Note: Descriptions are shown in the official language in which they were submitted.
2~.9~~H~
SPECIFICATION
DNA Fragment, Recombinant Vector.Containing the Same and
Method for Expressing Foreign Genes Using the Same
mRC'.HNICAL FIELD
The present invention relates to a novel DNA
fragment having function to promote expression of genes,
a vector -containing the same and a method for expressing
foreign genes using the same.
BACKGROUND ART
Promotion of expression of foreign genes is one of
the most important techniques in applying genetic
engineering processes to plants. One of the methods
therefor is utilization of a DNA having a nucleotide _
sequence which promotes expression of a gene.
Known nucleotide sequences which promote expression
of foreign genes include the intron of the catalase gene,
of castor bean (Japanese Laid-open Patent Application
(Kokai) No. 3-103182; Tanaka et al., Nucleic Acids Res.
18, 67'67-6770 (1990)). However, since there are wide
varieties of plants to be manipulated and since promotion
of expression of genes is required in each of the desired
growth stages ox tissues of organs, it is desired that
wide varieties of DNAs which promote expression of genes
can be utilized.
pjSCT,nSURF OF THE INVENTION
Accordingly, an object of the present invention is-
to provide a novel DNA, which can promote expression of
CA 02191624 2004-04-30
7
foreign genes and which has a nucleotide sequence
different from those of known DNAs that promote
expression of foreign genes.
The present inventors intensively studied to
discover introns of rice phospholipase D (hereinafter
also referred to as "PLD") gene by comparing a rice cDNA
and a rice genomic DNA, and discovered that one of the
introns has a function'to prominently promote expression
of the gene downstream thereof, thereby completing the
present invention.
That is, the present invention provides an isolated
DNA fragment having a nucleotide sequence shown in SEQ ID
NO. 1 in Sequence Listing or having a nucleotide sequence
which is the same as the nucleotide sequence shown in SEQ
ID N0. 1 in Sequence Listing except that one or a
plurality of nucleotides are added, inserted, deleted or
substituted, the latter nucleotide sequence having a
function to promote expression of a gene downstream
thereof. More specifically, the present invention provides
for an isolated DNA fragment having a nucleotide sequence
shown in SEQ ID NO. 1 in Sequence Listing or having a
nucleotide sequence, which hybridizes with said nucleotide
sequence shown in SEQ ID N0. 1 when the hybridization is
carried out in a hybridization solution containing 0.5M
sodium phosphate buffer (pH7.2), 7% SDS, 1 mM EDTA and 100
ug/ml of salmon sperm DNA, at 65°C for 16 hours, and then
washing the product twice with a washing solution
containing 0.5xSSC and 0.1o SDS at 65°C for 20 minutes
CA 02191624 2004-04-30
2a
each, the latter nucleotide sequence having a function to
promote expression of a gene downstream thereof in a plant.
The present invention also provides a recombinant vector
comprising the above-mentioned DNA fragment according to
the present invention and foreign gene to be expressed,
which is operably linked to the DNA fragment at a
downstream region of the DNA fragment. The present
invention further provides a method for expressing a
foreign gene comprising introducing the recombinant vector
according to the present invention into host cells and
expressing the foreign gene.
219162
As experimentally confirmed in the Example described
below, the DNA fragment according to the present -
invention largely promotes expression of the gene
downstream of the DNA fragment. Therefore, it_is
expected that the presentinvention will largely
contribute to expression of foreign-genes by genetic
engineering processes.
RRTEF DESCRIPTION F THE DRAWINGS
Fig. 1 shows the important part of a genetic map of
pBI221 into which the DNA fragment according to the
present invention is inserted, which was prepared in the
Example of the present invention.
BEST MODE FOR CARRING OUT THE INVENTION __
As mentioned above,-the DNA fragment according to
the present invention has a nucleotide sequence shown in
SEQ ID N0. I in the Sequence Listing. As will be
described in detail in the Example below, introns located
upstream of rice PLD gene were identified by comparing
the nucleotide sequence of the cDNA of rice PLD gene and
that of the rice genomic DNA. A fragment containing one
of these intron sequences having a size of 173 by located
at the 5'-flanking region was prepared by PCR and the DNA
fragment was inserted into an upstream site of a reporter
gene of an expression vector containing the reporter gene..
By checking the expression activity of the reporter gene,
it was confirmed that the DNA fragment has a function to
promote expression of the gene downstream thereof. The
~1~1s~~
nucleotide sequence of the DNA fragment according to the
present invention corresponds to 1661nt to 1843nt of the
nucleotide sequence of the rice genomic PLD gene, which
nucleotide sequence is shown in SEQ ID N0. 3 of the
Sequence Listing.
The nucleotide sequence of the above-mentioned
intron sequence having a size-of 173 bp, which is located
upstream of the rice PLD gene, is shown in SEQ ID NO. 4
in the Sequence Listing. Needless to say, the sequence
shown in SEQ ID N0. 4 also has a function to promote
expression of the gene downstream thereof. The
nucleotide sequence shown in SEQ ID NO. 4 corresponds to
1666nt to 1838nt of the-nucleotide sequence of the rice -
genomic PLD gene, which is shown in SEQ ID NO. 3 in the
Sequence Listing.
Since the DNA fragment according to the present
invention is an intron existing upstream of the rice PLD
gene, and since its nucleotide sequence was determined
according to the present invention, the DNA fragment may
easily be prepared by PCR using the rice genomic DNA as a
template. PCR-is a conventional technique widely used in
the field of genetic engineering and a kit therefor is
commercially available, so that those skilled in the art
can easily perform the PCR. One concrete example thereof
is described in detail in the Example below.
It is well-known in the art that there are cases
wherein the physiological activity of a physiologically
I 5
active DNA sequence is retained even if the nucleotide
sequence of the DNA is modified to a small extent, that
is, even if one or more nucleotides are added, inserted,
deleted or substituted. Therefore, DNA fragments having
the same nucleotide sequence as shown in SEQ ID NO. 1
except-that the DNA fragments have such modifications,
which have the function to promote expression of the gene
downstream thereof, are included within the scope of the
present invention. That is, the DNA fragments having the
same nucleotide sequence as shown in SEQ ID N0. 1 except
that one or more nucleotides are added, deleted or
substituted, which have the function to promote
expression of the gene downstream thereof, are included
within the scope of the present invention. Particularly,
in the nucleotide sequence shown in SEQ ID N0. 1, the 5
nucleotides at the 5'-end and the 6 nucleotides at the
3'-end are the nucleotides in the exon regions, so that
it is thought that the nucleotide sequences which do not
have these regions also have the function to promote gene
expression. Thus, these DNA fragments are within the
scope of the present invention.
Modification of DNA which brings about addition,
deletion or substitution of the amino acid sequence
encoded thereby can be attained by the site-specific
mutagenesis which is well-known in the art (e. g., Nucleic
Acid Research, Vol. 10, No. 20, p6487-6500, 1982). In
the present specification, "one or a plurality of
6 2~9~62~
nucleotides" means the number of nucleotides which can be
added, deleted or substituted by the site: specific
mutagenesis.
Site-specific mutagenesis may be carried out by, for
example, using a synthetic oligonucleotide primer
complementary to a single-stranded phage DNA except that
the desired mutation as follows. That is, using the
above-mentioned synthetic oligonucleotide.as a primer,-a
complementary chain is produced by a phage, and host
bacterial cells are transformed with the obtained double-
stranded DNA. The culture of the transformed bacterial _
cells is plated on agar and plaques are formed from a
single cell containing the phage. Theoretically, 50$ of -
the new colonies contain the phage having a single-
stranded chain carrying the mutation and remaining 50~ of -
the colonies contain the phage having the original
sequence. The obtained plaques are then subjected to
hybridization with a kinase-treated synthetic probe at a
temperature at which the probe is hybridized with the DNA
having exactly the same sequence as the DNA having the
desired mutation but not with the original DNA sequence.
that is not completely complementary with the probe.
Then the plaques in which the hybridization was observed
are picked up, cultured and the DNA is collected.
In addition to the above-mentioned site-specific ._-
mutagenesis, the methods for substituting, deleting or
adding one or more amino-acids without losing the
2191~~~
function include a method in which the gene is treated _..
with a mutagen and a method in which the gene is
selectively cleaved, a selected nucleotide is removed,
added or substituted and then the gene is ligated.
The DNA fragment according to the present invention
has a function to promote expression of the gene
downstream thereof. Therefore, by inserting the DNA
fragment according to the present invention into the
transcriptional region of a desired foreign gene to be
expressed, preferably into the 5'-end region of the
transcriptional region, expression of the foreign gene is
promoted. The method for expressing a foreign gene has
already been established in the field of genetic
engineering. That is, by inserting the desired foreign
gene into a cloning site of an expression vector,
introducing the resulting vector into host cells. and
expressing it, the foreign gene may be expressed.
According to the method of the present invention, the DNA
fragmeht-according to the present invention is inserted
at a site upstream of the foreign genein a manner such
that the DNA fragment is operably linked to the foreign _
gene, and the foreign gene is expressed. The term that
the DNA fragment according to the present invention is
"operably linked" to the foreign gene meansthat
expression of the foreign gene is delectably increased by
inserting the DNA fragment according to the present
invention when compared with the case wherein the DNA
21916~~
fragment according to the present invention is not _.
inserted. The DNA according to the present invention may
be inserted.into the site immediately upstream of the
foreign gene. Alternatively, another sequence may be
located between the DNA according to the present
invention and the foreign gene. Although the size of
this intervening sequence is not restricted, it usually
has a size of 0 - 1000- bp. A promoter sequence is
located upstream of the DNA fragment according to the
present invention. The DNA fragment according to. the
present invention may be inserted into the site ._
immediately downstream of the promoter, or another
sequence may be located between the promoter and the DNA
according to the present invention. Although the size of
this intervening sequence is not restricted, it is
usually 0 - 1000 bp. In summary, all recombinant vectors
with which the expression of the- foreign gene is
detectably increased by inserting the DNA fragment
according to the present invention when compared with the
case wherein the DNA fragment is not inserted, are within
the scope of the present invention.
Since the nucleotide sequence of the cloning site of
an expression vector is known, the DNA fragment according
to the present invention may easily be inserted into the
vector:.
Wide varieties of such an expression vectorare
well-known in the art and are commercially available.
9 2I916?4
These expression vectors contain at least a replication
origin for replication in the host cells, a promoter, a
cloning site giving a restriction site for inserting the
foreign gene, and a selection marker such as drug
resistance, and usually contain a terminator which stably
terminates transcription, and an SD sequence when the
host is a bacterium. In the method of the present
invention, any of these known expression vectors may be
employed.
Example
The present invention will now be described in more
detail by way of examples thereof.- However, the present
invention is not restricted to the examples.
1. pmr~~~~ation of PZD of Rice Bran _
For purification, a reference--(Takano et al.,
Journal of Japan Food-Industry Association, 34, 8-13
(1987) was referred. The enzyme activity was measured-by
employing phosphatidylcholine as a substrate and
quantifying the choline generated by the enzyme reaction
(Imamura et al., J. Biochem. 83, 677-680 (1978)). It
should be noted, however, the enzyme reaction was stopped
by heat treatment at 95°G for-5 minutes.
That is, to 100 g of bran of rice (Oryza sativa),
variety "KOSHIHIKARI", one liter of hexane was added and
the mixture was stirred for a whole day and night,
thereby defatting the rice bran. To the resultant, 10 g
of Polycral AT (trademark, polyvinylpyrrolidone,
X191624
commercially available from GAF Chemical) and 500 ml of
mM Tris-HC1 buffer (pH7) containing 1 mM CaCI2 and 5
mM 2-mercaptoethanol were added, and the resulting
mixture was stirred for 1 hour to extract the enzyme.
5 The extract was filtered through an 8-layered gauze and
the filtrate was centrifuged at 15,000 x g for 20 minutes,
followed by recovering the middle layer asa crude
extract. The crude extract was treated with ammonium
sulfate (65~ satuxation) and the generated precipitates
10 were collected by centrifugation (15,000 x g, 20 minutes),
followed by dialyzing the precipitates after dissolution
against the above-mentioned buffer. After the dialysis,
precipitates were eliminated by filtration to obtain
ammonium sulfate fraction.
The ammonium sulfate fraction was- applied to a
column (2.0 x IO cm) of DEAE-Cellulose (commercially
available from Whattman) equilibrated with buffer A (10
mM Tris-HCl, pH 7, 1 mM CaClz, I mM 2-mercaptoethanol).
After washing the column with about 100 ml of buffer A
containing 50 mM NaCl, elution was carried out with 120
m1 of buffer A having a linear gradient of NaCl
concentration from 50 mM to 350 mM. P2~D was eluted at a
NaCl concentration of about 0.2 M. The fraction having
PLD activity was collected as an eluted solution_(DEAE-
cellulose).
To the eluted solution (DEAE-cellulose), 3 M
ammonium sulfate was added in an amount attainingthe
CA 02191624 2004-04-30
1l
final concentration of ammonium sulfate of 1 M, and the
resulting mixture was applied to a Phenyl Sepharose
column (commercially available from Pharmacia, 2.6 x 10
cm) equilibrated with buffer A containing 1 M ammonium
sulfate. Elution was performed using 240 ml of buffer A
having a linear gradient of ammonium sulfate
concentration from 1.0 M to 0 M. PLD was eluted at a
concentration of ammonium sulfate of about 0.1 M. The
fraction having the activity was recovered and dialyzed
against buffer A to obtain an eluted solution (Phenyl
Sepharose).
The eluted solution (Phenyl Sepharose) was applied
to Mono Q column (anion-exchange column commercially
available from Pharmacia, 16 x 10 cm) equilibrated with
buffer A, and elution was performed using 150 ml of
buffer A having a gradient of NaCl concentration from 50
mM to 350 mM. PLD was eluted at NaCl concentration from
210 mM to 235 mM. The fraction having PLD activity was
recovered and dialyzed against buffer A to obtain an
eluted solution (Mono Q*lst).
The eluted solution (Mono Q 1st) was concentrated to
0.5 ml by ultrafiltration and applied to Superose 6
column (commercially available from Pharmacia, 1.0 x 30
cm equilibrated with buffer A containing 0.1 M NaCl and
elution was performed using the same buffer. The
molecular weight of PLD was estimated to be 78 kDa. The
fraction having PLD activity was recovered as an eluted
* Trademarks
219.16~~
i2
solution (Superose 6).
To the eluted solution (Superose 6), 2.5 ml of 40°s -
Carrier Ampholite (commercially available from Pharmacia,
pH4.0-6.0) and distilled water were added to attain a
final volume of 50 ml and isoelectric electrophoresis was
carried out using Rotofore (commercially available from -
Biorad). Electrophoresis was performed at 2°C with a
constant power of 12W for 4 hours. PLD activity was
observed at about pH 4.9. The fraction having PLD
activity was collected and dialyzed against buffer A to -
obtain an isoelectric electrophoresis fraction.
The isoelectric electrophoresis fraction was applied
to Mono Q column (commercially available from Pharmacia,
0.5 x 5 cm) and eluted with NaCl having a linear gradient
of concentration of 50 mM to 350 mM. PLD was eluted at _
NaCl concentrations of about 210 mM and about 235 mM.
The two fractions having PLD activity were recovered as
eluted solutions (Mono Q 2nd-I, II).
Purities of the eluted solutions (Mono Q 2nd-I, II)
were checked by SDS-polyacrylamide electrophoresis
(Laemmli (1970)) using 7.5$ acrylamide. After the
electrophoresis, the gel was stained with Coomassie
brilliant blue R-250. With either eluted solution, a
main band was observed at a position corresponding to a
molecular weight of 82 kDa. With the eluted solution
(Mono Q 2nd-II), only a single band was observed.
By the purification described above, the
219164
13
purification magnificati-ons of the eluted solutions (Mono
Q 2nd-I, II) were 380 times and 760 times, respectively,
with respect to the crude extract.
Properties of the enzymes contained in the two
fractions were determined. The results are shown in
Table 1. The buffer solutions used for the measurement
of the optimum pH were sodium acetate (pH 4-6), MES-NaOH
(pH 5.5-7.0) and Tris-HC1 (pH 7-9), all of which have a
concentration of 100 mM in all of the buffer solutions.
The pH stability means the pH range in which decrease in
the enzyme activity is not observed after leaving the
enzyme at the respective pH at 25°C for-30 minutes. The
temperature stability was measured by measuring the
remaining activity after leaving the enzyme to stand at
4°C, 25°C, 37°C or 50°C for 30 minutes. The
substrate
specificity was measured at a substrate concentration of
5 mM and expressed in terms of the relative activity
taking the enzyme activity to phosphatidylcholine as 100.
~4 219I6~~
Table 1
Mono Q 2nd-I Mono Q 2nd-II
Km Value 0.29 mM 0.29 mM
Optimum pH 6 6
pH Stability 7-8 7-8
Temperature Stability 4-37C 4-37C
Ca2+ Dependency not less not less than
than 20 mM 20 mM
Substrate Specificity
Phosphatidylcholine 100 100
Lysophosphatidylcholine 13 12
Sphingomyelin 6 4
2. Prnnf that Purified Protein is PLD _
Each of the eluted solutions (Mono Q 2nd-I, II) was-
subjected to SDS-polyacrylamide gel electrophoresis in
the same manner as in the purity test, and the obtained
patterns were transferred to PVDF membranes (commercially
available from Millipore), followed by staining the
membranes. The band of the protein having the molecular
weight of 82 kDa was cut out and the amino acid sequence
of the N-terminal region of the protein was determined by
a protein sequencer (commercially available from Shimazu
Seisakusho, PSQ-1). For both proteins, amino acid
sequence up to 10 residues from the N-terminal could be -
determined, and the determined sequences were identical.
The sequence was as follows.
Val Gly Lys Gly Ala Thr Lys Val Tyr Ser
Although the relationship between the proteins
CA 02191624 2004-04-30
l
having the molecular weight of 82 kDa contained in the
two fractions having the enzyme activity is not clear, it
is thought that they have high homology in their amino
acid sequences, so that it was judged that there would be
no problem even if a mixture of the fractions is used as
an antigen for preparing an antibody.
A mixture of the eluted solutions (Mono Q 2nd-I, II)
was subjected to SDS-polyacrylamide gel electrophoresis
using 7.50 acrylamide, and the gel was stained with
Coomassie brilliant blue R-250. The band of the protein
having the molecular weight of 82 kDa was cut. out and
recovered by electroelution (25 mM Tris, 192 mM glycine,
0.0250 SDS, 100V, 10 hours). Then SDS was removed by
electrodialysis (15 mM ammonium bicarbonate, 200 V, 5
hours) and the resultant was lyophilized. For the
electroelution and electrodialysis, BIOTRAP*(commercially
available from Schleicher & Schuell) was used.
The protein having the molecular weight of 82 kDa
highly purified by the above-described method was
administered to a rabbit in an amount of 50 )gig per time
at 7 days' intervals. Immunological titration test was
performed for the sera before the immunization and after
the third immunization. To the PLD solution containing
8.6 x 10 3 units of PLD, were added 0 - 50 ~1. of the
serum before the immunization or after the third
immunization, 50 u1 of 250 mM Tris-HC1 (pH~.O), 5 u1 of
50 mM CaCl2, 50 u1 of 0.2% Triton X-100 (trademark) and
* Trademark
21~16~~
16
water to a total volume of 250 u1, and the mixture was
left to stand at room temperature for 2_5 hours. To the
resultant, 200 u1 of Protein A Sepharose (commercially
available from Pharmacia) was added and the resulting
mixture was gently shaken at room temperature for 2 hours.
The mixture was then centrifuged (500 x g, 5 minutes) and
the enzyme activity in the supernatantwas measured.
Taking the measured enzyme activity in the case where the
serum was not added as 100$, the enzyme activities in
- cases where 20 p1 and 50 u1 of the serum before _
immunization were added were-95$ and 88$, respectively,
and the enzyme activities in cases where 20 u1 and 50 u1
of the serum after the third immunization were added were .
75$ arid 30$, respectively. These results prove that the
protein having the molecular weight of 82- kDa is PLD.
3. Determination of Aminn Acid sequence of Internal
Regions
The PLD protein was fragmentated in a gel (Cleveland
et al., J. Biol. Chem., 252, 1I02(I977)). The cut out
gel containing the PLD protein was inserted into a
stacking gel well on a 15$ acrylamide gel prepared for
separation of peptides, and Staphylococcus aureus V8
protease (commercially available from Wako Pure Chemical
Industries, Ltd) in an amount of 1/10 volume of the PLD
protein was overlaid, followed by starting
electrophoresis. The electrophoresis was stopped at the
time point at which the bromophenol blue reached the
219~62~
center:of the stacking gel and then restarted 30 minutes-
later.. After the electrophoresis, the pattern was
transferred to a PVDF membrane and the membrane was
stained. Clear bands were observed at the positions
corresponding to molecular weights of 20, I4, 13, 11 and
kDa. Each of the bands of the peptide fragments
having molecular weights of 20, 14 and 13 kDa were cut
out and their amino acid sequences were determined by a -
protein sequencer. The determined sequences are as
IO follows.
kDa Asn Tyr Phe His Gly Ser Asp Val Asn ? Val
Leu ? Pro Arg Asn Pro Asp Asp (Asp) ?
?
I1e
14 kDa Thr ? Asn Val Gln Leu Phe Arg Ser I1e Asp
15 G1y Gly Ala Ala Phe G1y Phe Pro Asp Thr Pro
Glu Glu Ala Ala Lys ? G1y Leu Va1 Ser Gly
13 kDa Ile Ala Met GIy Gly Tyr Gln Phe Tyr His Leu
Ala Thr Arg Gln Pro Ala Arg Gly Gln Ile His-
Gly Phe Arg Met Ala Leu ? Tyr Glu His Leu ...-
20 Gly Met Leu ? Asp Val Phe
(In the sequences, "?" means ino residue which
the acid
am
could not be d, nd ino residue in
identifie a the acid
am
parentheses means that the amino acid residue could not
be identified confidentially.
4. Preparation of cDNA Library of Rice Immature Seeds
Total RNAs were extracted from immature seeds
obtained after 5 days from flowering by the SDS-phenol
CA 02191624 2004-04-30
18
method, and prepared by the lithium chloride
precipitation. Poly(A)rRNA was prepared using Oligotex-
dT30 (commercially available from Takara Shuao) according
to the instructions provided by the manufacturer. For
the cDNA cloning, cDNA synthesis System Plus
(commercially available from Amersham) and cDNA Cloning
System ~.gtl0 (commercially available from Amersham) were
used. However, a,2APII*vector (commercially available
from Stratagene) was used as the cloning vector and XLl-
Blue was used as the host cells.
5. Preparation of Probes
Oligonucleotides corresponding to the amino acid
sequences of PLD were synthesized by a DNA synthesizer
(commercially available from Applied Biosystems). The
sequences thereof as well as the corresponding amino acid
sequences are as follows.
20KF 5' AAYTAYTTYCAYGG 3'
20KR1 5' RTCRTCRTCNGGRTT 3'
(In these sequences, "R" represents a purine base A or G;
"Y" represents a pyrimidine base T or G; and N represents
G, A, T or C. )
The 20KF is a mixture of 32 kinds of
oligonucleotides containing the DNA sequences encoding
the amino acid sequence of
Asn Tyr Phe His Gly
found in a peptide having a molecular weight of 20
kDa, and the 20KR1 is a mixture of 128 kinds of
* Trademarks
CA 02191624 2004-04-30
19
oligonucleotides containing complementary chains of
the DNA sequences encoding the amino acid sequence
of
Asn Pro Asp Asp(Asp)
found in the same peptide.
The cDNA synthesis was carried out using 10 ng
of Poly(A)+RNA, 0.3 ug of random hexamer (N6), 10 LI of
an RNase inhibitor (RNA Guard, commercially available
from Pharmacia), 1 mM each of dATP, dCTP, dGTP and dTTP,
1 x PCR buffer (commercially available from Takara Shuzo),
50 mM of magnesium chloride and 100 U of a reverse
transcriptase (M-MuLV RTase, commercially available from
BRL) in a total volume of 10 u1. The reaction was
carried out at 37°C for 30 minutes and the reaction
mixture was then heated at 95°C for 5 minutes, followed
by retaining the reaction mixture in ice.
Polymerase chain reaction (PCR) was performed using
the above-described cDNA as a template and 20KF and 20KR1
as primers. The reaction was performed using 10 u1 of
the cDNA synthesis reaction mixture, a mixture of 50 pmol
each of the primers, 200 uM each of dATP, dCTP, dGTP and
dTTP, 1 x PCR buffer (commercially available from TAKARA
SHUZO), and 2.5 U of AmpliTaq DNA polymerase
(commercially available from TAKARA SHUZO) in a total
volume of 50 u1. A cycle of temperature conditions of
94°C for 1 minute, 40°C for 1 minute and 72°C for 2.5
minutes was repeated 30 times in a DNA Thermocycler
* Trademark
CA 02191624 2004-04-30
(commercially available from Perkin Elmer Cetus).
The PCR product was separated on 2o agarose gel. A
small number of fragments were detected by the ethidium
bromide staining method. One of them had a size of 94 by
5 as expected.
The PCR fragment was cut out from the gel and
subcloned into pUCl9 plasmid. The DNA sequence of the
subcloned PCR fragment was determined by the dideoxy
method using T7 sequencing kit (commercially available
10 from Pharmacia). Between the two primers, a DNA sequence
encoding the expected amino acid sequence was observed.
The nucleotide sequence of the DNA between the primers
and the amino acid sequence encoded thereby are as
follows.
15 C TCT GAC GTG AAC TGT GTT CTA TGC CCT CGC
Ser Asp Val Asn Cys Val Leu Cys Pro Ar_g
Isotope 32P (commercially available from Amersham)
was incorporated into the oligonucleotide using a DNA 5'-
end labelling kit MEGALABEL*(commercially available from
20 Takara Shuzo) to obtain a radioactive oligonucleotide
probe.
6. Screening of PLD Gene-containing Clones
Using the radioactive oligonucleotide as a probe, a
cDNA library was screened. Hybridization solution
contained 0.5 M sodium phosphate buffer (pH 7.2), 7o SDS,
1 mM EDTA and 100 ug/ml of salmon sperm DNA, and
hybridization was performed after adding the probe to the
* Trademark
CA 02191624 2004-04-30
21
hybridization solution at 45°C for 16 hours. The washing
solution contained 0.3 M NaCl and 30 mM sodium citrate,
and washing was performed twice at 45°C for 20 minutes
each. Positive plaques were isolated and subcloned in
vivo into pBluescript* plasmid (commercially available
from Stratagene) in accordance with the instructions
provided by the manufacturer of a,2APII cloning vector.
The nucleotide sequence was determined by the dideoxy
method. As a result, a region encoding the internal
amino acid sequence determined in the "Section 3" existed.
7. Determination of Nucleotide Sequence of 5'-end
Re,~n
Since a clone containing the full length of cDNA
could not be isolated, a DNA fragment having the 5'-end
region was prepared by RACE method (Edwards et al.,
Nucleic Acids Res., 19, 5227-5232 (1991)). 5'-
AmpliFINDER RACE Kit (commercially available from
Clonetech) was used in accordance with the manual
attached to the product. An oligoDNA was synthesized
based on the nucleotide sequence of the cDNA determined
in "Section 6", and PCR was performed using the mRNA
prepared by the method described in "Section 4" as a
template. The PCR product was subcloned into a PCRII
vector (commercially available from Invitrogen) and the
nucleotide sequence was determined by the dideoxy method.
The thus determined nucleotide sequence of the cDNA of
rice PLD as well as the deduced amino acid sequence
* Trademarks
CA 02191624 2004-04-30
encoded thereby is shown in SEQ ID N0. 2 in the Sequence
Listing. It is thought that translation is initiated
from the 182nd nucleotide shown in SEQ ID NO. 2 since a
termination codon exists at 36 bases upstream thereof.
8. Isolation of PLD Genomic Clone Corresponding to PLD
cDNA and Identification of Promoter Re Qion
To isolate a genomic DNA clone having tree regulatory
sequence of the PLD gene corresponding to the PLD cDNA
determined in "section 6", which was cloned into
pBluescript plasmid, a genomic library of rice, variety
"KOSHIHIKARI" was prepared. This was carried out by
partially digesting DNAs from live leaves of KOSHIHIKARI
with Mbo I, purifying a fraction having a size of 16 - 20
kb by sucrose gradient centrifugation, and using lambda
DASH II (commercially available from Stratagene) and
GigapackII*Gold (commercially available from Stratagene).
The genomic library was screened with the PLD cDNA clone
as a probe. The screening was carried out in the same
manner as in "Section 6" except that hybridization was
performed at 65°C for 16 hours, the washing solution
contained 0.5 x SSC and 0.1o SDS, and that the washing
was performed twice at 65°C for 20 minutes each. The
nucleotide sequence of the hybridized genomic clone was
determined by the dideoxy method. As a result, a region
homologous to the cDNA sequence determined in "Section 6"
existed.
The transcription initiation site was determined by
* Trademarks
~19162~
z3
the method described in "Section 7". In the vicinity of
the transcription initiation site, a "TATA" consensus
sequence box was observed. The ATG translation -
initiation site was determined based on the determined
DNA sequence as the most upstream ATG codon in the
translation open reading frame of the clone and as the
ATG codon which is first accessible in the mRNA
synthesized in rice.
The DNA sequence of a-part of the genomic clone
hybridized with the cDNA clone is shown in SEQ ID N0. 3.
In the genomic DNA sequence, an open reading frame
starting from the ATG translation initiation codon, which
overlaps with the corresponding cDNA sequence has been
identified. The promoter region exists upstream of the
ATG translation initiation codon and starts from the sits
immediately upstream thereof.
9, rrlPnt;f;ration of Introns and Analysis of Functions
thereof on Expression of Genes
From comparison between the cDNA (SEQ ID N0. 2) and
the genomic DNA (SEQ ID N0. 3), it was proved that 3
introns exist in PLD gene. Among these, the intron
having a size of 173 by located at the 5'-flanking region
of the mRNA (i.e., the nucleotide sequence between 1666nt
and 1838nt of the nucleotide sequence shown in SEQ ID N0.
3, the sequence being shown in SEQ ID N0. 4) was tested _.
for its influence on expression of a gene in plant cells.
Primers of l5mer each of which contains 5 bases of.exon
,
2~9I6~~
24
region (5'-ACCCGGTAAGCCCAG-3', 3'-CCCCCGCGTCCATCC-5')
were synthesized and PCR was carried out using the
genomic clone as a template according to the method
described in the Section of "5. Preparation of
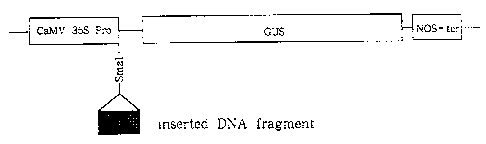
Probes". The PCR product was subcloned into PCRII
vector and a fragment was cut out with Eco RI. The _
fragment was blunted and inserted into the Sma I
site of a plasmid pBI221-(commercially available
from Toyobo) (see Fig. 1). The obtained recombinant
plasmid was introduced into rice cultured cells
(Baba et al., Plant Cell Physiol. 27, 463-471
(1986)) in accordance with the reported method
(Shimamoto et.al., Nature, 338, 274-276 (1989)) and
(3-glucuronidase (GUS) activity was measured. As
shown in Table 2, by introducing the intron, the GUS
activity was increased. Further, as shown in Table 3,
increase in the GUS activity was also observed in
the case where the intron was inserted in the
reverse direction. The direction of the intron was
determined based on the sizes of the fragments cut
out with Bg1 II and Bam HI, utilizing the Bg1 II
site existing in the intron sequence and the Bam HI
site existing in pBI221.
2191~2~
Table 2
Plasmid GUS Activity
pBI221 10.4
pBI22I + intron 105.7
(pmol MU/min./mg protein)
Table 3
Plasmid GUS Activity
pBI221 8.8
pBI22I + intron 79-4
pBI221 + intron (reverse direction) 54.2
(pmol MU/min./mg protein)
2I9~~~~
26
SEQUENCE LISTING
SEQ ID NO.1
SEQUENCE LENGTH: 183
SEQUENCE TYPE: Nucleic Acid
MOLECULE TYPE: Genomic DNA
ORIGINAL SOURCE
ORGANISM: Oryza sativa
SEQUENCE DESCRIPTION
ACCCGGTAAG CCCAGTGTGC TTAGGCTAAG CGCACTAGAG CTTCTTGCTC GCTTGCTTCT 60
TCTCCGCTCA GATCTGCTTG CTTGCTTGCT TCGCTAGAAC CCTACTCTGT GCTGCGAGTG 120
TCGCTGCTTC GTCTTCCTTC CTCAAGTTCG ATCTGATTGT GTGTGTGGGG GGGCGCAGGT 180
AGG 183
SEQ ID NO.: 2
SEQUENCE LENGTH: 3040
SEQUENCE TYPE: Nucleic Acid __
MOLECULE TYPE: cDNA to mRNA
ORIGINAL SOURCE
ORGANISM: Oryza sativa
SEQUENCE DESCRIPTION
AGTCTCTCTT CTCCCGCAAT TTTATAATCT CGATCGATCC AATCTGCTCC CCTTCTTCTT 60
CTACTCTCCC CATCTCGGCT CTCGCCATCG CCATCCTCCT CTCCCTTCCC GGAGAAGACG 120
CCTCCCTCCG CCGATCACCA CCCGGTAGGG CGAGGAGGGA GCCAAATCCA AATCAGCAGC 180
C ATG GCG CAG ATG CTG CTC CAT GGG ACG CTG CAC GCC ACC ATC TTC GAG 229
Met Ala Gln Met Leu Leu His Gly Thr Leu His Ala Thr Ile Phe Glu
1 5 i0 15
GCG GCG TCG CTC TCC AAC CCG CAC CGC GCC AGC GGA AGC GCC CCC AAG 277
Ala Ala Ser Leu Ser Asn Pro His Arg Ala Ser Gly Ser Ala Pro Lys
2191624
20 25 30
TTCATC CGC AAGTTT GTG GAGGGG ATT GAGGAC ACT GTG GGTGTC GGC 325
PheIle Arg LysPhe Val GluGly Ile GluAsp Thr Val GlyVal Gly
35 40 45
AAAGGC GCC ACCAAG GTG TATTCT ACC ATTGAT CTG GAG AAAGCT CGT 373
LysGly Ala ThrLys Val TyrSer Thr IleAsp Leu Glu LysAla Arg
50 55 60
GTAGGG CGA ACTAGG ATG ATAACC AAT GAGCCC ATC AAC CCTCGC TGG 421
ValGly Arg ThrArg Met IleThr Asn GluPro Ile Asn ProArg Trp
65 70 75 80
TATGAG TCG TTCCAC ATC TATTGC GCT CATATG GCT TCC AATGTG ATC 469
TyrGlu Ser PheHis Ile TyrCys Ala HisMet Ala Ser AsnVal Ile
85 90 95
TTCACT GTC AAGATT GAT AACCCT ATT GGGGCA ACG AAT ATTGGG AGG 517
PheThr Val LysIle Asp AsnPro Ile GlyAla Thr Asn IleGly Arg
100 105 110
GCTTAC CTG CCTGTC CAA GAGCTT CTC AATGGA GAG GAG ATTGAC AGA 565
AlaTyr Leu ProVal Gln GluLeu Leu AsnGly Glu Glu IleAsp Arg
115 120 125
TGGCTC GAT ATCTGT GAT AATAAC CGC GAGTCT GTT GGT GAGAGC AAG 613
TrpLeu Asp IleCys Asp AsnAsn Arg GluSer Val Gly GluSer Lys
130 135 140
ATCCAT GTG AAGCTT CAG TACTTC GAT GTTTCC AAG GAT CGCAAT TGG 661
IleHis Val LysLeu Gln TyrPhe Asp ValSer Lys Asp ArgAsn Trp
145 150 155 160
GCGAGG GGT GTCCGC AGT ACCAAG TAT CCAGGT GTT CCT TACACC TTC 709
AlaArg Gly ValArg Ser ThrLys Tyr ProGly Val Pro TyrThr Phe
165 170 175
za ~~9~ ~~~
TTC TCT CAG AGG CAAGGG TGC AAAGTT ACC TTGTAC CAA GAT GCTCAT 757
Phe Ser Gln Arg GlnGly Cys LysVal Thr LeuTyr Gln Asp AlaHis
180 185 190
GTC CCA GAC AAC TTCATT CCA AAGATT CCG CTTGCC GAT GGC AAGAAT 805
Val Pro Asp Asn PheIle Pro LysIle Pro LeuAla Asp Gly LysAsn
195 200 205
TAT GAA CCC CAC AGATGC TGG GAGGAT ATC TTTGAT GCT ATA AGCAAT 853
Tyr Glu Pro His ArgCys Trp GluAsp Ile PheAsp Ala Ile SerAsn
210 215 220
GCT CAA CAT TTG ATTTAC ATC ACTGGC TGG TCTGTA TAC ACT GAGATC 901
Ala Gln His Leu IleTyr Ile ThrGly Trp SerVal Tyr Thr GluIle
225 230 235 240
ACC TTG GTT AGG GACTCC AAT CGTCCA AAA CCTGGA GGG GAT GTCACC 949
Thr Leu Val Arg AspSer Asn ArgPro Lys ProGly Gly Asp ValThr
245 250 255
CTT GGG GAG TTG CTCAAG AAG AAGGCC AGT GAAGGT GTT CGG GTCCTC 997
Leu Gly Glu Leu LeuLys Lys LysAla Ser GluGly Val Arg ValLeu
260 265 270
ATG CTT GTG TGG GATGAC AGG ACTTCA GTT GGTTTG CTA AAG AGGGAT 1045
Met Leu Val Trp AspAsp Arg ThrSer Val GlyLeu Leu Lys ArgAsp
275 280 285
GGC TTG ATG GCA ACACAT GAT GAGGAA ACT GAAAAT TAC TTC CATGGC 1093
Gly Leu Met Ala ThrHis Asp GluGlu Thr GluAsn Tyr Phe HisGly
290 295 300
- TCT GAC GTG AAC TGTGTT CTA TGCCCT CGC AACCCT GAT GAC TCAGGC 1141
Ser Asp Val Asn CysVal Leu CysPro Arg AsnPro Asp Asp SerGly
305 310 315 320
AGC ATT GTT CAG GATCTG TCG ATCTCA ACT ATGTTT ACA CAC CATCAG 1189
219624
i
Ser IleVal Gln AspLeu Ser Ile SerThr Met PheThr His HisGln
325 330 335
AAG ATAGTA GTT GTTGAC CAT GAG TTGCCA AAC CAGGGC TCC CAACAA 1237
Lys IleVal Val ValAsp His Glu LeuPro Asn GlnGly Ser GlnGln
340 345 350
AGG AGGATA GTC AGTTTC GTT GGT GGCCTT GAT CTCTGT GAT GGAAGG 1285
Arg ArgIle Val SerPhe Val Gly GlyLeu Asp LeuCys Asp GlyArg
355 360 365
TAT GACACT CAG TACCAT TCT TTG TTTAGG ACA CTCGAC AGT ACCCAT 1333
Tyr AspThr Gln TyrHis Ser Leu PheArg Thr LeuAsp Ser ThrHis
370 375 380
CAT GATGAC TTC CACCAG CCA AAC TTTGCC ACT GCATCA ATC AAAAAG 1381
His AspAsp Phe HisGln Pro Asn PheAla Thr AlaSer Ile LysLys
385 390 395 400
GGT GGACCT AGA GAGCCA TGG CAT GATATT CAC TCACGG CTG GAAGGG 1429
Gly GlyPro Arg GluPro Trp His AspIle His SerArg Leu GluGly
405 410 415
CCA ATCGCA TGG GATGTT CTT TAC AATTTC GAG CAGAGA TGG AGAAAG 1477
Pro IleAla Trp AspVal Leu Tyr AsnPhe Glu GlnArg Trp ArgLys
420 425 430
CAG GGTGGT AAG GATCTC CTT CTG CAGCTC AGG GATCTG TCT GACACT 1525
Gln GlyGly Lys AspLeu Leu Leu GlnLeu Arg AspLeu Ser AspThr
435 440 445
ATT ATTCCA CCT TCTCCT GTT ATG TTTCCA GAG GACAGA GAA ACATGG 1573
Ile IlePro Pro SerPro Val Met PhePro Glu AspArg Glu ThrTrp
450 455 460
AAT GTTCAG CTA TTTAGA TCC ATT GATGGT GGT GCTGCT TTT GGGTTC 1621
Asn ValGln Leu PheArg Ser Ile AspGly Gly AlaAla Phe GlyPhe
219~62~
465 470 475 480
CCTGAT ACC CCTGAG GAG GCTGCA AAA GCTGGG CTT GTA AGCGGA AAG 1669
ProAsp Thr ProGlu Glu AlaAla Lys AlaGly Leu Val SerGly Lys
485 490 495
GATCAA ATC ATTGAC AGG AGCATC CAG GATGCA TAC ATA CATGCC ATC 1717
AspGln Ile IleAsp Arg SerIle Gln AspAla Tyr Ile HisAla Ile
500 505 510
CGGAGG GCA AAGAAC TTC ATCTAT ATA GAGAAC CAA TAC TTCCTT GGA 1765
ArgArg Ala LysAsn Phe IleTyr Ile GluAsn Gln Tyr PheLeu Gly
515 520 525
AGTTCC TAT GCCTGG AAA CCCGAG GGC ATCAAG CCT GAA GACATT GGT 1813
SerSer Tyr AlaTrp Lys ProGlu Gly IleLys Pro Glu AspIle Gly
530 535 540
GCCCTG CAT TTGATT CCT AAGGAG CTT GCACTG AAA GTT GTCAGT AAG 1861
AlaLeu His LeuIle Pro LysGlu Leu AlaLeu Lys Val ValSer Lys
545 550 555 560
ATTGAA GCC GGGGAA CGG TTCACT GTT TATGTT GTG GTG CCAATG TGG 1909
IleGlu Ala GlyGlu Arg PheThr Val TyrVal Val Val ProMet Trp
565 570 575
CCTGAG GGT GTTCCA GAG AGTGGA TCT GTTCAG GCA ATC CTGGAC TGG 1957
ProGlu Gly ValPro Glu SerGly Ser ValGln Ala Ile LeuAsp Trp
580 585 590
CAAAGG AGA ACAATG GAG ATGATG TAC ACTGAC ATT ACA GAGGCT CTC 2005
GlnArg Arg ThrMet Glu MetMet Tyr ThrAsp Ile Thr GluAla Leu
595 600 605
CAAGCC AAG GGAATT GAA GCGAAC CCC AAGGAC TAC CTC ACTTTC TTC 2053
GlnAla Lys GlyIle Glu AlaAsn Pro LysAsp Tyr Leu ThrPhe Phe
610 615 620
2I916~4
31
TGC TTG GGT AAC CGT GAG GTG AAG CAG GCT GGG GAA TAT CAG CCT GAA 2101
Cys Leu Gly Asn Arg Glu Val Lys Gln Ala Gly Glu Tyr Gln Pro Glu
625 630 635 640
GAA CAA CCA GAA GCT GAC ACT GAT TAC AGC CGA GCT CAG GAA GCT AGG 2149
Glu Gln Pro Glu Ala Asp Thr Asp Tyr Ser Arg Ala Gln Glu Ala Arg
645 650 655
AGG TTC ATG ATC TAT GTC CAC ACC AAA ATG ATG ATA GTT GAC GAT GAG 2197
Arg Phe Met Ile Tyr Val His Thr Lys Met Met Ile Val Asp Asp Glu
660 665 670
TACATC ATCATC GGT TCT GCAAAC ATC AACCAG AGG TCGATG GAC GGC 2245
TyrIle IleIle Gly Ser AlaAsn Ile AsnGln Arg SerMet Asp Gly
675 680 685
GCTAGG GACTCT GAG ATC GCCATG GGC GGGTAC CAG CCATAC CAT CTG 2293
AlaArg AspSer Glu Ile AlaMet Gly GlyTyr Gln ProTyr His Leu
690 695 700
GCGACC AGGCAA CCA GCC CGTGGC CAG ATCCAT GGC TTCCGG ATG GCG 2341
AlaThr ArgGln Pro Ala ArgGly Gln IleHis Gly PheArg Met Ala
705 710 715 720
CTGTGG TACGAG CAC CTG GGAATG CTG GATGAT GTG TTCCAG CGC CCC 2389
LeuTrp TyrGlu His Leu GlyMet Leu AspAsp Val PheGln Arg Pro
725 730 735
GAGAGC CTGGAG TGT GTG CAGAAG GTG AACAGG ATC GCGGAG AAG TAC 2437
GluSer LeuGlu Cys Val GlnLys Val AsnArg Ile AlaGlu Lys Tyr
740 745 750
TGGGAC ATGTAC TCC AGC GACGAC CTC CAGCAG GAC CTCCCT GGC CAC 2485
TrpAsp MetTyr Ser Ser AspAsp Leu GlnGln Asp LeuPro Gly His
755 760 765
CTCCTC AGCTAC CCC ATT GGCGTC GCC AGCGAT GGT GTGGTG ACT GAG 2533
21916~~
32
Leu Leu Ser Tyr Pro Ile Gly Val Ala Ser Asp Gly Val Val Thr Glu
770 775 780
CTG CCC GGG ATG GAG TAC TTT CCT GAC ACA CGG GCC CGC GTC CTC GGC 2581
Leu Pro Gly Met Glu Tyr Phe Pro Asp Thr Arg Ala Arg Val Leu Gly
785 790 795 800
GCC AAG TCG GAT TAC ATG CCC CCC ATC CTC ACC TCA TAGACGAGGA AGCACT 2633
Ala Lys Ser Asp Tyr Met Pro Pro Ile Leu Thr Ser
805 810
ACACTACAAT CTGCTGGCTT CTCCTGTCAG TCCTTCTGTA CTTCTTCAGT TTGGTGGCGA 2693
GATGGTATGG CCGTTGTTCA GAATTTCTTC AGAATAGCAG TTGTTACAGT TGTGAATCAT 2753
AAAGTAATAA GTGCAGTATC TGTGCATGGT TGAGTTGGGA AGAAGATCGG GGATGCAATG 2813
ATGCTTGTGA AGTTGTGATG CCGTTTGTAA GATGGGAAGT TGGGAACTAC TAAGTAATTG 2873
GCATGATTGT ACTTTGCACT ACTGTTTAGC GTTGTTGATA CTGGTTAACC GTGTGTTCAT 2933
CTGAACTTGA TTCTTGATGC AGTTTGTGGC ATTACCAGTT TATCATCGTT CTTCAGGAAA 2993
AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAA 3040
SEQ ID NO.: 3
SEQUENCE LENGTH: 2799
SEQUENCE TYPE: Nucleic Acid
MOLECULE TYPE: Genomic DNA
ORIGINAL SOURCE
ORGANISM: Oryza sativa
SEQUENCE DESCRIPTION
CAAGGGTGTA CATAGATTTG TCTCGTAAAA TAGTATTATA ATATTATAAA CTTATTACTC 60
TATCCGTTCT AAAATATAAG AACCTTATGA CTGGATGGAA CATTTCCTAG TACTACGAAT 120
CTGAACACAT GTCTAGATTC ATAGTACTAG GAAATGTCTC ATCGCGGTAC TAGGTTCTTA 180
TATTTTAGGA TGGAGGGAGT TTAATATAAA ACTAATGGTT AGAACTTTGA AAGTTTGATT 240
TTAAATGTCA AATATTTATG GCTGGAGGTA GTATAATATG TTTTTTTTGG GACGTAGACT 300
AGGTAGTATA ATATGTTTGG TTGTGTTTAG ATCCAATATT TGGATCCAAA CTTCAGTCAT 360
33-
TTTCCATCAC ATCAACTTGT CATATACACA TAACTTTTCA GTCACATCAT CCCCAATTTC 420
AACCAAAATC AAACTTTGCG CTGAACTAAA CACAACCTTT GGGCCCGTTT AGTTCCCCAA 480
TTTTTTTCCC AAAAACATCA CATCGAATCT TTGGACACAT GCATGAAGCA TTAAATATAG 540
ATAAAAAGAA AAACTAATTG CACAGTTATG GAGGAAATCG CGAGACGAAT CTTTTAAGCC 600
TAATTAGTCC GTGATTAGCC ATAAGTGCTA CAGTAACCCA ATTGTGCTAA TGACGGCTTA 660
ATTAGTCTCC ACAAGATTCG TCTCGCAGTT TCCAGGCGAG TTCTGAAATT AGTTTTTTCA 720
TTCGTGTCCG AAAACCCCTT CCGACATCCG GTCAAACGTT CGATATGACA CCCACAAATT 780
TTCTTTTCCC CAACTAAACA CACCCTTTAT CTCTTACCCT CTGGCTCTTT CAGTAGGCAT 840
ATCCAAGACA GCTGGTAATG CAGGCTCGGA CATAATTTGA CAGTTACGTT CATGTGACCG 900
ACGGTTGATG CTAGTGCAAC TGCAACATAC TGTTCAGATG GATGTCCCAA CGAGCTCAAA 960
ACAACTTAGG TGGCGCGTCG CGATTCATCA ATAACTCAAA TGGAAGCGCA AGTGCACGTA 1020
CGAAAATGAC AGCGAGTGAG GTGGCGAGCC TCACCTTGGT GATCCCAACC GGATAAGCTA 1080
TGCATCAGCC AGTTTCGTGG GGCTGCACAT TTCGTCGAAC ACCTGGAGTC CACGCCGCCG 1140
GCGACGTCGG CACAGCGCGC CCGCCCACCG CCCACGCACG CGCTTGACTC CACCCATGTT 1200
CTCCCTTCTC GACGCCCGCG AAGCCAGCGA ACCGATCCGA GGAAGTCAAG CCCCCACCGC 1260
CACTTGGACC GACCTCGGGA CGACGACGCC CCCGCGCTCT TCTAGACGCG CGGACGACGC 1320
GGGCGCTGGC TCCGCGACGC GACGTCGCGG TCATGGAGTA ACCGCGACGG ACAGATACTT 1380
CTACCCGTTT TTAACCTCGC CTCCTCCTCC TCCCGGCTCG AGATCCGTGG CCACGACGCG 1440
TGGTGGGAAA CCGGGAACGA CGTGCACGCA CGCACACAGG GCAAGTTTCA GTAGAAAAAT 1500
CGCCGGCATC CAGATCGGGA CAGTCTCTCT TCTCCCGCAA TTTTATAATC TCGCTCGATC 1560
CAATCTGCTC CCCTTCTTCT TCTACTCTCC CCATCTCGGC TCTCGCCATC GCCATCCTCC 1620
TCTCCCTTCC CGGAGAAGAC GCCTCCCTCC GCCGATCACC ACCCGGTAAG CCCAGTGTGC 1680
TTAGGCTAAG CGCACTAGAG CTTCTTGCTC GCTTGCTTCT TCTCCGCTCA GATCTGCTTG 1740
CTTGCTTGCT TCGCTAGAAC CCTACTCTGT GCTGCGAGTG TCGCTGCTTC GTCTTCCTTC 1800
CTCAAGTTCG ATCTGATTGT GTGTGTGGGG GGGCGCAGGT AGGGCGAGGA GGGAGCCAAA 1860
TCCAAATCAG CAGCC ATG GCG CAG ATG CTG CTC CAT GGG ACG CTG CAC GCC 1911
Met Ala Gln Met Leu Leu His Gly Thr Leu His Ala
1 5 10
34
ACC ATC TTC GAG GCG GCG TCG CTC TCC AAC CCG CAC CGC GCC AGC GGA 1959
Thr Ile Phe Glu Ala Ala Ser Leu Ser Asn Pro His Arg Ala Ser Gly
15 20 25
AGC GCC CCC AAG TTC ATC CGC AAG GTTCGGACCC TTCTCCTTAA TCTACTCGTC 2013
Ser Ala Pro Lys Phe Ile Arg Lys
30 35
TTTGCTCTTG CTCTTTTTCT TTTGTGTCCC TTTCTTGTGT GTGCGTTTGC ATGAGCCCGA 2073
ATTTGATCTG CTAGTGCACA GTACAGTCAG ATACACTGAA ACGATCTGGA AATTCTGGAT 2133
TATTAGGAAA AATAAAGAGG TAGTAGACAA GAATTGGAGA TACTTTCTAT CAAGATTGGT 2193
CTATTATGCT TGGCCATTTC TTGTTTGACC CAAGTACTTC TTTGAATCTA GAGTTTGCTG 2253
TGTGTGATGT GGTGTGTGTT TGTGTCACCA AAAATCTTCA TTAGCTAAAA CTGAAATTTT 2313
ATTTATTAAC TGACCTACTA AAAATGTAGA GTTCTCTGTG TGTGATGTGT GCTTGTGTCA 2373
CCAAAAATCT TGATTTGATA GAGTTTTTAT TTATTTATTA ACTGACCTAC TACAAATCTA 2433
TTGCTGTATG CTATGTGTGT CTGTATCACC TGAAATGCAA TGTCTTCTTC TTTGTTGTTC 2493
TTGATCTAAC ACGTGAGCTC ATGTCAACAG TTT GTG GAG GGG ATT GAG GAC ACT 2547
Phe Val Glu Gly Ile Glu Asp Thr
GTGGGT GTCGGC AAA GGCGCC ACC AAGGTG TAT TCT ACCATT GAT CTG 2595
ValGly ValGly Lys GlyAla Thr LysVal Tyr Ser ThrIle Asp Leu
50 55 60
GAGAAA GCTCGT GTA GGGCGA ACT AGGATG ATA ACC AATGAG CCC ATC 2643
GluLys AlaArg Val GlyArg Thr ArgMet Ile Thr AsnGlu Pro Ile
65 70 75
AACCCT CGCTGG TAT GAGTCG TTC CACATC TAT TGC GCTCAT ATG GCT 2691
AsnPro ArgTrp Tyr GluSer Phe HisIle Tyr Cys AlaHis Met Ala
80 85 90
TCCAAT GTGATC TTC ACTGTC AAG ATTGAT AAC CCT ATTGGG GCA ACG 2739
SerAsn ValIle Phe ThrVal Lys IleAsp Asn Pro IleGly Ala Thr
35
95 100 105
AAT ATT GGG AGG GCT TAC CTG CCT GTC CAA GAG CTT CTC AAT GGA GAG 2787
Asn Ile Gly Arg Ala Tyr Leu Pro Val Gln Glu Leu Leu Asn Gly Glu
110 115 120
GAG ATT GAC AGA 2799
Glu Ile Asp Arg
125
SEQ ID NO.: 4
SEQUENCE LENGTH: 173
SEQUENCE TYPE: Nucleic Acid
MOLECULE TYPE: Genomic DNA
ORIGINAL SOURCE
ORGANISM: Oryza sativa
SEQUENCE DESCRIPTION
GTAAGCCCAG TGTGCTTAGG CTAAGCGCAC TAGAGCTTCT TGCTCGCTTG CTTCTTCTCC 60
GCTCAGATCT GCTTGCTTGC TTGCTTCGCT AGAACCCTAC TCTGTGCTGC GAGTGTCGCT 120
GCTTCGTCTT CCTTCCTCAA GTTCGATCTG ATTGTGTGTG TGGGGGGGCG CAG 173