Note: Descriptions are shown in the official language in which they were submitted.
WO 95/28935 218 8695 PCTIEP95101546
PHARMACEUTICAL FORMULATIONS CONTAINING A BETA-LACTAMASE INHIBITING
PENEM IN COMBINATION WTCH A BETA-LACTAM ANTIBIOTIC AND THEIR USE IN THE
TREATIvIENT OF BACTERIAL INFEC7TONS
This invention relates to novel antibacterial formulations, in parricular to
formulations including 6-(substituted-methylene) penems and derivatives
thereof
having (3-lactamase inhibitory and antibacterial properties. The invention
also
relates to methods for the preparation of such formulations and to uses
thereof.
The compound ceftazidime, [6R-[6a,7R(Z)]1-1-[[7-[[(2-amino-4-
thiazolyl)[(1-carboxy-l-methylethoxy)imino]acetyl]amino]-2-carboxy-8-oxo-5-
thia-
1-azabicyclo[4.2.0]oct-2-en-3-yl] methyllpyridinium hydroxide inner salt, is a
known and much used cephalosporin antibiotic compound. Ceftazidime is normally
administered by injection as its pentahydrate. The term "ceftazidime" as used
herein includes all forms of ceftazidime including the free acid, hydrates,
salts and
ester thereof. Ceftazidime is susceptible to hydrolysis by (3-lactamase
enzymes, for
example those of B;fragilis, S.aureus and enterobacterfaceae producing
extended
spectrum (i-laactamases or elevated levels of Chiss 1 enzymes.
The compound cefotaxime, [6R-[6a,7R(Z)]]-3-[(acetyloxy)methyl]-7-[[(2-
amino-4-thiazolyl)(syn-methoxyimino)acetyl]amino-8-oxo-5-thia-l-
azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid, is a known and much used
cephalosporin antibiotic compound. Cefotaxime is normally administered by
injection as its sodium salt. The term "cefotaxime" as used herein includes
all
forms of cefotaxime including the free acid, salts and ester thereof.
Cefotaxime is
susceptible to hydrolysis by 0-lactamase enzymes, for example those of
B.fragilis,
Group 1 enzymes (typically encountered in Etuerobacter Citrobacter and
Pseudomonas), or showing mutational changes around the active site of the
Group 2
enzymes TEM-1 and SgIV-1 (typically encountered in E.coli and Klebsiella.
The compound atnoxycillin, 6-[D(-)-a.-amino-p-hydroxyphenyl-
acetamido]penicillanic acid, is a known and much used antibiotic compound.
Amoxycillin is normally administered orally in the form of amoxycillin
trihydrate,
or parenterally as sodium amoxycillin. The term "amoxycillin" as used herein
includes all forms of amoxyciIlin including the free acid, salts and ester
thereof.
Amoxycillin is hydrolysed by a broad range of (3-lactamase enzymes, and is
generally ineffective against organisms producing Group I and Group II(3-
lactamases. Therefore amoxyciIlin is often administered together with a(3-
lactamase inhibitor, for example clavulanic acid.
The compound piperacillin, 6-[[[[94-ethyl-2,3-dioxo- l-piperazinyl)
carbonyl]amino]phenylacetyl]amino]-3,3-dimethyl-7-oxo-4-thia-azabicyclo
[3.2.0]heptane-2-carboxylic acid, is a Imown and much used antibiotic
compound.
PiperaciIlin is normally administered parenterally as its sodium salt. The
term
"piperacillin" as used herein includes all forms of piperacillin including the
free
- - - - -
CA 02188695 2006-07-07
acid, salts and ester thereof. Piperacillin is hydrolysed by (3-lactamase
enzymes.
It is an object of this invention to provide novel combinations of 0-lactam
antibiotics with a0-lactamase inhibitor, having improved characteristics
compared
with known combinations.
According to the present invention, a pharmaceutical formulation
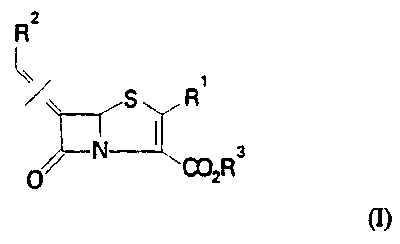
comprises, in combination with a penem of formula (I):
2
R
~ ~ S R 'a
0002R
in which:
R 1 is hydrogen or an organic substituent group;
R2 is a fused bicyclic heterocyclic ring system of general formula:
4
R P
PHy ~~_
N \N
s
R
wherein R4 and R5 are independently hydrogen or one or more substituents
replacing hydrogen atoms in the ring system shown; m is 2 or 3; p is zero, 1
or 2;
and R3 is hydrogen, a salt-forming cation or an ester-forming group; and the
symbol = / = indicates that the double bond may be in either the E or Z
configuration; and a pharmaceutically acceptable carrier; aP-lactam antibiotic
selected from the group consisting of cefotaxime, amoxycillin, piperacillin
and
ceftazidime, and their pharmaceutically acceptable derivatives including salts
and in
vivo hydrolysable esters.
Compounds of formula (I) are disclosed in W094/10178.
The compound of formula (I), its salts and esters, may exist in a number of
isomeric forms, all of which, including racemic and diastereoisomeric forms
are
encompassed within the scope of the formulations of the present invention.
Moreover, the compounds of formula (I) may exist in two isomeric forms
at the methylene group at the 8-position, ie the E- and Z- isomeric forms. The
Z-
isomer is generally preferred as generally being the more active form.
Consequently preferred forms of the compounds of the present invention
-2-
W O 95/28935 pCr/Ep95101546
i have the structure (IA):
2
R
H
~ R
0~ COZR
(IA)
In general formula (I), Rl denotes hydrogen or an organic group, which may
suitably be linked through a sulphur or carbon atom. For example, RI may
represent hydrogen or a group of formula -R5 or -SR5, where R5 denotes an
unsubstituted or substituted (Ci-10)hydrocarbon or heterocyclyl group.
Preferably, Rl represents hydrogen, (C1-10)a1ky1 or (C1-10)alkYlthio, or
substituted (C1-10)alkyl or substituted (C1-10)-alkYlthio, wherein the
substituent
may be hydroxy, (Cl-6)alkoxy, (CI-6)alkanoyloxy, halogen, mercapto,
(Cl-6)alkylthio, heterocyclylthio, amino, (mono or di)-(C1-6)alkylamino,
(C1-6)alkanoylamino, carboxy, or (Cl-6)alkoxycarbonyl.
Examples of suitable organic groups Rl include methyl, ethyl, propyl,
methylthio, ethylthio, methylsulphinyl, ethylsulphinyl, hydroxymethyl,
methoxymethyl, ethoxymethyl, acetoxymethyl, (1 or 2)-acetoxyethyl,
aminomethyl,
2-aminoethyl, acetamidomethyl, 2-acetamidoethyl, carboxymethyl,
2-hydroxyethylthio, methoxymethylthio, 2-methoxyethylthio, acetoxymethylthio,
2-aminoethylthio, acetanlidomethylthio, 2-acetamidoethylthio,
carboxymethylthio,
2-carboxyethylthio, aryl (especially phenyl), arylthio (especially
phenylthio),
pyridyl, pyrimidyl, isoxazolyl, pyrimidylthio, tetrazolylthio, and pyridylthio
groups.
In particular, Rl may be hydrogen.
Suitable groups R2 include: 2,3-dihydroimidazo[2,1-b]thiazol-6-yl, 2,3-
dihydro-l-(R,S)-oxoimidazo[2,1-b]thiazol-6-yl, 2,3-dihydro-1,1-
dioxoimidazo[2,1-
b]thiazol-6-yl, 6,7-dihydro-5H-imidazo[2,1-b]-thiazin-2-yl and 6,7-dihydro-8,8-
dioxo-5H-imidazo [2 ,1-b] [ 1, 3] thiazin-2-yl.
Examples of suitable substituents R4 and R5 include(Cl-6)alkanoyl,
(Cl-6)alkanoylozy, heterocyclyl, amino, (CI-6)allcanoylamino, (mono or
di)-(Cl-6)alkYlamino, hydroxy, (Cl-6)alkoxy, sulpho, mercapto, (Cl-
6)aikylthio,
(Cl-6)allcylsulphinyl, (Cl-6)alkyl-sulphonyl, heterocyclylthio, arylthio,
sulphamoyl,
carbamoyl, amidino, guanidino, nitro, halogen, carboxy, carboxy salts, carboxy
esters, arylcarbonyl, and heterocyclylcarbonyl groups, and also unsubstituted
or
substituted (C1-6)alkYl, (C2-6)alkenYl, (C2-6)alkYnYl, aryl, and azyl(C1-
6)albyl
groups.
Examples of suitable optional substituents for the above-mentioned
-3-
WO 95/28935 218U 6 95 PCT/Ep95/01546
(C1-6)alkyl, (C2-6)alkenYl, (C2-6)alkynYl, aryl and aryl(C1-6)alkyl
substitutents ~
include (CI-6)alkanoyl, (Cl-6)alkanoyloxy, heterocyclyl, amino,
(Cl-6)alkanoylamino, (mono or di)-(Cl-6)alkylamino, hydroxy,
(Cl-6)alkylsulphinyl, (Cl-6)atkYlsulphonyl, heterocyclylthio, arylthio,
sulphamoyl,
carbamoyl, amidino, guanidino, nitro, halogen, carboxy, carboxy salts, carboxy
esters,arylcarbonyl and heterocyclylcarbonyl groups.
Suitably R4 and R5 may both be hydrogen.
Suitable pharmaceutically acceptable salts of the 3-carboxylic acid group of
the (3-lactam antibiotic or the compound of formula (1) or of other carboxylic
acid
groups which may be present as optional substituents include those in which R3
is a
metal ion e.g. aluminium salts, allcati metal salts (e.g. sodium, lithium or
potassium
salts), alkaline earth metal salts (e.g. calcium or magnesium salts), ammonium
salts,
and substituted ammonium salts, for example those with lower alkylamines (e.g.
triethylamine), hydroxy-lower alkylamines (e.g. 2-hydroxyethylamine),
di(2-hydroxyethyl)amine tri(2-hydroxyethyl)amine), bis-(2-hydroxy ethyl)amine,
tris-(2-hydroxyethyl)amine, lower-alkylamines (e.g. dicyclohexylanrine), or
with
procaine, dibenzyhunine, jq,N-dibenzyl- ethylenediamine, 1-ephenamine, jq-
methylmorpholine, bl-ethylpiperidine, jq-benzyl-b-phenethylamine,
dehydroabietylamine, ethylenediamine, Z(,bl'-bishydroabietylethylenediamine,
bases
of the pyridine type (e.g. pyridine, collidine and quinoline), and other
amilles which
have been or can be used to form quatemary ammonium salts with penicillins.
Pharmaceutically acceptable salts may also be acid addition salts of any
amino or substituted amino group(s) that may be present as optional
substituents on
the compound of formula (1), or of any heterocyclic group ring nitrogen atoms.
Suitable salts include for example hydrochlorides, sulphates, hydrogen
sulphates,
acetates, phosphates etc. and other pharmaceutically acceptable salts will be
apparent to those sldlled in the art. Suitable addition salts are the
hydrochlorides
and hydrogen sulphates. Preferred salts are sodium salts.
When R3 is an ester-forming group it may be a carboxylate protecting
group or a pharmaceutically acceptable in-vivo hydrolysable ester.
Suitable ester-forming carboxyl-protecting groups are those which may be
removed under conventional conditions. Such groups for R3 include benzyl,
p-methoxybenzyl, benzoylmethyl, p-nitrobenzyl, 4-pyridylmethyl,
2,2,2-trichloroethyl, 2,2,2-tribromoethyl, 1-butyl, 1-antyl, allyl,
diphenylmethyl,
triphenylmethyl, adamantyl, 2-benzyloxyphenyl, 4-methylthiophenyl,
tettahydrofur-2-yl, tetrahydropyran-2-yl, pentachlorophenyl, acetonyl,
g-toluenesulphonylethyl, methoxymethyl, a silyl, stannyl or phosphorus-
containing
group, an oxime radical of formula -N=CHR6 where R6 is aryl or heterocyclyl,
or
an jn vivo hydrolysable ester radical such as defined below.
-4-
WO 95/28935 218 8 6 9 5
PCT/EP95101546
= A carboxyl group may be regenerated from any of the above esters by usual
methods appropriate to the particular R3 group, for example, acid- and base-
catalysed hydrolysis, or by enzymically-catalysed hydrolysis, or by
hydrogenolysis
under conditions wherein the remainder of the molecule is substantially
unaffected.
Examples of suitable pharmaceutically acceptable 'Lir vivo hydrolysable ester
groups include those which break down readily in the human body to leave the
parent acid or its salt. Suitable ester groups of this type include those of
part
formulae (i), (ii), (iii), (iv) and (v):
R
I
-COZCH-O.CO.Rb
Ra
/
-C02R' - N~
R GA
-CO2 CH2-ORf t'Ih]
R'
I ~Q-CO-H-R9
-COa -CHOCO---1 I
NH2
RkOC _ Rj - -- - _ -
-CO2Ri
Rh
wherein Ra is hydrogen, (CI4 alkyl, (C3-7) cycloaikyl, methyl, or
phenyl, Rb is (Cl-6) alkyl, (CI-6) alkoxy, phenyl, benzyl, (C3-7) cycloalkyl,
(C3-
7) cycloalkyloxy, (C1-6) allcyl (C3-7) cycloalkyl, 1-amino (Cl-6) alkyl, or 1-
(Cl-6
alkyl)amino (Cl-6) alkyl; or Ra and Rb together form a 1,2-phenylene group
optionally substituted by one or two methoxy groups; Rc represents (C1-6)
allcylene
optionally substituted with a methyl or ethyl group and Rd and Re
independently
represent (C1-6) alkyl; Rf represents (C1-6) alkyl; Rg represents hydrogen or
phenyl opdonally substituted by up to three groups selected from halogen, (C1-
6)
alkyl, or (C1-6) alkoxy; Q is oxygen or NH; Rh is hydrogen or (C1-6) allcyl;
Rt is
hydrogen, (C1-6) alkyl optionally substituted by halogen, (C2-6) allcenyl, (Cl-
6)
alkoxycarbonyl, aryl or heteroaryl; or Rh and Rl together form (C1-6)
alkylene; Ri
represents hydrogen, (Cl-6) alkyl or (C1-6) alkoxycarbonyl; and Rk represents
(Cl-
-5-
w0 95/28935 2188695 pCl'/Ep95/01546
8) alkYl, (C1-8) alkoxy, (C1-6) alkoxy(CI-6)aIkoxy or aryl. =
Examples of suitable in vivo hydrolysable ester groups include, for
example, acyloxyalkyl groups such as acetoxymethyl, pivaloyloxymethyl,
a-acetoxyethyl, a-pivaloyloxyethyl, 1-(cyclohexylcarbonyloxy)prop-l-yl, and
(1-aminoethyl) carbonyloxymethyl; alkoxycarbonyloxyalkyl groups, such as
ethoxy
carbonyloxymethyl, a-ethoxycarbonyloxyethyl and propoxycarbonyloxyethyl;
diaIlcylaminoalkyl especially di-lower alkylamino alkyl groups such as
dimethyl
aminomethyl, dimethyl aminoethyl, diethylaminomethyl or diethylaminoethyl;
2-(alkoxy carbonyl)-2-alkenyl groups such as 2-(isobutoxycarbonyl)pent-2-
enyland
2-(ethoxycarbonyl)but-2-enyl; lactone groups such as phthalidyl and dimethoxy
phthalidyl; and esters linked to a (second) (3-lactam antibiotic or (3-
lactamase
inhibitor.
A further suitable pharmaceutically acceptablejll vivo hydrolysable ester
group is that of the formula:
-C02aHZR k
C~C
0
wherein Rk is hydrogen, C1-6 alkyl or phenyl.
When used herein the term 'aryl' includes phenyl and naphthyl, each
optionally substituted with up to five, preferably up to three, groups
selected from
halogen, mercapto, (Cl-6) alkyl, phenyl, (C1-6) alkoxy, hydroxy(Cl-6)alkyl,
mercapto(C1-6)alkyl, halo(CI-6) allcyl, hydroxy, amino, nitro, carboxy, (C1-6)
alkylcarbonyloxy, alkoxycarbonyl, formyl, or (C1..6) alkylcarbonyl groups.
The terms 'heterocyclyl' and 'heterocyclic' as used herein include aromatic
and non-aromatic, single and fused, rings suitably containing up to four
hetero-atoms in each ring selected from oxygen, nitrogen and sulphur, which
rings
may be unsubstituted or substituted by, for example, up to three groups
selected
from halogen, (Cl{)aIlcy1, (C1-6)alkoxy, halo(Cl-6)aikyl, hydroxy, carboxy,
carboxy salts, carboxy esters such as (Cl4alkoxycarbonyl, (Cl-
6)alkoxycarbonyl(C1-6)alkyl, aryl, and oxo groups. Each heterocyclic ring
suitably
has from 4 to 7, preferably 5 or 6, ring atoms. The term 'heteroaryl' refers
to
heteroaromatic heterocyclic ring or ring system, suitably having 5 or 6 ring
atoms in
each ring. A fused heterocyclic ring system may include carbocyclic rings and
need
include only one heterocyclic ring. Compounds within the invention containing
a
heterocyclyl group may occur in two or more tautometric forms depending on the
nature of the heterocyclyl group; all such tautomeric forms are included
within the
scope of the invention.
-6-
W O 95/28935 218869[ p~ygp95l01546
When used herein the terms 'allcyl', 'allcenyl', 'alky ny1J' and 'alkoxy'
include straight and branched chain groups containing from 1 to 6 carbon
atoms,
such as methyl, ethyl, propyl and butyl. A particular alkyl group is methyl.
When used herein the term 'halogen' refers to fluorine, chlorine, bromine
and iodine.
It will be appreciated that also included within the scope of the invention
are formulations which utilise salts and carboxy-protected derivatives,
including in
vivo hydrolysable esters, of any carboxy groups that may be present as
optional
substituents in compounds of formula (I).
Certain compounds of formula (I) may include an amino group which may
be protected. Suitable amino protecting groups are those well known in the art
which may be removed under conventional conditions if required without
disruption
of the remainder of the molecule.
Examples of amino protecting groups include (C1-6) aikanoyl; benzoyl;
benzyl optionally substituted in the phenyl ring by one or two substituents
selected
from (C1-4) alkyl, (C1-4) alkoxy, trifluoromethyl, halogen, or nitro; (C1-4)
alkoxycarbonyl; benzyloxycarbonyl or trityl substituted as for benzyl above;
allyloxycarbonyl,trichloroethoxycarbonyl or chloroacetyl.
Some compounds of formula (1) and (IA) may be crystallised or
recrystallised from solvents such as organic solvents. In such cases solvates
may be
formed. This invention includes within its scope stoichiometric solvates
including
hydrates as well as compounds containing variable amounts of solvents such as
water that may be produced by processes such as lyophilisation. Compounds of
formula (I) and (IA) may be prepared in crystalline form by for example
dissolution
of the compound in water, preferably in the minimum quantity thereof, followed
by
admixing of this aqueous solution with a water miscible organic solvent such
as a
lower aliphatic ketone such as a di-(C1-6) alkyl ketone, or a(Cl-6) alcohol,
such as
acetone or ethanol.
The compounds of formula (I) and (IA) are (i-lactamase inhibitors and/or
antibiotics and are intended for use in pharmaceutical compositions. Therefore
it
will readily be understood that they are preferably each provided in
substantially
pure form, for example at least 60% pure, more suitably at least 75 b pure and
preferably at least 85 % pure, especially at least 95 % pure particularly at
least 98 %
pure (% are on a weight for weight basis). Impure preparations of the
compounds
may be used for preparing the more pure forms used in the pharmaceutical
compositions; these less pure preparations of the compounds should contain at
least
1%, more suitably at least 5% and preferably from 10 to 59 % of a compound of
the
formula (I) or (IA) or ester or salt thereof.
Compounds of formula (I), and in particular of formula (IA) are believed to
-7-
WO95/28935 2188695 PCl'/EP95/01546
be active (i-lactamase inhibitors, and to have the further advantage of
improved
pharmacokinetics.
Accordingly, specific compounds of formula (I) include the following
pharmaceutically acceptable salts:
Sodium (5R)-6-[(Z)-(2,3-dihydroimidazo[2,1-b]thiazol-6-yl) methylene]-
penem-3-carboxylate.
Sodium (5R)-6-[(Z)-(2,3-dihydro-1(R,S)-oxoimidazo[2,1-b]thiazol-6-
yl)methylene]penem-3-carboxylate.
Sodium (5R)-6-[(Z)-(2,3-dihydro-l,l-dioxoimidazo[2,1-b]thiazol-6-
yl)methylene]penem-3-carboxylate.
Sodium (5R)-6-[(Z)-(6,7-dihydro-5H-imidazo[2,1-b][1,3]thiazin-2-
yl)methylene]penem-3-carboxylate.
Sodium (5R)-6-[(Z)-(6,7-dihydro-8,8-dioxo-SH-imidazo[2,1-b][1,3] thiazin-
2-yl)methylene] penem-3-carboxylate.
Compounds of formula (I) as defined above may be prepared by subjecting
a compound of the formula (II):
z
2 X
R ~R
N I
x
O C02R -
(11)
wherein Rl and R2 are as defined in formula (I) above, Rx is a carboxy-
protecting group, X is a halogen atom, and Z denotes a halogen atom, a hydroxy
group, a substituted hydroxy group, an -S(O)qR7 group or an -Se(O)rR7 group
where q denotes 0,1 or 2, r denotes 0 or 1, and R7 denotes a hydrogen atom, a
hydrocarbon group or a heterocyclyl group, to a reductive elimination reaction
to
eliminate the elements of the group X and Z,
and thereafter if necessary or desired:
(i) converting the group Rx to a different group Rx such as a
substituent R3,
('ri) converting the group R2 into a different group R2,
('iii) converting the group OR5 into a different group OR5.
(iv) converting the compound into a pharmaceutically acceptable salt.
The reductive elimination reaction may be carried out in a manner known
per se for such elimination reactions for example as described in EP 0232966A.
The elimination may for example be carried out by reaction with a metal, for
example zinc, magnesium, aluminium, or iron, in the presence of an acid (for
example, acetic acid or a mineral acid), or by reaction with a
triorganophosphorus
-8-
2188695
WO 95/28935 PCT1EP95101546
compound, for example triphenyiphosphine, suitably at a temperature within the
range of from -20 C to +40'C, preferably from 0 C to 20 C. The reaction may
be carried out in the presence of a polar or non-polar, protic or aprotic,
organic
solvent, for example dioxane, dimethoxyethane, or tetrahydrofuran.
The product of this reaction is generally a mixture of isomers of the E and
Z isomers of formula (1). The desired isomer of the general formula (I) may be
isolated and purified in conventional manner for example by known
crystallisation
or chromatographic techniques. Moreover, the carboxy group -COORx may be
deprotected, that is to say, converted to a free carboxy, carboxy salt or
carboxy
ester group -COOR3 in a conventional manner, for example as described in
EP0232966A.
When it is desired to obtain a free acid or salt of the preferred penem
isomer of the formula (I) from such an isomeric mixture, this may be effected
by
chromatographic separation of the product followed by deprotection of the
desired
isomer to give the corresponding free acid or salt. In some cases, however, it
has
been found particularly convenient first to deprotect the isomeric mixture to
give an
isomeric mixture of the free acid or salt of formula (1), followed by
fractional
recrystallisation to give the desired acid or salt isomer.
Compounds of formula (II) in which Z is a hydroxy group may be prepared
by the reaction of known (see EP0232966) compounds of formula (III):
z
RZ X S Rl
N "
O ~'OZR
where X, Rl and Rx are as defined in formula (II), with an aldehyde of
formula (IV):
R2-CHO - - - (IV)
where R2 is as defined in formula (II), thereby forming the corresponding
halohydrin of formula (II).
The reaction between the compound (III) and the aldehyde (IV) may
suitably be carried out in the presence of a base, preferably a non-
nucleophilic base,
and preferably a strong base. Suitable bases include, for example, Hthium
amide
bases, for example lithium bistrimethyl silylamide, lithium dicyclohexlamide,
lithium diisopropylamide, lithium 2,2,6,6-tetramethylpiperidide, lithium
diphenylarnide, and butyllithium.
-9-
Llt~b by~
WO 95/28935 PCTIEP95/01546
Suitable solvents for the reaction are aprotic organic solvents (which may =
be polar or non-polar), for example tetrahydrofuran, toluene, dimethoxyethane,
dimethylformamide, and mixtures of two or more such solvents.
The reaction may suitably be carried out at a temperature within the range
of from -100 C to ambient temperature, preferably from -85 C to 0 C,
especially
from -85 C to 40 C.
The aldehyde of the general formula (IV) and the base may be added to the
halo-penem (III) in either order. If it is desired to isolate the halohydrin-
penem of
the general formula (II) in which Z denotes a hydroxy group, the reaction
mixture
may conveniently be quenched by adding a protic reagent, for example an acid,
such as acetic acid or citric acid, or water.
Aldehydes of formula (IV) may be prepared from known (e.g. Reuben G
Jones, CA: (45) 7153e, US Patent 2, 541, 924) compounds of formula (V):
SH
~N
Rs COZR
(V)
where R is alkyl, e.g. (CI-6) alkyl, and R5 is as defined above, by reaction
with known compounds of formula (VI):
X-(CH2)m-Y
(VI)
where m is as defined above, and X and Y are halogen, preferably chlorine
or bromine. Preferably one of X or Y is chlorine and the other is bromine. A
compound of formula (VII) is formed:
s-cH2L it
'
'
H N
R CO2R
(Vil)
The reaction between compounds (V) and (VI) may be carried out in an
organic solvent e.g. DMF, in the presence of a base, such as triethylamine.
The compound (VII) may be cyclised, for example by treatment with an
alkali metal hydride such as sodium hydride in a solvent such as THF, to form
a
-10-
_
WO 95/28935 218 8 6 9 5 PCT/M5/01546
~ compound (V1II):
s
PHA
N N
R CO2R
(VIII)
Compounds of formula (VIII) may then be converted to compounds of
formula (IV) by various procedures.
For example the CO2R group of the compound (VIII) may be reduced,
using for example di-isobutylaluminium hydride to form the corresponding
aldehyde
(IV) having p as zero. The corresponding aldehyde (IV) having p as 1 or 2 may
then be prepared by oxidation of the S atom using a peroxy acid such as
chloroperbenzoic acid.
Alternatively for example the compound (VIII) may be treated with a
peroxy acid, e.g. as above, to oxidise the S atom and form the sulphoxide or
sulphone analogue of compound (VIII), followed by reduction of the CO2R group
to an aldehyde group e.g. as above to form an aldehyde (IV) having p as 1 or
2.
Alternatively for example the CO2R group of compound (VIII) may be
partly reduced to form the corresponding hydroxymethyl compound (M:
s
kUH~
N N
R* CH2-0H
OIX)
for example using lithium aluminium hydride. The hydroxymethyl
compound (IX) may then for example be further oxidised, e.g. using Mn(IV),
e.g.
Mn02, to form the corresponding aldehyde (IV) having p as zero, and which may
then be further oxidised using a peroxy acid to form an aldehyde (IV) having p
as 1
or 2.
Alternatively the hydroxymethyl compound (IIQ may be oxidised using a
peroxy acid, e.g. as above to form the corresponding sulphoxide or sulphone
(IX),
and this sulphoxide or sulphone may then be further oxidised, e.g. using Mn
(IV) as
above to convert the hydroxymethyl group of (IX) to an aldehyde group, to form
an
aldehyde (IV) having p as 1 or 2.
Alternatively for example the hydroxymethyl compound (IX) may be
-il-
WO 95l28935 21" " 695 PCT/EP9S/01546
acylated to form a compound (X): =
s
(Cxz
N
~z-0~ -
R
(X)
where A is an acyl group, for example a(C1_6) acyl group such as acetyl.
Acylation may be by the use of an acylating derivative of A, for example an
acyl
halide or an acid anhydride. The compound (X) may then be oxidised using a
peroxy acid to form the corresponding sulphoxide or sulphone. The
hydroxymethyl
group may then be regenerated, e.g. by treatment with methanolic ammonia,
followed by oxidation of the hydroxymethyl group e.g. using Mn (IV) as above
to
form the corresponding aldehyde group in an aldehyde (IV).
Compounds of formula (II) in which Z is a substituted hydroxy group or a
group of formula -S(O)qR7 or -Se(O)rR7 may be prepared from compounds of
formula (II) in which Z is hydroxy by known methods, for example as described
in
EP 0232966A.
When Rx is a carboxylate protecting group, such as 4-methoxybenzyl, these
protecting groups may be removed to form the parent acid by methods well known
in the art, for example in the case of 4-methoxybenzyl treatment with a Lewis
acid
such as ethyl aluminium dichloride or aluminium chloride. Pharmaceutically
acceptable salts may be prepared from such acids by treatment with a base,
after a
conventional work-up if necessary. Suitable bases include sodium hydrogen
carbonate to form sodium salts.
Crystalline forms of the compounds of formula (I) may for example be
prepared by dissolving the compound (I) in the minimum quantity of water,
suitably
at ambient temperature, then adding a water miscible organic solvent such as
a(C1-
6) alcohol or ketone such as ethanol or acetone, upon which crystalisation
occurs
and which may be encouraged for example by cooling or trituration.
The compounds of formula (I) have (3-lactamase inhibitory and antibacterial
properties. Compounds of formula (I) provide (i-lactam antibiotics with
protection
against the P-lactamase enzymes of such microorganisms as B.fragilis,
S.aureus,
and strains of Enterobacteriaceae producing extended spectrum (3-lactamases,
strains producing high levels of Group 1(3-lactamase, K.pneumoniae, and Ent.
Cloacae.
Compounds of formula (I) provide amoxycillin with protection against most
of the medically important Group I and Group 11 (3-lactamase producing
organisms
-12-
R'O 95/28935 2188695 pC'1J&P95101546
= at in vitro concentrations as low as 0.25 g/ml. Protection is also observed
against
problem (3-lactamase producing organisms such as high level (3-lactamase
producing
strains of Ent.Cloacae P99 and E.coli JT4 (Group IIb). Synergy between
compounds of formula (I) and amoxycillin is also observed against B;fragilis,
Q-
lactamase producing S.aureus, and against most Gram negative producing Group
II
or inducible Group I(i-lactamases, and against organisms producing high levels
of
Group I(i-lactamase. Protection against the (3-lactamases produced by
Pseudomonas aeruginosa is also observed.
The pharmaceuticai formulations of the invention are useful for the
treatment of infections in animals, especially mammals, including humans, in
particular in humans and domesticated (including farm)animals. The
formulations
of this invention may be used, for example, for the treatment of infections
for
which a(3-lactam antibiotic is normally administered, for example of, inter
alia, the
respiratory tract, urinary tract, and soft tissues, especially in humans. The
formulations of this invention may be used for the treatment of infections
caused by
strains of, for example, the organisms mentioned above.
Some compounds of formula (I), for example sodium (5R)-6-[(Z)-(2,3-
dihydroimidazo[2,1-b]thiazol-6-yl)methylene]penem-3-carboxylate, appear to
have
an advantageously long serum half life. The compound of formula (I) or (IA)
and
the p-lactam antibiotic can be administered separately or in the form of a
single
formulation containing both active ingredients as discussed in more detail
below.
The compounds of formula (I) or (IA) may be formulated for
administration in any convenient way for use in human or veterinary medicine,
by
analogy with other antibiotics. The compounds of formula (1), particularly
(IA) are
pardcularly suitable for parenteral administration.
The formulation may be formulated for administration by any mute, such
as oral, topical or parenteral. The compositions may be in the form of
tablets,
capsules, powders, granules, lozenges, creams or liquid preparations, such as
oral
or sterile parenteral solutions or suspensions.
The topical formulations of the present invention may be presented as, for
instance, ointments, creams or lotions, eye ointments and eye or ear drops,
impregnated dressings and aerosols, and may contain appropriate conventional
additives such as preservatives, solvents to assist drug penetration and
emollients in
ointments and creams.
The formulations may also contain compatible conventional carriers, such
as cream or ointment bases and ethanol or oleyl alcohol for lotions. Such
carriers
may be present as from about 1% up to about 98 % of the formulation. More
usually they will form up to about 80% of the formulation.
-13-
- - - -
WO 95/28935 PGTIEP95/01546
~
218$05
Tablets and capsules for oral administration may be in unit dose
presentation form, and may contain conventional excipients such as binding
agents,
for example syrup, acacia, gelatin, sorbitol, tragacanth, or
polyvinylpyrollidone;
fillers, for example lactose, sugar, maize-starch, calcium phosphate, sorbitol
or
glycine; tabletting lubricants, for example magnesium stearate, talc,
polyethylene
glycol or silica; disintegrants, for example potato starch; or acceptable
wetting
agents such as sodium lauryl sulphate. The tablets may be coated according to
methods well known in normal pharmaceutical practice. Oral liquid preparations
may be in the form of, for example, aqueous or oily suspensions, solutions,
emulsions, syrups or elixirs, or may be presented as a dry product for
reconstitution
with water or other suitable vehicle before use. Such liquid preparations may
contain conventional additives, such as suspending agents, for example
sorbitol,
methyl cellulose, glucose syrup, gelatin, hydroxyethyl cellulose,
carboxymethyl
cellulose, aluminium stearate gel or hydrogenated edible fats, emulsifying
agents,
for example Iecithin, sorbitan monooleate, or acacia; non-aqueous vehicles
(which
may include edible oils), for example almond oil, oily esters such as
glycerine,
propylene glycol, or ethyl alcohol; preservatives, for example methyl or
propyl
p-hydroxybenzoate or sorbic acid, and, if desired, conventional flavouring or
colouring agents.
Suppositories will contai;7 conventional suppository bases, e.g. cocoa-butter
or other glyceride.
For parenteral administration, fluid unit dosage forms are prepared utIlizntg
the compound of formula (I), R-lactam and a sterile vehicle, water being
preferred.
These compounds, depending on the vehicle and concentration used, can be
either
suspended or dissolved in the vehicle. In preparing solutions these compounds
can
be dissolved in water for injection and fiiter sterilised before filling into
a suitable
vial or altipoule and sealing.
Advantageously, agents such as a local anaesthetic, preservative and
buffering agents can be dissolved in the vehicle. To enhance the stability,
the
formulation can be frozen after filling into the vial and the water removed
under
vacuum. The dry lyophilized powder is then sealed in the vial and an
accompanying vial of water for injection may be supplied to reconstitute the
liquid
prior to use. Parenteral suspensions are prepared in substantially the same
manner
except that the compound is suspended in the vehicle instead of being
dissolved and
sterilization cannot be accomplished by ffitration. The compound can be
sterilised
by exposure to ethylene oxide before suspending in the sterile vehicle.
Advantageously, a surfactant or wetting agent is included in the composition
to
facilitate uniform distribution of the compound.
The formulations may contain from 0.1 % by weight, preferably from
-14-
_
WO 95/28935 PCr1EP95101546
10-60% by weight, of the active materials, cpend ng 9 on5the method of
administration. Where the formulations comprise dosage units, each unit will
preferably contain from 50-500 mg of the active ingredient. The dosage as
employed for adult human treatment will preferably range from 100 to 3000 mg
per
day, for instance 1500 mg per day depending on the route and frequency of
administration. Such a dosage corresponds to 1.5 to 50 mg/kg per day. Suitably
the dosage is from 5 to 20 mg/kg per day.
No toxicological effects are indicated when a formulation in accordance
with the invention is administered in the above-mentioned dosage range.
A formulation according to the invention may comprise a single (i-lactam
antibiotic and a compound of formula (I) or (IA) as the sole active
ingredients or
therapeutic agents, or it may also comprise one or more additional active
ingredients
or therapeutic agents, for example a second 0-lactam antibiotic, or pro-drug
thereof.
Ceftazidime may be used in the form of the free acid, e.g as its pentahydrate.
Cefotaxime may be used in the form of the free acid or its pharmaceutically
acceptable salts, for example its sodium salt.
Amoxycillin may be used in the form of amoxycillin trihydrate or its
pharmaceutically acceptable salts, for example its sodium salts.
Alternatively,
amoxycillin may be used in the form of fine particles of its zwitterionic form
(generally as amoxyciIlin trihydrate) for use in an injectable or infusable
suspension,
for example, in the manner hereinbefore described. Amoxycillin in the form of
its
sodium salt or the trihydrate is particularly preferred for use in synergistic
compositions according to the invention.
Piperacillin may be used in the form of its pharmaceutically acceptable salts,
for example its sodium salts, in an injectable or infusable suspension, for
example,
in the manner hereinbefore described. PiperaciIIin in the form of its sodium
salt is
particularly preferred for use in synergistic formulations according to the
invention.
A compound of formula (1) or (IA) may be administered to the patient in a
synergistically effective amount, together with R-lactam antibiotic.
The compounds of formula (I) or (IA) may suitably be administered to the
patient at a daily dosage of from 0.7 to 50 mg/kg of body.weight. For an adult
human (of approximately 70 kg body weight), from 50 to 3000 mg, preferably
from
100 to 1000 mg, of a compound according to the invention may be adminstered
daily, suitably in from 1 to 6, preferably from 2 to 4, separate doses..
Higher or
lower dosages may, however, be used in accordance with clinical practice.
When the compositions according to the invention are presented in unit
dosage form, each unit dose may suitably comprise from 25 to 1000 mg,
preferably
from 50 to 500 mg, of a compound of formula (I). Each unit dose may, for
example, be 62.5, 100, 125, 150, 200 or 250 mg of a compound of formula (1).
-15-
WO 95/28935 2 1 Q Q6(1 5 PCT/EP95/01546
The ratio of the amount of the compound ofvfovrmuIla (1) to the amount of
lactam antibiotic(s) may vary within a wide range. The said ratio may, for
example, be from 100:1 to 1:100; more particularly, it may, for example, be
from
2:1 to 1:30.
The amount of (3-lactam antibiotic administered in a formulation according to
the invention, i.e in unit doses or the total amount per day will normally be
approximately similar to the amount in which it is conventionally used = =,.
The amount of cefotaxime in a formulation according to the invention will
normally be approximately similar to the amount in which it is conventionally
used
= m, for example 1-2g intraveneously or intraveneously every 8 hours, up to a
maximum of 12g daily.
The amount of amoxycillin in a formulation according to the invention will
normally be for example from about 50 mg, advantageously from about 62.5 mg,
to
about 3000 mg per unit dose, more usually about 125, 250, 500, 625, 875 or
1000
mg per unit dose up to the normal maximum with or daily dose of amoxycillin.
The present invention provides a formulation as described above for use as
a therapeutic agent.
The present invention further provides a formulation as described above for
use in the treatment of bacterial infections.
The present invention includes a method of treating bacterial infections in
humans and animals which comprises the administration of a therapeutically
effective amount of a formulation as described above.
The present invention also includes the use of formulation as described
above, in the manufacture of a medicament for the treatment of bacterial
infections,
either alone or in combination.
The following Examples illustrate compounds of formula (I), intermediates
in their preparation, and synergistic effect of these with (3-lactam
antibiotics.
-16-
WO 95/28935 2188695 PCTIEP95101546
Preparation 1.
2,3-D'ehydroimidaxo[2,1-b]thiazole-6-carboxaldehyde
Method 1
a) Ethy12,3-dihydroimidazo[2,1-b]thiazole-6-carboxylate - Ethy12-
mercaptoimidazole-4(or 5)-carboxylate (1.27g, 10mmol) was dissolved in the
minimum volume of N,IV-dimethylformamide (DMF) and treated with triethylamine
(1.11g, 1lmmol). This solution was added dropwise to a rapidly-stirred
solution of
1,2-dibromoethane (9.4g, 50mmo1) in DMF (5m1). After 0.5h, the reaction
mixture was poured into a mixture of ethyl acetate (100m1) and water (50m1).
The
organic phase was washed with water (5 x 50m1), dried over anhydrous magnesium
sulphate and evaporated to dryness under reduced pressure to give an orange
oil.
Chromatography on silica gel, eluting with mixtures of ethyl acetate in
hexane, gave
ethy12-(2-bromoethylthio)imidazole-4(or 5)-carboxylate as a white solid (1.2g,
4.3mmol; 61 %).
The above white solid was added portionwise to a stirred suspension of
sodium hydride (206mg of a 50% dispersion in oil, 4.3mmol) in dry, redistilled
tetrahydrofuran (THF) under argon at room temperature. After 0.5h, the
reaction
mixture was treated carefully with water (5m1) and the mixture filtered
through
Celite. The filtrate was evaporated to dryness under reduced pressure, re-
evaporated (2x) with ethanol and purified by chromatography on sifica gel,
eluting
with ethyl acetate, to give the title compound as a white solid (0.72g, 81 %),
mp
107-109 C (dichloromethane-hexane) (Found: C, 48.25; H, 4.87; N, 14.17; S,
16.34%; M+ 198.0465. C8H10N202S requires C, 48.48; H, 5.05; N, 14.14; S,
16.16%; 198.0463); nmax (CH202) 1722, 1703, 1270 and 1260cm-1; dH
(250MHz; CD3OD) 1.33 (3H, t, J 7Hz), 3.92 (2H, t, J 7Hz), 4.24-4.38 (4H, m),
7.81 (1H, s).
b) 2,3-Dihydro-6-hydroxymethylimidazo[2,1-b]thiazole - Lithium aluminium
hydride (280mg, 7.3mmol) was suspended in dry, redistilled THF (20m1) under
argon and treated dropwise with a solution (THF, 20m1), of ethy12,3-
dihydroimidazo[2,1-b]thiazole-6-carboxylate (1.32g, 6.7mmol). After 2h, water
was added carefully until effervescence ceased when the mixture was filtered
through Celite, the filter pad washed with TFF and water and the filtrate and
washings combined and evaporated to dryness under reduced pressure. The
residue
was evaporated twice from ethanol to give the title compound as a white solid
(1.03g, 100%); dH (250MHz; CD3OD) 3.73-3.95 (2H, m), 4.06-4.30 (2H, m),
4.42 (2H, s), 7.04 (1H, s).
-17-
WO 95/28935 21 88695 PGT/EP95/01546
c) 2,3-Dihydroimidazo[2,1-b]thiazole-6-carbl oxaldehyde - 2,3-Dihydro-6- =
hydroxymethylimidazo[2,1-b]thiazole (1.47g, 9.4mmol) was dissolved in
acetonitrile (30m1) by addition of.water (minimum volume). Manganese dioxide
(4.41g, 3wt. equivalents) was added and the niixture stirred at ambient
temperatures
for 1.5h. The mixture was filtered through Keiselguhr, the filter pad washed
with
water and the combined filtrate and washings evaporated to dryness under
reduced
pressure. The residue was triturated under diethyl ether, the solid collected
by
filtration and dried in air (1.33g, 92%); nmax (CH2C12) 1685, 1528, 1272, 1260
and 1152cm-1; dH (90MHz; CD30D) 3.84-4.10 (2H, m), 4.20-4.50 (2H, m), 7.97
(1H, s), 9.52 (1H, s).
Method 2
2,3-Dihydroimidazo[2,1-b]thiazole-6-carboxaldehyde - Ethyl 2,3-
dihydroimidazo[2,1-b]thiazole-6-carboxylate (4.2g; 21.21mmo1) was dissolved in
=dry dichloromethane (150m1) and cooled to -70 C under a stream of dry argon.
This solution was treated with a solution of diisobutylaluminium hydride in
toluene
(1.5M, 26.9m1, 2 equivalents) over 40 minutes at -70 C. The reaction mixture
was
stirled at -70 C for a further 0.5h. Water (10m1) was added and the mixture
stirred
at ambient temperature for 0.5hr. The nrixture was acidified with 5M HCI,
filtered
through a celite pad and the pad further washed with dichloromethane. The
combined organic extracts were dried over anhydrous magnesium sulphate and
evaporated to dryness. Chromatography on silica gel, eluting with ethyl
acetate,
gave the title compound (1.4g, 43 %).
Preparation 2
2,3-Dihydroimidazo [2,1-b]tiliazole-6-carboxaldehyde-1(R, S)-oxide
Method 1
2,3-Dihydroimidazo[2,1-b]thiazole-6-carboxaldehye-1(R,S)-oxide - 2,3 -
Dihydroimidazo[2,1-b]thiazole-6-carboxaidehyde (154mg, lmmol) was dissolved in
dichloromethane (minimum volume) and the solution cooled to 0-5 C. m-
Chloroperbenzoic acid (604b pure, 287.6mg, lmmol) was added and the mixture
stirred at 0-5 C for 0.5h. Diethyl ether was added, which dissolved the
existing
precipitate and eventually produced a new precipitate. This new precipitate
was
collected by filtration, washed with diethyl ether and dried in air (128mg,
75%)
(Found: M+ 170.0149. C6H6N202S requires M 170.0150); nmax (CH2C12)
1697, 1268 and 1259cm-1; dH (250MHz; CD3OD) 3.69-3.88 (IH, m), 3.94-4.11
(IH, m), 4.50-4.90 (2H, m), 8.20 (IH, s), 9.81 (IH, s).
-18-
2188695
WO 95/28935 PCTlEP95101546
~ Method 2
a) 2,3-Dihydro-6-hydroxymethylimidazo[2,1-b]thiazole-1(R, S)-oxide.
2,3-Dihydro-6-hydroxymethylimidazo[2,1-b]thiazole (1.5g, 10mmo1) in
dichloromethane (500m1) was cooled to 0-5 C and treated with m-
chloroperbenaoic
acid (60 % pure, 2.88g, lOmmol). After 15 minutes, the volatiles were removed
under reduced pressure and the residue triturated with diethyl ether. The
solvent
was decanted and the process repeated twice. The residual solid was dissolved
in
methanol (minimum volume), filtered and the filtrate evaporated to dryness
under
reduced pressure to yield an off-white foam (1.64g, 99%) (Found: M+ 172.0308.
C6H8N2O2S requires M 172.0306); dH [250MHz; (CD3)2S0] 3.58-3.67 (IH, m),
3.89-4.01 (1H, m), 4.39-4.63 (4H, m), 5.14 (1H, t, J 6Hz), 7.41 (1H, s).
b) 2,3-Dihydroimidazo[2,1-b]thiazole-6-carboxaldehyde-1(R,S)-oxide.
2,3-Dihydro-6-hydroxymethylimidazo[2,1-b]thiazole-1(R,S)-oxide (376mg,
2.19mmol) was suspended in acetonitrile (lOml) and water was added to obtain a
clear solution. Manganese dioxide (1.13g, 3wt. equivalents) was added and the
mixture stirred vigorously at ambient temperatures for 24h. More manganese
dioxide (lg) was added and the mixture stirred for a further 24h. The reaction
mixture was filtered through Celite, the filter pad washed with water and the
filtrate
evaporated to dryness under reduced pressure to give a white solid (340mg, 91
%).
Preparation 3
2,3-Dihydroimidazo[2,1-b]thiazole-6-carbozaldehyde-1,1-dioxlde
a) 6-Acetoxymethyl-2,3-dihydroimidazo[2,1-b]thiazole
2,3-Dihydro-6-hydroxymethylimidazo[2,1-b]thiazole (312mg, 2mmol) was
suspended in dichloromethane (lOml) and treated with pyridine (174mg, 2.2mmol)
and acetic anhydride (224mg, 2.2mmo1). 4-Dimethylaminopyridine (10mg) was
added and the mixture stirred at ambient temperature for 4h. The volatiles
were
removed by evaporation to dryness under reduced pressure and the residue was
triturated under hexane, the hexane decanted (2x) and the residue
chromatographed
on silica gel, eluting with mixtures of ethyl acetate and hexane to give the
product
(374mg, 94%) as a white solid; (Found: M+ 198.0465. C8H10N202S requires M
198.0463); nmax (CH2C12) 1734 and 1258cm-1; dg(250MHz; CDC13) 2.08 (3H,
s), 2.80 (2H, t, J 7Hz), 4.15 (2H, t, J 7Hz), 4.97 (2H, s), 7.11 (1H, s).
-19-
WO 95/28935 2 I88 U 9 5
PCTIEP95/01546
b) 6-Acetoxymethyl-2,3-dihydroimidazo[2,1-b]thiazole-l,l-dioxide
6-Acetoxymethyl-2,3-dihydroimidazo[2,1-b]thiazole (358mg, 1.81mmo1)
was dissolved in dichloromethane (lOml) and treated at room temperature with m-
chloroperbenzoic acid (60% pure, 936mg, 3.78mmol). After the initial
sulphoxidation was complete, the reaction mixture was heated under reflux for
4h
then left to stand at ambient temperature for 72h. The volatiles were removed
under reduced pressure and the residue triturated under diethyl ether and the
solvent
decanted. This process was repeated (2x) and the residual white solid
dissolved in
methanol and adsorbed onto silica gel. Chromatography on silica gel, eluting
with
mixtures of ethyl acetate and hexane, gave the title compound as a white solid
(305mg, 73%) (Found: M{' 230.0361. C8HION204S requires M 230.0361);
nmax (CH2C12) 1739, 1336, 1272, 1264 and 1258cm-1; dH (250MHz; CDC13)
2.08 (3H, s), 3.94 (2H, t, J 6Hz), 4.55 (2H, t, J 6Hz), 5.07 (2H, s), 7.16
(IH, s).
c) 2,3-Dihydro-6-hydroxymethylimidazo[2,1-b]thiazole-l,l-dioxide
6-Acetoxymethyl-2,3-dihydroimidazo[2,1-b]thiazole-1,1-dioxide (305mg,
1.33mmol) was treated with methanolic ammonia (prepared by saturating methanol
(20m1) with ammonia gas then diluting with more methanol (20m1)) at room
temperature. After 2.5h, the volatiles were removed under,reduced pressure and
the residue triturated with diethyl ether and the resulting solid collected by
filtration,
washed with diethyl ether and dried in air (207mg, 83%) (Found: M+ 188.0256.
C6H8N203S requires M 188.0256); nmax (nujol) 3354, 1377, 1325 and 1133cm-1;
dH [25oMHz; (CD3)2S0] 4.14 (2H, t, J 6Hz), 4.40 (2H, d, J 6Hz), 4.54 (2H, t, J
6Hz), 5.20 (1H, t, J 6Hz, exchangeable), 7.36 (IH, s).
d) 2,3-Dihydrobnidazo[2,1-b]thiazole-6-carboxaldehyde-l,l-dioxide
2,3-Dihydro-6-hydroxymethylimidazo[2, 1-b]thiazole- 1, 1-dioxide (207mg,
l.lmmol) was dissolved in acetonitrile (minimum volume) and treated with
manganese dioxide (621mg, 3wt. equivalents) and the mixture stirred vigorously
at
ambient temperature. After lh, more manganese dioxide (621mg) was added and
the mixture sitrred for a further 18h. The mixture was filtered through
Celite, the
filter bed washed with acetonitrile, the filtrate and washings combined and
evaporated to dryness under reduced pressure. The residue was triturated under
dichloromethane and the resulting solid collected by filtration, washed with
dichloromethane and dried in air (108mg, 53%) (Found: M+ 186.0103.
C6H6N203S requires M 186.0099); nmax (Nujol) 1691, 1320 and 1132cm-1; dH
[250MHz; (CD3)2S0] 4.25 (2H, t, J 7Hz), 4.68 (2H, t, J 7Hz), 8.32 (1H, s),
9.81
(1H, s).
-20-
2188695
WO 95/28935 PCT(EP95101546
= PrBparatlon 4
6, 7-Dihydro-SH-imidazo [2,1-b] [1, 3]thiazine-2-carboxaldehyde
a) Ethyl6,7-dihydro-5H-imidazo[2,1-b][1,3]thiazine-2-carboxylate
Ethyl 2-mercaptoimidazole-4(or 5)-carboxylate (860mg, 5mmol) was
dissolved in DMF (minimum volume) containing triethylamine (555mg, 5.5mmo1).
This solution was added dropwise to rapidly-stirred 1,3-dibromopropane (5m1).
After 0.5h the reaction mixture was partitioned between ethyl acetate and
water.
The phases were separated and the organic phase washed with water (3x),
saturated
brine, dried over anhydrous magnesium sulphate and evaporated to dryness under
reduced pressure. Chromatography on silica gel, eluting with 253G ethyl
acetate-
hexane gave the intermediate ethy12-(3-bromo-l-propylthio)imidazole-4(or 5)-
carboxylate, which was dissolved in dry, redistilled TT-IF (minimum volume)
and
added dropwise to a stirred suspension of sodium hydride (60% dispersion in
oil,
240mg, 6mmol) in dry, redistilled TFF (20m1) under argon. After 10 nrinutes,
water was carefully added to the reaction mixture, which was filtered through
Celite. The filter bed was washed with T'fH' and the filtrate and washings
combined
and evaporated to dryness under reduced pressure. Chromatography on silica
gel,
eluting with 50% ethyl acetate in hexane, gave the title compound as a white
solid
(635mg, 60%) mp 99-100 C (dichloromethane-hexane) (Found: C, 50.86; H, 5.74;
N, 13.14; S, 15.07%; M+ 212.0619. C9H12N202S requires C, 50.94; H, 5.66;
N, 13.21; S, 15.09% 212.0619); nmax (CH2C12) 1720, 1212 and 1198cm-1; dH
(250MHz; CDC13) 1.34 (3H, t, J 7Hz), 2.29-2.38 (2H, m), 3.13-3.17 (2H, m),
4.09 (2H, t, J 6Hz), 4.33 (2H, q, J 7Hz), 7.53 (1H, s).
b) 6,7-Dihydro-SH-imidazo[2,1-b][1,3]thiaziae-2- carboxaldehyde
Ethyl 6,7-dihydro-5H-imidazo[2,1-b][1,3]thiazine-2-carboxylate (2.12g,
10mmo1) was dissolved in dry dichloromethane (40m1) under argon and cooled to -
70%. Diisobutylaluminium hydride (1.SM in toluene, 12m1, 18mmo1) was added
below -68 C and the reaction mixture stirred at -70 C for lh. Water was added
carefully and the cooling removed. The reaction mixture was stirred vigorously
at
ambient temperature for 15 minutes and Celite (2g) added. The mixture was
filtered through Celite, the filter bed washed with dichloromethane and water,
the
filtrate and the washings combined and evaporated to dryness under reduced
pressure. The residue was evaporated from ethanol (2x) to give the title
compound
as a white solid (1.31g, 78%); nmax (CH2C12) 1685, 1543 and 1453cm 1; dH
(250MHz; CDC13) 2.34-2.43 (2H, m), 3.20 (2H, t, J 6Hz), 4.17 (2H, t, J 6Hz),
7.58 (1H, s), 9.75 (iH, s).
-21-
2188695
WO 95/28935 PCI'/EP951O1S46
Preparation $ =
6,7-Dihydro-5H-imidazo[2,1-b] [1,3]thiazine-2-carboxaldehyde-8,8-
dioxide
a) Ethyl 6,7-dihydro-5H-imidazo[2,1-b][1,3]thiazine-2-carboxylate-8,8-
dioxide
Ethyl 6,7-dihydro-5H-imidazo[2,1-b][1,3]thiazine-2-carboxylate (212mg,
immol) in dichloromethane (20m1) was treated with m-chloroperbenzoic acid (50%
pure, 690mg, 2mmol). The initial sulphoxidation was rapid and exothermic and,
when the sulphoxidation reaction was complete, the mixture was heated under
reflux
for 2h. The volatiles were removed under reduced pressure and the residue
triturated under diethyl ether. The resulting white solid was collected by
filtration,
washed with diethyl ether and dried in air (226mg, 93 %) (Found: M+ 244.0521.
C9H12N204S requires M 244.0518); nmax (CH2C12) 1735, 1717, 1331, 1270,
1257, 1218, 1198, 1167 and 1120cm-1; dH (250MHz; CDC13) 1.36 (3H, t, J
7Hz), 2.71-2.80 (2H, m), 3.54-3.59 (2H, m), 4.28-4.42 (4H, m), 7.65 (1H, s).
b) 6,7-Dihydro-SH-imidazo[2,1-b][1,3]thiazine-2-carboxaldehyde-8,8-
dioxide
Ethy16,7-dihydro-5H-imidazo[2,1-b][1,3]thiazine-2-carboxylate-8, 8-dioxide
(200mg, 0.82mmol) was dissolved in dry dichloromethane (minimum volume)
under argon and cooled to -70 C. Diisobutylaluminium hydride (1.5M in toluene,
lml, 1.5mmo1) was added at <-70 C and the mixture stirred at -70 C until thin
layer chromatography and infra-red spectroscopy showed little or no starting
material remaining. Water (5m1) was added carefully, cooling removed and the
mixture stirred at ambient temperature for lh. Celite was added to the mixture
and
the resulting mixture filtered through a Celite pad. The Celite pad was washed
with
dichloromethane and water and the filtrate and washings combined and
evaporated
to dryness under reduced pressure. The residue was re-evaporated with ethanol
(2x), triturated under diethyl ether and the product collected by filtration,
washed
with diethyl ether and dried in air (274mg, 30%) (Found: M+ 200.0256.
C7H8N203S requires M 200.0253); nmax (Nujol) 1678, 1316, 1161 and 1191cni
1; dH [250MHz; (CD3)2S0] 2.50-2.57 (2H, m), 3.81-3.85 (2H, m), 4.31 (2H, t, J
6Hz), 8.27 (1H, s), 9.80 (1H, s).
-22-
WO 95128935 2188695 pC('(g,p95101546
= Example 1
Sodium (5R)-6-[(Z)-(2,3-dihydroimidazo[2,1-b]thiazol-6-yl) methylene]-
penem-3-carboxylate
a) 4-Methoxybenzyl [5R,6RS,8RS]-6-[acetoxy(2,3-dihydrobnidazo[2,1-
b]thiazol-6-yl)methyl]-6-bromopenem-3-carboxylate
Diphenylamine (604mg, 3.57mmol) was dissolved in dry, redistilled TfF
(35m1) under argon and cooled to -20 C. n-Butyllithium (1.48M in hexane;
208mg, 3.25mmol) was added and the mixture stirred at ambient temperature for
10
minutes. The mixture was cooled to -70 C and treated dropwise with a solution
of
4-methoxybenzyl (5R,6R)-6-bromopenem-3-carboxylate (1.2g, 3.25mmo1) in dry,
redistilled THF (lOml). The resulting mixture was stirred at -70 C for 10
minutes
then treated with a solution of 2,3-dihydroimidazo[2,1-b]thiazole-6-
carboxaldehyde
(500mg, 3.25mmol) in dry DMF (5m1). The resulting mixture was stirred at -70 C
for 20 minutes then treated with acetic anhydride (331mg, 3.25mmo1) and 4-
dimethylaminopyridine (100mg). When all the bromohydrin intermediate had been
converted to the title compound, the reaction mixture was concentrated to low
volume under reduced pressure and partitioned between dichloromethane and
water.
The organic phase was separated and washed repeatedly with water (5x), dil.
aq.
sodium hydrogencarbonate, water, saturated brine, dried (MgSO4) and evaporated
to dryness under reduced pressure to give a brown oil. Chromatography on
silica
gel, eluting with 50% ethyl acetate in hexane, gave the title compound as a
brown
foam (1.Og, 55%); nmax (CH2CI2) 1801, 1753 and 1715cm-1.
b) 4-Methoxybenzyl (5R)-6-(2,3-dihydroimidazo[2,1-b]thiazol-6-yl)-
methylene]penem-3-carboxylate
4-Methoxybenzyl [5R,6RS,8RS]-6-[acetoxy(2,3-dihydroimidazo[2,1-
b]thiazol-6-yl)methyl]-6-bromopenem-3-carboxylate (930mg, 1.65mmo1) was
dissolved in THF (20m1) and treated with N,N,N',N'-tetramethylethylenediamine
(TMEDA, 478mg, 4.1mmo1) followed by zinc powder (269mg, 4.lgm atoms).
The mixture was stirred vigorously and treated with glacial acetic acid
(247mg,
4.1mmo1). After 10 minutes, more glacial acetic acid (247mg, 4.lmmol) was
added and, after a further 10 minutes, the reaction mixture was partitioned
between
ethyl acetate and water and the resulting mixture filtered through Celite. The
phases were separated and the organic phase washed with IM aq. potassium
hydrogensulphate (3x), saturated brine, saturated, aq. sodium
hydrogencarbonate
(2x), saturated brine, dried (MgSO4) and evaporated to dryness under reduced
pressure. The residue was chromatographed on silica gel, eluting with 50%
ethyl
acetate in hexane, to give the product as a yellow foam (459mg, 65%); [a]D25 +
522 (c = 0.1% in acetonitrile); nmax (CH202) 1773, 1709, 1252 and 1232cm-1;
-23-
WO 95/28935 2188 6 9 5 PCl'/EP95/01546
dH [250MHz; (CD3)2C0] 3.79 (3H, s), 3.93 (2H, t, J 7Hz), 4.34 (2H, t, J 7Hz),
~
5.16 (2H, ABq, J 12.5Hz), 6.55 (IH, d, J 1Hz), 6.91-6.96 (3H, m), 7.40 (2H, d,
J7Hz), 7.45 (IH, s), 7.61 (IH, s).
c) Sodium (5R)-6-[(Z)-(2,3-dihydroimidazo[2,1-b]thiazol-6-yl) methyl-
ene]penem-3-carboxylate
Anisole (1.52m1, 14mmo1) was dissolved in dry dichloromethane (2m1)
under argon and the solution cooled to -20 C. Ethyl aluminium dichloride (1.8M
in
toluene, 147mg, 1.16mmo1) was added and the mixture stirred at -20 C for 10
minutes before cooling to -70 C. This mixture was treated, dropwise, with a
solution of 4-methoxybenzyl (5R)-6-[(Z)-(2,3-dihydroimidazo[2,1-b]thiazol-6-
yl)methylene]penem-3-carboxylate (166mg, 0.39mmo1) in dry dichloromethane
(5ml). After 15 minutes at -70 C, the mixture was treated with an excess of
O.SM,
aqueous trisodium citrate and cooling removed. When the reaction mixture had
regained room temperature, it was treated with diethyl ether, acetone and
water
until two clear phases were obtained with very little material on the
interface. The
phases were separated and the organic phase extracted with dilute, aqueous
sodium
hydrogencarbonate. The combined aqueous extracts were acidifed to pH 2 with SM
hydrochloric acid in the presence of ethyl acetate and the phases separated.
The
aqueous phase was further extracted with ethyl acetate and the combined
extracts
washed repeatedly with water (Sx). The washed organic phase was stirred in the
presence of water and the pH of the aqueous phase adjusted to 6.6 by addition
of
dilute aqueous sodium hydrogencarbonate and the phases separated. The organic
phase was further extracted with water and the aqueous extracts combined and
freeze-dried. The resulting, orange powder was purified by chromatography on
Diaion HP20SS resin, eluting with mixtures of THF in water, to give, after
freeze-
drying, the title compound as a yellow solid (56.2mg, 44%); nmax (KBr) 1741,
1670, 1597, 1394, 1304 and 1268cm-1; 1max (H20) 325 (e dm3 mol-Icm-1
13,514) and 237 (9768) nm; dH (250MHz; D20) 3.86 (2H, d, J 7Hz), 4.22 (2H, t,
J 7Hz), 6.46 (1H, s), 6.86 (1H, s), 7.01 (IH, s), 7.47 (1H, s). -
-24-
2188695
WO 95/28935 pCT1Ep95101546
= Example 2
Sodium (5R)-6-[(Z)-(2,3-dihydro-1(R,S)-oxoimidazo[2,1-bjthiazol-6-
yI)methylene] penem-3-carboxylate
a) 4-Methoxybenzyl (5R)-6-[(Z)-(2,3-dibydro-1(RS)-oxoimidazo[2,1-
bjthiazol-6-yl)methylenejpenem-3-carboxylate
Diphenylamine (372mg, 2.2mmol) was dissolved in dry, redistilled THF
(10mi) under argon and cooled to -20 C. n-Butyllithium (2.5M in hexane, 128mg,
2mmol) was added and the mixture stirred at ambient temperatures for 10
minutes.
The resulting reaction mixture was cooled to -70 C and treated dropwise with a
solution of 4-methoxybenzyl (5R,6R)-6-bromopenem-3-carboxylate (740mg,
2mmol) in dry, redistilled THF (10m1). After twenty minutes at -70 C, the
reaction mixture was treated with a solution of 2,3-dihydroimidazo[2,1-
b]thiazole-6-
carboxaldehyde-1(RS)-oxide (340mg, 2mmol) in dry DMF (5m1), stirred at -70 C
for 0.5h then treated with acetic anhydride. Cooling was removed and the
mixture
stirred at ambient temperatures for lh before being partitioned between ethyl
acetate
and water. The organic phase was washed well with water (5x), saturated brine,
dried (MgSO4) and evaporated to dryness under reduced pressure. The residue
was
purified by chromatography on silica gel, eluting with ethyl acetate, to give
the
intermediate 4-methoxybenzyl [5R,6RS,8RSj-6-[acetoxy(2,3-dihydro-I(RS)-
oxoimidazo[2,1-b]thiazol-6-yl)methylJ-6-bromopenem-3-carboxylate (527mg, 45%,
0.9mmol).
The above mixture of bromoacetates (0.9mmol) was dissolved in TEF
(10m1) and treated with TMEDA (263mg, 2.3mmol) followed by zinc powder
(148mg, 2.3g atoms). Glacial acetic acid (136mg, 2.3mmol) was added and the
m'vcture stirred vigorously for 10 minutes before more glacial acetic acid
(136mg,
2.3mmol) was added. After a further 10 minutes the mixture was diluted with
ethyl
acetate and water and filtered through Celite. The phases in the filtrate were
separated, the aqueous phase further extracted with ethyl acetate, the
extracts
combined, washed with 1M, aqueous potassium hydrogensulphate (3x), saturated
brine, saturated, aqueous sodium hydrogencarbonate (2x), saturated brine,
dried
(MgSO4) and evaporated to dryness under reduced pressure. The residue was
chromatographed on silica gel, eluting with ethyl acetate then mixtures of
ethanol in
ethyl acetate, to give a mixture of (E) and (Z)-isomers, plus pure Z-isomer.
The
mixture of isomers was rechromatographed on silica gel and the two fractions
of
pure (Z)-isomer combined (236mg, 27%); [aJDU + 409 (c = 0.1% in
acetonitrile); nmax (KBr) 1772, 1703, 1233 and 1057cm-1; dH [250MHz;
(CD3)2C0] 3.67-3.76 (1H, m), 3.81 (3H, s), 4.00-4.14 (1H, m), 4.62-4.87 (2H,
2m), 5.18 (2H, s), 6.60 (IH, d, J IHz), 6.65 (1H, d, J 1Hz); 6.91-6.97 (2H,
m),
-25-
WO 95/28935 2183695 PCT/EP95/01546
=
7.14 (1H, s), 7.38-7.43 (2H, m), 7.51 and 7.52 (1H, 2s), 7.89 and 7.90 (1H,
2s);
m/z (F.A.B., positive ion xenon, NOBA sodium) 482 (MNa+).
b) Sodium (5R)-6-[(Z)-(2,3-dihydro-1(RS)-oxoimidazo[2,1-b] thiazol-6-
yl)methylene]penem-3-carboxylate
Anisole (499mg, 4.6mmol) was dissolved in dry dichloromethane (0.5m1)
under argon and treated with aluminium trichloride (61.5mg, 0.45mmo1). When a
complete solution had been obtained, the mixture was cooled to -40 C and
treated
with a solution of 4-methoxybenzyl (5R)-6-[(Z)-(2,3-dihydro-1(RS)-
oxoimidazo[2,1-
b]thiazol-6-yl)methylene]penem-3-carboxylate (68mg, 0.15mmo1) in dry
dichloromethane (2m1). After 15 minutes at -40 C, 0.5M trisodium citrate
(10m1)
was added and cooling was removed. The mixture was stirred at ambient
temperature for 15 minutes and the phases separated. The aqueous phase was
washed with dichloromethane then acidified to pH 2 with 5M hydrochloric acid
in
the presence of ethyl acetate. The phases were separated, the aqueous phase
further
extracted with ethyl acetate, the extracts combined, washed with water (5x)
then
stirred vigorously in the presence of water while the pH of the aqueous phase
was
adjusted to 6.8 with dilute, aqueous sodium hydrogencarbonate. The phases were
separated, the organic phase extracted with water, the extracts combined and
freeze-
dried to give the product (23mg, 43%); 1max (H20) 370.5 (e dm3 mol-lcm-1
1761) and 301.5 (18,005) nm; nmax (KBr) 1751, 1598, 1383, 1268, 1139, 1090
and 1047cm-1; dH (250MHz; D20) 3.83-3.91 and 4.01-4.18 (each 1H, 2m), 4.57-
4.66 (1H, m), 6.55 and 6.60 (each 1H, 2d, J 1H), 7.00 (1H, s), 7.09 (1H, s),
7.77
and 7.80 (each 111, 2s).
Example 3
Sodium (5R)-6-[(Z)-(2,3-dihydro-1,1-dioxoimidazo[2,1-b]thiazol-6-
yqmethylene]penem-3-carboxyiate
a) 4-Methoxybenzyl (5R)-6-[(Z)-(2,3-dihydro-l,l-dioxoimidazo[2,1-
b]thiazol-6-yl)methylene]penem-3-carboxylate
Diphenylamine (372mg, 2.2mmol) was dissolved in dry, redistilled TBF
(10m1) under argon, cooled to -20 C and treated with n-butyllithium (2.5M in
hexane, 128mg, 2mmol). The mixture was stirred at ambient temperature for 10
minutes then cooled to -70 C. A solution of 4-methoxybenzyl (5R,6R)-6-
bromopenem-3-carboxylate (740mg, 2mmol) in dry, redistilled THF-(5m1) was
added dropwise and, after a further 10 minutes at -70 C, a solution of 2,3-
dihydroimidazo[2,1-b]thiazole-6-carboxaldehyde-1,1-dioxide (372mg, 2mmol) in
dry DMF (5m1) was added to the reaction mixture. This mixture was stirred at -
70 C for 0.5h then treated with acetic anhydride (204mg, 2mmol). Cooling was
-26-
WO 95/28935 2j8;O 6Q5 PGTlEP95l01546
= removed and the mixture stirred at ambient temperture for 1.25h before being
partitioned between ethyl acetate and water. The organic phase was washed with
water (4x), saturated brine, dried(MgSO4) and evaporated to dryness under
reduced pressure to yield a brown foam. Chromatography on silica gel, eluting
with mixtures of ethyl acetate in hexane, gave the bromoacetate intermediate
as a
mixture of diastereoisomers (504mg, 0.84mmo1).
The diastereoisomeric mixture of bromoacetates (504mg, 0.84mmol) was
dissolved in THF (5ml) and treated with TMEDA (216mg, 1.9mmol). Zinc powder
(121mg, 1.9gm atoms) was added, the mixture stirred vigorously and treated
with
glacial acetic acid (112mg, 1.9mmo1). After 10 minutes, more glacial acetic
acid
(112mg, 1.9mmol) was added and after a further 0.5h, the mixture was
partitioned
between ethyl acetate and water, filtered through Celite and the phases
separated.
The organic phase was washed with 1M aqueous potassium hydrogensulphate (3x),
saturated brine, saturated aqueous sodium hydrogencarbonate, saturated brine,
dried
(MgSO4) and evaporated to dryness under reduced pressure. Chromatography of
the residue on silica gel, eluting with mixtures of ethyl acetate in hexane,
gave the
title compound (250mg, 27%); [a]DU + 464 (c = 0.146 in acetonitrile); nman
(CH202) 1770, 1714, 1274 and 1256cm-1; dH [250MHz; (CD3)2C01 3.81 (3H,
s), 4.18 (2H, t, J 7Hz), 4.87 (2H, t, J 7Hz), 5.19 (2H, brs), 6.57 (1H, s),
6.95
(2H, d, J 8Hz), 7.41 (2H, d, J 8Hz), 7.65 (1H, s), 8.39 (1H, s); m/z (F.A.B.,
+ve ion xenon, NOBA sodium) 482 (MNa+).
a) Sodium (5R)-6-[(Z)-(2,3-dihydro-1,1-dioxoimidazo[2,1-b] thiazol-6-
yl)methylene]penem-3-carboxylate
Anisole (1.8g, 16.3mmol) was dissolved in dry dichloromethane (2m1) under
argon and treated with aluminium trichloride (218mg, 1.63mmo1). When a
complete solution had been obtained, the mixture was cooled to -40 C and
treated
with a solution of 4-methoxybenzyl (5R)-6-[(Z)-(2,3-dihydro-l,l-
dioxoimidazo[2,1-
b]thiazol-6-yl)methylene]penem-3-carboxylate (250mg, 0.54mmo1) in dry
dichioromethane (2m1). The resulting mixture was stirred at -40 C for 10
minutes
then treated with 0.5M aqueous trisodium citrate (15m1) and the cooling
removed.
After a further 15 minutes, the mixture was diluted with diethyl ether,
acetone and
water until two clear phases were obtained. The phases were separated, the
aqueous
phase washed with diethyl ether then acidified to pH 2 with 5M hydrochloric
acid in
the presence of ethyl acetate. The phases were separated, the aqueous phase
further
extracted with ethyl acetate, and the extracts combined. The combined extracts
were
washed well with water (4x) then stirred vigorously in the presence of water,
while
the pH of the aqueous phase was adjusted to 6.8 with dilute sodium
hydrogencarbonate. The phases were separated, the organic phase further
extracted
-27-
WO95128935 2188695 PCT/Ep95101546
with water, the combined aqueous extracts freeze-dried then purified by
chromatography on Diaion HP20SS resin, eluting with mixtures of THF in water,
to give the title compound (1 14mg, 58%); lmyx (H20) 370 (e dm3mol-1cm 1 2127)
and 296.5 (25,942) nm; nmax (KBr) 1755, 1599, 1389, 1322, 1269 and 1136cm-1;
dH (KBr) 1755, 1599, 1389, 1322, 1269 and 1136cm-1; dH (250MHz; D20) 4.20
(2H, t, J 7Hz), 4.66 (2H, t, J 7Hz), 6.47 (111, d, J 1Hz), 6.98 (1H, s), 7.04
(1H,
s), 7.64 (111, s).
Example 4
Sodium (5R)-6-[(Z)-(6,7-dihydro-5H-imidazo[2,1-b][1,3]thiazin-2-
yl)methylene]penem-3-carboxylate
a) 4-Methoxybenzyl (5R,6RS,SRS)-6-[acetoxy(6,7-dihydro-SH-imidazo[2,1-
b] [1,3]thiazin-2-yl)methyl]-6-bromopenem-3-carboxylate
Diphenylamine (589mg, 3.5mmo1) was dissolved in dry, redistilled THF
(20m1) under argon and the solution cooled to -20 C. n-Butyllithium (2.5M in
hexane, 203mg, 3.2mmol) was added and the mixture stirred at ambient
temperature for 10 minutes before being cooled to -70 C. A solution of 4-
methoxybenzyl (5R,6R)-6-bromopen:-,n-3-carboxylate (1.17g, 3.2mmol) in dry,
distilled TEEIF' (lOml) was added dropwise at -70 C and the resulting mixture
stirred
at -70 C for 10 minutes. A solution of 6,7-dihydro-5H-imidazo[2,1-
b][i,3]thiazine-2-carboxaldehyde (532mg, 3.2mmol) in dry, redistilled THF
(20m1)
was added dropwise at -70 C and the resulting mixture stirred at -70 C for 20
minutes. Acetic anhydride (323mg, 3.2mmol) then 4-dimethylantinopyridine
(20mg) were added and the cooling removed. After lh at ambient temperature,
the
volatiles were removed under reduced pressure and the residue partitioned
between
ethyl acetate and water. The organic phase was washed with saturated aqueous
sodium hydrogencarbonate, water, saturated brine, dried (MgSO4) and evaporated
to dryness under reduced pressure. The residue was purified by chromatography
on
silica gel, eluting with mixtures of ethyl acetate in hexane, giving the title
compound as a light-brown foam (1.04g, 56%); nmax (CH2C12) 1801, 1749,
1716cm-1.
b) 4-Methoxybenzyl (5R)-6-[(Z)-(6,7-dihydro-5H-imidazo[2,1-b][1,3]-
thiazin-2-yl)methylene]penem-3-carboxylate
4-Methoxybenzyl (5R,6R4,8RS)-6-[acetoxy(6,7-dihydro-SH-ilnidazo[2,1-
b][1,3]thiazin-2-yl)methyl]-6-bromopenem-3-carboxylate (1.04g, 1.79mmo1) was
dissolved in THF (20m1) and treated sequentially with TMEDA (521mg,
4.48mmol), zinc powder (293mg, 4.48gm. atom) and glacial acetic acid (296mg,
4.48mmo1) with vigorous stirring. After 10 minutes, more glacial acetic acid
-28-
WO 95/28935 2 18 8 6 g 5 pCF1EP95101546
(269mg, 4.48mmo1) was added and the mixture stirred vigorously for a further
10
minutes. The reaction mixture was partitioned between ethyl acetate and water
and
filtered through Celite. The phases were separated and the organic phase
washed
with IM aqueous potassium hydrogensulphate (3x), saturated brine, saturated
aqueous sodium hydrogencarbonate, saturated brine, dried (MgSO4) and
evaporated
to dryness under reduced pressure. The product was obtained by chromatography
on silica gel, eluting with mixtures of ethyl acetate in hexane, as a yellow
foam
(532mg, 67%); nmy x (CH2C12) 1773, 1710, 1270 and 1232cm-1; dH [250MHz;
(CD3)2C0] 2.30-2.42 (2H, m), 3.22-3.33 (2H, m), 3.80 (3H, s), 4.20 (2H, t, J
6Hz), 5.16 (2H, brs), 6.55 (IH, d, J 11Hz), 6.88-6.97 (3H, m), 7.38-7.53 (4H,
m).
c) Sodium (SR)-6-[(Z)-(6,7-dibydro-5H-imidazo[2,1-b][1,3]thiazin-2-
yi)methylene]penem-3-carboxylate
Anisole (2.02g, 18mmol) was dissolved in dry dichloromethane (2m1) under
argon and treated with aluminium trichloride (248mg, 1.8mmol). When complete
solution had been obtained, the mixture was cooled to -40 C and treated
dropwise
with a solution of 4-methoxybenzyl (5R)-6-[(Z)-(6,7-dihydro-5H-imidazo[2,1-
b][1,3]thiazin-2-yl)methylene]penem-3-carboxylate in dry dichlommethane
(10ml).
After 10 nrinutes at -40'C, the reaction nrixture was treated with O.SM
aqueous
trisodium citrate (15m1) and cooling was removed. After 15 minutes at ambient
temperature, the mixture was diluted with diethyl ether, water and acetone
until two
clear phases were obtained. The phases were separated, the aqueous phase
washed
with diethyl ether and acidified to pH 2 with SM hydrochloric acid in the
presence
of ethyl acetate. The phases were separated, the aqueous phase further
extracted
with ethyl acetate, the extracts combined and washed well with water (4x). The
ethyl acetate extract was stirred vigorously in the presence of water and the
pH of
the aqueous phase adjusted to pH 6.8 with dilute, aqueous sodium
hydrogencarbonate. The phases were separated and the aqueous phase freeze-
dried.
The freeze-dried residue was chromatographed on Diaion HP20SS resin, eluting
with mixtures of TEF in water, to give the title compound as a bright-yellow,
freeze-dried solid (79.5mg, 37%); lmax (H20) 328 (e dm3mol-1cm-1 14122) and
247.5 (12142) nm; nmax (KBr) 1742, 1672, 1597cm-1; dH (250MHz; D20) 2.18-
2.23 (2H, m), 3.17 (2H, t, J 6Hz), 4.04 (2H, t, J 6Hz), 6.44 (IH, s), 6.86
(1H, s),
6.98 (1H, s), 7.35 (1H, s); m/z (F.A.B., +ve ion xenon, glycerol) 366 (MNa+)
and 344 (MH+).
Example 5
Sodium (5R)-6-[(Z)-(6,7-dihydro-8,8-dioxo-5H-imidazo[2,1-b][1,3]
-29-
WO 95/28935 21V t1 U/5 PGT/SP95/01546
thiazin-2-yl)methylene] penem-3-carboxylate.
a) 4-Methoxybenzyl (5R,6RS,8RS)-6-[acetoxy (6,7-dihydro-8,8- dioxo-5H-
imidazo[2,1-b][1,3]thiazin-2-yl)methyl]-6- bromopenem-3-carboxylate.
Diphenylamine (186mg, 1.1 mmol) was dissolved in dry, redistillied THF
(10m1) under argon and cooled to -200C. A solution of n-butyllithium in hexane
(2.45M, 410 ml, lmmol) was added and cooling removed. After 10 minutes, the
mixture was cooled to -700C and treated with 4-methoxy-benzyl (5R,6R)-6-
bromopenem-3-carboxylate (370mg, Immol) in dry, redistilled THF (5m1). The
resulting mixture was stirred at -700C for 10 minutes then treated at -700C
with a
solution of 6,7-dihydro-5H-imidazo [2,1-b][1,3]thiazine-2-carboxaldehyde-8,8-
dioxide (200mg, 1 mmol) in dry THF (2m1). This mixture was stirred at -700C
for
minutes before being treated with acetic anhydride (102 mg, Immol) and 4-
dimethylamino- pyridine (10mg). Cooling was removed and the reaction mixture
stin-ed at ambient temperature for lh. After lh, the volatiles were removed
under
15 reduced pressure and the residue partitioned between ethyl acetate and
water. The
organic phase was washed with water (4x), saturated aq. sodium
hydrogencarbonate, water, saturated brine, dried (MgSO4) and evaporated to
dryness under reduced pressure. The residue was purified by chromatography on
silica gel, eluting with niixtuns of ethyl acetate in hexane, to give the
title
20 compound (229.6mg, 37.5 %); nmax (CH2C12) 1802, 1758, 1716, 1330, 1275,
1216 and 168cm-1.
b) 4-methoxybenzyl (5R)-6-[(Z)-(6,7-dihydro-8,8-dioxo-SH-
-midazo[2,1-b][1,3] thiazin-2-yl) methylene] penem-3- carboxylate.
4-Methoxy (5R,6RS,8RS)-6-[acetoxy (6,7-dihydo-8,8-dioxo-5H-imidazo
[2,1-b][1,3] thiazin-2-yl) methyl]-6-bromopenem-3-carboxylate (410mg, 0.7
mmol)
was dissolved in THF (IOml) and treated sequentially with TMEDA (195mg, 1.67
mmol), zinc powder (109mg, 1.67 g atoms) and glacial acetic acid (101 mg, 1.67
mmol). After 10 minutes more glacial acetic acid (101mg, 1.67 mmol) was added
and the resulting mixture stirred for a further 10 minutes. The reaction
mixture was
partitioned between ethyl acetate and water and the organic phase washed with
IM
aq. sodium hydrogen sulphate (3x), saturated brine, saturated aq. sodium
hydrogen
carbonate, saturated brine, dried (MgSO4) and evaporated to dryness under
reduced
pressure. Chromatography on silica gel, eluting with ethyl acetate, gave the
title
compound as a bright yellow foam (201 mg, 63%); [a]D25 +4460 (c = 0.1Y6 in
acetonitrile); Imax (EtOH) 302.5 (e dm3 mol-Icm-1 30,087), 227 (19,073) and
202
(24,890) nm; nmax (CH2CI2) 3134, 1777, 1732, 1711, 1330 and 1235cm-1; dH
[250MHz; (CD3)2C0)] 2.68-2.77 (2H,m), 3.67-3.72 (2H, m), 3.81 (3H, s), 4.46
(2H, t, J 6Hz), 5.18 (2H, s), 6.59 (IH, d, J 1Hz), 6.94 (2H, d,J 9Hz), 7.11
(IH,
-30-
WO 95/28935 2=1 $ 8C '( [ pC'1'/Ep9S101546
d, J 1Hz), 7.41 (2H, d, J 9Hz), 7.50 (1H, s), 7.74 (1H,iJS)7; Jm/z (NH3DCI)
474
(MH+) and 491 (MNH4{').
c) Sodium (SR)-6-[(Z)-(6,7-dihydro-8,8-dioxo-SH-imidazo [2,1-
b][1,3]thiazin-2-yl) methylene] penem-3-carboxylate.
Anisole (1.2g, 11.4 mmol) was dissolved in dry dichloromethane (lml)
under argon and the resulting solution treated with aluminium trichloride
(152mg,
1.14mmo1). When a complete solution had been obtained, the solution was cooled
to -40oC and treated at <-30oC with a solution of 4-methoxybenzyl (5R)-6-[(Z)-
(6,7-dihydro-8,8-dioxo-SH-imidazo [2,1-b][1,3] thiazin-2-yl) methylene] penem-
3-
carboxylate (180 mg 0.38 mmol) in dry dichloromethane (5m1). After 10 minutes,
0.5M aq. trisodium citrate (lOmi) was added, cooling was removed and the
mixture
allowed to regain room temperature. The reaction mixture was diluted with
diethyl
ether, water and acetone until two, clear phses had been obtained. The phases
were
separated and the aqueous phase wahed with diethyl ether then acidified to pH
2
with 5M hydrochloric acid in the presence of ethyl acetate. The phases were
separated and the aqueous phase further extracted with ethyl acetate and the
extracts
combined, washed with water (5x) then stirred with water. The pH of the
aqueous
phase was adjusted to 6.8 by addition of dilute, aq. sodium hydrogen carbonate
and
the phases separated. The aqueous phase was freeze-dried and the resulting
orange
powder purified by chromatography on HP20SS resin, eluting with water, to give
the title compound as a bright orange powder, (54.2mg, 38%); lmax (1120) 298
(e
dm3mol-lcm-1 22,425)nm; nmax (KBr) 1750, 1597, 1385, 1317 and 1165 cm-1;
dH (250MHz; D20) 2.60-2.77 (2H, m), 3.76-3.80 (2H, m), 4.27 (2H, t, J 7Hz),
6.84 (IH, s), 6.96 (1H, s), 7.01 (IH, s), 7.56 (1H, s); m/z (F.A.B., +ve ion
xenon, glycerol) 376 (MH+) and 398 (MNa+).
Example 6.
Sodium (5R)-6-[(Z)-(2,3-dihydroimidazo [2,1-b] thiazol-6-yl) methylene]-
penem-3-carboxylate (448mg; 1,36 mmol) was dissolved in the minimum volume of
water at ambient temperature and acetone was added until the solution became
turbid. The mixture was left to stand for 24 hours at 40C and the resulting
yellow
microcrystalline solid was collected by filtration, washed with acetone and
dried
under reduced pressure (327 mg; 67% recovery).
-
Example 7.
Sodium (5R)-6-[(Z)-(2,3-dihydroimidazo [2,1-b] thiazol-6-yl) methylene
penem-3-carboxylate (100mg; 0.3 mmol) was dissolved in the minimum volume of
water at room temperature and diluted with ethanol until the solution became
turbid.
-31-
WO 95/28935 21886 9 5 PGT/EP95/01546
Trituration gave bright orange crystals, which were collected by filtration,
washed
with a little ethanol and dried under reduced pressure (42 mg; 42% recovery).
Example 8.
4-methoxybenzyl (5R)-6-[(Z)-(2,3-dihydrohnidazo[2,1-b]thiazol-6-yq-
methylene]penem-3-carboxylate
A solution of diphenylanune (2.52g; 14.85mmol) dissolved in dry, distilled
tetrahydrofuran FTHF] (50mis) was cooled to -20 C with stirring and treated
with a
solution of n-butyl lithium (5.7m1s of 2.6M solution in hexanes). The solution
was
stirred at -20 C for 10 minutes, then cooled to <-70 C whereupon a solution of
4-
methoxybenzyl 6a-bromopenem-3- carboxylate (5g; 13.5mmol) dissolved in dry
distilled THF (601nls) was added dropwise, keeping the reaction temperature <-
65 C. Stirring at this temperature was maintained for 15 nrinutes, whereupon a
solution of 2,3- dihydroimidazo[2, 1-b]thiazole-6-carboxaldehyde (2.29g;
14.85mmol) dissolved in dry dimethylformamide (approx 25m1s) was added over
2-3 mins. Stirring at <-65 C was maintained for 30 minutes before the addition
of
acetic anhydride (1.34m1s, 14.2mmol). The cooling bath was removed and the
reaction vessel transfered to an ice bath. Stirring was maintained for 30
minuntes
whereupon zinc powder (1.34g; 20.6mmol), glacial acetic acid (2.32m1s;
40.5mmo1) and N,N,N ,N -tetramethylethyl- enediamine (3mls; 20.2mmol) were
added and the reaction allowed to regain ambient temperature over approx. 1
hour.
The reaction mixture was then diluted with ethyl acetate (ca 500 mls) and
washed
with water (4 x 500mis) followed by brine (1 x 250m1s) before drying over
magnesium sulphate. Filtration and evaporation gave a residue which was
chromatographed on silica gel. Elution with a gradient 50% 3A > 75 % ethyl
acetate/hexane gave the title compound identical in an analytical aspects to
that
described in example lb, as a yellow foam (4.Olg, 69.5%).
Sodium (5R)-6-[(Z)-[(2,3-dihydroimidazo[2,lb]thiazol-6-yl)methylene]- penem-
3-carboxylate
A solution of anisole (59.7g; 60mis; 0.55mo1) dissolved in dry dichloro-
methane [DCM] (60mis) was cooled to -20 C with stirring and treated with a
solution of ethylaluminium dichloride (39mls of 1.8M solution in toluene;
70.2mmol). After stirring for 5 minutes, the reaction was cooled to <-50 C and
treated with a solution of 4-methoxybenzyl (5R)-6-[(Z)-(2,3-dihydroimidayo[2,1-
b]-
1-b]-
thiazol-6-yl)methylene]penem-3-carboxylate (lOg; 23.4mmol) dissolved in dry
DCM (100nils) added dropwise, keeping the reaction temperature below -50 C.
After stirring for a further 15 minutes, a solution of aqueous trisodium
citrate
(5001nls of 0.5M soln) was added and the cooling bath removed. Water (5001nls)
-32-
WO 95/28935 2188695 PCTlEP95101546
= was added and the pH of the reaction mixture adjusted to 7.2 with aqueous
sodium
hydrogen carbonate. Diethyl ether (500mis) was added and the phases separated.
The organic phase was further extracted with water, (2 x 100m1s), the combined
aqueous solution was washed with diethyl ether (2 x 250m1s) before evaporating
briefly to remove residual organic solvent. The pH of the aqueous soln. was
further
adjusted to 7.5 before chromatography on Dianion HP20SS eluting with water.
The
fractions were combined and reduced in volume by reverse osmosis to give,
after
freeze drying, the title compound as a yellow solid, having identical
analytical
properties to those of the compound described in example lc (4.98g, 65%). The
compound was crystallized under similar conditions to that in example 6.
Example 9.
The in-vftro synergistic activity of the compound of Example 1 above (i.e
sodium (5R)-6-[(Z)-(2,3-dihydroimidazo[2,1-b]thiazol-6-yl)methylene]penem-3-
carboxylate) with ceftazidime was investigated.
The presence of l g/ml of the compound of Example I was fbund to
reduce the MIC of ceftazidime against B. fragilis from around 7 g/ml to
around 4
g/mI. Similarly the presence of l g/ml of the compound of Example I was found
to reduce the MIC of ceftazidime against a strain of Enterobacter producing
extended spectrum [i-lacatmase enzymes from around 7 g/ml to around 2 g/ml.
In a population of E. cloacae growing in the presence of ceftazidime at a
concentration of 0.5 MIC a population of cells showing high-level resistance
to
ceftazidime was rapidly selected in vitro. The presence of the compound of
Example
1 reduced the rate of emergence of resistant isolates dramatically. In
addition the
final level of resistance seen with the combination of ceSazidime and the
compound
of Example 1 was much lower than that observed with ceftazidime alone.
Example 10
The in-vitro synergistic activity of the compound of Fxample 1 above (i.e
sodium
(5R)-6-[(Z)-(2,3-dihydroimidazo[2,1-b]thiazol-6-yl)methylene]penem-3-
cxrboxylate) with cefotaxime was investigated.
Effective synergy with cefotaxime was observed with l g/ml of this
compound against B. fragilis, and strains of Enterobacteriaceae producing
extended
spectrum (3-lactamases. .
The MIC (minimum inhibitory concentration) of cefotaxime against one
TEM-3 producing strain of Kpneumoniae was measured. In the absence of the
compound of Example 1 the MIC of cefotaxime was 64 g/ml. Concentrations of
the compound of Example 1 as low as 0.25 g/ml reduced the MIC of cefotaxime to
below l g/ml. When a similar titration was carried out against a strain of
-33-
2188695
WO 95/28935 - _ PCT/EP95101546
Ent.cloacae producing high levels of Class 1-0-lactamases constitutively, a
cefotaxime MIC < 1 g/ml was achieved in the presence of l g/ml of the compound
of Example 1.
Fxample 11
Preliminary experimental infection studies have demonstrated the in vivo
efficacy of the compound of Example 1, when coadministered parenterally with
cefotaxime, against infections caused by different bacterial pathogens that
produce
(i-lactamase, the compound protecting cefotaxime (Table 1) from inactivation
by a
variety of important 0-lactamases in an experimental model of intraperitoneal
infection.
In these studies, mice were infected intraperitoneally with a lethal challenge
inoculum of strains of either Klebsiella pneuntoniae (520), producing extended-
spectrum TEM type 0-lactamase,(TEM-3) or Enterobacter cloacae (4593)
producing high levels of derepressed class (AmpC) (3-lactamase. Virulence of
all
strains was enhanced by suspending the bacteria in hog gastric mucin prior to
infection.
Mice were dosed subcutaneously at 1 and 5 hours post-infection, with
either cefotaxime alone or coadministered with the 0-lactamase inhibitor at
lmg/kg
or 5mg/kg.
Table 1
Efficacy of Cefotaxime Alone and with Example 1
in Mouse Intraperitoneal Infections
Cefotaxime CD501(m k )
Organism Alone + +
lmg/kg 5mg/kg
Example 1 Example 1
K.pnewnoniae Test 1 42 36 13.6
Test 2 54 26 8
E. cloacae 4593 Test la 240 170 48
Test 2a 400 170 32
1 CD50 (dose protecting 5096 of animals from letrtal infection) calculated
from
groups of 5 animals per test, subjected to a range of 4 dose levels (4-fold
serial
dilutions)
Efficacy was assessed by the number of animal surviving at 4 days after
infection,
and the total dose that protected 50% of treated animals (CD50 value) was
-34-
WO95l28935 218869PGT/EP95101546
calculated. The results of these studies showed consistent protection of
animals
intraperitoneally infected with cefotaxime-resistant strains of K.pneumoniae
(520)
and E. cloacae (4593), which received cefotaxime and the compound of Example I
compared with those receiving cefotaxime alone.
Example 12.
Table 2 shows the in-vitro synergistic activity of the compound of Example 1
above
(i.e sodium (5R)-6-[(Z)-(2,3-dihydroimidazo[2,1-b]thiazol-6-yI)methylene]penem-
3-
carboxylate) with amoxycillin, expressed in terms of minimum inhibitory
concentration (MIC).
Table 2
AmoxycHlin
Alone +
Example 1 at
Organism (3-lactamase 0.25 1
g/nil mi
Ent.cloacae Nl 1 >256 16 4
(inducible expression)
Ent.cloacae P99 1 >256 256 32
(high constitutive
expression)
S.aureus Russell 2a 128 1 -
E.coli 2b (low expression) >256 4 4
JT39
P.coli JT4 2b (high expression) >256 8 2
P.mirabilis C889 2c >256 64 16
E. coli P91 2d > 256 - 8
P. vulgaris C 2e 32 0.5 0.5
Example 13
Preliminary experimental infection studies have demonstrated the in vivo
efficacy of the compound of Example 1, when coadministered pa.renteraily with
amoxycillin against infections caused by different bacterial pathogens that
produce
(3-lactamase, the compound protecting amoxycillin (Table 3) from inactivation
by a
variety of important 0-lactamases in an experimental model of intraperitoneal
infection.
-35-
WO 95128935 - - - 21*6 95 pC'P/tp95101546
In these studies, mice were infected intraperitoneally with a lethal challenge
inoculum of strains of Escherichia coli (E96), producing TEM-1 (3-lactamase,
for
tests to protect amoxycillin. Virulence of all strains was enhanced by
suspending
the bacteria in hog gastric mucin prior to infection.
Mice were dosed subcutaneously at 1 and 5 hours post-infection, with
either amoxyciIlin alone or coadministered with the (3-lactamase inhibitor at
2mg/kg.
Table 3
Efficacy of amoxyciliin alone and co-administered with Example 1
against an Intraperitoneal Infection of E.coli E96 (TEM-1) in mice.
CDSOl(mg/kg) of
Amox ciIlin
Alone + 2 mg/kg
Exam le 1
Test 1 > 1000 1.0
Test 2 300 < 1.5
Efficacy was assessed by the number of animal surviving at 4 days after
infection,
and the total dose that protected 50% of treated animals (CD50 value) was
calculated. The results of two studies showed amoxycillin to be markedly more
effective in protecting those animals that received coadministration with the
compound of Example 1, than those that received amoxycillin alone (Table 3).
-36-