 1.53(s,3H),
1.98(t,lH,J=6.6Hz), 3.25(d,lH,J=15.5Hz),
3.46-3.60(m,2H), 3.59(d,lH,J=15.5Hz),
5.15(s,lH), 6.96(s,lH)
IR(cm-1): 3640, 3432, 2956
Mass: 308(M')
EXAMPLE 9: Synthesis of 4 6-Di-t-butyl-5-hydroxy-2-methyl
2-(4,8-dimethylnona-3(E) 7-dienyl)-2 3-dihydrobenzothiophene
1) Synthesis of 4,6-Di-t-butyl-5-hydroxy-2-methyl-2-(4,8-
- 45 -
- 2187315
dimethyl-2-p-toluenesulfonylnona-3(E),7-dienyl)-2,3-
dihydrobenzothiophene:
In 12 ml of a 4:1 mixture of tetrahydrofuran and
hexamethylphosphoric triamide was dissolved 1.52 g of 3,7-
dimethyl-1-(p-toluenesulfonyl)-2(E),6-octadiene prepared
according to Gosselin, P. et al., Synthesis, 876 (1984).
To the resulting solution was added dropwise 3.42 ml of
a 1.6M n-pentane solution of n-butyl lithium at -78°C,
followed by stirring for 2 hours. Then, a solution of
2.0 g of 5-acetoxy-4,6-di-t-butyl-2-iodomethyl-2-methyl-
2,3-dihydrobenzothiophene obtained in Example 7-1) in 10. m1
of tetrahydrofuran was added thereto dropwise, followed by
stirring for 4 hours. After completion of the reaction,
a saturated ammonium chloride aqueous solution was added
to the reaction mixture, and the mixture was extracted
with ethyl acetate. The organic layer was washed with
a saturated sodium chloride aqueous solution, dried over
anhydrous magnesium sulfate, and concentrated. The con-
centrate was purified by silica gel chromatography (10~
ethyl acetate in n-hexane) to provide 0.5 g of 4,6-di-t-
butyl-5-hydroxy-2-methyl-2-(4,8-dimethyl-2-p-toluene-
sulfonylnona-3(E),7-dienyl)-2,3-dihydrobenzothiophene
(a mixture of diastereomers) as a pale yellow oil.
1H NMR (270 MHz, CDC13)
8ppm: 1.12-1.73(m,l2H), 1.37(s,4.5H), 1.38(s,4.5H),
1.50(s,9H), 1.89-2.08(m,4H), 2.42(s,l.5H),
2.44(s,l.SH), 2.69-2.76(m,2H), 3.26-3.51(m,2H),
3.95-4.06(m,lH), 5.03-5.07(m,2H), 5.11(s,0.5H),
- 46 -
2187315
5.12(s,0.5H), 6.89(s,0.5H), 6.90(s,0.5H),
7.25-7.32(m,2H), 7.64-7.76(m,2H)
IR(cm-1): 3636, 2920
Mass : 582 ( M' )
2) Synthesis of 4,6-Di-t-butyl-5-hydroxy-2-methyl-2-(4,8
dimethylnona-3(E),7-dienyl)-2,3-dihydrobenzothiophene:
In 4 ml of tetrahydrofuran was dissolved 0.5 g of
4,6-di-t-butyl-5-hydroxy-2-methyl-2-(4,8-dimethyl-2-p-
toluenesulfonylnona-3(E),7-dienyl)-2,3-dihydrobenzothiophene
under a nitrogen atmosphere, and 48 mg of [1,4-bis(diphenyl-
phosphono)butane]palladium chloride prepared according to
Sugi, Y. et al., Chem. Lett., 1331 (1982) was added thereto
at 0°C. Then, 3.2 ml of a 1M tetrahydrofuran solution of
lithium triethylborohydride was added thereto dropwise,
followed by stirring at -20°C for 24 hours. After the
reaction, a saturated ammonium chloride aqueous solution
was added to the reaction mixture, and the mixture was
extracted with ethyl acetate. The organic layer was washed
with a saturated sodium chloride aqueous solution, dried
over anhydrous magnesium, and concentrated. The concentrate
was purified by silica gel chromatography (2% ethyl acetate
in n-hexane) to give 0.15 g (41°s) of 4,6-di-t-butyl-5-
hydroxy-2-methyl-2-(4,8-dimethylnona-3(E),7-dienyl)-2,3-
dihydrobenzothiophene as a colorless oil.
1H NMR (270 MHz, CDC13)
8ppm: 1.39(s,9H), 1.47(s,3H), 1.52(s,9H), 1.59(s,3H),
1.61(s,3H), 1.67(s,3H), 1.70-1.88(m,2H), 1.94-
2.18(m,6H), 3.31(d,lH,J=15.2Hz), 3.39(d,lH,J=15.2Hz),
- 47 -
2187313
5.06-5.19(m,2H), 5.10(s,lH), 6.97(s,lH)
IR( cm-1 ) : 3644, 2960
Mass : 428 ( M+ )
EXAMPLE 10: Synthesis of 4 6-Di-t-butyl-5-hydroxy 2 methyl
2-(4.8-dimethylnonyl)-2 3-dihydrobenzothiophene
In 20 ml of a 9:1 mixture of ethyl acetate and
acetic acid was dissolved 0.1 g of 4,6-di-t-butyl-5-
hydroxy-2-methyl-2-(4,8-dimethylnona-3(E),7-dienyl)-2,3-
dihydrobenzothiophene obtained in Example 9. To the
solution was added 0.5 g of 10% palladium-on-carbon,
followed by stirring for 24 hours under a hydrogen
atmosphere. Palladium-on-carbon was removed by filtration,
and the filtrate was concentrated. The concentrate was
purified by silica gel chromatography (4% ethyl acetate in
n-hexane) to give 0.09 g (91%) of 4,6-di-t-butyl-5-hydroxy-
2-methyl-2-(4,8-dimethylnonyl)-2,3-dihydrobenzothiophene as
a colorless oil.
1H NMR (270 MHz, CDC13)
sppm: 0.86(dd,9H,J=6.6Hz,J=3.OHz), 1.05-1.35(m,l2H),
1.39(s,9H), 1.45(s,3H), 1.52(s,9H), 1.56-1.83(m,2H),
3.30(d,lH,J=15.2Hz), 3.36(d,lH,J=15.2Hz), 5.10(s,lH),
6.97(s,lH)
IR(cm-1): 3648, 2952
Mass: 432(M')
EXAMPLE 11: Synthesis of 4 6-Di-t-butyl-5-hydroxy 2 methyl
2-(4,8,12-trimethytrideca-3(E) 7(E) 11-trienyl) 2 3
dihydrobenzothiophene
The title compound was obtained in the same manner as
- 48 -
218731 a
in Example 9.
1H NMR (270 MHz, CDC13)
8ppm: 1.39(s,9H), 1.47(s,3H), 1.52(s,9H), 1.59(s,6H),
1.61(s,3H), 1.68(s,3H), 1.71-1.88(m,2H),
1.90-2.19(m,lOH), 3.31(d,lH,J=15.2Hz),
3.38(d,lH,J=15.2Hz), 5.00-5.16(m,3H),
5.10(s,lH), 6.97(s,lH)
IR( cm-1 ) : 3644, 2960
Mass: 496(M+)
EXAMPLE 12: Synthesis of 4 6-Di-t-butyl-5-hydroxy 2 methyl
2-(4.8,12-trimethyltridecyl)-2 3-dihydrobenzothiophene
The title compound was obtained in the same manner as
in Example 10.
1H NMR (270 MHz, CDC13)
sppm: 0.83-0.88(m,l2H), 0.99-1.28(m,l8H), 1.39(s,9H),
1.45(s,3H), 1.52(s,9H), 1.55-1.75(m,3H),
3.30(d,lH,J=15.2Hz), 3.36(d,lH,J=15.2Hz),
5.10(.s,lH), 6.97(s,lH)
IR( cm-1 ) : 3648; 2952
Mass : 502 ( M+ )
The following Test Examples 1 to 3 will prove the
compounds of the invention excellent as an antioxidant.
[Test Case 1]
Amount of TBARS
Rabbit LDL was prepared in accordance with the method
of Havel et al. [Havel, R.J. et al., J. Clin. Invest., 34,
1345 (1955)]. After adding 5 uM of Cuz+, the mixture was
warmed until a thiobarbituric acid reactive substance
- 49 -
2181315
(THARS) was produced. The test compounds were evaluated
for their anti-oxidative action with the amount of TBARS
being used as an index.
TBARS produced =
TBARS produced when sample was added X 100 ( o)
TBARS produced in solvent
The results are shown in Table 1.
Table 1
THARS produced
($)
Compound at 10-6M of at 10-5M of
com ound compound
1 89.6 4.3
2 31.7 3.5
3 16.9 3.1
4 92.7 37.6
5 9.8 2.8
[Test Case 2]
Effect Against Lipid Peroxidation by Autoxidation of
Linoleic Acid
Using a cypridina luciferin analog (2-methyl-6-
(p-methoxyphenyl)-3,7-dihydroimidazo[1,2-a]pyrazin-3-one:
MCLA) as a sensitizer for lipid peroxyl radicals, the test
compounds were evaluated for their inhibitory effect against
the generation of lipid peroxyl radicals by autoxidation of
linoleic acid. A n-butanol solution (0.5 ml) containing
MCLA (0.2 uM) and linoleic acid (10 mM) was used in a
chemiluminescence measuring vial and the intensity of
- 50 -
217315
chemiluminescence due to the autoxidation of linoleic
acid was measured in a thermostatic bath at 37°C.
MCLA =
Change in chemiluminescence intensity
when sample was added x 100 (~)
Change in chemiluminescence intensity
when solvent was added
The results are shown in Table 2.
Table 2
MCLA ($)
Compound at 2 x 10-5M at 2 x 10-4M
of compound of com ound
1 7.2 0.9
2 26.8 1.1
3 9.4 0.9
4 73.3 19.7
17.7 0.7
[Test Case 3]
Effect Against Fluorescence Generation of Rabbit LDL
by AAPH
Using 2,2'-azobis(2-aminodipropane)hydrochloride
(AAPH) which was a radical initiator for a lipid peroxida-
tion that was not mediated by active oxygen (see Sato, K.
et al., Arch. Biochem. Biophys., 279, 402 (1990)], the
test compounds were evaluated for their inhibitory effect
against fluorescence generation in rabbit LDL. Rabbit LDL
was prepared in accordance with the method of Havel [Havel,
R.J. et al., J. Clin. Invest., 34, 1345 (1955)]; after
- 51 -
2187315
addition of AAPH (2 mM), the mixture was warmed at 37°C
for 24 h and LDL fraction was separated by gel-permeation
chromatography. The fluorescence intensity of LDL fraction
was measured by fluorometry at an excitation wavelength of
360 nm and at
an emission wavelength of 430 nm.
AAPH =
Fluorescence intensity of LDL fraction
when sample was added x 100 (~)
Fluorescence intensity of LDL fraction
when solvent was added
The results are shown in Table 3.
Table 3
Compound AAPH ( $ ) at 10-4M
of com ound
1 29.0
2 11.9
3 13.1
4 70.5
15.3
The results of Test Cases 1 - 3 obviously show that
the tested compounds of the invention had an excellent anti-
oxidative activity. The active oxygen induced by Cu2' in
the TBARS experimental model in Test Case 1 is believed
to be a direct radical initiator, so even a water-soluble
active oxygen scavenger will be effective in that model.
It should, however, be stressed that the tested compounds
of the invention also proved to be effective in the AAPH
- 52 -
- 2i873i5
using experimental model in Test Case 3 and, hence, it
became clear that those compounds also suppressed the chain
reaction for lipid peroxidation due to carbon-centered
radicals which could not be inhibited by water-soluble
active oxygen scavengers. This fact suggests that the
compounds of the invention show effective anti-oxidative
actions in LDL oxidation or lipid peroxidation.
Industrial Applicability
The compounds represented by formula (I) exhibit
an inhibitory action on the oxidative modification of
LDL and are useful as therapeutics of arteriosclerosis..
Further, the compounds represented by formula (II) are
useful as intermediates for synthesis of the compounds
of formula (I).
- 53 -