Note: Descriptions are shown in the official language in which they were submitted.
2~ 901 q4
A XYLANASE OBTAINED FROM AN ANAEROBIC FUNGUS
FIELD OF THE INVENTION
The present invention relates to the field of molecular biology. In particular, the
invention relates to genes encoding xylanases obtained from strains of the anaerobic
fungus Neocallimastixpatriciarum.
BACKGROUND OF THE INVENTION
Endo-xylanases are enzymes that randomly cleave the ,B(1-4) linkages between
xylose residues making up the backbone of xylans, a prevalent form of hemicellulose
found predominantly in plant primary and secondary cell walls. If this complex plant
cell wall poly~accl1d~ide is hydrolyzed with xylanases, it can be exploited as a rich
source of carbon and energy for the production of livestock and microo,yd"i:,",s.
Enzymatic disruption of plant cell walls also increases the efficiency of a number of
industrial processes such as juice extraction, retting of flax fibres and pulp production.
As discussed in greater detail herein, it will be applt:cidL~:d that plant cell walls are
highly variable structures containing several forms of hemicell~ se. Thus, the need
exists to identify and produce novel xylanases that are efficient at degrading this
complex polysaccharide.
The plant cell wall is a highly variable, complex and resilient structure encasing
essentially every cell of higher plants. It ,~ st" ,l~ a rich store of carbon and energy
for herbivores as well as an important renewable resource utilized by the pulp and
paper, lumber, food, and phal" ,aceutical industries. The plant cell wall consists largely
of polysac~il ,alicles and contains lesser amounts of lignin (phenolic esters) and protein.
The primary polysaccharide components of plant cell walls are cellulose (a hydrogen-
bonded ,B(1-4)-linked D-glucan), hemicellulose, and pectin (McNeil et al., 1984). Fibrils
of cellulose t:",bedded in a matrix of pectin, hemicellulose (comprising various ~-xylan
polymers), phenolic esters and protein produce a protective structure resistant to
dehydration and p~ lldLiol1 by phytopathogens through mechanical and enzymatic
" ,echal1i~" ,:,.
Hemicellulose, the second most prevalent polysac-,l ,al ide in many plant cell walls
is composed mainly of xyloglucan or xylan polymers. Xyloglucans consist of a
~ 21 qol 94
backbone of ~-4-linked-D-glucosyl residues sllhstitlltPd with a-linked D-xylosyl side
chains, some of which are extended by fucose, galactose or arabinose residues (McNeil
et al., 1984). Xylans have a backbone structure of ~(1-4)-linked xylose residues. The
structure of xylan is co",, ' ~' by the dlldulll"~"l of various side chains (e.g., acetic
5 acid, arabinose, coumaric acid, fenulic acid, glucuronic acid, 4-~methylglucuronic acid)
to the xylose residues (McNeil et al., 1984). The strands of hemicellulose are hydrogen
bonded to cellulose fibrils to form a strong interconnected lattice.
Cell wall collll,Oailion varies with plant species, variety, tissue type, growthconditions, and age. DiKerences in cell wall composition have been reported between
10 dicotyledonous and monocotyledonouos plants (Chesson et al., 1995). The primary cell
walls of all dicots and many monocots contain greater amounts of xyloglucan thanarabinoxylan. In contrast, plants belonging to the family Gramineae (e.g., grasses and
cereal) have primary walls in which only cellulose is more abundant than arabinoxylan.
Higher pectin cu"c~ ldliOlls are found in the exterior wall or middle lamellae than in the
15 primary or secondary cells walls. Finally, as cells age, cell walls may become more
lignified and resistant to microbial attack.
The complexity of the plant cell wall is related not only to cu" ,~.. ,al variation
but also to the high degree of interaction between constituent cellulose, hemicellulose
and pectin molecules. Dual i"lt:l",e~l,i"g networks of polysaccharides, comprising
20 cellulose fibrils crosslinked with hemicellulose and pectic polysacchraides linked by
calcium bridges, not only produce a resilient primary cell wall but are of direct relevance
to enzymatic dey,dddliol1 (Chesson et al., 1995).
Digestion of the plant cell wall is further cu",, I -' by the structure of
polysau~,ha,ides. Cellulose is a simple unsubstituted polymer of ~(1-4)-linked glucose
25 and requires an endoglucanase and cellobiase for complete degradation. In
cu",~-ali:,on, highly sl Ih~t:~ If''d arabinoxylan requires up to seven different enzymes for
complete degradation. An endo-xylanase randomly cleaves the xylan backbone into
xyloc'~, ',arides which are 5llh~Pt~ ntly degraded to xylose by a xylosidase.
Substituents are cleaved from the xylan backbone with arabinofuranidase, acetylxylan
30 esterase and a-glucuronidase. Ferulic and ~coumaric acid crosslinks are degraded
by feruloyl and ~coumaryl esterases. If complete degradation of the arabinoxylan is
not required, fewer enzymes may be needed I iquef~ tirn of arabinoxylan requires
~ 2~ 9Cl q4
only the ~I,oll~l,i"g of the xylan polymers. Consequently, this objective may beachieved by the production o~ XYIOC' Jnl ' ,arides through the action of a single endo-
xylanase. The choice of enzymes is dependent upon the substrate to be degraded.
The known a,, ' ~s of xylanases are numerous. For instance, the treatment
5 of forages with xylanases (along with cellulases) to increase the rate of acid production
thus ensuring better quality silage and improvement in the subsequent rate of plant cell
wall digestion by numinanst has been described. Xylanases can be used to treat rye,
and other cereals with a high arabinoxylan content to improve the digestibility of cereal
by poultry and swine. Xylanases can be used in bioconversion involving the hydrolysis~0 of xylan to xyi ' ~ hal i.les and xylose which may serve as growth substrates for
uorydni~llls. This could involve simultaneous saccharification and ~ullt:llldliom
Xylanases can be used in biopulping to treat cellulose pulps to remove xylan impurities
to produce pulps with different characteristics. In some cases they can be applied to
reduce the amount of chlorine needed to bleach the pulp and reduce the energy of15 refining pulp. Further, xylanases are useful in the retting of flax fibres, the clarification
of fruit juices, the plt~,ua~dtion of dextrans for use as food thickeners and the production
of fluids and juices from plant materials.
Some cha, d~ , of an endo-xylanase from N. patriciarum strain 27 (from the
Agriculture and Agri-Food Canada Lethbridge culture collection) have been reported
20 previously (Tamblyn Lee et al., 1993). Tamblyn Lee et al. described the isolation of a
6.~-EcoRI fragment containing a gene encoding an endo-xylanase. The N. patriciarum
strain 27 was not disclosed or made publicly available. The location of the xylanase
gene was narrowed down to a 3.6-kb EcoRI Sall fragment. Expression of the
endo-xylanase gene in E. coli produced at least three proteins (51, ~8 and 68 kDa)
25 having xylanase activity. This study did not fully ~;l ,al dl,l~ the N. patriciarum strain
27 endo-xylanase gene. No attempt was made to detemmine the nucleotide sequence
of the gene. Nucleotide sequence data is required to create an efficient fusion
construct between the endo-xylanase gene and the sequences of a heterologous
t~x,ui~ssiun system. Without this i"~ul" IdliUI 1, the large DNA fragments of Tamblyn Lee
30 et al. would not be useful for the construction of a functional gene fusion. This effort
would be hampered by a lack of detailed infommation about the stnucture of the gene
and the location of useful restriction sites. The large DNA fragments identified by
Tamblyn Lee et al. are not useful for~om~merlial enzyme production. Specifically, if
these large DNA fragments were cloned into efficient uX~ 5~iull systems, translation
of the resulting Lldl ,s.;,i,ul~ transcribed from a strong heterologous promoter would not
be possible as translation would be 1~l " ,in dl~d at one of the multiple stop codons found
in AT rich sequences upstream from the endo-xylanase gene. Further ~il ,aldul~ dlion,
isolation and nucleotide sequencing of the N. patriciaNm strain 27 endo-xylanase gene
would be required if it were to be of col"",el~ial importance.
In light of the many industrial a; ,:' -us for xylanases, the need for new
xylanases is apparent. Accordingly, it would be of great importance to obtain genes
encoding xylan-degrading enzymes from novel sources which may be brought to
e,.~ Iu55iOI1 in other, high-producing microbial or eukaryotic ex~,ussiol1 systems.
SUMMARY OF THE INVENTION
In ac~;ou~dnce with the present invention, DNA sequences encoding novel and
useful xylanases derived from anaerobic fungi are provided. As used herein and in the
claims, the term "xylanase" means an enzyme having xylan degrading activity.
A xylanase gene (xynC) from Neoc~"U~a~ patriciaNm strain 27 from the
Agriculture and Agri-Food Canada culture collection at Lethbridge, Alberta, Canada has
been cloned and sequenced, and the nucleotide sequence of a DNA fragment including
xylanase encoding region (CDS) of the xynC gene is provided in SEQ ID NO. 1.
Escherichia coli strain DH5a (pNspX-04), canying the xynC gene was deposited
November 8, 1996 with the American Type Culture Collection (12301 Parklawn Drive,
Rockville, Maryland, 20852-1776, as ATCC98249) .
The invention extends to DNA sequences which encode xylanases and which
are capable of hybridizing under stringent conditions with all or part of the xynC gene
sequence. As used herein and in the claims, "capable of hybridizing under stringent
condilio,1~" means annealing to a subject nucleotide sequence, or its complementary
strand, under standard conditions (ie. high temperature and/or low salt content) which
tend to disfavor annealing of unrelated sequences. As used herein and in the claims,
~conditions of low stringency" means hyulidi~dliol1 and wash conditions of 40 - 50~C,
6 X SSC and 0.1 % SDS (indicating about 50 - 80% homology). As used herein and in
the claims, "~iu~ Idiliul~s of medium stringency~ means hybl idi~dlion and wash conditions
219~194
of 50 - 65~C, 1 X SSC and 0.1% SDS (indicating about 80 - 95% homology). As usedherein and in the claims, "conditions of high stringency" means hybridi~dlioll and wash
conditions of 65 - 68~C, 0.1 X SSC and 0.1% SDS (indicating about 95-100%
homology) .
A method for identifying other nucleic acids having xylanase activity is also
provided wherein nucleic acid molecules are isolated from an organism and nucleic acid
hybl idi~dliu n is perfommed with the nucleic acid molecules and a labelled probe having
a nucleotide sequence that includes all or part of nucleotide sequence SEQ ID NO. 1.
By this method, xylanase genes similar to the xynC gene may be identified and isolated
from other anaerobic fungi.
The invention extends to purified and isolated xylanases obtained from strains
of Neocr"'"a:,li,~ patriciaNm, particularly Neor~"'"a:,li,~ patriciaNm strain 27. A
preferred xylanase has the amino acid sequence shown in SEQ ID NO. 2.
The invention extends to ~ iol l constructs constituting a DNA having a
coding region encoding a xylanase of the present invention operably linked to control
sequences capable of directing ~ ,sion of the xylanase in a suitable host cell. The
control sequences may be homologous to or h~l~lulogous to the xylanase encoding
region. As used herein and in the claims, the temm "homologous" DNA refers to DNA
originating from the same species as the host cell or control sequences, as the context
requires. For example, Aspergillus nigermay be transformed with DNA from A. niger
to improve existing properties without introducing properties that did not exist previously
in the species. As used herein and in the claims, Lheterologous" DNA refers to DNA
originating from a diflerent species. For example, the N. patriciaNm strain 27 xynC may
be cloned and e,~ ssed in E. coli.
The invention further extends to host cells which have been lldn~fulllled with,
and express DNA encoding a xyianase of the present invention, and to methods of
producing such transformed host cells. As used herein and in the claims, "host cell"
includes animal, plant, yeast, fungal, protozoan and prokar,votic host cells.
The invention further extends to lldnsy~l ,iu plants which have been transfommedwith a DNA encoding a xylanase of the present invention so that the transfommed plant
is capable of ~ ssi"g the xylanase and to methods of producing such transfommed
plants. As used herein and in the claims, "Lldllsg~,lic plant" includes lldnsge,,ic plants,
2~ 9~ 7~
plant tissues and plant cells. In a prefenred t", Ibodi, "~"1, the transformed plant is of the
species Brassica napus (canola).
The present invention also extends to oleosin-xylanase fusion proteins, DNA
sequences encoding oleosin-xylanase fusion proteins, and 11 dl ,sge"ic plants, preferably
S B. napus, which have been llan:~lurllled to express such oleosin-xylanase fusion
proteins. Surprisingly, these oleosin-fusion proteins have been discovered to retain
xylanase activity. When B. napus is transformed with a DNA sequence of the present
invention encoding an oleosin-fusion protein, the oleosin-fusion protein may
i"""~ Hn the IlltnllL)ldlle sunrounding the oil-bodies found in the B. napusseeds.
Iû The canola oil is extracted from the seeds by, for instance, crushing, leaving a solid
fraction and an oil fraction. Disruption of oil-body membranes in the oil fraction leaves
the oil-body ",e",l,,dnes forming a gum which can be separated from the oil. The gum
contains the oleosin-xylanase fusion protein. The gum is then added to the soildfraction during production of canola meal. Canola meal is a low-cost animal feed15 supplement which is high in protein. Canola meal made from lldllSg~lliC B. napus
l,dn~ ",ed with a DNA sequence encoding an oleosin-xylanase fusion protein
therefore also provides an excellent source of su~ " ,e, lldl xylanase for the animal.
Xylanases of the present invention are useful in a wide variety of a;, ':c " :lsinvolving the degradation of xylan. Accordingly, the invention extends to feed
20 su~ ",~"l:, containing a xylanase of the present invention. Such feed supplements
may also contain other enzymes, such as, proteases, ~:PI~ CPC, phytases and acidpho~ dldses. The xylanase may be added directly to an untreated, pelletized, or
otherwise pluce~ ed feedstuff, or it may be provided separately from the feedstuff in,
for instance, a mineral block, a pill, a gel fommulation, a liquid fommulation, or in drinking
25 water.
BRIEF DESCRIPTION OF THE DRAWINGS
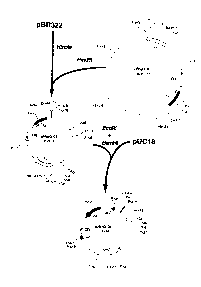
Figure 1 depicts a constnuction pathway of plasmid constructs carrying an endo-
xylanase gene cloned from N. patriciaNm 27.
Figure 2 is a schematic ,~p,~se"ldlion of the deletion analysis in which the
30 location of the endo-xylanase gene cloned from N. patriciarum 27 was determined. E
= EcoRI; H = Hindlll; P = Pvull; B = Bglll.
2t 9~1 ~4
Figure 3 is the nucleotide sequence of a fragment containing the endo-xylanase
gene (xynC) cloned from N. patriciarum 27. The predicted amino acid sequence is
shown beneath the nucleotide sequence. The CDS is located from nucleotide 301 tonucleotide 1755.
Figure 4 is a schematic ~ sel lldliun of: A) the structure of the xynC coding
region. The signal sequence is followed by the sequence encoding for the catalytic
domain of the enzyme (solid box) and a repeated peptide (shaded box). The 3' end of
the gene codes for a region of unknown function; and B) the oleosin-xylanase C
t9Xp~5~iul I constnuct. The endo-xylanase gene was ligated between the oleosin gene
(promoter plus coding region) and the temminator of nopaline synthetase (NOS).
Figure 5 is a schematic representing the N. patriciarum endo-xylanase gene
(xynC) fragments cloned into pGEX-4T-3. (Pvu - Pvull)
DETAILED DESCR~r~llON OFTHE INVENTION
The present invention provides purified and isolated DNA sequences of
anaerobic fungal origin, which encode xylanases and genetic variants thereof. The
DNA sequence preferably includes the xylanase-encoding region (CDS, protein coding
sequence). Genetic variants include hybrid DNA sequences containing the xylanaseCDS fused to regulatory regions such as promoter, leader peptide and terminator
signals, originating from hollloloyuus or h~l~lulo!Juus sources. Genetic variants also
include DNA sequences encoding mutant xylanase proteins and degenerate DNA
sequences wherein the xylan-degrading activity of the enzyme is retained. The present
invention provides the starting material for the construction of "second generation''
xylanases, i.e., mutant xylanases with properties that difler from those of the enzymes
isolated herein, or DNA sequences (encoding the xylanase CDS) altered to reflect the
degeneracy of the genetic code or cross-species variation. Genes can be readily
mutated by procedures known in the art (e.g., chemical, site directed, random
polymerase chain reaction mutagenesis~ thereby creating gene products with altered
properties (e.g., temperature or pH optima, specific activity or substrate specificity). The
xylanase gene of the present invention can be used also in htlleluloyous hyl~lidi~dlion
and polymerase chain reaction ~ Jel i" ,t:"l~, directed to isolation of xylanase-encoding
genes from other natural sources.
21 901 94
Screening o,yani:,",s for endo-xylanase activity may be acomplished by a
number of assays methods not critical to the present invention. These include visual
assays such as the incorporation of xylan (e.g., oat spelt xylan, rye arabinoxylan) or
.,hlulllogullic substrates (e.g., remazoi brilliant blue xylan or RBB-xylan) into agar
media. Hydrolysis of the xylan will be indicated by the presence of zones of clearing
around isolates with endo-xylanase activity. Staining of the medium with Congo red
(Teather and Wood,1982) allows visualization of the hydrolytic activity on solid medium
containing non-"l " u" ,ogt:",c substrates such as oat spelt xylan.
Once a xylanase of interest has been identified, the DNA sequence encoding
such a xylanase may be cloned from the organism which naturally produces the
xylanase by a variety of methods. Gene libraries (genomic DNA and/or cDNA) are
constructed by standard methods (Ausubel et al., 1990; Sambrook et al., 1989) and
screened for the desired gene. In the case of eukaryotic olyd"i~",s and inducible
xylanase exp,t,s:,iol1, it may be advantageous to construct cDNA libraries with mRNA
isolated from the organism, which naturally produces the xylanase, following cultivation
in an inducing medium (e.g., a medium containing straw or xylan as the soie carbon
source). Clones carrying the desired xylanase gene may be isolated by screening the
library with enzyme activity assays (Teather and Woods, 1982), heterologous probes,
or results generated during purification of the gene product, such as N-terminal and
internal amino acid sequence data and a, ltiL,o.lies.
Using Congo red detection, a A'~ r " lldstixpatricianum strain 27 genomic DNA
library was screened for lambda clones possessing xylanase activity (Tamblyn Lee et
al.,1993). A xylanase positive clone carrying a 6.5-kb EcoRI insert was identified and
confinmed by Southem blot hybridization to have originated from N. patriciarum strain
27.
Plasmid DNA extracted from the newly isolated clone and introduced into E. coli
cells by t~dl1~ul'''dlion produced ampicillin resistant, xylanase positive colonies.
Zymogram analysis of cell extracts from E. coR DH5a cells canying a 3.5-kb EcoRI Sall
DNA fragment isolated from the original 6.5-kb EcoRI fragment showed active bands
of 68, 58, and 51 kDa. The gene (xynC), encoding the observed xylanase activity in
r~combillalll E. coli clones, was identified by deletion and nucleotide sequence
2t 901 94
analysis. The nucleotide sequence and deduced amino acid sequence are shown in
Figure 3, and further illustrate that the cloned sequence encoded a xylanase.
It is known in the biological arts that certain amino acid ~llhstitl~tiQns can be
made in protein sequences without affecting the function of the protein. Generally,
5 conservative amino acid sl Ihstitl ~tions are tolerated without affecting protein function.
Similar amino acids can be those that are similar in size and/or charge properties, for
example, aspartate and glutamate and isoleucine and valine are both pairs of similar
amino acids. Similarity between amino acid pairs has been assessed in the art in a
number of ways. For example, Dayhoff et al. (1978) in Atlas of Protein Sequence and
Structure, Volume 5, Supplement 3, Chapter 22, pages 345-352, which is i"col~.olaled
by reference herein, provides frequency tables for amino acid sl Ih~tih Itions which can
be employed as a measure of amino acid similarity. Dayhoff et al.'s frequency tables
are based on comparisons of amino acid sequences for proteins having the same
function from a variety of evolutionarily different sources.
It is also known that often less than a full length protein has the function of the
complete protein, for example, a truncated protein lacking an N-terminal, intemal or a
C-terminal portion of the protein often has the biological and/or enzymatic activity of the
complete natural protein. Those of ordinary skill in the art know how to make truncated
protein and proteins with intemal deletions. In the present invention, the function of a
20 truncated xylanase protein or an intemally deleted xylanase protein can be readily
tested using the xylanase assay described ht:l~il Ibelu.. and in view of what is generally
known in the art.
Sl~hstitllt~d and truncated xylanase derivatives which retain subbldlllially thesame the enzymatic activity of the xylanase ~,c " 'Iy disclosed herein are con:,ide, ~d
equivalents of the t~ ",, ' 'icd xylanase and are within the scope of the present
invention, particularly where the specific activity of the c, Il- It~d or truncated xylanase
derivative is at least about 10% of the specifically ~x~",, ' 'icd xylanase. The skilled
artisan can readily measure the activity of a tnuncated or .sl Ih~tih It~d xylanase using the
assay procedures taught herein and in view of what is generally known in the art.
This invention includes structurally variant xylanases derived from a xylanase
obtained from an anaerobic fungi, particularly those derived from a xylanase ~e~ ic..ll"
disclosed herein, that are suL,:,Ldllli~lly functionally equivalent to that xylanase as
~1 901 74
assayed as described herein in view of what is generally known in the art. Structurally
variant, functional equivalents of the xylanases of this invention include those xylanases
of an anaerobic fungi having a contiguous amino acid sequence as in the xylanaseamino acid sequence disclosed herein (SEQ ID NO. 2), particularly those variant
5 xylanases which have a contiguous amino acid sequence of a xylanase of an anaerobic
fungi that is a contiguous sequence at least about 25 amino acids in length.
As with other genes, it is possible to use the characterized xylanase-coding
sequences from anaerobic fungi in a variety of ~X,ul'~55iOI1 systems for co"""ell.idl
protein production. Application of ,~col"bi"al,l DNA I~:u-l",ology has enabled enzyme
10 manufacturers to increase the volume and efficiency of enzyme production, and to
create new products. The original source organism need no longer limit the production
of co"""e,uial enzymes. Genes encoding superior enzymes can be lldl1~ u~d from
organisms such as anaerobic fungi, typically i" ,,ul dUliCdl for commercial production, into
well .;I,ald.;l~ d industrial microbial production hosts (e.g., Aspergillus, Pichia,
15 Trichoderma, Bacillus spp.). As well, these genes may be transferred to novel plant
and animal ~ iu" systems.
Industrial strains of ",i.;,uo~yani:,ll,s (e.g., Aspergillus niger, Aspergillus ficcum,
Aspergillus awamori, Aspergillus oryzae, Tri,,l,od~""d reesei, Mucor miehei,
Kluyveromyces lactls, Pichia pastoris, Saccharomyces cerevisiae, Escherichia coll,
20 Bacillus subtilis or Bacillus li,,l,enlru""i~) or plant hosts (e.g., canola, soybean, corn,
potato) may be used to produce xylanases. All systems employ a similar approach to
gene exp,u:,:,iu". An t~AIn~ssioll construct is assembled to include the protein coding
sequence of interest and control sequences such as promoters, enl1al,ce,:, and
~ l lil ldIul :~. Other sequences such a signal peptide sequences and selectable markers
25 may be included. To achieve extracellular ~,UI~:~Sioll of xylanase, the ex~ ssiol1
construct of the present invention utilizes a secretory signal peptide sequence. The
signal peptide sequence is not included on the ~-,ul~ssion construct if cytoplasmic
expression is desired. Tldnsc,i,uIiol1dl tellllilldIul~ are included to ensure efficient
transcription. Ancillary sequences enhancing ~pl~5~iU" or protein purification may
30 also be included in the e~ s~ion construct. The promoter, enhancer, signal peptide
and terminator elements are functional in the host cell and provide for efficient
expression and secretion of the xylanase.
21 901 94
The xylanase-coding sequences are obtained from anaerobic fungal sources
sources. Various promoters (l~d~,,,i,uliollal initiation regulatory region) may be used
according to the present invention. The selection of the appropriate promoter isd~,uel1~ld"l upon the proposed ~,~,ul~SSiO11 host. The promoter may be ho",ologous or
S heterologous to the cloned protein coding sequence. Examples of heterologous
promoters are the E. coli tac and trc promoters (Brosius et al., 1985), Bacillus subtilis
sacB promoter and signal sequence (Wong, 1989), aox1 and aox2 from Pichia pastoris
(Ellis et al., 1985), and oleosin seed specific promoter from Brassica napus (van
Rooijen and Moloney, 1 995a). Promoter selection is also d~,ut,l ~dt"l upon the desired
10 efficiency and level of peptide or protein production. Inducible promoters such tac and
aox1 are often employed in order to dldllldlically increase the level of protein~A,ul~s~ium OVel~,UI~;OI1 of proteins may be harmful to the host cells.
Consequently, host cell growth may be limited. The use of inducible promoter systems
allows the host cells to be cultivated to acceptable densities prior to induction of gene
15 expression, thereby, facilitating higher yields of product. If the xylanase-coding
sequence is to be integrated through a gene I~Jlac~ l ll (omega insertion) event into
a target locus, then promoter selection may also be influenced by the degree of
homology to the target locus promoter.
Various signal peptides may be used according to the present invention. A
20 signal peptide sequence which is ho",ologous to the xylanase-coding sequence to be
expressed may be used. Alternatively, a signal peptide sequence which has been
selected or designed for improved secretion in the ~,~,ul~ion host may also be used.
For example, B. subtilis sacB signal peptide for secretion in B. subtilis, the
Saccharomyces cerevisiae a-mating factor or P. pastoris acid phosphatase phol signal
25 sequences for P. pastoris secretion. A signal peptide sequence with a high degree of
homology to the target locus may be required if the xylanase-coding sequence is to be
integrated through an omega insertion event. The signal peptide sequence may be
joined directly through the sequence encoding the signal peptidase cleavage site to the
xylanase-coding sequence, or through a short nucleotide bridge consisting of usually
30 fewer than ten codons.
Elements for ~"hal1ui,lg t"~,u~SSiO11 lldl1scli,uliol1 (promoter activity) and
lldl1sldliu" have been identified for eukaryotic protein expression systems. For
1 1
2~ 901 ~4
example, the po~iliuni"g the Cauliflower Mosaic Virus (CaMV) promoter 1000 bp oneither side of a h~ uloyùus promoter may elevate transcriptional levels by 10 to 400
fold. The expression construct should also include the app,uplidLu translationalinitiation sequences. Mo-liricdlion of the tl~,ultlssiùll construct to include the Kozak
5 consensus sequence for proper l~dnsldliollal initiation may increase the levei of
llallsldlioll by 10 fold.
Elements to enhance purification of the protein may also be included in the
u.~p,us~iu" construct. The product of oleosin gene fusions is a hybrid protein containing
the oleosin gene joined to the gene product of interest. The fusion protein retains the
10 lipophilic properties of oleosins and is illcoi,uoldlt:d in the oil body membranes (van
Rooijen and Moloney, 1995a). Association with the oil bodies may be exploited tofacilitate the purification of the ,uco",ui"a"l oleosin fusion proteins (van Rooijen and
Moloney, 1995a).
A selection marker is usually employed, which may be part of the ex,ulussio
15 construct or separate from the C:A,UI~S~ioll construct (e.g., carried by the ~X,u~ iOIl
vector). The selection marker may be used as an alternative target locus for t:~,u~ssion
construct illlUyldliOIl. Tldll:~ulllldliull of the host cells with the lucollluilldlll DNA
molecules of the invention is monitored through the use of selectable markers.
Examples of these are markers that confer resistance to antibiotics (e.g., bla confers
20 resistance to ampicillin for E. coli host cells, nptll confers kanamycin resistance to B.
napus cells) or that pemmit the host to grow on minimal medium (e.g., HIS4 enables P.
pastoris GS115 His cells to grow in the absence of histidine). Sel~ctAhle markers are
usuually associated with lldllscli~uliullal and l~dnsldlional initiation and IUIIIIilldliOIl
regulatory regions different from the tlh~ ssion construct in order to allow for25 independent t~x,u~s:~ioll of the marker. Where antibiotic resistance is employed, the
conce"l~dliull of the antibiotic for selection will vary depending upon the antibiotic,
generally ranging between 10 and 500 ,ug of the alllibiulic/lllL of medium.
The tl~,u~s:,iu~, constnuct is assembled by employing known recombinant DNA
techniques. Restriction enzyme digestion and ligation are the basic steps employed to
30 join two fragments of DNA. The ends of the DNA fragment may require Illodi~icdliull
prior to ligation and this may be accu" ,,u li~hed by filling in overhangs, deleting terminal
portions of the fragment(s) with nucleases (e.g., Exolll), site directed mutagenesis, and
12
~1 9~1 ~4
adding new base pairs by the Polymerase Chain Reaction (PCR). Polylinkers and
adaptors may be employed to facilitate joining of select fragments. The ex,u,~ssioll
construct is typically as~e" ~uled in stages employing rounds of restriction, ligation and
lldl1~iulllldliunofE coli. Therearenumerouscloningvectorsavailableforconstnuction
S of the t:X,~ ssiu" constnuct and the particular choice is not critical to this invention. The
selection of cloning vector will be influenced by the gene transfer system selected for
introduction of the ~,u~ussioll contruct into the host cell. At the end of each stage, the
resulting construct may be analyzed by restriction, DNA sequence, hyblidi~dIiu,, and
PCR analyses.
Thet,,~p,us~iu,,constructmaybel,d,,~ul,,,edintothehostasthecloningvector
construct, either linear or circular, or may be nemoved from the cloning vector and used
as is or introduced onto a delivery vector. The delivery vector facilitates the introduction
and "..~;.,t~,ndnce of the expression construct in the selected host cell type. The
~cu~t:s~iu~ I constnuct is introduced into the host cells by employing any of a number of
15 genetransfersystems(e.g.,naturalco",,u~tu"ce,~l,u",ica'!ymediatedl~d~ ul",dliul"
protoplast lldll~rulllldlioll, eleuIIupo,dlioll, biolistic lldl1~iUIIIIdIiOI1, Ird~ .Iiul1, or
conjugation) and is dependent upon the host cells and vector systems used.
For instance, the expression construct can be introduced into P. pastoris, cellsby protoplast I,dn:,~u", IdtiO n or t:le~.IIupol..tion. Electroporation of P. pastoris is easily
2û accol"ul;~l,ed and yields Ildll~ulllldIiull efficiencies co",,ualdble to spheroplast
transformation. Pichia cells are washed with sterile water and resuspended in a low
conductivity solution (e.g., 1 M sorbitol solution). A high voltage shock applied to the
cell suspension creates transient pores in the cell membrane through which the
I~dn~uilllillg DNA (e.g., e~ lussion construct) enters the cells. The expnession25 construct is stably "IdillIdil,ed by integration, through homologous It~collluilldIiull~ into
the aoxl (alcohol oxidase) locus.
Al~ cly, an ~,~,ul~ iu" construct, comprising the sacB promoter and signal
sequence operably linked to the protein coding sequence, is carried on a plasmid,
pUB110, capable of autonomously replicating in B. subtilis cells. The resulting plasmid
30 construct is introduced into B. subtilis cells by lldn~UlllldliUII. Bacillus subtilis cells
develop natural co, Il,UUlt:l ,ce when grown under nutrient poor conditions.
13
21 901 94
Host cells carrying the t:A,u,~s~iun construct (i.e., l,dll~lu~ ed cells) are identified
through the use of the selectable marker carried by the ~dA,UI ~S::~iOIl construct or vector
and the presence of the gene of interest confimmed by a variety of techniques including
h~,bl-idi~dliol1~ PCR, and antibodies.
Trdrl~ullllad microbial cells may be grown by a variety of techniques including
batch and continuous 1~" "~"laliOIl on solid or semi-solid media. Tldn~ilul " ,ad cells are
propagated under conditions optimized for maximal product to cost ratios. Product
yields may be d~ dl 11 " 'Iy increased through the manipulation of cultivation parameters
such as temperture, pH, aeration and media composition. Careful manipulation andmonitoring of the growth conditions for recombinant hyper-~x~ i"g E. coli cells may
result in culture biomass and protein yields of 150 g (wet weight) of cells/L and 5 g of
insoluble protein/L, respectively. Low ~:ullc~lllldliolls of a protease inhibitor (e.g.,
phenylmethylsulfonyl fluoride or pepstatin) may be employed to reduce proteolysis of
the over-expressed peptide or protein. Altematively, protease deficient host cells may
be employed to reduce or eliminate deyldddliull of the desired protein.
Following ~""enldtion, the microbial cells may be removed from the medium
through du~ d,,l processes such as centrifugation and filtration. If the desiredproduct is secreted, it can be extracted from the cell free nutrient medium. Altematively,
the culture or cell free medium may be used directly or concel Illdl~d (e.g., ullld~illldliol1,
dehydration, Iyu~ h ' 1) and used in an ~ 1 requiring xylanase activity. In the
case of intracellular production, the cells may be harvested and used directly or
ruptured (e.g., rl ,e.;l Idl ,ical forces, ultrasound, enzymes, chemicals, high pressure). The
resulting Iysate may be used as in an a,),' " n requiring xylanase activity or
subjected to further p, uces:~il ,g.
In a third example, Brassica napus celis are l~dn~r~,l",ed by Agrobacterium
mediated l~dn~ul,,,atioll. The t~X~ ssioll construct is inserted onto an binary vector
capable of replication in A. h""~rdciensand ", ' " 1 into plant cells. The resulting
contruct is lldl~iulllled into A. tu",~ld,,iel7s cells carrying an attenuated Ti or ~helper
"plasmid. When leaf disks are infected with the ,~cu,,,ui, ,anl A. t~ rdcie"~ cells, the
~x,ul~:~sioll construct is lldll~rt~ d into B. napus leaf cells by conjugaHI l ' :" " n of
the binary veuLul..ex~ iu" construct. The ~A~ iOIl constnuct integrates at random
into the plant cell genome.
14
2~ 9~1 94
After selection and screening, lldl1a~u~ ed plant cells can be regenerated into
whole plants and varietal lines of Irdnsgel1ic plants developed and cultivated using
known methods.
Xylanase may be extracted from harvested portions or whole plants by grinding,
S homogenization, and/or chemical treatment. The use of seed specific lipophilicoleo~i"..g~"e fusions can facilitate purification by partitioning the oleosin fusion protein
in the oil fraction of crushed canola seeds and away from the aqueous proteins (van
Rooijen and Moloney, 1995a).
Expression of xylanases of the present invention in Brassica napus (canola) is
useful, particularly as the enzyme will be expressed in every seed of the plant. Canola
is an important agricultural crop due to its high oil content. There are many uses for
canola oil, including such diverse a" I " ns as lubricating oils and oils for human
consumption. The non-oil fraction remaining after the oil is extracted from canola seeds
by techniques such as cnushing may be described as canola meal. Canola meal is
typically used as an animal feed supplement due to its high protein content, which may
be as high as 40-50~/0. Canola meal makes an ideal feed supplement as it is
sub~ldnlially less expensive than alternatives such as soybean meal. Furthemmore,
canola meal also contains higher conce, IlldliUI ,:j than soybean meal of nutrients such
as carbohydrates.
The oil in the seeds of B. napus is found within oil-bodies surrounded by an oil-
body rl 1~ ~ Ibldl 1~1 which functions to contain the oil. Oleosin proteins are located in the
rllt:lllbldlle surrounding the oil body. Oleosins (oil-body proteins) are structural proteins
found in the seeds of all higher plants investigated to date (monocots, dicots and
g~"""o~,uel",s). They are highly liophilic with a unique secondary structure which
permits their central core to be embedded in oil-bodies while the more hydrophilic N-
and C-temmini reside on the cytoplasmic side. Their role appears to be primarily that of
stabilizing triacylglyceride-containing oil-bodies as discrete organelles (van Roijen and
Moloney, 1 995a). The hydrophilic N- and C-temmini of the oleosin protein may provide
dlldUI Illlt~l 11 sites for forming fusions with other proteins.
In a preferred embodiment of the present invention, B. napus is 11 dl l~ UI 11 ,ed with
an ~p~ iull construct containing a nucleotide sequence encoding a xylanase of the
present invention llal1sldliu,,,~lly fused to a nucleotide sequence encoding an oleosin
21 901 q4
.
protein to provide seed oil body t!X,u~u~siol1 of the xylanase, as described in the
examples which follow.
The oleosin-xylanase fusion protein is immobilized in the seed oil-body
membrane and remains with the canola meal portion during oil extraction. As
5 d~ on~ lud in the examples which follow, the oleosin-xylanase fusion proteins retain
xylanase activity. Canola meal produced from the transgenic B. napus of the present
invention thus provides an ideal source of xylanase when the canola meal is used as
a feed supplement (protein source) in animal diets. Su,upl~ll ,e, lldl xylanase in animal
diets degrades cell wall components in the animal feed, resulting in increased feed
10 digestion and a reduction in pollution from animal wastes.
If necessary, various methods for purifying the xylanase, from microbial
~t~ullt:llldlion and plant extracts, may be employed. These include pl~-;i,uildliol1 (e.g.,
ammonium sulfate pl~ui,uildliun)~ ulllullldluyldplly (gel filtration, ion exchange, affinity
liquid ~,hlullldluyldplly), ulll "" " 1, elt:ul,uphoresis, solvent-solvent extraction (e.g.,
15 acetone precipitation), cc." ~bi~ IdliOlls thereof, or the like.
All or a portion of the microbial cultures and plants may be used directly in
a;, ' " 15 requiring the action of a xylanase. Various formulations of the cnude or
purified xylanase p,t,pal ~s may also be prepared. The xylanase can be stabilized
through the additions of other proteins (e.g., gelatin, skim milk powder) and chemical
20 agents (e.g., glycerol, polyethylene glycol, reducing agents and aldehydes). Enzyme
su~,u~ ,iulls can be cu"c~"l, dl~d (e.g., tangential flow ull, d~ill, dliu") or dried (spray and
drum drying, Iyupll' " 1) and formulated as liquids, powders, granules and gels
through known p,uce~es.
Formulations of the desired product may be used directly in A!,' " 15
25 requiring the action of a xylanase. Liquid cunce"l,dlu~, powders and granules may be
added directly to reaction mixtures and ~ m ~ dliUI 1::~. The fommulated xylanase can be
administered to animals in drinking water. It may be mixed also with, sprayed on or
pelleted with other feed stuffs through known processes. Alternatively, the xylanase
gene may be introduced into an animal, thereby el;. "i, Idlil 19 the need for the addition
30 of extraneous xylanase.
In another fommulation, the xylanase of the present invention may take the form
of viable microbial feed inoculants. Cultures of ",i,_,uolyani:,",s expressing a xylanase
16
2t ~01 9~
.
gene such as N. patriciarum strain 27 or, ucu" ,L i"a"l " ,iu, uo, ydl li~>l l l5 expressing the
xylanase CDS are grown to high Cul lC~ ldliOl)s in fenmentors and then harvested and
cu"cu"lldlt:d by ct"~ 9~qtion. Food-grade whey and/or other cryop,ul~uld"l~ are then
admixed with the cell concentrate. The resulting mixture is then cryogu,)ically frozen
5 and freeze dried to preserve xylanase activity by standard Iyopl ,- - 1 procedures.
The freeze-dried culture may be further p,ucessed to fomm finished product by such
further steps as blending the culture with an inert carrier to adjust the strength of the
product.
All or a portion of the microbial cultures and plants as produced by the presentlû invention may also be used in a variety of industrial plucesses requiring the action of
a xylanase.
Examples of such - r r~ 5 are in the production of feed i"y, udit:"l:, and feed
additives for livestock production the retting of flax fibres the cld~ of fruit juices
the pl~pdldliol1 of dextrans for use as food Illiukul1elx and the production of fluids and
15 juices from plant materials. Xylanases can be used also in the bioconversion involving
the hydrolysis of xylan to xyloc --- ,arides and xylose and biopulping to treat
cellulose pulps to remove xylan impurities to produce pulps with different
Chal dutul i~lics.
2û EXAMPLES
Example 1. Cloning an endo-xylanase gene from Neocallimastixpatriciarum
N~,---"'"a:,lixpatriciarum strain 27 was cultivated anaerobically at 39~C in a
modified semi-defined medium (Lowe et al. 1985) containing either Whatman No. 1
filter paper or 0.15% glucose as a carbon source. Cells were harvested by
25 centrifugation after 4 d growth resuspended in extraction buffer (25 mM Tris-HCI pH
8.0; 10 mM EDTA; 50 mM glucose) and stored at-70~C overnight. The preparation
was thawed at room temperature and ho",oy~ni~ed until all cells were resuspended.
Sodium dodecyl sulfate (SDS} and diethylpylucdllJol1dl~ (DEPC} were added to a final
col1celllldliol1 of 0.5~/O (w/v) and 25 mM respectively. The suspension was incubated
3û at 37~C for 1 h. Flutt:i,,ase K (0.1 mg/mL) was added and the mixture was incubated
for 1 h at 55~C extracted twice with phenol and twice with phenol/ul ,lol U~UI " ,. The DNA
21 901 ~4
was precipitated with ethanol and the resulting DNA pellet resuspended in TE (10 mM
Tris, pH 8.0;1 mM EDTA) buffer.
Neocallimastix patriciarum strain 27 genomic DNA was partially digested by
EcoRI. Agarose gel purified 4- to 7-kb EcoRI fragments were ligated (overnight, 4~C)
S to EcoRI-cut and dt,lJho~,uholylated AgtWESAB amms at a molar ratio of 1:2. The ligated
DNA was packaged with a A DNA in vitro packaging kit. The phage library was
amplified on TN plates (10 g Bacto-tryptone, 5 g NaCI per litre containing 0.2% maltose
and 10 mM MgCI2, pH 7.5) with Escherichia coli ED8654 as the host bacterium.
R~oll II,il ,anl phage were screened for xylanase activity by overlaying plaqueswith 0.7~/O (wlv) agarose co"ldi"i"g 0.1-0.25% (w/v) water soluble oat spelt xylan
dissolved in 25 mM potassium phosphate buffer (pH 6.5). The plates were incubated
at 39~C for 3 -18 h and stained with a 0.1~/O (w/v) aqueous solution of Congo red and
destained with 1 M NaCI. Xylanase-producing plaques were surrounded by a yellow
halo visible against the red background. Two positive clones were recovered after
screening 50,000 plaques. Positive plaques were picked and resuspended in SM buffer
(Sambrooket al., 1989). The plaques were purified three successive times by isolation
from agar plates.
Examole 2. Chalduleli~dliQll of positive endo-xylanase clones
Phage stocks and DNA were prepared according to methods described by
Sambrook et al. (1989). Restriction analyses detenmined that the two positive clones
canried an identical 6.5-kb EcoRI insert. This clone was desiy"dled ANspX-101. The
location of the endo-xylanase gene on the 6.5-kb EcoRI fragment was narrowed down
by subcloning fragments of the 6.5-kb EcoRI fragment onto a number of cloning vectors
including pBR322 (Bethesda Research l ~ ie:, - BRL, M -llg~, ON), pUC18
(BRL) or pBluescriptllSK+(Stratagene Cloning Systems, La Jolla, CA). [~,,l,e"~l,ia coli
HB101 andDH5a(BRL)wereusedasthehostbacteriaforthevariouscloningvectors.
Tldn:,~ul,,,ed cells were p,.,paydl~d at 37~C in Luria-Bertani (LB) medium (Sambrook
et al., 1989). Ampicillin (100 ,ug/mL) was illco,,uo,dl~d into media used to culture
plasmid-bearing E. coiistrains. Tldlla~UIIIIdlll colonies were cultivated on LB medium
containing 0.1-0.28% oat spelt xylan. Xylanase activity was detected by staining plates
with Congo red.
18
~7 ~01~4
The 6.5-kb EcoRI insert from ANspX-101 was subcloned into pBR322 yielding
plasmid pNspX-01 (Figure 1). The 1.7- and 2.2-kb Hindlll fragments from pNspX-01were cloned in the correct orit~ alion into pBR322 to produce pNspX-02. The 4.3-kb
BamHI EcoRI fragment from pNspX-02 was ligated with BamHI EcoRI digested pUC18.
S The resulting plasmid was desiylldl~d pNspX-04 (Figure 1). Deletion of the Sall
fragment from pNspX-04 produced pNspX-06. Further truncation of the 3.6-kb EcoRISallfragmentcontainedonpNspX-06wasacco,,,l,1ic,l,edbysubcloningtheEcoRlPvull
or EcoRI Hindlll fragments on to pBluescriptSKII+ (Stratagene Cloning Systems).
Xylanase activity was observed only for the clone carrying the EcoRI Pvull fragment
thereby su,, ,g that the Hindlll site was located within the endo-xylanase gene
(Figure 2).
The origin of the endo-xylanase gene was confirmed by Southem blot
hybl idi~dliUIl using the 3.5-kb Clal fragment from pNspX-02, labelled with [a-~P]-dCTP,
as a probe (Tamblyn Lee et al.,1993).
ExamPle3. Bk,ch~",;cdlul~ald~ liu:,oftheclonedNe- ""a~ patriciaNmendo-
xvlanase
The cloned endo-~-1,4-xylanase was secreted into the pel i~ld~ i.; space of hostEscherichia coli (pNspX-06) cells. The biochemical ullaldult~ ,s of the cloned
enzyme were dt~ l"illed using cnude extracts containing periplasmic endo-xylanase
released by osmotic shock (Table 1, Tamblyn Lee et al., 1993). Xylanase activity was
d~le",li"ed by measuring the amount of reducing sugars released from substrates
according to the method of Nelson-Somogyi (Somogyi, 1952). The N. patriciarum
endo-xylanase hydrolyzed oat spelt xylan and birch wood xylan almost equally well, but
exhibited very low activity on arabinoxylan. The pH and temperature optima for the
periplasmic endo-xylanase activity were 6.2 and 40~C, I~ e-,tiicly, and the Km for oat
spelt xylan hydrolysis was 0.89 mg/mL. SDS-PAGE followed by zymogram analysis
showed active bands of 68, 58, and 51 kDa. The isoelectric point, d~lell"i,led by
isoelectric focusing combined with zymogram analysis, was 3.6.
19
~19019~
Table 1. General biochemical properties of the N. patriciarum 27 xylanase
Property
Molecularweight (kDa) 68158/51
Isoelectric point 3.6
pH optimum 6.2
Temperature optimum 40~C
Substrate specificity
Oat spelt xylan +++++
Birch wood xylan +++++
Rye flour arabinoxylan +/-
Carboxymethylcellulose ++
Acid-swollen cellulose +/-
Barley ~-glucan +/-
Lichenan
Km (mg oat spelt xylan/ml) 0.89
Example 4. Nuclçotide seauence and structural analyses of the Neocallimastix
patriciarum endo-xylanase gene
The 3.4-kb EcoRI - Bglll fragment of pNspX-06 was sequenced in both strands.
Samples were prepared for DNA sequence analysis on an Applied Biosystems Model
373A DNA sequencing system (Applied Biosystems, Inc., l\~ ~ --'19A, ON) by usinga Taq DyeDeoxyTM Tenminator Cycle Sequencing Kit (Applied Biosystems, Inc.).
Template DNA was extracted from ovemight cultures of E. coli DH5~ (pNspX-06) with
the WizardsTM minipreps DNA purification system (Promega Corp., Madison, Wl).
Overlapping sequences were generated by primer walking. The DNA sequence data
was analyzed using MacDNASlS DNA software (Hitachi Software Engineering Co., Ltd.,
San Bruno, CA).
DNA structural analysis identified a single 1458 bp open reading frame (ORF),
designated xynC, overlapping the Hindlll and Pvull sites of the 3.4 kb EcoRI - Bglll
insert and large enough to encode a 51 kDa endo-xylanase (Figure 3). Translation of
the ORF would result in the ~ SSiOI1 of a 485 amino acid polypeptide with a predicted
molecular weight of 50.4 kDa. Further analyses identified a putative signal sequence
(nu~:leotirl~ 301 - 360, Figures 3 and 4) followed by a catalytic domain. The N-tenminal
catalytic domain is followed by a putative proline rich, highly reiterated linker region and
a region of unknown function.
~ 2~ ~al 94
The extent of the endo-xylanase catalytic domain was determined by deletion
analysis. The coding sequence of xynC encoding sequence less the first 41
n~ (nt 301 - 341 Figure 3) was amplified by PCR with oligonucleotide primers
Xl(ATCTCTAGAATTCAACTACTCTTGCTCMAG;SEQlDNO.3)andXll(GGG
5 TTG CTC GAG ATT TCT AAT CAA m AT; SEQ ID NO. 4). The oligonucleotides
were designed to place an EcoRI site at the 5~ end and a Xhol site at the 3 end of the
PCR product. This enabled the xynC PCR product (Figure 4) to be cloned as a
I,dnsldliol1al fusion into EcoRI Xhol digested pGEX~T-3 (Phammacia Biotech Inc. Baie
D Urie PQ). C;",1,ari,,hi~ colicells lldnb~ulllled with this construct named pGEXxynC,
I0 produced endo-xylanase activity. A series of 3 deletions to xynCwas also constructed
in pGEX-4T-3 (Figure 5). Fusion proteins (glu ,iu"e S-ll dl ,~r~, dSt,..,~ylanaSe c) were
expressed and aflinity purified on glutathione Sepharose 4B according to the GST gene
fusion system manual (Phammacia Biotech Inc.). Bound fusion protein was either eluted
and used directly in xylanase assays (GST-fusions) or cleaved with thrombin to release
15 only the xyn~encoded peptides. The specific activities of the purified GST-fusions and
cleaved xylanase C peptides were d~l~""i"ed. Protein conce~ dliu" was measured
with a BioRad (BioRad Ldbul .ies Canada Ltd. 1\~ I~R, ON) protein assay kit.Protein samples were added to a 50 mM potassium phosphate buffer (pH 6.5)
containing 1.5 % oat spelt xylan. Samples were incubated at 40~C. Xylanase activity
20 was determined by measuring the amount of reducing sugar released from the
substrate (Somogyi 1952). All truncated proteins tested (Figure 5, Table 2) had lower
specific acitivities (Table 2) indicating that the full length xylanase C is required for
maximal activity. The reduction in activity was particularly pronounced in the case of
the Pvu fusion protein. This construct displayed only 1.2% of the activity of the full
25 length GST-XynC fusion protein. By comparison the cleaved Pvu xylanase C protein
retained three quarters of the full length protein activity (Table 2).
The specific activity of cleaved affinity purified xylanase C was determined to be
555 units of endo-xylanase activity/mg of protein. One unit of endo-xylanase activity
was defined as one umol of reducing sugar equivalents released per minute.
~1 q~l 94
.
Table 2. Relative xylanase activity (~/O) of truncated xylanase C proteins.
Treatment ConstructRelative xylanase activity
Cleaved XynC 100.0
l~co47 80.2
Hae 85.3
Pvu 74.8
GST-fusion XynC 100.0
Pvu 1. 2
Example 5. O~,e,~x~,tssiol1 of the Neo,- " "a~li,rpatriciarum endo-xylanase qeneIsolation and chald~ dliol1 of xynC from N. patriciarum 27 enables the large
scale production of Xylanase C in any of a number of prokaryotic (e.g., E. co/i and B.
subtilis) or eukayotic (e.g., fungal - Pichia, Saccharomyces, Aspergillus, Trichoderma;
plant - Brassica, Zea, Solanum; or animal - poultry, swine or fish) expression systems
using known methods. For example, general teachings for the construction and
~ ssion of xynC in E. coli, P. pastoris, and B. napus are provided below. Similar
approaches may be adopted for expression of the N. patriciarum 27 endo-xylanase in
other prokaryotic and eukaryotic organisms.
A. Cloning of the Neo " "astix patriciarum xvnC in an Escherichia coli- specific~yl~siol1 construct
An expression construct is constructed in which the region encoding the the fulllength xylanase C, less amino acids 1 -14 (Figure 3) is lldns~i,i,uliul1ally fused with the
tac promoter (Brosius et al., 1985). The promoter sequences may be replaced by those
from other promoters that provide for efficient ~ SSioll in E. coli. The expression
construct is introduced into E. colicells by lldl1~r~llllldliull.
I. Construction of the E. co/iexl.l~s~ 1 vector
A number of E. coli ~ ssion vectors based on the tac or related promoters are
co"""~,~;i..lly available. The constnuct may be prepared with pKK223-3 available from
Pharmacia Biotech Inc. The region of xynC encoding the XynC protein (less amino
acids 1-14) is amplified with oligonucleotide primers Xlll (SEQ ID NO.5 - GC GM TTC
35 ATG TCA ACT CTT GCT CM AGT TTC) and XIV (SEQ ID NO. 6 - GCC TGC AGT
GAT TTC TM TCA ATT TAT). The oligonuclPoticleS Xlll (SEQ ID NO. 5) and XIV
~ 9~ 9~
(SEQ ID NO. 6) were designed to insert suitable restriction sites at the PCR product's
termini to allow direct assembly of the amplified product with pKK223-3. The region of
xynCamplified with Xlll (SEQ ID NO.5) and XIV (SEQ ID NO. 6) is digested with EcoRI
and Pstl and ligated into similarly cleaved pKK223-3.
S li. Transformation of E. coliand XylanaseC ~,~,,,tssion
The pKK223-3::xynC ligation mix is used to transform competent E coli cells.
Strains suitable for high levels of protein ~c,u~t:SSiO11, such as SG13009, CAG926 or
CAG929 (carrying laclon a plasmid such as pREP4), will be employed. T~d~lulll,edcells are spread on LB agar containing ampicillin (100 ,ug/mL) and incubated overnight
10 at 37~C. Ampicillin resistant colonies are screened for the presence of the desired
pKK223-3::xynCconstruct by extracting pDNA and subjecting the pDNA to agarose gel
electrophoresis and restriction analysis. Positive clones may be further characterized
by PCR and nucleotide sequence analysis.
Expression of the N. patriciarum 27 xylanase by 1, dl l~ UU 1 ,ed E. coli cells is tested
15 by growing the cells under vigorous aeration at 37~C in a suitable liquid medium (e.g.,
LB or 2xYT) containing the d,l)UlU,UI idl~ antibiotic selection until the optical density (600
nm) is between 0.5 and 1Ø The tac promoter is induced by adding isopropyl-~-D-thiogAl~cto~ifle (IPTG) to a final concentration between 0.1 and 2 mM. The cells are
cultivated for an additional 2 to 4 h and harvested by centrifugation. Protein ~ sion
20 is monitored by SDS-PAGE, and western bloVimmunodetection techniques.
The expressed XynC may be extracted by breaking (e.g., sonication or mechanical
disruption) the E. coR cells. Protein inclusions of XynC may be harvested by
centrifugation and solllb" ' with 1 to 2 ~/O SDS. The SDS may be removed by
dialysis, electroelution or ulll "" dliOn. The xylanase activity of prepared cell extracts
25 may be assayed by standard methods described in Example 4.
B. Cloning of the Neocallimastix ~atriciarum endo-xylanase in a Pichia pastoris-specific ~,~,u,t,s~ion construct
An expression construct is constructed in which the region encoding the the fulllength xylanase C, less amino acids 1 - 14 (Figure 3) is lldnsldliul.al',l fused with the
30 secretion signal sequences found on P. pastoris ex,~ iu" vectors (Pichia Expression
Kit Instruction Manual, Invitrogen Corporation, San Diego, CA) in order to express the
N. patriciarum xylanase as a secreted product. The promoter and secretion signal
23
21 90~ 9~
sequences may be replaced by those from other promoters that provide for efficient
expression in Pichia. The expression construct is introduced into P. pastoris cells by
lld~ UIllldliUII.
i. Construction of the P. pastoris exp~ iu" vector
A number of P. pastorls ex~ ,ion vectors based on the aox1 promoters and a-
Factor or phol signal sequences are colllll,e,.;i.~'ly available. The construct may be
prepared with pPlCaB available from Invitrogen Corporation. The region of xynC
encoding the XynC protein (less amino acids 1-14) is amplified with oligonucleotide
primers Xl (SEQ ID NO.3) and Xll (SEQ ID NO. 4). The oligonucleotides Xl (SEQ IDNO. 3) and Xll (SEQ ID NO. 4) were designed to insert suitable restriction sites at the
PCR product's termini to allow direct assembly of the amplified product with pPlCaB.
The region of xynC amplified with Xl (SEQ ID NO. 3) and Xll (SEQ ID NO. 4) is
digested with EcoRI and Xhol and ligated into similarly cleaved pPlCaB.
ii. Tldll~ull I IdliUn of P. pastoris and XynC ~AUI t:SSiOIl
The pPlCaB::xynC ligation mix is used to transform competent E. coli DH5a
cells. Tldll~u,,,,edcellsarespreadonLBagarcontainingampicillin(1ûO,ug/mL)and
incubated ovemight at 37~C. Ampicillin resistant colonies are screened for the
presence of the desired pPlCaB::xynCconstruct by extracting pDNA and subjecting the
pDNA to agarose gel el~ulluplloresis and restriction analysis. Positive clones are
further ~;l Idl dul~ d by PCR and DNA sequence analysis. Plasmid DNA is preparedfrom a 1 L culture of an E. coliclone carrying the desired pPlCaB::xynC construct. The
pDNA is digested with Pmel and analyzed by agarose gel elt:~.l,u~,l,ol~ , to confirm
complete digestion of the vector. The digested pDNA is extracted with
phenol:chloroform, ethanol plt:~i,uildl~d and resuspended in sterile distilled H20 to a
final C(JI~C~31111dliUII of 1 ,ug/,uL. In pl~:lpdldlioll for ildll~ UIllldliUll, P. pastorisGS115 or
KM71 cells are grown for 24 h at 30~C in YPD broth. Cells from a 100 ,uL of culture are
harvested by centrifugation and resuspended in 100 ,uL of 1, dn~ UI 11 Idliul I buffer (0.1 M
LiCI, 0.1M dillliullllt~ilul, 45~/O polyethylene glycol 4000) containing 10 ,ug salmon spemm
DNA and 10 ,ug of linearized pPlCaB::xynC. The mixture is incubated for 1 h at 37~C,
spread on YPD agar containing zeocin (1009 ,ug/ml) and incubated for 2 to 5 d at 30~C.
Colonies growing on the selective medium are streaked for purity and analyzed for the
presence of the integrated xynC by PCR and Southern blot hybridization.
24
21 901 q4
Expression of the N. patriciarum 27 xylanase by transfommed P. pastoris cells istested by growing the cells at 30~C and under vigorous aeration in a suitable liquid
medium (eg., buffered complex glycerol media such as BMGY) until a culture optical
density (600 nm) of 2 to 6 is reached. The cells are harvested and resuspended to an
S OD~ of 1.0 in an inducing medium (e.g., buffered complex methanol medium, BMMY)
and incubated for a further 3 to 5 days. Cells and cell free culture supennatant are
collected and protein ~ ssi.,n is monitored by enzyme assay, SDS-PAGE, and
westem blot/immullod~ ,liol1 techniques.
C. Clonina of the Neocallimastix patriciarum endo-xylanase in a Brassica rlaPus seed
10 - sPecific t"~pl~5iOI1 construct
Transfommation and gene ~ plt~ iOIl methods have been developed for a wide
variety of monocotyledonous and dicotyledonous crop species. In this example, a N.
patriciarum 27 xylanase ~ ssi~n constnuct was constructed in which the region
encoding the full length xylanase C, less amino acids 1 - 14 (Figure 3) is lld~lsldli~Jnally~5 fused with an oleosin coding sequence in order to target seed oil body specific
ssion of the N. patriciarum xylanase. The promoter and/or secretion signal
sequences may be replaced by those from other promoters that provide for efficient
~xp~s~io" in B napusoranyotherl~dl~ ""ableplantspecies in ordertoachievethe
same goal as is the objective of this invention. The ~ 5:,;0n constnuct is introduced
20 into B. napus cells by Agrobacterium mediated transfommation.
L constructiQn of the B, naDus ~ s~ion vector
A number of ~X~J,t,ssiol1 vectors functional in B. napus are described in the
literature (Gelvin et al., 1993). To constnuct a oleosin-xylanase gene fusion, the oleosin
and l~colllbil1dll1 xynCgenes were first cloned into pBluescriptllKS+ (pBS) to creat an
25 illL~Illledidl~ plasmid. The construct pCGYOBPGUSA (van Rooijen and Moloney,
1995b) was digested with Pstl and BamHI to isolate the 1608-bp fragment containing
the oleosin promoter and oleosin coding region. The xynC coding region was obtained
by the digestion of pGEXxynC with BamHI and Xhol. These two fragments were cloned
into pBS previously digested with Pstl and Xhol. The resulting plasmid was de:,iy, Idl~d
30 as pBSOleXyn. To obtain a nopaline synthetase (NOS) temminator sequence flanked
by Xbal and Xhol restriction sites, a BamHI and Hinalll fragment from pCGYOBPGUSA
was subcloned into pBS to make an i"l~l"~edidl~ plasmid pBSNos. Digestion of this
~1 9~1 9~
constnuct with Xba I and Xhol liberated a Xbal Xhol flanked fragment containing the
NOS terminator. The oleosin-xylanase C t~,ult:55iOIl construct was assembled in
pCGN1559 (McBride and Summerfelt 1990). The oleosin-xynCgene fusion was cut
out of pBSOleXyn with Pstl and Xhol. This fragment was ligated with the Xbal Xhol
5 flanked NOS fragment from pBSNos and Pstl Xhol digested pCGN1559. This plasmid was named pCGOleXyn.
ii. Trdl i~ionl ,aliu" of B. napus and stablç xylanase Ç ~Xu~55iOIl
Transgenic B. napus were prepared as described by van Rooijen and Moloney
(1995a; 1995b). Agrobacterium tumefaciens strain EHA101 was transfommed by
10 ~ Iu,uoldtiùll with pCGOleXyn. Cotyledonary petioles of B. napuswere tldll~iul",ed
with A. t~",~r~ "s EHA101 (pCGOleXyn). Transgenic plants were regenerated from
explants that rooted on hormone-free MS medium containing 20 ug/mL kanamycin.
Young plants were assayed for NPTII activity grown to maturity and allowed to self
pollenate and set seed. Seeds from individual lld":,~unllalll~ were pooled and the
l5 presence of ~¢ynC was confirmed by PCR and Southem blot hyl ri- i~dlion. XynCproduction was confimmed by westem blot immu, lodt~l~ulioll with polyclonal antibodies
specific for this protein. Part of the seed sample was assayed for xylanase activity, and
compared to seeds from u"l, dl ,:,~u, " ,ed plants (Table 3). Oil bodies were isolated from
mature dry seeds by the method described in van Rooijen and Moloney (1 995a). Oil
20 bodies were suspended in 50 mM potassium pho:,uhdl~ buffer (pH 4.5). Xylanaseassays were perfommed as described in Example 4. Transgenic plants carrying the
oleosin-xylanase C constnuct produced 10 to 50 times higher levels of xylanase activity
than the wild type control plants.
The -rr~ ~- 1 of l,dnsg~,,icoil-bodiesasani""": dmatrixwastestedas
25 described in van Rooijen and Moloney (l 995a). Oil-bodies carrying oleosin-xylanase
C fusion protein (0.1 mL) were mixed with 0.2 mL of substrate mix containing 0.5~/O
RBB-xylan in a 50 mM potassium-phosphate buffer (pH 6.5). The reaction mixtures
were incubated for 60 min at 40~C. After each incubation, the reaction mix was
centrifuged to separate oil-bodies and substrate. The ~u~ mdldl ,I was then removed
30 and the oil pad was recycled in a new reaction through the addition of fresh substrate.
The ulllt:llldldlll was assayed for RBB-xylan digestion by the method of Biely et al.
(1988). Absolute ethanol (0.8 mL) was added to the u"l~i, laldn t samples to stop the
26
21 9~ ~4
xylanase reactions. The samples were allowed to stand at room temperature for 30 min
and centrifuged for 5 min in a microfuge to remove the precipitated substrate. The
absoi bal1ces of the resulting suu~" Idld~ were measured at 595 nm. The transgenic
oil-bodies retained their endo-xlanase activity through four rounds of recycling (Table
5 4). These results clearly cl~lllon~LIdl~d the stability and potential of lldl15yt"~ic oil-
bodies as an i""" ' ~ enzyme matrix.
Table 3. Xylanase activity of ~, . ., "1,; "~ oil-bodies extracted from transgenic canola carrying
the oleosin-xylanase C expression construct.
Transgenic line Xylanase activity
nmol/min/mg (standard error)
Wild type 0.413 (0.31)
Tl 18.78 (2.63)
T4 29.19 (8.39)
T7 2Z.85 (0.42)
T13 6.68 (0.76)
T18 12.23 ( I .74)
T23 5.86 (0.84)
Table 4. Xylanase activity of recycled oil-bodies
Number of cycles Relative xylanase activity
100.0
2 152.5
3 1540
4 148.5
Example 6. Ide, I~; icdliùl I of Related Xylanase Genes in Other Microorganisms
To identify a xylanase gene related to xynC, hylJIidi~dliùll analysis can be used
35 to screen nucleic acids from other organisms of interest using xynC (SEQ ID NO. 1 ) or
portions thereof as probes by known techniques (Ausubel et al., 1990, Sambrook et al.,
1989). Related nucleic acids may be cloned by employing techniques known to those
skilled in the art. R~l;uisu~ .es (i.e., 32p) may be required when screening olyani~",s
with complex genomes in order to increase the sensitivity of the analysis. Polymerase
40 Chain Reaction (PCR) a" ,, ' " ~ .1 may also be used to identify genes related to xynC.
27
2190194
Related sequences found in pure or mixed cultures are p~ tn it;all~l amplified by PCR
(and variations of such as Reverse Transcription - PCR) with oligonucleotides primers
designed using SEQ ID NO. 1. Amplified products may be visualized by agarose gelelectrophoresis and cloned using techniques know to those skilled in the art.
A variety of materials, including cells, colonies, plaques, and extracted nucleic
acids (e.g., DNA, RNA), may be examined by these techniques for the presence of
related sequences.
All pll' I ,~ ",el,lioned in this specification are indicative of the level of skill
of those skilled in the art to which this invention pertains. All p~' I " ,s are herein
illcollJoldl~d by reference to the same extent as if each individual publication was
:".e.;i~ica'ly and individually indicated to be incorporated by reference.
Although the foregoing invention has been described in some detail by way of
illustration and examples for purposes of clarity and of understanding, it will be obvious
that certain changes and mo~ ns may be practised within the scope of the
appended claims.
2 1 90 1 94
REFCRCrlCES
Ausubel, F.A., R. Brent, R.E. Kingston, D.D. Moore, J.G. Sneidman, J.A. Smith, and K.
Struhl. (eds.) 1990. Current protocols in molecular biology. Green Publishing and
Wiley-l"l~,~ciel1ce, New York.
Biely, P., D. Mislovicova and R. Toman. 1988. Remazol brilliant blue-xylan: a soluble
cl ll umoyt:l lic substrate for xylanases. Methods Enzymol. 160:536-542.
Brosius, J., M. Erfl and J. Storella. 1985. Spacing of the -10 and -35 regions in the tac
promoter. J. Biol. Chem. 260:3539-3541.
I0 Chesson, A., C.W. Forsberg, and E. Grenet. 1995. Improving the digestion of plant cell
walls and fibrous feeds. In: M. Joumet, E. Grenet, M-H. Farce, M. Theriez, C.
Demarquilly (eds) Recent develo,ul "enl~ in the nutrition of herbivores. P, ucde.li, ,y~
of the IVth l"l~" IdliUI Idl Symposium on the Nutrition of Herbivores. INRA Editions,
Paris. pp249-277.
Ellis, S.B., P.F. Brust, P.J. Koutz, A.F. Waters, M.M. Harpold, and R.R. Gingeras.1985.
Isolation of Alcohol oxidase and two other methanol regulated genes from the
yeast, Pichia pastoris. Mol. Cell. Biol. 5:1111-1121.
Gelvin, S.B., R.A. Schilperoort, and D.P.S. Verma. (eds.).1993. Plant Molecular Biology
Manual. Kluwer Academic Publishers, Boston, MA.
Hodgson J. (1994) The changing bulk biocatalyst market. Bio/Technology 12: 789-790
Lowe, S.E., M.K. Theodorou, A.P. Trinci, and R.B. Hespell. (1985) Growth of anaerobic
fungi on defined and semi-defined media lacking rumen fluid. J. Gen. Microbiol.
131 :2225-2229.
McBride, K.E. and K.R. Summerfelt.1990. Improved binary vectors for Aylvba~luli~lm
mediated plant llal1~ulllldlium Plant Mol. Biol. 15:269-276.
McNeil M., A.G. Darvill, S.C. Fry and P. Albersheim. (1984) Structure and function of
the primary cell wall of plants. Ann Rev Biochem 53:625-663
Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular clonin~. A laboratory
manual. 2nd. edn. Cold Spring Harbor Laboratory Press. Cold Spring Harbor, NY.
Somogyi, M.J. 1952. Notes on sugar determination. J. Biol. Chen. 195:19-23.
29
Tamblyn Lee, J.M., Y. Hu, H. Zhu, K.-J. Cheng, P.J. Krell and C.W. Forsberg. 1993.
Cloning of a xylanase gene from the ruminal fungus NeQ. - " "a:,l;X patriciarum 27
and its ex~ ," in Es~,l,oli,,l)id coli. Can J. Microbiol. 39:134-139.
Teather, R.M. and P.J. Wood.1982. Use of Congo red -poly~ac.,l,a~ ld~;liolls in
S enumeration and chald~ dlion of cellulolytic bacteria from the bovine rumen.
Appl. Environ. Microbiol. 43:777-780.
van Rooijen, G.J.H. and M. M. Moloney. 1995a. Plant seed oil-bodies as carriers for
foreign proteins. Bio/Technology 13:72-77.
van Rooijen G.J.H. and M.M. Moloney. 1995b. Structural requirements of oleosin
domains for sl Ihcelll llAr targeting to the oil body. Plant Physiol 109:1353-1361
Wong, S.-L. 1989. Development of an inducible and enhancible expression and
secretion system in Bacillus subtilis. Gene 83:215-223.
2 ~ 9 4
.
SEQUENCE LISTING
(1) GENERAL INFORMATION:
(i) APPLICANT: Cheng, Kuo-Joan
Selinger, Leonard B.
Liu, Jin-Hao
Hu, Youji
Forsberg, Cecil W.
Moloney, Maurice M.
(ii) TITLE OF INVENTION: A xylanase obtained irom an
anaerobic ~ungus
(iii) NUMBER OF SEQUENCES: 6
(iV) ~KK~UN~N~ ADDRESS:
(A) AnnR~ T~T~: McKay-Carey & Company
(B) STREET: 2125 Commerce Place, 10155 - 102nd Street
(C) CITY: Edmonton
(D) STATE: Alberta
(E) COUNTRY: Canada
(F) ZIP: TSJ 4G8
(v) COMPUTER READABLE FORM:
(A) MEDIUM TYPE: Floppy disk
(B) COMPUTER: IBM PC compatible
(C) OPERATING SYSTEM: PC-DOS/MS-DOS
(D) SOFTWARE: PatentIn Release #1.0, Version #1.30
(vi) CURRENT APPLICATION DATA:
(A) APPLICATION NUMBER:
(B) FILING DATE:
(C) CLASSIFICATION:
(viii) ATTORNEY/AGENT INFORMATION:
(A) NAME: McKay-Carey ~ Company,
(ix) TRT~R~nMMTTT\TTcATIoN INFORMATION:
(A) TELEPHONE: (403) 424-0222
(B) TELEFAX: (403) 421-0834
(2) INFORMATION FOR SEQ ID NO:1:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 2058 base pairs
(B) TYPE: nucleic acid
(C) STRAT~TnRnN~ double
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(iii) ~Y~ lCAL: NO
(iv) ANTI-SENSE: NO
30~
21 9ûl 94
.
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Neocallimastix patriciarum
(B) STRAIN: 27
(vii) IMMEDIATE SOURCE:
(A) LIBRARY: genomic DNA library
(B) CLONE pNspX-06
(ix) PEATURE:
(A) NAME/KEY: CDS
(B) LOCATION: 301 1755
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:1:
ATATTATAAT AATTGTTCAA AAAAAGTAAT AA~AAAAAAA AAATTTTTTT T1L111L111 60
GGGAAAATTG AGTATAaATA ~lll~ r TACCTTTTTT ~lllll~l TTATTCTTTA 120
TAAAGTTAAT TGTTTAATAA TTATTGGTGG AAATATTTAA AAGTTGTATA TATATTTTAA 180
TATTTATTGG AATTATTTAC TTTCACTGGT ~.~.AAA~AAA~ ATTAATAGTG ~A~AA~A~AT 240
TATTAGAAAA A~.AAAAAAAA AAATTATTAC AATTAATTAC ~A~AAA~AAA ATAGTTAAaA 300
ATG AaA TTT TTA CAA ATT ATT CCT GTA TTA TTA TCT TTA ACT TCA ACT 348
Met Lys Phe Leu Gln Ile Ile Pro Val Leu Leu Ser Leu Thr Ser Thr =~
5 10 15
ACT CTT GCT CAA AGT TTC TGT AGT TCA GCT TCT CAC TCT GGA CAA AGT 396
Thr Leu Ala Gln Ser Phe Cys Ser Ser Ala Ser His Ser Gly Gln Ser
20 25 30
GTA AAG GAA ACC GGC AAC AAG GTT GGA ACT ATT GGT GGT GTT GGT TAC 444
Val Lys Glu Thr Gly Asn Lys Val Gly Thr Ile Gly Gly Val Gly Tyr
35 40 45
GAA TTA TGG GCT GAT AGT GGT AAT AAC AGT GCT ACT TTC TAT TCT GAT 492
Glu Leu Trp Ala Asp Ser Gly Asn Asn Ser Ala Thr Phe Tyr Ser Asp
50 55 60 '-
GGT TCC TTC TCA TGT ACT TTC CAA AAT GCT GGG GAT TAC TTA TGT CGT 540
Gly Ser Phe Ser Cys Thr Phe Gln Asn Ala Gly Asp Tyr Leu Cys Arg
65 70 75 80
AGT GGT CTT TCT TTC GAT AGT ACT AAG ACC CCA TCT CAA ATT GGT CGT 588
Ser Gly Leu Ser Phe Asp Ser Thr Lys Thr Pro Ser Gln Ile Gly Arg
85 90 95
ATG AAG GCT GAT TTC AaA CTT GTC AaA ACA AAA TAT TTC CAA TGT TGG 636
Met Lys Ala Asp Phe Lys Leu Val Lys Thr Lys Tyr Phe Gln Cys Trp
100 105 110 =.
TTA TTC CTA TGT TGG TGT TTA CGG TGG ACT AGA AGT CCA CTT GTC GGA 684
Leu Phe Leu Cys Trp Cys Leu Arg Trp Thr Arg Ser Pro Leu Val Gly
115 120 125
3o~
2~ 90~ 94
.
ATA CTA CAT GTC GAT AAT TGG CTT AGT CCA TCC CCA CCA GGT GAT TGG 732
Ile Leu His Val Asp Asn Trp Leu Ser Pro Ser Pro Pro Gly Asp Trp
130 135 140
GTT GGT AAC AAG AAG CAT GGT TCT TTC ACT ATT GAT GGT GCT CAA TAC 780
Val Gly Asn Lys Lys His Gly Ser Phe Thr Ile Asp Gly Ala Gln Tyr
145 150 155 160
ACT GTT TAT GAA AAC ACT CGT ACT GGT CCA TCT ATT GAT GGT AAT ACC 828
Thr Val Tyr Glu Asn Thr Arg Thr Gly Pro Ser Ile Asp Gly Asn Thr
165 170 175
ACC TTC AAA CAA TAC TTT AGT ATT CZT CAA CA~ GCT CGT GAT TGT GGT 876
Thr Phe Lys Gln Tyr Phe Ser Ile Arg Gln Gln Ala Arg Asp Cys Gly
180 185 190
ACC ATT GAT ATT TCT GCT CAC TTT GAT CAA TGG GAA AAG CTT GGT ATG 924
Thr Ile Asp Ile Ser Ala His Phe Asp Gln Trp Glu Lys Leu GIy Met
195 200 205
ACT ATG GGT AAA TTA CAT GAA GCC AAG GTT TTA GGT GAA GCC GGT AAC 972
Thr Met Gly Lys Leu His Glu Ala Lys Val Leu Gly Glu Ala Gly Asn
210 2~5 220
GGT AAC GGT GGT GTC AGT GGT ACT GCT GAT TTC CCA TAC GCA AAG GTT 1020
Gly Asn Gly Gly Val Ser Gly Thr Ala Asp Phe Pro Tyr Ala Lys Val
225 230 235 240
TAC ATT GGT GAT GGA AAT GGT GGT GGT GCT TCT CCA GCT CCA GCT GGT 1068
Tyr Ile Gly Asp Gly Asn Gly Gly Gly Ala Ser Pro Ala Pro Ala Gly ::
245 250 255
GGC GCT CCA GCA GGC GGC GCT CCA GCC GGT AAC GAC CAA CCA CAA GGA 1116
Gly Ala Pro Ala Gly GIy Ala Pro AIa Gly Asn Asp Gln Pro Gln Gly ~ ~~ 260 265 270
CCA CAA GGT CAA CAA CCA CCA CAA GGT CAA CAA CCA CCA CAA GGT CAA 1164
Pro Gln Gly GIn Gln Pro Pro Gln Gly Gln Gln Pro Pro Gln Gly Gln
275 280 285 ~:
CAA CCT CCA CAA GGC CAA CAA CCA CCA CAA GGC CAA CAA CCA CCA CAA 1212
Gln Pro Pro Gln Gly Gln Gln Pro Pro Gln Gly Gln Gln Pro Pro Gln
290 2g5 300
GGT AAC GAT CAA CAA GGA CAA CAA CCA CCA CAA GGC CAA CAA CCA CCA 1260
Gly Asn Asp Gln Gln Gly Gln Gln Pro Pro Gln Gly GIn GIn Pro Pro ::
305 310 = 315 320
CAA GGT AAC GAT CAA CAA CAA GGA CAA CAA CCA CCA CAA CCA C~A GGA 1308
Gln Gly Asn Asp Gln Gln Gln Gly Gln Gln Pro Pro Gln Pro Gln Gly
325 330 335
CCA CAA GGA GGT AAC CCA GGT GGT TCT GAT TTT AAC AAC TGG AAC CAA 1356
Pro Gln Gly Gly Asn Pro Gly Gly Ser Asp Phe Asn Asn Trp Asn Gln '~
340 345 350
3c c
21 901 94
.
GGT GGT AGT CCA TGG GGT GGT AAT CAA GGT GGT AGT CCA TGG GGA GGT 1404
Gly Gly Ser Pro. Trp Gly Gly Asn Gln GIy Gly Ser Pro Trp Gly Gly
355 360 365
AAC CAA GGC GGT AAT CCA TGG GGA GGA AAC CAA GGT GGT AGC CCA TGG 1452
Asn Gln Gly Gly Asn Pro Trp Gly Gly Asn GIn Gly Gly Ser Pro Trp
370 375 380 :
GGT GGT AAC CAA GGT GGC AGT CCA TGG GGT CAA GGT AAC CAA GGC GGT 1500
Gly Gly Asn Gln Gly Gly Ser Pro Trp Gly Gln Gly Asn Gln Gly Gly
385 390 395 . 400
AAT CCA TGG GGA GGA AAC CAA GGT GGT AGC CCA TGG GGT GGT AAC CAA 1548
Asn Pro Trp Gly Gly Asn Gln Gly Gly Ser Pro Trp Gly Gly Asn Gln
405 410 415
GGT GGT AAT CCA TGG GGT GGT AAT CAA TGG GGT GCT CCA CA~ AAT GCT 1596
Gly Gly Asn Pro Trp Gly Gly Asn Gln Trp Gly Ala Pro Gln Asn Ala
420 425 430
GCT GCT CCA CAA AGC GCT GCT GCT CCA CAA AAC GCT TCT GAT GGT GGT 1644
Ala Ala Pro Gln Ser Ala Ala Ala Pro Gln Asn Ala Ser Asp Gly Gly
435 440 445
AAC TGT GCT TCT CTT TGG GGT CAA TGC GGT GGA CAA GGT TAT AAT GGT 1692
Asn Cys Ala Ser Leu Trp Gly Gln Cys Gly Gly Gln Gly Tyr Asn Gly
450 455 460
CCA TCT TGC TGT TCC GAA GGT TCC TGT AAG CCA ATT AAC GAA TAC TTC 1740
Pro Ser Cys Cys Ser Glu Gly Ser Cys Lys=Pro Ile Asr Glu Tyr Phe
465 470 475 480
CAC CAA TGT CAA AAA TAAATTGATT AGAAATCATT ATCAACCCAT ATTTATTTTG 1795
His Gln Cys Gln Lys
485
TAGATTAAAA TAATAAA~.AA AAAAAAAAAA AL1LL1L1L~ lLl~LLLlLL LLLLl~ 1855
CAATTAATAA ATCATTAAAA TAGATCATTA ATATAATTAT TTATTTTCAT ;L L 1 L LLLll 1915
TTTAATTAAT ACGTAAATGT AAATGTAAAT TTAAACAATT TATTTAATAT TTAATATTTT 1975
ATAAAA~.ATA CTATTTTAAT AAAATTATAA AAAAAAAATA ~ATAAAAAAA AAATATAAAA 2035
AAAAAAAAAA TATTAATGAA AGT 2058
(2) INFORNATION FOR SEQ ID NO:2:
(i) SEQUENCE CHARACTERISTICS: =
(A) LENGTH: 485 amino acids
(B) TYPE: aAlino acid
(D) TOPOLOGY: linear
(ii) NOLECULE TYPE: protein
3o ~
21 901 't~
(xi) SEQ~7ENCE DESCRIPTION: SEQ ID No:2:
~et Lys Phe Leu Gln Ile Ile Pro Val Leu Leu Ser Leu Thr Ser Thr
~hr Leu Ala Gln Ser Phe Cy6 Ser Ser Ala Ser His Ser Gly Gln Ser
Val Lys Gru Thr Gly Asn Lys Val Gly Thr Ile Gly Gly Val Gly Tyr
Glu Leu Trp Ala Asp Ser Gly Asn Asn Ser Ala Thr Phe Tyr Ser Asp
Gly Ser Phe Ser Cys Thr Phe Gln Asn Ala Gly Asp Tyr Leu Cys Arg
~er Gly Leu Ser Phe Asp Ser Thr Lys Thr Pro Ser Gln Ile Gly Arg
~et Lys Ala Asp Phe Lys Leu Val Lys Thr Lys Tyr Phe Gln Cys Trp
100 105 ~ 110
Leu Phe Leu Cys Trp Cys Leu Arg Trp Thr Arg Ser Pro Leu Val Gly
115 120 125
Ile Leu His Val Asp Asn Trp Leu Ser Pro Ser Pro Pro Gly Asp Trp
130 135 140
Val Gly Asn Lys Lys His Gly Ser Phe Thr Ile Asp Gly Ala Gln Tyr
145 150 155 160
~hr Val Tyr Glu Asn Thr Arg Thr Gly Pro Ser Ile Asp Gly Asn Thr
165 170 175
~hr Phe Lys Gln Tyr Phe Ser Ile Arg Gln Gln Ala Arg Asp Cys Gly
180 185 190
Thr Ile Asp Ile Ser Ala His Phe Asp Gln Trp Glu Lys Leu Gly Met
195 200 205
Thr Met Gly Lys Leu His Glu Ala Lys Val Leu Gly Glu Ala Gly Asn
210 215 220
Gly Asn Gly Gly Val Ser Gly Thr Ala Asp Phe Pro Tyr Ala Lys Val
225 230 235 2g0
~yr Ile Gly Asp Gly Asn Gly GIy Gly Ala Ser Pro Ala Pro Ala Gly
245 250 255
~ly Ala Pro Ala Gly Gly Ala Pro Ala Gly Asn Asp Gln Pro Gln Gly
260 265 ~ 270
Pro Gln Gly Gln Gln Pro Pro Gln Gly Gln Gln Pro Pro Gln Gly Gln
275 280 285
3~ F
21 9~1 9~
Gln Pro Pro Gln Gly Gln Gln Pro Pro Gln Gly Gln Gln Pro Pro Gln
290 295 300
Gly Asn Asp Gln Gln Gly Gln Gln Pro Pro Gln Gly Gln Gln Pro Pro r
305 310 315 320
~ln Gly Asn Asp Gln Gln Gln Gly Gln Gln Pro Pro Gln Pro GIn Gly
325 330 335
~ro Gln Gly Gly Asn Pro Gly Gly Ser Asp Phe Asn Asn Trp Asn Gln
340 345 350
Gly Gly Ser Pro Trp Gly Gly Asn Gln Gly Gly Ser Pro Trp Gly Gly
355 360 365
Asn Gln Gly Gly Asn Pro Trp Gly Gly Asn GIn Gly Gly Ser Pro Trp
370 375 380
Gly Gly Asn Gln Gly Gly Ser Pro Trp Gly Gln Gly Asn Gln Gly Gly
385 390 395 400
~sn Pro Trp Gly Gly Asn Gln Gly Gly Ser Pro Trp Gly Gly Asn Gln
405 410 415
~ly Gly Asn Pro Trp Gly Gly Asn Gln Trp Gly Ala Pro Gln Asn Ala
420 425 430
Ala Ala Pro Gln Ser Ala Ala Ala Pro Gln Asn Ala Ser Asp GIy Gly
435 440 445
Asn Cys Ala Ser Leu Trp Gly Gln Cys Gly Gly Gln Gly Tyr Asn Gly
450 455 460
Pro Ser Cys Cys Ser Glu Gly Ser Cys Lys Pro rle Asn GIu Tyr Phe
465 470 475 480
~is Gln Cys Gln Lys
485
~2) INFORMATION FOR SEQ ID NO:3:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 31 base pairs
(B) TYPE: nucleic acid
(C) sTR~mFn~Fc~: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: other nucleic acid
(A) DESCRIPTION: /desc = "oligonucleotide XI~'
(iii) HYPOTHETICAL: NO
(iv) ANTI-SENSE: NO
30 F
21 901 ~
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:3:
ATCTCTAGAA TTCAACTACT CTTGCTCAAA G -31
(2) INFORMATION FOR SEQ ID NO:4:
(i) SEQUENCE ~TTARA~TERT.CTICS:
(A) LENGTH: 29 base pairs
(B) TYPE: nucleic acid
(C) s~RANnEnNT~sc: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: other nucleic acid
(A) DESCRIPTION: /desc = "oliyonucleotide XII"
(iii) ~Y~u~ CAL: NO
(iv) ANTI-SENSE: NO
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:4:
;LL\~ C~; AGATTTCTAA TC~TTTAT 293 : :
(2) INFORMATION FOR SEQ ID NO:5:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 32 base pairs
(B) TYPE: nucleic acid
(C) s~RAT\TnEnTTEcs single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: other nucleic acid
(A) DESCRIPTION: /desc = ~oligonucleotide XIII" :~
(iii) ~Y~ CAL: NO
(iv) ANTI-SENSE: NO
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:5:
GCGAATTCAT GTCAACTCTT GCTCAAAGTT TC .. . 32
(2) INFORMATION FOR SEQ ID NO:6:
(i) SEQUENCE ~TTARA~TERTCTICS:
(A) LENGTH: 27 base pairs
(B) TYPE: nucleic acid
(C) sTRAT\TnEnT\T~cc: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: other nucleic acid
30 Gi
2~ 90~ q4
(A) DESCRIPTION: /desc = "oligonucleotide ~rv"
(iii) ~YS~~ CAL: NO
(iv) ANTI-SENSE: NO
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:6:
GCCTGCAGTG ATTTCTAATC AATTTAT 27
~0 ~