Note: Descriptions are shown in the official language in which they were submitted.
- 21 96727
FIELD OF THE INVENTION
The invention relates to compounds and compositions for treatment of disease,
disorder or abnormal physical state which downregulate or inactivate p27KiP1 andreverse or prevent resistance to anticancer drugs. Tumors are made more sensitive to
5 drugs which increases the effectiveness of chemotherapeutic treatments. The
invention also involves the use of these compounds and compositions as
chemosensitizers and regulators of drug resistance of solid tumors. The invention also
relates to screening chemicals to identify anti-cancer agents which are preferentially
active against slowly dividing cells or chemicals which act as chemosensitizers.10 BACKGROUND OF THE INVENTION
One of the major problems of current chemotherapy treatments of cancer is
that tumor cells develop resistance to drugs. Chemotherapy is an effective treatment
for certain malignancies (eg. those of the haematopoietic system), however, the
success of current cytotoxic treatment modalities is severely compromised due to15 either intrinsic (de novo) or acquired resistance to anti-cancer agents. This is
particularly true in the case of solid tumors.
It would be very useful if specific chemosensitizers were found which could
effectively prevent or reduce the occurrence of tumour cell resistance to
chemotherapy. There is an intense world-wide effort to discover and use drugs in20 cancer patients which are themselves non toxic to tumour cells but which can
significantly augment the sensitivity of tumours to conventional cytotoxic drugs or
irradiation. In some cases such drugs may also be used to reverse acquired drug
resistance, or delay its emergence in treated tumours. At present there is no widely
accepted or used chemosensitizer. Hence if one could be found, the commercial
25 potential would be enormous given the number of cancer patients receiving
chemotherapy and/or irradiation around the world. The benefits would include an
improved success rate for treatment of cancer as well as treatments which require
lower dosages of cytotoxic drugs and which could be administered at less frequent
intervals. Even if the use of a chemosensitizer was restricted to one or two types of
30 cancer, eg. breast or ovarian carcinoma, the commercial potential would be
considerable.
- 2I q~127
The ways in which drug resistance is thought to dcvelop in patients with solid
tumours is based largely on the use of two common experimental approaches: (i) the
derivation of stable and high level drug resistant mutants after long-term exposure of
monolayer cell cultures to cytotoxic anti-cancer drugs, and (ii) the use of "liquid"
5 tumour models in vivo, such as leukemias or ascites tumours, for experimental
therapeutic studies. These approaches tend to emphasise "(uni)cellular" resistance
mechanisms of drug resistance but ignore, or mask, the possible important
contributions of multicellular structure and cell-cell contact on the expression of drug
resistance and the ability to reverse or prevent it with chemosensitizers. This could
10 help explain, for example, why P-glycoprotein multi-drug resistance reversal agents
work well in monolayer tissue culture systems in vitro, or in various liquid tumour
models in vivo, but have failed to demG"~l,ale thus far any significant clinical benefit in
randomized clinical trials involving patients with solid tumours.
A major factor thought to contribute to de novo resistance of tumour cells is a
15 low proliferating fraction (many tumours have a heterogeneous proliferation rate). The
rate of cell division is important because the majority of anti-cancer agents in current
use are preferentially active against rapidly dividing cells 1,2. Part of the reason for this
is that historically, many of the in vivo screening methods used to uncover new drugs
employed rapidly growing mouse tumours such as the L1210 and P388
20 leukaemias 1'3. Thus, conventional methods of identifying potential new drugsselectively focused on chemicals effective against rapidly dividing cells despite the fact
that most solid tumour cells grow at a rate similar to or slower than normal mitotically
active cells in the body (eg. gut mucosal and bone marrow cells). Thus, these
chemicals fail to overcome resistance in the low proliferating fraction of a tumour
25 because they do not target the slowly dividing tumour cells.
The therapeutic window for administering chemotherapy is usually very small
and the amount of therapy that can be administered is limited by normal tissue toxicity.
This is because most solid tumors contain a heterogeneous population of both
proliferating and non-proliferating tumor cells, while certain normal tissue types (eg gut
30 mucosal and bone marrow cells) are ",itGtically active~ 245. The low growth fraction
found in solid tumors is puzling in view of the fact that the genetic alterations found in
21 9612~
advanced stage cancer cells, such as colorectal carcinoma, would be expected to
provide them with a strong growth advantage over their normal cell counterparts.There is a need for compounds which target the slowly dividing de novo resistanttumour cells and leave healthy tissue unaffected. There is also a need to dcvelop
5 effective treatments which increase the fraction of prolirerdti"g tumour cells in order to
increase chemosensitivity.
We have studied the impact of multicellular growth and cell-cell adhesion on
acquired or intrinsic resistance to alkylating agents (and more recently taxol and
~irradiation) in solid tumours. These studies led to the discovery of "acquired
10 multicellular drug resistance," which means that solid tumours selected for resistance
to various types of anti-cancer drugs in vivo sometimes fail to express their resistance
properties in tissue culture unless grown as three dimensional multicellular tumour
spheroids79. This resistance is frequently accompanied by major increases in theadhesivity of the drug-resistant tumour spheroids in culture7 9.
Thus, a need exists to overcome acquired resistance in tumour cells by
developing (1) a method of identifying anti-cancer agents which are preferentially
active against slowly dividing tumour cells, (2) anti-cancer agents which are
preferentially active against slowly dividing tumour cells.
Progression through the cell cycle is governed by cyclins and their partners the20 cyclin dependent kinases (CDKs)12 Recently, two families of CDK inhibitors (CKls)
have been described which inhibit cell cycle progression by binding to and inactivating
cyclin/CDK complexes'3. The INK family, composed of four members to date p15, p16,
p18 and p19, all share a highly conserved ankyrin motif. Members of the INK family
are thought to act primarily on the D-cyclins complexed with cdk4 or cdk6. Mutations
25 are prevalent in p15 and p16 suggesting that they may normally act as tumour
suppressor genes (reviewed by Hirama, T. and Koeffler H.P. 14). The second family is
currently comprised of three members, p21Waf', p27KiP' and p57Kip2. Although p21Waf1
and p27KiP1 are considered universal inhibitors since they can inhibit many cyclin/CDK
complexes in vitro 15-18, transfection studies suggest that their in vivo activity is
30 restricted to the G1 cyclin/CDK complexes '7~'9. Unlike INK family members,
mutations in p21Waf1 and p27KiP1 appear to be surprisingly rare in tumours 20-26. This
21 96727
finding coupled with the observation that p27KiP1 is upregulated by contact-inhibition in
normal cells in culture 27-30, stimulated us to examine expression of p27KiP1 in relation
to the adhesion-dependent kinetic drug resistance of tumour cells growing in a solid
tumour-like context.
The p27KiP1 kinase inhibitor is well known in the literature and there are several
studies which investigate its role in cell proliferation. Prior art which proposes
regulating p27KiP1 in treatment of hyperproliferative disorders, such as cancer,recommends that intracellular p27KiP1 conce~ dlion may be increased in order to
decrease the rate of cell division in tumours. For example, WO9602140 teaches the
use of agents which increase p27KiP1 concentration in order to inhibit cyclin E-Cdk2
complexes in the treatment of cancer.
WO9518824 describes a host vector system which is useful for the production
of p27KiP1. The system is used in assays to identify agents which enhance or inhibit
the ability of p27KiP1 to inhibit the activation of a cyclin E-Cdk2 (cyclin-dependent
kinase) complex. This application does not disclose the use of this protein in
conjunction with chemotherapeutic drugs to treat cancer or other diseases.
There is no known chemosensitizer which safely and effectively reverses or
prevents tumour resistance to chemotherapy. There is also a lack of knowledge of the
molecular basis of drug resistance and the role of p27KiP1 in regulating cell metabolism
and cell survival. Once the role of p27KiP1 is further characterized, it would lead to the
possibility of development of an assay which closely simulates de novo and acquired
tumour cell resistance and better predicts the effectiveness of drugs in vivo. It would
also permit the development of pharmaceutical compositions which regulate
intracellular levels of p27KiP1 to increase chemosensitivity to anti-cancer drugs.
Since drug resistance is often related to the presence of a low growth fraction in
a tumour, regulation of the concenl~dlio,1 of intracellular or extr~cellul~r chemicals
which regulate cell-cell adhesion and the rate of cell proliferation could provide a
means of increasing chemosensitivity. Recruitment of cells into the cell cycle to
enhance the effficacy of anti-cancer therapy is an established concept that contributed
to the development of current hyperfractionated radiation treatment schedules2.
Results from several experimental and clinical studies also demonstrated the utility of
21 96727
combining chemotherapy with hormonal or growth factor stimulation for the treatment
of breast or haematopoietic malignancies. However, the results were variable (see
Conte P.F. et al.34 and Gore S.D. et al.35 and references therein). Part of the reason
for this variability is that hormones or growth factors often have plciollopic effects on
5 cells and in addition to stimulating growth may also, for example, suppress apoptosis
3~38, Strategies used to increase the proliferative fraction of tumour cells are limited by
the fact that for many types of cancer, such as those of the gastrointestinal tract, the
hormones or growth factors regulating tumour cell growth remain relatively
uncharacterised 39. The prior art teaches that a better under~tdnding of the molecula
10 machinery regulating growth in solid tumours may offer new avenues for therapeutic
intervention40, and could potentially be exploited to overcome resistance to
conventional cancer treatments.
Regulation of cyclin dependent kinase inhibitors could offer a new approach to
recruiting cells into the cell cycle and increasing the effectiveness of chemotherapy.
15 Before this invention, no person proposed to inactivate or decrease intracellular
p27KiP1 levels to decrease cell-cell adhesion, increase cell proliferation, or increase
susceptibility to undergoing spontaneous or drug induced cell death in order to
increase chemosensitivity or reverse or prevent cell resistance to anti-cancer agents.
The invention of compounds' which chemosensitize tumour cells would produce new,20 safer and more effective therapeutic treatments for cancer. It would also have the
potential to be applied to a widespread variety of tumours because similar metabolic
pathways regulate cell proliferation in different types of cells.
SUMMARY OF THE INVENTION
Our own studies identified the molecular basis of tumour cell resistance. We
25 have been studying the cellular "sociology" of drug resistance, and more specifically,
the molecular and cellular basis of multicellular drug resistance, and ~ssessingwhether it can be prevented or reversed by non-toxic "anti-adhesive" agents We have
shown the following: (i) multicellular resistance can be rapidly induced in a transient
fashion by a single exposure to a chemotherapeutic drug56; (ii) a form of multicellular
30 resistance can also account for some examples of intnnsic drug resistance to agents
such as cyclophosphamide (CTX) and rirradiation,8 conri""..,g earlier studies by
21 96727
others. For example, if tumour cells are exposed to highly toxic concentrations of CTX
in monolayer culture, the massive clonogenic cell death that normally ensues can be
substantially suppressed by growing the drug exposed cells as multicellular
aggregates for a period of a few days8; (iii) this multicellular "rescue" effect can be
5 aborted if the cells are prevented from aggregating by the presence of an anti-
adhesive agent such as the enzyme hyaluronidase882. Thus, hyaluronidase can, in
effect, function as a chemosensitizing agent in a three-dimensional solid tumourcontex~, evidence for this in vivo has also been obtained, using an ascites tumour
model in which breast cancer cells grow as aggregated clumps, or spheroids, after
10 intraperitoneal injection8.
Our most recent studies dealt with how cell adhesion and multicellular growth
can function to reduce the toxic effects of anti-cancer DNA damaging agents. We
obtained findings which implicate an important relationship between increased cell
adhesion, reduced cell cycle kinetics and altered expression of the cyclin dependent
15 kinase inhibitor known as p27KiP1. For example, growth as multicellul~r spheroids
results in a significantly reduced fraction of cycling cells, and which mimics the slow
growth of many human solid tumours in vivo. Hyaluronidase disaggregation of
compact spheroids can prevent this cell cycle "shutdown"882. Because ~-irradiation,
and most chemotherapeutic drugs, have a preferential toxic effect on dividing vs20 quiescent cells, the chemosensitizing effects of hyaluronidase in vivo may beexplained, in part, by recruitment of a higher fraction of cells into the cycling pool8 82.
There is an issue of why cell-cell adhesion results in contact inhibition of cell
growth even in tumour cells and whether it can be prevented. We found82 that growth
as multicellular spheroids of over a dozen different human tumour cell lines results in a
25 remarkable upregulation of p27KiPl. As explained above, p27KiP1 is a so-called
universal cyclin-dependent kinase inhibitor which can block cell cycle progression and
is known to be upregulated by close cell-cell contact in non-transformed cell lines. The
question was asked, therefore, whether specific antisense oligodeoxynucleotide
(ODN) mediated downregulation of p27KiP1 expression would chemosensitize tumour
30 spheroids to CTX82 By using a new generation of 'C5 propyne' modified ODNs it was
found that, indeed~ the "rescue" effect of multicellular growth can be largely
21 967Z7
suppressed. Moreover, antisense p27KiP1 ODN treatment was also found to exert both
an anti-adhesive and cell growth promoting effect in multicellular spheroids82. These
results implicate the properties of cell adhesion and upregulation of p27KiP1 astherapeutic targets for increasing chemosensitivity and reversing drug resistance in
5 solid tumours. They also provide a generic explanation for the relatively slow growth
of many types of solid tumours in humans. Spheroid tumour cell cultures also provide
a new way of screening drugs which are preferentially active agai~st slowly dividing
cells or which chemosensitize solid tumours.
Accordingly, the invention relates to an antisense oligonucleotide molecule for
10 treatment of a disease, disorder or abnormal physical state which inhibits p27KiP1
having at least 60% homology to the sequence selecting from a group consisting of 5'-
GCGUCUGCUCCACAG-3' and 5'-UGGCUCUCCUGCGCC-3'. The antisense
oligonucleotide molecule has at least a portion of the sequence selecting from a group
consisting of 5'-GCGUCUGCUCCACAG-3' and 5'-UGGCUCUCCUGCGCC-3'. The
15 antisense oligonucleotide molecule is a C5-propyne modified oligonucleotide.
The invention also relates to an inhibitor of p27KiP1 for l,eal"~ent of a disease,
disorder or abnormal physical state which performs a function selected from a group
consisting of decreasing intracellular p27KiP1 concentration, suppressing p27KiP activity
or abolishing p27KiP activity. This inhibitor may be either a p27KiP1 antisense
20 oligonucleotide molecule or a small molecular weight peptide antagonist of p27KiP1.
The antisense oligonucleotide molecules and other inhibitors of this invention
may be used in gene therapy. The antisense oligonucleotide molecules and other
inhibitors of the invention may also be used for modulating cell proliferation,
modulating cell adhesion, modulating cell susceptibility to undergoing cell death,
25 modulating cellular drug resistance, modulating cell chemosensitivity and modulating
cell cycle perturbations.
It is another object of the invention to provide a pharmaceutical composition
comprising an inhibitor of p27KiP1 for decreasing intracellular p27KiP1 concentration and
a pharmaceutically acceptable carrier, auxiliary or excipient. The inhibitor in the
30 pharmaceutical composition is either a p27KiP1 antisense oligonucleotide molecule an
antagonist of p27KiP1 . The antisense oligonucleotide molecule in the pharmaceutical
21 96727
composition has at least 60% homology to the sequence selected from a group
consisting of 5'-GCGUCUGCUCCACAG-3' and 5'-UGGCUCUCCUGCGCC-3'. The
antisense oliogonucletide molecule also has at least a portion of the sequence
selected from a group consisting of 5'-GCGUCUGCUCCACAG-3' and 5'-
5 UGGCUCUCCUGCGCC-3'. The pharmaceutical compositions of the invention may
be used in gene therapy. The pharmaceutical compositions may also be used for
modulating cellular activity, wherein the cellular activity is selected from a group
consisting of cell proliferation, cell adhesion, cellular drug resistance, cell
chemosensitivity and cell cycle perturbations.
Another object of the invention includes a kit for the treatment of chemotherapy-
resistant cells and cells likely to become chemotherapy-resistant, comprising the
pharmaceutical compositions of the invention. The kit for the detection of
chemotherapy resistant cells and cells likely to become chemotherapy-resistant, may
comprise the oligonucleotide and a control.
The invention also includes a method of treating a disease, disorder or
abnormal physical state in a mammal by decreasing intracellular p27K~P1 conce~ dlion
or suppressing or abolishing p27KiP1 function. According to this method, the
intracellular concentration of p27KiP1 is decreased by a p27KiP~ inhibitor. The p27KiP1
inhibitor is a p27KiP1 antisense oligonucleotide in one embodiment of the invention. In
20 an alternate embodiment, the p27KiP1 inhibitor is a small molecular weight peptide
antagonist of p27KiP1. In this method, the antisense oligonucleotide has a sequence
having at least 60% homology to the sequence selected from a group consisting of 5'-
GCGUCUGCUCCACAG-3' and 5'-UGGCUCUCCUGCGCC-3'. The antisense
oligonucleotide may also have at least a portion of the sequence selected from a25 group consisting of 5'-GCGUCUGCUCCACAG-3' and 5'-UGGCUCUCCUGCGCC-3'.
In one embodiment of this invention, the method consists of administering the
pharmaceutical compositions of the invention to the mammal. An alternate
embodiment consists of the steps of 1) administering to the r,lamnlal the
pharmaceutical compositions of the invention and 2) administering to the mammal a
30 chemotherapeutic drug.
21 q6727
Another object of the invention is to provide a method for identifying anti-cancer
agents which are preferentially active against slowly dividing cells. This method
consists of introducing a chemical to a slowly dividing spheroid tumor cell culture, and
determining whether the spheroid tumor cell culture is adversely affected by the5 presence of the chemical.
It is also an object of the invention to provide a method for identifying anti-
cancer agents which chemosensitize solid tumors, which consists of: 1) combining a
first chemical with a spheroid tumor cell culture, 2) combining an anti-cancer agent
with the first chemical and the spheroid tumor cell culture, and 3) determining whether
10 the spheroid tumor cell culture is more adversely affected by the presence of the first
chemical and the anti-cancer agent than by the presence of the first chemical alone.
An additional object of the invention is to provide a method for
chemosensitizing tumor cells by treating the tumor cells with hyaluronidase.
FIGURES
15 The invention will now be described in relation to the figures in which:
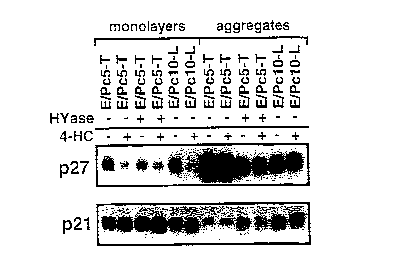
Figure 1. Increase in p27KiP1 but not p21Waf~ protein levels in adherent EMT-6
cells grown as three-dimensional aggregates ~Western blots). A. ElPc5-T (tight) and
E/Pc10-L (loose) cells were collected from either exponentially growing monolayer
cultures or 3 day old three-dimensional cultures. Hyaluronidase (HYase) disrupted
20 intercellular adhesion of E/Pc5-T cells under these conditions, converting them to a
"loose" E/Pc10-L-like morphology. Compact spheroids in three-dimensional cultureconsistently displayed higher levels of p27KiP1 but not p21Waf1 compared to loose
aggregates and monolayer cultures. Treatment with 6,uM 4-HC led to a decrease inp27KiP1 but not p21Waf1 in monolayer, but not in three-dimensional culture. B. Western
25 blot of E/Pc5-T cells showing that p27KiP1 protein levels increase while p21Waf1 protein
levels decrease with time in three-dimensional (3-D) culture. Control cells (time 0)
were taken from exponentially growing monolayer cultures.
Figure 2. p21Waf1 and p27KiP1 protein levels in various mouse and human
breast, colon and ovarian tumour cell lines in monolayer (2D) or three-dimensional
30 (3D) culture. A. Western blot of human breast (MCF-7, BT-549, MDA 468, HBL100)
and colon (SW480.7, HT29) carcinoma cell lines. Note that p27KiP1 protein levels are
21 967~7
consistently elevated in three-dimensional culture where as the effect on p21Waf'
varied between cell lines. B. Western blot of breast (EMT-6, MDA 435) and ovarian
(A2780) cell lines, and their drug selected variants (E/CTX, E/DDP, 435/TO.3,
2780/PDD). p27KiP~ protein levels are elevated in the drug resistant variants compared
5 to their parental counterparts, but only in three-dimensional culture. In contrast,
p21Waf1 protein levels are only slightly elevated in two of the four drug resistant
variants in three-dimensional culture.
Figure 3. Intense p27KiP1 immunostaining through out cross-sections of E/Pc5-
T spheroids. A~ter 3 days of growth in three-dimensional culture spheroids were
10 collected, paraffin embedded, and cross-sections were stained using a p27KiP1monoclonal antibody. Note the prominent nuclear staining in the majority of cells and
the presence of a few non-labelled cells in the outer rim and near the necrotic core.
Mitotic cells were also non-stained.
Figure 4. Antisense oligonucleotides inhibit p27KïP1 protein expression and
15 stimulate growth of E/Pc5-T cells. Cells treated with 7.5-60 nM of either antisense or
mismatch oligonucleotides in monolayer culture were placed into three-dimensional
culture in complete medium and 48 hours later pulsed with 3H-thymidine. In a
separate experiment (inset) cells treated with 5-30nM of oligonucleotides were
collecled for western blot analysis after 48 hours of growth in three-dimensional
20 culture. The Y-axis represents the relative fold decrease in p27KiP1 protein levels over
cytofectin treated controls as measured by densitometry of Western blot analysis.
Antisense treated cells showed up to a 2.5 fold decrease in p27KiP1 expression over
controls.
Figure 5. p27KiP1 antisense oligonucleotides abolish tight intercellular
25 adhesion of E/Pc5-T cells. Cells growing for 48 hours on either polyhema coated U-
bottom 96-well plates (top panel) or 6-well tissue culture plates coated with 1%SeaPlaque agarose (bottom panel) were photographed under 40X magnification.
Even when forced together in the U-bottom wells, antisense treated cells did notcompact into a spheroid but instead formed a wide loose sheet of cells, 5-10 cell
30 layers thick. (Bar = 0.5mm.).
- 10 -
~1 96~27
Figure 6. Effect of antisense p27KiP1 oligonucleotides on cell cycle changes
following exposure to ~-IR and 4-HC. E/Pc5-T cells treated with antisense or
missense p27KiP1 oligonucleotides were grown in three-dimensional culture for two
days, exposed to 20Gy ~-IR or 2011M 4-HC and 24 hours later, cells were collected
5 and DNA profiles were analyzed by flow cytometry.
Figure 7. Tumorigenicity assay of antisense or missense p27KiP1 treated
E/Pc5-T cells following exposure to 4-HC. After 2 days of growth in three-dimensional
culture oligonucleotide treated cells were exposed to 10 or 1511M 4-HC and 24 hours
later 5x105 cells were injected into Balb/c mice. Tumour volume = average i SE.
10 Tumour take is indicated in the legend.
DETAILED DESCRIPTION OF THE INVENTION
To determine the existence of acquired multicellular resistance, we showed
that acquired resistance in tumour cells can, in some cases, be accompanied by adecreased proliferating fraction. Specifically, when the mouse EMT-6 mammary
15 carcinoma cell line was made resistant to alkylating agents in vivo 6 it acquired new in
vitr~ properties that were detected only when analyzed in three-dimensional culture; ie
as multicellular tumour spheroids 7-9. Under these conditions we observed an increase
in intercellular adhesion (or compaction), a decrease in the rate of proliferation, and
maintenance of the in vivo drug resistance phenotype which was lost when cells were
20 grown in monolayer culture. Acquired multicellular resistance is only manifested at
the tumour population or proto-tissue level so it may be grossly under appreciated by
current research, at least in the case of solid tumours, due to the almost exclusive use
of monolayer culture systems for the study of drug resistance mechanisms 10~11. Since
the drug resistance properties of these cells correlated with a compact morphology,
25 we hypothesised a causal relationship between these two phenotypes. Adhesive
interactions at the cell surface, therefore, may be regulating cell proliferation which in
turn may regulate tumour cell resistance to anticancer agents most active against
rapidly dividing cells 8. In support of this view, we also found that addition of the
enzyme hyaluronidase simultaneously abolished intercellular adhesion, stimulated30 tumour cell proliferation and sensitized EMT-6 tumour cells to an activated form of
cyclophosphamide, 4-hydroperoxycyclophosphamide (4-HC) 8 and ~-irradiation (~-IR).
- 11 -
21 ~6727
Furthermore, hyaluronidase sensitized EMT-6 tumour bearing mice to
cyclophosphamide in vivo by a mechanism which appears to be unrelated to
increased drug penetration or microenvironmental influences3. Thus, an i"lil"aterelationship may exist between tumour cell adhesion, proliferation and sensitivity to
5 DNA damaging agents both in three-dimensional culture and in solid tumour masses
in vivo.
In the present study, we were interested in examining these relationships by
determining the molecular basis of either de novo or acquired adhesion-dependent(kinetic) resistance to anti-cancer agents.
We found that p27KiP1 (but not p21Waf' ) was consistently elevated in all mouse
and human breast, colon and ovarian carcinoma cell lines analyzed in three-
dimensional culture compared to the same cells grown in monolayer culture (Example
1). Furthermore, increased p27KiP1 levels correlated with acquisition of a drug
resistance phenotype (Example 2). By using antisense oligonucleotides to
15 downregulate p27KiP1 we found that this treatment sensitized tumour cell spheroids to
the cytotoxic effects of 4-HC (Example 4). Treatment with p27KiP1 antisense
oligonucleotides also caused disaggregation of spheroids and rendered tumour cells
sensitive to drug or radiation induced cell cycle perturbations (Examples 3 and 4).
These results implicate p27KiP1 as a possible major regulator of the adhesion-
20 dependent (kinetic) resistance of solid tumour cells to anti-cancer therapy, and
suggests that antagonists of p27KiP1 induction may be useful sensitizers in combination
with current treatment modalities.
E~.r~ ssio.. of p27~P1 is regulated by cellular topology
A solid tumour mass, by definition, contains cells in close proximity to one
25 another. p27KiP1 is a cyclin dependent kinase inhibitor that is known to be upregulated
in normal cells by contact inhibition 27-30. Our studies demonslrate that some tumour
cells, like normal cells, can also maintain a certain degree of contact-dependent
growth inhibition and retain functionally active growth inhibitory molecules like p27KiP1.
In three-dimensional culture, where the growth of tumour cells is more spatially30 restricted than in standard monolayer culture 41,42, we consistently observed an
increase in the level of p27KiP1. This increase in p27KiP1 appears to be regulated by at
2 1 9~7~7
least two different factors. First, growth in suspension under conditions of reduced
intercellular adhesion, as noted in our hyaluronidase treated E/Pc5-T cells, resulted in
a 6-7 fold increase in p27KiP1 levels over that in monolayer culture. This is consistent
with a recent report showing a similar increase in p27KiP1 (~4-fold) in both normal and
5 lldn~forl-1ed fibroblasts in suspension culture 43. The signals regulating this increase
in p27KiP1 could be related to a change in cell shape or a change in the cells ability to
adhere to, and receive signals from an extracelluar matrix. The second factor
upregulating p27K~P1 levels appears to be the degree of intercellular adhesion, as
shown in our E/Pc5-T cell line, grown in the absence of hyaluronidase, and our drug-
10 selected tumour cell variants also displaying a tightly adhesive spheroid morphology.
Taken together, these results suggest p27KiP1 levels in tumour cells can be stronglyupregulated by intercellular adhesion.
Also of interest was our finding that human tumour cell lines previously selected
for resistance in vitro to certain drugs, such as cisplatin or taxol, show an even greater
15 relative increase in p27KiP1 expression in three-dimensional culture than their
respective parental (drug-sensitive) counterparts. This observation may help to
explain the phenomenon of "reverse transformation" of drug resistant sublines in vivo
44'45. This refers to the reduced tumorigenic or growth rate of drug resistant sublines
that is often observed in vivo but not in vitro. Thus, in some cases the expression of
20 acquired resistance in solid tumours may be modulated, in part, by reduced growth
rates of the surviving cells due to increased p27KiP1 expression making such cells less
sensitive to various cytotoxic drugs.
Increased levels of p27KiP1 in adherent cells in three-dimensional culture
suggests that p27KiP1 is a critical downstream target of intercellular adhesion.25 Conversely, enforced p27KiP1 downregulation by antisense oligonucleotides resulted in
increased growth and reduced intercellular adhesion suggesting that the reverse is
also true; ie. that intercellular adhesion is also a downstream target of the cell cycle
machinery. In this regard, it is of note that cells need to round up and detach when
they divide. The molecules responsible for decreased adhesion at mitosis are
30 unknown, however, in human fibroblasts hyaluronic acid synthesis appears to be
necessary for cells to complete rounding and mitosis. The cell surface molecules
- 1 3 -
- 21 96727
mediating intercellular adhesion of our EMT-6 variants are also unknown, although
hyaluronic acid and its receptors are likely to be involved 8.
The relationship beh~een p27~ ex~.ression, cell cycle kinetics and tumour cell
death induced by DNA damage
Cells in compact spheroids expressing high p27KiPl levels fail to arrest in G2/Mfol'~w;ng exposure to 4-HC or ~-IR. By increasing the proliferative fraction, antisense
but not missense oligonucleotides to p27KiP1 sensitized cells to DNA damaging agents,
with resultant arrest in G2/M. Importantly, this failure to arrest in G2/M appears to be
specific to tightly adherent cells growing in three-dimensional context 8,46-48. The
reason for this may be simply that cells are growing at a maximum rate in subconfluent
monolayer cultures, and consequently, hyaluronidase or antisense p27KiP1 treatment
does not increase the proportion of cycling cells under these conditions. However,
when restrictions on cell growth are put in place, determined in our studies largely by
the levels of intercellular adhesion and p27KiP1 in three-dimensional culture, then cells
become resistant to cell cycle perturbations induced by DNA damaging agents.
Caution is necessary, therefore, when using monolayer culture systems for studying
cell cycle changes following drug exposure ". Even when exposed to increasing
concenl,dlions of 4-HC and observed for extended periods of time, cells of compact
spheroids never arrested significantly in G2/M 8. Since the G2/M arrest is predictive of
drug induced cell death 8 monitoring cell cycle alterations and p27KiP1 levels in tumour
cells following chemo- or radiation therapy in three-dimensional culture may be of
greater importance than was heretofore appreciated. A number of recent reports
using serial biopsy specimens, particularly with breast cancer patients, also showed a
similar correlation between G2/M arrest and tumour response to chemotherapy in the
clinic. 49-52.
Over the past two decades, many reports demonstrated that cells in three-
dimensional (multicellular) culture are intrinsically more resistant to cell-cycle specific
drugs than those grown in monolayer 41,42. Although barriers to drug penetrationand/or microenvironmental influences can sometimes account for this resistance, in
other cases they clearly cannot. This led to the proposal that another mechanism,
which Sutherland and Durand called the "contact effect", may be responsible for this
- 14 -
2 1 (t G 7 2 7
increased intrinsic resistance 41,42,53. Similarly, the term "confluence dependent
resistance" is used to describe drug resistance found in confluent versus subconfluent
monolayer cultures 54'55. Based on our present studies, we propose that an
upregulation of p27KiP1 may be responsible, at least in part, for each of these forms of
5 resistance. It is unclear why a p27KiP1 dependent accumulation of cells in G0/G1 is
associated with resistance to DNA damaging agents. One likely explanation for this
type of resistance is an enhanced capacity of G0/G1 cells to repair DNA lesions
through a process known as potentially lethal damage repair (PLDR). PLDR occurs in
confluent monolayer cultures, spheroids or tumours which are exposed to various
10 DNA damaging agents and then left intact for a period of time before stimulating their
growth by plating cells in a colony fon~dliol1 assay 8~56~7. Similarly, the experiments of
this study involved a 24 hour recovery period before cell survival was monitored.
Interestingly, PLDR may be modulated by changes in p27K~P1 expression. In other
words, inducing tumour cells to proliferate by decreasing p27KiP1 may limit their ability
15 to repair drug or radiation induced DNA damage. Another possibility is that adherent,
slowly proliferating spheroid cells may be less susceptible to DNA damage due toaltered DNA"packaging'~2.
Targeting p27K'P~ as a strdtey~ to enhance efficacy of anticancer therapy
Our antisense results demonstrate that p27KiP1 is essential for the adhesion
20 dependent resistance of solid tumour cells to DNA damaging agents. One recentreport suggests that an increase in p21Waf~ is associated with chemoresistance in
patients with AML 70. In this sense p27KiP1 and p21Waf~ may be viewed as survival
genes. Hence, localized downregulation rather than upregulation of p27~P~ would be a
desirable goal while administering cytotoxic therapy to tumours. This represents a
25 potential paradigm shift; high expression of cell cycle inhibitors may allow clonogenic
tumour cells to escape cell kill by chemo- or radiation therapy. Thus, downregulation
of p27KiP1 and perhaps p21Waf1 in some tumour types, represents a new potential
strategy for augmenting anticancer therapy. Of concern, however, is the potential for
a simultaneous increase in normal tissue toxicity. If normal tissues retain other intact
30 CKls such as p15 and p16 which can compensate for the loss of other inhibitors, then
downregulation of a single CKI, p27KiP1, may be minimally toxic. Therefore, the
- 1 5 -
2~ q6727
potential for achieving enhanced specificity using this type of treatment (ie. an
increased therapeutic index), may depend on tumour cells having lost some CKl's,such as the INK family members but retaining others, such as p21 Waf1 and p27taP1. An
analysis of tumor biopsies suggests that this may occur in some cases2~267~.
Importantly, p21Waf~ and p27KiP1 knockout mice are viable and do not manifest any
gross morphological abnormalities, and with the exception of benign pituitary
adenomas (reported only in the p27 homozygous knockout mice) do not appear to
have an increased propensity for developing tumors72~74. The present results suggest
that the anti-adhesive agent hyaluronidase may also be exerting its chemosensitizing
effects on EMT-6 cells in vivo 8 indirectly, by downregulating p2PaP1. Unfortunately, the
ability of hyaluronidase to sensitize tumours may be limited due to fact that it may
affect only a subset of the many different adhesion molecules associated with various
tumours types. More promising perhaps, may be the tissue specific targeting of p27KïP1
through an antisense approach in vivo75'76, or through other low molecular weight
pharmacological inhibitors of p27KiP1.
With respect to the clinical relevance and application of our results, one couldreasonably ask if it would be ethical to increase tumour cell growth to augment anti-
cancer therapy. We believe it may be, for the following reasons. First, results from
clinical trials which combined chemotherapy with hormonal or cytokine induction
showed either some or no improvement in response, but patients generally did not do
worse than would be expected if given chemotherapy alone34 35. Secondly, a series of
reports relating cell cycle parameters with drug or radiation sensitivity, primarily in
breast cancer patients, showed that tumours with a high S-phase fraction almost
always respond better than those with slower growing tumors4951 75~79. Finally, the
tumour types that are considered curable by chemotherapy, such as childhood
tumours, some Iymphomas, choriocarcinoma and testicular carcinoma tend to grow
very rapidly' 2.
Spheroid tumour cultures as an assay for new anti-cancer agents and
che",~sensitizers
As described above, there are many problems with the use of monolayer cell
cultures and liquid tumour models in detecting drugs which will work well in vivo. Drug
- 16 -
- ~1 96727
resistance is clearly related to cell-cell adhesion, so there are problems with
experimental approaches which emphasise unicellular resistance mechanisms of
resistance. Spheroid tumour cell cultures are able to more closely simulate the in vivo
response of tumours to chemicals. This is because spheroid cultures replicate the
5 effects of multicellular structure and cell-cell contact on the expression of drug
resistance and the ability to reverse or prevent it with chemosensitizers. Spheroid
cultures may be used in assays to screen for anti-cancer agents which are
preferentially active against slowly dividing cells. They may also be used in assays to
screen for chemicals which chemosensitize solid tumours to anti-cancer drugs.
In summary, our studies demonstrate that adherent tumour cells generally
express high levels of p27~P~, but not necessarily p21 Waf1, in three-dimensional
culture. This may help to explain why most solid tumour types, despite harbouring
multiple oncogenes and tumour suppressor genes, do not grow demonstrably faster
than some mitotically-active normal cells in the body such as gut mucosal or certain
15 bone marrow derived cells. Furthermore, cell lines with acquired resistance to
chemotherapeutic agents show an increase in both intercellular adhesion and p27KiP1
levels, but again only when grown as three-dimensional aggregates. Enforced p27KiP1
downregulation resulted in decreased intercellular adhesion, increased cell growth,
and altered cell cycle kinetics following drug and radiation treatment. Downregulation
20 of p27KiP1 also sensitized tumour cells to 4-HC implicating p27KiP1 as a mediator of both
acquired and intrinsic resistance to anticancer agents. Thus antagonists of cyclin
dependent kinases represent a novel class of chemosensitizers in the rational
treatment of solid tumours with anticancer therapy. Spheroid tumour cell cultures
represent a novel tool to screen chemicals which are anti-cancer agents or
25 chemosensitizers.
EXAMPLE 1
Overexpression of p27K'P' in tumour cells grown as multicellular aggleylates
Our previous studies suggested that an acquired adhesion-dependent
decrease in cell proliferation may be necessary for the resistance of tumour cells to
30 cytotoxic agents most active against rapidly dividing cells. In an allen1pt to understand
the molecular basis of adhesion-dependent cell proliferation we analyzed levels of
21 q6727
p21 Waf1 and p27WP1 in our clonally derived variants of the EMT-6 mammary carcinoma
cell line. The two variants used, E/Pc1 0-L and E/Pc5-T, were clones that
spontaneously formed either loosely or tightly adherent aggregates, respectively,
when grown in three-dimensional culture. We also took advantage of the fact that5 compact intercellular adhesion can be disrupted in E/Pc5-T cells by treatment with
hyaluronidase.
p21 Waf1 and p27KiP1 protein levels were relatively constant in non-synchronizedmonolayer cultures, regardless of which clone was analyzed or whether or not
hyaluronidase was added to the medium (Figure 1 a). In sharp contrast, large changes
10 were observed, particularly with respect to p27KiP1 expression, when cells were
analyzed after three days of growth in three-dimensional culture. In all cases an
increase in the level of p27KiP1 was observed in suspension culture; however, the
increase was most pronounced in cells of the tightly adherent clone, E/Pc5-T. Bydensiton,etric analysis of p27KiP1 protein on western blots, this clone showed a ~10-1
15 fold increase in p27KiP1 protein. In contrast, the loosely adherent E/Pc1 1-L clone and
the hyaluronidase treated E/Pc5-T clone, displayed a much more moderate increase(6-7 fold) when transferred from two- to three-dimensional culture. The increase in
p27KiP1 expression in three-dimensional culture was gradual and paralleled an
increase in aggregate compaction. As shown in figure 1b, after 8 hours in suspension
20 p27KiP1 levels were unchanged but by 24 hours an increase in p27KiP1 was apparent.
The level of p27KiP1 continued to increase until 96 hours after which p27KiP1 protein
levels appeared to plateau. In contrast, p27KiP1 expression in hyaluronidase treated
cells increased rather slowly and never reached the same level as in adherent E/Pc5-
T cells (data not shown). Unlike p27KiP1, p21 Waf1 levels were relatively high in
25 monolayer and were undetectable by 96 hours in cells grown as aggregates (figure
1b) Thus, increased levels of p27KiP1, but not p21Waf1, correlated with increased
cellular compactness and a greater proportion of cells which show a 2N DNA content
in three-dimensional culture.
We also analyzed p21 Waf1 and p27KiP1 protein levels 24 hours following
30 exposure to a low concentration (6,uM) of 4-HC (Figure 1a). In rapidly proliferating
monolayer cultures, a decrease in p27KiP1 level was evident following drug exposure.
- 18-
21 96727
This decrease coincided with a massive G2/M arrest 8 In three-dimensional culture,
p27KiP1 levels were relatively unaffected by exposure to 4-HC.
In order to determine how general this p27KiP1 increase in response to three-
dimensional culture might be, we analyzed the expression of p27KiP1 in a variety of
human breast, colon and ovarian tumour cell lines after plating cells into suspension
(figure 2a). In every cell line tested, we observed an increase in p27KiP1 levels (~1.2-
15 fold) in three-dimensional culture. Again, no such correlation was noted for p21Waf'
expression except in the case of the SW480.7 colon carcinoma which showed a
significant increase in response to three-dimensional culture. We also undertookimmunohistochemistry on cross-sections of E/Pc5-T spheroids to determine the
geographical pattern of p27KiP1 staining. Intense nuclear staining of p27KiP1 was found
in cells distributed through out the spheroid (Figure 3). Only a few non-stained cells
were observed and these were found either in the outermost rim of proliferating cells
or in the centre of the spheroid near the necrotic core. The layer of tightly packed cells
directly beneath the outermost proliferating rim stained most intensely. This pattern is
consistent with a role for intercellular adhesion, in regulating p27KiP1 production.
EXAMPLE 2
.12iLK'p1 levels are elevated in cell lines with an acquired
drug resistance phenot,vpe
We compared the levels of p21Waf~ and p27KiP1 in cell lines which had been
selected either in vitro or in vivo for resistance to a variety of chemotherapeutic agents
(Figure 2b). In both the MDA 435 breast cancer cell line and its in vitro derived taxol
resistant variant, 435/TO.3, very little p27KiP1 was noted in monolayer culture. In three-
dimensional culture, however, both cell lines showed an increase in p27KiP1 although
435/TO.3 showed a much higher level than the parental MDA 435 cell line. The same
pattern was also observed for the human ovarian carcinoma cell line A2780 and its
cisplatin resistant variant A2780/PDD, also selected in vitro. The in vivo derived
cyclophosphamide and cisplatin resistant variants of the EMT-6/P cell line, E/CTX and
E/DDP also showed a greater increase in p27KiP1 when transferred to three-
dimensional culture than did the parental cell line. Notably, all of these drug resistant
variants also displayed an obvious increase in intercellular adhesion, growing as
- 19-
21 96727
compact multicell~ r aggregates compared to their looser parental counterparts7
(unpublished observations). The relatively high level of p27KiP1 observed in the EMT-
6/P cell line grown in three-dimensional culture, may be due to the fact that this is a
heterogeneous population composed of clones capable of forming both loose or tight
5 aggregates 8. In this regard, the loose clones derived from this population displayed a
much lower level of p27KiP1 by western blot when compared to the tight clones in three-
dimensional culture (Figure 1a). Although a slight increase in p21Waf1 was evident in
two human drug resistant varia"l~ compared to their parental counterparts in three-
dimensional culture, the same pattern was not observed in two resistant variants of the
10 murine EMT-6/P cell line (figure 2b). Thus, acquisition of a drug resistance phenotype
correlated with a consistent increase in the level of p27KiP1 and an increase inintercellular adhesion, but only when assayed in three-dimensional culture.
EXAMPLE 3
A"lise.,se p27WP~ oligonucle Dtides decrease i"l~rcellular adhesion
and i"cr~ase cell proliferation
In order to determine if the increase in p27KiP1 was responsible for the
decreased growth kinetics of these cells under three-dimensional culture conditions,
we employed an antisense approach to downregulate p27KiP1. For this purpose we
utilized a new generation of oligonucleotides which contain C5-propyne modified
20 bases 31~32 Due to an enhanced afffinity for complementary RNA, lower concenl~dliol1s
of these modified oligonucleotides can be used, making them potentially more specific
than conventional phosphorothioates 31. Cells were treated with antisense or
missense p27KiP1 oligonucleotides in monolayer culture and then placed into three-
dimensional culture. As shown in figure 4, the most significant reduction in the level of
25 p27K~P1 protein (~2.5fold over control) was noted in the antisense treated cells at 48
hours. Our finding that a 2.5 fold reduction in p27KiP1 protein levels was suffficient to
profoundly alter the cell cycle profile (see below) is consistent with the observation that
only a slight increase in p27KiP1 expression is necess~ to saturate cyclinE/cdk2 33.
These differences were obvious but less pronounced at 24 hours, perhaps due to a30 lower endogenous level of p27KiP1 overall (unpublished observations). As expected,
- 20 -
21 ~61Z7
the expression of p21Waf~ protein was unaltered by the antisense oligonucleotides
(Figure 4).
When we exposed the E/Pc5-T tightly adherent clone to antisense p27KiP1
oligonucleotides, we immediately noticed a massive reduction in the level of
intercellular aggregation in three-dimensional culture (Figure 5). This morphological
change appeared to be specific as it occurred with two independent antisense
oligonucleotides but was not apparent in two mismatch controls (data not shown). As
shown in Figure 4, the same two independent antisense oligonucleotides caused a
marked dose-dependent increase in thymidine incorporation. The two mismatch
sequences had a negligible effect a low concentrations (30nM) but caused a slight
increase in 3H-thymidine uptake at higher (60nM) concentrations. Since non-specific
effects were minimal at 30nM of oligonucleotide, this concentration was chosen for the
subsequent drug sensitivity assays. Not surprisingly, the effect of antisense p27KiP1 on
DNA synthesis was transient reaching a maximum at about 48 hours and decreasing
to a barley detectable level by about 96 hours. Notably, the effect on adhesion was
also most pronounced at 48 hours and was diminished by 96 hours. Thus, treatmentwith C5-propyne modified antisense oligonucleotides against p27KiP1 simultaneously
released E/Pc5-T cells from adhesion induced G1 arrest and decreased intercellular
adhesion in three-dimensional culture.
EXAMPLE 4
Antisense p27~P' oli~onucleotides alter cell cycle kinetics
and sensitize tumour cells to 4-HC
Previously we observed that loose aggregates of hyaluronidase treated E/Pc5-
T cells arrested in G2/M follcw;"g exposure to 4-HC8 or ~-IR (unpublished
observations) in three-dimensional culture. In contrast, untreated E/Pc5-T cellsgrowing as tight spheroids failed to arrest in G2/M fcl'~J.;,lg drug treatment. In order to
determine whether or not the level of p27KiP1 affected these cell cycle perturbations,
we treated E/Pc5-T cells with antisense or mismatch p27K7P1 oligonucleotides and then
evaluated cells for drug or radiation induced cell cycle changes (Figure 6). In this
experiment, cells were treated with oligonucleotides in monolayer culture, placed into
three-dimensional culture, and 48 hours later exposed to 4-HC or ~-IR. After an
21 ~6727
additional 48 hours in suspension, cells were collected and analyzed by flow
cytometry. As expected, 48 hours following treatment with oligonucleotides, a much
larger proportion of the p27~aP1 antisense treated cells were in S-phase than in the
mis,nalch treated control (44.5% vs 28.4% -see figure 6). This effect disappeared by
5 96 hours, at which time point 15.1% of the antisense ll~:aled cells were in S-phase
compared to 14.5% in the mis,nalch control. Forty-eight hours after exposure to 4-HC,
a much larger proportion of cells were arrested in G2/M in the antisense treated group
than in the misl"atcl1 control. A similar pattern was also observed following exposure
to~-lR.
In a parallel experiment E/Pc5-T cells, treated with oligonucleotides, were
placed back into monolayer culture instead of spheroid culture. After 48 hours, cells
were then exposed to 4-HC or ~-IR and after an additional 48 hours analyzed for cell
cycle changes. In this case, no difference in cell cycle distribution was observed in
cells treated with antisense or mismalch oligonucleotides alone (54.9% and 52.6% of
cells in S-phase respectively). After exposure to either 4-HC or ~-IR a majority of the
cells in monolayer culture were arrested in G2/M with no obvious difference between
the antisense or mismatch treated groups (unpublished observations). Thus, p27~aP1
levels had a prominent effect on cell cycle perturbations observed following exposure
to DNA damaging agents, but only in three-dimensional culture.
To determine whether or not p27~GP1 may play a causative ro!e in resistance to
anti-cancer agents, we used our antisense strategy to downregulate p27~aP1 and then
assayed tumour cells for drug sensitivity. To measure drug resistance, a
tumorigenicity assay which we previously developed was employed8. In this assay,E/Pc5-T tumour cells treated with oligonucleotides were plated into three-dimensional
culture and 48 hours later exposed to 4-HC. Twenty-four hours after drug exposure,
cells were harvested and injected s.c. into syngeneic Balb/c mice and their tumorigenic
ability was evaluated. As shown in figure 7, no significant difference in tumour growth
was observed in cells treated with antisense or missense oligonucleotides alone.When 4-HC was added to the medium, however, obvious differences in vivo became
apparent. When exposed to 10 or 15~1M 4-HC, antisense treatment led to a
significantly longer in vivo latency period than in the corresponding mismatch treated
- 22 -
21 96727
cells. As well, at the higher concentration, tumour take was lower in the antisense
than in the mismatch treated group. As expected, control cells without oligonucleotide
behaved similarly to mismatch treated cells (data not shown). Our previous studies
suggest that reduced drug access is unlikely to be a major mechanism of resistance
5 since cells exposed to 4-HC in monolayer and then aggregated in three-dimensional
culture retain levels of resistance which correspond to their respective adhesive
properties 8 Taken together, these results demonstrate that p27KiP1 is a critical
component required for the adhesion-dependent (kinetic) resistance of tumour cells to
4-HC.
Example 5
Therapeutic Downre~ulation of p27K'P~
We downregulate p27KiP1 in solid tumour cells growing in experimental mice to
produce a better anti-tumour response after exposure to conventional
chemotherapeutic drugs such as cyclophosphamide or taxol. These studies are
15 initiated using a mouse breast cancer cell line called EMT-6 grown intraperitoneally in
Balb/c mice as an "ascites" tumour in which the tumour cells grow as small spheroidal
aggregates in the fluid filled peritoneal cavity. Tumour-bearing mice are injected with
antisense oligonucleotides to the p27KiP1 gene incorporated into cationic lipids to
increase selective delivery and uptake into tumour cells. This is followed by injection
20 of a cytotoxic drug such as cyclophosphamide or taxol. The injections are performed
in a manner similar to that described by St. Croix et al8 except that antisense
oligonucleotides are used as the chemosensitizing agent rather than hyaluronidase.
Controls include animals injected with cyclophosphamide alone, antisense
oligonucleotides alone, or no treatment. Evidence for a synergistic chemosensitizing
25 effect of the antisense oligonucleotides is closely evaluated and compared with control
"mismatch" oligonucleotide preparations as described in reference 82. If a beneficial
effect is achieved, evidence for a therapeutic downregulation of p27KiP1 in the
antisense treated tumour cells in vivo is dete"l~i.,ed. Evidence fo~ the possibility of
increased toxicity to normal cells and tissues which express p27KiP1 is evaluated. In
30 addition to using a cytotoxic drug for such experiments, such as cyclophosphamide,
-- 21 C~61~
we study ~-irradiation therapy, the effect of which we wish to enhance by injection of
antisense oligonucleotides to p27KiP1.
Further experiments are undertaken to evaluate the possibility that antisense
oligonucleotide-mediated downregulation of p27KiP~ in small microscopic tumours may
increase the beneficial effect of conventional cytotoxic drugs in an "adjuvant therapy"
setting, ie. in situations where animals have minimum residual (occult) disease which
has a good chance of eventually growing into life threatening metastatic tumours. For
example, this could be done by intravenous inoculation of EMT-6 or HEY cells into
mice in order to establish small microscopic deposits of tumours in the lungs ("lung
colonies"). The animals are then injected with a cytotoxic drug alone, eg.
cyclophosphamide, antisense oligonucleotides to p27KiP1 alone, or the two agentstogether. The number and size of lung colonies can then be determined at definedpoints after the therapy, as can overall survival times.
We undertake experiments to determine whether the therapeutic
("chemosensitizing") effect of hyaluronidase, as described above, is due in part to an
induced downregulation of p27KiP1 expression in ascitic EMT-6 spheroids. For
example, the relative expression of p27KiP~ is determined in non-dispersed versus
dispersed EMT-6 cells removed from animals which were not treated, or treated, with
hyaluronidase, respectively.
We undertake experiments in which the levels of p27KiP~ in specimens of
human solid tumours, especially breast and ovarian cancer, are evaluated. This type
of information should be important in indicating what types of tumours might be
worthwhile as targeting for therapeutic downregulation of p27KiP~.
The last set of experiments we undertake is aimed at reproducing in human
tumour cell lines the chemosensitizing results we obtained with p27KiP~ antisense
oligonucleotides and mouse mammary EMT-6 cells6. In other words, the sensitivity of
human breast or ovarian cells to a cytotoxic drug such as cyclophosphamide or taxol
is increased by co-treatment of the cells with antisense p27KiP1 oligonucleotides. In
these experiments we also use various cell lines that are previously selected for high
levels of resistance to taxol or cisplatin as well as their drug sensitive parental
- 24 -
21 96127
counterparts. These experiments indicate whether the levels of acquired drug
resistance can be reversed to some extent by downregulating p27KiP~.
MATERIALS AND METHODS
Cell lines and culture con~ . The human MCF-7 breast carcinoma and the
5 murine EMT-6 mammary carcinoma (EMT-6/P) and its in vivo derived alkylating-agent
resistant variants E/CTX and E/DDP6 were a gift of Dr. Beverly Teicher (Dana-Farber
Cancer Institute, Boston, MA). E/Pc5-T, E/Pc7-T and E/Pc10-L are "tightly"(T) or"loosely"(L) adherent clones of the EMT-6/P cell line which were described previously8.
All EMT-6 cells were cultured in Waymouths MB 752/1 medium, supplemented with
10 10% FBS. For hyaluronidase treatment of E/Pc5-T cells, 2mg/ml of bovine testicu!ar
hyaluronidase (Sigma) was added to the culture medium at the time of cell plating and
cells were collected and assayed three days later. The human MDA 435 breast
carcinoma cell line and its taxol resistant variant 435/TO.3, a gift of Dr. Dalia Cohen
(Sandoz Research Institute, Fast Hanover, NJ), were grown in DMEM supplemented
15 with 10% FBS, 0.1mg/ml Sodium Pyruvate and 0.1mM non-essential amino acids.
The human ovarian A2780 carcinoma and its cisplatin resistant variant A2780/PDD,donated by Dr. Tom Hamilton (Fox Chase Cancer Center, Philadelphia, PA) were
grown in RPMI 1640 supplemented with 10% FBS. All other cell lines were purchased
from ATCC and were grown in DMEM with 10% FBS except BT549 and HBL100
20 which were grown according to the suppliers recommended protocol.
Immunoblotting. Cells from monolayer culture were harvested in exponential growth
phase while three-dimensional cultures were grown for three days before harvesting.
Cells were treated and collected after 3 days or growth in hyaluronidase containing
medium or 24 hours following treatment with 0 or 6 ~M 4-
25 Hydroperoxycyclophosphamide (4-HC). After collecting, cells were rinsed with PBS
and stored as cell pellets at -70~C until ready for use. Cells were Iysed in ice-cold NP-
40 Iysis buffer (1% NP-40, 10% glycerol, 20mM Tris-HCI pH7.5, 137mM NaCL,
100mM NaF, 1mM sodium vanadate, 1mM phenylmethyl sulphonyl fluoride (PMSF)
and 0.02mg/ml each of aprotinin, leupepsin and pepstatin). The Iysates were
30 sonicated and clarified by centrifugation. Protein was quantified by Bradford analysis
and 30mg/lane was resolved by SDS polyacrylamide gel electrophoresis (PAGE) and
- 25 -
21 96727
blotted onto Immobilon-P-membranes (Millipore Corporation,Bedford, MA). To control
for loading and transfer, membranes were stained with 0.1 % Naphtol Blue Black
(Sigma, St. Louis, MO). The membranes were blocked by TBST (0.25 % Tween)
containing 10% dry milk and incubated for 1h at room temperature with a mouse
5 monoclonal p27KiP1 antibody (Transduction Laboratories, Lexington, KY) diluted1 :1000 or a rabbit polyclonal p21 Waf1 antibody (Santa Cruz Biotechnology, Santa Cruz,
CA) diluted 1:500 in TBST containing 5% dry milk. After washing in TBST, the
immunoreactive proteins were visualized using horseradish peroxidase conjugated
antimouse-lgG (Promega Corporation, Madison, Wl) diluted 1:5000 and the ECL
10 Western blotting detection system (Kirkegaard & Perry Laboratories, Gaithersburg,
MD)
Immunohistocl-e.~-ical staining of p27~P' E/Pc5-T spheroids grown in three-
dimensional culture for 3 days were carefully removed from agarose coated 24-well
dishes using a pasture pipette and placed into a 1.5 ml eppendorf tube. After gently
15 spinning for 5 minutes (800 rpm) spheroids were rinsed once with PBS, and then 10%
formalin solution (in PBS) was overlaid onto pelleted spheroids, which were later
embedded in parafffin. The paraffin embedded sections were deparafffinized with
xylenes, rehydrated, and microwaved for 10 minutes in citrate buffer (pH 6.0).
Sections were blocked for endogenous peroxidase with 3% H2O2 in methanol then
20 blocked for non-specific staining with immunoglobulins from normal horse serum (1 :20
dilution). After removing excess blocking serum, sections were incubated overnight
with monoclonal p27KiP1 antibody (Transduction Laboratories, Lexington, KY) diluted
1:1000 (0.25mg/ml) in PBS, followed by incubation with biotin labelled anti-mouse
secondary antibody. The sections were incubated with preformed avidin-biotin-
25 peroxidase complex (Vector Laboratories, Burlingame, CA). The metal enhanceddiaminobenzidine (DAB) substrate (Pierce, Rockford, IL) was added that develops into
a dark brown precipitate in the presence of Horse Radish Peroxidase. The sections
were then counterstained with hematoxylin, dehydrated and mounted with permount.Oligonucleotide l,e~l,ent The sequences of the antisense (AS) and mismatch
30 (MSM) p27KiP1 C-5-propyne modified phosphorothioates utilized in the experiments
shown, (designated AS1 and MSM1 in the 3H-thymidine incorporation assay) were 5'-
- 26 -
21 q6727
GCGUCUGCUCCACAG-3' and 5'-GCAUCCCCUGUGCAG-3', respectively. The
alternative oligonucleotides labelled AS2 and MSM2 in the 3H-thymidine assay were
5'-UGGCUCUCCUGCGCC-3' and 5'-UCCCUUUGGCGCGCC-3', respectively. The
specificity of precisely the same set of oligonucleotides for p27~aP1 has been shown
5 previously30. For maximum and uniform delivery, all treatments were performed on
rapidly growing monolayer cultures. For transfection, C5-propyne modified
oligonucleotides (Gilead Sciences, Foster City, CA) at 20X the final conce,llldlion in
excell-300 medium (pH7.2) were heated (65~C) for 5 minutes to denature secondarystructure, and then mixed at room temperature with a 20X solution of GS2888
10 cytofectin (Gilead Sciences). After 10-15 minutes, oligo/cytofectin complexes were
diluted to 1X conce"lldlion in excell-300 and overlaid onto cells. Final concentrations
used were 2mg/ml for GS2888 and 5-60nM for oligonucleotides. After 5 hours of
incubation with oligonucleotides, cells were rinsed with PBS, harvested with trypsin,
and then plated into various assays in complete medium containing 10% FBS.
15 Proliferation assay A thymidine incorporation assay was used to measure
proliferation in E/Pc5-T cells. A single cell suspension was prepared from cells that
had been treated with antisense or Illis,,,atch oligonucleotides and 5000 cells/well in
100ml of complete medium was added to 96-well U-bottom plates (Nunc). After 48
hours of incubation, 211Ci of 3H-thymidine was added in 50ml to each well and plates
20 were pulsed for 4 hours. Labelled cells were frozen at -70~C and later harvested onto
filtermats using a Titertek cell harvester 530. Radioactive filtermats were then counted
using a 1205 beta plate liquid scintillation counter (Fisher Scientific Ltd. Nepean, ON).
The rate of DNA synthesis of the antisense or mismatch treated groups were
c~lclll~ted as a fraction of the control counts obtained from cells treated with cytofectin
25 alone. To prevent attachment of cells to the bottom of 96-well plates, a heated (56~C)
solution of 2% poly(2-hydroxyethylmethacrylate) (polyhema) (Aldrich Chemical
Company, Inc. Milwaukee Wl) in ethanol was briefly added to and then removed from
the plate leaving behind a thin film in each well.
Monitoring cell cycle changes following exposure to 4-HC or ~-IR Antisense or
30 mismatch treated cells from monolayer culture were harvested and plated into three-
dimensional culture using the liquid overlay technique previously described 7. After 48
21 ~b727
hours of growth in suspension, cells were treated with either 20 ~M 4-HC or 20Gy y-IR
and left for an additional 48 hours before collecting. For DNA analysis, cells were
dissaggregated with trypsin, rinsed with PBS, fixed in 70% ethanol, filtered through
30mm mesh, and then stained with Pl solution (50mg/ml propidium iodide, 10mg/ml
5 RNAaseA in PBS). Cells were analyzed using the Lysisll software on a Facscan flow
cytometer (Beckton-Dickinson, San Francisco, CA). Cell cycle phase distributionswere calculated using Cell Fit software.
Tumorigenicity assay E/Pc5-T cells treated with oligonucleotides in monolayer
culture were harvested and plated into three-dimensional culture using the liquid
10 overlay technique 7. After two days of growth in suspension, tumor cells were exposed
to 10 or 15~LM 4-HC and 24 hours later, rinsed with PBS, typsinized and 5x105 cells
were injected subcutaneously into syngeneic Balb/c mice. Tumor volume was
calculated using the formula T=d2D/2 where d= the smallest tumor diameter (mm) and
D= the largest tumor diameter (mm).
All publications, patents and patent applications are herein incorporated by
reference in their entirety to the same extent as if each individual publication, patent or
patent application was specifically and individually indicated to be incorporated by
20 reference in its entirety.
The present invention has been described in terms of particular embodiments
found or proposed by the present inventors to comprise preferred modes for the
practice of the invention. It will be appreciated by those of skill in the art that, in light of
the present disclosure, numerous modifications and changes can be made in the
25 particular embodiments exemplified without departing from the intended scope of the
invention. All such modifications are intended to be included within the scope of the
appended claims.
- 28 -
21 q6727
References:
1. Tannock, I. Cell kinetics and chemotherapy: a critical review. Cancer TreatRep.
62:11 17-1133 (1978).
2. Tannock, I.F. Principles of cell proliferation: cell kinetics. in Current Cancer
Therapeutics 3-13 (Princeton Academic Press, 1994).
3. Grindey, G.B. Current status of cancer drug development: failure or limited
success? Cancer Cells 2:163-171 (1990).
4. Webb, A.& Cunningham, D. Curing gastric cancer - hone the scalpel with
magic? Br.J. Cancer 73:418419 (1996).
5. Lipkin, M., Bell, B.& Sherlock, P. Cell proliferation kinetics in the gastrc..,testi,lal
tract of man. I. Cell renewal in colon and rectum. J.Clin.lnvest 42:767-776 (1963).
6. Teicher, B.A. et al. Tumor resistance to alkylating agents conferred by
mechanisms operative only in vivo. Science 247:1457-1461 (1990).
7. Kobayashi, H., Man, S., Kapitain, S.J., Teicher, B.A.& Kerbel, R.S. Acquired
multicellular-mediated resistance to alkylating agents in cancer.
Proc.Natl.Acad.Sci.(USA) 90:3294-3298 (1993).
8. St.Croix, B. et al. Reversal by hyaluronidase of adhesion dependent drug
resistance in mammary carcinoma cells. J.Natl.Cancerlnst in press:(1996).
9. Kerbel, R.S. et al. Multicellular resistance: a new paradigm to explain aspects of
acquired drug resistance of solid tumors. Cold Spring Harbor Symposium on
Quantitative Biology: Molecular Genetics of Cancer59:661-672 (1994).
- 29 -
21 96727
10. Hoffman, R.M. In vitro assays for chemotherapy sensitivity.
Crit.Rev.Oncol.Hematol. 15:99-111 (1993).
11. Hoffman, R.M. The three-dimensional question: can clinically relevant tumor
drug resistance be measured in vitro? Cancer Metastasis Rev. 13:169-173 (1994).
12. Morgan, D.O. Principles of CDK regulation. Nature 374:131-134(1995).
13. Sherr, C.J.& Roberts, J.M. Inhibitors of mammalian G1 cyclin-dependent
kinases. Genes & Develop. 9:1149-1163 (1 995).
14. Hirama, T.& Koeffler, H.P. Role of the cyclin-dependent kinase inhibitors in the
development of cancer. Blood86:841-854 (1995).
15. Harper, J.W., Adami, G.R., Wei, N., Keyomarsi, K.& Elledge, S.J. The p21
Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. CeR
75:805-816 (1993).
16. Xiong, Y. et a/. p21 is a universal inhibitor of cyclin kinases. Nature
366:701-704 (1993).
17. Polyak, K. et a/. Cloning of p27KiP~, a cyclin-dependent kinase inhibitor and a
potential mediator of extracellular antimitogenic signals. Cell 78:59-66 (1994).
18. Toyoshima, H.& Hunter, T. p27, a novel inhibitor of G1 cyclin-Cdk protein
kinase activity, is related to p21. Cell78:67-74 (1994).
19. Harper, J.W. et a/. Inhibition of cyclin-dependent kinases by p21. Mol.Biol.Cell
6:387400 (1995).
- 30 -
21 q6727
20. Piete,)pol, J.A. et al. Assignment of the human p27K~P1 gene to 12p13 and its
analysis in leukemias. CancerRes. 55:1206-1210 (1995).
21. Ponce-Castaneda, M.V. etal. p27K'P~: chromosomal mapping to 12p12-12p13.1
and absence of mutations in human tumors. CancerRes. 55:1211-1214 (1995).
22. Kawamata, N. et al. Molecular analysis of the cyclin-dependent kinase inhibitor
gene p27A<ip1 in human malignancies. CancerRes. 55:2266-2269 (1995).
23. Shiohara, M. et al. Absence of WAF1 mutations in a variety of human
malignancies. Blood84:3781-3784 (1994).
24. Morosetti, R. et al. Alterations of the p27KiP1 gene in non-Hodgkin's Iymphomas
and adultT-cell leukemia/lymphoma. Blood86:1924-1930 (1995).
25. Ferrando, M. et al. Mutational analysis of the human cyclin dependent kinaseinhibitor p27kiP~ in primary breast carcinomas. Hum. Genet. 97:91-94 (1996).
26. Spirin, K.S. et al. p27/Kip1 mutation found in breast cancer. Cancer Res.
56:2400-2404 (1996).
27. Slingerland, J.M. et al. A novel inhibitor of cyclin-Cdk activity detected in
transforming growth factor b-arrested epithelial cells. Mol.Cell.Biol. 14:3683-3694
(1994).
28. Hengst, L., Dulic, V., Slingerland, J.M., Lees, E.& Reed, S.l. A cell
cycle-regulated inhibitor of cyclin-dependent kinases. Proc.Natl.Acad.Sci.(USA)
91 :5291 -5295 (1994).
29. Polyak, K. et al. p27KiP1, a cyclin-Cdk inhibitor, links transforming growth factor-b
and contact inhibition to cell cycle arrest. Genes & Develop. 8:9-22 (1994).
-31 -
21 96727
30. Poon, R.Y.C., Toyoshima, H.& Hunter, T. Redistribution of the CDK inhibitor
p27 between different cyclin-CDK complexes in the mouse fibroblast cell cycle and in
cells arrested with lovastatin or ultraviolet irradiation. Mol.Biol. Cell 6: 1197-1213 (1995).
31. Wagner, R.W. et al. Antisense gene inhibition by oligonucleotides containingC-5 propyne pyrimidines. Science 260:1510-1513 (1993).
32. Wagner, R.W. Gene inhibition using antisense oligodeoxynucleotides. Nature
372:333-335 (1994).
33. Reyniskottir, I., Polyak, K., lavarone, A.& Massague, J. Kip/Cip and Ink4 Cdk
inhibitors cooperate to induce cell cycle arrest in response to TGF-b. Genes &
Develop. 9: 1831 -1845 (1995).
34. Conte, P.F. et al. Conventional versus cytokinetic polychemotherapy with
estrogenic recruitment in metasld~ic breast cancer: results of a randomized
cooperative trial. J. Clin. Oncol. 5:339-347 (1987).
35. Gore, S.D. et al. Impact of in vivo administration of interleukin 3 on proliferation,
differentiation, and chemosensitivity of acute myeloid leukemia. Clin.Cancer Res.
1 :295-303 (1995).
36. Williams, G.T., Smith, C.A., Spooncer, E., Dexter, T.M.& Taylor, D.R.
Haemopoietic colony stimulating factors promote cell survival by suppressing
apoptosis. Nature 343:76-79 (1990).
37. Lotem, J.& Sachs, L. Hematopoietic cytokines inhibit apoptosis induced by
transforming growth factor b1 and cancer chemotherapy compounds in myeloid
leukemic cells. Blood 80: 1750-1757 (1992).
- 32 -
21 '~6727
38. Teixeira, C., Reed, J.C.& Pratt, M.A.C. Estrogen promotes chemotherapeutic
drug resistance by a mechanism involving Bc1-2 proto-oncogene expression in human
breast cancer cells. Cancer Res. 55:3902-3907 (1995).
39. Morris, D.L., Watson, S.A., Durrant, L.G.& Harrison, J.D. Hormonal control of
gastric and colorectal cancer in man. Gut 30:425429 (1989).
40. Karp, J.E.& Broder, S. Molecular foundations of cancer: new targets for
intervention. Nature Medicine 1:309-320 (1995).
41. Sutherland, R.M. Cell and environment interactions in tumor microregions: the
multicell spheroid model. Science 240:177-184 (1988).
42. Olive, P.L.& Durand, R.E. Drug and radiation resistance in spheroids: cell
contact and kinetics. CancerMet~st?~si~ Rev. 13:121-138 (1994).
43. Fang, F., Orend, G., Watanabe, N., Hunter, T.& Ruoslaht, E. Dependence of
cyclin E-Cdk2 kinase activity on cell anchorage. Science 271:499-502 (1996).
44. Biedler, J.L.& Spengler, B.A. Reverse transformation of multidrug-resistant
cells. Cancer Metastasis Rev. 13:191 -207 (1994).
45. Kerbel, R.S., Kobayashi, H.& Graham, C.H. Intrinsic or acquired drug
resistance and melastasis: are they linked phenotypes? J.Cell Biochem. 56:37-47
(1994).
46. Sano, Y., Hoshino, T., Bjerkvig, R.& Deen, D.F. The relative resistance of
non-cycling cells in 9L multicellular spheroids to spirohydantoin mustard. Eur.J.Cancer
Clin.Oncol. 19:1451-1456 (1983).
21 ~6727-
47. Deen, D.F., Hoshino, T., Williams, M.E., Nomura, K.& Bartle, P.M. Response of
9L tumor cells in vitro to spirohydantoin mustard. Cancer Res. 39:44364340 (1979).
48. Hinz, G.& Dertinger, H. Increased radioresistance of cells in cultured multicell
spheroids. Radiat.Environ.Biophys. 21:255-264 (1983).
49. Remvikos, Y. et al. Cell cycle modifications of breast cancers during
neoadjuvant chemotherapy: a flow cytometry study on fine needle aspirates.
Eur.J. Cancer 29A: 1843-1848 (1993).
50. Briffod, M. et a/. Evaluation of breast carcinoma chemosensitivity by flow
cytometric DNA analysis and computer assisted image analysis. Cytometry
13:250-258 (1992).
51. Spyratos, F. et al. Sequential cytopunctures during preoperative chemotherapy
for primary breast carcinoma. Cancer69:470475 (1992).
52. Wennerberg, J. et al. Cell cycle perturbations in hel~r~ nsplanted
squamous-cell carcinoma of the head and neck after mitomycin C and cisplatin
treatment. Int.J.Cancer33:213-222 (1984).
53. Durand, R.E.& Sutherland, R.M. Effects of intercellular contact on repair ofradiation damage. Exp.CellRes. 71:75-80 (1972).
54. Dimanche-Boitrel, M.-T., Garrido, C.& Chauffert, B. Kinetic resistance to
anticancer agents. Cytotech. 12:347-356 (1993).
55. Dimanche-Boitrel, M. et al. Confluence-dependent resistance in human colon
cancer cells: role of reduced drug accumulation and low intrinsic chemosensitivity of
resting cells. Int.J.Cancer50:677-682 (1992).
- 34 -
21 q67~7
56. Graham, C.H. et al. Rapid acquisition of multicellular drug resistance after a
single transient exposure of ",a",r"ary tumor cells to alkylating agents.
J.Natl.Canc.lnst 86:975-982 (1994).
57. Barcellos-Hoff, M.H., Marton, L.J.& Deen, D.F. Differential drug sensitivityconferred by growth status detected in a mixed population of cycling and noncycling
cells. CancerRes. 50:3551-3555 (1990).
58. Mendonca, M.S., Rodriguez, A.& Alpen, E.L. Differential repair of potentially
lethal damage in exponentially growing and quiescent 9L cells. Rad.Res. 122:3843
(1 99o)
59. Rodriguez, A., Alpen, E.L., Mendonca, M.& DeGuzman, R.J. Recovery from
potentially lethal damage and recruitment time of noncycling clonogenic cells in 9L
confluent monolayers and spheroids. Rad.Res. 114:515-527 (1988).
60. Kwok, T.T.& Twentyman, P.R. The response to cytotoxic drugs of EMT6 cells
treated either as intact or disaggregated spheroids. Br.J. Cancer 51 :211 -218 (1985).
61. Sano, Y., Takao, H., Barker, M.& Deen, D.F. Response of 9L rat tumor
multicellular spheroids to single and fractionated doses of
1,3-bis(2-chloroethyl)-1 -nitrosourea. Cancer Res. 44:571 -576 (1984).
62. Twentyman, P.R. Response to chemotherapy of EMT6 spheroids as measured
by growth delay and cell survival. Br.J.Cancer42:297-304 (1980).
63. Kwok, T.T.& Twentyman, P.R. The relationship between tumour geometry and
the respones of tumour cells to cytotoxic drugs - an in vitro study using EMT-6
multicellular spheroids. IntJ. Cancer 35:675-682 (1985).
- 35 -
21 Y6727
64. Hahn, G.M., Rockwell, S., Kallman, R.F., Gordon, L.F.& Frindel, E. Repair ofpotentially lethal damage in vivo in solid tumor cells after X-irradiation. Cancer Res.
34:351-354 (1974).
65. Little, J.B., Hahn, G.M., Frindel, E.& Tubiana, M. Repair of potentially lethal
radiation damage in vitro and in vivo. Radiology 106:689-694 (1973).
66. Hahn, G.M., Ray, G.R., Gordon, L.F.& Kallman, R.F. Response of solid tumor
cells exposed to chemotherapeutic agents in vivo: cell survival after 2- and 24-hour
exposure. J.Natl.Canc.lnst. 50:529-533 (1973).
67. Little, J. B. Repair of sub-lethal and potentially lethal radiation damage in
plateau phase cultures of human cells. Nature 224:804-806 (1969).
68. Zhang, W. et al. High levels of constitutive WAF1tCip1 protein are associated
with chemoresistance in acute myelogenous leukemia. Clin.CancerRes. 1:1051-1057
(1995).
69. Sheaff R.J. and J.M. Roberts. Lessons in p16 from phylum falconium Current
Biol. 5(1):28-31 (1995).
70. Deng, C., Zhang, P., Harper, J.W., Elledge, S.J.& Leder,- P. Mice lacking
p21C~P~NVAF~ undergo normal development, but are defective in G1 checkpoint control.
Cell 82:675-684 (1995).
71. Nakayama, K. et al. Mice lacking p27kiP~ display increased body size, multiple
organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell 85:707-732 (1996).
72. Fero, M.L. et al. A syndrome of multiorgan hyperplasia with features of
gigantism, tumorigenesis, and female sterility in p27kiP~-deficient mice. Cell 85:733-744
(1996).
- 36 -
21 96727
73. Wagner, R.W. The state of the art in antisense research. Nature Medicine
1:1116-1118 (1995).
74. Tonkinson, J.L.& Stein, C.A. Antisense oligodeoxynucleotides as clinical
therapeutic agents. Cancerlnvest. 14(1):54-65 (1996).
75. Hietanen, P. et al. Do DNA ploidy and S-phase fraction in primary tumour
predict the response to chemotherapy in metaslalic breast cancer. Br.J.Cancer
71:1029-1032 (1995).
76. Remvikos, Y. et al. Prognostic value of the S-phase fraction of breast cancers
treated by primary radiotherapy or neoadjuvant chemotherapy. Ann.N.Y.Acad.Sci.
193-203 (1993).
77. O'Reilly, S.M., Camplejohn, R.S., Rubens, R.D.& Richards, M.A. DNA flow
cytometry and response to preoperative chemotherapy for prima~ breast cancer.
Eur.J.Cancer213:681-683 (1992).
78. Remvikos, Y. et al. Correlation of pretreatment proliferative activity of breast
cancerwith the response to cytotoxic chemotherapy. J.Natl.Cancerlnst. 81:1383-1387
(1989).
79. Sulkes, A., Livingston, R.B.& Murphy, W.K. Tritiated thymidine labelling index
and response in human breast cancer. J.Natl.Canc.lnst. 62:513-515 (1979).
80. Coats, S., Flanagan, W.M., Nourse, J.& Roberts, J.M. Requirement of p27
for restriction point control of the fibroblast cell cycle. Science 272:877-880 (1996).
81. Kerbel, R.S., St. Croix, B., Rak, J., and Graham, C. (1995) Is there a role for
"anti-adhesives" as chemosensitizers in the treatment of solid tumors by
chemotherapy? Bulletin de L'lnstitut Pasteur, 92: 248-256.
- 37 -
2 ~ ~ 6 7 2 7
82. St. Croix, B., Florenes, V.A., Rak, J.W., Flanagan, M., Bhattacharya, N.,
Slingerland, J.M., and Kerbel, R.S. Impact of the cyclin dependent kinase inhibitor
p27kiP1 on adhesion-dependent resistance of tumor cells to anticancer agents. Nature
Medicine, 2~ 1204-1210 (1996).
- 38 -