Note: Descriptions are shown in the official language in which they were submitted.
W O 95132197
i.
,_.,';2~ X0851
-I-
PCTIUS95f05506
TTTLE OF THE INVENTION
PALLADIUM CATALYZED RING CLOSURE OF TRIAZOLYL
TRYPTAMINE
BACKGROUND OF THE INVENTION
1. Field of the Invention
The present invention relates to the preparation of a class of
5-heterocyclic substituted tryptamines, e.g., 5-(1, 2, 4-triazol-1-yl)
to tryptamine, compounds, therapeutically active as anti-migraine agents.
The invention concerns an improved process for producing these 5-
heterocyclic substituted tryptamine derivatives which involves a
palladium-catalyzed coupling and ring closure.
is 2. Brief Descril?tion of Disclosures in the Art
The complex physiological and pathophysiological
processes of the neurotransmitter serotonin (5-HT) are becoming
increasingly elucidated.l (Superscripted References are listed in the
back). In one role, serotonin acts as a vasoconstrictor in the brain and,
20 hereby, displays beneficial properties in migraine therapy. Its potential
as a pharmaceutical agent, however, is limited due to its rapid
metabolism in vivo. Over the past few years an extensive effort has been
devoted to the development of N,N-dialkyltryptamines as 5-HTID
receptor agonists to achieve the desired activity and selectivity for the
as treatment of migraine. Sumatriptan is the first of this class of drugs to
be
approved for this purpose.2 MK-0462 (developed by Merck & Co.), is
described in USP 5,298,520 and is also a potent 5-HT1D receptor agonist
that is undergoing clinical studies.
R'O 95132197 PCTlUS95/05506
v~'~190851
-2-
NH NMe2
2 S02NHMe
HO / I I
s \ I N I H
H
Serotonin (5-HT) Sumatriptan
io
y,~N ~NMe2
C02H
\ I NI / ~
is H \
MK-462 (1)
Generally, this class of compounds is made by the Fisher
indole reaction for the preparation of the N,N-dimethyltryptamine
framework. Application of this methodology to the synthesis of MK-
20 0462, however, is ineffective and low-yielding due to the instability of
the benzyl triazole moiety to the reaction conditions, which generally
leads to polymerization of the triazole moiety, producing oligomers.
What is desired in the art is a highly efficient method for the preparation
of the N,N-dimethyltryptamine, MK-0462 (1~ which eliminates the
2s desirable tendency of triazole polymerization.
Larock et al., have shown that coupling of an iodoaniline
species with an internal acetylene using palladium catalysis gives 2,3-
disubstituted indoles in good-to-excellent yields.3 Smith et al., have also
demonstrated this for 4-pyrimidinyl and pyridinyl derivatives of indol-3-
ao yl_allcyl piperazines as in published EPO 548 831 A1. Two other
applications of this methodology have been demonstrated in the
syntheses of hetero-condensed pyrroles4a and tryptophans4b. However,
all of these methods require triphenylphosphine, as part of the catalyst
system, which is an environmental hazard.
W O 95132197 ~ PCTlUS95105506
~' si': ..
-3-
The application of palladium-catalyzed coupling
methodology to the specific synthesis of the 5-triazolyl N,N-
dimethyltryptamine ring system, has not been reported previously.
s SUMMARY OF TI-IE INVENTION
We have found that MK-462 can be synthesized in high
yield by the palladium-catalyzed coupling/ring closure of a 3-iodo-4-
aminobenzyltriazole with a suitably protected butynol derivative to the
corresponding tryptophol, followed by conversion of the hydroxyethyl
to moiety to a dimethylaminoethyl. The advantages of this new process are -
that it does not require the use of triphenylphosphine, and also
tetrabutylammonium chloride and lithium chloride and it also eliminates
the tendency of triazolyl polymerization as experienced in the Fischer
Indole Synthesis. In general, the process can be used to prepare 5-
is substituted tryptamines where the 5-substituent is triazolyl, triazolyl
methyl, imidazolyl, imidazolylinethyl, tetrazolyl, or tetrazolylmethyl.
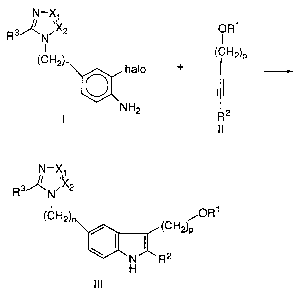
By this invention there is provided a process comprising the
step of contacting a compound of Structure I with a compound of
Structure II to form a compound of Structure lII:
25
wO 95132197 PCT/US95I05506
1 .- r~.;_" . ~~
2?90851
-4-
IN!-X~
Rs~ .X2 ORS
N I
(CH2)n (CH2)P
halo + --
ii
NH2 2
l ll
1 o N-X~
I
Rs~N.X2
I
(CH2)~ FOR
I (CH2)a
is
R2
2o said process being carried out in a dry inert organic solvent for
Structures
I and II, at a temperature in the range of about 70 to 120°C, in
the
presence of a palladium catalyst, soluble in said solvent, present in an
amount of 0.5 to 5 mole percent relative to I, and in the presence of an
inorganic or organic amine compound which functions as a proton
2s acceptor, i.e., acid scavenger, and does not chemically react with said
catalyst,
wherein:
X1 and X2 are independently ring nitrogen or carbon atoms;
halo represents Br or I;
so a is an integer from 0-l; ,
p is an integer from 1-4; ,
R3 is H or linear or branched Cl-C4 alkyl; ,
R 1 is H or a radical which functions as a hydroxy protecting group,
which is removable, under mild acid hydrolyses, for example, by
R'O 95/32197 PC1YUS95105506
.;2190851
i ~ .'., s_, , ; ,
-s-
contacting with a mixture of HCl/MeOH, e.g. 1:1 2N HC1/MeOH,
at 0-30°C;
R2 is a radical which functions as a terminal acetylene carbon protecting
group, which is removable by mild acid hydrolysis, for example, by
contacting with a mixture of HCI/MeOH, e.g. 1:1 2N HCl/MeOH at
0-30°C.
DESCRIPTION OF THE INVENTION AND PREFERRED
EMBODIMENTS
to The synthesis of MK-462 (~ is illustrated in the following
Scheme 1 below.
A key element of the synthesis is the production of the
tryptophol precursor 7, which can be prepared by coupling of 3-iodo-4-
aminobenzyltriazole ~ with a suitably protected butynol derivative ~.
is
/ N~ OR'
I
20 'N N a ~N + ~BH2~P
b
/I / ~
\. NH2 \ I NHz II
R2
R
25 ~' $:R=H;4_:R=I
WO 95132197 PCT/US95/05506 .
r , ? ~'
'21'90851
-6-
N-
H
c
d
w n
m
H
1 o N-
Z
NMe2 NMe2
C02H
a
i
is H H \
MK-462 (1)
aReaction Conditions: a) ICI, CaC03, MeOH-H20; b) 2 mol %
Pd(OAc)2, Na2C03, DMF, 100°C; c) MeOH-HCI; d) i. MsCI, Et3N,
2o THF; ii. 40% HNMe2; e) Benzoic acid, isopropanol, RT.
The synthesis of MK-462 (~ begins with the preparation of
the iodoaniline ~. 4-Aminobenzyltriazole ~ is available in 3 steps and
>90% overall yield from 4-nitrobenzyl bromide and 4-amino-1,2,4-
25 triazole using a modified literature procedures Reaction of _2 with iodine
monochloride in the presence of CaC03 in aqueous methanol furnishes
the 4-triazolyliodoaniline ,~ in 91 % yield; some over-iodination occurs to
provide 1-3% of the diiodoaniline 4. The over-iodination is not difficult
to control since it occurs much more slowly.
ao The palladium-catalyzed coupling/ring closure between the '
iodoaniline ~ and the butynol ~ was found to proceed smoothly in
surprisingly high yield in the absence of the standard required reagents
triphenylphosphine, tetrabutylammonium chloride, and lithium chloride,
and in the absence of any triazolyl-induced polymerization.
W O 95!32197 PCT/US95/05506 .
The coupling reaction between the iodoaniline ~ and various
derivatives of 3-butyn-I-of was intimately studied in detail (Table I
~ below). In order to prevent coupling at the terminal carbon of the
acetylene, it was found that silyl protection was necessary.3 The silyl
groups were incorporated by formation of the dianion with BuLi followed
by quenching with two equivalents of the silyl chloride. In the case of
the TBDMS-protected (tertiary butyl-dimethylsilyl) alkyne, the bis-
silylation unexpectedly did not go to completion; rather, a 1:1 mixture of
Sri and Sg resulted.It was found that alternative O-protection could be
to carried out by selective hydrolysis of the O-silyl group; for example,,
was converted to ~ in quantitative yield using dilute HCl in aqueous
methanol. The hydroxy group of ~c_ can then be protected with the
TBDMS or THP group to afford the alkynes Sf and,5~, respectively, in
quantitative yields.
is
TABLE 1
effect o_f Bu~mol Protection on Yield of Couplinga
2o Entry Acety lenes Yields of Indoles
1 5a R',R2 = SiEt3 (TES) 6a + 6b (80%)
2 5b R~ = H, RZ = SiEt3 6b (74%)
3 5c R' = H, R2 = SiMe3 6c (56%)
4 5d R~ R2 = TBDMS 6d (78%)
,
25 5 5e R' = H, R2 = TBDMS 6e (60%)
g 5f R' = TBDMS, R2 = TMS 6f (77%)
7 5g R' = THP, R2 = TMS 6g (79%)
aConditions: 2 mol% Pd(OAc)2, Na2C03, DMF, 100°C; Ratio of 3/5 =
30 1:1.05-1.2. Me = methyl, Et = ethyl, TBDMS = t-butyldimethylsilyl,
TMS = trimethylsilyl, TES = triethylsilyl, THP = tetrahydropyranyl.
The simplest derivative ~ couples with the iodoaniline ,~ to
afford the 2-TMS-indole ør in 56% yield.6 The undesired regioisomer ~
(5%) is also formed (See below in Chart 1). Other undesired impurities
WO 95!32197 PCTIUS95105506
y, :.~~~19~851
are also formed. The TMS group is believed to be responsible for these
byproducts and the low yield. We surprisingly found that the more stable
TES and 1'BDMS groups on the alkyne give indoles ~ and ,~A,
respectively, in higher yields (Entries 1 and 4). Although the more stable
s C-protection gives better results, the bullcy TBDMS butyne couples
considerably slower; therefore we found that TES is a particularly useful
protecting group in this specific synthesis because it offers a suitable rate
of coupling and stability.
t o CHART 1 _ _
N
is CH2
I OH
\ N
H
a° ;3
Desilylation of the combined indoles 6a and ~b in MeOH-
HCl affords tryptophol 7g in 70-80% overall yield after work-up and
crystallization (Scheme 1). Desilylation of 2-silylated-indoles can also be
2s carried out with other acids such as alkanoic acids, A1C13,
methanesulfonic acid and other sulfonic acids. However, we found that
the system MeOH-HCl is decidedly more useful and convenient to use
particularly from an environmental standpoint. Conversion of 3_ and Sa to
7 is carried out directly without isolation of C. In the crystallization of 7
30 ~e regioisomer 9_ (6%) is removed in the mother liquor.
Conversion of the tryptophol 7 to MK-0462 (1) involves the
formation of mesylate from tryptophol 7 followed by the dimethylamine
displacement to afford MK-0462 free base R_g in 79% yield. We
unexpectedly found that the mesylate is prone to polymerization from
intermolecular alkylation by the triazole; therefore, the mesylate is treated
w0 95132197 PCT'/US95105506
_.~ ~.1 ~_z, 9085 ~
-9-
directly with 40% dimethylamine. The isolated tryptamine is then
purified by addition of a solution of benzoic acid to the free base to afford
the MK-0462 as a benzoate salt in 95% yield.
This new synthesis of MK-0462 ( 1_) featuring a palladium-
s catalyzed coupling of the iodoaniline 3 and the bis-TES-butynol ,5~ to
form the indole ring is an efficient process amenable to scale up that
despite formation of several impurities, unexpectedly requires no
chromatographic purifications as contrasted to the standard Fischer
Indole Synthesis.
io
REFERENCES AND NOTES
1. Glennon, R. A.; Darmani, N. A.; Martin, B. R. Life Sciences
1991, 48, 2493.
is 2. (a) Feniuk, W.; Humphrey, P. P. A. Drug Dev. Res. 1992,
26, 235; (b) Hopkins, S. J. Drug of Today 1992, 28, 155.
3. Larock, R. C; Yum, E. K. J. Am. Chem. Soc. 1991, 113,
6689.
4. (a) Wensbo, D.; Eriksson, Jeschke, T.; Annby, U.;
2o Gronowitz, S.; Cohen, L. A. Tetrahedron Lett. 1993, 34,
2823. (b) Wensbo, D.; Eriksson, Jeschke, T.; Annby, U.;
Gronowitz, S.; Cohen, L. A. Tetrahedron Lett. 1993, 34,
6471.
5. Astleford, B. A, Goe, G. L; Keay, J. G.; Scriven, E. F. V. J.
2s Org. Chem. 1989, 54, 731-732.
6. (a) Sc was purchased from Farchan Laboratories. (b)
Pd(OAc)2 was purchased from Johnson-Matthey.
8. All new compounds were characterized by 1 H NMR, 13C
NMR and elemental analysis. Selective data (1H NMR at
$ 0 250 MHz, 13C NMR at 62.5 MHz):
Jndole 6b: 1H NMR (CDCI3) 8 0.90 (m, 15 H), 1.60 (t, J =
5.2 Hz, 1 H), 3.09 (t, J = 7.9 Hz, 2H), 3.85 (dt, J = 7.9, 5.2
Hz, 2H), 5.40 (s, 2H), 7.10 (dd, J = 8.3, 1.4 Hz, 1H), 7.35 (d,
J= 8.3 Hz, 1H), 7.60 (d, J= 1.4 Hz, 1H), 7.92 (s, 1H), 7.98
W O 95132197 PCTIUS95/05506
. : ~,v ~ ,;219 0 8 51
-lo-
(s, 1H), 8.10 (s, 1H); 13C NMR (MeOH-d4) b 152.1, 144.5,
140.5, 134.0, 130.3, 126.2, 123.0, 122.3, 119.9, 112.7, 64.5,
55.3, 30.9, 7.9, 4.6; Anal. Calcd for C19H27NSOSi: C,
64.18; H, 7.66; N, 15_76. Found: C, 63.81; H, 7.87; N,
s 16.15.
Tr~t_ophQl 7: mp 131-132°C; 1H NMR (DMSO-d6) 8 2.81
(t, J = 7.4 Hz, 2H), 3.63 (dt, J = 7.4, 5.3 Hz, 2H), 4.65 (t, J =
5.3 Hz, 1H), 5.43 (s, 2H), 7.00 (dd, J= 8.4, 1.4 Hz, 1H),
7.15 (d, J= 2.0 Hz, 1H), 7.51 (s, 1H), 7.94 (s, 1H), 8.62 (s,
io 1H), 10.85 (s, 1H); 13C NMR (DMSO-d6) 8 151.3, 143.6,
135.7, 127.3, 125.8, 123.6, 121.1, 118.3, 111.7, 111.4, 61.5,
53.0, 28.7; Anal. Calcd for C13H1qN4O: C, 64.44; H, 5.82;
N, 23.12. Found: C, 64.38; H, 5.85; N, 23.28.
Tr~ptamine 8: mp 120-121°C; 1H NMR (DMSO-d6) 8 2.34
1s (s, 6H), 2.63 (m, 2H), 2.93 (m, 2H), 5.43 (s, 2H), 7.05 (m,
2H), 7.31 (d, J = 8.3 Hz, 1H), 7.56 (s, IH), 7.97 (s, 1H), 7.99
(s, 1H), 8.49 (s, 1H); 13C NMR (CDCI3) 151.7, 142.8,
136.4, 127.7, 124.5, 123.1, 121.9,.119_1, 113.9, 112.0, 60.2,
54.6, 45.3, 23.5; Anal. Calcd for C15H19N5: C, 66.89; H,
20 7.I1; N, 26.00. Found: C, 66.89; H, 7.20; N, 26.04.
The above described specific synthesis of MK-462 can also
be extended to other active analogs containing an imidazole, triazole or
tetrazole in the indole 5-position attached through a ring nitrogen atom,
2s trough a methylene group, or attached directly to the 5-position of the
indole ring as illustrated in the following flow scheme:
WO 95J32197 PCTlUS95105506
i.
~''.~~1 90851
-u-
FLOW SCHEME
N-X~ /N-X~
Ra~ .Xz Ra~N.Xz
N I
I (C Hz)n
(CHz)n ~ ~ halo
NH
NHz z
to la I
/N/-Xi
Ra~N.X2 ORS
I (CHz)
15 (CHz)n halo 'E'
NHz Rz
N-X~
,I ,
Rs~ N.~z
I
(CHz)n / (CHz)P OR
N Rz
H
w0 95132197 PCT/US95105506
. -~.~190851 1
-12-
N-X~
R3 ~ N.~z
I
(CHz)n (CHz) FOR
O R
O ~~~R2
H
N-X~
1o Ra~N.Xz
I
(CHz)n / (CHz)P OH
N
H
IV
N-X~
Rs~N ~C2
I
(CHz)n / (CHz)PN(C1-C4)2
N
H
V
In the beginning step of the process, Structure Ia is reacted
with a halogenating agent to form Structure I at a temperature of about
-10 to 10°C in a suitable solvent and in the presence of a suitable
proton
acceptor.
so The halogenating agent can be, for example, iodine
monochloride, N-iodosuccinimide, N-bromosuccinimde, and the like. By
the term "halo" as used herein is meant Br or I.
The values for "n" are 0, 1 and the values for "p" are 1, 2, 3
and 4.
WO 95131197 PCTlUS95105506
'~y;;;:~21.90851
-13-
The solvent in this step can be MeOH, MeOH-H20, EtOH,
THF-H20; CH2Cl2, and the like, and a useful solvent is 95% MeOH-
H20.
Useful proton acceptors which can be used include: CaC03,
s K2C03, lVa2C03, Li2C03, LiOH, KOH, NaOH, NaHC03, and the like.
When using N-bromosuccinimide or N-iodosuccinimide, a separate
proton acceptor is not required.
A useful set of reaction conditions for the halogenating step
is MeOH-H20 (95%), as the solvent, a temperature of about 0°C,
to whereby the reaction is carried out at atmospheric pressure under an inert
atmosphere, e.g., dry N2, in the presence of calcium carbonate.
Structure II, being the protected I-alkynol, is prepared by
reacting the starting 1-alkynol IIa; which can be selected from 2-propyn-
1-0l (propargyl alcohol), 3-butyn-1-ol, 4-pentyn-I-ol, and S-hexyn-I-ol:
is
H = (CH2)P OH ----~ R2 = (CH2)P ORS
Ila II
where p = 1-4
in a suitable inert organic ether solvent, e.g., tetrahydrofuran, dioxane,
diethyl ether, 1,2-dimethoxyethane, and the like, under a dry atmosphere,
e.g., dry N2, at a temperature of -50°C to -10°C with a slight
excess of n-
2s butyllithium, being about 2.1 moles per mole of alkynol for a sufficient
time, e.g., 2-8 hours to completely generate the dilithium anion of the
alkynol. Then the protecting group is attached by adding a precursor,
e.g., chlorotrimethylsilane, in also a slight excess of about 2.1 moles per
mole of the lithiumdianion of the alkynol and allowed to stir for 1-4
' 3o hours, to complete the reaction. The reaction is worked up by
conventional procedures to yield the diprotected alkynol II.
The R 1 protecting group can be selectively removed by mild
acid hydrolysis, e.g., stirring in about 1:1 by volume 2N HCl/MeOH at
below 30°C, e.g., 0-30°C, and recovering the product. The
resulting
alcohol can be selectively protected with another protecting group, R 1
R'O 95132197 PC1'/ITS95f05506
,.. ,;.~~~_2T 90851
-14-
following the above-described protecting procedure to derive II, where
R1 and R2 are different protecting groups.
The silylating agents which can be used are generally
halogenated trihydrocarbyl silanes, e.g., chloro-triethylsilane.
s The tetrahydropyranyl, THP, protecting group can be
applied by using the starting compound dihydropyran as a precursor, in
the presence of an acid catalyst e.g., p-CH3PhS020H, to convert the
alkynol to the THP ether.
Structure I is then coupled with Structure II to form
to Structure III via a palladium-catalyzed reaction in a dry inert organic
solvent containing a soluble palladium catalyst and in the presence of a
proton acceptor, being an aromatic amine, alkylamine or inorganic base,
which is not a catalyst poison, at a temperature of about 70 to 120°C.
In the Structure IB, R3 is H or C1-C4 linear or branched
is alkyl, including methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl,
isobutyl and t-butyl.
R 1 is H, or a hydroxy protecting group selected from:
the silyl ligand SiRa3, where each Ra is independently selected from
linear or branched C1-C4 alkyl (as described above) or phenyl; and
2o tetrahydropyranyl.
Representative examples of SiRa3 radicals include
trimethylsilyl, triethylsilyl, tributylsilyl, triphenylsilyl, dimethyl-t-
butylsilyl, dimethylphenylsilyl, diphenylmethylsilyl, triisopropylsilyl,
and the like.
25 R2 acts as a protecting group for the terminal acetylene
carbon and has the same structure SiRa3 as described above,
Both R1 and R2 are removable by mild acid hydrolysis, e.g.,
contacting with about a 1:1 by volume 2N HCl/MeOH solvent mixture at
about 0-30°C for 1-24 hours to completely remove the R1, R2 radicals. .
3o The organic solvent useful in this coupling/ring closure step
must be one in which Structure I, Structure II and the palladium-catalyst
are soluble and compatible and is chemically inert under the reaction
conditions.
W O 95/32197 PCTIUS95105506
i , ' '
2190851
-ls-
Classes of solvents useful in this reaction are N,N-di(CI-
Cø)Cl-C2 alkanoamides, Cq-Cg linear ethers, C4-C( cyclic mono or
diethers, di C1-Cq. alkoxyethanes, C(-Clp aromatic hydrocarbons, mono
or dichlorinated Cl-C4 allcanes, alkylnitriles, and the like, or mixtures
s thereof.
Representative solvents include: dimethylformamide,
dimethylacetamide, diethylether, dipropyl ether, tetrahydrofuran,
dioxane, 1,2-dimethoxyethane, benzene, toluene, o-xylene, m-xylene, p-
xylene, acetonitrile, propionitrile and the like, or mixtures thereof.
to The temperature is earned out in the range of 70 to 120°C.
A useful temperature is in the range of about 90-110°C. Generally,
the
reaction is earned out under dry N2 at atmospheric pressure.
The palladium catalyst useful in the reaction can be selected
for example, from the following classes: Pd alkanoates, Pd acetonates,
is pd halides, Pd halide complexes, Pd-benzylidene acetone complexes, and
the like. Representative examples include: Pd ()I) acetate, Pd (II)
acetylacetonate, Pd (O) bis-dibenzylidene acetone, Pd (II) bromide, Pd
(II) chloride, Pd (II) iodide, Pd (II) sulfate, Pd (II) trifluoroacetate, Pd
(II)
CI2(CH3CN)~ and the like. A useful catalyst is palladium acetate.
20 ~e palladium catalyst is used in an amount of about 0.5 to s
mole per cent based on the iodoaniline I and a useful range is about 2 to 3
mole percent of soluble palladium catalyst based on the iodoaniline, I.
The proton acceptor useful in this step is a basic compound
which can be organic or inorganic and acts as a proton acceptor and is not
2s a "catalyst poison". By the term "catalyst poison" is meant interaction
with the catalyst to inhibit its catalytic activity and prevent the
coupling/ring closure between Structure I and II from occurring.
Suitable classes of proton acceptors include allcylamines,
aromatic amines, heterocyclic amines, Group I alkali metal and Group II
ao alkaline earth carbonates, bicarbonates, phosphates, bisphosphates, and
the like.
Representative compounds include lithium carbonate
sodium carbonate, potassium carbonate, sodium bicarbonate, potassium
WO 95!32197 PCT/US95/05506 .
190-851
.,
-16-
bicarbonate, calcium carbonate, triethylamine, diisopropylethylamine,
pyridine, N,N-dimethylaniline, 4-dimethylaminopyridine, and the like.
The removal of the protecting groups R I and R2 from III is
usually accomplished by mild acid hydrolysis without the separate
s isolation of IIL Where the coupling/ring closure is complete, the solvents
are generally removed under reduced pressure. A mixture of about 1:1
by volume 2N HCl/MeOH is added to the concentrate of III at room
temperature and the mixture allowed to stir at below 30°C, e.g., 0-
30°C,
for about 2-4 hours to completely remove both R 1 and R2 protecting
to groups to obtain IV.
The replacement of hydroxyl in IV with dialkylamine to
produce V is generally carried out as a two-step reaction in one reaction
vessel.
The alcohol IV can be reacted with mesyl chloride,
is (CF3 S02)2 O, and the like, in a dry inert organic solvent, e.g.,
tetrahydrofuran, dioxane, diethyl ether, 1,2-dimethoxyethane,
dichloromethane, and the like, at about -30 to -10°C, under a dry N2
atmosphere, in the presence of a proton acceptor, being a soluble
aliphatic or aromatic amine, e.g., triethylamine, pyridine, diethylinethyl-
20 a diisopropylethylamine, tributylamine, 4-dimethylaminopyridine,
and the like to form the intermediate mesylate, or sulfonate, in situ.
The dialkylamine analog V can then prepared by simply
adding the dialkylamine to the mesylate reaction vessel contents, and
allowing to stir at room temperature for 1 hour.
2s The obtained Structure V can be isolated as is or reacted
with an appropriate pharmaceutically acceptable acid, e.g., HCI, H2SOq.,
benzoic acid, succinic acid; lactic acid, malefic acid, and the like, to form
the corresponding acid addition salt.
Representative examples of Structure V that can be
so produced by the process are:
W O 95/32197 PCl'1US95105506
'2-x190851
-17-
N,N-Dimethyl-2-[5-(1,2,4-triazol- I -ylmethyl)- I H-indol-3-yl]ethylamine
N,N-Dimethyl-2-[5-( 1,3-imidazol-I -ylmethyl}-1 H-indol-3-yl]ethylamine
N,N-Dimethyl-2-[5-(5-methyl-I ,2,3,4-triazol-I -ylmethyl)-1 H-indol-3-
yl]ethylamine
' N,N-Dimethyl-2-[5-(1,3,4-triazol-I-ylmethyl)-IH-indol-3-yl]ethylamine
N,N-Dimethyl-2-[5-( 1,3,4-triazol-1-yl)-1 H-indol-3-yl]ethylamine
N,N-Diethyl-2-[5-( 1,2,4-triazol-1-ylmethyl)-1 H-indol-3-yl]ethylamine
N,N-Diethyl-2-[5-( 1,3-imidazol-1-ylmethyl)-1 H-indol-3-yl]ethylamine
N,N-Diethyl-2-[5-(5-methyl-1,2,3,4-tetrazol-1-ylinethyl)-I H-indol-3-
to yl]ethylamine
N,N-Diethyl-2-[5-( 1,3,4-triazol-1-ylmethyl)-I H-indol-3-yl]ethylamine
N,N-Diethyl-2-[5-( 1,3,4-triazol-I -yl)- I H-indol-3-yl]ethylamine
N,N-Dimethyl-[5-( 1,2,4-triazol-I -ylinethyl)-1 H-indol-3-yl]methylamine
N,N-Dimethyl-[5-( 1,3-imidazol-1-ylinethyl)-1 H-indol-3-yl]methylamine
is N,N-Dimethyl-[5-(5-methyl-1,2,3,4-tetrazol-I-ylmethyl)-1H-indol-3-
yl]methylamine
N,N-Dimethyl-[5-( 1,3,4-triazol-1-ylmethyl)- I H-indol-3-yl]methylamine
N,N-Dimethyl-[5-( 1,3,4-triazol-1-yl)-I H-indol-3-yl]methylamine
N,N-Diethyl-3-[5-( 1,2,4-triazol- I -ylinethyl)-1 H-indol-3-yl]propylamine
2o N~N-Dimethyl-3-[5-(1,3-imidazol-I-yl)-IH-indol-3-yl]propylamine
N,N-Diethyl-3-[5-(5-methyl-1,2,3,4-tetrazol-1-ylmethyl)- I H-indol-3-
yl]propylamine
N,N-Dimethyl-3-[5-( 1,3,4-triazo I- I -ylmethyl)-1 H-indol-3-yl]propyl-
amore
2s N,N-Diethyl-3-[5-(1,3,4-triazol-1-yl)-1H-indol-3-yl]propylamine
N,N=Dimethyl-4-[5-(3-methyl-I ,2,4,5-tetrazol-I -ylmethyl)-1 H-indol-3-
yl]butylamine
N,N-Dimethyl-4-[5-(2-ethyl-1,3-ethyl-imidazol-1-ylmethyl)-1 H-indol-3-
yl]butylamine
3o N,N-Dimethyl-4-[5-(5-ethyl-1,2,3,4-triazol-1-ylmethyl)-IH-indol-3-
yl]butylamine
N,N-Dimethyl-4-[5-(2-methyl-1,3,4-triazol-1-ylmethyl) 1 H-indol-3-
yl]butylamine
N,N-Dimethyl-4-[5-(2-ethyl-1,3,4-triazol-I -yl)-1 H-indol-3-yl]butylamine
W O 95132197 PCTIUS95105506
,.. ,'; 4. i ',
-18-
Also included are the alcohol analogs of the above amines,
including, e.g., _
2-[5-( 1,2,4-triazol-1-ylmethyl)-1 H-indol-3-yl]ethylalcohol
2-[5-( 1,3-imidazol-1-ylmethyl)-1 H-indol-3-yl]ethylalcohol
2-[5-(5-methyl-1,2,3,4-tetrazol-1-yhnethyl)-1H-indol-3-yl]ethylalcohol
2-[5-( 1,3,4-triazol-1-ylmethyl)-1 H-indol-3-yl]ethylalcohol
2-[5-(1,3,4-triazol-1-yl)-1 H-indol-3-yl]ethylalcohol
[5-(1,2,4-triazol-1-ylinethyl)-1H-indol-3-yl]-methylalcohol
3-[5-( 1,3-imidazol-1-ylinethyl)-1 H-indol-3-yl]propylalcohol
l0 4_[g_(5-methyl-1,2,3,4-tetrazol-1-ylinethyl)-1H-indol-3-yl]butylalcohol
2-[5-(2-methyl-1,3,4-triazol-1-ylmethyl)-1 H-indol-3-yl]ethylalcohol
2-[5-(5-methyl-1,3,4-triazol-1-yl)-1 H-indol-3-yl]ethylalcohol
The following examples are illustrative of the invention as
is contemplated by the inventors and should not be construed to limit the
scope or spirit of the instant invention.
EXAMPLE 1
2o Step 1: preparation of Iodoaniline 3 _ _
Iodine monochloride ~ N
N' FW 162.36 N'
CaC03 / I
25 I FW 100.09
NH MeOH ~ NH
2 z
2
FW 174.21 FW 300.10
W O 95132197 PCTIUS95105506
°:i~'~ ' ~.'
'2190851
-19-
Mat ri c Amount .MQI 11~
Aniline ~ 30.0 g 0.17 174.21
Iodine monochloride 30.3 g 0_19 162.3
s Calcium carbonate 34.0 g 0.34 100.09
Methanol 240 mL
Ethyl acetate 350 mL
To a mixture of powdered calcium carbonate (34 g, 0.34
to mol) and aniline ~ (30.0 g, 0.17 mol) in methanol (240 mL) and water (12
mL) at 0°C under nitrogen is added a solution of iodine monochloride
(30.3 g, 0.19 mol) in methanol (120 mL) over 0.5 h.
The mixture is warmed to room temperature and quenched
with half saturated sodium thiosulphate solution (5 mL). The mixture is
is stirred for 30 min. 'The solids are filtered and washed with ethyl acetate
(100 mL).
The filtrate is concentrated in vacuo to 100 nlL, diluted with
ethyl acetate (250 mL), washed with half saturated sodium thiosulphate
(200 mL), dried with magnesium sulfate and concentrated to 100 mL.
2o Hexanes is added to precipitate the iodoaniline 3_ as a pale-tan solid
(4$.5
g, 91 %).
Recrystallization of the iodoaniline ~ (24 g.) from ethanol
affords the iodoaniline ~ (14.5 g, 60% recovery) as a white powder: mp
114-115°C.
Step 2: Protection of Butvnol as bis-Triethylsilyl-butvnol Sa
1. n-BuLi
2. TESCI
3o H~OH FW 150.7 TES- =~~R2
THF
,5~ R2=TES, FW 298
FW 70.09 ~, RZ-H, FW 184
R'O 95132197 PCTIUS95I05506
;', ,~2~1:.~0~851 i1
-20-
Math ~ ~ ~W
3-Butyn-I-of 958.5 g 13.68 70.09
n-BuLi (1.6 M in Hexane) 17.1 L 27.36 64.06
s Chlorotriethylsilane 4.218 kg 28.04 150.73
THF 15.9 L
Heptane 20 L
Sodium Carbonate 90 g ~ 0.85 105.99
as 1 % (w/w) aqueous solution
to Water 30.4 L
Dry tetrahydrofuran (15.9 L) is charged to a flask fitted with
a mechanical stirrer and thermocouple under a nitrogen atmosphere and
3-butyn-1-of (958.5 g, 13.68 rnol) is charged to the flask. The mixture is
is cooled to -30°C and n-BuLi (17.1 L, 27.36 mol) is added dropwise
over 4
h, keeping the temperature below -20°C.
The mixture is aged at -20°C for I.2 h. Chlorotriethylsilane
(4.218 kg, 28.04 mol) is added dropwise over 55-60 min, keeping the
reaction temperature below -10°C. The mixture is then allowed to warm
2o to room temperature. The reaction is complete after 1.5 h at
approximately 22°C.
The solution is cooled to -10°C and 1 % (w/w) Na2C03 (8.4
L) is added over 25 min at <0°C. Heptane ( 10 L) is added and the
layers
are partitioned. The aqueous layer is extracted with heptane (10 L). The
as combined organic layers are washed with water (22 L) and concentrated
to a pale orange-yellow oil to afford product Sa (98.1 % yield, 93.8 wt%
purity).
R'O 95!32197 PCTlUS95105506
~:t,~;':'~i 9as~~
-21 -
Step 33: ~'alladium-catalyzed Coupligg to Prepare Try~tonhol 7
s ~ N
N OTES pd(OAc)2
I + I I Na2C03, DMF
TES 100°C . 4~1
N H2
io
FW 300.10 FW 298
ORd N~
is ~ I I 1. MeOH-HCI
2. Na2C03
SiEt3 --
H
~: Rd = H; ~: Rd = SiEt3 H
FW 242
- 30
W O 95132197 PCTIUS95105506
~.2:~ 90851
i ,,: j:, x,11,
-22-
aterials _ _ . _ Amount mMol MW
Iodoaniline ,~ 9 g 30 - 300.10
bis-TES-butynol 5~, (40 W%) 24.5 g 31.5 298
s Palladium acetate 134.4 mg 0.6 224.5
Sodium carbonate (powdered) 15.9 g 150.0 105.9
Dimethylformamide 120 mL
Solka-Floc 2 g
Isopropyl acetate 365 mL
to Water 100 mL
Methanol 35 mL
2N HCl 30111L
Heptane 70 mL
Saturated Na2C03 24 mL
1 s Darco G-60 0.5 g
IPAc/Heptane ( 1:2) 36 mL
To dry dimethylformamide (12 mL) is charged a solution of
bis-TES-butynol ~ in heptane (24.5 g, 31.5 mmol, 40% by wt). The
20 texture is concentrated under vacuum to a volume.of 22 mL. To this
concentrate is charged dimethylfonnamide (78 mL), iodoaniline 3_ (9 g,
30 mmol), and powdered sodium carbonate (15.9 g, 0.15 mol). The
mixture is degassed with vacuum/nitrogen purges.
Palladium acetate (134.4 mg, 0.6 mmol) is added and the
25 mixture is heated at 100°C for 4 h.
The product mixture is cooled to room temperature and
filtered through Solka-Floc. The cake is washed with dimethyl-
formamide (30 mL). The combined filtrate and wash are distilled at 26
mmHg (bp for DMF: 67°C) to ~25 mL to remove --100 mL of distillate.
so Isopropyl acetate (IPAC) (150 mL) and water (50 mL) are added to the
distillation residue. The resultant mixture is filtered through 2 g of
Solka-Floc and the cake is washed with isopropyl acetate (15 mL). The
combined filtrates are washed with water (50 mL) and concentrated to 50
mL.
W O 95/32197 PC'fIUS95105506
w~ ~ X0851
-23-
. The above concentrate is diluted with methanol (35 mL)
and 2N HCl (30 mL, 2 eq) is added over 20 min, keeping the reaction
temperature below 30°C. The mixture is aged at room temperature for 2
hours or until the reaction is complete.
s Heptane (36 mL) is added and the heptane-isopropyl acetate
layer is separated. The methanol-water layer containing the product Z is
concentrated irc vacuo to 65 mL with the removal of methanol (20 mL).
Isopropyl acetate (50 mL) is added to the mixture. The
mixture is cooled to 18°C followed by the addition of saturated aqueous
to sodium carbonate (24 mL) over 10 min. Isopropyl acetate (50 mL) is
added to the mixture. The aqueous layer is separated and extracted with
isopropyl acetate (100 mL). The combined organic solution (200 mL) is
treated with Darco G-60 (0.5 g). The mixture is stirred for 5 h and
filtered. The filtrate is concentrated to 100 mL to give a thin slurry,
is followed by the addition of heptane (34 mL). The slurry is aged at room
temperature for 1 h. The solid is filtered and washed with
heptane/isopropyl acetate (2:1; 36 mL). The product is dried to afford the
tryptophol 7 (5.5 g, 75%). The NMR data and C, H, N analytical data is
presented above on "References and Notes".
25
R'O 95132197 PCT/US95105506
._ :_<i:~~1~9:0851
-24-
SteD 4: MK-0462 Free Base
g 1. MsCI, FW 114.55
Et3N, FW 101.19
2. 40% HNMe2
H FW 45.09
Z
FW 242
N
NMe2
H
MK-462 FreeBase (8)
FW 269.35
Materials amount I~1 ~y
Tryptophol Z 4.878 0.0201 242.28
Methanesulfonyl chloride 230 g 0.0201 114.55
z5 Triethylamine 264 g 0.0261 101.19
Tetrahydrofuran 97 mL
Aqueous dimethylamine (40%w/w) 49 mL 0.39 45.09
Aqueous potassium carbonate (saturated)15 mL
Isopropyl acetate 100 mL,
3 o Darco G-60 0.48 g
Heptane 64 mL
-, W095I32197 PCTIUS95105506
-25-
The tryptophol 7 (4.87 g) is slurried in dry tetrahydrofuran
(97 mL) and sieve-dried triethylamine (2.64 g, 26.1 mmol) is added. The
slurry is cooled to -20°C and methanesulfonyl chloride (2.30 g, 20.1
nunol) is added at <-15°C over 45 min. The reaction mixture is aged for
s 30 min, at -20°C.
The slurry is filtered at <-15°C and the filter cake is washed
with cold, dry tetrahydrofuran (25 mL).
Aqueous dimethylamine (40% w/w, 49 mL, 0.39 mol) is
added to the combined filtrates. The reaction mixture is allowed to
i o warm to room temperature.
Most of the THF is removed by distillation under vacuum at
<30°C (final volume 60 mL). Isopropyl acetate (50 mL) and saturated
aqueous potassium carbonate (5 mL) are added. The layers are well-
mixed and separated. The aqueous layer is extracted with isopropyl
is acetate (50 mL).
The combined organic layers are washed with saturated
aqueous potassium carbonate (10 mL). Isopropyl acetate (20 mL) is
added to the diluted organic layer and the product solution is dried by
heating under reflux over a Dean/Stark trap. The solution is cooled and
2 o treated with Darco G60 (0.5 g) for 60 min, and the mixture is filtered.
The filtrates are concentrated to 20 mL by distillation under vacuum,
seeded and then allowed to crystallize for >1 h. Heptane (64 mL) is
added to the seed bed over 1 hour and the slurry is cooled to 0°C.
After
a 1 hour age the slurry is filtered. The product is washed with cold 4:1
2s heptane-isopropyl acetate (2 X 10 mL) and dried in a vacuum at 40°C.
The free base of MK-0462 (8) is obtained as a cream-colored solid (4.30
g, 73% yield). The NMR and C,H,N analytical data is presented above
on "References and Notes".
W 0 95132197 PCT1US95105506
;;;w~21v0851
-26-
Step 5: Formation of MK-0462 Benzoate _ _
N~ COzH N
~N~N NMe2 / NMe2
s ~ ~ C02H
FW 122.12 /
N
H H
to Free Base ($) Benzoate MK-462 (i)
FW 269.35 FW 391.47
Materials - Amount - mMol . MW -
is MK-0462 Free base (89 wt% purity) 10 g 33.0 269.35
Benzoic Acid 4.5 g 36.8 122.12
Isopropanol 80 mL
Isopropyl acetate 30 mL,
2o To a solution of MK-0462 free base (10 g, 89 wt% pure) in
isopropyl alcohol (80 mL) at room temperature is added a solution of
benzoic acid (4.5 g, 36.8 mmol) in isopropyl acetate (20 mL) over 10
min. The mixture is aged at room temperature for 0.5 h, cooled to 0-5°C
and filtered. The cake is washed with isopropyl acetate (10 mL) and
2s dried to give crude MK-0462 benzoate salt (13.1 g, 95 wt% pure, 96%
assay yield). Recrystallized from EtOIi to yield pure solid material. The
elemental analysis and analytical spectra were consistent with the
proposed structure.