Note: Descriptions are shown in the official language in which they were submitted.
~ -- 21 9331~
HOECHSTAKTIENGESELLSCHAFT HOE 95/F 314 DR.LV/as
5 Das~ i~tiGI I
Suppo, l6d catalyst system a process for itS preparation and its use for the
poly",eri~tion of olefins
10 The pres~n~ invention relates to a SUppGI led catalyst system having a high activity
and to an economical and enviro. ""entally friendly process for pr~paring the
catalyst system and also to the use of the catalyst system for poly",eri~dlion.
rr~c~sses for prepari.)g polyolefins with the aid of soluble ho-nogenous catalyst
15 systems cGI~ risi~9 a lfansiliGn metal col"ponent of the met~llocene type and a
yst col;"~onent seleGled from the group consisting of alumir,oxanes Lewis
acids or ionic cGI"pounds have been des~ ibed. These catalysts have a high activity
and give ho."opolymers and copolymers having a narrow molerul^- weight
distribution.
When soluble (hol"Ggeneous) met~locene/methylaluminoxane catalyst systems are
used in processes in which the polymer for",ed is obtained as a solid heavy
deposits on the reactor walls and agiL~lor are frequently formed. These deposits are
fol",ed by agglomeration (Polymer Commun. (1991) 32 58) of the polymer particles25 when the metallocene or alu",i"oxane or both are presenl in solution in the
suspensio. . medium. Such deposits in the rea~tor systems have to be removed
regularly since they rapidly reach considerable thicknesses have a high al(en~Jth
and hinder heat ll ansrer to the cooling medium.
30 To prevent these deposits it is advant~geous to use metallocenes in suppol led
form.
WO-A 94/28034 discloses a supported catalyst system in which a preactivated (at
room te"~peralure) solution of metallocene and the coc~t~lyst methylaluminoxane
21 9331 9
(MAO) is applied to a porous support material (preferably silica gel). The preliminary
r~a~ti~n l~tween MAO and metallocene takes between 1 and 60 minutes
pr~fetably 10 minutes.
EP-A 0 518 092 ~isclQses a suppo, led catalyst system in which the prea~;tivatedmetallocene/MAO solution is al.plied to porous polymers as support ",alerials. The
preactivation take~ from 1 to 120 minutes preferably from 10 to 100 minutes. Thepreacti~ation takes place at te,.,peralures betwecn 0 and 50C.
The catalyst systems des~ ibed have a low productivity based on the suppo, led
system in kg of polymer per g of catalyst when compared with cl ~ssi~l Ziegler-
Natta systems.
The catalyst systems les~ il,ecJ also have high catalysts costs which are esse, Itially
~us~ by the fact that the e~e"sive co~t~lyst c~",ponent methylaluminoxane has
to be used in excess.
It is an object of the prese- Il invention to provide a SUppGI led catalyst system having
a high activity and an econG"~ical and envi~..."entally friendly process for prepa.ing
the catalyst system.
The object of the present invention is achieved by a SUppGI led catalyst system
having a high activity which is obtainable by
a) bringing at least one met- 'locene com~onent in at least one solvent
into conlact with at least one cocPt~lyst w",pone"t
b) adding the soluble product to the support material and
c) removing the solvent.
The advantage of the catalyst system of the invention is a significant increase in the
activity and the productivity for the same co,l,posilion of the catalyst system where
in particular the ratio of cocAt~lyst to metallocene is not altered.
A prefer.ed e,nbodiment of the invention is a supported catalyst system where
21 9331 9
-
d) the SUppGi led catalyst system is isolated and
e) prepoly",eri~ed with at least one olefinic ",G"G,oer.
The metalloce"e cG~"~Gnenl of the catalyst system of the invention can in principle
5 be any metallocene. The metallocene can be either l ridged or u,)bridgeJ and have
identical or Ji~rent ii,~dl nls. Preference is given to mePIloc6l ,es of group IVb of the
Periodic Table of the El~lllents, for example titanium, zirconium or hafnium.
The metallocene is prdferably a met-'locene of the formula (I) below. Particular10 prefarence is given to ~i, conocel ,es which bear indenyl and tetrah~Jro.ndenyl
derivatives as ligands.
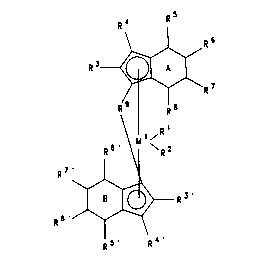
R4 RS
R3~ 6
~ ~R I ( I )
R \ R
~ ~
/ B >--
6~
Rs~ R4
25 where
M1 is a metal of group IVb of the Periodic Table of the Elements,
R1 and R2 are ideutical or dirrerent and are each a hydrogen atom, a
C1-C10-alkyl group, a C1-C10-alkoxy group, a C6-C20-aryl group, a C6-C10-aryloxygroup, a C2-C10-alkenyl group, an OH group, an NR122 group, where R12 is a C1- to
30 C2-alkyl group or a C6-C14-aryl group, or a halogen atom,
R3 to R8 and R3 to R8 are identical or dirrere"t and are each a hyJ~-,ge" atom,
a C1-C40-hyd~ocarL,on group which may be linear, cyclic or bré."~;l,eJ, e.g. a C1-C10-
alkyl group, a C2-C10-alkenyl group, a C6-C20-aryl group, a
21 933 1 9
-
C7-C40-arylalkyl group, a C7-C40-alkylaryl group or a C8-C40-arylalkenyl group, or
c~, It radicals R4 to R8 and/oder R4 to R8 togetl ,er with the atoms
con- ,~clin~ them form a ring system,
R9 is a bridge, preferal,ly
s
R10 R10 R10 R10 R1o
_ o _ ~2_ O-- --C-- --o_ ~2-- --C--~2_
R11 R11 R11 R11 R11
R1o Rl Rl R10 R10 Rl Rl Rl
_ ~,~2_ --C--C_ _ ~t C ~,~2_
R l ' , R l ~ R l l , R l l R l l , R ' l R l l R l l ,
-- -- x
R10 R10 Rl
--C--C--C--
R 1 1 R 1 1 R
~ ~10, >~ . - G ~ - . - O - . - S - . ~5 0, ~5 0 ~ , ~C O . ~ or ~11 ( 0 ) ~ ~ ,
where R10 and R11 are iWentical or differenl and are each a hydroge" atom, a
halogen atom or a C1-C40-group such as a C1-C20-alkyl group, a C1-C10-fluoroalkyl
group, a C1-C10-. 'koxy group, a C6-C14-aryl group, a C6-C10-fluoroaryl group, a C6-
C10-aryloxy group, a C2-C10-alkenyl group, a C7-C40-arylalkyl group, a C7-C40-
alkylaryl group or a C8-C40-arylalkenyl group, or R10 and R1 1 together with theatoms co, In6~Ain!3 them form in each case one or more rings, and x is an integer
from zero to 18,
M2 is silicon, ger")anium or tin and the rings A and B are identical or difrerent,
saturated or unsaturated.
R9 can also link two units of the formula I to one another.
219331~
In formula 1 it is particularly pr~fer,~d that
M1 is zirconium or hafnium
R1 and R2 are idel ltical and are methyl or chlorine in particular chlorine and R9 =
M2R10R11, where M2 is silicon or germanium and R10 and R11 are each a C1-C20-
hydloc~ ol~ group such as C1-C10-alkyl or C6-C14 aryl
The indenyl or tetrahydroindenyl ligands of the met-llQcenes of the formula I are
~,r~rer~ly substituted in the 2 2 4 4 7 2 6 2 4 6 2 5 6
2 4 5 6 or 2 4 5 6 7 posilions in particular in the 2 4 positions. Pleferled
substituents are C1-C4-alkyl groups such as methyl ethyl or isopropyl or C6-C10-aryl groups such as phenyl naphtl ,yl or mesityl. The 2 position is preferably
s~ ~hstit~n~d by a C1-C4-alkyl group such as methyl or ethyl.
If the 2 and 4 positions are s~ ~hstihlted then
R5 and R5 are preferably ide"tical or difrerent and are each a C6-C10-aryl group a
C7-C10-arylalkyl group a C7-C40-alkylaryl group or a C8-C40- arylalkenyl group.
The following numb~ri,)g is used here for the substitution position:
5~
7 /R9
25 Of particular i,npG"ance are also metallocenes of the formula I in which the
substituents in the 4 and 5 positions of the indenyl radicals (R5 and R6 as well as
R5 and R6 ) tog~tl .er with the atoms cc"necting them form a ring system pre~erably
a six~"e"~bered ring. This condensed ring system can likewise be substituted by
radicals having the ,..eanings of R3 - R3. An example which may be mentioned of
30 such cGI"~ounds I is din.etl,ylsilanediylbis(2-methyl4 5-benzoindenyl)zirconium
dichloride.
Particular pr~fere"ce is given to those compounds of the formula I which bear a C6-
21 9331 9
-
C20-aryl group in the 4 positiGn and a C1-C4-alkyl group in the 2 position. An
example of such a cGn"~ound of the formula I is dimetl,ylsilanediylbis(2-methyl4-
phenylindenyl)zirconium dichloride.
5 Examples of the met~'locene cori"~onenl of the catalyst system of the invention are:
dimethylsilanediylbis(indenyl)zirconium di~l,loride
dimethylsilan~li~rlLis(4~aph~1,ylindenyl)zirconium dichloride
dimethylsilan~diylbis(2-methyl~"~oindenyl)zirconium dichloride
dimethylsilanediylbis(2-methy,indenyl)~irconium dichloride
10 dimethylsilan~i~lLis(2-methyl4-(1~,aphthyl)indenyl)~irconium dichloride
dimethylsilanediylbis(2-methyl4-(2-"aphthyl)indenyl)zirconium dichloride
dim~th~lsilanediylbis(2-methyl4-phenylindenyl)zirconium dichloride
dirnethylsila"~liylbis(2-methyl4-t-butylindenyl)zirconium dichloride
dimethylsilanediyll.is(2-methyl4-isol~ropylindenyl)zirconium dichlorid
15 dimethylsilan~diylbis(2-methyl-4-ethyli"denyl)~ircG"ium dichloride
dimethylsilanediylbis(2-methyl4ff-acena~htl,indenyl)zirconium dichloride
dimethyl ~ nediylLis(2 4-dimethylindenyl)zirconium dichloride
dimethylsilanediylLis(2-ethylindenyl)zirconium dichloride
dimethylsil~"ediylbis(2-ethyl4-ethylindenyl)zirconium di~,loride
20 dimethylsil~ lbis(2-ethyl4-phenylindenyl)~i,wnium dichloride
dimethylsilanedi~bis(2-methyl4 5-benzoindenyl)zirconium dichloride
dimethylsila"ediylbis(2-methyl4 6-diisopropylindenyl)~ ium dichloride
dimethylsilaneJi~lbis(2-methyl4 5-diisop,opylindenyl)zirconium dichloride
dil "etl ,ylsilanediylbis(2 4 6-l, i" ,e~l ,ylindenyl)~i, conium dichloride
25 dimethylsilanediylbis(2 5 6-t,i"~e~lIylindenyl)zirconium dichloride
dimethylsilanedi~lbis(2 4 7-trimethylindenyl)zirconium dichloride
dimethylsilanediylbis(2-methyl-5-isobutylindenyl)zirconium dichloride
dimethylsilanediylbis(2-methyl-5-t-butylindenyl)zirconium dichloride
dimethylsilanediylbis(2-methyl4-,c 1 ,enantl u ylindenyl)~irconium dichloride
30 dimethylsilanediylbis(2-ethyl4-phenanthrylindenyl)zirconium dichloride
methyl(phenyl)silanediylbis(2-methyl4-phenylindenyl)zirconiumdichlorid
methyl(phenyl)silanediylbis(2-methyl4 6-diisopropylindenyl)zirconium dichloride
methyl(phenyl)silanediylbis(2-methyl4-isopropylindenyl)zirconium dichloride
21 9331 9
-
methyl(phenyl)sila"edi~lbis(2-methyl-4 S-l,en~oindenyl)~i,conium dichloride
methyl(phenyl)silanediylbis(2-methyl-4 5-(methyll.e"~o)-indenyl)~irconium dichloride
methyl(phenyl)sil~,)edi~lbis(2-methyl4 5-(tetr~" ,eli ,ylL,er,~o)indenyl)zirco-
nium dichloride
methyl(phenyl)silanediylbis(2-methyl-4-a-acenaphtl ,i"clenyl)zirconium dichloride
methyl(phenyl)sila"eJ;ylL,is(2-methylindenyl)~irconilJm dichloride
methyl(phenyl)sila"eJiylbis(2-methyl-5-isobutylindenyl)zirconium dichloride
methyl(phenyl)silanedi~lbis(2-methyl4-pl)enarltl " yl;ndenyl)~irconium dichloride
methyl(phenyl)silanediylbis(2-ethyl4-phena, Ithlylindenyl)zirconium dichloride
1 2~th~n~i~1bis(2-methyl-4~henylindenyl)~ircon. ~m dichloride
1 4-bulanediylbis(2-methyl-4-phenylindenyl)zirconium dichloride
1 2 eti .aneJi~lbis(2-methyl-4 6-diisopropylindenyl)~irconium dichloride
1 4-butan~d;ylbis(2-methyl-4-isopr~pylindel "~ ir~n ~rn dichloride
1 4-butsinedi~rlbis(2-methyl-4 5-beri~oindenyl)~irconium dichloride
1 2~ti.~.)eJiylbis(2-methyl-4 5-benzoindenyl)zirconium dichloride
1 2~tl.a,leJiylbis(2 4 7-tri",eU,ylindenyl)zirconium dichloride
1 2~hanedi~1bis(2-methylindenyl)~irconium dichloride
1 4-butanedi~rlbis(2-methylindenyl)~irconium dichloride
bis(butylcyclo,.)entadienyl)Zr+CH2CHCHCH2B~(C6F5)3
bis(methylindenyl)Zr+CH2CHCHCH2B~(C6F5)3
dimethylsilal ,e Jiybis(2-methyl4,5-benzoindenyl)Zr+CH2CHCHCH2B~(C6F5)3
1 2~tl,anediylbis(2-methyli"denyl)Zr+CH2CHCHCH2B~(C6F5)3
1 ,4-butaned;~lL,is(2-methylindenyl)Zr+CH2CHCHCH2B~(C6F5)3
dimethylsilanediylbis(2-methyl-4 6~iisopropylindenyl)-
Zr+CH2CHCHcH2B (C6F5)3
dimethylsilaneJi~ll,is(2-ethyl-4-phenylindenyl)Zr+CH2CHCHCH2B~(C6F5)3
dimethylsilanediylbis(2-methyl-4-phenylindenyl)Zr+CH2CHCHCH2B~(C6F5)3
methyl(phenyl)silandiylbis(2-methyl-4-phenylindenyl)-
Zr+cH2cHcHcH2B (C6F5)3
dimethylsilanedi~rlbis(2-methylindenyl)Zr+CH2CHCHCH2B~(C6F5)3
dimethylsilanediylbis(indenyl)Zr+CH2CHCHCH2B~(C6F5)3
dimethylsilandiyl(tert-butylamido)(tetramethylcyclopentadienyl)-
zirconium dichloride
21 9331 9
[tris(p6, Itanuoro~l ,eny-l)(cyclopenladienylidene)L,orateo](cyclopenladienyl)-
1 2 3 4-tetra~l ,e"ylbuta-1 3-dienyl ~i, conium
dimethylsilanJiyl [tris(penldlluoropl)eny-l)(2-methyl-4-phenylindenylidene)-
boraleo](2-methyl4-phenylindenyl)-1 2 3 4-tetra~l,enylbuta-1 3-dienyl zirconium
5 dimethylsilandiyl-[tris(trifluoromethyl)(2-methylber,~indenylidene)-
borateo](2-methyl~,kindenyl-)-1 2 3 4-tet, apl ,eny-lbuta-1 3-dienyl~irconium
dimethylsilan~iyl [tris(pent~lluoropl)el,y-l)(2-methylindenylidene)boraleo]
(2-methylindenyl)-1 2,3 4-tetra,c l ,enylbuta-1 3-dienyl ~ircGI)ium
dimethylsila. ,eJiylbis(indenyl)dimethyl~ircG"ium
1 0 dimethylsilaneJiylbis(4-naphlhylindenyl)dimethylzirconium
dimethylsila"ediylbis(2-methylLe, I~Gil ,Jenyl)dimethykirconium
dimethylsilan~Jiylbis(2-methylindenyl)dimethyl~ircGnium
dimethylsilaned;ylbis(2-methyl-4-(1 -naphll ,yl)indenyl)di,nethykirconium
dimethylsilaneJiylbis(2-methyl-4-(2-nap ht hy-l)indenyl)dimeth-ykirco~ ,ium
15 dimethylsilanediylbis(2-methyl4-phenylindenyl)di",ell,ykirconium
dimethylsilan6Jiylbis(2-methyl-4-t-butylinJenyl)dimethykirconium
dimethylsilanedi~lbis(2-methyl-4-isopropylindenyl)dir"etl ,ykirconium
dimethylsilanediylbis(2-methyl-4-ethylindeny-l)dimethyl~irconium
dimethylsilaneJiylbis(2-methyl-4~-acena,chtl ,indenyl)dimethyl~i, cGnium
20 dimethylsilan~Jiylbis(2 4-di" ,etl ,ylindenyl)di" ,etl ,~kirconium
dimethylsilanediylbis(2-ethylindenyl)di" ,e~l ,yl~ir~nium
dimethylsilanediylbis(2-ethyl4-ethylindenyl)dimethyl~ir~nium
dimethylsilaneJiylbis(2-ethyl-4-phenylindenyl)dimethyl~i, conium
dimethylsilaned;ybis(2-methyl4 5-be"~oindenyl)dimetl,ykirconium
25 di",etl ,yl~ilanediylbis(2-methyl4 6-diisopropylindenyl)dil "eth~l~irconium
dimethylsilaneJiylbis(2-methyl45~iisopropylindenyl)di,netl,yl irconium
di. "etl ,ylsilanediylbis(2 4 6-t, i,netl ,ylindenyl)dimethykirconium
dimethylsilanediylbis(2 5 6-l,imetl,ylindenyl)dimethylzirconium
dimethylsilanediylbis(2 4 7-l, i."ethylindenyl)dimeth~l~irconium
30 dimethylsilaneJiylbis(2-methyl-5-isobutylindenyl)dirnethylzirconium
dimethylsilanediylbis(2-methyl-5-t-butylindenyl)dimethykirconium
dimethylsilanediylbis(2-methyl4-phenanlhl ylindenyl)dimethylzirconium
dimethylsilanediylbis(2-ethyl4-phe"anll ,rylindenyl)dimethykirconium
21 9331 9
methyl(phenyl)silan6Jiylbis(2-methyl4-phenylindenyl)dimethykir~"ium
methyl(phenyl)silanediylbis(2-methyl4 6- diisopropylinJenyl)dimethylzirconium
methyl(phenyl)silanediyll,is(2-methyl4-isopropyli. ,de,)yl)dimethyl~irco"ium
methyl(phenyl)silanedi~lbis(2-methyl-4 5-benzoindenyl)dimethykircol ,ium
5 methyl(phenyl)sila"eJ;~ll,is(2-methyl4 5-(methylbenzo)indenyl)dirnell,ykirconium
methyl(phenyl)silaneJiylbis(2-methyl-4 Sttet, ar"ell "~I~"~o)indenyl)-
dimethylzirconium
methyl(phenyl)sila"6Jiyll,is(2-methyl-4~-acenaphtl ,indenyl)d,. "ethyl~ir~. ,iummethyl(phenyl)silanediylbis(2-methylir,-Jenyl).li",ethykir~nium
10 methyl(phenyl)silane.Jiylbis(2-methyl-5-isobutylindenyl)dimethyki, conium
methyl(phenyl)silan6diylbis(2-methyl-4-phenanthrylindenyl)dimethyl- ~ircol ,ium
methyl(phenyl)silanediylbis(2-ethyl4-~1 ,enan U "ylindenyl)dimethyl~irconium
1 2~U,aneJiyll,is(2-methyl4-pheny,i. ,Jenyl)dimethykirconium
1 4-butan~ lbis(2-methyl4-phenylindenyl)dimethykirconi~m
1 2~th 2. aJ;ylbis(2-methyl4 6-diisop, opylindenyl)dimethyki, conium
1 4but ,~Ji~1L.is(2-methyl q iso~ropylindenyl)dimethyl~irconium
1 4-butan~iylbis(2-methyl4 5-benzoindenyl)dimethyl~irconium
1 2~U ,~n~liylL.is(2-methyl4 5-benzoindenyl)dimethykirconium
1 2~U,aneJi~lbis(2 4 7-~,i",etl"~lindenyl)dimethyl,irconium
20 1 2~U ,a"eJ;yll,is(2-methylindenyl)dimeU ,ykir~"ium
1 4-butaneJ;ylbis(2-methylindenyl)dimethykir~nium
Particular pr~ference is given to:
dimethylsil~nediylbis(2-methylinden~l)zirconium dichloride
25 dimethylsila"eJiylbis(2-methyl4-(1-naphthyl)indenyl)zirconium dichloride
dimethylsilanediylbis(2-methyl4-phenylindenyl)~irconium dichloride
dimethylsilanediylbis(2-methyl44~-acenaphthindenyl)zirconium dichloride
dimetl,ylsilanediylbis(2-ethyl4-phenylindenyl)zirconium dichloride
dimethylsilaned;ybis(2-methyl4 5-benzoindenyl)zirconium dichloride
30 dimethylsila"ediylbis(2-methyl4 6-diisop,opylindenyl)zirconium dichloride
di. "etl ,ylsilanediylbis(2-methyl4-phenanthrylindenyl)zirconium dichloride
dimethylsilanediylbis(2-ethyl4-pl)e"a, IU " ylindenyl)zirconium dichloride
2 1 933 1 ~
methyl(phenyl)silanediylbis(2-methyl4~henar,ll"ylindenyl)~ir~"ium dichloride
methyl(phenyl)silaneJiylbis(2-ethyl4-pl ,ena. ni llyli"denyl)~irconium dichloride
Methods of ~,r~paring metallocenes of the formula I are des~ibed for example in
S Joumal of O(gano",etallic Chem. 288 (1985) 63 - 67 and the documents cited
tl,erei.~.
The catalyst system of the invention preferably further comprises at least one
~ yst.
The cocatalyst c~mpGI ,ent which can according to the invention be present in the
catalyst system is at least one co",pound selscted from the group consisting of
aluminoAanes or Lewis acids or ionic co",pounds which react with a met~llocene to
convert the latter into a calionic co",pound.
Aluminoxanes which are preferably used are co",pounds of the formula ll
(R Al)n (Il)
Alumir,oxanes can for example be cyclic as in formula lll
--O A I
-- _
or linear as in formula IV
R\ R /R
A I--O A I O " A I ~ I V )
R/ \R
2 1 933 1 9
11
or of the cluster type as in formula V, as are desa ibed in recent literature; cf. JACS
117 (1995), 6485-74, OrganG",et~lics 13 (1994), 2957-2969.
I~
~ "~ t ~l )
1~
The radicals R in the formulae (Il), (Ill), (IV) and (V) can be identical or dirrerenl and
each be a C1-C20-hydlo~,bon group such as a C1-C6-alkyl group, a C6-C18-aryl
group, benzyl or hydl ogen, and p is an integer from 2 to 50, preferably from 10 to
35.
The radicals R are preferably identical and are methyl, isobutyl, n-butyl, phenyl or
benzyl, particularly prererably methyl.
If the radicals R are difreren~, they are prererably methyl and hydlogen, methyl and
isobutyl or methyl and n-butyl, with hyd~gen, isobutyl or n-butyl preferably being
pr~sent in a propG, lion of 0.01 - 40% (number of rrdic~ls R).
25 The aluminoxane can be prepared in various ways by known methods. One of the
melhod~ is, for exd"~ple, reacting an aluminum hydrocarbon compound and/or a
hydlidoaluminum hydlocarl,on cG",,,~ound with water (grseous, solid, liquid or bound
- for example as water of cryslr'li~lion) in an inert solvent (e.g. toluene). Toprepare an alum noxane having difrerent alkyl groups R, two difrerenl
30 trialkylaluminums (AIR3 + AIR'3) corresponding to the desired composition andreactivity are reacled with water (cf. S. Pasynkiewicz, Polyhedron 9 (1990) 429 and
EP-A 302 424).
2 1 933 1 ~
_
12
Regardless of the ~ tl)od of preparation, all aluminoxane solutions have in cGi"r.,on
a varying cGntent of unreacled aluminum slal ling cornpound which is present in free
form or as ~duct
5 As Lewis acid, pre~er~nce is given to using at least one Grganoboron or
or~,anoaluminum cGr"~ound containing C1-C20-groups such as L ranched or
unL,an.;l.~.J alkyl or haloalkyl, e.g. methyl, propyl, isopropyl, isobutyl and
trifluor~.,etl,yl or unsaturated groups such as aryl or haloaryl, e.g. phenyl, tolyl,
benzyl, p-fluorophenyl,
3,5-difluGru~Jhe"~l, ~.lta*.loropî,enyl, pentalluoropl,enyl, 3,4,5-trifluoropl,ei"~l and
3,5-di(trifluo~o.netl ,yl)phenyl.
Particular preference is given to o(ga.,oboron cG.."~ounds.
Examples of Lewis acids are trifluoroborane, triphenyll,orane,
15 tris(4-fluoro~l ,enyl)l,ora. .e, tris(3,5-difluoropl ,enyl)l,orane,
tris(4-fluor~..,etl .yl~hel ,yl)boral .e, tris(pentt~nuorophenyl)borane,
tris(tolyl)l,Gr~.,e, tris(3,5-dimethylphenyl)borane,
tris(3,5-difluoro~l .enyl)borane and/or tris(3,4,5-trifluoropl ,enyl)borane.
In particular, pr~ference is given to tris(penlalluoropl)enyl)bo~ne.
lonic co,~r'~lysts which are preferably used are cGmpounds containing a non-
coordinating anion, for example tetrakis(pentan.lorophenyl)borates,
tet~,cl .enylL,orales, SbF6-, CF3S03- or Cl04-. As cationic counterion, use is made of
Lewis bases such as methylamine, aniline, dimethylamine, diethylamine, N-
25 methylaniline, diphenylamine, N,N~i."ethylaniline, l,i",ethylamine, triethylamine, tri-
n-butyla"~ine, methyldiphenylamine, pyridine, p-bromo-N,N~i,.,etl,ylaniline, p-nitro-
N,N-dimethyla"iline, triethylphosphine, triphenylphos~l,ine, diphenylphospl)ine,tetrahydrothiophene and triphenyl~lbenium.
30 Examples of such ionic compounds according to the present invention are:
triethylal, .r. IGnium tetra(phenyl)borate,
tributyla,.".,oni ~m tetra(phenyl)borate,
l, i..,etl ,yla,nmonium tetra(tolyl)borate,
2193319
13
tributyla"",~onium tetra(tolyl)borale
tributyla"""onium tetra(~,enldlluoropl ,enyl)borate
tributyl8,."~0nium tetra(penldlluoropl ,ei .yl)aluminate
lripr~pyl~,..,.~onium tstra(dimethylphenyl)borate
5 tributyla" " "onium tetra(trifluoror"ell ,ylphenyl)borale
tributyl~r"",onium tetra(4-fluorophenyl)borale
N N-dimethylanilinium tetra(phenyl)borale
N N-diethylanilinium tetra(phenyl)borale
N N-dimethylanilinium tetrakis(pentafluorophenyl)borate
10 N N-dimethylanilinium tetrakis(pentdlluorophenyl)aluminate
di(propyl)ammonium tetrakis(,l~entdlluorophel~yl)borate
di(cyclol ,exyl)ammonium tetrakis(penlanuorophenyl)bor~le
t, i~l ,enylpl ,ospl onium tetrakis(phenyl)borate
triethyl~l os~l ,onium tetrakis(phenyl)bora~e
15 diphenylpl)ospl.o,nium tetrakis(phenyl)borate
tri(methyl~ "yl)pl .ospl .onium t~trakis(phenyl)borate
tri(dimethylphenyl)~l .os~l ,onium tetrakis(phenyl)borale
l, i~l ,enylc~- ~nium tetrakis(pe, tdlluorophenyl)borate
l ,enyl~, t,enium tetrakis(pentanuoropl)enyl)aluminate
20 b ipl .enyl~, Len. un tetrakis(phenyl)aluminate
fe"~c~nium tetrakis(penldlluorophenyl)borate and/or
fe"oeenium tetrakis(pentdiluoropl,e"yl)aluminate.
rre~ere"ce is given to lri,c I ,enylcarbenium tetrakis(pentafluorophenyl)borate and/or
N N-dimethylanilinium tetrakis(~,e, .tdlluo, o~l ,enyl)borate.
25 It is also possible to use mixtures of at least one Lewis acid and at least one ionic
cGI"pound.
Also illl~lldl)t as cocAI~lyst col"ponents are borane or ca,l,orane co""~ounds such
as 7 8-dicarbaunde~ rane(13)
30 undeca~,ydrido-7 8~ir"etl,yl-7 8-dicarbaunde~Ahorane
d~le¢~hydrido-1-phenyl-1 3-dicarbanonaborane
tri(butyl)a",-noniumundecahydrido-8-ethyl-7,9-dicarbaundec~horate,
4~rl,~nona~orane( 1 4)-bis(tri(butyl)a" ,r"onium) nonaborate
2193319
14
bis(tri(butyl)ammonium) u.,dec~l~orate
bis(tri(butyl)ammonium) d~JecAhorate
bis(tri(butyl)a..,..,onhm) decacl1lorodecaborale
tri(butyl)ammonium 1~,bAclecAhorates
5 tri(butyl)ammonium 1~.bAdoJe~horates
tri(butyl)ammonium 1 -b imdt h~lsilyl-1~, b~decAhorates
tri(buyl)ammonium bis(nonahydrido-1 3-dica. L,onnonal~rate)cob~t~te(lll)
Tri(butyl)ammonium bis(u..~ecal)ydrido-7 8-dicarbaunde-~horate)-fer,dle(lll).
10 The support co",ponenl of the catalyst system of the invention can be any Grga"i~
or inor~anic inert solid in particular a porous support such as talc inorganic oxides
and finely di~ided polymer powders (e.g. polyolefins).
Suitable inorganic oxides are those of elem6nts of groups 2 3 4 5 13 14 15 and
15 16 of the Periodic Table of the Elements. Examples of oxides prefer,ed as supports
include silicon dioxide aluminum oxide and also mixed oxides of the two ele,.,ents
and cor, e5PGI .ding oxide mixtures. Other inorganic oxides which can be used alone
or in combination with the last-named prefer, ed oxidic supports are for exampleMgO ZrO2 TiO2 or B2O3 to name only a few.
The support ,.,aterials used have a specir,c surface area in the range from 10 to
1000 m2/g a pore volume in the range from 0.1 to 5 ml/g and a mean particle size of
from 1 to 500 IJm. P~eference is given to supports having a specir,c surface area in
the range from 50 to 500 m2/g a pore volume in the range between 0.5 and 3.5 ml/g
25 and a mean particle size in the range from 5 to 350 um. Particular preferel)ce is
given to supports having a specific surface area in the range from 200 to 400 m2/g
a pore volume in the range from 0.8 to 3.0 ml/g and a mean particle size of from 10
to 200 ~m.
30 If the support material used naturally has a low moisture contenl or residual solvent
cGnlent dehydration or drying before use can be omitted. If this is not the case as
when using silica gel as support material dehydration or drying is advisable. The
weight loss on ignition (LOI) should be 1% or less. The thermal dehydration or
- -- 21 9331 9
drying of the support ",alerial can be carried out under rerluced pressure and
simultaneous ~lanl;eting with inert gas (e.g. nilrogen). The drying te"~peralure is in
the range between 100 and 1000C preferably between 200 and 800C. In this
case the pressure is not decisive. The duration of the drying process can be
5 between 1 and 24 hours. Shorter or longer drying times are possible provided that
under the cG-~-liliGns selected e~ librium can be established with the hydroxyl
groups on the support surface which normally requires betwocn 4 and 8 hours.
Dehyd~dliG., or drying of the support ",aterial can also be achieved by chemical10 metl l~ds by r~acting the adsG, Led water and the hydroxyl groups on the surface
with suitable s~ sl~)ces for making the surface inert. The reaclion with the reagent
for making the surface inert can convert the hydroxyl groups completely or partially
into a form which does not lead to any adverse inlera.tion with the catalytically
active cen~ers. Suitable s~ nces for making the surface inert are for example
15 silicon halides and silanes e.g. silicon teba~;l.loride chlorol,i"~ethylsilane
dimethylaminotrichlorosilane and o~anometallic cG"")ounds of aluminum boron
and "~s~ esium e.g. I,i",~ll"~laluminum triethylaluminum triisobutylaluminum
triethyll,orane dibutyl~"agnesium. The chemical dehydration or making inert of the
support ",at6(ial is carried out for example by reac~ing with exclusion of air and
20 moisture a suspension of the support material in a sl~-t~h!e solvent with the reagent
for makin~ the surface inert in pure form or dissolved in a suitable solvent. Suitable
solvents are for example alipha~ic or aro",atic hydrocarl.ons such as penlane
I ,exane heptane toluene or xylene. The reaclion for making the surface inert iscarried out at tel"peratures between 25 C and 120 C preferably between 50 and
25 70C. Higher and lower tel"peral-Jres are possible. The reaction time is between 30
minutes and 20 hours preferably from 1 to 5 hours. After the chemical dehydl alio"
has proceeded to completion the support material is isolated by filtration under inert
condiliGns, washed one or more times with suitable inert solvents as have been
desu ib~J above and finally dried in a stream of inert gas or under reduced
30 pressure.
Organic support materials such as finely divided polyolefin powders (e.g.
polyethylene polypropylene or polystyrene) can also be used and should likewise
219331~
16
be freed of a~ . ing moisture, residu~l solvent or other impurities by appropri~le
pUIifiC~tiGn and drying operatiG,.s before use.
To prepar~ the SlJ~JpGI led catalyst system, at least one of the above- des~ ibed
S mePIloc~.,e cG",ponenls is brought into contact with the coc~PIyst cG""~onenl in a
suitable solvent so as to obtain a soluble reaction product.
The soluble r eactiG" product is then added to the support material which has been
dehyJ~ ated or made inert, the solvent is removed and the resulting SUp,)GI l~d
."ct-'lcc~ catalyst system is dried to ensure that all or most of the solvent isremoved from the pores of the support material. The SUppGI led catalyst is ol,tained
as a froe flowing powder.
A process for prepdri,~ a free-flowing and, if desired, prepolym6ri~6.J SUppGI led
catalyst system comprises the following steps:
a) pre~J~IdtiGll of a preacti.lated met~llocene/cocA~A~yst mixture in a suitable
solvent, where the metallocene con"~onent has one of the structures
desu ibeJ above,
b) ~p~ ion of the preacli~ated met~llocene/co~lalyst solution to a porous,
generally inorganic dehydrated support,
c) removal of the major part of solvent from the resulting mixture,
d) isol-tion of the suppo. led catalyst system,
e) if desi~d, a prepoly",eri~alion of the resulting supported catalyst system
using one or more olefinic monomer(s), in order to obtain a prepoly",~ri~ed
SUppGI l~d catalyst system.
Plefer.ed solvents for the preparalion of the preactivated metallocene/coc~l~lyst
mixture are hydrocarl,ons and hydrocarL,on mixtures which are liquid at the selected
- - 21 q331 9
17
reaction te",~rat.lre and in which the individual co,nponents preferably dissolve.
However the solubility of the individual COrnpGI ,enls is not a prerequisite if it is
ensured that the rea-tiGn product of met~llocene and co~l~lyst COmpGi ,ent is
soluble in the sale~t6J solvent. Examples of suitable solvents include alkanes such
aspenlane isopenta"e l,exane hep~ane octaneandnG,)ane;cycloalkanessuch
as cyclopenlane and cyclohexane; and arol"dtics such as ber,~el,e toluene
ethyl~n~ne and diethyll~"~ene. It is also possible to use halogel)ate-i
hydroca,L.ons such as methylene chloride or ar~",atics such as fluorobe"~ene or o-
dichlor~ , .e. Very particular preference is given to toluene.
The amounts of alu."ino~cane and metallocene used in the preparalion of the
suppGIt~d catalyst system can be varied over a wide range. r~ference is given tousing a molar ratio of aluminum to the transition metal in the metallocene of from
10:1 to 1000:1 very particularly prererably a ratio of from 50:1 to 500:1.
In the case of methylaluminoxane preferel)ce is given to using 30% sll en~tl,
solutions in toluene but the use of 10% strength solutions is also possi~le.
For the preacli~ation the metallocene in the form of a solid is dissolved in a solution
of the aluminoxane in a suit~'e solvent. It is also possible to dissolve the
",et~'l~ne separalely in a suitable solvent and s~hse~uently to col,lbil,e this
solution with the aluminoxane solution. rleference is given to using toluene.
EP-A 0302424 des~ ibes the preacti-/ation of metallocenes for use in unsupportedform in poly",eri~tiol,s. The preactivation time is from 5 minutes to 100 hours
preferably from 5 to 60 minutes in particular from 10 to 20 minutes. A significantly
longer prea~ /ation is possible but it normally neither increases the activity nor
Je~eases the activity although it can be quite useful for storage pu".oses. Theprea~i-/ation is carried out at a temperalure of from -78 to 1 00C preferal)ly from 0
to 70C.
Sul ~risi, Igly it has now been found that in the case of sterically demanding
metallocenes such as rac-dimethylsilanediylbis(2-methyl4-phenylindenyl)zirconiumdichloride a distinctly longer preactivation time leads to a significant increase in
21 933 1 9
.
18
activity. In the case of sterically demanding met~locenes the preactivation time is
pr~fo, aL,ly from 30 minutes to 200 hours in particular from 1 to 50 hours especi~'ly
from 5 to 20 hours.
5 The preacti~ation can take place at room te,nperal-Jre (25 C). The use of higher
te",~eratures can in some cases shG, ~n the time necess~y for preactivation and
effect an ~ tional inc(ease in activity. In this case Uhigher tel"peraluren means a
ran~e between 50 and 1 00C. Even at higher temperatures the prefer,ed
,~,rea~ti~ation time in the case of the metr"ccenes prefer,ed accordi,1g to the
10 invention is d;slinctly above the preacti.ration time known from the prior art.
The preacti-/ated solution is s~hse~uently combined with an inert support ",ale,ial
usually silica gel which is in the form of a dry powder or as a suspe"sio" in one of
the abov~",enlioned solvents. The powder used is preferably silica gel. Any order of
15 addition can be used. The preactivated met~llocene/coc~'AIyst solution can be the
support ",alerial or else the support ",aterial can be introd~Ged into the solution.
The volume of the preacli~ated solution can exceed 100% of the total pore volumeof the support ",alerial used or else can be up to 100% of the total pore volume.
20 The te",perat~lre at which the preactivated solution is brought into cGnlac~ with the
support ",alerial can vary in the range between 0 and 1 00C. However lower or
higher temperat.lres are also possible. After co",b..)ing the support material and
solution the mixture is maintained at this te",peral-Jre for a further period of from
about 1 minute to 1 hour prereral,ly 5 minutes.
All or most of the solvent is suhse~uently removed from the suppol led catalyst
system; for this procedure the mixture can be stirred and if desired also heated.
rl~erably both the visible propo, lion of solvent and also the solvent in the pores of
the support material are removed. The removal of the solvent can be carried out in a
30 conventional ",a"ner using reduced pressure and/or flushing with inert gas. During
the drying procedure the mixture can be heated until the free solvent has been
removed which usually takes from 1 to 3 hours at a telnperalure which is pre~erably
selected so as to be between 30 and 60C. The free solvent is the visible proportion
21 93319
19
of solvent in the mixture. For the pl" ~,oses of the present invention, residual solvent
is the propG, liGI ~ which is ~, Iclosed in the pores. As an allel "ali~/e to complete
removal of the solvent, the SUIJpGl led catalyst system can also be dried only to a
certain r~sid~' solvent contenl, with the free solvent having been completely
5 removed. The SU,upGI led catalyst system can subsequently be washed with a low-
boiling hyd~ l l,on such as penlane or I ,exane and dried again.
The SU~pGI t~i catalyst system prepared according to the invention can either beused directly for the poly-"eri~-dlion of olefins or be prepoly"~eri~ed with one or more
10 olefinic ~G~GI~ers before use in a poly..,eri~dlion process. The procedure for
prepoly..~ri~atio.. of suppG-led catalyst systems is described, for example, in
WO 94/28034.
The prese. It invention also provides a process for preparing a polyolefin by
polymeri~tion of one or more olefins in the presence of the catalyst system of the
invention colr~prisin~ at least one l-~nsilion metal col.,pG.,ent of the formula 1. For
the pu.~oses of the presenl invention, the term poly",eri~3tion refers to either a
l,G",opoly"~eri~dtion or a copoly",eri~alion.
Preference is given to polymerizing olefins of the formula Rm-CH=CH-Rn, where Rmand Rn are iJ6ntical or Jif~renl and are each a h~JI ogen atom or a carL,on-
containin~ radical having from 1 to 20 carbon atoms, in particular from 1 to 10
cal bGI I atoms and Rm and Rn to~ether with the atoms connectin~ them can form one
or more rings. Examples of such olefins are 1-olefins having 2 - 40, preferably from
2 to 10 ca. I,Gn atoms, for example ethene, p~pene, 1 -butena, 1 -pentene, 1 -hexene,
4-methyl-1-pentene or 1-octene, styrene, dienes such as 1,3-but~diene,
1 ,4-hexadiene, vinylnorbG. "ene, no, bG" ,adiene, ethylnorbor"adiene and cyclicolefins such as no, bor"ene, tetracyclododecene or methylnorbornene. In the
p~cess of the invention, preference is given to ho",Gpoly")eri~i"g ethene or
~ rop~ne, or copoly",eri~in~ ethene with one or more 1-olefins having from 3 to 20
carbon atoms, e.g. propene, and/or one or more dienes having from 4 to 20 carbonatoms, e.g. 1,4-butadiene, nGrbG",adiene or ethylno,l,Grnadiene. Examples of such
copolymers are ethene-propene-copolymers or ethene-propene-1,4-hexadiene
2 1 933 1 9
terpolymers.
The poly",eri~atiG" is ca"ieJ out at a te",perature of from ~0 to 300 C preferably
from 50 to 200C. The pressure is from 0.5 to 2000 bar pre~erably from 5 to 64 bar.
The poly",eri~dtion can be carried out in solution in bulk in suspension or in the
gas phase continuously or batchwise in one or more stages.
The ca~alyst system prepared according to the invention can be used as sole
10 catalyst CGlllpGI ,ent for the polymeri~a~iGn of olefins having from 2 to 20 ca, l,on
atoms or pre~erably in combi. IdtiG, I with an aluminum alkyl or an alu",inoxane. The
soluble aluminum celllpGnent is added to the ,nonomer and serves to free the
mG,)G,ner of s~ lA~s which can impair the catalyst activity. The amount of
aluminum cefi,pon6nt added .lepe"ds on the quality of the ",onor"el:i used.
If, .ec~ss~ hyJ~ o9el l can be added as mcl~c~ weight regul~'or and/or to
inerease the activity.
The invention is illusba~ed by the following exa",ples.
Examples
General
The preparation and handling of the organGI"etallic cG",pounds were carried out
with excl- ~siQn of air and moisture under argon (Schlenk technique). All necess~ry
solvents were freed of air and moisture before use by boiling for a number of nours
over a suitable desiccant and s~ ~hse~uent distillation under argon.
The met~locenes used were charactel i~ed by 1 H-NMR 1 3C-NMR and IR
spe-;t~oscopy.
2193319
21
D~rl,~ilions;
PP = polypropylene
MC = ",et~'lc~ne
cat = SUppG~ catalyst system
h= hour
VN = viscosity number in cm3/g
Mw = weight average molar mass in g/mol
M~JMn = molar mass distribution deter",ined by gel per",edliGn chromatoy,d~l,y
BD = bulk density in g/dm3 and
mp. = ",elt;ng point in C deter"~ined by dirrere,ltial scanning calorimetry
(DSC 2ndl,eatiny)
2193319
22
Example 1
Prepa~tion of the SU~JpGI led catalyst system
444 mg (0.71 mmol) of rac~li",etl,ylsilanediylbis(2-methyl4-phenylindenyl)~i,~i,ium
dichloride were dissolved at room te",perature in 35.5 ml (128 mmol of Al) of 30%
st,e,.yth methylaluminoxane solution in toluene1). For the prea.;ti~ation the mixture
was allowed to stand at 25C for 18 h while being prote~;ted from light. The
mePIlocene/MAO solution thus prepareJ was s~ ~hse~uently diluted with 86.9 ml oftoluene to a total volume of 122.4 ml. 30.6 9 of SiO22) were slowly introd~ ~ced into
this solution. The ratio of the volume of the solution to the total pore volume of the
support material was 2.5. After ~ddi~i~n was cG"~plete the mixture was stirred for 5
minutes at room ~e",perat.lre. Subsegl~ently the mixture was evaporaled to dryness
under red~ pressure at 40C over a period of 2 h and the residue was dried for 5h at 25C and 10~3 mbar. This gave 39.5 9 of a free-flowing pink powder which
according to ~le."enlal analysis conlained 0.15% by weight of Zr and 8.0% by
weight of Al.
Poly" ,~ri~dtion
A dry 16 dm3 reactor which had been flushed first with nil,ogen and s~lhse~uently
with pr~p~ne was filled with 10 L of liquid propene. 8 ml of 20% strength
triethylaluminum sol! ~tion in Varsol (Witco) were added as scavenger and the
mixture was stirred for 15 minutes at 30 C. S! ~hse~uently a suspension of 1.2 9 of
the su~ J ",et~llocene catalyst in 20 ml of a dearomatized petroleum fraction
having a boiling point range from 100C to 120C was added to the reactor the
mixture was heated to the poly"~eri~alion temperature of 65 C and the
poly",eri~ation system was held at 65C for 1 h. The poly",eri~ation was sl~pped by
addition of 20 ml of isGpropanol the excess monomer was vented and the polymer
obtained was dried under reduce~ pressure. This resulted in 2.4 kg of polypropylene
powder.
The catalyst activity was 193 kg of PP/(g of MC x h) or 2 kg PP/(g of cat x h)
2193319
23
The isot~tic polypropylene prepared had the following prope"ies:
mp. 147C; Mw = 901,000 g/mol, MW/Mn = 2.3, VN = 603 ml/g, BD = 370 g/dm3
5 CG",parati~e Example 1
r, epar~tion of the S~lppGI le.J catalyst system
460 mg (0.73 mmol) of rac-dimethylsilanediylbis(2-methyl-4-
10 phenylin~l~"yl)~i,cGnium dichloride were dissolved at room te",pera~ure in 36.8 ml
(133 mmol of Al) of 30% sll enytl l methylaluminoxane solution in toluene1). Themixture was diluted with 93 ml of toluene and stirred for 10 min at 25C. 33.2 9 of
SiO22) were slowly introdt~ced into this solution. The ratio of the volume of the
solution to the tobl pore volume of the support ",alerial was 2.5. After addition was
15 complete, the mixture was stirred for 5 minutes at room tei"perature. S! Ihseg~ ~ently,
the mixture was evaporaled to dryness under reduced pressure at 40C over a
period of 2 h and the residue was dried for 5 h at 25C at 10~3 mbar. This gave 44 9
of a free-flowing, pink powder which, according to ele,~enl31 analysis, contained
0.16% by weight of Zr and 8.0% by weight of Al.
Poly",eri~ation
The poly",eri~atiGn was carried out using a Ille~llod similar to Example 1. Thisresulted in 1.4 kg of polypropylene powder.
The catalyst activity was 109 kg of PP/(g of MC x h) or 1.2 kg PP/(g of cat x h)
The isot~ctic polypropylene prepared had the following properties:
mp. 146C; Mw = 1,010,000 g/mol, MW/Mn = 2.3, VN = 643 mllg, BD = 380 g/dm3
1) AlL,e",arle CGr~,oration, Baton Rouge, l oui~i^na, USA
2) Silica grade MS 948, W.R. Grace, Davison Chemical Division, Baltimore,
Maryland, USA, pore volume 1.6 ml/g, calcined at 800 C.
2193319
.
24
Example 2
r, eparaliG" of the SUppG~ ~ed catalyst system
198 mg (0.31 mmol) of rac~imethylsilanediylbis(2-methyl4-phenylindenyl)~ircG"iumdichloride were ~issolvcd at room te""~erature in 17.4 ml (68 mmol of Al) of 30%st~r,.Jtl, methylalumir,oxane sol~ ~tion in toluene3). For the preacti~/ation the mixture
was allow~d to stand at 25C for 18 h while being prot~ct~d from light. The
met^'lcc~ne/MAO solution was s~hse~uently diluted with 44.6 ml of toluene to total
volume of 62 ml. 15.5 9 of sio24) were slowly intro~uce~ into this solution. The ratio
of the volume of the soMtion to the total pore volume of the support material was
2.5. After A-l~lition was complete the mixture was stirred for 5 minutes at roomt6",~rature. S! Ihse~l ~ently the mixture was evaporaled to dryness under re~luce~l
pressure at 40C over a period of 2 h and the residue was dried for 5 h at 25 C and
10~3 mbar. This gave 20.2 9 of a free-flowing pink powder which accord;ng to
cle."ental analysis contained 0.12% by weight of Zr and 7.9% by weight of Al.
Poly",~ ation
The poly",6ri~tion was carried out under the same conditions as in Example 1 butusing 1.6 9 of catalyst and 8 ml of 20% stren!Jtl, triethylaluminum in Varsol (Witco).
This res~lte~ in 1.7 kg of polypropylene powder.
The catalyst activity was 128 kg of PP/(g of MC x h) or 1.06 kg of PP/(g of cat x h)
Th~ isot~ctic polypropylene prepared had the following properties:
mp. 148 C; Mw = 1.300.000 g/mol M~,JMn = 2.8 VN = 762 ml/g BD = 290 g/dm3
Co""~arati~e Example 2
PreparatiG" of the supported catalyst system
203 mg (0.32 mmol) of rac-dimethylsilanediylbis(2-methyl4-phenylindenyl)~i,cGnium
2 1 933 1 9
dichloride were d;ssolvcd at room te",peralure in 17.8 ml (70 mmol of Al) of 30%ab~rl!Jtll methylalumir,oxane solution in toluene3). The mixture was diluted with 45
ml of toluene and stirred for 10 minutes at room te",perat, re.15.7 9 of SiO2~) were
slowly intro~l~c~ into this solution. The ratio of the volume of the solution to the
total pore volume of the support material was 2.5. After addition was complete, the
mixture was stirred for 5 minutes at room te",peral,Jre. Subsequently, the mixture
was evaporated to dryness under red~ ~ced pressure at 40 C over a t,eriod of 2 h
and the residue was dried for 5h at 25C and 10-3 mbar. This gave 20.0 9 of a free-
flowing, pink powder which, according to elen,enlal analysis, contained 0.12% byweight of Zr and 8.0% by weight of Al.
Poly"~e, i~lion
The poly.,~eri~tion was car- ied out under the same conditions as in Example 1, but
using 2.2 9 of catalyst and 8 ml of 20% strength triethylaluminum in Varsol (Witco).
This res~ ~lted in 0.8 kg of polypropylene powder.
The catalyst activity was 60 kg of PP/(g of MC x h) or 0.5 kg of PP/(g of cat x h)
The iSQt~CtiC polypropylene prepared had the following prope, lies:
mp. 148C; MW = 1,300,000 g/mol, MW/Mn = 3.0, VN =698 mllg, BD = 310 g/dm3
3) Witco GmbH, Bergkamen, Federal Republic of Germany
4) Silica grade MS 948, W.R. Grace, Davison Chemical Division, Baltimore,
Maryland, USA, pore volume 1.6 mllg, calcined at 600C
Exa",ple 3
r, eparatiGn of the SUppOI led catalyst system
23 mg (0.040 mmol) of rac-di",etl,ylsilanediylbis(2-methyl4,5-
ben7r..)denyl)~ir~i)ium dichloride were dissolved in 1.8 ml (7.1 mmol of Al) of 30%
-~;tlt:lnJtll methylaluminoxane solution in toluene3) and the mixture was allowed to
21 9331 9
26
stand at 25C for 18 h while being prote~;ted from light. S~ ~hseg! ~ently the
metallocene/MA0 solution thus prepared was diluted with toluene to a total volume
of 4 ml. 6.5 9 of PP powder5) were then slowly introd~ ~ce~ into this solution. A
vacuum (0.1 mbar) was briefly applied to remove the gas prese"l in the pores of the
support and the solution was thus completely soaked up. After slir~ing inlensively for
a further 10 minutes a l,G",ogeneous finely divided and free-flowing powder was
obtained.
Poly",eri~ation
The poly",eri~ation was carried out under the same conditions as in Example 1 but
using the total amount of catalyst powder from Example 3 and 10 ml of 20% slren.Jtl,
triisobutylaluminum in Varsol (Witco). This resulted in 2.5 kg of polypropylene
powder.
The ~talyst activity was 110 kg of PP/(g of MC x h)
The isot~-~1ic polypropylene prepared had the following properties:
mp. 146 C; Mw = 350.000 g/mol MWJMn = 2.2 VN = 250 ml/g BD = 280 g/dm3
5) AccurellD -PP (sieve h~-;tion ~ 200 ,um) from Akzo, freed of impurities by
exl,detion with toluene in a Soxhlet exl, actor under inert conditions s~ ~hse~uently
dried for 5 h at 25C and 2 x 104 mbar and flushed with argon
CG"~parati~e Exslllple 3
Preparation of the su~,po, led catalyst system
23 mg (0.040 mmol) of rac-d;metl ,~lsilanediylbis(2-methyl4 5-
benzoindenyl)~irconium dichloride were dissolved in 1.8 ml (7.1 mmol of Al) of 30%
-~l, er,!Jth methylaluminoxa"e solution in toluene3) and diluted with toluene to a total
volume of 4 ml. This mixture was stirred for 10 minutes at 25C. 6.5 g of PP
powder5) were slowly introduced into the metallocene/MA0 solution thus prepared.
- 2193319
27
A vacuum (0.1 mbar) was briefly applied to remove the gas present in the pores of
the support and the solution was thus completely soaked up. After stirring
intensively for a further 10 minutes, a hG"~Ggeneous, finely divided and free-flowing
powder was ol)tained.
Poly,Y)eri~ation
The polyll,eri~dliG" was csr.ied out under the same conditions as in Exa",ple 1, but
using the total amount of the catalyst powder from Col"parali~e Exal.,~le 3 and 10
ml of 20% st~n~tl) triisobutylaluminum in Varsol (Witco). This resulted in 1.8 kg of
polypropylene powder.
The catalyst activity was 78 kg of PP/(g of MC x h)
The i50~iC polypropylene prepared had the following properties:
mp. 146C; Mw = 330.000 g/mol, Mw/Mn = 2.3, VN = 244 ml/g, BD = 290 g/dm3
5) Accurell -PP (sieve f~ dCliOn < 200 ,um) from Akzo, freed of impurities by
e~ ~-,tion with toluene in a Soxhlet e,~l, actor under inert conditions, sl ~hse~uently
dried for 5 h at 25 C and 2 x 10~ mbar and flushed with argon
Example 4
rl eparalion of the suppo-led catalyst system
200 mg (0.32 mmol) of rac~;,netl,~lsilanediylbis(2-methyl4-phenylindenyl)zirconium
dichloride were dissolved at room temperature in 17.6 ml (69 mmol of Al) of 30%
sl,eny~l, methylalumir,oxa,)e solution in toluene3). For the preactivation, the mixture
was allowed to stand at 25C for 18 h while being protected from light. The
30 metallocene/MAO solution thus prepared was s~lhse~uently diluted with 103 ml of
toluene to a total volume of 120.6 ml. 16.1 9 of SiO26) were slowly introduced into
this solution. The ratio of the volume of the solution to the total pore volume of the
support male,ial was 2.5. After addition was complete, the mixture was stirred for 5
- 21 9331 9
28
minutes at room te")perature. Subsequently, the mixture was evaporaled to dryness
under redl ~c~ pressure at 40 C over a period of 2 h and the residue was dried for
5 h at 25C and 10~3 mbar. This gave 21.1 9 of a free-flowing, pink powder which,
according to elefi,en~al analysis, contained 0.14% by weight of Zr and 8.6% by
5 weight of Al.
6) Grade MS 3030 manuf^~tllred by PQ Cor~ oration, Valley Forge, Pennsylvania,
USA, pore volume: 3 ml/g, calcined at 300 C
10 Poly"~e,i~lion
The polyn,eri~tio" was cs"ied out under the same conditions as in Example 1, butusing 1.6 9 of catalyst and 8 ml of 20% triethylaluminum in Varsol (Witco). Thisresulted in 1.3 kg of polypropylene powder.
The catalyst activity was 83 kg of PP/(g of MC x h) or 0.8 kg of PP/(g of cat x h)
The isot~ic polypropylene pr~parad had the following prope, lies:
mp. 147C; Mw = 910,000 g/mol, MW/Mn = 2.3, VN = 610 ml/g, BD = 380 g/dm3
Co"",a(ali~e Exsr"ple 4
rl apar~tion of the sL~ Jo~ led catalyst system
198 mg (0.31 mmol) of rac-dimethylsilanediylbis(2-methyl4-phenylindenyl)zirconium
dichloride were dissolved at room temperature in 17.6 ml (69 mmol of Al) of 30%
s~ra"5Jtl, methylal~""i.,o~ane solution in toluene3) The mixture was diluted witn 106
ml of toluene and stirred for 10 minutes at 25C.16.5 9 of SiO26) were slowly
30 introduced into this solution. The ratio of the volume of the solution to the total pore
volume of the support material was 2.5. After addition was complete, the mixturewas stirred for 5 minutes at room te",,l~eral.lre. Subsequently, the mixture wasevaporated to dryness under red~ ~ced pressure at 40C over a period of 2 h and the
2193319
29
residue was dried for 5 h at 25C and 10-3 mbar. This gave 21.3 9 of a free-flowing
pink powder which according to ele",ental analysis contained 0.13 % by weight ofZr and 8.7% by weight of Al.
Poly",eri~alion
The polyn,ericalion was ca"ied out using a method similar to Example 1 but using1.6 9 of the catalyst from CG",,,~ara~i~e Example 4. This resulted in 0.8 kg of
polypropylene powder.
The catalyst activity was 56 kg of PP/(g of MC x h) or 0.5 kg of PP/(g of cat x h)
The isol~ic polypropylene prepared had the following prope, lies:
mp. 147C; Mw = 1 000 000 g/mol MW/Mn = 2.2 VN = 640 ml/g BD = 390 g/dm3
Example 5
r~pa~atiG" of the suppo, led catalyst system
109 mg (0.17 mmol) of rac4i",ethylsilanediylbis(2-methyl4-phenylindenyl)~irconium
dichloride were dissolved in 9.7 ml (38 mmol of Al) of 30% sllenylil
methylalumir,oxane solution in toluene3) and the mixture was stirred for 5 h at 60C.
The metallocene/MAO solution was s~ ~hsequently cooled to 25C and diluted with
55.6 ml of toluene to a total volume of 65.3 ml. 8.7 9 of sio27) were slowly
introduced into the solution thus prepared. The ratio of the volume of the solution to
the total pore volume of the support material was 2.5. After addition was complete
the mixture was stirred for 5 minutes at room temperalure. Subsequently the
mixture was evaporaled to dryness under reduced pressure at 40 C over a period
of 2 h and the residue was dried for 5 h at 25C and 10-3 mbar. This gave 12.8 g of
a free-flowing pink powder which according to ele,nental analysis contained 0.12%
by weight or Zr and 8.0% by weight of Al.
21933t9
7) Grade MS 3030 manuf~ctl~red by PQ CGr~,oralion, Valley Forge, Pennsylvania,
USA, pore volume: 3 ml/g, calcined at 400 C
Poly",~, i~alion
The poly",6ri~aliG" was cslli~J out under the same conditions as in Example 1, but
using 1.6 9 of the catalyst from Example 5 and 8 ml of 20% triethylaluminum in
Varsol (Witco). This res~ ~ted in 1.8 kg of polypropylene powder.
The catalyst activity was 133 kg of PP/(g of MC x h) or 1.1 kg of PP/(g of cat x h)
The isot~ctic polypropylene prepared had the following prope, lies:
mp. 148C, Mw = 1,200,000 g/mol; MW/Mn = 2.4; VN = 680 ml/g, BD = 390 gldm3
Co."pdr~ti./e Example 5
rreparalion of the suppo. leJ catalyst system
123 mg (0.20 mmol) of rac-dimethylsilanediylbis(2-methyl-4-phenylindenyl)~irconium
dichloride were dissolved in 10.9 ml (43 mmol of Al) of a 30% strength
methylaluminoxane solution in toluene3) and the mixture was stirred for 5 h at 25C.
The ",dl^'lccene/MAO solution was s~ ~hse~uently diluted with 65 ml of toluene to a
total volume of 75.9 ml. 10.1 9 of sio27) were slowly introduGed into the solution
thus pre~ared. The ratio of the volume of the solution to the total pore volume of the
support ",ale,ial was 2.5. After addi~iG" was complete, the mixture was stirred for a
further 5 minutes at 25C. Sl ~hseguently, the mixture was evaporaled to drynessunder re~hlced pressure at 40C over a period of 2 h and the residue was dried for
5 h at 25C and 10~3 mbar. This gave 14.8 9 of a free-flowing, pink powder which,
according to ele."ental analysis, contained 0.12% by weight of Zr and 7.8% by
weight of Al.
21 93319
31
Poly" ,~ri~dtiG, ~
The poly-.,eri~alion was carried out using a ",eU,od similar to Example 1 but using
1.6 9 of the catalyst from Col"parali~/e Example 5. This resulted in 1.1 kg of
5 polypropylene powder.
The catalyst activity was 85 kg of PP/(g of MC x h) or 0.7 kg of PP/(g of cat x h)
The isQl^clic polypropylene prepareJ had the following propel lies:
mp. 149C; Mw = 1 250 000 g/mol MW/Mn = 2.3 VN = 678 mllg BD = 380 g/dm3
The chara~t6ri~tic data of the respective catalyst systems and the polymers
obtained measured in the Exampl~s and Co",parali~/e Examples 1 to 5 are to
15 provide a better overview sullllllali~ed in Tables 1 and 2 below.
32 21 933 1 9
Table 1
Catalyst system
E~)C#) Metallocene T i m e T e m p . %Zr %AI Acta)
lminl rcl
E1 I*) 1080 25 0.15 8.0 193
C1 I*) 10 25 0.16 8.0 109
E2 I*) 1080 25 0.12 7.9 128
C2 I*) 10 25 0.12 8.0 60
E3 Il**) 1080 25 n.d. n.b. 110
C3 Il**) 10 25 n.d. n.b. 78
E4 I~) 1080 25 0.14 8.6 83
C4 I*) 10 25 0.13 8.7 56
E5 I*) 300 60 0.12 8.0 133
C5 I*) 300 25 0.12 7.8 85
15 +) Examples
#) C~.""~ara~i~/e examples
a) Catalyst activity in kg of PP/(g of MC x h)
*) rac~imethylsilanediylbis(2-methyl4-phenylindenyl)zirconium dichloride
**) rac~imethylsilanediylbis(2-methyl-4,5-benzoindenyl)zirconium dichloride
Table 2
Polymers
E~l C#) mp. Mw Mw/Mn VN BD
r~ [gmol] lmlg1l[gldm3]
E1 147901,000 2.3 603 370
C1 1461,010,000 2.3 643 380
E2 1481,300,000 2.8 762 290
C2 1481,300,000 3.0 698 310
E3 146350,000 2.2 250 280
C3 146330,000 2.3 244 290
E4 147910,000 2.3 610 380
C4 1471,000,000 2.2 640 390
E5 1481,200,000 2.4 680 390
C5 1491,250,000 2.3 678 380