Note: Descriptions are shown in the official language in which they were submitted.
- 21 ~4561
Technetium-Sulfonamide Complexes, Their Use,
Pharmaceutical Agents Containing the Latter,
as well as Process for the Production of the Complexes and Agents
The invention relates to the object characterized in the
claims, i.e., chelating agents containing new sulfonamide groups,
their metal complexes, pharmaceutical agents containing these
compounds, their use in radiodiagnosis and radiotherapy, process
for the production of these compounds and agents, as well as
conjugates of these compounds with substances, especially
peptides, selectively accumulating in diseased tissue.
The use of radiopharmaceutical agents for diagnostic and
therapeutic purposes has long been known in the area of
biological and medical research. In particular,
radiopharmaceutical agents are used to visualize certain
structures such as, for example, the skeleton, organs or tissues.
The diagnostic application requires the use of such radioactive
agents, which, after administration, accumulate specifically in
the structures in patients which are to be examined. These
locally accumulating radioactive agents can then be traced,
plotted or scintiscanned with suitable detectors, such as, for
example, scintillation cameras or other suitable imaging
processes. The distribution and relative intensity of the
detected radioactive agent identifies the point of a structure at
which the radioactive agent is present and can display the
presence of anomalies in structure and function, pathological
changes, etc. In a similar way, radiopharmaceutical agents can
2 2194561
be used as therapeutic agents to irradiate specific pathological
tissues or areas. Such treatment requires the production of
radioactive therapeutic agents, which accumulate in certain
structures, tissues or organs. By concentration of these agents,
the therapeutic radiation is brought directly to the pathological
tissue.
Generally, metallic radionuclides are used as diagnostic
agents or therapeutic agents, whereby the metal can be present in
free form, as ions or in the form of a metal complex. Examples
of metallic radionuclides, which can form complexes, are
technetium-99m and various rhenium isotopes. The former is used
in diagnosis, the latter in therapy. Besides the metal
(complex), in general the radiopharmaceutical agents contain
additional suitable vehicles and additives, which allow an
injection, inhalation or ingestion by the patient, such as, e.g.,
physiological buffers, salts, etc.
The radionuclide most often used for nuclear-medicine
problems is technetium-99m, which is especially well-suited as
radioisotope for in vivo diagnosis because of its advantageous
physical properties (no corpuscular radiation, 6 hours of
physical half-life, 140 KeV of gamma radiation) and the small
radiation exposure following from it. Technetium-99m can be
obtained problem-free from nuclide generators as pertechnetate
and can be used in this form directly for the produc~ion of kits
for routine clinical demand.
The production of radiopharmaceutical agents first requires
the synthesis of a suitable ligand. In clinical practice, the
2 1 945~ 1
complex is then produced immediately before use from the
respective complexing agent (also named ligand or chelating agent
below) and the desired radionuclide ~labeling). In this respect,
the complexing agent, which is already present in the form of a
freeze-dried kit, is reacted under conditions of complex
formation with a solution containing the radionuclide. If, for
example, the production of technetium-99m radiopharmaceutical
agent is desired, the ligand produced with addition of a suitable
reducing agent is mixed with a pertechnetate solution, and the
corresponding technetium complex is produced under suitable
reaction conditions. These complexes are then administered to
the patient in a suitable way by injection, inhalation or
ingestion.
The solution containing the radionuclide can, as in the case
of technetium-99m, be obtained from a commercially available Mo-
99/Tc-99m nuclide generator, or -- as in the case of rhenium-
186 -- be obtained directly from a manufacturer. The complex-
formation reaction is performed under suitable temperatures
(e.g., 20~-100~C) within a few minutes to several hours. To
assure a complete complex formation, a larger excess (more than
100 times the excess) of the ligand produced and an amount of
reducing agent (e.g., SnCl2, S204, etc.) sufficient for a complete
reduction of the radionuclide used are necessary.
Since technetium can be present in a series of oxidation
stages (+7 to -1), which can greatly change the pharmacological
properties by changes of the charge of a complex, it is necessary
to provide chelating agents that can bond the technetium in a
4 2194561
stable manner in a defined oxidation stage to prevent an
undesirable biodistribution from taking place by redox processes
or technetium releases from the corresponding radiodiagnostic
agents occurring in vivo, which makes more difficult a reliable
diagnosis of corresponding diseases.
The efficiency of radionuclides in in vivo diagnosis and
therapy depends on the specificity and the selectivity of the
labeled chelates in the target cell. An improvement of these
properties can be achieved by coupling chelates to biomolecules
according to the "drug-targeting" principle. As biomolecules,
antibodies, their fragments, hormones, growth factors and
substrates of receptors and enzymes present themselves. Thus, in
British Patent Application GB 2,109,407, the use of
radioactively-labeled monoclonal antibodies to tumor-associated
antigens is described for in vivo tumor diagnosis. Also, direct
protein labeling with donor groups (amino, amide, thiol, etc.) of
the protein (Rhodes, B. A. et al., J. Nukl. Med. 1986, 27, 685-
693) or by the introduction of complexing agents (US 4,479,930
and Fritzberg, A. R. et al., J. Nucl. Med. 1986, 27, 957) with
technetium-99m is described. These experimental methods,
however, are not available for clinical use, since, on the one
hand, the selectivity is too low and, on the other hand, the
"background activity" in the organism is too high to make
possi~le an in vivo imaging.
As suitable complexing agents for technetium and rhenium
isotopes, there are, e.g., cyclic amines, as they are described
by Volkert et al. (Appl. Radiol. Isot. 1982, 33; 891) and
21 94561
Troutner et al. (J. Nucl. Med. 1980, 21; 443), but which have the
drawback that starting from a pH > 9, they are only able to bond
technetium-99m in good yields.
N2O2 systems are present in clinical use, but they are
affected by the drawback that the corresponding metal complexes
in vivo are not very stable. According to investigations of
Pillai and Troutner, the complexes lose up to 30% of complexed
metal in the plasma as early as after 1 hour (Pillai, M. R. A.,
Troutner, D. E. et al.; Inorg. Chem. 1990, 29; 1850).
Noncyclic N4 systems, such as, e.g., the HM-PA0, have their
low complex stability as a great drawback. Because of its
instability (Ballinger, J. R. et al., Appl. Radiat. Isot. 1991,
42; 315), Billinghurst, M. W. et al., Appl. Radiat Isot. 1991,
42; 607), Tc-99m-HM-PA0 must be administered within 30 minutes
after its labeling, so that the portion of decomposition
products, which have a different pharmacokinetics and
precipitation, can be kept small. Such radiochemical impurities
make more difficult the detection of diseases to be diagnosed. A
coupling of these chelates or chelating agents to other
substances selectively accumulating in foci of disease cannot be
achieved with simple means, so that the latter are generally
dispersed unspecifically in the organism.
N2Sz chelating agents (Bormans, G. et al.; Nucl. Med. Biol.
1990, 17; 499), such as, e.g., ethylene dicysteine (EC;
Verbruggen, A. M. et al.; J. Nucl. Med. 1992, 33; 551), meet the
requirement of sufficient stability of the corresponding
technetium-99m complex, but radiodiagnostic agents with a purity
2 1 9456 1
of greater than 69~ form first starting from a pH of the
complexing medium > 9.
The previously known N3S systems (Fritzburg, A.; EP 0 173
424 and EP 0 250 013) form stable technetium-99m complexes, but
they have to be heated to temperatures of about 100~C to form a
homogeneous radiopharmaceutical agent.
In recent years, the need for radiodiagnostic agents
accumulating specifically in diseased tissues has risen. This
can be achieved if complexing agents can be coupled easily to
selectively accumulating substances and in this case not lose
their advantageous complexing properties. Since it very often
results, however, that after coupling of a complexing agent with
use of one of its functional groups to such a molecule, a
weakening of the complex stability is observed, the present
attempts to couple chelating agents to selectively accumulating
substances appear hardly satisfactory, since a diagnostically
intolerable portion of the isotope is released in vivo from the
conjugate (Brechbiel, M. W. et al; Inorg. Chem. 1986, 25, 2772).
It is therefore necessary to produce bifunctional complexing
agents that carry both functional groups for bonding the desired
metal ion and a (different, several) functional group for bonding
the selectively accumulating molecule. Such bifunctional ligands
make possible a specific, chemically defined bond of technetium
or rhenium isotopes to the most varied biological materials, even
though a so-called pre-labeling is performed.
In EP 0 247 866, EP 0 188 256 and EP 0 200 492, several
chelating agents, which are coupled to monoclonal antibodies or
7 2 1 9456 1
fatty acids, are described. As chelating agents, however, the
already-mentioned N2S2 systems are used, which are hardly
suitable based on their low stability. Since both the
selectively accumulating substances in their properties and also
the mechanisms, according to which they are concentrated, are
very different, it is further necessary to vary the couplable
chelating agents and to be able to adapt to the physiological
requirements of the coupling partner with respect to its
lipophilia, membrane permeability, etc.
The object of the invention is therefore to find complexes
or complexing agents which overcome the drawbacks of the prior
art, i.e., which
-- can be produced at a physiological pH from the
corresponding complexing agent and the respective metal
oxide/salt,
-- can be produced at low temperatures, preferably at room
temperature, from the corresponding complexing agent
and the respective metal oxide/salt,
-- show a high complex stability even under in vivo
conditions,
-- show a high selectivity or tissue-/organ-specificity.
Furthermore, the complexes must meet the requirements which
generally are to be imposed on pharmaceutical agents, such as,
e.g., good compatibility (i.e., no side effects), good solubility
and complete precipitation.
The object is achieved by this invention.
8 2194561
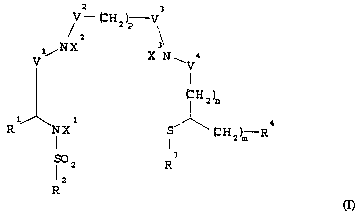
It has been found that compounds of general formula I
~H2)p V~
NX X N 4
~H2)n
Rl,~NXl S l ~H2)m R
I ~ 2 R
R
in which
V1, V2, V3, *, independently of one another, stand for a
carbonyl, >CH(COOH) or -CH2 group,
X1 stands for a hydrogen atom, a C1-C12 alkyl radical
optionally substituted with a carboxyl, an amino or a
thiocyanate group, or a metal ion equivalent of a
radioactive metal ion of an element of atomic number
43, 45, 46, 75, 82 or 83,
X2, X3, independently of one another, stand for a hydrogen
atom or a metal ion equivalent of a radioactive metal
ion of an element of the above-mentioned atomic
numbers,
n, m, p stand for numbers o or 1, whereby m t n - 1 holds
true,
R1 stands for a hydrogen atom, a carboxyl group or a group
--U--Z,
2194561
in which U stands for a direct bond, a straight-chain
or branched, saturated or unsaturated Cl-C20 alkylene
radical, which optionally contains a maleimide radical,
a succinimide radical, a phenyl radical optionally
substituted by 1 to 5 fluorine atoms, an amino or nitro
group, one or two imino, phenylene, phenylenoxy,
phenylenamino, amide, hydrazide, carbonyl, ureido,
thioureido, thioamide, ester group(s), 1 to 2 oxygen,
sulfur and/or nitrogen atom(s) as well as optionally 1
to 5 hydroxy, mercapto, oxo, thioxo, carboxy,
alkylcarboxylic acid, ester, thiocyanate and/or amino
groups, and Z stands for a hydrogen atom, a radical of
an amino acid, a peptide, a polynucleotide or a
steroid, or a functional group with which optionally
the radical of an amino acid, a peptide, a
polynucleotide or a steroid is bound,
R2 stands for a straight-chain or branched C1-C10 alkyl
radical, which optionally contains a -COOH group, a C7-
C12 aralkyl.radical or an aromatic compound, which
optionally is substituted with a chlorine or bromine
atom; a thiocyanate, a methyl, ethyl, carboxyl and/or
methoxy group,
R4 stands for a hydrogen atom or a carboxyl group or if R1
means a hydrogen atom or a carboxyl group, it stands in
addition for a group -U-Z, in which U and Z have the
indicated meanings,
lo 21 94561
R3 stands for a hydrogen atom, a metal ion equivalent of
an element of the above-mentioned atomic numbers; a
trifluoroacetate, acetate, benzoate, C1-C6 acyl, a
benzoyl, a hydroxyacetyl, an acetamidomethyl radical, a
benzoic acid radical optionally substituted with a
chlorine or bromine atom, a methyl, ethyl, carboxyl
and/or methoxy group, a p-methoxybenzyl radical, an
ethoxyethyl radical, an SH protective group, a
~ radical, or
if X2, X3 stand for a hydrogen and X1 stands for a
hydrogen or an optionally substituted C1-C1z alkyl
radical, for a radical of formula II
I 2
RVNX1 S~ H2)m ~ R
H 2 )n
X ~N
V ~H 2 )p V
in which V1 v2 V3, V4, X1, X2, X3, n, m, p, R1, R and
R4 have the indicated meanings,
11 21 ~4561
whereby at least one and at most two radicals V1, V2, V3, V4 stand
for a carbonyl group, are very well suited as radiodiagnostic
agents and radiotherapeutic agents or for the production of
radiodiagnostic agents and radiotherapeutic agents.
The complexing agents (chelating agents) according to the
invention, i.e., compounds of general formula I with X1, X2, X3
and R3 in the indicated meanings with the exception of a metal
ion equivalent, meet the above-mentioned requirement profile.
They are distinguished more particularly in that they complex the
respectively desired metal quickly at the physiological pH and
low temperatures. They are therefore especially suitable for
routine use in clinical practice.
During the complexing process, monomeric metal complexes of
formula I with R3 meaning a metal ion equivalent are from the
dimeric chelating agents with R3 meaning a radical of general
formula II.
As metal ions, radioactive metal ions of the elements of
atomic numbers 43, 45, 46, 75, 82 or 83, such as, e.g., the
radioisotopes technetium-99m, rhodium-103, palladium-109,
rhenium-186, lead-212 and bismuth-212, are used, whereby the
selection of the metal isotope depends on the desired field of
use. According to the invention, metal complexes of the elements
technetium and rhenium are preferred.
If the complexes of general formula I according to the
invention contain isotopes that emit ~y-radiation, such as, e.g.,
Tc-99m, the latter can be used in single-photon emission
tomography (SPECT).
12 21 94561
If the complexes of general formula I according to the
invention contain isotopes that emit ~-particles, such as, e.g.,
Bi-211, Bi-212, Bi-213, Bi-214 or B-emitting isotopes, such as,
e.g., Re-186 or Re-188, the latter can be used in radiotherapy.
According to the invention, compounds of formula I, in which
V1 and V4 each stand for a carbonyl group, v2 and V3 each for a
-CH2 group and p for number 0, are preferred.
As radical R1, hydrogen or a carboxylic acid group and
especially a group -U-Z in which Z is a hydrogen atom are
suitable, but it preferably stands for the radical of a
biomolecule with tissue- or structure-specific properties or a
functional group optionally present in activated form with which
optionally such a biomolecule can be bound.
As examples of biomolecules, there can be mentioned radicals
of an amino acid, a peptide, or a steroid, such as, e.g., the
known steroid hormones (androgens, gestagens, estrogens,
cholesterol, cholic acid derivatives, pregnanes, etc.) as well as
polynucleotides such as RNA or DNA.
As examples of functional groups with which optionally the
bond of a biomolecule can take place, a -COOH, an -SCN, an -OH, a
-Cl or an -NH2 group can be mentioned. Such groups can also be
present in their activated form, e.g., as succinimide ester or
acid chloride.
U can stand for a direct bond but preferably a straight-
chain or branched, saturated or unsaturated C1-C20 alkylene
radical, which optionally contains a succinimide radical, a
phenyl radical optionally substituted by 1 to 5 fluorine atoms,
13 2 1 9456 1
..
an amino or a nitro group, one or two imino, phenylene,
phenylenoxy, phenylenamino, amide, hydrazide, carbonyl, ureido,
thioureido, thioamide, ester group(s), 1 to 2 oxygen, sulfur
and/or nitrogen atom(s) as well as optionally 1 to 5 hydroxy,
mercapto, oxo, thioxo, carboxy, alkylcarboxylic acid, ester,
thiocyanate and/or amino groups.
As examples of U, if Z stands for a biomolecule, there can
be mentioned a -CH2-C6H4-O-CH2-C6H4-~ -cH2-c6H4 O CO C15 30 ' 2
O (CH ) COO- -CH2-C6H4-O-cH2-cOO-c6H4 NH ~ 2 6 4
COO-, - (CH2) 5-COO-, or a -CH2-C6H4-O-(CH2) 2-~- (CH2) 2-~-
group.
As radicals -U-Z with Z meaning hydrogen, there can be
mentioned a -CH2-C6H4-O-CH2-cOO-c6F5~ -CH2-C6H4 ~ CH2 6 5 ~ 2 6 4
3, CH2 C6H4 O-C6H3, -CH2~c6H4~0-c12H2s or a -CH2-
C6H4 O CO C15H31 group.
As radicals -U-Z with Z meaning an optionally activated
functional group, there can be mentioned a -CH2-(CH2) 4-NCS, -CH2-
O ( CH ) -COOH -CH2C6H4-O-CH2-COO-C6H4 NH2 ~ 2 6 4
COO-C6H4-NO2 ~ -CH2-C6H4 NCS, O ~
CH2~30~H.~oo._N~ ---N~3
-CH2-C6H4-O-(CH2)2-O-(CH2)2-OH or a -CH2-C6H4-O-CH2-COOH group, i.e.,
o
Z stands for an -NCS, -COOH, -NH2, -NO2, -OH or a ~oO_N
group.
14 21 94561
As examples of radicals RZ, there can be mentioned a -C6H4-
NCS or a -C6H4-COOH group, but especially a -CH3 group or a phenyl
ring.
As radicals R3, there can be mentioned as examples,
a) in the case of complexing agents: -SH protective
groups, such as, e.g., a -CO-CH3 , o ~ CO C6H5 ~ CO CF3
or a -C(C6H5) 3 group ~ ~>
or a radical of general formula II, in which V1, V2, V3, V4, X1,
X2, X3, n, m, p, R1, R2 and R4 have the indicated meanings,
b) in the case of the complexes according to the
invention: one of the previously mentioned radicals, with the
exception of a radical of general formula II: but in addition,
R3 can also stand for a metal ion equivalent of a radioactive
metal ion of an element of the above-mentioned atomic numbers.
In the case of complexing agents, X2, X3 stand for a
hydrogen atom and X1 stands for a hydrogen atom or an optionally
substituted C1-C12 alkyl radical, in the case of complexes,
depending on the oxidation stage of the metal in the complex, at
least 2 of radicals x1, X2, X3 or R3 have the meaning of a metal
ion equivalent.
R4 stands for hydrogen or a carboxyl group, but R4 can also
stand for a group -U-Z with U and Z in the previously-indicated
meanings, whereby it always holds true that at most one of two
radicals R1 or R4 means a group -U-Z. According to the
invention, compounds with R4 meaning hydrogen are preferred.
Indices m and n stand for numbers O or l. Since isomeric
compounds can accumulate in the synthesis of complexing agents
21 9456 1
according to the invention, it holds true that the sum of m and n
is always 1.
The invention also relates to the process for the production
of complexing agents and complexes according to the invention,
whereby suitably different reaction paths are followed in the
synthesis of the complexing agent as a function of the desired
target structure. Some typical syntheses are described as
examples below. Other complexing agents can be produced
analogously to the synthesis methods described.
1. If ligands are to be produced, which contain an -O-C6H4-
CH2 group in R1, a start is advantageously made from a tyrosine
ester, whose amino group first is protected in the way known to
one skilled in the art by reaction with a reagent Z1-Cl, in which
Z1 stands for any amino protective group, preferably for a
benzyloxycarbonyl group (also named Z group below). Then, the
phenolic hydroxyl group is alkylated with an alkyl iodide in a
way known in the art, the amino protective group is cleaved off
in acidic form and then tosylated, e.g., with toluenesulfonic
acid chloride. The thus obtained intermediate product is reacted
in an aminolysis of the ester function with ethylenediamine.
Then, it is reacted with 2-acetylmercaptosuccinic anhydride to
the complexing agents according to the invention, whereby both
isomers are always obtained in the last reaction step.
2. The production of ligands containing an -o-CO-CH2-O-C6H4-
CHz group in R1 according to the invention is carried out by
first, as described above, the corresponding Zl-protected
compound -- starting from tyrosine ester -- being produced. The
16 2194561
phenolic hydroxyl group is then alkylated with bromoacetic acid-
t-butyl ester, and the amino protective group is cleaved off in a
way known in the art. Tosylation and aminolysis of the ester
function with ethylenediamine are carried out as described under
1, then it is reacted, however, with chloroacetyl chloride.
Chlorine is substituted in a way known in the art by reaction
with potassium thioacetate or sodium thioacetate, and the t-butyl
ester is acidically saponified. Optionally, the resulting
carboxylic acid group can be activated with hydroxysuccinimide
and then reacted with the respectively desired biomolecule.
3. Ligands according to the invention, in which R1 stands
for an SCN-C6H4-CH2 radical, can be obtained by first the amino
group of a p-nitrophenylalanine ester being tosylated in a way
known in the art. Then, the nitro group is hydrogenated, and the
resulting aromatic amino group is protected, e.g., by reaction
with benzyl chloroformate. The aminolysis of the ester function
with ethylenediamine follows this reaction step, as described in
the preceding cases, followed by a reaction with S-protected
mercaptoacetic acid. The cleavage of the benzyloxycarbonyl group
follows the latter. The introduction of the isothiocyanate group
is carried out by reaction with thiophosgene.
4. The production of the ligands according to the invention
with x1 meaning an alkyl radical is carried out by first the
amino group of a glycine ester being tosylated with
toluenesulfonyl chloride. Then, the ester function is reacted in
an aminolysis with ethylenediamine. The resulting free amino
group is protected with a tert-butoxycarbonyl group (designated
17 2194561
below as a BOC group) and the nitrogen substituted with a tosyl
radical is N-alkylated with an alkyl iodide in a way known in the
art. After the cleavage of the BOC group, the reaction with 2-
acetylmercaptosuccinic anhydride follows, whereby again a mixture
of isomers is obtained.
5. Ligands according to the invention with R1 meaning an
aminobutyl radical can be obtained by the BOC-protected lysine
ester on a nitrogen atom first being tosylated on the remaining
unprotected primary amino group and then being reacted with
ethylenediamine in an aminolysis. Below, it is reacted with 2-
acetylmercaptosuccinic anhydride (whereby a mixture of isomers
results) and the amino protective group is cleaved off according
to known methods.
6. The production of ligands, according to the invention,
that contain an isothiocyanatophenyl radical as R2 is carried out
by reaction of a glycine methyl ester with nitrobenzenesulfonyl
chloride, subsequent reduction of the nitro group and protection
of the resulting amino group. The thus obtained intermediate
compound is reacted in an aminolysis with ethylenediamine and
then with S-protected mercaptoacetic acid. The subsequent
cleavage of the (BOC) protective group is carried out in a known
way. The free amino group is then reacted with thiophosgene to
the corresponding isothiocyanate group.
The production of the metal complexes of general formula I
according to the invention with at least two radicals Xl, x2, X3
and/or R3 in the meaning of a metal ion equivalent is carried out
in a way known in the art, by the complexing agents according to
18 2 1 9456 1
the invention (which can be obtained as previously described)
being dissolved in aqueous medium with the addition of a reducing
agent, preferably tin(II) salts, such as tin chloride or tin
tartrate -- and optionally with the addition of the additives
usual in galenicals, such as physiologically harmless buffers
(e.g., tromethamine), small additions of electrolytes (e.g.,
sodium chloride), stabilizers (e.g., gluconate, phosphates or
phosphonates), etc. -- and then sterilized by filtration. In the
case of short-lived metal isotopes, such as, e.g., Tc-99m, this
solution is reacted immediately before administration with an
aqueous solution of the respectively desired metal ion. In the
case of long-lived isotopes, the reaction with the metal
salt/oxide can even be performed by the manufacturer of
radiopharmaceutical agents. To assure a complete complexing of
the metal isotope, the complexing agent is generally added in at
least a 100-fold excess, i.e., besides the desired metal complex,
the agents according to the invention contain in addition the
metal-free complexing agent, which is added advantageously in the
form of its potassium salt.
Reaction solutions containing the above-mentioned described
metal complex can basically be administered immediately without
further working-up.
Since the technetium in particular can be present in a
series of oxidation stages (+7 to -1), it is advisable to attach
stabilizers to the complexing agents. The latter keep the
radionuclide in a stable form, until it has reacted completely
with the complexing agent (ligand). The stabilizers, which are
19 21 94561
~ .
known as transfer or auxiliary ligands, first complex the metal
in an exactly defined oxidation stage and then provide it to the
target ligands (complexing agents) in a ligand exchange reaction.
Examples of auxiliary ligands are gluconoheptonic acid, tartaric
acid, citric acid (including their salts) or other substances
known to one skilled in the art.
Optionally, the obtained metal complexes are mixed with
pharmacologically acceptable radiological vehicles. This
radiological vehicle should exhibit advantageous properties for
the administration of the radiopharmaceutical agent in the form
of an injection, inhalation or ingestion. As vehicles there can
be mentioned as examples HSA, aqueous buffer solutions, such as,
e.g., tris(hydroxymethyl)aminoethane (or their salts), phosphate,
citrate, bicarbonate, etc., sterile water, physiological common
salt solution, isotonic chloride or dicarbonation solutions or
normal plasma ions such as Ca2+, Na+, K+, and Mg2+.
In nuclear-medicine in vivo use, the agents according to the
invention are dosed in amounts of 10-5 to 5 x 104 nmol/kg of body
weight, preferably in amounts between 10-3 to 5 x 102 nmol/kg of
body weight (relative to the metal complex). Starting from an
average body weight of 70 kg, the required amount of
radioactivity for diagnostic uses is between 1.85 MBq and 1.85
GBq per administration. The administration is carried out
normally by intravenous, intraarterial, peritoneal or
intratumoral injection of 0.1 to 2 ml of a solution of the agents
according to the invention. Intravenous administration is
preferred.
2 1 ~456 1
The metal complexes according to the invention with metal
ions of the above-mentioned elements and the pharmaceutical
agents prepared from them are distinguished by good compatibility
and high stability in vivo. The complexing agents according to
the invention are distinguished by an easy labeling capability,
i.e., they complex the desired metals in high yields at room
temperature and neutral pH.
The following examples are used for a more detailed
explanation of the object of the invention, without intending
that they be limited to this object.
21 2 1 9456 1
Example 1
a) N-Benzyloxycarbonyl-tyroqine methyl ester
A suspension of 97.6 g (0.5 mol) of tyrosine methyl ester in
500-1000 ml of dichloromethane is mixed at room temperature with
a solution of 86 g of benzyl chloroformate in 100 ml of
dichloromethane. Then, 51 g of triethylamine is slowly added in
drops while being cooled, and it is stirred overnight. The
mixture is concentrated by evaporation on a rotary evaporator at
40~C, the residue is taken up in 750 ml of EtOAc, and the
undissolved material is filtered out. The filtrate is washed
three times saturated common salt solution, dried and
concentrated by evaporation and recrystallized.
Yield: 72%
Cld: C 65.64~ H 5.82~ N 4.25% O 24.29%
Fnd: C 65.47% H 5.98% N 4.02%
b) N-Benzyloxycarbonyl-2-t~4-hexyloxy)benzyl]-2-aminoacetic
acid methyl e~ter
1.12 g of potassium-tert-butylate (10 mmol) is added to a
solution of 3.29 g of the compound produced according to Example
la) (10 mmol) in 50 ml of DMF at room temperature, and then the
solution of 2.12 g of hexyl iodide (10 mmol) in 10 ml of DMF is
added in drops and heated for 3 hours to 110~C. After cooling to
room temperature, it is poured onto ice, extracted with
dichloromethane, washed several times with water, dried, filtered
and concentrated by evaporation. After chromatography on a
silica gel column (CH2Cl2/EE 19:1), 1.8 g of a pale yellow oil remains.
22 21 94561
Yield: 44%
Cld: C 69.71% H 7.56% N 3.39% o 19.35%
Fnd: C 69.54% H 7.69% N 3.13%
c) 2-[(4-Hexyloxy)benzyl]-2-aminoacetic acid methyl ester
4.14 g (10 mmol) of the Z1-protected compound produced
according to Example lb) is hydrogenated with hydrogen in 50 ml
of ethyl acetate in the presence of 1.5 g of palladium on
activated carbon (10%) at 50~C. After the reaction is completed,
the catalyst is filtered out, and the solvent is drawn off. 2.7
g of colorless oil remains.
Yield: 97%
Cld: C 68.79% H 9.02% N 5.01% 0 17.18%
Fnd: C 68.64% H 9.11% N 5.09%
d) N-p-Toluenesulfonyl-2t(4-hexyloxy)benzyl]-2-aminoacetic acid
methyl ester
8.38 g of the amine (30 mmol) produced according to Example
lc) is dissolved in 50 ml of dichloromethane and mixed at 0~C
with 5.72 g of toluenesulfonic acid chloride in 30 ml of
dichloromethane. While being stirred intensively, 3.0 g of
triethylamine is added in drops at 0~C and stirred for 1 hour at
room temperature. After the reaction is completed, it is mixed
with ice water and extracted several times with dichloromethane.
The combined organic extracts are washed twice with cold 10% HCl,
three times with 10% NaHC03 solution and twice with saturated
23 2 1 9456 1
,
NaCl solution. After drying, the solvent is drawn off and
chromatographed (silica gel, CH2Cl2).
Yield: 60%
Cld: C 63.72% H 7.21% N 3.23~ O 18.45% S 7.40%
Fnd: C 63.48% H 7.52% N 3.02% S 7.25%
e) N-p-Toluenesulfonyl-2t~4-hexyloxy)benzyl]-2-aminoacetic
acidtN-(2-amino-ethyl)]amide
A solution of 433 mg of tosyl glycine ester (1 mmol)
produced according to Example ld) in 1 ml of anhydrous
dichloromethane is slowly added in drops to a solution of 1.2 g
of ethylenediamine (20 mmol) in 1 ml of anhydrous dichloromethane
and refluxed. After the reaction is completed, it is
concentrated by evaporation in a vacuum, and the residue is
chromatographed (silica gel, MeOH).
Yield: 42~
Cld: C 62.45% H 7.64~ N 9.10% O 13.864% S 6.95%
Fnd: C 62.36~ H 7.81% N 8.94% S 6.80%
f) N-p-Toluenesulfonyl-2[(4-hexyloxy)benzyl]-2-aminoacetic
acid-N-{2-Nt~3-carboxy-2-mercaptoacetyl-1-oxopropyl)]-
aminoethyl}amide
0.38 g (2.2 mmol) of 2-acetylmercaptosuccinic anhydride is
added to a solution o~ 1.00 g of the amine (2.2 mmol) produced
according to Example le) in 10 ml of pyridine/DMF (50:50) and
stirred for 4 hours at room temperature. Then, the solvent is
drawn off, the residue is taken up in 0.5N HCl and extracted with
2 1 9456 1
24
CH2Cl2. After flash chromatography, 420 mg of a yellow oil
remains.
Yield: 31%
Cld: C 56.67~ H 6.50% N 6.61% 0 20.13% S 10.09%
Fnd: C 56.41% H 7.01% N 6.43% S 9.90%
g) Tc-99m complex of N-p-toluenesulfonyl-2[~4-hexyloxy)benzyl]-
2-aminoacetic acid-N-{2-Nt~3-carboxy-2-mercaptoacetyl-1-
oxopropyl)]aminoethyl}amide
1 mg of the compound produced according to Example lf) is
dissolved in 100 ~l of EtOH. 50 ~l of this solution is added to
250 ~l of a 0.1 M phosphate buffer of pH 8.5 and then mixed with
100 ~l of a 99m-Tc-gluconate solution and allowed to stand for 15
minutes. The labeling yield is determined with the aid of HPLC.
Example 2
a) N-Benzyloxycarbonyl-2-[(4-methyloxy)benzyl]-2-aminoacetic
acid methyl ester
1.12 g of potassium-tert-butylate (10 mmol) is added to a
solution of 3.29 g of the alcohol (10 mmol) produced according to
Example la) in 50 ml of DMF at room temperature and then the
solution of 1.42 g of methyl iodide (10 mmol) in 10 ml of DMF is
added in drops and heated to 110~C. After the reaction is
completed, it is allowed to cool to room temperature, poured onto
ice, extracted with dichloromethane, washed several times with
water, dried, filtered and concentrated by evaporation. After
21 94561
column chromatography (silica gel, CH2Cl2/EE 19:1), 1.92 g of an
oil remains.
Yield: 56%
Cld: C 66.46~ H 6.16% N 4.08% 0 23.30%
Fnd: C 66.18% H 6.41% N 3.97%
b) 2-t(4-Nethyloxy)benzyl]-2-aminoacetic acid methyl ester
3.43 g (10 mmol) of the Z1-protected compound produced
according to Example 2a) is hydrogenated with hydrogen in 50 ml
of ethyl acetate in the presence of 1.5 g of palladium on
activated carbon (10%) at room temperature. After the reaction
is completed, the catalyst is filtered out, and the solvent is
drawn off.
Yield: 98%
Cld: C 63.14% H 7.23% N 6.69% 0 22.94%
Fnd: C 62.91% H 7.36% N 6.50%
c) N-p-Toluenesulfonyl-2t(4-methyloxy)benzyl]-2-aminoacetic
acid methyl ester
6.28 g of the amine (30 mmol) produced according to Example
2b) is dissolved in 50 ml of dichloromethane and mixed at 0~C
with 5.72 g of toluenesulfonic acid chloride in 30 ml of
dichloromethane. While being stirred intensively, 3.0 g of
triethylamine is added in drops at 0~C and stirred for 1 hour at
room temperature. After the reaction is completed, it is mixed
with ice water and extracted several times with dichloromethane.
The combined organic extracts are washed twice with cold 10% HCl,
26 2 1 9456 1
three times with 10% NaHCO3 solution and twice with saturated
NaCl solution. After drying, the solvent is drawn off.
Yield: 76%
Cld: C 59.49% H 5.82% N 3.85% O 22.01% S 8.82%
Fnd: C 59.32% H 6.03% N 3.66% S 8.64%
d) N-p-Toluenesulfonyl-2t(4-methyloxy)benzyl]-2-aminoacetic
acid-~N-~2-aminoethyl)]amide
A solution of 363 mg of the ester (1 mmol) produced
according to Example 2c) in 1 ml of anhydrous dichloromethane is
slowly added in drops to a solution of 600 mg of ethylenediamine
(10 mmol) in 1 ml of anhydrous dichloromethane and refluxed.
After the reaction is completed, it is concentrated by
evaporation in a vacuum, mixed with ethanolic HCl and stirred for
2 hours at room temperature. The solvent is drawn off, and the
residue is recrystallized.
Yield: 49%
Cld: C 58.29% H 6.44% N 10.73% O 16.35% S 8.19%
Fnd: C 57.89% H 6.84% N 10.01% S 7.52%
e) N-p-Toluenesulfonyl-2t(4-methyloxy)benzyl]-2-aminoacetic
acid-N-{2-N[~3-carboxy-2-mercaptoacetyl-l-
oxopropyl)]aminoethyl}amide
0.38 g (2.2 mmol) of 2-acetylmercaptosuccinic anhydride is
added to a solution of 941 mg of the amine (2.2 mmol) produced
according to Example 2d) in 10 ml of pyridine/DMF (50:50), and it
is stirred for 4 hours at room temperature. Then, the solvent is
27 21 94561
.
drawn off, the residue is taken up in 0.5N HCl and extracted with
CH2Cl2. After flash chromatography, 905 mg of a yellow oil
remains .
Yield: 73%
Cld: C 53.08% H 5.52% N 7.43% O 22.63% S 11.34%
Fnd: C 52.31% H 5.45% N 7.06% S 11.61%
f) Tc-99m complex of N-p-toluene~ulfonyl-2t~4-
methyloxy)benzyl]-2-aminoacetic acid-N-{2-N[(3-carboxy-2-
mercaptoacetyl-l-oxopropyl)]aminoethyl}amide
1 mg of the compound produced according to Example 2e) is
dissolved in 100 ~1 of EtOH. 50 ~1 of this solution is diluted
with 250 ~1 of EtOH, mixed with 50 ~1 of a 0.1 M phosphate buffer
of pH 8.5 and then mixed with 100 ~1 of a 99m-Tc-gluconate
solution and allowed to stand for 15 minutes. After filtration,
the labeling yield is determined with the aid of HPLC.
Example 3
a) N-Benzyloxycarbonyl-2-~(4-dodecyloxy)benzyl]-2-aminoacetic
acid methyl ester
1.12 g of potassium-tert-butylate (10 mmol) is added to a
solution of 3.29 g of the alcohol (10 mmol) produced according to
Example la) in 50 ml of DMF at room temperature, and then the
solution of 3.55 g of dodecyl iodide (12 mmol) in 10 ml of DMF is
added in drops and heated for 3 hours to 110~C. Then, it is
allowed to cool to room temperature, poured onto ice, extracted
with dichloromethane, washed several times with water, dried,
21 94561
28
filtered and concentrated by evaporation. After SC tcolumn
chromatography] (silica gel, CHzCl2/EE 19:1), 2.2 g of the
desired substance remains.
Yield: 44%
Cld: C 72.40% H 8.71% N 2.81% 0 16.07%
Fnd: C 72.08% H 8.85~ N 2.68%
b) 2-[(4-Dodecyloxy)benzyl]-2-aminoacetic acid methyl ester
4.98 g (10 mmol) of the Z1-protected compound produced
according to Example 3a) is hydrogenated with hydrogen in 50 ml
of ethyl acetate in the presence of 1.5 g of palladium on
activated carbon (10%) at 50~C. After the reaction is completed
(4 hours), the catalyst is filtered out, and the solvent is drawn
off.
Yield: 91%
Cld: C 72.69% H 10.26% N 3.85% 0 13.20
Fnd: C 72.53% H 9.98% N 3.82%
c) N-p-Toluenesulfonyl-2[l4-dodecyloxy)benzyl]-2-aminoacetic
acid methyl e~ter
10.91 g of the amine (30 mmol) produced according to Example
3b) is dissolved in 50 ml of dichloromethane and mixed at 0~C
with 5.72 g of toluenesulfonic acid chloride in 30 ml of
dichloromethane. While being stirred intensively, 3.0 g of
triethylamine is added in drops at 0~C and stirred for l hour at
room temperature. After the reaction is completed, it is mixed
with ice water and extracted several times with dichloromethane.
29 2 1 9456 1
The combined organic extracts are washed twice with cold 10% HCl,
three times with 10% NaHC03 solution and twice with saturated
NaCl solution. After drying, the solvent is drawn off and
chromatographed (silica gel, CHzCl2).
Yield: 75%
Cld: C 67.2B% H 8.37% N 2.71% 0 15.45% S 6.19%
Fnd: C 66.96% H 8.26% N 2.60% S 6.11%
d) N-p-Toluenesulfonyl-2t~4-dodecyloxy)benzyl]-2-aminoacetic
acidtN-(2-aminoethyl)]amide
A solution of 3.3 g of the ester (6.38 mmol) produced
according to Example 3c) in 10 ml of anhydrous dichloromethane is
slowly added in drops-to a solution of 1.92 g of ethylenediamine
(31.9 mmol) in 20 ml of anhydrous dichloromethane and refluxed
for 4 hours. After the reaction is completed, it is concentrated
by evaporation in a vacuum, and the residue is recrystallized.
2.3 g of white crystals remains.
Yield: 80%
Cld: C 66.02% H 8.68% N 7.70% 0 11.73% S 5.86%
Fnd: C 65.88% H 8.80% N 7.59% S 5.76%
e) N-p-Toluenesulfonyl-2[t4-dodecyloxy)benzyl]-2-aminoacetic
acid-N-{2-Nt(3-carboxy-2-mercaptoacetyl-1-oxopropyl)~-
aminoethyl}amide
0.32 g (1.8 mmol) of 2-acetylmercaptosuccinic anhydride is
added to a solution of 1.00 g of the amine (1.8 mmol) produced
according to Example 3d) in 10 ml of pyridine/DMF (50:50), and it
2 1 9456 1
.,.
is stirred for 4 hours at room temperature. Then, the solvent is
drawn off, the residue is taken up in 0.5N HCl and extracted with
CH2Cl2. After flash chromatography (silica gel, EtOAc/MeOH 9:1),
420 mg of a yellow oil remains.
Yield: 32%
Cld: C 60.06% H 7.42% N 5.84~ O 17.78% S 8.91%
Fnd: C 59.76% H 7.61% N 5.77% S 8.78%
f) Tc-99m complex of N-p-toluene~ulfonyl-2t(4-
dodecyloxy)benzyl~-2-aminoacetic acid-N-{2-Nt(3-carboxy-2-
mercaptoacetyl-l-oxopropyl)]aminoethyl}amide
1 mg of the compound produced according to Example 3e) is
dissolved in 100 ~1 of EtOH. 50 ~1 of this solution is added to
250 ~1 of a 0.1 M phosphate buffer of pH 8.5 and then mixed with
100 ~1 of a 99m-Tc-gluconate solution and allowed to stand for 15
minutes. The labeling yield is determined with the aid of HPLC.
Example 4
a) N-p-Toluenesulfonyl-2[(4-benzyloxy)benzyl]-2-aminoacetic
acid-N-{2-Nt~3-carboxy-2-mercaptoacetyl-1-
oxopropyl)]aminoethyl}amide
The solution of 4.68 g (10 mmol) of the amino compound
produced according to Example 16b) in 150 ml of THF is slowly
added in drops to a solution of 3.48 g of acetylmercaptosuccinic
anhydride (20 mmol) in 50 ml of THF at room temperature and
stirred overnight. Then, it is allowed to crystallize out at
-20~C. 2.8 g of white crystals remains.
31 2194561
r
Yield: 44%
Cld: C 58.02% H 5.50% N 6.55% O 19.95% S 9.99%
Fnd: C 57.80% H 5.72% N 6.51% S 9.85%
b) Tc-99m complex of N-p-toluenesulfonyl-2t~4-
benzyloxy)benzyl]-2-aminoacetic acid-N-{2-N[~3-carboxy-2-
mercaptoacetyl-1-oxopropyl)]aminoethyl}amide
1 mg produced according to Example 4a) is dissolved in 100
~l of EtOH. 50 ~l of this solution is added to 250 ~l of a 0.1 M
phosphate buffer of pH 8.5 and then mixed with 100 ~1 of a 99m-
Tc-gluconate solution and allowed to stand for 15 minutes. The
labeling yield is determined with the aid of HPLC.
Example 5
a) N-p-Toluenesulfonyl-~4-nitrophenyl)alanine methyl e~ter
7.56 g of 4-nitrophenylalanine methyl ester (30 mmol) is
suspended in 30 ml of water and mixed at 0~C with 5.72 g of
toluenesulfonic acid chloride in 20 ml of diethyl ether. While
being stirred intensively, 3.0 g of anhydrous sodium carbonate is
added in portions at 0~C within one hour, and it is stirred
overnight at room temperature. It is mixed with water and
extracted several times with ethyl acetate. The combined organic
extracts are washed twice with cold 10% HCl, three times with 10%
NaHCO3 solution and twice with saturated NaCl solution. After
drying, the solvent is drawn off. 8.6 g of yellowish crystals
remains.
Yield: 76%
32 21 94561
Cld: C 53.96% H 4.80% N 7.40% O 25.37% S 8.47%
Fnd: C 53.79% H 4.99% N 7.29% S 8.30%
b) N-p-Toluenesulfonyl-~4-aminophenyl)alanine methyl ester
The solution of 2.0 g of N-p-toluenesulfonyl-(4-
nitrophenyl)alanine methyl ester in methanol is added to the
suspension of 500 mg of Pd/C (10%) in 25 ml of methanol, and it
is hydrogenated at 55~C with hydrogen. After filtration and
concentration by evaporation, 1.4 g of whitish crystals remains.
Yield: 76%
Cld: C 58.60% H 5.79% N 8.04% O 18.37% S 9.20%
Fnd: C 58.09% H 5.99% N 8.80% S 9.01%
c) N-p-Toluenesulfonyl-{4-tN-(tert-
butyloxycarbonyl)laminophenyl}-alanine methyl ester
A solution of 3.78 g (10 mmol) of the amino compound
produced according to Example 5b) and 3 g of triethylamine (30
mmol) in 50 ml of dioxane is mixed in one portion with 7.4 g of
di-tert-butyl-dicarbonate (34 mmol). (Gas generation!) The
solution is stirred for 3 hours at room temperature, then poured
onto ice water and extracted three times with ethyl acetate. The
combined organic phases are washed three times with water and
once with saturated common salt solution, dried (MgSO4),
concentrated by evaporation and recrystallized.
Yield: 89%
Cld: C 58.91% H 6.29% N 6.25% O 21.40 S 7.15%
Fnd: C 58.12% H 6.69% N 6.21% S 6.59%
21 94561
d) N-p-Toluenesulfonyl-{4-tN-(tert-
butyloxycarbonyl)]aminophenyl}-alanine[N-~2-amino-
ethyl)]amide
The solution of 700 mg of N-p-toluenesulfonyl-{4-[N-(tert-
butyloxycarbonyl)]aminophenyl}-alanine methyl ester (1.6 mmol) in
1 ml of anhydrous dichloromethane is added in drops to a solution
of 450 mg of ethylenediamine (7.5 mmol) in 5 ml of anhydrous
dichloromethane and refluxed for 8 hours. After the reaction is
completed, it is concentrated by evaporation in a vacuum, and the
residue is chromatographed (silica gel, ethyl acetate/MeOH 9:1).
Yield: 85%
Cld: C 57.97% H 6.77% N 11.76% O 16.79% S 6.73%
Fnd: C 57.08% H 6.12% N 12.33% S 6.41%
e) N-p-Toluene~ulfonyl-{4-lN-~tert-
butyloxycarbonyl)]aminophenyl}-alaninetN-
(chloroacetylaminoethyl)]amide
The solution of 1.24 g of chloroacetyl chloride (11 mmol) in
5 ml of dichloromethane is added in drops to a solution of 4.77 g
of the amine (10 mmol) produced according to Example 5d in 10 ml
of dichloromethane at 0~C, then the solution of 2.02 g of
triethylamine in 5 ml of dichloromethane is slowly added in drops
(attention: strong heat build-up) and stirred overnight. Then,
it is mixed with water, extracted with EtOAc, washed neutral with
water, dried and concentrated by evaporation. 2.77 g of white
crystals remains.
Yield: 50%
21 94561
Cld: C 54.29% H 6.01% N 10.13% O 17.38% S 5.80~
Fnd: C 53.76% H 5.78% N 9.66% S 5.92%
f) N-p-Toluene~ulfonyl-{4-~N-~tert-
butyloxycarbonyl)]aminophenyl}-alanine~N-~2-
~acetylmercapto)acetylaminoethyl)]amide
In 5 ml of anhydrous DMF under nitrogen atmosphere, a
solution of 238 mg of potassium thioacetate (2 mmol) in anhydrous
DMF is added in drops to a solution of 553 mg of the derivative
(1 mmol) produced according to Example 5e and a catalytic amount
of Nal and heated for 3 hours to 110~C. The hot solution is
allowed to cool to room temperature, and it is poured into lN HCl
(50 ml). Then, it is extracted with EtOAc, washed neutral with
water, dried, concentrated by evaporation and chromatographed
(silica gel, EtOAc/MeOH 9:1).
Yield: 35~
Cld: C 54.71% H 6.12% N 9.45% O 18.90~ S 10.82%
Fnd: C 54.40% H 6.45% N 9.25% S 10.04%
g) N-p-Toluenesulfonyl-{4-aminophenyl}-alanine-[N-(2-
(acetylmercapto)acetylaminoethyl)]amide, hydrochloride
85 mg of N-p-toluenesulfonyl-{4-[N-(tert-
butyloxycarbonyl)]aminophenyl}-alanine[N-(2-
(acetylmercapto)acetylaminoethyl)]amide is dissolved in 2 ml of
3 M HCl in ethyl acetate and stirred for 6 hours at room
temperature. After the solvent is drawn off, 70 mg of white
crystals remains.
3521 94561
,
Yield: 100%
Cld: C 49.95% H 5.53%N 10.59% 0 15.12% S 12.12%
Fnd: C 49.32% H 5.76%N 10.20% S 11.77%
h) N-p-Toluene~ulfonyl-{4-isothiocyanatophenyl}-alanine-tN-~2-
(acetylmercapto) a cetylaminoethyl)]amide
The solution of 11.5 mg of thiophosgene in a little
dichloromethane is added in drops with exclusion of moisture to a
solution of 53 mg of N-p-toluenesulfonyl-{4-aminophenyl}-alanine-
[N-(2-(acetylmercapto)acetylaminoethyl)]amide, hydrochloride and
20 ~l of triethylamine in 4 ml of dichloromethane, and it is
stirred for 4 hours at room temperature. After the solvent is
drawn off, 45 mg of yellowish crystals remains.
Yield: 84%
Cld: C 51.67% H 4.90% N 10.48% O 14.96% S 17.99%
Fnd: C 50.99% H 5.15% N 10.21% S 18.56%
Example 6
a) N-p-Toluene~ulfonyl-{4-tN-~tert-
butyloxycarbonyl)]aminophenyl}-alanine-tN-~2-
(benzoylmercapto)acetylaminoethyl)]amide
2 mmol of S-benzoyl-2-mercaptoacetic acid (394 mg) and 4
mmol of NEt3 (560 ~l) and 2 mmol of N-p-toluenesulfonyl-{4-[N-
(tert-butyloxycarbonyl)]aminophenyl}-alanine-tN-(2-
aminoethyl)]amide (953 mg) are mixed with 5 ml of dichloromethane
and cooled to 10~C. Then, 2 mmol (509 mg) of 1-
benzotriazolyloxy-tris-(dimethylamino)-phosphonium-
36 21 9456 1
hexafluorophosphate-chloride (designated below as BOP-Cl) is
added and stirred for 4 hours while being cooled in water, then
it is mixed with water and brought to pH 1-1.5 with 4N HCl.
Then, it is extracted with dichloromethane, the combined organic
extracts are washed with NaHCO3 and water, dried, concentrated by
evaporation and chromatographed.
Yield: 60%
Cld: C 58.70% H 5.85% N 8.56% O 17.10% S 9.79%
Fnd: C 58.04% H 6.03% N 8.39% S 9.30%
b) N-p-Toluenesulfonyl-t4-aminophenyl}-alanine-~N-~2-~benzoyl-
mercapto)acetyl-aminoethyl)]amide, hydrochloride
A freshly produced solution of 3 M HCl in EtOAc (10 ml, 30
mmol) is added to a solution of 654 mg of the protected amine
produced according to Example 6a) (1 mmol) in S ml of EtOAc. It
is stirred for 30 minutes at room temperature. The solvent is
drawn off, and the residue is dried in a vacuum at room
temperature.
Yield: 98%
Cld: C 54.86 H 5.29% N 9.48% O 13.53% S 10.85%
Fnd: C 53.90% H 5.52% N 9.09% S 9.97%
c) N-p-Toluenesulfonyl-t4-isothiocyanatophenyl}-alanine-~N-~2
(benzoylmercapto)-acetylaminoethyl)]amide
The solution of 115 mg of thiophosgene in a little
dichloromethane is added in drops with exclusion of moisture to a
solution of 296 mg of the title compound of Example 6b) (0.5
37 21 94561
mmol) and 200 ~1 of triethylamine in 4 ml of dichloromethane, and
it is stirred for 4 hours at room temperature. After the solvent
is drawn off, a residue remains, which is taken up in chloroform
and washed with 0.1% citric acid, washed twice with NaHC03
solution and once with water. After drying and concentration by
evaporation, 45 mg of yellowish crystals remains.
Yield: 85%
Cld: C 56.36 H 4.73% N 9.39% 0 13.14% S 16.12%
Fnd: C 56.31% H 4.98% N 9.08% S 17.01%
Example 7
a) N-(3-Nitrobenzenesulfonyl)glycine methyl ester
25.11 g of glycine methyl ester (0.2 mol) is dissolved in
500 ml of dichloromethane and mixed drop by drop with 44.3Z g of
3-nitrobenzenesulfonic acid chloride in 100 ml of dichloromethane
at 0~C. While being stirred intensively, 40 g of triethylamine,
50 ml of dichloromethane are added in drops at 0~C and stirred
for 1 hour at room temperature. After the reaction is completed
(TLC control), it is mixed with ice water and extracted several
times with dichloromethane. The combined organic extracts are
washed twice with cold 10% HCl, three times with 10% NaHC03
solution and twice with saturated NaCl solution. After drying,
the solvent is drawn off.
Yield: 82%
Cld: C 39.42% H 3.68% N 10.22% O 35.00% S 11.69%
Fnd: C 40.31% H 3.37% N 9.89% S 11.04%
21 94561
38
b) N-(3-Aminobenzenesulfonyl)glycine methyl ester
2.74 g (10 mmol) of the nitro compound produced according to
Example 7a) is hydrogenated with hydrogen in 50 ml of glacial
acetic acid in the presence of 1.5 g of palladium on activated
carbon (10%) at room temperature. After the reaction is
completed, the catalyst is filtered out, and the solvent is drawn
off.
Yield: 90%
Cld: C 44.26% H 4.95% N 11.47% 0 26.20% S 13.13%
Fnd: C 44.01% H 5.11% N 12.08% S 12.76%
c) N-t~3-Aminobenzenesulfonyl)]glycyl-tN~-(2-aminoethyl)]amide
- A solution of 1 g of the N-(3-aminobenzenesulfonyl)-glycine
methyl ester (3.6 mmol) in 2.5 ml of anhydrous dichloromethane is
slowly added in drops, refluxed, to a solution of 2.2 g of
ethylenediamine (36 mmol) in 2.5 ml of anhydrous dichloromethane.
After the reaction is completed, it is concentrated by
evaporation in a vacuum, and the residue is chromatographed
(silica gel, CH2Cl2/MeOH 9:1).
Yield: 76%
Cld: C 44.11% H 5.92% N 20.57% O 17.63% S 11.78%
Fnd: C 43.09% H 6.41% N 21.82% S 10.68%
d) N-~(3-Aminobenzenesulfonyl)~glycyl-{N'-
[~acetylmercapto)acetyl~2-aminoethyl)}amide
250 mg (1.08 mmol) of SATA (SIGMA Chemie [SIGMA Chemistry],
1994; A 9043) in THF is slowly added in drops under argon and
39 2 1 9456 1
,
with exclusion of moisture to a solution of 272 mg of amine (1
mmol) produced according to Example 7c) in anhydrous THF at 0~C,
and it is stirred for 2 hours at 0~C, then for 12 hours at room
temperature. The solvent is drawn off, and the residue is
chromatographed (silica gel, CH2Cl2/MeOH 9:1).
Yield: 60%
Cld: C 43.29% H 5.19% N 14.42% O 20.59% S 16.51%
Fnd: C 42.45% H 5.23% N 15.64% S 15.36%
e) N-t(3-Isothiocyanatobenzenesulfonyl)]glycyl-{N'-
1(acetylmercapto)acetyll-~2-aminoethyl)}amide
The solution of 65 mg of thiophosgene in a little
dichloromethane is added in drops to a solution of 200 mg of N-
[(3-aminobenzenesulfonyl)]glycyl-{N'-[(acetylmercapto)-acetyl]-
(2-aminoethyl)}amide and 20 ~l of triethylamine in 4 ml of
dichloromethane with exclusion of moisture, and it is stirred for
4 hours at room temperature. After the solvent is drawn off, a
residue remains, which is taken up in chloroform and washed with
0.1% citric acid, washed twice with NaHCO3 solution and once with
water. After drying and concentration by evaporation, 197 mg of
yellowish crystals remains.
Yield: 89%
Cld: C 41.85% H 4.21% N 13.01% O 18.58% S 22.35%
Fn~: C 40.44% H 3.93~ N 12.57% S 23.66%
21 94561
Example 8
a) N-Benzyloxycarbonyl-0-t-butyloxycarbonylmethyl-~tyro~ine
methylester)
3.29 g of the Z1-protected tyrosine methyl ester produced
according to Example la) is dissolved in anhydrous DMF and mixed
with 1.12 g of potassium-t-butylate. After 30 minutes, the
solution of 1.95 g of bromoacetic acid-t-butyl ester is added in
drops and heated for 4 hours to 110~C, then it is stirred for 4
more hours at room temperature. The mixture is added to water,
extracted with CH2Cl2, washed, dried and concentrated by
evaporation. After column chromatography (silica gel, CHzCl2/EE
19:1), 2.5 g of yellow oil remains.
Yield: 57%
Cld: C 65.00% H 6.59% N 3.16% 0 25.25%
Fnd: C 64.65% H 6.82% N 3.09%
b) 0-(t-Butyloxycarbonylmethyl)-tyro~ine methyl ester
1.5 g (3.4 mmol) of the Z1-protected compound produced
according to Example 8a) is hydrogenated with hydrogen in 50 ml
of ethyl acetate in the presence of 1.5 g of palladium on
activated carbon (10%) at 50~C. After the reaction is completed,
the catalyst is filtered out, and the solvent is drawn off. 1.0
g of colorless oil remains.
Yield: 99%
Cld: C 62.12% H 7.49% N 4.53% 0 25.86%
Fnd: C 61.88% H 7.67% N 4.47%
41 2 1 9456 1
c) N-Toluenesulfonyl-O-t-butyloxycarbonylmethyl-tyrosine methyl
ester
The solution of 570 mg of toluenesulfonyl chloride in CH2Clz
is added in drops to a solution of 920 mg of O-(t-
butyloxycarbonylmethyl)-tyrosine methyl ester in CH2Cl2 at 0~C,
and it is then slowly mixed with 300 mg of triethylamine and
stirred overnight. It is poured onto ice water, extracted with
CH2Cl2, the organic phase is washed twice with 10% HCl, twice
with 10% NaHCO3 and twice with saturated common salt solution,
dried and concentrated by evaporation. After treatment with
ether, 900 mg of white crystals is obtained.
Yield: 65%
Cld: C 59.60% H 6.31% N 3.02% O 24.16% S 6.92%
Fnd: C 59.38% H 6.55% N 3.08% S 6.66%
d) N-Toluenesulfonyl-O-t-butyloxycarbonylmethyl-N-(2-
aminoethyl)tyrosinamide
The solution of 3.0 g of N-toluenesulfonyl-O-t-
butyloxycarbonylmethyl-tyrosine methyl ester (6.5 mmol) in CH2Clz
is added in drops to a solution of 5 g of ethylenediamine in 50
ml of CHzCl2. Then, it is refluxed for 4 hours. After cooling,
it is mixed with water, the organic phase is separated and
extracted several times with CH2Clz. The combined organic
extracts are washed, dried and concentrated by evaporation.
After column chromatography (silica gel NeOH), l.S g of colorless
oil remains.
Yield: 47%
42 2 1 9456 1
Cld: C 58.64% H 6.77% N 8.55% O 19.53% S 6.52%
Fnd: C 58.39% H 6.91% N 8.38% S 6.56%
e) N-Toluenesulfonyl-O-carboxymethylt-N-~2-
aminoethyl)tyrosinamide]
2 ml of trifluoroacetic acid is added at room temperature to
a solution of 491 mg of the tert-butyl ester (1 mmol) produced
according to Example 8d) in 25 ml of dichloromethane, and it is
stirred at room temperature. After the reaction is completed,
the trifluoroacetic acid is drawn off in a vacuum, the residue is
taken up in chloroform, washed with water, dried and concentrated
by evaporation.
Yield: 83%
Cld: C 55.16% H 5.79% N 9.65% O 22.04% S 7.36%
Fnd: C 53.88% H 5.64% N 10.02% S 7.44%
f) N-Toluenesulfonyl-O-carboxymethyl-tyrosine-[N-~2-
~piperonylmercapto)-acetylaminoethyl)]amide
350 mg of piperonylmercaptoacetic acid-N-
hydroxysuccinimidoester (1.08 mmol) in THF is slowly added in
drops under argon and with exclusion of moisture to a solution of
435 mg of amine (1 mmol) produced according to Example 8e) in
anhydrous THF at 0~C, and it is stirred for 2 hours at 0~C, then
for 12 hours at room temperature. The solvent is drawn off, and
the residue is chromatographed (silica gel, CH2Cl2/MeOH/HOAC
9:1:0.1) .
Yield: 58%
2 1 9456 1
Cld: C 55.98% H 5.17% N 6.53% O 22.37% S 9.96%
Fnd: C S5.38% H 5.04% N 6.68% S 9.09%
g) N-Toluenesulfonyl-O-carboxymethyl-tyrosine-{N-t2-
~mercaptoacetyl)-aminoethyl]}amide
644 mg of the protected S-compound (1 mmol) produced
according to Example 8f) and a trace of anisole are added to 10
ml of trifluoroacetic acid at room temperature and heated briefly
to boiling. After the reaction is completed, the trifluoroacetic
acid is drawn off in a vacuum, the residue is taken up in a
suitable solvent, washed with water, dried and concentrated by
evaporation and chromatographed.
Yield: 34%
Cld: C 51.85% H 5.34% N 8.25% O 21.98% S 12.59
Fnd: C 51.62% H 5.53% N 8.65% S 13.61%
h) Tc-99m complex of N-toluenesulfonyl-O-carboxymethyl-
tyrosine-{N-t2-(mercaptoacetyl)aminoethyl]}amide
1 mg of the compound produced according to Example 8g) is
dissolved in 100 ~l of EtOH. 50 ~l of this solution is added to
250 ~l of a 0.1 M phosphate buffer of pH 8.5 and mixed with 50 ~l
of a citrate solution (50 mg of trisodium citrate in 1 ml of
water) and 2.5 ~l of a tin(II) chloride solution (5.0 mg of
tin(II) chloride in 1 ml of 0.lN HCl). Then, it is mixed with
100 ~l of a 99m-Tc generator eluate and allowed to stand for 15
minutes. The labeling yield is determined with the aid of HPLC.
44 21 ~4561
Example 9
a) Cholesteryl diethylene glycol ~DEG-cholesterol)
A solution of 54.1 g of cholesteryl toluenesulfonate (100
mmol) and 106 g of diethylene glycol in 250 ml of anhydrous
dioxane is refluxed under nitrogen atmosphere (TLC control).
After the reaction is completed, it is mixed with water and
extracted with CH2Cl2. After column chromatography (silica gel
CH2Cl2/MeOH 9:1), a colorless solid remains.
Yield: 81%
Cld: C 78.43% H 11.46% 0 10.11%
Fnd: C 76.99% H 12.32%
b) Cholesteryl-2-chloroethyl(ethylene glycol)
A solution of 4.75 g of DEG-cholesterol (10 mmol) produced
according to Example 9a) in 50 ml of anhydrous carbon
tetrachloride is mixed under nitrogen atmosphere with 3.14 g of
pulverized triphenylphosphine (12 mmol) and refluxed. After
cooling, it is diluted with 50 ml of petroleum ether or hexane
and stored for some time at -20~C. The precipitate is suctioned
off, and the procedure is again as above until no more
precipitate settles. Then, it is dried and concentrated by
evaporation. After column chromatography (silica gel CH2Cl2/MeOH
9:1), 3.70 g of an oil remains.
Yield: 75~
Cld: C 75.49% H 10.83% 0 6.49%
Fnd: C 74.73% H 11.74%
2 1 9456 1
c) N-Benzyloxycarbonyl-2-t(4-cholesteryldiethylene-
glycolyl)benzyl]-2-aminoacetic acid methyl ester
The solution of 493 mg of the cholesterol derivative (1
mmol) (produced according to Example 9b) in 10 ml of toluene is
added to a boiling solution of 295 mg of the N-benzyloxycarbonyl-
tyrosine methyl ester (l mmol) (produced according to Example la)
and 183 mg of potassium carbonate (1 mmol) in 10 ml of toluene,
and it is refluxed for 6 hours. After the reaction is completed,
it is allowed to cool to room temperature, concentrated by
evaporation and chromatographed (silica gel petroleum ether/ethyl
acetate 1:1)
Yield: 29%
Cld: C 73.46% H 9.78% N 1.86% 0 14.89%
Fnd: C 73.28% H 10.01% N 1.70%
d) 2-[(4-Cholesteryldiethyleneglycolyl)benzyl~-2-aminoacetic
acid methyl ester hydrochloride
7.52 g (10 mmol) of the Z-protected compound produced
according to Example 9c) is dissolved in 50 ml of 3 M HCl in
ethyl acetate and stirred for 6 hours at room temperature. After
the solvent is drawn off, 6.02 g of white crystals remains.
Yield: 93%
Cld: C 71.53% H 9.66% N 2.04% 0 11.62%
Fnd: C 71.82% H 9.39% N 2.01%
46 2 1 9456 1
e) N-p-Toluenesulfonyl-2[~4-
cholesteryldiethyleneglycolyl)benzyl]-2-aminoacetic acid
methyl ester
19.53 g of the amine (30 mmol) produced according to Example
9d) is dissolved in 50 ml of dichloromethane and mixed at 0~C
with 5.72 g of toluenesulfonic acid chloride in 30 ml of
dichloromethane. While being stirred intensively, 3.0 g of
triethylamine is added in drops at 0~C, and it is stirred for 1
hour at room temperature. After the reaction is completed, it is
mixed with ice water and extracted several times with
dichloromethane. The combined organic extracts are washed twice
with cold 10% HCl, three times with 10% NaHC03 solution and twice
with saturated NaCl solution. After drying, the solvent is drawn
off and chromatographed (silica gel, CH2Cl2).
Yield: 77%
Cld: C 71.52% H 8.88% N 1.74% 0 13.89% S 3.98%
Fnd: C 70.89% H 9.02% N 1.66% S 3.70%
f) N-p-Toluenesulfonyl-2[~4-
cholesteryldiethyleneglycolyl)benzyl]-2-aminoacetic acidtN-
(2-aminoethyl)]amide
A solution of 806 mg of the tosylglycine ester (1 mmol)
produced according to Example 9e in 1 ml of anhydrous
dichloromethane is slowly added in drops to a solution of 1.2 g
of ethylenediamine (20 mmol) in 1 ml of anhydrous
dichloromethane, and it is refluxed. After the reaction is
2 1 9456 1
,
completed, it is concentrated by evaporation in a vacuum, and the
residue is chromatographed (silica gel, MeOH).
Yield: 85%
Cld: C 70.55% H 9.06% N 5.04% O 11.51% S 3.84%
Fnd: C 69.24% H 9.31~ N 4.83% S 3.71%
g) N-p-Toluenesulfonyl-2tt4-cholesteryldiethyleneglycolyl)-
benzyl]-2-aminoacetic acid-N-{2-Nt(3-carboxy-2-
mercaptoacetyl-l-oxopropyl)]aminoethyl}amide
0.38 g (2.2 mmol) of 2-acetylmercaptosuccinic anhydride is
added to a solution of 1.83 g of the amine (2.2 mmol) produced
according to Example 9f) in 10 ml of pyridine/DMF (50:50), and it
is stirred for 4 hours at room temperature. Then, the solvent is
drawn off, the residue is taken up in 0.5N HCl and extracted with
CH2Cl2. After flash chromatography, 887 mg of a yellow oil
remains.
Yield: 40%
Cld: C 65.51% H 8.10% N 4.17% O 15.87% S 6.36%
Fnd: C 63.66% H 7.58% N 3.99% S 7.41%
h) Tc-99m complex of N-p-toluenesulfonyl-2t~4-
cholesteryldiethyleneglycolyl)-benzyl]-2-aminoacetic acid-N-
{2-Nl(3-carboxy-2-mercaptoacetyl-1-oxopropyl)]-
aminoethyl}amide
1 mg of the ligand produced according to Example 9g is
dissolved in 100 ~l of ethanol. 50 ~l of this solution is added
to 250 ~l of a 0.1 M phosphate buffer (pH 8.5) and then mixed
48 2 1 9456 1
with 50 ~l of a citrate solution (50 mg of trisodium citrate in
1.0 ml of water) and 2.5 ~l of a tin(II) chloride solution (5.0
mg of tin(II) chloride in 1 ml of O.lN HCl). Then, it is mixed
with 50 ~l of a Tc-99m generator eluate and allowed to stand for
15 minutes. The labeling yield is determined with the aid of
HPLC.
Example 10
a) l-Tosyl-1,4,7-triazaheptan-3-one
A solution of 25.73 g of tosylglycine methyl ester (0.1 mol)
in anhydrous dichloromethane is slowly added in drops to a
solution of 30 g of ethylenediamine (0.5 mol) in 50 ml of
anhydrous dichloromethane. It is stirred for 12 hours at room
temperature and then concentrated by evaporation in a vacuum. 24
g of a yellow oil remains.
Yield: 88%
Cld: C 48.69% H 6.32% N 15.49% 0 17.69% S 11.82%
Fnd: C 47.23% H 6.67% N 15.34% S 11.54%
b) 7-N-tert-Butoxycarbonyl-1-tosyl-1,4,7-triazaheptan-3-one
A solution of 27 g (100 mmol) of the amino compound produced
according to Example lOa) in 500 ml of dioxane is mixed in one
portion with 74 g of di-tert-butyl-dicarbonate (340 mmol). The
solution is stirred for 3 hours at room temperature, ~hen poured
onto ice water and extracted three times with ethyl acetate. The
combined organic phases are washed three times with water and
once with saturated common salt solution, dried (MgSO4),
49 2194561
concentrated by evaporation and recrystallized. 27.9 g of white
crystals remains.
Yield: 75%
Cld: C 51.74~ H 6.78% N 11.31% 0 21.54% S 8.63%
Fnd: C 51.41% H 6.96% N 11.12% S 8.91%
c) 7-N-t-Butyloxycarbonyl-1-n-hexyl-1-tosyl-1,4,7-triazaheptan-
3-one
5 g of potassium carbonate is added to a solution of 3.71 g
of the sulfonamide (10 mmol) produced according to Example lOb)
in 50 ml of DMF at room temperature, and then the solution of
2.54 g of hexyl iodide (12 mmol) in 10 ml of DMF is added in
drops and heated for 3 hours to 110~C. After the reaction is
completed, it is allowed to cool to room temperature, poured onto
ice, extracted with CH2Cl2, washed several times with water,
dried, filtered and concentrated by evaporation. The residue is
recrystallized from ethyl acetate.
Yield: 89%
Cld: C 58.00% H 8.19% N 9.22% 0 17.56% S 7.04%
Fnd: C 57.37% H 7.87% N 9.01% S 6.95%
d) l-n-Hexyl-1-tosyl-1,4,7-triazaheptan-3-one
456 mg of the sulfonamide (1 mmol) produced according to
Example lOc) is added to 10 ml of trifluoroacetic acid at 0~C,
and then it is stirred for 2 hours at room temperature. After
the reaction is completed, it is poured onto ice, weakly
2 1 9456 1
alkalized (NaHCO3), extracted, concentrated by evaporation and
dried.
Yield: 94%
Cld: C 57.44% H 8.22% N 11.82% O 13.50% S 9.02%
Fnd: C 57.96% H 8.01% N 11.63% S 8.75%
e) 10-Acetyl-9-carboxymethyl-1-n-hexyl-1-to~yl-10-thia-1,4,7-
triazadecane-3,8-dione
The solution of 3.55 g (lO mmol) of the amino compound in 10
ml of DMF is slowly added in drops to a solution of 1.91 g of
acetylmercaptosuccinic anhydride (20 mmol) in 20 ml of DMF at
room temperature, and it is stirred overnight. Then, it is
poured onto semiconcentrated HCl, extracted with CH2Cl2 and
chromatographed (silica gel, EtOAc/MeOH from 9:1 to 1:1).
Yield: 66%
Cld: C 52.16% H 6.66% N 7.93% O 21.15% S 12.11%
Fnd: C 52.85% H 6.87% N 7.52% S 11.66%
f) Tc-99m complex of 10-Acetyl-9-carboxymethyl-1-n-hexyl-1-
tosyl-10-thia-1,4,7-triazadecane-3,8-dione
1 mg of N,N thexyl-p-toluenesulfonyl]glycyl-N-[(3-carboxy-2-
mercaptoacetyl-1-oxopropyl)]aminoethyl}amide is dissolved in 100
~l of EtOH. 50 ~l of this solution is diluted with 100 ~l of
EtOH and mixed with 100 ~l of a 0.1 M phosphate buffer at pH 7.5
and 100 ~l of a Tc-99m-gluconate solution. The labeling yield is
> 95% (silica gel, 95% EtOH)
2 1 ~4561
51
Example 11
a) N-~-Toluenesulfonyl-N-~-tert-butyloxycarbonyllysine methyl
ester
1.91 mg of toluenesulfonyl chloride is added in portions to
a solution of 2.60 g of N-~-tert-butyloxycarbonyllysine methyl
ester (10 mmol) in 50 ml of anhydrous pyridine at 0~C, and it is
allowed to stand for 24 hours at 4~C. Then, it is poured onto
ice water, and the precipitate is separated and recrystallized.
Yield: 78%
Cld: C 55.05% H 7.30% N 6.76% 0 23.16% S 7.74%
Fnd: C 54.67% H 7.42% N 6.64% S 7.54%
b) N-~-Toluenesulfonyl-N-~-tert-butyloxycarbonyllysine-[N-~2-
aminoethyl)]amide
A solution of 415 mg of the tosyllysine ester (1 mmol)
produced according to Example lla) in 1 ml of anhydrous
dichloromethane is slowly added in drops to a solution of 600 mg
of ethylenediamine (10 mmol) in 1 ml of anhydrous dichloromethane
and refluxed. After the reaction is completed, it is
concentrated by evaporation in a vacuum, and the residue is
recrystallized.
Yield: 59%
Cld: C 54.28% H 7.74% N 12.66% 0 18.08% S 7.25%
Fnd: C 54.54% H 7.57% N 12.18% S 7.06%
21 94561 -
52
c) N-~-Toluenesulfonyl-N-~-tert-butyloxycarbonyllysine-[N-(2-
~piperonylmercapto)-acetylaminoethyl)]amide
350 mg of piperonylmercaptoacetic acid-N-
hydroxysuccinimidoester (1.08 mmol) in THF is slowly added in
drops under argon and with exclusion of moisture-to a solution of
443 mg of amine (1 mmol) produced according to Example llb) in
anhydrous THF at 0~C, and it is stirred for 2 hours at 0~C, then
for 12 hours at room temperature. The solvent is drawn off, and
the residue is chromatographed (silica gel, CH2Cl2/MeOH 9:1).
Yield: 54%
Cld: C 55.37% H 6.51% N 8.61% 0 19.67% S 9.85%
Fnd: C 55.22% H 6.85% N 8.41% S 9.66%
d) N-~-Toluenesulfonyl-lysine-[N-(2-
(piperonylmercapto)acetylaminoethyl)]amide
651 mg of N-~-toluenesulfonyl-N-~-tert-
butyloxycarbonyllysine-[N-(2-(piperonylmercapto)-
acetylaminoethyl)]amide is dissolved in 5 ml of 3 M HCl in ethyl
acetate, and it is stirred for 6 hours at room temperature.
After the solvent is drawn off, 530 mg of white crystals remains.
Yield: 96%
Cld: C 54.63% H 6.05~ N 10.19% 0 17.46% S 11.67
Fnd: C 54.40% H 6.47~ N 10.01% S 11.84%
2194561
Example 12
a) N-~-Toluenesulfonyl-N-~-tert-butyloxycarbonyllysine-tN-~2-
benzoylmercapto)-acetylaminoethyl)]amide
316 mg of benzoylmercaptoacetic acid-N-
hydroxysuccinimidoester (1.08 mmol) in tetrahydrofuran is slowly
added in drops under argon and with exclusion of moisture to a
solution of 443 mg of amine produced according to Example llb) in
anhydrous tetrahydrofuran at 0~C, and it is stirred for 2 hours
at 0~C, then for 12 hours at room temperature. The solvent is
drawn off, and the residue is chromatographed (silica gel,
CH2Clz/MeOH 9:1).
Yield: 65%
Cld: C 56.11% H 6.50% N 9.03% 0 18.04% S 10.33%
Fnd: C 55.98% H 6.86% N 8.88% S 10.05%
b) N-~-Toluenesulfonyl-lysine-tN-(2-
benzoylmercapto)acetylaminoethyl)lamide
621 mg of the title compound of Example 12a) is dissolved in
5 ml of 3 M HCl in ethyl acetate, and it is stirred for 6 hours
at room temperature. After the solvent is drawn off, 500 mg of
white crystals remains.
Yield: 90%
Cld: C 51.74% H 5.97% N 10.06% 0 14.36% S 11.51%
Fnd: C 51.40% H 6.14% N 10.10% S 11.48%
54 21 q4561
c) Tc-99m complex of N-~-toluenesulfonyl-lysine-lN-~2-
~benzoylmercapto)-acetylaminoethyl)]amide
10 mg of the amide produced according to Example 12b) is
mixed with 500 ~1 of lN NaOH and allowed to stand for lS minutes.
50 ~1 of this solution is added to 250 ~l of a 0.1 M phosphate
buffer of pH 8.s and mixed with 50 ~l of a citrate solution (So
mg of trisodium citrate in 1.0 ml of water) and 2.5 ~1 of a
tin(II) chloride solution (5.0 mg of tin(II) chloride in 1.0 ml
of 0.lN HCl). Then, it is mixed with 50 ~1 of a Tc-99m generator
eluate and allowed to stand for 15 minutes. The labeling yield
is determined with the aid of HPLC (> 95%).
Example 13
a) N-t3-~N-tert-butyloxycarbonyl)aminobenzenesulfonyl]-glycine
methyl ester
A solution of 2.12 g (10 mmol) of the amino compound
produced according to Example 7b) and 3 g of triethylamine (30
mmol) in 50 ml of dioxane are mixed in one portion with 7.4 g of
di-tert-butyldicarbonate (34 mmol). (Gas generation!) The
solution is stirred for 30 minutes at room temperature, then
poured onto ice water and extracted three times with ethyl
acetate. The combined organic phases are washed three times with
water and once with saturated common salt solution, dried
(MgSO4), concentrated by evaporation and recrystallized.
Yield: 85%
Cld: C 48.83% H s.85% N 8.13% o 27.88% S 9.31%
Fnd: C 47.44% H 5.93% N 8 . 57% S 9 . 66%
21 94561
b) N-t3-(N-tert-butyloxycarbonyl)aminobenzenesulfonyl]-glycyl-
[N'-(2-aminoethyl)amide
A solution of 12.11 g of the glycine ester produced
according to Example 13a) (35 mmol) in 250 ml of anhydrous
toluene/dioxane or optionally only dioxane is slowly added in
drops to a boiling solution of 42 g of ethylenediamine (700 mmol)
in 100 ml of anhydrous toluene, and it is refluxed. After the
reaction is completed, it is concentrated by evaporation in a
vacuum, and the residue is taken up in chloroform and washed with
water.
Yield: 78%
Cld: C 44.06% H 6.16% N 13.70% O 19.56% S 7.84%
Fnd: C 45.44% H 6.63% N 14.57% S 7.66%
c) N-~3-(N-tert-Butyloxycarbonyl)aminobenzenesulfonyl]-glycyl-
~N'-tt8-benzoylmercapto)acetyl]-(2-aminoethyl)}amide
5 mmol of the S-benzoylmercaptoacetic acid (0.98 g) and 10
mmol of NEt3 (1.4 ml) and 5 mmol of the amine produced according
to Example 13b) (1.86 g) are mixed with 50 ml of dichloromethane
and cooled to 10~C. Then, 5.5 mmol (1.375 g) of BOP-Cl is added
and stirred while being cooled in water. (After 10-20 minutes, a
clear solution is obtained.) Then, it is stirred, stirred for 1
more hour, mixed with water and brought to pH 1-1.5 with 4N HCl.
Yield: 62%
Cld: C 52.16% H 5.84% N 10.14% O 20.27% S 11.60%
Fnd: C 53.44% H 6.63% N 10.57% S 11.66%
2 1 9456 1
56
d) N-[3-Aminobenzenesulfonyl]-glycyl-{N'-t(S-
benzoylmercapto)acetyl]-~2-aminoethyl)}amide
A freshly prepared solution of 3 M HCl in EtOAc (10 ml, 30
mmol) is added to a solution of 552 mg of the protected amine
produced according to Example 13c) (1 mmol) in 5 ml of EtOAc. It
is stirred for 30 minutes at room temperature. The solvent is
drawn off, and the residue is dried in a vacuum at room
temperature.
Yield: 93%
Cld: C 46.86% H 4.76% N 11.51% O 16.43% S 13.17%
Fnd: C 45.44~ H 5.33% N 10.57% S 13.66%
e) N-[3-Isothiocyanatobenzene~ulfonyl]-glycyl-{N'-t(S-
benzoylmercapto)acetyl]-(2-aminoethyl)}amide
The solution of 65 mg of thiophosgene in a little
dichloromethane is added in drops with exclusion of moisture to a
solution of 200 mg of N-[(3-aminosulfonyl)]glycyl-{N'-
[(acetylmercapto)-acetyl](2-aminoethyl)}amide produced according
to Example 13d) and 20 ~l of triethylamine in 4 ml of
dichloromethane, and it is stirred for 4 hours at room
temperature. After the solvent is drawn off, a residue remains,
which is taken up in chloroform and washed with 0.1% citric acid,
washed twice with sodium chloride solution and once with water,
dried, concentrated by evaporation and chromatographed.
Yield: 66%
Cld: C 48.77% H 4.09% N 11.37% O 16.24% S 19.53%
Fnd: C 48.44% H 4.63% N 11.75% S 20.06%
2 I q456 1
57
Example 14
a) N-Toluenesulfonyl-O-methoxycarbonylmethyl[-N-(2-
~minoethyl)tyro~inamide]
HCl gas is introduced until saturation in a solution of4.35 g of amine produced according to Example 8e) (10 mmol) in
100 ml of anhydrous methanol, and it is stirred overnight. After
the solvent is removed, a crystalline residue remains.
Yield: 92%
Cld: C 51.90% H 5.81% N 8.65% 0 19.75% S 6.60%
Fnd: C S1.59% H 5.93% N 8.47% S 6.49%
b) N-Toluenesulfonyl-0-methoxycarbonylmethyltyro~ine-[N-(2-
tritylmercapto)-acetylaminoethyl)]amide
4.66 g of S-tritylmercaptoacetic acid-N-
hydroxysuccinimidoester (10.8 mmol) in tetrahydrofuran is slowly
added in drops under argon to a solution of 4.35 g of amine
produced according to Example 14a) (10 mmol) and 2.02 g of
triethylamine in tetrahydrofuran/water at 0~C, and it is stirred
for 2 hours at 0~C, then for 12 hours at room temperature. The -
solvent is drawn off, and the residue is chromatographed (silica
gel, CH2C12)-
Yield: 68%
Cld: C 65.86% H 5.66% N 5.49% 0 14.62% S 8.37%
Fnd: C 65.66% H 5.71% N 5.36% S 8.21%
58 2194561
c) N-Toluenesulfonyl-O-carboxymethyltyrosine-tN-(2-
tritylmercapto)acetylaminoethyl)]amide
The solution of 766 mg of the methyl ester produced
according to Example 14b) (1 mmol) in 2.5 ml of methanol is added
to a methanolic KOH solution and stirred at room temperature.
After the reaction is completed, the solvent is drawn off, the
potassium salt is taken up in water, weakly acidified and
extracted with chloroform. The organic extract is washed with
water, dried and concentrated by evaporation.
Yield: 63%
Cld: C 65.49% H 5.50% N 5.59% O 1490~ S 8.53%
Fnd: C 65.26% H 5.72% N 5.43% S 8.43%
d) N-Toluenesulfonyl-~(HOOC-Trp-Ile-Ile Asp-Leu-His Gly-NH)-
carbonylmethyltyrosine-tN-(2-
tritylmercapto)acetylaminoethyl)]amide
206 mg of dicyclohexylcarbodiimide (1 mmol) dissolved in 2
ml of dimethylformamide is added in drops to a solution of 1 mmol
of the acid (980 mg) produced according to Example 14c) and 115
mg of N-hydroxysuccinimide (1 mmol) in 2 ml of anhydrous
dimethylformamide at 0~C within 5 minutes. It is stirred first
for another 30 minutes at 0~C, then the solution of 1 mmol of the
peptide (853 mg) H2N-Gly-His-Leu-Asp-Ile-Ile-Trp (produced
analogously to Barany and Merrifield, The Peptides: Analysis,
Biology, Academic Press New York, 1980; Steward and Young, Solid
Phase Peptides Syntheses, 2nd ed.; Pierce Chemical W., Rockford,
II, 1984) and 304 mg (3 mmol) of triethylamine in 10 ml of
59 2194561
anhydrous dimethylformamide is added under argon atmosphere
within 30 minutes. It is stirred for 12 more hours at room
temperature, 200 ~l of glacial acetic acid is added, stirred
again for 30 minutes, the product is filtered off from N,N'-
dicyclohexylurea, and the residue is concentrated by evaporation.
After stirring up with diethyl ether, a white residue remains,
which is recrystallized from dimethylformamide/ether.
Yield: 35%
Cld: C 62.07% H 6.29% N 4.04% O 16.13% S 4.04%
Fnd: C 61.77% H 6.52% N 4.32% S 4.34%
e) N-Toluenesulfonyl-[(HOOC-Trp-Ile-Ile Asp-Leu-Hi~ Gly-
NH)carbonyl]-methyltyroqine-[N-
(mercapto)acetylaminoethyl)]amide
530 mg of the protected S-compound produced according to
Example 14d) (0.3 mmol) and a trace of anisole are added to 10 ml
of trifluoroacetic acid under argon atmosphere at room
temperature and heated briefly to boiling. Then, the
trifluoroacetic acid is drawn off in a vacuum, the residue is
taken up in tetrahydrofuran, concentrated by evaporation and
chromatographed.
Yield: 59%
Cld: C 56.67% H 6.32~ N 13.42% O 18.87% S 4.73%
Fnd: c 56.14~ H 6.48~ N 12.98~ S 4.81%
2 1 9456 1
f) Tc-99m complex of N-toluenesulfonyl-[~HOOC-Trp-Ile-Ile Asp-
~eu-Hi-Q Gly-NH)carbonylmethyltyro-Qine-tN-
(mercaptoacetyl)aminoethyl)]amide
1 mg of the above-named compound (Example 14e) is dissolved
in 100 ~l of EtOH/water 1:1. 50 ~l of this solution is mixed
with 100 ~l of a 0.1 M phosphate buffer of pH 9.5 and 100 ~1 of a
Tc-99m-gluconate solution. The labeling yield is > 95% (HPLC,
LiChrospher RP18, H2O/MeCN + 0.1% TFA).
Example 15
a) N-Toluenesulfonyl-O-t-butyloxycarbonylmethyltyrosine-tN-(2-
benzoylmercapto)-acetylaminoethyl)]amide
2 mmol of S-benzoyl-2-mercaptoacetic acid (394 mg) and 4
mmol of NEt3 (560 ~1) and 2 mmol (982 mg) of the compound
obtained according to Example 8d) are mixed with 5 ml of
dichloromethane and cooled to 10~C. Then, 2 mmol (509 mg) of
BOP-Cl is added and stirred for 12 hours while being cooled in
water, then mixed with water and brought to pH 1-1.5 with 4N HCl.
Then, it is extracted with dichloromethane, the combined organic
extracts are washed with NaHCO3 and water, dried, concentrated by
evaporation and chromatographed.
Yield: 76%
Cld: C 59.18% H 5.87% N 6.27% O 19.11% S 9.58%
Fnd: c 60.88~ H 5.89% N 5.63% S 8.74%
61 2194561
b) N-Toluenesulfonyl-O-carboxymethyltyrosine-tN-(2-
benzoylmercapto)acetyl-aminoethyl)]amide
644 mg of the protected S-compound produced according to
Example 15a) (1 mmol) is added to 10 ml of trifluoroacetic acid
at room temperature, and it is stirred at room temperature.
After the reaction is completed, the trifluoroacetic acid is
drawn off in a vacuum, the residue is taken up in ethyl acetate,
washed with water, dried and concentrated by evaporation and
chromatographed.
Yield: 77%
Cld: C 56.76% H 5.09% N 6.85% 0 20.86% S 10.45%
Fnd: C 57.14% H 5.68% N 7.23% S 11.31~
c) N-Toluenesulfonyl-[(HOOC-Trp-Ile-Ile Asp-~eu-(D-Trp)-
NH)carbonylmethyl-tyrosine-[N-(2-
benzoylmercapto)acetylaminoethyl)1amide
206 mg of dicyclohexylcarbodiimide (1 mmol) dissolved in 2
ml of tetrahydrofuran is added in drops to a solution of 1 mmol
of acid (614 mg) produced according to Example 15b) and 115 mg of
N-hydroxysuccinimide (1 mmol) in 2 ml of anhydrous
tetrahydrofuran at 0~C within 5 minutes. First, it is stirred
for another 30 minutes at 0~C, then the solution of 1 mmol of the
peptide (845 mg) of H2N-(D-Trp)-Leu-Asp-Ile-Ile-Trp (produced
analogously to Barany and Merrifield, The Peptides: Analysis,
Biology, Academic Press, New York, 1980; Steward and ~oung, Solid
Phase Peptides Syntheses, 2nd ed.; Pierce Chemical W., Rockford,
Il 1984) and 304 mg (3 mmol) of triethylamine in 10 ml of
62 2 1 9456 1
anhydrous dimethylformamide are added under argon atmosphere
within 30 minutes. It is stirred for 12 more hours at room
temperature, 200 ~l of glacial acetic acid is added, stirred
again for 30 minutes, the product is filtered off from N,N'-
dicyclohexylurea, and the residue is extracted twice with boiling
tetrahydrofuran. The combined filtrates are evaporated to
dryness and chromatographed (silica gel, CH2Cl2).
Yield: 40%
Cld: C 60.86% H 6.23% N 10.69% O 17.77% S 4.45%
Fnd: C 60.77% H 6.42% N 10.32% S 4.65%
d) Tc-99m complex of N-toluene~ulfonyl-t(HOOC-Trp-Ile-Ile Asp-
Leu-(D-Trp)-NH)carbonylmethyltyro~ine-tN-(2-
mercaptoacetyl)aminoethyl)]amide
2 mg of the compound produced according to Example 15c) is
dissolved in 100 ~l of EtOH and mixed with 100 ~l of lN NaOH.
After 15 minutes, 50 ~l of this solution is added to 250 ~l of a
0.1 M phosphate buffer of pH 8.5 and mixed with 50 ~l of a
citrate solution (50 mg of trisodium citrate in 1.0 ml of water)
and 2.5 ~l of a tin(II) chloride solution (5.0 mg of tin(II)
chloride in 1.0 ml of 0.lN HCl). Then, it is mixed with 50 ~l of
a Tc-99m generator eluate and allowed to stand again for 15
minutes. The labeling yield (> 95%) is determined with the aid
of HPLC.
63 2194561
...
Example 16
a) N-Tosyl-tyro~inemethyle~ter-4-benzyl ether
A solution of 32.58 g (171 mmol) of p-toluenesulfonic acid
chloride in 100 ml of pyridine is added in drops to 50 g (155.37
mmol) of tyrosinemethylester-4-benzyl ether hydrochloride in 300
ml of pyridine at 0~C, and it is stirred for 3 hours at 0~C. It
is evaporated to dryness in a vacuum, and the residue is
dissolved in 500 ml of methylene chloride. The organic phase is
shaken out twice with 300 ml of 5N hydrochloric acid. The
organic phase is dried on magnesium sulfate and concentrated by
evaporation in a vacuum. The residue is recrystallized from a
little methanol. 68.29 g of a colorless crystalline powder is
obtained.
Yield: 93%
Cld: C 65.58% H 5.73% N 3.19% S 7.29%
Fnd: C 65.30% H 5.81% N 3.02% S 7.18%
b) 2-t4-Benzyloxybenzyl)-1-tosyl-1,4,7-triazaheptan-3-one
45 g (102.38 mmol) of the title substance of Example 16a) is
introduced in 1 l of 1,2-diaminoethane within 1 hour and then
stirred for 3 hours at 80~C. The residue is evaporated to
dryness, and the residue is absorptively precipitated in 200 ml
of water. The precipitate is suctioned off and rewashed with a
great deal of water. Then, it is dried overnight in a vacuum at
60~C. 46.91 g of a cream-colored, amorphous powder is obtained.
Yield: 98%
Cld: C 64.22% H 6.25% N 8.95% S 6.86%
64 21 94561
Fnd: C 64.05% H 6.17% N 9.05% S 6.78%
c) 9-Chloro-2-(~-benzyloxybenzyl)-1-tosyl-1,4,7-triaza-nonane-
3,8-dione
lO g (21.39 mmol) of the title compound of Example 16b) is
dissolved in 100 ml of chloroform, and 2.38 g (23.53 mmol) of
triethylamine is added. At 0~C, 2.66 g (23.53 mmol) of
chloroacetyl chloride in 20 ml of chloroform is added in drops
within 30 minutes. It is stirred for 30 minutes at 0~C. 200 ml
of lN hydrochloric acid is added and shaken vigorously. The
organic phase is separated, dried on magnesium sulfate and
concentrated by evaporation in a vacuum. The residue is
recrystallized from a little methanol. 10.36 g of a cream-
colored, crystalline solid is obtained.
Yield: 89%
Cld: C 59.61% H 5.56% N 7.72% S 5.89% Cl 6.52%
Fnd: C 59.50% H 5.69% N 7.55% s 5.71% Cl 6.38%
d) 10-Benzoyl-2-~4-benzyloxybenzyl)-1-tosyl-10-thia-1,4,7-
triazadecane-3,8-dione
9 g (16.54 mmol) of the title compound of Example 16c) is
dissolved in 100 ml of chloroform, and 1.67 g (16.54 mmol) of
triethylamine is added. Then, 2.29 g (16.54 mmol) of thiobenzoic
acid is added and refluxed for 10 minutes. It is cooled to room
temperature and shaken out once with 2N hydrochloric acid and
once with a 5% sodium carbonate solution. The organic phase is
dried on magnesium sulfate and concentrated by evaporation in a
2 1 9456 1
vacuum. The residue is recrystallized from a little methanol.
9.61 g of a colorless, crystalline powder is obtained.
Yield: 90%
Cld: C 63.24% H 5.46% N 6.51% S 9.93%
Fnd: C 63.15% H 5.57% N 6.40% S 9.81%
Example 17
a) 10-Acetyl-2-~4-benzyloxybenzyl)-1-to~yl-10-thia-1,4,7-
triazadecane-3,8-dione
9 g (16.54 mmol) of the title compound of Example 16c) is
dissolved in 100 ml of chloroform, and 1.67 g (16.54 mmol) of
triethylamine is added. Then, 1.28 g (16.54 mmol) of thioacetic
acid is added and refluxed for 10 minutes. It is cooled to room
temperature, shaken out with 2N hydrochloric acid and then with a
5% sodium carbonate solution. The organic phase is dried on
magnesium sulfate and concentrated by evaporation in a vacuum.
The residue is recrystallized from a little acetone. 8.20 g of
cream-colored crystals is obtained.
Yield: 85%
Cld: C 59.67% H 5.70% N 7.20% S 10.98%
Fnd: C 59.51% H 5.81% N 7.05% S 10.80%
Example 18
a) lo-Trifluoroacetyl-2-~4-benzyloxybenzyl)-1-tosyl-lo-thia-
1,4,7-triazadecane-3,8-dione
9 g (16.54 mmol) of the title compound of Example 16c) is
dissolved in 100 ml of chloroform and 1.67 g (1654 mmol) of
66 2 1 9456 1
trifluoromethylthioacetic acid, and it is refluxed for 10
minutes. It is cooled to room temperature, shaken out with 2N
hydrochloric acid and then with a 1% sodium carbonate solution.
The organic phase is dried on magnesium sulfate and concentrated
by evaporation in a vacuum. The residue is recrystallized from a
little acetone/ether. 8.75 g of colorless crystals is obtained.
Yield: 83%
Cld: C 54.62% H 4.74% N 6.59% S 10.05% F 8.94%
Fnd: C 54.47% H 4.61% N 6.50% S 9.90% F 8.81%
Example 19
a) N-Mesyl-tyrosinemethylester-4-benzyl ether
19.58 g (171 mmol) of methanesulfonic acid chloride is added
in drops to 50 g (155.37 mmol) of tyrosinemethylester-4-benzyl
ether hydrochloride in 300 ml of pyridine at 0~C, and it is
stirred for 3 hours at 0~C. It is evaporated to dryness in a
vacuum, and the residue is dissolved in 500 ml of methylene
chloride. The organic phase is shaken out twice with 300 ml of
5N hydrochloric acid. The organic phase is dried on magnesium
sulfate and concentrated by evaporation in a vacuum. The residue
is recrystallized from a little methanol. 53.64 g of a
colorless, crystalline powder is obtained.
Yield: 95%
Cld: C 59.49% H 5.82% N 3.85% S 8.82%
Fnd: C 59.30% H 5.95% N 3.71% S 8.70%
67 2 1 9456 1
b) 2-(4-Benzyloxybenzyl)-1-mesyl-1,4,7-triazaheptan-3-one
37.2 g (102.38 mmol) of the title substance of Example l9a)
is introduced in 1 liter of 1,2-diaminoethane within 1 hour and
then stirred for 3 hours at 80~C. The residue is evaporated to
dryness, and the residue is absorptively precipitated in 200 ml
of water. The precipitate is suctioned off and rewashed with a
great deal of water. Then, it is dried overnight in a vacuum at
60~C. 37.68 g of a cream-colored, amorphous powder is obtained.
Yield: 97%
Cld: C 56.97% H 6.64% N 11.07% S 8.45%
Fnd: C 56.81% H 6.72% N 10.93% S 8.32%
c) 9-Chloro-2-(4-benzyloxybenzyl)-1-mesyl-1,4,7-triaza-nonane-
3,8-dione
10 g (26.35 mmol) of the title compound of Example l9b) is
dissolved in 100 ml of chloroform, and 2.93 g (28.99 mmol) of
triethylamine is added. At 0~C, 3.27 g (28.99 mmol) of
chloroacetyl chloride in 20 ml of chloroform is added in drops
within 30 minutes. It is stirred for 30 minutes at 0~C. 200 ml
of lN hydrochloric acid is added and vigorously shaken. The
organic phase is separated, dried on magnesium sulfate and
concentrated by evaporation in a vacuum. The residue is
recrystallized from a little methanol. 10.93 g of a cream-
colored, crystalline solid is obtained.
Yield: 91%
Cld: C 52.68% H 5.75% N 9.22% S 7.03% Cl 7.78%
Fnd: C 52.51% H 5.82% N 9.13% S 6.90% Cl 7.68%
68 21 94561
....
d) 10-Benzoyl-2-(4-benzyloxybenzyl)-1-mesyl-10-thia-1,4,7-
triaza-decane-3,8-dione
7.54 g (16.54 mmol) of the title compound of Example 16c) is
dissolved in 100 ml of chloroform, and 1.67 g (16.S4 mmol) of
triethylamine is added. Then, 2.29 g (16.54 mmol) of thiobenzoic
acid is added and refluxed for 10 minutes. It is cooled to room
temperature and shaken out once with 2N hydrochloric acid and
once with a 5% sodium carbonate solution. The organic phase is
dried on magnesium sulfate and concentrated by evaporation in a
vacuum. The residue is recrystallized from a little methanol.
8.11 g of a colorless, crystalline powder is obtained.
Yield: 88%
Cld: C 58.15% H 5.60% N 7.53% S 11.50%
Fnd: C 58.03% H 5.71% N 7.61% S 11.38%
Example 20
a) 9-Chloro-2-~4-hydroxybenzyl)-1-tosyl-1,4,7-triazanonane-3,8-
dione
20 g (36.76 mmol) of the title compound of Example 16c) is
dissolved in 200 ml of methylene chloride, and 2 ml of glacial
acetic acid is added. Then, 3 g of palladium catalyst (10% Pd/C)
is added and hydrogenated overnight. The catalyst is filtered
out, and it is evaporated to dryness in a vacuum. 16.52 g of an
amorphous solid is obtained.
Yield: 99%
Cld: C 52.92% H 5.33% N 9.26% S 7.06% Cl 7.81%
Fnd: C 52.81% H 5.26% N 9.11% S 6.93% Cl 7.72%
69 2194561
b) 9-Chloro-2-(4-palmitoyloxybenzyl)-l-tosyl-1,4,7-
triazanonane-3,8-dione
10 g (22.03 mmol) of the title compound of Example 20a) is
dissolved in 100 ml of chloroform, and 2.45 g (23.53 mmol) of
triethylamine is added. At 0~C, 6.66 g (24.23 mmol) of palmitic
acid chloride is added in drops within 10 minutes, and it is
stirred for 2 more hours at this temperature. It is shaken out
with 200 ml of 2N hydrochloric acid, the organic phase is dried
on magnesium sulfate and concentrated by evaporation. The
residue is chromatographed on silica gel (mobile solvent:
methylene chloride/acetone = 20:1). 11.90 g of a waxy solid is
obtained.
Yield: 78%
Cld: C 62.45% H 7.86% N 6.07% S 4.63% Cl 5.12%
Fnd: C 62.28% H 7.70% N 5.89% S 4.54% Cl 4.98%
c) lO-Benzoyl-2-~4-palmitoyloxybenzyl)-l-tosyl-lO-thia-1,4,7-
triazadecane-3,8-dione
8 g (11.55 mmol) of the title compound of Example 20b) is
dissolved in 100 ml of chloroform and 1.17 g (11.55 mmol) of
triethylamine is added. Then, 1.60 g (11.55 mmol) of thiobenzoic
acid is added and refluxed for 10 minutes. It is cooled to room
temperature and shaken out once with 2N hydrochloric acid and
once with a 5% sodium carbonate solution. After drying on
magnesium sulfate, the organic phase is concentrated by
evaporation in a vacuum, and the residue is chromatographed on
silica gel (mobile solvent: methylene chloride/hexane/
2 1 9456 1
acetone:20:10:1). 8.53 g of colorless flakes (of ether) is
obtained.
Yield: 93%
Cld: C 65.04% H 7.49% N 5.29% S 8.07%
Fnd: C 64.90% H 7.55% N 5.17% S 7.91%
Example 21
a) Phenolester of 3-cholesterol ~uccinic acid semi-e~ter with
9-chloro-2-~4-hyd~oxybenzyl)-1-tosyl-1,4,7-triazanonane 3,8-
dione
10 g (22.03 mmol) of the title compound of Example 20a) and
10.72 g (22.03 mmol) of cholesterol succinic acid semi-ester are
dissolved in 49 ml of dimethylformamide and cooled to 0~C. Then,
300 mg of 4-dimethylaminopyridine and 5.45 g (26.44 mmol) of
dicyclohexylcarbonyl are added and stirred for 3 hours at 0~C.
It is allowed to stir at room temperature overnight. 50 ml of
ether is added and precipitated urea is filtered out, the
filtrate is diluted with 250 ml of ethyl acetate and shaken out 5
times with 200 ml of water. The organic phase is dried on
magnesium sulfate and concentrated by evaporation in a vacuum.
The residue is chromatographed on silica gel (mobile solvent:
methylene chloride/hexane/ethyl acetate = 10:5:1). 17.60 g of a
waxy solid is obtained.
Yield: 78~
Cld: C 66.39% H 7.87% N 4.55% S 3.48% Cl 3.84%
Fnd: C 66.28% H 7.95% N 4.47% S 3.31% Cl 3.70%
71 21 ~4561
b) Phenolester of cholesterol succinic acid semi-ester with
10-benzoyl-2-(4-hydroxybenzyl)1-tosyl-10-thia-1,4,7-
triazadecane-3,8-dione
6 g (6.50 mmol) of the title compound of Example 21a) is
dissolved in 50 ml of chloroform and 0.66 g (6.50 mmol) of
triethylamine is added. Then, 0.9 g (6.50 mmol) of thiobenzoic
acid is added and refluxed for 10 minutes. It is cooled to room
temperature and shaken out once with 2N hydrochloric acid and
once with a 5% sodium carbonate solution. The organic phase is
separated and dried on magnesium sulfate. After concentration by
evaporation in a vacuum, the residue is recrystallized from
methyl-tert-butyl ether. 6.06 g of waxy flakes is obtained.
Yield: 91%
Cld: C 68.01% H 7.58% N 4.10% S 6.26%
Fnd: C 67.85% H 7.43% N 3.98% S 6.17%
Example 22
a) 7-N-~tert-Butoxycarbonyl)-2-(4-benzyloxybenzyl)-1-tosyl-
1,4,7-triazaheptan-3-one
20 g (42.77 mmol) of the title compound of Example 16b) is
dissolved in 200 ml of chloroform, and 4.76 g (47.05 mmol) of
triethylamine is added. At 0~C, a solution of 10.27 g (47.05
mmol) of di-tert-butyl dicarbonate in 50 ml of chloroform is
added in drops and stirred for 30 minutes at 0~C. Then, it is
stirred for 5 hours at room temperature. It is shaken out three
times with 5% sodium carbonate solution, the organic phase is
dried on magnesium sulfate and concentrated by evaporation in a
72 2 1 9456 1
vacuum. The residue is recrystallized from a little methanol.
22.34 g of colorless crystals is obtained.
Yield: 92%
Cld: C 63.47% H 6.57~ N 7.40% S 5.65%
Fnd: C 63.31% H 6.42% N 7.45% S 5.49%
b) 7-N-tert-Butoxycarbonyl-2-(4-hydroxybenzyl)-1-tosyl-1,4,7-
triazaheptan-3-one
21 g (36.99 mmol) of the title compound of Example 22a) is
dissolved in 300 ml of methylene chloride and 4 g of palladium
catalyst (10% Pd/C) is added. It is hydrogenated overnight. The
catalyst is filtered out, and the filtrate is evaporated to
dryness. 16.39 g of a vitreous foam is obtained, which
solidifies after a short time.
Yield: 99%
Cld: C 57.84% H 6.54% N 8.80% S 6.71%
Fnd: C 57.70% H 6.61% N 8.69% S 6.54%
c) 7-N-tert-Butoxycarbonyl-2-t4-(benzyloxycarbonylmethyloxy)-
benzyl]-1-tosyl-1,4,7-triazaheptan-3-one
15 g (33.51 mmol) of the title compound of Example 22b),
7.68 g (33.51 mmol) of bromoacetic acid benzyl ester and 13.8 g
(100 mmol) of potassium carbonate are refluxed in 300 ml of
acetonitrile for 24 hours. The salts are filtered out, and the
filtrate is evaporated to dryness. The residue is dissolved in
200 ml of methylene chloride and shaken out twice with 100 ml of
water. The organic phase is dried on magnesium sulfate and
73 21 94561
concentrated by evaporation in a vacuum. The residue is
chromatographed on silica gel (mobile solvent: methylene
chloride/hexane/acetone: 20/10/1). 10.69 g of a colorless oil
is obtained.
Yield: 51%
Cld: C 61.42% H 6.28~ N 6.72% S 5.12%
Fnd: C 61.27% H 6.09% N 6.68% S 5.03%
d) 1-Tosyl-2-(4-~benzyloxycarbonylmethyloxy)-benzyl)-1,4,7-
triazaheptan-3-one (a~ trifluoroacetate salt)
10 g (15.98 mmol) of the title compound of Example 22c) is
stirred for 1 hour in 100 ml of trifluoroacetic acid at room
temperature. It is evaporated to dryness in a vacuum. 10.22 g
of a vitreous foam is obtained, which solidifies with standing.
Yield: 100%
Cld: C 54.45% H 5.04% N 6.57% S 5.01% F 8.91%
Fnd: C 54.51% H 5.10% N 6.43% S 4.89% F 9.15%
e) 9-Chloro-2-~4-(benzyloxycarbonylmethyloxy)-benzyl)-1-tosyl-
1,4,7-triazanonane-3,8-dione
10 g (15.63 mmol) of the title compound of Example 22d) and
4.75 g (46.90 mmol) of triethylamine are dissolved in 200 ml of
chloroform. At 0~C, 1.94 g (17.19 mmol) of chloroacetyl chloride
is added in drops within 30 minutes and then stirred for 2 hours
at 0~C. The organic phase is shaken out twice with 5%
hydrochloric acid and twice with water, dried on magnesium
sulfate and evaporated to dryness in a vacuum. The residue is
~ ~ q456 1
74
chromatographed on silica gel (mobile solvent: methylene
chloride/ethyl acetate = 20:1). 7.62 g of a waxy solid is
obtained.
Yield: 81%
Cld: C 57.85% H 5.36% N 6.98% S 5.32% Cl 5.89%
Fnd: C 57.70% H 5.49% N 6.82% S 5.25% Cl 5.78%
f) 9-Chloro-2-(4-(carboxymethyloxy)-bensyl)-1-tosyl-1,4,7-
triazanonane-3,8-dione
7 g (11.63 mmol) of the title compound of Example 22e) is
dissolved in 150 ml of methylene chloride and mixed with 2 g of
palladium catalyst (10% Pd/C). It is hydrogenated overnight.
The catalyst is filtered out, and the filtrate is evaporated to
dryness in a vacuum. 5.89 g of a vitreous solid is obtained.
Yield: 99%
Cld: C 51.61% H 5.12% N 8.21% S 6.26% Cl 6.92%
Fnd: C 51.45% H 5.03% N 8.13% S 6.11% Cl 6.79%
g) 10-Acetyl-2-(4-(carboxymethyloxy)-benzyl)-1-tosyl-10-thia-
1,4,7-triazadecane-3,8-dione
5 g (9.77 mmol) of the title compound of Example 22f) is
dissolved in 80 ml of chloroform and 1.98 g (19.53 mmol) of
triethylamine is added. 0.74 g (9.77 mmol) of thioacetic acid is
added and refluxed for lo minutes. The solution is poured into
200 ml of ice-cooled 5% hydrochloric acid and stirred vigorously.
The organic phase is separated, dried on magnesium sulfate and
evaporated to dryness in a vacuum. Chromatographic purification
2 1 9456 1
on silica gel (mobile solvent: hexane/ethyl acetate = 3:1)
yields 4.47 g of the title compound as vitreous solid.
Yield: 83%
Cld: C 52.26% H 5.30% N 7.62% S 11.62%
Fnd: C 52.11% H 5.39% N 7.50% S 11.49%
h) N-Hydroxysuccinimide~ter of 10-acetyl-2-(4-
~carboxymethyloxy)-benzyl)-l-tosyl-10-thia-1,4,7-
triazadecane-3,8-dione
4 g (7.25 mmol) of the title compound of Example 22g), 1.65
g (7.98 mmol) of dicyclohexylcarbodiimide, 30 mg of 4-
dimethylaminopyridine and 0.92 g (7.98 mmol) of N-
hydroxysuccinimide are dissolved at 0~C in 20 ml of chloroform
and stirred for 1 hour at this temperature. Then, stirring is
continued for 24 hours at room temperature. 20 ml of ether is
added, the precipitated dicyclohexylurea is suctioned off, and
the filtrate is concentrated by evaporation in a vacuum. The
residue is chromatographed on silica gel (mobile solvent:
methylene chloride/dioxane = 10:1). 4.09 g of a colorless solid
is obtained.
Yield: 87%
Cld: C 51.84% H 4.97~ N 8.64~ S 9.88%
Fnd: C 51.68% H 4.80% N 8.53% S 9.68%
76 2 1 9456 1
Example 23
a) 4-Nitrophenolester of 10-acetyl-2-(4-(carboxymethyloxy)-
benzyl)-1-tosyl-10-thia-1,4,7-triazadecane-3,8-dione
4 g (7.25 mmol) of the title compound of Example 22g), 1.11
g (7.97 mmol) of 4-nitrophenol, 30 mg of 4-dimethylaminopyridine
and 1.65 g (7.97 mmol) of dicyclohexylcarbodiimide are dissolved
at 0~C in 20 ml of chloroform and stirred for 3 hours at this
temperature. Then, it is stirred for 24 hours at room
temperature. 20 ml of ether is added, settled precipitate is
suctioned out, and the filtrate is concentrated by evaporation in
a vacuum. The residue is chromatographed on silica gel (mobile
solvent: methylene chloride/dioxane = 15:1). 3.85 g of a cream-
colored solid is obtained.
Yield: 79%
Cld: C 53.56% H 4.79% N 8.33% S 9.53%
Fnd: C 53.41% H 4.63% N 8.17% S 9.38%
Example 24
a) Pentafluorophenolester of 10-acètyl-2-(4-(carboxymethyloxy)-
benzyl)-1-tosyl-10-thia-1,4,7-triazadecane-3,8-dione
4 g (7.25 mmol) of the title compound of Example 22g), 1.47
g (7.97 mmol) of pentafluorophenol, 30 mg of 4-
dimethylaminopyridine and 1.65 g (7.97 mmol) of
dicyclohexylcarbodiimide are dissolved at 0~C in 20 ml of
chloroform and stirred for 3 hours at this temperature. Then, it
is stirred for 24 hours at room temperature. 20 ml of ether is
added, settled precipitate is suctioned out, and the filtrate is
77 2l ~ 456l
concentrated by evaporation in a vacuum. The residue is
chromatographed on silica gel (mobile solvent: methylene
chloride/dioxane = 15:1). 3.95 g of a colorless solid is
obtained.
Yield: 76%
Cld: C 50.20% H 3.93% N 5.85% S 8.93% F 13.24%
Fnd: C 50.05% H 3.87% N 5.69% S 8.71% F 13.03%
Example 25
a) 7-N-tert-Butoxycarbonyl-2-(4-benzyloxybenzyl)-1-me~yl-1,4,7-
triazaheptan-3-one
16.23 g (42.77 mmol) of the title compound of Example l9b)
is dissolved in 200 ml of chloroform and 4.76 g (47.05 mmol) of
triethylamine is added. At 0~C, a solution of 10.27 g (47.05
mmol) of di-tert-butyldicarbonate in 50 ml of chloroform is added
in drops and stirred for 30 minutes at 0~C. Then, it is stirred
for 5 hours at room temperature. It is shaken out three times
with 5% sodium carbonate solution, the organic phase is dried on
magnesium sulfate and concentrated by evaporation in a vacuum.
The residue is recrystallized from a little methanol. 20.19 g of
a foamy solid is obtained.
Yield: 96%
Cld: C 58.64% H 6.77% N 8.55% S 6.52%
Fnd: c 58.48% H 6.59% N 8.41% S 6.42
78 21~4561
b) 7-N-tert-Butoxycarbonyl-2-(4-hydroxybenzyl)-l-me5yl-1,4,7-
triazaheptan-3-one
20 g (40.68 mmol) of the title compound of Example 25a) is
dissolved in 300 ml of methylene chloride, and 4 g of palladium
catalyst (10% Pd/C) is added. It is hydrogenated overnight. The
catalyst is filtered out, and the filtrate is evaporated to
dryness. 16.17 g of a vitreous foam is obtained, which
solidifies after a short time.
Yield: 99%
Cld: C 50.86% H 6.78% N 10.47% S 7.99%
Fnd: C 50.70% H 6.69% N 10.31% S 7.78%
c) 7-N-tert-Butoxycarbonyl-2-(4-benzyloxycarbonylmethyloxy)-
benzyl)-l-mesyl-1,4,7-triazaheptan-3-one
15 g (37.36 mmol) of the title compound of Example 25b),
8.56 g (37.36 mmol) of bromoacetic acid benzyl ester and 13.8 g
(100 mmol) of potassium carbonate are refluxed in 300 ml of
acetonitrile for 24 hours. The salts are filtered out, and the
filtrate is evaporated to dryness. The residue is dissolved in
200 ml of methylene chloride and shaken out twice with 100 ml of
water. The organic phase is dried on magnesium sulfate and
concentrated by evaporation in a vacuum. The residue is
chromatographed on silica gel (mobile solvent: methylene
chloride/hexane/acetone = 20/10/1). 10.06 g of foamy solid is
obtained.
Yield: 49%
79 2 1 9456 1
Cld: C 56.82~ H 6.42% N 7.64% S 5.83%
Fnd: C 56.65% H 6.35% N 7.51% S 5.72%
d) 2-(4-(Benzyloxycarbonylmethyloxy)-benzyl)-1-mesyl-1,4,7-
triazaheptan-3-one (as trifluoroacetic acid salt)
10 g (18.19 mmol) of the title compound of Example 25c) is
stirred for 1 hour in 100 ml of trifluoroacetic acid at room
temperature. It is evaporated to dryness in a vacuum. 9.95 g of
a vitreous foam is obtained, which solidifies in the case of
standing.
Yield: 97%
Cld: C 49.02% H 5.01% N 7.46% S 5.69% F 10.11%
Fnd: C 48.91% H 4.90% N 7.30% S 5.51% F 9.96%
e) 9-Chloro-2-(4-(benzyloxycarbonylmethyloxy)-benzyl)-1-mesyl-
1,4,7-triazanonane-3,8-dione
9 g (15.97 mmol) of the title compound of Example 25d), 1.78
g (17.57 mmol) of triethylamine are dissolved in 200 ml of
chloroform. At 0~C, 1.98 g (17.57 mmol) of chloroacetyl chloride
is added in drops within 30 minutes and then stirred for 2 hours
at 0~C. The organic phase is shaken out twice with 5%
hydrochloric acid and twice with water, dried on magnesium
sulfate and evaporated to dryness in a vacuum. The residue is
chromatographed on silica gel (mobile solvent: methylene
chloride/ethyl acetate = 20:1).
Yield: 83%
- - -
2 1 9456 1
Cld: C 52.52% H 5.37% N 7.99% S 6.09% Cl 6.74%
Fnd: C 52.37% H 5.43 N 7.81 S 5.93% Cl 6.58%
f) 9-Chloro-2-(4-(carboxymethyloxy)-benzyl)-1-mesyl-1,4,7-
triazanonane-3,8-dione
6.5 g (12.36 mmol) of the title compound of Example 25e) is
dissolved in 150 ml of methylene chloride and mixed with 2 g of
palladium catalyst (10% Pd/C). It is hydrogenated overnight.
The catalyst is filtered out, and the filtrate is evaporated to
dryness in a vacuum. 5.33 g of a vitreous solid is obtained.
Yield: 99%
Cld: C 44.09% H 5.09% N 9.64% S 7.36% Cl 8.13%
Fnd: C 43.93% H 4.95% N 9.52% S 7.22% Cl 8.03%
g) 10-Acetyl-2-(4-(carboxymethyloxy)-benzyl)-1-mesyl-10-thia-
1,4~7-triazadecane-3,8-dione
5 g (11.47 mmol) of the title compound of Example 22f) is
dissolved in 80 ml of chloroform, and 1.98 g (19.53 mmol) of
triethylamine is added. 0.74 g (9.77 mmol) of thioacetic acid is
added and refluxed for 10 minutes. The solution is poured into
200 ml of ice-cooled 5% hydrochloric acid and stirred vigorously.
The organic phase is separated, dried on magnesium sulfate and
evaporated to dryness in a vacuum. Chromatographic purification
on silica gel (mobile solvent: hexanetethyl acetate = 3;1)
yields 4.64 g of the title compound as vitreous solid.
Yield: 85%
81 21 94561
Cld: C 45.46% H 5.30% N 8.84% S 13.48%
Fnd: C 45.28% H 5.17% N 8.61% S 13.38%
h) N-Hydroxysuccinimidester of 10-acetyl-2-(4-
(carboxymethyloxy)-benzyl)-1-mesyl-10-thia-1,4,7-
triazadecane-3,8-dione
4 g (8.41 mmol), 4 g (7.25 mmol) of the title compound of
Example 25g), 1.91 g (9.25 mmol) of dicyclohexylcarbodiimide, 30
mg of 4-dimethylaminopyridine and 1.06 g (9.25 mmol) of N-
hydroxysuccinimide are dissolved at 0~C in 20 ml of chloroform
and stirred for 1 hour at this temperature. Then, stirring is
continued for 24 hours at room temperature. 20 ml of ether is
added, precipitated dicyclohexylurea is suctioned out, and the
filtrate is concentrated by evaporation in a vacuum. The residue
is chromatographed on silica gel (mobile solvent: methylene
chloride/dioxane = 10:1). 3.47 g of a cream-colored solid is
obtained.
Yield: 72%
Cld: C 46.15% H 4.93% N 9.78% S 11.20%
Fnd: C 46.03% H 4.83% N 9.64% S 11.05%
Example 26
a) 3-Glycerol-ester of 10-acetyl-2-(4-(carboxymethyloxy)-
benzyl)-1-mesyl-10-thia-1,4,7-triazadecane-3,8-dione with
glycerol-1,2-dipalmitate
2 g (5.27 mmol) of the title compound of Example 25g), 20 mg
of 4-dimethylaminopyridine, 3.30 g (5.80 mmol) of glycerol-1,2-
82 21 q4561
dipalmitic acid ester and 1.20 g (5.80 mmol) ofdicyclohexylcarbodiimide are dissolved at 0~C in 5 ml of
chloroform and stirred for 3 hours at this temperature. Then, it
is stirred for 24 hours at room temperature. 20 ml of ether is
added, and precipitate is filtered out. The filtrate is
evaporated to dryness in a vacuum, and the residue is
chromatographed on silica gel (mobile solvent: hexane/ethyl
acetate = 30:1). 3.35 g of a waxy solid is obtained.
Yield: 62%
Cld: C 62.02% H 8.94% N 4.09% S 6.25%
Fnd: C 61.91% H 8.75% N 3.91% S 6.18%
Example 27
a) 4-Benzyl ether of N-mesyl-tyrosine
10 g (27.5 mmol) of the title compound of Example l9a) and
5.50 g (137.6 mmol) of sodium hydroxide are refluxed in a mixture
of 50 ml of water/150 ml of ethanol for 3 hours. It is
evaporated to dryness, the residue is taken up in 200 ml of 3N
hydrochloric acid, and it is stirred for 2 hours at room
temperature. The precipitated acid is suctioned off, washed with
water and dried in a vacuum at 70~C. 9.42 g of cream-colored
solid is obtained.
Yield: 98%
Cld: C 58.44% H 5.48% N 4.01% S 9.18%
Fnd: C 58.28% H 5.37% N 3.91% S 9.02%
83 2 1 9456 1
b) 6~6~-Bis-tl-N-tert-butoxycarbony~ 4-diaza-hexan-3-one]
disulfide
10 g (65.67 mmol) of cystamine, 11.50 g (65.67 mmol) of N-
boc-glycine and 14.90 g (72.24 mmol) of dicyclohexylcarbodiimide
are dissolved at 0~C in 50 ml of tetrahydrofuran and stirred for
2 hours at this temperature. Then, it is stirred for 12 hours at
room temperature. 50 ml of ether is added, settled precipitate
is suctioned out, and the filtrate is evaporated to dryness. The
residue is chromatographed on silica gel (mobile solvent:
hexane/acetone = 6:1). 20.84 g of a vitreous solid is obtained.
Yield: 68%
Cld: C 46.33% H 7.34% N 12.01% S 13.74%
Fnd: C 46.15% H 7.28% N 11.93% S 13.67%
c) 6,6'-Bi~-[1,4-diaza-hexan-3-one]-disulfide
20 g (42.86 mmol) of the title compound of Example 27b) is
dissolved in 100 ml of trifluoroacetic acid, and it is stirred
for 2 hours at room temperature. It is evaporated to dryness,
the residue is taken up with 300 ml of 10% sodium carbonate
solution and extracted 6 times with 50 ml of chloroform. The
combined chloroform phases are dried on magnesium sulfate and
concentrated by evaporation in a vacuum. 10.96 g of a slightly
yellow-colored solid is obtained.
Yield: 969~
Cld: C 36.07% H 6.81% N 21.03% S 24.07%
Fnd: C 35.91% H 6.90% N 20.89% S 23.89%
2 1 9456 1
84
d) 9,9'-Bis[2-(4-benzyloxybenzyl)-1-mesyl-1,4,7-triazanonane-
3,6-dione]disulfide
9 g (25.76 mmol) of the title compound of Example 27a),
3.43 g (12.87 mmol) of the title compound of Example 27c) and
6.19 g (30 mmol) of dicyclohexylcarbodiimide are dissolved at 0~C
in 40 ml of tetrahydrofuran and stirred for 2 hours at this
temperature. Then, it is stirred for 12 hours at room
temperature. 30 ml of ether is added, and precipitate is
suctioned out. The filtrate is evaporated to dryness in a
vacuum, and the residue is chromatographed on silica gel (mobile
solvent: methylene chloride/acetone = 20:1). 5.59 g of a cream-
colored solid is obtained.
Yield: 48% [relative to 27c)]
Cld: C 53.08% H 5.79% N 9.28% S 14.17%
Fnd: C 52.93% H 5.84% N 9.13 S 14.02%
Example 28
a) 4-Benzyl ether of N-mesyl-tyrosinamide
20 g (55.03 mmol) of the title compound of Example l9a) is
dissolved in 300 ml of tetrahydrofuran, and an ammonia stream is
introduced at 0~C for 3 hours. It is evaporated to dryness, and
the residue is recrystallized from methanol. 18.6 g of colorless
flakes is obtained.
Yield; 97~
Cld: C 58.60% H 5.79% N 8.04% S 9.20%
Fnd: C 58.47% H 5.88% N 7.91% S 9.05%
2 1 ~456 1
b) 2-~4-Benzyloxybenzyl)-1-mesyl-1,4-diazabutane
18 g (51.66 mmol) of the title compound of Example 28a) is
dissolved in 200 ml of tetrahydrofuran, and 310 ml of 1 M
diborane in THF is added under nitrogen atmosphere. It is
refluxed for 24 hours. It is cooled to 0~C in an ice bath, and
70 ml of concentrated hydrochloric acid is added. Then, it is
refluxed for 5 hours. It is evaporated to dryness, and the
residue is taken up with 300 ml of saturated sodium carbonate
solution. It is extracted 3 times with 100 ml of methylene
chloride, the combined phases are dried on magnesium sulfate and
evaporated to dryness in a vacuum. The chromatographic
purification is carried out on silica gel (mobile solvent:
methylene chloride/ethanol = 10:1). 15.38 g of a cream-colored
solid is obtained.
Yield: 89%
Cld: C 61.05% H 6.63% N 8.38% S 9.59%
Fnd: C 60.91% H 6.54% N 8.27% S 9.41%
c) 4-N-Butoxycarbonyl-2-(4-benzyloxybenzyl)-1-me~yl-1,4,7-
triazaheptan-5-one
15 g (44.85 mmol) of the title compound of Example 28b) is
dissolved in 200 ml of chloroform, and 5 g (49.34 mmol) of
triethylamine and 12.84 g (49.34 mmol) of N-tert-butoxycarbonyl-
glycine-N-hydroxysuccinimide ester are added at 0~C. It is
stirred for 12 hours at room temperature. It is extracted twice
with cold 5% hydrochloric acid and once with water. The organic
86 2194561
phase is dried on magnesium sulfate and evaporated to dryness in
a vacuum. 20.51 g of an amorphous solid is obtained.
Yield: 93%
Cld: C 58.64% H 6.77% N 8.55% S 6.52%
Fnd: C 58.47% H 6.85% N 8.43% S 6.41%
d) 2-(4-Benzyloxybenzyl)-1-mesyl-1,4,7-triazaheptan-5-one
20 g (40.68 mmol) of the title compound of Example 28c) is
dissolved in 100 ml of trifluoroacetic acid and dissolved for 5
hours at room temperature. It is evaporated to dryness in a
vacuum, the residue is taken up in 200 ml of saturated sodium
carbonate solution and extracted 3 times with 100 ml of
chloroform. The organic phases are dried on sodium sulfate and
concentrated by evaporation in a vacuum. 15.61 g of a vitreous
solid is obtained.
Yield: 98%
Cld: C 58.29% H 6.44% N 10.73% S 8.19%
Fnd: C 58.13% H 6.60~ N 10.61% S 8.05%
e) 9-Chloro-2-(4-benzyloxybenzyl)-1-mesyl-1,4,7-triazanonane-
5,8-dione
10 g (25.54 mmol) of the title compound of Example 28d) and
2.58 g (25.54 mmol) of triethylamine are dissolved in 200 ml of
chloroform, 2.88 g (25.54 mmol) of chloroacetyl chloride is
added in drops at 0~C within 30 minutes. It is stirred for 3
hours at 0~C. It is poured onto 200 ml of 5% cold hydrochloric
acid and stirred vigorously. The organic phase is separated,
-' ' 21~4561
87
dried on magnesium and evaporated to dryness in a vacuum. The
residue is recrystallized from methanol. 11.36 g of a cream-
colored solid is obtained.
Yield: 95%
Cld: C 53.90% H 5.60% N 8.98~ S 6.85% Cl 7.58~
Fnd: C 53.80% H 5.71% N 8.91% S 6.73% Cl 7.44%
f) 10-Benzoyl-2-~4-benzyloxybenzyl)-1-mesyl-1,4,7-triaza-10-
thiadecane-5,8-dione
5 g (10.68 mmol) of the title compound of Example 28e),
1.08 g (10.68 mmol) of triethylamine and 1.48 g (10.68 mmol) of
thiobenzoic acid are refluxed in 50 ml of chloroform for 10
minutes. It is evaporated to dryness, and the residue is
chromatographed on silica gel (mobile solvent: methylene
chloride/acetone = 15:1). 4.93 g of a cream-colored, amorphous
solid is obtained.
Yield: 81%
Cld: C 59.03% H 5.48% N 7.38% S 11.26%
Fnd: C 58.87% H 5.31% N 7.25% S 11.04%
Example 29
a) 9,9'-Bis-[2-~4-benzyloxybenzyl)-1-mesyl-1,4,7-triazanonan-3-
one]-disulfide
2 00 g (13.17 mmol) of 1,6-dichloro-3,4-dithiahexane
(dissolved in 20 ml of acetonitrile) is added in drops to 10 g
(26.35 mmol) of the title compound of Example l9b) and 10.92 g
(79 mmol) of potassium carbonate in 100 ml of acetonitrile in the
88 2194561
boiling heat within 1 hour. It is refluxed for 12 hours. The
salts are filtered out, the filtrate is evaporated to dryness in
a vacuum, and the residue is chromatographed on silica gel
(mobile solvent: methylene chloride/isopropanol = 10:1). 2.19 g
of a slightly yellow-colored crystalline powder is obtained.
Yield: 19% (relative to 1,6-dichloro-3,4-dithiahexane)
Cld: C 54.77% H 6.43% N 9.58% S 14.62%
Fnd: C 54.61% H 6.53% N 9.41% S 14.51%
Example 30
a) 6-Chloro-2-(4-benzyloxybenzyl)-1-mesyl-1,4-diazahexan-5-one
10 g (25.54 mmol) of the title compound of Example 28b) is
dissolved together with 3.03 g (29.90 mmol) of triethylamine in
200 ml of chloroform. 3.38 g (29.90 mol) of chloroacetyl
chloride is added in drops at 0~C, and it is stirred for 2 hours
at this temperature. It is poured onto 200 ml of 5% cold
hydrochloric acid and stirred well. The organic phase is
separated, dried on magnesium sulfate and evaporated to dryness
in a vacuum. The residue is recrystallized from methanol. 11.67
g of colorless crystals is obtained.
Yield: 95%
Cld: C 55.54% H 5.64% N 6.82% S 7.80% Cl 8.63%
Fnd: C 55.38% H 5.71% N 6.67% S 7.63% Cl 8.51%
89 21 94561
b) 9,9'-Bis-[2-(4-benzyloxybenZyl)-l-mesyl-1,4,7-triaZanonan-5-
one]-disulfide
10 g (24.34 mmol) of the title compound of Example 30a),
1.52 g (10 mmol) of cystamine and 8.29 g (60 mol) of potassium
carbonate are refluxed in 150 ml of tetrahydrofuran for 8 hours.
The salts are filtered out, the filtrate is evaporated to dryness
in a vacuum, and the residue is chromatographed on silica gel
(mobile solvent: methylene chloride/ethanol = 15:1). 2.02 g of
a slightly yellowish solid is obtained.
Yield: 23% (relative to cystamine)
Cld: C 54.77% H 6.43% N 9.58% S 14.62%
Fnd: C 54.61% H 6.52% N 9.47% S 14.48%
Example 31
a) Cystamine-bis-~chloroacetamide)
10 g (65.67 mmol) of cystamine and 13.29 g (131.34 mmol) of
triethylamine are dissolved in 100 ml of chloroform at 0~C.
14.38 g (131.34 mol) of chloroacetyl chloride is added in drops,
and it is stirred for 3 hours at 0~C. The solution is poured
into 200 ml of cold 5% hydrochloric acid and stirred vigorously.
The organic phase is dried on magnesium sulfate and concentrated
by evaporation in a vacuum. The residue is recrystallized from a
little acetone. 17.04 g of a cream-colored solid is obtained.
Yield: 85%
Cld: C 31.48% H 4.62% N 9.18% S 21.01% Cl 23.23%
Fnd: c 31.27% H 4.51% N 9.09% S 20.93% Cl 23.12%
-
2194561
b) 9,9'-Bist2-~4-benzyloxybenzyl)-1-mesyl-1,4,7-triazanonan-6-
one]-di~ulfide
5 g (16.38 mmol) of the title compound of Example 31a),
13.69 g (40.95 mmol) of the title compound of Example 28b) and
20.73 g (150 mmol) of potassium carbonate are refluxed in 200 ml
of ethanol for 8 hours. 200 ml of methylene chloride is added,
salts are suctioned out, and the filtrate is concentrated by
evaporation in a vacuum. The residue is chromatographed on
silica gel (mobile solvent: methylene chloride/ethanol = 15:1).
4.74 g of a vitreous solid is obtained.
Yield: 33
Cld: C 54.77% H 6.43% N 9.58% S 14.62~
Fnd: C 54.65% H 6.37% N 9.41~ S 14.53%
Example 32
a) 2-t4-Benzyloxybenzyl]-l-me~yl-1,4,7-triazaheptane
5 g (13.18 mmol) of the title compound of Example l9b) is
dissolved in 50 ml of tetrahydrofuran, and 80 ml of 1 M diborane
solution (1 M in THF) is added. It is refluxed for 24 hours. It
is cooled to 0~C, and 20 ml of concentrated hydrochloric acid is
added. Then, it is refluxed for 5 hours. The solution is
evaporated to dryness and taken up with 200 ml of saturated
sodium carbonate solution. Then, it is extracted 5 times with
loo ml of chloroform. The combined organic phases are dried on
sodium sulfate and concentrated by evaporation in a vacuum. The
residue is chromatographed on silica gel (mobile solvent:
91 21 94561
methylene chloride/isopropanol = 8:1, +2% NH40H). 4.19 g of a
vitreous solid is obtained.
Yield: 87%
Cld: C 59.15% H 7.45% N 11.50% S 8.77%
Fnd: C 59.03% H 7.28% N 11.37% S 8.61%
b) 9,9'-Bi~-t2-~4-benzyloxybenzyl)-1-mesyl-1,4,7-triazanonan-8-
one]-disulfide
4 g (10.94 mmol) of the title compound of Example 32a), 0.67
g (3.65 mmol) of 2,2'-dithiodiacetic acid and 2.48 g (12.04 mmol)
of dicyclohexylcarbodiimide are dissolved at 0~C in 20 ml of
tetrahydrofuran and stirred for 3 hours at this temperature.
Then, it is stirred for 24 hours at room temperature. 20 ml of
ether is added, precipitate is filtered out, and the filtrate is
concentrated by evaporation in a vacuum. The residue is
chromatographed on silica gel (mobile solvent: methylene
chloride/ethanol = 15:1). 0.99 g of an amorphous solid is
obtained.
Yield: 31% (relative to dicarboxylic acid)
Cld: C 54.77% H 6.43% N 9.58% S 14.62%
Fnd: C 54.58% H 6.38% N 9.47% S 14.51%