Note: Descriptions are shown in the official language in which they were submitted.
WO 96/3920Q''' '' PCT/US96/09202
2196778
1
SUSTAINED F~ELEAS1E BIOCIDAL COMPOSITIONS AND THEIR USES
Background of the Invention
The present invention relates generally to methods
of using a biocidal composition that releases chlorine
dioxide upon being activated.
Chlorine dio~s:ide (C102) is a superior oxidizing
agent widely used as a bleach, disinfectant, fumigant or
deodorizer. It ca.n penetrate the cell wall or membrane
and cytoplasm of mold spores, bacteria and other
microbiological contaminants at concentrations below one
part per million a.nd destroy them.
The incorporation of chlorine dioxide or sodium
chlorite in food packaging has prompted studies to
determine whether residual levels of such preservatives
result in a significant genetic or carcinogenic hazard
to humans. :Meier et al. studied the effect of
subchronic a:nd acute oral administration of chlorine,
chlorine dioxide, sodium chlorite and sodium chlorate on
the induction of chromosomal aberrations and spermhead
abnormalities in mice [Environ. Mutagenesis, 7, 201
(1985)). Only the :highly reactive hypochlorite resulted
in a weak positive effect for mutagenic potential. The
other compounds, including chlorine dioxide and sodium
chlorite, failed to induce any chromosomal aberrations
or increased numbers of micronuclei in the bone marrow
of mice. Vilagines et al. attribute the relatively
innocuous effect of chlorine dioxide to its inability to
produce halomethanes, unlike hypochlorite and chlorine
[Proc. AWWA :Disinfect. Semin., 24 pp. (1977); Chem. Abs.
93, 173513f). Recently, Richardson et al. reported that
an extensive study of the reaction of chlorine dioxide
with water b~~rne organics by the Environmental
Protection A~~ency confirmed this observation [Environ.
Sci. Technol., 28, '.~92 (1994)].
WO 96/39200 PCT/US96/09202
~ ~ 9~ 77~
2
Japanese Kokai Nos. 63/296,758, 63/274,434, and
57/168,977 describe deodorants containing chlorine
dioxide incorporated in a polymer, ceramic beads, or
calcium silicate wrapped in nonwoven cloth,
respectively. Gels that generate chlorine dioxide for
use as topical applications for disinfection are
disclosed by Kenyon et al., Am. J. Vet. Res., 45(5),
1101 (1986). Chlorine dioxide generating gels are
generally formed by mixing a gel containing suspended
sodium chlorite with a gel containing lactic acid
immediately prior to use to avoid premature chlorine
dioxide release. Chlorine dioxide releasing gels have
also been used in food preservation.
Encapsulation processes have also been used in
preparing sources of chlorine dioxide. Canadian Patent
No. 959,238 describes generation of chlorine dioxide by
separately encapsulating sodium chlorite and lactic acid
in polyvinyl alcohol and mixing the capsules with water
to produce chlorine dioxide.
Tice et al., U.S. Patent No. 4,585,482 describe
gradual hydrolysis of alternating polyvinyl methyl
ether-malefic anhydride) or poly(lactic-glycolic acid) to
generate acid that can release chlorine dioxide from
sodium chlorite. A polyalcohol humectant and water are
encapsulated with the polyanhydride or polyacid in a
nylon coating. After sodium chlorite is diffused into
the capsule through the nylon wall, an impermeable
polystyrene layer is coacervated around the nylon
capsule. Solvents are required for reaction and
application of the capsules. The capsules can be coated
onto surfaces to release chlorine dioxide. Although the
capsules are said to provide biocidal action for several
days to months, chlorine dioxide release begins
immediately after the capsules are prepared. The
batchwise process used to prepare the capsules also
involves numerous chemical reactions and physical
;.
3
processes, some of which involve~environmental disposal
problems.
There is a ns~ed for an inert composition that can be
easily activated to in~_tiate chlorine dioxide release in use.
A composition that is composed of and generates only FDA
f
approved substances, or those generally recognized as safe, is
particularly needed for food packaging and other applications
where the substances coin be ingested by or in contact with
humans.
Summary of the Invention
Among the objects of the invention, therefore, may
be noted the provision of a composition that releases a
concentration of chlorine dioxide sufficient to eliminate
bacteria, fungi, molds and viruses; the provision of such a
composition that releases such chlorine dioxide concentrations
after activation for a period of up to several months; the
provision of such a composition that can be easily blended
with other ingredients prior to application; the provision of
such a composition that increases the release rate of chlorine
dioxide in proportion to increased temperature which promotes
mold and bacteria growth; the provision of such a composition
that only releases substances approved for human exposure or
ingestion; and the provision of an inexpensive composition
that does not adversely affect the appearance or mechanical
properties of a substrate to which it is applied.
64725-687
.. 2~9g778
3a
According to one aspect on the present invention
there is providedi a method of retarding bacterial, fungal, and
viral contamination and growth of molds in meat, poultry or
seafood comprising:
exposing a surface of meat, poultry or seafood to
chlorine dioxide gas for a period of time sufficient to reduce
bacterial, fungal, and viral contamination and growth of molds
on the surface of and within the meat, poultry or seafood.
According to a further aspect of. the present
invention there s provided a method of sterilizing a medical
device, instrument or ~cupply comprising:
applying a first composition to an outer surface of a
first component , the f i.rst composit ion being inert in the
absence of moisture;
applying a second composition to an inner surface of a
second component, the ~;econd composition being inert in the
absence of moisture; and
contacting the first and second compositions on the
surfaces of the first and second components to form a
composite;
exposing the composite to moisture to initiate the
release of chlorine dioxide gas from the composite into the
atmosphere surrounding the medical device, instrument or
supply to sterilize they medical device, instrument or supply.
The present invention is also directed to a method
of retarding bacterial, fungal, and viral contamination and
growth of molds on the surface of a material and/or
64725-687
.. 2196778
3b
deodorizing the Material by exposing a surface of a material
to a composite treat does not release chlorine dioxide in the
absence of moisture, and exposing the surface to moisture to
release chlorine dioxide from the composite into the
atmosphere surrounding the material
64725-687
CA 02196778 2002-06-19
64725-687
4
to retard bacterial, fungal, and viral contamination and
growth of molds on the surface of the material and/or'
deodorize the material.. The material is pet foods, d.ry
foods, cereals, grains, laundry detergent, bar soap,
medical supplies, paper documents, paint, shoes,
disposable footwear, or disposable or nondisposable
personal care products.
Another embodiment of the preaent invention is
directed to method of inhibiting fungal infection and
growth on a surface of a human foot o:r fingernail by
contacting a surface of a human foot, a human fingernail
or an artificial fingernail with a composite that does
not release chlorine dioxide in the absence of moisture,
and exposing the surface to moisture to release chlorine
1~~ dioxide from the composite into the atmosphere
surrounding the surface to inhibit fungal infection and
growth on the surface.
Another embodiment of the invention is directed to
a method of retarding bacterial, fungal, and viral
2G contamination and growth of molds on a surface of meat,
poultry or seafood by exposing a surface of meat,
poultry or seafood to a composite which does not release
chlorine dioxide in the absence of moisture, and
exposing the composite to moisture to release chlorine
2~~ dioxide from the composite into the atmosphere
surrounding the surface of the meat., poultry or seafood
to reduce bacterial, fungal, and vi.ra.l contamination and
growth of molds on the surface and within the meat,
poultry or seafood.
3f In another embodiment the invention provides a method
of retarding bacterial, fungal, and viral contamination and
growth of molds in rrveat, poultry o:r seafood comprising:
exposing a surface of meat, poultry or seafood to a
composite releasing chlorine dioxide gas during
refrigeration for a period of time sufficient to reduce
bacterial, fungal, and viral contamination and growth of
CA 02196778 2002-06-19
64725-687
4a
molds on the surface of and within the mE=at, poultry or
seafood, wherein the cc:>mposite does not release chlorine
dioxide gas in the absence of moisture.
Another embodiments of the invention is directed to
a method for retarding mycotic growth on seeds by
exposing a surface of a seed to a composite that does
not release chlorine dioxide in the absence of moisture,
planting the seed in soil. after the seed has been
exposed to the composite, and exposing the composite to
moisture to release chlclrine dioxide from the composite
WO 96/39200 PCT/US96/09202
2 ? ~E;~~g
into the atmosphere surrounding the seed to retard
myotic growth on the surface of the seed.
Yet another embodiment of the invention is directed
to a method of deodorizing carpeting by contacting
5 carpeting with a composite that does not release
chlorine dioxide in the absence of moisture, and
exposing the carpE~t.ing to moisture to release chlorine
dioxide from the cc>mposite into the atmosphere
surrounding the carpeting to deodorize the carpeting.
Yet another embodiment of the invention is a method
of sterilizing a rne~dical device, instrument or supply by
applying a first composition to an outer surface of a
first component, t:he first composition being inert in
the absence of moisture, applying a second composition
to an inner surfac:e~ of a second component, the second
composition being inert in the absence of moisture, and
contacting the first and second compositions on the
surfaces of the first and second components to form a
composite. When t:h.e composite is exposed to moisture,
the release of chlorine dioxide is initiated from the
composite into the atmosphere surrounding the medical
device, instrument: or supply to sterilize the medical
device, instrument: or supply.
Other object's and advantages of the invention will
be apparent from t:he following detailed description.
Brief Descri tion of the Drawings
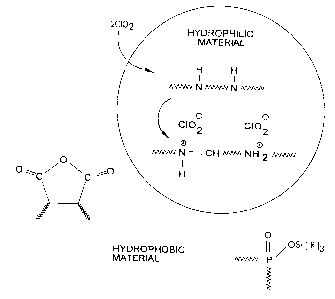
FIG. 1 is a ~>chematic that illustrates conversion
of an amine precursor to an iminium chlorite;
FIG. 2 illustrates hydrolysis of an acid anhydride
in a hydrophobic phase and migration of hydronium ion to
the iminium chlorite to release chlorine dioxide gas;
FIGS. 3a, 3b and 3c are schematics of multilayered
composites for prc>viding sustained release of chlorine
dioxide;
~WO 96/39200 ~ PCT/US96/09202
~'~ 9 67 7
6
FIG. 4 is a plot of chlorine dioxide release rates
for several powder compositions;
FIG. 5 is a plot of chlorine dioxide release rates
for a layered composite;
FIG. 6 is a plot of chlorine dioxide release rates
in relation to atmospheric temperature and humidity;
FIG. 7 is a plot of chlorine dioxide release rates
for a layered composite;
FIGS. 8 and 9 are plots of chlorine dioxide release
rates in relation to atmospheric temperature and
humidity; FIG. 10 is a plot of maximum chlorine
dioxide concentration as a function of leakage from a
container; and
FIG. 11 is a plot of chlorine dioxide concentration
as a function of time.
Detailed Description of the Preferred Embodiments
In accordance with the present invention, it has
been discovered that sustained release of chlorine
dioxide can be generated from a composite containing
chlorite anions when the composite is exposed to
moisture. The composite comprises a hydrophilic
material and a hydrophobic material. The composite may
be, for example, a dispersion composed of hydrophilic
and hydrophobic phases, or a mechanical combination of
the hydrophilic and hydrophobic materials, such as
powders and adjacent films. The powder has a
hydrophobic core embedded with chlorite containing
particles. Adjacent films comprise separate layers of
the hydrophilic or hydrophobic materials.
Preferably, the composite comprises between about
5.0 wt.~ and about 95 wt.o hydrophilic material and
between about 5.0 wt.~ and about 95 wt.~ hydrophobic
material, more preferably between about 15 wt.~ and
about 95 wt.~ hydrophilic material and between about 15
wt.$ and about 95 wt.~ hydrophobic material. If the
_ _._... ,
WO 96/39200
2' y 6 > > 8 PCT/US96/09202
7
composite is a dispersion, either material can form the
continuous phase. The continuous phase constitutes
between about 15 wt.% and about 95 wt.% of the
dispersion and the= dispersed phase constitutes between
about 5 wt.~; and <about 85 wt.% of the dispersion, and
preferably, the continuous phase constitutes between
about 50 wt.% and about 95 wt.% of the dispersion and
the dispersed phase constitutes between about 5 wt.% and
about 50 wt.% of t:he dispersion.
The hydrophobic material of the composite can be
composed entirely o~f an acid releasing agent or can
comprise the acid releasing agent in combination with a
diluent and/or a plasticizer. Any acid releasing agent
that is capable of: being hydrolyzed by ambient moisture
is acceptable for purposes of the present invention.
Preferably, the acid releasing agent does not react with
the hydrophilic material, and does not exude or extract
into the environment. The hydrophobic material
comprises between about 10 wt.% and about 100 wt.% of
the acid releasincr agent, up to about 80 wt.% diluent,
and up to about 60 wt.% plasticizer, and preferably,
between about 40 wt.% and about 100 wt.% of the acid
releasing agent, between about 20 wt.% and about 80 wt.%
diluent, and up to about 20 wt.% plasticizer.
The hydrophilic material of the composite can be
composed entirely of a source of chlorite anions or can
comprise the chlorite anion source in combination with
another hydr~~philic material. The hydrophilic material
preferably c~~ntains an amine, amide or an alcohol, or a
compound containing amino, amido or hydroxyl moieties
and having a high hydrogen bonding density. A source of
chlorite anions is :incorporated in the hydrophilic
material and preferably constitutes between about 2 wt.%
and about 40 wt.% o:E the hydrophilic material in the
form of chlorite anions and counterions, and more
preferably, between about 8 wt.% and about 10 wt.% of
WO 96/39200 PCT/US96J09202
X1987 78
8
the hydrophilic material. When the chlorite source is a
chlorite salt, the salt dissociates in the hydrophilic
material such that the hydrophilic material in the
composite will include chlorite anions and counterions.
However if the hydrophilic material is an amine and the
chlorite source is chlorine dioxide gas, the chlorine
dioxide reacts with the amine to form iminium chlorite
in situ, if the oxidation potential of the amine is
sufficiently low for the amine to be oxidized.
It has been found that the acid releasing agent
within the hydrophobic material is hydrolyzed by
adsorbed moisture, releasing acid and hydronium ions
that diffuse from the hydrophobic material to the
hydrophilic material containing chlorite anions. The
hydronium ions react with the chlorite anions in the
hydrophilic material, releasing chlorine dioxide gas
that diffuses out of the composite into the surrounding
atmosphere for a period of up to about six months in
order to prevent the growth of bacteria, molds, fungi
and viruses on a treated surface.
The hydrophobic and hydrophilic materials are
substantially free of water to avoid significant release
of chlorine dioxide prior to use of the composite. For
purposes of the present invention, a hydrophilic
material, a hydrophobic material, or a dispersion
thereof is substantially free of water if the amount of
water in the composite does not provide a pathway for
transmission of hydronium ions from the hydrophobic
material to the hydrophilic material. Generally, each
of the hydrophilic and hydrophobic materials can include
up to about 0.1 wt.~ water without providing such a
pathway for interdiffusion between the materials.
Preferably, each material contains less than about 1.0 X
10-3 wt.~ water, and, more preferably, between about 1 X
10'' wt.~ and about 1 X 10-' wt.~ water. Insubstantial
amounts of water can hydrolyze a portion of the acid
r
WO 96/39200
9 6 % ~ ~ PCT/US96/09202
9
releasing anent to produce acid and hydronium ions
within the composite. The hydronium ions, however, do
not diffuse into t=he hydrophilic material until enough
free water is present for transport of hydronium ions.
The chlorite anions generally do not react with the
hydrophilic material, but are surrounded by hydrogen
bonds contributed by the nitrogen or hydroxide within
the hydrophilic material. Suitable chlorite sources
that can be incorporated into the composite of the
present invention include alkali metal chlorites such as
sodium chlorite or' potassium chlorite, alkaline-earth
metal chlorites such as calcium chlorite, or chlorite
salts of a transition metal ion or a protonated primary,
secondary, tertiary or quaternary amine. Many chlorite
sources, such as sodium chlorite, are stable at
processing t~~mperat~ures in excess of about 100 °C,
allowing for processing at relatively high temperatures.
FIG. 1 :illustrates preparation of a composite
containing iminium chlorite. The amine hydrophilic
material is :in contact with a hydrophobic acid releasing
agent (both lzydroly:~ed P-O-Si and malefic anhydride are
shown in FIG. 1). Chlorine dioxide (ClOZ) is reduced by
extracting an,electron from the amine, forming an
aminium radical ca n on (not shown) and a chlorite
counterion (C:102~). The aminium cation quickly converts
to an iminiurn cati~on by loss of a proton from an
adjacent carbon atorn and oxidation by another chlorine
dioxide molecule. ~fhe mechanism for above reaction in
an aqueous s~~stem .is described by Rosenbatt et al., J.
Org. Chem., a'.8, 27'9() (1963); J. Amer. Chem. Soc. 89(5),
1158, 1163 0.967 ) .
High ch~.orine dioxide to chlorite conversions are
obtained if the chlorite anion and/or iminium cation
that is generated by the initial electron transfer from
the amine arE~ rapidly complexed and stabilized by a
hydrophilic molecule. In some formulations, uncomplexed
WO 96/39200 PCT/US96/09202
~~ ~8
chlorite anion may be depleted by subsequent reactions
with the iminium counterion at temperatures above about
60 °C. Chlorites are also subject to disproportionation
into chloride and chlorate. An amine with a high pKa is
5 preferred because it reacts more rapidly with chlorine
dioxide and acts as a more effective proton sink,
maintaining the basic pH required for chlorite ion
stability.
FIG. 2 illustrates the mechanism for release of
10 chlorine dioxide from iminium chlorite when moisture
contacts the composite. Hydrolysis of the acid
releasing agent provides hydronium cations (H30') that
react with iminium chlorite to release chlorine dioxide
gas. The decomposition products of the reaction are an
aminium cation ( shown as ~~~~""z~ in FIG . 2 ) , a
carboxylate (COO-, not shown in FIG. 2), and C1-. These
products are retained within the composite.
It has been found that, in some instances, iminium
chlorite may decompose if the composite is exposed to
temperatures exceeding about 60 °C, reducing the
available chlorite concentration for conversion to
chlorine dioxide. In order to maximize chlorine dioxide
release from the composite, it has been discovered that
the chlorite source can be omitted from the composite
until the composite is applied to a surface when the
hydrophilic material in the composite is an amine.
After application, the composite is exposed to chlorine
dioxide gas that either reacts with the amine to form
iminium chlorite in situ or dissolves in the amine to
provide chlorite anions. The composite is then
activated in the presence of moisture to release
chlorine dioxide. This method enables the composite to
be exposed to elevated temperatures during processing,
storage and application as compared to the temperatures
at which the iminium chlorite decomposes, because the
WO 96/39200
2 ~ 9 E) 7 7 8 PCT/US96/09202
11
hydrophilic material does not contain uminium chlorite
or any chlorite anions. The method also precludes
premature release of chlorine dioxide from the
composite. Chlorine dioxide can be provided on site by
passing the composite through a chlorine dioxide
generator.
Conventional chlorine dioxide generators generate
an atmosphere of chlorine dioxide that is saturated with
water. Chlorine dioxide that comes into contact with
the composite of t:he invention must first be dissolved
into a material that does not absorb water such as a low
melting hydrocarbon wax or chlorohydrocarbon wax.
Alternatively, chlorine dioxide is dried with a
desiccant. Chlorine dioxide is thus delivered from a
wet industrial process into the composite without
exposing the composite to water.
In order for an amine to form iminium chlorite in
neat form or in the presence of a plasticizer, the amine
must be sufficiently electron rich and the amine
nitrogen must be locally mobile. Electron withdrawing
groups should be separated from the amine center by at
least two methylene groups in order for the chlorine
dioxide to e:~ctract an electron from the amine. Movement
of the bonds about the nitrogen center of the amine is
required for aminium formation. If the amine is frozen
into a glass~r matri:~c, the amine nitrogen will not be
mobile and the amine will not convert to iminium
chlorite. A glassy amine can be softened to increase
mobility by adding at least about 10 wt.~ of a
plasticizer, such a:; a low molecular weight amide, to
the amine to lower class transition temperature below
the reaction temperature. Other suitable plasticizers
are well knovm in 'the polymer art .
The rate of chlorine dioxide release from a
composite care be a:Lt:ered by changing the viscosity of
the hydrophilic and hydrophobic materials, changing the
WO 96/39200 PCT/US96/09202
12
dispersibility of the hydrophilic and hydrophobic
materials, changing the temperature of the composite,
changing the concentration of acid releasing agent in
the composite, adding a desiccant or humectant to the
composite to control release of chlorine dioxide from
the composite once it is exposed to moisture, or
changing the volume fractions of the hydrophilic and
hydrophobic materials to produce continuous or discrete
phases within a dispersion.
Maximum chlorine dioxide release from a composite
can also be achieved by stabilizing the chlorite anion.
Iminium chlorite is unstable to nucleophilic attack by
the chlorite anion. It has been discovered that the
room temperature lifetime of chlorite anion is
substantially extended when a strong base, such as a
metal alkoxide, is present in the hydrophilic material
containing the iminium chlorite. The mechanism of
alkoxide stabilization of the chlorite counterion is
shown below.
R" ONa
2O ~ R 'zN=CRZ ] + C I Oz- ~ R '2N-CRZ-OR" + NaC I Oz
wherein R'2 and Rz are groups that correspond to those of
the selected amine and R' is an alkyl or hydrogen group.
In the absence of water, the iminium ion is immediately
decomposed into an a-amino ether and a more stable
sodium chlorite salt. If water is present during the
oxidation of the tertiary amine, an unstable oc-amino
alcohol is formed that can attack the chlorite anion
unless the chlorite anion has been effectively complexed
by the hydrophilic solvent. Addition of water after
solvation of the chlorite ion is not as deleterious.
Acceptable strong bases for use in stabilizing the
chlorite include metal alkoxides such as sodium,
potassium or calcium methoxides, ethoxides, propoxides
or butoxides, metal oxides such as aluminum oxide, or
WO 96/39200 ~ PCT/US96/09202
13
sodium oxide, metal ions such as Na', trialkyl ammonium
salts of alkoxide~:, ammonium salts of alkoxides,
acetates such as ;odium acetate, substituted acetates,
or other materials that would generate a strong basic
reaction to attack: the nitrogen center of iminium
chlorite.
In a hydrophilic material containing a tertiary
amine (dimet~zylaminoacrylamide), N-methylacetamide and
urea, an a-amino ether and chlorite salt is formed when
the iminium ~~hlorite is stabilized. Any monomeric or
oligomeric amide substituted plasticizer, such as
succinamide, formam:ide, or N-methyl formamide, can be
substituted :'or N-:mE=thylacetamide in order to soften the
amine. Formamide and N-methyl formamide are toxic and
would not be preferred in applications involving human
contact. If the amine center is sufficiently mobile,
the addition of a ;plasticizer is unnecessary. Urea
improves the chlorine dioxide uptake and release
efficiency of: the hydrophilic material because it has a
high hydrogen bonding density and will not react with
the acid releasing agent. Compounds having a high amide
concentration can also be used to improve hydrophilic
material efficiency. Preferably, the composite
comprises between about 5 wt.~ and about 95 wt.~ of the
hydrophilic material. and between about 5 wt.o and about
95 wt.~ of the hydrophobic material. The hydrophilic
material com~~rises between about 5 to about 30 wt.o of
an amine and between. about 70 and about 95 wt.~ of a
hydrophilic solvent: including between about 35 and about
55 wt.~ urea, between about 35 wt.o and about 55 wt.~
plasticizer and about 10 wt.% base. It has been found
that not more than about 0.5 moles of chlorine dioxide
per mole of amine should be added to the hydrophilic
material or the stability of the material could be
compromised.
WO 96/39200 PCT/US96/09202
14
Preferred amides for use as the hydrophilic
material include formamide, acrylamide-
isopropylaczylamide, copolymers of formamide and
acrylamide-isopropylacrylamide, and copolymers of
acrylamide, isopropylacrylamide or N,N-methylene
bisacrylamide and a primary amine or a secondary amine.
Such amides can be useful vehicles for film casting
prior to exposure to chlorine dioxide, which does not
react with polymerizable, electron deficient alkenes
such as acrylamide.
Suitable amines for use as the hydrophilic material
include primary amines, secondary amines, and tertiary
amines having pendant hydrogen bonding groups. An amine
substituted with electron donating groups that donate
electrons to convert chlorine dioxide to chlorite is
preferred. Electron withdrawing groups concentrate
electron density at such groups such that it is
difficult for the chlorine dioxide to extract an
electron from the amine. Tertiary amines having non-
hydrogen bonding pendant groups that are dissolved in a
hydrophilic solvent are also acceptable. Representative
amines include: alkanolamines; copolymers of
aminoalkanes and alkene bisacrylamides;
alkylaminopyridine; alkene diamines; alkylamino
cycloalkanes; alkylamino-carboxyamido alkanes dissolved
in a diluent ; amines having the formula R3_xNHx
R1RZNCHzCH2C (O)NHZ; solubilized N(CH2CHZOH) 3_XHX~
R3N I NCHzCH2C ( O ) NHz ) z , ( CH3 ) 2N ( CHZ ) ZN ( CH3 ) 2 ,
RSR6N ( CHZ ) ZNHC ( O ) NH2 ,
0 ,
CR4-CHZ-CHZ-IC-NH~CHz
3 0 N ( CHzCH2NHC ( O ) NHz ) 3 , 2 , or
/ Ii Ii ~
~NH-[CHZ~~NH-CHZCHZC-NHCH2NHCCH2CH2~ . NN NH .
\\ ~ n
WO 96/39200 PCT/US96/09202
2196778
H2N-[ CHZ] 3-N~N-[ CHZ] 3 NH2
0
- ~ ~
CHZCH -C-NH-[ CHZ]ZN-[ CH3] 2J-'T
Or
HzN
N N
0 ~ ~NH
~0
HzN wherein: R substituents are,
independently, - (CHzCH20)YH,
5 -C ( CH3 ) z ( CHz ) ZOH , - ( CHz ) ZNH ( CHzCH20 ) ZH , -CH ( CH3 ) z ,
-[CHz]2 N~~ --[CH2]2-N~NH
alkyl, cycloalkyl,
benzyl, acrylamide, or pyridyl; R" Rz, R5, and R6 are
alkyl; R3 is straight chain C6 to Clz alkyl; R4 is
cycloalkyl or ben~:yl; m is 1-100; n is 2 or 3; x is 0, 1
10 or 2; y is 1 or 2; and z is 1-6. Generally, the above
compounds can be ~;olubilized in formamide,
isopropylacrylamicle-acrylamide or other conventional
plasticizers.
Preferred amines include monoethanolamine,
15 diethanolamine, triethanolamine, a copolymer of 1,3-
diaminopropane or 1,2-diaminoethane and N,N-methylene
bisaczylamide, 4-d'.i:methylaminopyridine, tetramethylene
ethylene dia:mine, N,N-dimethylamino cyclohexane,
solubilized 1-(N-d.ipropylamino)-2-carboxyamido ethane or
1-(N-dimethylamino)-2-carboxyamido ethane, a primary
amine having the formula R,NHz, a secondary amine having
the formula :R,R,NH, N(CH,CH~OH)~,
WO 96/39200 PCT/US96/09202
16
N
0 ~ ~NH
~0
HN NH
H2N
HzN-[ CHZ] 3-N N-[ CHZ] 3 NHZ
i
so lubi 1 i z ed NRSR6R, , ( CH3 ) 2NCHZCHzN ( CH3 ) Z , RBR9NCHZCHzC ( O )
NHz ,
RloN ( NCHZCHZC ( O ) NHz ) z , RmRizN ( CHZ ) 3NHC ( O ) NHz ,
N ( CHZCHzNHC ( O ) NHz ) s ,
0
~R,~-CHZ-CH2-IC-NH-f-CHz
2
O 0
~NH-[ CH2] ~NH-CHzCH2IC-NHCH2NHCICH2CHZ~
Or
0
~CH2CH~IC-NH-CH2CHzCHzN-[ CH3] z~
/m
wherein: R1 is -CHzCH20CH2CH20H, -C (CH3) zCHzOH,
-CH CH NHCH CH OH -CH CH -CH CH OH [ cHZ] z N~~ or
z z 2 2 ~ ( 3)2~ 2 z
-[CHZ]z-N NH
U ; RZ and R3 are, independently, hexyl,
benzyl, n-propyl, isopropyl, cyclohexyl, acrylamide, or
-CHzCH20H; R4 is cyclohexyl or benzyl; RS and R6 are
methyl; R, is cyclohexyl or Q-pyridyl; Re and R9 are,
independently, methyl, n-propyl or isopropyl; R,o is n-
C6H,3 or n-ClzHzS % R" and R12 are, independent 1y, methyl ,
H2N
~N
WO 96/39200 219 6 7 7 ~ PCT/US96/09202
17
ethyl, n-propyl or isopropyl; m is an integer from 1 to
100; and n is 2 or 3. Suitable diluents include
formamide or acry7_a.mide-isopropyl acrylamide.
Oligomeric or pol~nr~eric secondary amines converted to
acrylamide substituted tertiary amines by Michael
reaction with acrylamides are also suitable because the
amide group does not react with the acid releasing
agent.
Hydroxylic compounds, including ethylene glycol,
glycerin, methanol., ethanol, methoxyethanol,
ethoxyethanol or other alcohols, can be used as the
hydrophilic material. However, chlorine dioxide release
can occur very rapidly when a hydroxylic compound is
incorporated in the composite and can limit the
applications for such composites to rapid chlorine
dioxide releasing systems.
Suitable acid. releasing agents include carboxylic
acids, esters, anhydrides, acyl halides, phosphoric
acid, phosphate esters, trimethylsilyl phosphate esters,
dialkyl phos~~hates, sulfonic acid, a sulfonic acid
esters, sulf~~nic acid chlorides, and phosphosilanes of
glycerol based esters. Examples of such acid releasing
agents inclu~3e an anhydride or phosphate ester blended
with or grafted to polypropylene, polyethylene or
polystyrene, or trimethylsilyl phosphate esters of the
formulae
-''-
o-:~,._ ~ 0
,, ~ ,,
/P~\0 ~ /P~\0
-S(-0 - SI-0
or
WO 96/39200 ~ PCT/US96/09202 I
18
(CH3) 3SiOP (O) (OR) z wherein R is a non-hydrogen bonding
group, alkyl or aryl.
Linear or star like oligomers (e. g., a micelle like
molecule with a lipid wall and a P-O-Si core), such as a
phosphosilane of a glycerol based ester, are preferred
acid releasing agents because they can be melt- or
solvent- processed with the option of being crosslinked
after processing to provide film stability. A preferred
phosphosilane of a glycerol based ester is known as
LPOSI and has the formula
0
II
GO-P-OG
I
0 0
I II
0-St-0-P-OG
I I I
0-P-0 0 0
I I II
OG 0-S i-0-P-OG
I I I
[G0'2 I~ OC2H5 OG
0
0
0
OH
wherein G has the formula off .
A free radical polymerizable alkene or condensable group
on the terminal end of a lipid is a representative
oligomer.
Acid anhydrides are also preferred acid releasing
agents and include organic acid anhydrides, mixed
organic acid anhydrides, homopolymers of an organic acid
anhydride or a mixed inorganic acid anhydride, and
copolymers of an organic acid anhydride or a mixed
inorganic acid anhydride with a monomer containing a
double bond. Preferred mixed inorganic acid anhydrides
contain a phosphorus-oxygen-silicon bond. Preferred
anhydrides include copolymers of malefic anhydride,
WO 96/39200 PCT/US96/09202
2 ~ 96778
19
methacrylic anhydride, acetic anhydride, propionic
anhydride, or succ:inic anhydride, and vinyl, styrene or
an alkene, such a=> malefic anhydride-styrene copolymers,
or grafts thereof with olefins such as polypropylenes,
polyethylenes, or polystyrenes. Copolymers of acid
anhydrides and esters of lactic or glycolic acids can
provide a rapid initial chlorine dioxide release rate
followed by a slow release rate.
The hydrophobic material can further include a
diluent such as atactic polypropylene, hydrocarbon wax,
chlorinated wax, polyethylene wax, low molecular weight
polyolefins, polyesters, derivatized polyolefin
copolymers, or mixt,ares thereof. Diluents can be
included in t=he hyd:rophilic material as well.
Plasticizers can also be incorporated in either the
hydrophobic or hydrophilic materials as is known in the
art. Genera:Lly, formamide and isopropylacrylamide-
acrylamide are acceptable plasticizers.
A moisture scavenger, such as sodium sulfate,
calcium sulfate, silica gel, alumina, zeolites, and
calcium chloride can be added to the composite to
prevent prem~~ture :hydrolysis of the acid releasing
agent. Conventional. film forming additives can be added
to the hydrophobic and hydrophilic materials as needed.
Such additive's include crosslinking agents, flame
retardants, emulsi:Eiers and compatibilizers.
The composites of the present invention can be
formulated in various ways to accommodate a wide range
of end use aF~plicati.ons . The composite can be
formulated a~~ an eat.rudate, such as a film or pellets,
or as a powder using conventional extrusion and spray
drying methoc.s, respectively. When the composite is
formulated as a powder, chlorite containing particles
are formed by dissolving a chlorite source in a
hydrophilic solvent: and extruding the solution through
nozzles of a spray dryer. Once the solution is
WO 96/39200 PCTNS96/09202
transformed into spray dried particles, the particles
can be routed to a cyclone separator to isolate small
particles preferably having a diameter of between about
5 and about 150 microns. The particles can then be
5 stored in a dry atmosphere. Once the chlorite particles
are made, they are fed into a fluidized bed. The
hydrophobic material containing the acid releasing agent
is aerosolized by passing the material through small
diameter nozzles into the chamber of the fluidized bed
10 where it can impinge upon the fluidized chlorite
containing particles. Upon contact with the fluidized
particles, the chlorine dioxide releasing powder is
formed as the hydrophobic material solidifies to form a
hydrophobic core having a layer of chlorite particles
15 embedded in the outer surface thereof. Aggregation is
minimized because the chlorite particles are hard
inorganic materials. The particles can then be packaged
in a dry sealed container.
In forming the chlorine dioxide releasing powder,
20 anhydrous particles, such as anhydrous sodium sulfate,
calcium sulfate, magnesium sulfate, or a moisture
depleted silica gel, can be included in the fluidized
bed to form a mixture of chlorite particles and
anhydrous particles. The anhydrous particles delay
release of chlorine dioxide that is catalyzed by
atmospheric moisture. The anhydrous particles can also
be post-mixed with the chlorine dioxide releasing powder
to delay chlorine dioxide release.
Although the hydrophilic and hydrophobic materials
can be formulated as described above for the composite,
it is preferred that the chlorite powder contains an
alkali or alkaline-earth chlorite. The hydrophobic
material preferably contains a low melting hydrocarbon
wax, chlorohydrocarbon wax, atactic polypropylene,
polyethylene wax, a low molecular weight polyolefin,
derivatized polyolefin copolymer, or mixtures thereof.
~~~~ll
WO 96/39200 PCT/US96/09202
21
An acid releasing wax, such as a hydrocarbon solution of
a phosphorylated l.ipoglycerol reacted with silicon
alkoxides to produce mixed anhydride P-O-Si bonds, is
preferred as the hydrophobic material. LPOSI is a
particularly suitable acid releasing wax for use in
preparing the chlorine dioxide releasing powder.
If the acid releasing wax is extruded at a
viscosity between about 10 and about 1000 cP through
nozzles of between about 1 and about 10 mil diameter, a
fine spray of molten wax between about 5 and about 400
microns in diameter is generated.
In addition to formation of powdered composites,
the composites of t:he present invention can be
formulated in solvents to allow for film casting or
other application m~sthods. The composite can be applied
as a film by using well known hot melt, dip coat, spray
coat, curtain coat, dry wax, wet wax, and lamination
processes.
The composites can also be provided as a layer 12
composed of <~ microdispersed hydrophobic and hydrophilic
material as 3hoWn 1I1 FIG. 3b, or as a multilayered
composite 14 including a separate hydrophobic layer 16
and a separat=e hydrophilic layer 18 as shown in FIG. 3a.
The hydrophobic and hydrophilic layers can be applied by
casting the hydrophilic layer onto a substrate 20 and
then casting the hydrophobic layer onto the hydrophilic
layer, as il_LustratE.d in FIG. 3a. The multilayered
composite or single layer can be applied in conjunction
with moisture regulating layers 22 to control the rate
of moisture ingress into the hydrophilic material or
hydrophobic rlaterial to control chlorine dioxide release
from the mult:ilaye:rE:d composite when activated by
moisture.
In order to generate chlorine dioxide in a
controlled fashion it is useful to limit the access of
water to the hydrophobic layer containing the acid
WO 96/39200 ~ PCT/US96/09202
22
releasing agent and to control the surface area of
contact between the layer releasing the hydronium ion
and the hydrophilic layer containing chlorite. Such
controlled release can be obtained by casting the
hydrophobic and hydrophilic materials 16, 18 as separate
layers with an intermediate boundary layer 24 that
regulates hydronium ion transport between the materials
as shown in FIG 3c.
The layered composites of the present invention are
intended to maintain a desired rate of chlorine dioxide
release (moles/sec/cm~ of film) in the presence of
atmospheric moisture at a surface for a length of time
required for chlorine dioxide to absorb onto the surface
and kill bacteria or other microbiological contaminants.
However, leakage from a container or exposed surface
reduces the chlorine dioxide concentrations at the
surface because of chlorine dioxide diffusion into the
atmosphere. The chlorine dioxide concentration released
from the film for a chosen time period can be calculated
given the leakage rate and the rate of absorbance at a
surface. Thus after measuring the leakage rate, the
composite is formulated so that it contains a large
enough reservoir of chlorite reacting at a speed
sufficient to compensate for the leakage rate for the
desired time period of sustained release.
Therefore, design of a chlorine dioxide releasing
composite suitable for controlled release and biocidal
action within a container must take into account several
aspects, namely, the chlorine dioxide production rate
from the controlled release film, the partitioning of
chlorine dioxide between the phases within the container
(e. g. gas, liquid and solid phases) in a reversible
(absorbed) or irreversible (reacted) fashion, and the
leakage rate of gas from the container. Design of such
a composite is described in Example 15.
WO 96/39200 2 ~ 9 6 l l 8 PCT~S96/09202
23
A preferred extended release system of the present
invention conserve:> the chlorite reservoir by emitting a
series of periodic pulsed releases timed to coincide
with the su~~pected times of bacterial, viral or fungal
contamination or t=he typical incubation time for the
biological of interest. The system design can be
optimized to maint:a.in the desired kill concentration for
the requisite time at the atmospheric chlorine dioxide
leakage rates imposed by the specific application.
A typical controlled release multilayered composite
includes water swellable films A and B of a thickness of
about 5 mil with a hydrophobic layer A and a hydrophilic
layer B as descrif>ed above for the composite. The
hydrophobic layer A contains an acid releasing agent
such as an anhydride and hydronium ions generated by
anhydride hydrolysis. The hydrophilic layer B contains
chlorite ani~~ns as ;provided, for example, by dissolving
sodium chlorite or another chlorite source in a
hydrophilic solvent. The hydrophobic and hydrophilic
layers are separated by a water swellable intermediate
layer C having a thickness 1 (typically about 5 mil) and
diffusion constant, D. The effective ion concentrations
applied to the boundaries of intermediate layer C by the
layers A and B are a strong function of the water
transport properties of layer C.
The intE~rmediat:e layer C can be composed of a wide
variety of materials since chlorine dioxide can diffuse
equally well in both hydrophobic and hydrogen bonded
matrices. Such materials include polyionomers such as
protonated and neutralized, sulfonated, or
phosphorylated oligo- or poly-alkenes such as
polyethylene, polypropylene, alkyl acrylates and
copolymers thereof. Lipid substituted polyhydroxy
alcohol phosphates and phosphosilicates and their
mixtures with alkene~ polymers and oligomers are also
preferred. Finely divided anhydrous salts or desiccants
WO 96/39200 ~ ~ PCT/US96/09202
24
may be added to any of the layers to retard the reaction
to chlorine dioxide that is catalyzed by water.
It has been discovered that construction of a
multilayered composite wherein the arrangement of the
layers in the composite is defined by the formula
C(ACB)~C (wherein n represents the desired number of
pulses) provides periodic pulsed release of high
concentrations of chlorine dioxide over several weeks or.
months. Such pulsed release can be coordinated to the
growth, incubation and contamination of viruses, molds,
fungi and bacteria. The cycle time and peak
concentrations of chlorine dioxide would be controlled
by the layer thickness, chlorite and anhydride loading,
and the water and ionic permeation characteristics of
layers A, B and C. Pulsed release occurs as each layer
(ACB)I is successively penetrated by water vapor and
hydronium ions. Structures of type CDC can also be made
where D is a mixture or emulsion of A and B of a phase
size of between about 0.2 and about 100 microns. The
materials of construction for the CDC composites can be
the same as those used in making the C(ACB)"C composites.
Additionally, a multilayered composite C(DCD)"C can be
made in order to provide pulsed release as described
above.
Pulsed releases of chlorine dioxide that vary from
about one day to over about 200 days can be achieved for
5 mil thick A, B and C films by separating the
hydrophobic layer A containing the acid releasing agent
from the hydrophilic layer B containing chlorite anions
by an intermediate layer C capable of supporting varying
hydronium ion transport rates.
The pulsed release capabilities of a multiple
layered film can be calculated as provided in Example
16.
Applications for the composites are numerous. The
composites can be used in most any environment where
WO 96/39200 ~ ~ ~ b % 7 8 PCT/US96/09202
exposure to moisture will occur. The composites can be
used to prevent the growth of molds, fungi, viruses and
bacteria on the surface of a material and/or deodorize
the material by trE~ating the surface with a composite
5 that does not release chlorine dioxide in the absence of
moisture, anal exposing the treated surface to moisture
to release chlorine dioxide from the composite into the
atmosphere surrounding the material. The treated
surface is generally a portion of a container or is part
10 of a substrate placed within the container.
The biocidal atmosphere generated within the
container ca.n be used in storing food products including
blueberries, raspberries, strawberries, and other
produce, ground beef patties, chicken filets, and other
15 meats, enhanced foods, pet foods, dry foods, cereals,
grains, or most any food subject to bacterial
contamination or mold growth. Bar soap, laundry
detergent, stored paper documents, clothing, paint, and
seeds can be protected from mold growth. Medical
20 instruments, devices and supplies as well as disposable
or nondisposable personal care products can be
sterilized to prevent microbial contamination. Medical
or biological wast:e~ can also be sterilized to kill
microbials within the waste. Odors from athletic shoes,
25 disposable footwear, and refuse can also be minimized
when they are contained within a treated container.
Conventional containers can be used such as
paperboard or cont:ainerboard boxes, corrugated,
nonwoven, plastic, or polymeric multilaminate
containers, cellulosic, plastic or paper bags, seed
packets, or waste containers.
The treated surface can be a reusable or disposable
mat or sheet including a dental tray covering, a
surgical tray covering, a shower mat, nonwoven bandage
material, a meat cutting board, a liner for drawers or
shelves, an insert. for athletic bags or gym lockers, a
WO 96/39200 PCT/US96109202
26
food wrapper, a paper sheet for separating hamburger
patties, a meat packaging tray, an overpouch such as
those used in packaging intravenous bags, a fresh fruit
separator or box liner, an absorbent pad for poultry,
meat, seafood or produce, or an absorbent layer for use
in diapers. Such mats or sheets are typically made from
paper, cellulosic, polymeric, woven fabric or nonwoven
materials.
Such a method can also be used to coat the surface
of a seed to protect the seed from molds and fungi
during storage and to protect against mycotic growth
when the seed is planted. The coating, when activated
by moisture, creates a microatmosphere of chlorine
dioxide in the soil in the vicinity of the seed and
inhibits mycotic growth that normally would impede seed
germination. This coating has no effect upon the
germination of the seeds. Seeds in storage do not have
to be physically coated to be protected but rather can
be in a closed container containing the active material
as a packet, "tea bag" or coating on the container.
Paper impregnated with the composite generates
sufficient chlorine dioxide to protect the seeds.
Although any seeds can be protected by the coating,
edible seeds such as corn kernels, sunflower seeds, or
soybeans, remain fit for human consumption once they are
coated. Thus, the coated seeds can be provided for
planting or for human consumption after they have been
coated.
The surface can be treated with any of the
composites of the present invention by conventional
coating, extrusion, lamination and impregnation methods
well known in the art.
Another embodiment of the invention is a method of
preventing the growth of fungi, bacteria or molds on a
surface and/or deodorizing the surface by treating the
surface with a composite that does not release chlorine
WO 96/39200 PCT/US96/09202
216778
27
dioxide in the ab~~ence of moisture, and exposing the
treated surface tc> moisture to release chlorine dioxide
from the composite into the atmosphere surrounding the
surface.
A preferred application includes a foot powder for
preventing athlete's foot and other fungi. The powder
can be applied directly on the surface of the foot or
can be incorporated into a shoe insert. The composite
can be applied between the cloth covering and foam pad
of the shoe insert, impregnated within the foamed pad,
or impregnated or coated on a shoe counter or upper
lining. Chl~~rine dioxide generated from moisture within
the shoe diffuses from the composite into the atmosphere
to kill fungus and deodorize the shoe. The powder can
be blended with conventional ingredients such as talc,
cornstarch, Fragrance, miconazole nitrate, tolnastate
silica, boric acid, aluminum chlorhydrate, salicylic
acid, and ce:Llulose. The powder can also be blended
with other ingredients and used in bath powders or
powders used in treating jock itch.
The powder can also be applied to carpeting to
remove odors from the carpet. Ingredients commonly
incorporated in powdered carpet deodorizers or cleaners
can be blendE~d with the powder of the present invention.
The compositE: can also be formulated in microcapsules
that break aj'_ter being stepped on and are then activated
by moisture. Such microcapsules can be impregnated in
floor, shower or bat=h mats or can be used in carpet
deodorization .
Another use for the composites is in providing self
sterilizing packaging, which is particularly useful in
the medical :industry. The composite can be coated onto
tubing, connectors, fitments or other components as
separate layers of t:he hydrophobic or hydrophilic
material on separate components that are activated upon
being pressure fitted together. Tubing fitments used
WO 96/39200 ~ PCT/US96/09202
28
with intravenous bags, for example, can be treated such
that a surface of one tube fitment is coated with a
hydrophobic film containing acid releasing agent, a
surface of another tube fitment is coated with a
hydrophilic film containing chlorite, and the treated
surfaces of the fitments are interconnected in the
presence of moisture to initiate the release of chlorine
dioxide from the treated surfaces into the atmosphere
surrounding the material. Fitments for in-dwelling
catheters, needles, peritoneal dialysis, percutaneous
devices, percutaneous access, colostomy bags and other
medical devices can also be treated in accordance with
this method. Additionally, closures on a package can be
so treated to provide self sterilizing packaging for
medical devices, instruments and supplies.
The composite of the present invention was expected
to kill bacteria on the surface of meats. However, it
was not expected to penetrate a ground beef patty. It
has been discovered that chlorine dioxide evolved from
paper treated with the composite can effectively
penetrate the full thickness of a patty and kill
bacteria such as E. coli and Salmonella that result from
contamination during meat processing. E. coli 0157: H7
in tainted meat has caused death and severe illness and
appears to be especially resistant to cooking,
fermenting and drying. In a typical operation producing
meat patties for commercial consumption, meat is ground,
extruded and formed into patties that are separated by
sheets of coated paper that prevent adhesion of the
individual patties. After packaging, the ground meat
can be exposed to chlorine dioxide over a period of time
when in refrigerated storage to kill and inhibit the
growth of the bacteria.
The following examples are presented to describe
preferred embodiments and utilities of the present
invention and are not meant to limit the present
WO 96/39200 PCT/US96/09202
2196778
29
invention unless otherwise stated in the claims appended
hereto.
EXAMPLE 1
A hydrophilic. material was made which contained a 7
wt.~ solution of sodium chlorite in an amide mixture
composed of 33 wt..% formamide, 33 wt.o acrylamide, and
33 wt.o isopropylaczylamide. A hydrophobic material
consisting of a 40o solution of a copolymer composed of
33 moleo malefic anhydride and 66 moles styrene in
ethylbenzene plast:icizer was then made. The hydrophobic
material was vortex mixed with the hydrophilic material.
The resultant white mixture of the two disperse
materials started a sustained release of chlorine
dioxide in the ab:>ence of added water within five
minutes at room temperature. Interphase diffusion of
water within the dispersion initiated hydrolysis of the
anhydride. Hydronium ions formed during hydrolysis
reacted with chlorite anions to release chlorine
dioxide. The release rate could be slowed by cooling
the mixture to 0°C or by increasing the viscosity of the
materials.
EXAMPLE 2
1-(N-dipropyla:mino)-2-carboxyamidoethane (DPACAE)
was made by reacting 0.2 mole di(n-propyl)amine with 0.1
mole acrylamide in. the presence of a small amount of
acetic acid as a 10 wt.~ solution in methanol. The
reaction was carried out for 3 hours at 70°C. After
vacuum evaporation of the excess amine and
crystallization in the presence of pentane, a white low
melting solid was obtained (Tm = 60°C) which tended to
lose amine a:nd form acrylamide upon prolonged heating
above the melting point.
1-(N-Dimethylamino)-2-carboxyamidoethane (DMACAE)
was made by :reacting 0.2 mole dimethylamine (as a 40
wt.$ solution in water) with 0.1 mole acrylamide as a 10
wt.~ solution in mev:.hanol. The reaction was carried out
WO 96/39200 PCT/US96/09202
for one hour at room temperature. After vacuum
evaporation of excess amine, methanol and water, the
DMACAE was taken up in methylene chloride, dried with
magnesium sulfate and isolated as a low melting (Tm =
5 45°C) hydroscopic solid.
Both DPACAE and DMACAE crystallized only slowly and
thus could be studied in the liquid state at room
temperature. Neither neat liquid formed iminium
chlorite. However, 10-30~ wt.~ solutions in formamide
10 or acrylamide-isopropyl acrylamide readily formed
iminium chlorite when exposed to chlorine dioxide.
EXAMPLE 3
The amine-chlorine dioxide reaction was studied by
layering the requisite amount of 6.0X10-5 molar solution
15 of chlorine dioxide in pentane onto about 3.0X10-9 mole
of amine, either in neat form or dissolved 10-30 wt.o in
formamide or isopropyl acrylamide-acrylamide melt. The
chlorine dioxide-pentane solution was prepared by
reacting stoichiometric sodium chlorite with potassium
20 persulfate in a small amount of water in the presence of
pentane with vortex stirring in ice water. The
supernatant pentane layer was then removed and kept dry
in a sealed container over magnesium sulfate.
The formation of chlorite was detected by
25 acidification of the reaction product and the
observation of the odor and color of chlorine dioxide by
UV/Vis spectroscopy after exposure to dilute HC1. In
some cases the presence of chlorite was further verified
by observation of the IR spectrum. Characteristic IR
30 absorbance of chlorite at 830 cm-1 verified its presence.
The following neat primary amines formed chlorite
when exposed to chlorine dioxide:
HZNCHzCH20CH2CH20H, HzNC (CH3) ZCH20H, H~NCHzCH~NHCHzCHzOH,
HZNCH ( CH3 ) 2 , HzNCH~CH~OH ,
WO 96/39200 PCT/US96/09202
2196778
31
HzPJ[ CH2] 2--N~ H2N[ CH2] z-N~NH
and
H2N-[ CH2] 3-N"N-[ CHZ] 3 NHz
Chlorite was also formed by neat secondary amines
having the formula RzR3NH wherein RZ and R~ are,
independently, he~yl, benzyl, n-propyl, isopropyl,
cyclohexyl, acryla~mide, or -CH2CHZOH. These amines also
formed chlorite wren the amine was in formamide solvent.
The following' secondary amines yielded chlorite
when plasticized with formamide or isopropylacrylamide-
acrylamide:
0
CR4-CHZ-CHZ-IC--NH~CHz
z wherein R4 is cyclohexyl or
benzyl, and
0 0
~NI-+-[CHZ]~PJH-CHZCHZIC-NHCHzNHCICHZCHz
wherein
n is 2 or
3. The isopropylacrylamide-acrylamide and amine were
also prepolymerized and film formed by heating to 60-70°
C in the presence of about 0.01% azobisisobutyronitrile
initiator, providing chlorite so long as the film
temperature exceeded the glass transition temperature.
A hydrogen bonded amine having the formula
ReR9NCH2CH2C (O) NHz wherein Re is methyl and R9 is n-propyl
when in form~~mide or. isopropylacrylamide-acrylamide
solvent yielded chlorite. However, when R8 and Ra were
isopropyl groups, the neat amine did not yield chlorite.
A neat hydrogen bonded amine of the formula N(CHZCH,OH)3
yielded chlorite, which was also formed when the amine
was in formamide or isopropylacrylamide-acrylamide
solvent.
WO 96/39200 ~ PCT/US96/09202
32
To determine whether hydrogen bonding was
necessary, a Michael addition process was used to
provide a reaction product of 2-propenenitrile and (i-
C3H~ ) NHCHZC6H5 such that the amine portion of the product
did not have any hydrogen bonding and the nitrile
portion was very polar. Polarity was not sufficient to
generate stable chlorite when the neat amine or the
amine solvated in formamide was exposed to chlorine
dioxide. The nitrile group blocked formamide so that
the chlorite back attacked the amine and decomposed the
chlorite into a form that could not be reconverted to
chlorine dioxide. Thus, it was discovered that amines
in apolar environments react with chlorine dioxide but
the chlorite ion is unstable in such an environment.
Non-hydrogen bonded tertiary amines of the formula
NR5R6R~ wherein R5 and R6 are methyl and R~ is cyclohexyl
or 4-pyridyl were solubilized in formamide or
isopropylacrylamide-acrylamide and formed a stable
chlorite. Amines wherein R5 is benzyl, R6 is cyclohexyl
and R, is dodecyl or wherein R5, R6 and R, are n-butyl or
ethyl groups were insoluble in formamide and could not
form any chlorite . (CH3 ) ZNCHZCHZN (CH3 ) z was soluble in
formamide and yielded chlorite, but did not yield
chlorite in isopropylacrylamide-acrylamide although it
was solubilized by the solvent; the amine when neat or
in acetonitrile did not yield chlorite.
Thus, it was discovered that an amine having a
nitrogen of sufficiently high pKa solvated by a
hydrophilic material or substituted by hydrogen bonding
groups, such as hydroxylic, amide, primary amine or
secondary amine substituents, forms chlorite by reaction
with chlorine dioxide.
The amine-chlorine dioxide reaction as described
above was repeated wherein the amine was dissolved in
various solvents to determine the effect of the solvent
on reaction efficiency. All chlorine dioxide was
WO 96/39200 b PCT/US96/09202
33
released in ~Nater. More chlorine dioxide was released
in glycerin or ethylene glycol than was released in
methanol, acE~tonitrile, methoxyethanol, ethanol or
ethoxyethano=L. Chlorite suspended or dissolved in a
hydrophobic rnaterial, as a dilute solution in toluene or
benzene, and exposed to chlorine dioxide reacted with
chlorine dio5cide but: only released a minor amount of
chlorine dio~;ide wihen acidified. Many of these
solvents, such as ethanol, will not retain chlorite
counterion for long term storage unless iminium chlorite
is stabilized with a strong base to retain the chlorite
counterion.
EXAMPLE 4
Amines that are monosubstituted with short apolar
groups , such as ( CH3 ) zNCHzCHzC ( O ) NH2 , ( n-
C3H~ ) ZNCHZCH2C ( O ) NHz , and ( i -C3H~ ) zNCHzCH2C ( 0 ) NHZ , formed
stable chlorite in formamide. Amines that were
substituted with short apolar groups, namely
( CH3 ) zNCHzCH2C ( O ) NH ( i ~-C3H, ) , ( n-C3H~ ) NCHZCH2C ( O ) NH ( i -
C3H, )
and i-C3H,N(C~i2CHZC(O;INHz)z, did not form stable chlorites.
However, those with linear alkane lengths greater than
or equal to s ix, such as n-C6H13N ( CHzCHzC ( O ) NHz ) z and
n-Cl2HzsN(CHZCHZC (O) NH>) 2, did form stable chlorite in
formamide. It is possible that once the apolar chain
length had achieved a certain length, a microphase
separation into micelles with discreet hydrophobic
regions surrounded by continuous hydrophilic regions
took place. The destabilizing apolar phase was thus
removed from the reaction environment.
EXAMPLE 5
The following polymers were synthesized,
characterized usincr NMR techniques, and evaluated to
determine physical properties and ability to uptake (and
release) chlorine dioxide:
3 5 [ -CH,CH~N' ( CH,CH,C.'H ; ) - ) "
[ -OCHzCH ( CH: N ( C:H, ) ; ) - ] "
WO 96/39200 PCT/US96/09202
34
~ -CHZCH ( OCHZCHZN ( CH3 ) 2 ) - ]
-CHZCH ( C ( O ) N ( H ) CHZCHZCHZN ( CH3 ) z ) -
Of these polymers, the last polymer has the most
flexible amine containing side group and exhibited the
most efficient uptake and release of chlorine dioxide in
formamide that is a substantial improvement over that
demonstrated with in-chain amines. The polymer was also
soluble in molten urea.
EXAMPLE 6
The following compounds containing an N-amido
linkage and a tertiary amine center were synthesized in
pure form from the corresponding primary or secondary
amine, sodium cyanate, and hydrochloric acid as
described by J. March, "Advances in Organic Chemistry:
Reaction Mechanisms and Structure, 4th Ed., John Wiley,
N.Y., p. 903 (1992).
MeZN ( CHZ ) 3NHC ( O ) NHz
HNMR: 1.5, 2.1, 2.2, 2.95, 5.5, 6.2
N ( CHzCH2NHC ( O ) NHZ ) 3
HNMR: 2.4, 3.0, 5.65, 6.25
H2N ~--~
N/ \N
0 ~ ~NH
~0
H2N
HNMR: 2.35, 3.2, 5.6, 6.05 ppm
Each of these compounds reacted with chlorine dioxide
and later released it upon acidification in formamide,
indicating that tertiary amine compounds with N-amido
substitution of their primary and secondary amines can
complex chlorine dioxide, when dissolved in a suitable
hydrophilic solvent. Addition of urea to the formamide
clearly improved the uptake and release efficiency.
WO 96/39200
PCT/US96/09202
EXAMPhE 7
Up to _'>0 wt.'o of the tertiary amine
dimethylaminoacry:lamide (DMAA) was added to hydrophilic
solvent containing 50 wt.o urea and 50 wt.~ n-
5 methylacetamide (1~11~iA) solvent at 50°C and quickly cooled
to room tem~~eraturE~. The solution remained single phase
indefinitely at room temperature. The same behavior was
noted for th.e add~_tion of 20 wt.% DMAA to a solvent
containing 33 wt.s's urea, 33 wt.~ NMA and 33 wt.~ sodium
10 acetate, a solvent. containing 35 wt.o urea, 55 wt.o NMA
and 10 wt.o sodium methoxide, and a solvent containing
70 wt.o urea and .'.0 wt.~ sodium acetate.
The above mi~aures were exposed to a solution of
chlorine dioxide i.n pentane and were observed to rapidly
15 uptake (one :minute) one chlorine dioxide for every two
amine groups before the reaction slowed substantially.
The final pH of th.e hydrophilic material remained on the
basic side. A slight cloudiness was seen in the 50 wt.o
urea/50 wt.~ NMA-D~M,A.A mixture and the 33 wt.o urea/33
20 wt.o NMA/33 vNt.~ sodium acetate - DMA.A mixture while the
DMAA - 35 wt.~ urea/55 wt.~ NMA/10 wt.o sodium methoxide
mixtures remained c:Lear.
Upon acidification by 0.1N HC1 (pH<5), complete
release of chlorine dioxide from all three mixtures was
25 observed up t:o 30 :m:mutes after formation of the
chlorite salt:. The release of chlorine dioxide was
estimated by referring to the color of solutions
containing known amounts of chlorine dioxide. After
this time dif=ferent behavior was observed. For example,
30 after two hours, tlhe 50 wt.~ urea/50 wt.o NMA - DMAA
mixture released no chlorine dioxide. The 33 wt.o
urea/33 wt.~ NMA/33 wt.~ sodium acetate completely
released chlorine dioxide after two hours at room
temperature. However, only one third of the chlorine
35 dioxide was r~eleasE~d after 24 hours at 5°C, with no
WO 96/39200 PCT/US96/09202
36
chlorine dioxide being yielded after an additional 24
hours at room temperature.
35 wt.o urea/55 wt.o NMA/10 wt.o sodium methoxide
exhibited the greatest chlorite salt stability in that
complete release was noted after three days storage at
5°C. Complete release was also noticed after 24 hours
at room temperature. The presence of a strong inorganic
base greatly improves the stability of the chlorite salt
in urea based solvents.
A 20~ DMAA - 35 wt.~ urea/55 wt.o NMA/10 wt.o
sodium methoxide melt was examined at 60°C for up to one
hour in 300 MHz proton NMR to see if any DMAA
decomposition occurred. From the toxicological point of
view any decomposition of the DMAA into secondary amine
and toxic acrylamide would be highly undesirable.
No decomposition was observed over the one hour
heating period. Acrylamide alkene resonances were
expected between 6-4 ppm yet none were seen. Some
polymerization of the urea was revealed by the broad
band under a sharp urea band at 6-7 ppm. The NMR
obtained after heating at 120°C for two hours, much
above the 50°C at which the DMAA was mixed into the urea
based solvent, revealed extensive polymerization of the
urea that was evident from the increase in line width
and the complication in the urea resonance between 8 and
6 ppm. However, no alkene acrylamide resonances were
seen. Thus, the 20~ DMAA - 35 wt.o urea/55 wt.o NMA/10
wt.% sodium methoxide system produced no toxic alkene
products.
To avoid variability in chlorite stability from
incomplete drying of the solvent, 40 wt.o of carefully
dried urea (vacuum dried: 80°C, 18 hours, 0.1 torr) and
60 wt.$ NMA (Ca0 overnight reflux and distilled) were
mixed and heated for 18 hours at 120°C. Alkoxides were
first isolated as dry powders by reacting the required
amount of clean sodium metal with the alcohol and
WO 96/39200 2 ~ 9 6 l 7 8
PCT/US96/09202
37
isolating the product by washing with diethyl ether.
All mixing was carried out under dry nitrogen
atmosphere. Predrying of the urea/NMA mixture resulted
in room temperature stability of the iminium chlorite
salts for at least one week at room temperature.
The desired amount of alkoxide was then dissolved
in the urea/:NMA solvent using minimal heating followed
by DMAA to form a clear viscous liquid at room
temperature. The results of the chlorine dioxide uptake
and release ~~f several urea/NMA/DMAA/sodium alkoxide
hydrophilic material composites are presented in Table
1. Release ~~haract~~ristics are based on a relative
scale rangin~~ from, excellent (9) to poor (1).
WO 96/39200 PCT/US96/09202
38
O O O t!lc-IM Lflr1 lD O1
O O O l0 c-1M lD N l0 01
(CS
O O O l0 ~-ILf1l~ N t~ 01
O O O CW -It11[~
M
'y O O O t~ c-I~O CO d~ pp a1
N N
N .~ S-I
1~
O O O (~ ~ (~ Op [~ 0p 01 .
( ~ ~ N
U
~ t~
~
w
O 'd~~ d T5 f~ _
O ~ ~ O
d 0 N 00 01 00 01 01 N N
R~ N ~ b1
a
.~ a o
o rt3 U
Ea ~ o~ ~ o~ a~ M o~ o~ o~ o~ ~ U 23 b
~
(~ O N
i.-1
~w
~ U
r Lf1tI1Lf1Ln C'If1t!7L!1111I ,~ N O
l
~
,.,
. . . . . . . . 1
CT'-lo o 0 0 0 0 0 0 o I
W r>3 S.-I ~I
U
~ O 3
0
O O O N O L~ O M I
o N N N N N N M N I
M I O ~O
b
.~ ~ ~ ~ 3
.
x
0 0 ~ u, M o o ,-I~ o
r1 N M N M c-IM
~"~
11
Ao - r1 r-I
o '~
~ ~
Cr
Q,
~
J-1
N O N
r'I
b I M M d~ d~ O ~y ~
~ ~ ~
I r-i..IN N V U U U Wn
x V ,~ U
1 S
x
I V U (, U I I I I 1
I w -~ ~ u -1_
U7 N
~
x . ~ ~ ~ ~ ~ E.
-
O~
2 ~ l: 'O
tf~ O U1
WO 96/39200
PCT/US96/09202
39
The pre=_sence of an alkoxide promotes long term
iminium chlorite stability. However, the addition of
more than 0.5 mole chlorine dioxide per mole of amine
substantial:Ly decrc=ased iminium chlorite stability.
ExcellE~nt long term stability was found at room
temperature for them phases containing 23~ sodium
ethoxide, 3:L~ sodium isopropoxide or 30~ sodium t-
butoxide, in that at least 600 of the chlorine dioxide
was released upon acidification of the phase after three
weeks storage in diy, dark conditions. Since no change
in the chlorine dioxide release was noted after one
week, these phase's were considered indefinitely stable
after one week.
EXAMPLE 8
In order to make a hydrophobic acid releasing wax,
hydrocarbon wax ('rm =60°C) or atactic polypropylene (APP)
was first melted at 70°C under nitrogen with stirring.
An equivalent weight of glycerol monostearate or
glycerol di~~tearat~e was then dissolved in the molten wax
or APP. Two equiv<~l.ents (based upon phosphorous) of
powdered phosphorous pentoxide per three equivalents of
glycerol compound hydroxyl functions was slowly added to
the melt to avoid clumping. After stirring the melt an
additional two hour's at 80°C, one equivalent of
tetraethylorthosi~'_icate was added and the immediate
evolution of ethanol was detected. Stirring was
continued for an additional four hours while slowly
raising the temperature to 100°C and purging the mixture
of ethanol with a lOcc/minutes flow of nitrogen. The
reaction flask wa=> subsequently evacuated at 100°C to
remove any remaining ethanol or tetraethoxysilicate,
filled with nitrogen and cooled. Softening of the wax-
acid releasing agent (LPOSI) started at about 60-70°C.
The viscosity of the wax was 100 cP at 100°C.
WO 96/39200
PCT/US96/09202
The process for preparing LOPSI can be summarized
as follows. When hydrolyzed, silicon dioxide and a
phospholipid are formed.
0
0
P40~o + 3 OH
OH
60-80C
0 0
HO-IP-0 0 0
OH 0~0 0
II
0 OH O-P-0
I
0 0
0
0
SI(OC2H5]4
100'C
0
-C2HSOH II
GO-P-OG
I
0 0
II
0-S I-0-P-OG
I
0- P-O 0 0
I I~
OG 0-5 i -0-P-OG
~ I
(GO]z-P OCzHs OG
H z 0 IO
0
0 O
( RO] 2-P-OH + S f OZ G = OH
OH
5 Chlorite powder was prepared by first dissolving
commercial sodium chlorite in dry methanol at 3~ by
weight filtering and the resultant solution to remove
WO 96/39200 09202
PCT/US96/
41
sodium carbonate impurity. The chlorite solution was
then extruded into an anhydro spray drier in dry
nitrogen at 100°C through a self siphoning extrusion
head with co-axia7_ fluid and nitrogen flow. After
routing to a cyclone separator to isolate small sodium
chlorite particles of about 5 microns in diameter, the
powder was stored in a dry atmosphere.
Neat sodium chlorite powder or mixtures of sodium
chlorite powder and anhydrous sodium sulfate in a ratio
of 1:1 and 1:2 by weight was fluidized in the bottom of
a nitrogen filled container. A stream of acid releasing
wax was then directed into the fluidized bed through a
nozzle of 7 :mil in. diameter with a nitrogen back
pressure of 30-80 l:bs/inz to produce wax particles
encapsulated with chlorite and sulfate particles
(indicated a;s 1:1 pre and 2:1 pre in FIG. 4). The
freely flowing powders were then stored in a dry
atmosphere. In soma cases anhydrous sodium sulfate was
postmixed with the chlorite-wax particles (i.e., 1:1
post and 2:1 post in FIG. 4).
FIG. 4 :shows the chlorine dioxide release rate from
200mg of sevE=ral powder composites placed in a Petri
dish of approximately 62cc volume with a leakage of 2X10-
moles/sec. Controlled release over several days is
accomplished at about 75°F and 40o relative humidity.
EXAMPhE 9
A hydrophobic acid releasing wax was made as
described in Example 8. The controlled release layer
for an immediate release system was formulated by melt
coating approximate=_7.y 5 mil of acid releasing wax in a
low melting riydroc<~rbon wax (60°C=Tm) onto both sides of
a piece of paperboard. Next, approximately a 5 mil
thick layer of 10~ by weight, methanol recrystallized,
sodium chlorite in the low melting wax was melt coated
onto the acid rele<~~;ing layer. Another acid releasing
layer of about 5 mil thickness was then coated onto the
WO 96/39200 PCT/US96/09202
42
chlorite containing layer. The total volume of
controlled release material was 0.25 cc.
Two chlorine dioxide measuring sensors (0-10 ppm
and 0-100 ppm) were interfaced with a computer so that
chlorine dioxide concentration was recorded as a
function of time over a two week period automatically
along with humidity and temperature. Both sensor ends
were exposed to the chlorine dioxide atmosphere in a
closed Petri dish through two small holes drilled into
the top cover of the Petri dish. The humidity and
temperature in the room were close to that measured in
the Petri dish because the Petri dishes were of the
"breathable" type where the cover made contact with the
base at a serrated edge and no effort was made to
insulate the Petri dish from its surroundings.
In this configuration, the acid releasing layer was
placed in direct contact with the chlorite containing
phase and immediate release of chlorine dioxide was
observed as soon as the film was placed in the Petri
dish. The chlorine dioxide gas concentration dropped
from a high of 13 ppm to 1 ppm at 5-6 days in an
exponential fashion as shown in FIG. 5 (note that
detector error of ~ 0.5-1.0 ppm resulted in less than
zero concentration). However, surprisingly, the
concentration peaks that were superimposed upon this
exponential behavior, were correlated with the
temperature and not the relative humidity as shown in
FIG. 6.
Three mold species, Chaetomium globosum (CG),
Aspergillus terreus (AT), and Aspergillus niger (AN),
were grown in mineral loaded, but nutrient free agar
slants using paperboard as a nutrient. All growth
studies were carried out in accord with TAPPI standard
method T 487 pm-85 entitled "Fungus Resistance of Paper
and Paperboard."
WO 96/39200
7 PCT/US96/09202
43
Six samples ware tested for fungus resistance over
two weeks at room temperature in duplicate.
Photographic. comparisons showed considerable growth
after two wE~eks on the control samples, while no growth
showed on the controlled release films. The
effectiveness of chlorine dioxide in killing these three
molds was e~Tident i=rom the two week study.
EXAMPLE 10
In a delayed release system one side of a piece of
paperboard was co<~t:ed with an acid releasing layer
separated from a chlorite layer by an intermediate wax
layer. The 5 mil thick hydrophilic phase in the
chlorite layer waa a transparent blend containing 10
wt . o sodium chlorite, 50 wt . o (NHZC (O) CHzCH20CH2CH2 ) z0 and
40 wt.~ formamide.. The chlorite layer was separated
from the acid releasing LPOSI wax of about 5 mil
thickness by an urunodified wax layer of about 5 mil
thickness. The total volume of controlled release
material was about. 0.25 cc.
A delay in chlorine dioxide release was noted when
the acid releasing layer was separated from the chlorite
containing layer by an intervening wax layer. In this
case, a peak in the release was noted after one day as
shown in FIG. 7. Individual concentration peaks
superimposed on the averaged behavior were again
correlated with th.e temperature and not with the
humidity as shown i:n FIG. 8.
The three mold species tested for in Example 9 were
grown in mineral loaded, but nutrient free agar slants
using paperboard as a nutrient in accord with TAPPI
standard method T 487 pm-85.
Six sam~~les were tested for fungus resistance over
two weeks at room tE=_mperature in duplicate. The results
are presented in Table 2. Photographic comparisons
showed considerable growth after two weeks on the
control samp:Les, while most of the controlled release
WO 96/39200 ~ ~ ~ ' PCT/US96/09202
44
films showed no growth. In the few cases where mold did
grow on the controlled release films, only a single
nucleus was responsible. Invariably, this nucleus was a
large clump of mold spores where some self protective
effect was generated by the aggregate structure.
TABLE 2
CG Mold AT Mold AN Mold
Control Growth Growth Growth
Lawns' Growth from No growth No growth
single mold
spore
Soak2 Growth from No growth Growth from single
single mold mold spore (trial 1),
spore No growth (trial 2)
1 Agar covered with mold spores
2 Paper soaked in mold spores
EXAMPLE 11
The porous paper used throughout these examples had
one untreated side and one side that appeared glossy.
The chlorine dioxide release coatings were applied to
the untreated side of the paper with the chlorine
dioxide releasing composite sheets assembled with the
glossy side out. Consequently, only the glossy side of
the paper had contact with the meat. Sheets
approximately 3 ft. x 8 in. were cut to facilitate
handling during the coating process. The original paper
weight was 5 mg/cm2.
LPOSI acid releasing wax was applied to the porous
substrate paper in a nitrogen filled dry box containing
a large dish of stirred phosphorus pentoxide using a wax
coater operating at approximately 190 °F. If multiple
coatings were used, the paper was allowed to cool prior
~~- WO 96/39200
PCT/US96/09202
to applying subsequent layers. Once the paper was
coated, it was sealed in a dry atmosphere suitable for
storage.
The chlorite containing paper was applied from
5 methanol solution using a coater operating at room
temperature. A typical coating solution was prepared by
first dissolving 25 grams of poly N-vinyl pyrrolidinone
(PVNP, 1.7x106 M.W.) in 500 ml of methanol followed by 15
grams of sodium chlorite (technical grade). The
10 homogeneous solution was used immediately. If multiple
coatings were desired on a single substrate, the coating
was allowed to dr~~ between applications. The chlorite
containing paper was then sealed in dry atmosphere for
storage.
15 Immediately prior to use, the chlorite containing
film was compression molded at room temperature with the
LPOSI containing film to form a chlorine dioxide
releasing bilayer composite. Pressures under 10,000
lbs/inz were sufficient to induce cold flow and adhesion
20 of the wax t~o the chlorite containing film.
Samples of each individual sheet of coated
substrate bilayer were randomly set aside during the
pressing operation :in order to quantify the chlorite and
wax loadings. These sheets were cut, measured and
25 weighed, then compared with data obtained from uncoated
paper as sho~nrn in Table 3. Calculations of the
theoretical <acid output based on phosphorous pentoxide
and the relation:
5 C102 ~ + 4 H' ~ 4 C102 + 2 Hz0 + C1-
30 indicate a ratio of approximately 0.148 NaC102/g wax for
optimum C10~ utili~:ation.
WO 96/39200 ~ PCT/US96/09202 I
46
TABLE 3
Sample No. of NaC102 No. of Wax g NaClO,
( C C 102 / ( mg / Wax ( mg / g Wax
10z PV P cmz ) coatings cmZ )
: coatings
Wax)
1:1 1 0.44 1 2.9 0.15
3:6 6 1.6 3 6.3 0.25
2:4 4 1 2 5 0.21
2:2 2 0.45 2 4.7 0.096
The chlorine dioxide concentration released from
the films along with humidity and temperature was
monitored in a Petri dish under atmospheric conditions
using the sensor system and gas leakage rate previously
described in Example 9. Samples were monitored over
several days. FIG. 9 shows a typical plot generated
from data acquired from a sample composed of sheets with
two coats of each phase (2:2). Samples were monitored
at several different loading levels. All samples showed
an immediate maximum release of 10-20 ppm chlorine
dioxide within the first 2-3 hours followed by a very
gradual reduction in release over the next several days.
Higher loadings served to increase the maximum initial
concentration and prolong the release.
EXAMPLE 12
2:2 loaded papers were used as separators between
ground meat patties packed to different densities that
were loaded initially with high loadings of colony
forming units (CFU) of E. coli bacteria. Substantial
reductions in bacterial growth were noted as shown in
Table 4. In loosely packed patties, the chlorine
dioxide gas had access to the interior of the patty,
resulting in a more complete kill throughout.
2i96~~~
WO 96/39200 PCT/US96/09202
47
TABLE 4
Bacterial Load % Reduction in
Ground lKeat (CFU/patty) E. coli bacteria
Loosely packed 1.7X10' >99.g
Densely packed 5.0X10' 99.5
[
EXAMPLE 13
Escheri.chia coli ATCC (American Type Culture
Collection) #26 way; grown in Tryptic Soy Broth (Difco
0370-17-3) to a log phase activity with an optical
density of 0.8 at 600nm containing one billion colony
forming units per ml of culture. The concentration was
verified using plate counts on three separate dilutions.
Uniform dispe'r'sal of the bacteria was assured in
densely packed meat by the following inoculation method.
Chili-ground. sirloin purchased six hours before use and
stored at 8°C weic~h.ing two kilograms was placed in a pan
and pressed down into an even sheet. Five holes were
punched into the meat with a glass rod, and 0.1 ml of
the bacterial culture was pipetted into each hole. The
meat was then kneaded to disperse the bacteria evenly.
This was repeated three more times, with at least a
minute of vigorou~~ kneading each time. Since the two ml
of an inoculum with a culture concentration of 109 cfu
per ml was added t.o the meat, a concentration of one
million cfu/gram was introduced into the meat.
The meat was then reground to a fine texture on a
bench-mounted, har.~d-cranked sausage grinder and formed
into patties by replacing the meat in the pan and
cutting patties ou.t with a piece of tubing to form
positive control (i.e., added E. coli bacteria) patties.
The negative control (i.e., no added bacteria) ground
sirloin from the same source was ground first in the
uncontaminated grinder to prevent its own contamination.
WO 96/39200 ~ ~ ~ PCT/US96/09202
48
The patties were prepared in duplicate and consisted of
negative controls tested at 0 and 60 hours, positive
controls tested at 0, 4, 24 and 60 hours, and test
samples (i.e., patties exposed to a chlorine dioxide
releasing film of the present invention) at 0, 4, 24 and
60 hours.
The patties were placed between either unmodified
paper or the papers coated with a 2:2 chlorine dioxide
releasing film (as described in Example 11) in 10 cm
diameter plastic Petri dishes with covers. Two Petri
dishes containing duplicate samples were then put in
recloseable plastic bags and stored for the required
time at 4°C in a common refrigerator.
Two samples were taken from each patty, one from
the upper surface, T, contacted either by the unmodified
paper or by the test paper with the chlorine dioxide
releasing film, or from the middle one third of the
patty, M. Samples were obtained with angle tipped
forceps by either pinching across the surface to obtain
a small scraping of the meat, or by digging down and
exposing the middle third thickness region. The forceps
were sterilized between samples by dipping in
isopropanol and flaming.
Ten ml sterile water blanks in screw capped test
tubes were tared to zero on a sensitive electronic
scale, and roughly one gram samples added to the tubes
and the weights recorded. The tubes were then capped
and shaken vigorously to disperse the meat and release
the bacteria.
0.1 ml of the supernatant was plated onto Tryptic
Soy Agar (Difco 0369-17-6) in duplicate and spread with
a glass triangle on a turntable. The glass spreader was
sterilized between platings with isopropanol and flamed.
The viable bacterial content of the samples was
visualized by inverting the plates after 24 hours
incubation at 37°C.
WO 96/39200 219 6 l ~ ~ PCT~S96/09202
49
Uninoculated negative controls showed the normal
amount of bacteria commonly seen in ground sirloin with
no substantial growth noted over 60 hours at 4°C.
Inoculated ~~ositlVE'. controls showed large amounts of
bacterial growth fc>r all times with very minor
differences betweE~rA the top and middle samples. If the
unmodified diaper had an antimicrobial effect, it was
minor.
When tr.e colony counts of chlorine dioxide exposed
test samples were compared, a 50-100X kill was noted for
the surface sample as compared to the interior test
sample and the positive control samples, except for the
reduced surface kill on the sample contacted with the
weakly releasing i=elm. As for the four hour exposed
test samples, surface colony growth was 50-100X less
than the interior test sample or the positive controls.
The surprising observation made on the 60 hour sample
was the high kill in both the interior and surface
sections of the a};posed samples when compared to the
positive control :samples.
Because the positive control plates were expected
to be overloaded, a direct comparison for quantitation
purposes was not accurate, although a rough count
revealed anywhere between 50-200 fold reduction in
colony count. As an alternative the test plate counts
were compared to t:he confirmed inoculum titer instead.
A rough comparison may be made between the Ccfu and
the inoculum figure (corrected for sampling dilution).
This is termed the ratio to inoculum (RTI), which is
intended to compare the viability of the treated sample
and the maximum possible cfu count. RTI's were
calculated for the 60 hour plates on the basis of the
Ccfu count.
The average F;TI for the top samples of the plates
for the patties treat were exposed to chlorine dioxide
and tested for 60 hours was roughly 170, which would
- WO 96/39200 PCT/US96/09202
represent a 170 fold decrease in viability. The average
RTI for the interior of these patties was roughly 50.
At 60 hours, however, large reductions in the
bacterial viability in the center of the patty were
5 seen. Cooking the patties that were exposed to chlorine
dioxide and tested for 60 hours yielded a normal looking
hamburger with no unusual odors being noted.
EXAMPLE 14
Loosely packed 0.75 inch thick, ground sirloin
10 patties with approximately 25 cm2 top surface area were
formed by hand immediately after mixing and grinding in
of E. coli ATCC #26 broth (105-106 cfu/gram). The
initial inoculum was grown up to a slightly lesser
extent than the inoculum used in Example 13. The loose
15 packing was employed to help the penetration of chlorine
dioxide through interconnected air passages.
The patties then were placed between either 2:4 or
3:6 chlorine dioxide releasing papers as described in
Example 11, and covered with a Petri dish cover that was
20 enclosed in a recloseable plastic bag. The samples were
then stored at 4°C for 3.5 days. After this exposure
time the meat in contact with the 3:6 papers showed no
bacterial growth from either a surface or interior
sample when plated as described in Example 13. The
25 interior of the patty exposed to the lower chlorine
dioxide concentration (2:4) showed no bacterial growth
from either surface or middle samples when plated.
When compared to the results of Example 13, these
results confirm the deep penetrating biocidal action of
30 chlorine dioxide when released in a controlled fashion
over 2.5-3 days at 4°C. Clearly, the biocidal action is
more effective for a porous meat structure.
An additional experiment using chicken breasts was
also performed. A filet of chicken breast was
35 repeatedly dipped in undiluted E. coli ATCC #6 broth
(108-10° cfu/ml), placed between 2:2 chlorine dioxide
o
°
' WO 96/39200 PCT/US96/09202
51
releasing films amd then closed inside a Petri dish that
was placed in a recloseable plastic bag and placed in a
refrigerator at 4°C for 3.5 days. The surface of the
meat was then swabbed and plated to get an indication of
bacteria kill. P.gain no bacterial growth was noted
after incubation.
EXAMPhE 15
Design of a chlorine dioxide releasing film
suitable fo:r controlled release and biocidal action
within a container is described herein. The equation
describing 'the con~~entration of chlorine dioxide in a
coating of ~~hickness, Q, (0<x<Q) which is covering the
inside of a permeable container of total thickness Q +
a, where 'a' is the gas space thickness (Q<x<Q+a), above
the coating is shown below. Chlorine dioxide is
generated b~~ means of a completely permeable thin film
of infinite;~imal thickness that lies on top of the
coating at :~c=Q .
t
~ Qcx~ze b'cos (oc~X) Jeb''~,e-s~'d~,
C(x,t) - E p
n=0 ,~~ (h-k'0(.n2)2+(Q+k' )OCR'+1'1~COS(OC~Q )
where,
b=D°OCn2, k' =4!P % P, h=D9/ ( ID°)
The terms, a~, in the infinite series above are
roots of them equation:
atan ( ocj! ) =h-k' (x2
D'= Diffusion constant of chlorine dioxide (cm2/sec)
in
coat inch
D9= Diffusion constant of chlorine dioxide (cm2/sec)
in
gas phase
1 - Phenomenological length (cm) f leakage pore
o
P = Coat (x=1 ) /C9as (x=1 ) =Henzy'constant for
s law
partit_~~on of t:he chlorine ide between the
diox
coating and the gas phase
WO 96/39200 PCT/US96109202 I
52
Q = chlorine dioxide generation constant from
controlled release film (mole/cm~/secz)
k = a, the total thickness of the gas layer
s - inverse of the time of maximum release rate of
chlorine dioxide from the controlled release film
C(x,t) is evaluated for a given set of diffusion
constants, leakage rate, h, phase partitioning and
dimensional constant, k' chlorine dioxide release rate,
Q, and inverse relaxation time for release, s, by
plotting C (oc) vs oc at t=s-1 . As an example, C (Q , t ) is
calculated for a Petri dish of 62 cmz cross-sectional
area of 1cm total thickness which includes 0.8cm gas
space and 0.2cm Agar. Since the biologicals are
introduced at x=Q and grow in the Agar it is important
to calculate this concentration. This calculation is
necessitated by the strong partitioning of chlorine
dioxide into the liquid phase once it is generated by
the controlled release film. At the release rates
generated by a test film the gas phase concentration was
so low (<0.1 ppm) it could not be measured by the
detector.
In order to complete the calculation Q, s, P, D9, D'
and 1 must be assigned or measured. Since Agar is 900
water it is assumed that P=40 can be used [J. J. Kaczur
and D.W. Cawlfield, Kirk-Othmer Encycl. Chem. Tech. (4th
Ed. ) , 5, 971 ( 1993 ) ] . D~=1 . 5 x 10-5 cmz/sec and D9=0 .12
cm2/sec are reported in the Handbook of Chem. and Phys.,
52nd Ed., F47 (1971). In actuality D9 appears in the
model only in conjunction with 1 since for the purposes
of the calculation C9 is assumed to be uniform in
Q <x<Q +a .
The leakage flux constant, D9/1, is evaluated by
injecting a small quantity (about 10 ppm) of chlorine
dioxide into the Petri dish containing no Agar and
measuring the chlorine dioxide concentration as a
function of time. The Petri dish employed will leak
WO 96/39200 PCT/US96/09202
2196178
53
relatively rapidly because of the serrated edges of the
bottom dish that :i~; employed to assure good gas exchange
necessary for bio_Lc>gical growth.
(D9/1)= 0.154 cm/sec
When the source function of the form Qte-b' is
integrated from 0 to infinite time,
j Qte~b'dt - Q/bz - total moles of chlorine dioxide
available
For the purposes of the calculation the controlled
release film of density 0.8 gram/cm3 and total volume
0.315 cm3 contains .L5 wt.o sodium chlorite of molecular
weight 90.44 g mole or 3.35 x 104 mole available chlorine
dioxide (assuming complete reaction of 5 moles of C102-'
to 4 moles chlorine dioxide) and shows a maximum release
rate at one day or s-'=86,400 sec. This release maximum
is typical of an acid releasing film separated from the
chlorite containing film by an intermediate wax layer.
Q is thus calculated as 7.23 x 10-16 mole/cm2/sec2
over a 62 cmZ base area Petri dish where the area release
rate is assw:ned to :have no lateral dependence over the
entire surfa~~e of the dish. This is a valid assumption
since, even 'though the controlled release patch occupies
a smaller area than the total cross-sectional area of
the dish, bo~~h the gas and Agar diffusion rates of the
chlorine dio:Kide arc. large in comparison to the time
scale of the release rate.
The con~:entration in the gel phase C(Q,t) as a
function of time is then calculated for a range of
leakage rates, h as shown in Fig. 10. At fast leakage
rates (105<h<10-' cm-') , the release rate maximizes at t=s-
and the maximum concentration is proportional to h. In
essence the concentration at any time significantly
greater than the half time for leakage is simply some
constant factor multiplied times the source generation
rate. However as 'the leakage rate decreases lOw<h<10~',
WO 96/39200 ~ ~ PCT/US96/09202 I
54
the maximum concentration is generated only at
considerably longer time. Of course at h=0, no leakage
occurs, the maximum concentration is approached
asymptotically, and a total of 3.36 x 10-4 moles chlorine
dioxide (e.g. Qs-2x62 cm2) is distributed between the 0.2
cm thick gel phase and the 0.8 cm thick gas phase.
For the purposes of estimating how closely the h=0
concentration is approached at h=8.31 x 10-4 cm-', the
concentration in the gel phase at t=6.0 x 105 sec, x=Q
(2.4 x 10-5 mole/cm') is used to calculate the total
amount of chlorine dioxide in the Petri dish.
[0.8cm(62cmZ)Q/40)+(0.2cm) (62cm2) ] (2.4 x 10-
5mole/cm')
3.27 x 10-4mole
This value is very close to that expected for h=0.
For the leakage rate measured for the Petri dish in
which the biological growth experiments are carried out,
a maximum concentration of 2.5 ppm is expected in the
gel phase at x=Q with a concentration of 0.06 ppm in the
gas phase. About 0.25 ppm is required to kill mold
spores.
A slightly more complicated environment would be a
box of the same dimension as the Petri dish but with its
gas space filed with absorbing particles packed with a
volume fraction, ~=0.5 cm3/cm3. The diffusion of gas
through such a composite media has been studied [R. M.
Barrer and D.M. Grove, Trans. Far. Soc., 47, 826, 837
(1951); R. Ash and D.M. Grove, Trans. Far. Soc., 56,
1357 (1960) ] .
The diffusion constant D9 of a gas flowing through a
porous media must be replaced by:
D9P=D9 / [ 1 + ( 2 KS / r ) ]
--- WO 96/39200 219 6 7 7 8 PCT~S96/09202
where 1~S= Surf:ace Henry's law coefficient in the
relationship
CS ~ -KSC,a
where CS' is the number of moles of gas absorbed/cm2 of
5 surface and C9 is the gas phase concentration in
mole/cm3, r is the equivalent pore radius for a set of
axially directed capillaries within a solid having
porosity E ~~nd internal surface, A (cm2/cm3) , r=2E/A.
For the purposes of the calculation of surface
10 concentration of chlorine dioxide within the porous
media, the particles are considered to be small enough
so that the concentration of chlorine dioxide throughout
the particles' th:ic:kness is equilibrated with the gas
concentration. For- the purposes of this calculation,
15 the entire particle concentration is concentrated in the
particle surface.
In thin> case t:he surface Henry's law coefficient is
related to t:he bu:Lk; coefficient, KP, by
CP ( 1-E ) /A=C6' _ [ ( 1-E ) /A] KpC9
20 KS= ( 1-a ) Kp/A
D9~,=D9 / [ 1 + ( 1- F: / E ) KP ]
At a porosity of 0.5 and a partition coefficient of
40 into the particles, the diffusion constant for flow
through the absorbing porous media would be reduced by a
25 factor of 0.0244. This substantial reduction of
apparent ga~~ phased diffusion constant proportionally
reduces the leakage rate, h, resulting in a proportional
increase in the concentration expected at any time.
The amount, placement and controlled release
30 characteristics required for a biocidal film are
estimated where the film is protecting a small 62 cm'
particle filled box: that is assumed to leak at the same
rate as the Petri dish, h=8.3 x 103 cm-' (a rather good
assumption for a typical loosely sealed box). A pallet
35 of well packed, folded (unpacked) boxes might be an
analogous case. :>ince mold spore kill is guaranteed at
WO 96/39200 ~ ~ ~ PCT/US96/09202
56
an exposure of 1 ppm chlorine dioxide for a few minutes,
any strategy must generate at least this concentration
in a pulsed release in moist regions of the box
preferably after several days delay. Destruction of
growing mold requires only 0.1-0.5 ppm for a few
minutes. The destruction of the growth mechanisms of
the cell is so complete that strains with a natural
immunity to chlorine dioxide cannot develop. '
Conveniently, these concentrations are below the human
olfactory detection limit of about 10 ppm.
Practically, since such a short exposure is
required, a film that released chlorine dioxide in a
pulsed fashion would be the ideal system. Of course,
depending on the storage environment, this behavior
would assure that the initial mold spore infection
(originating inside and outside the box) and any
subsequent infections (originating outside the box) were
destroyed before any growth could occur. A continuous
release of 1 ppm thus wastes about 98~ of the available
chlorite. The preparation of such a film is discussed
in Example 16.
Figure 11 shows the release characteristics
expected for a controlled release film with a maximum
release at 10 days, 3.35 x 10-9 mole available chlorine
dioxide (0.33 cm3 film, 15o wt.~ sodium chlorite), placed
in a 0.5 porosity box with a bulk Henry's law
coefficient of 40 vs air leaking with h = 202.76 cm-1.
A maximum concentration of 10.4 ppm is reached
after 10 days and at least 1 ppm is generated for 0.4
day<t<46 days. Approximately 0.31 cm' of controlled
release material is needed for this purpose. At a
materials cost of $1.00/1b, the controlled release
material cost required to do the job would be about
0.056 cents. Thus, a box containing 1.1 liters of
material could be protected for 1 cent with the above
listed parameters.
~196~7~
WO 96/39200 PCT/US96/09Z02
57
EXAMPLE 16
The pulsed release capabilities of a multiple
layered com)~osite can be calculated as follows to
determine whether the composite will provide the desired
sustained r~=lease :rates for a particular application.
The time recxuired Eor complete cation exchange can be
predicted from the mobile ion concentration in each
layer, Ci , ~nrherein i is A, B, or C . In order to
determine such a tame period, hydronium ion transport
across the :intermediate layer C is considered to be the
rate contro:Lling step, and the diffusion constant and
effective mobile ion concentration for hydrogen ion are
considered the same in layers A, B and C. Chlorite ion
is considerE~d to bE~ relatively immobile and the reaction
of chlorite to chlorine dioxide is considered to occur
instantaneously once a hydrogen ion enters the
hydrophilic layer B.
Hydronium ion mobility in intermediate layer C can
be estimated by using experimental data reported by J.L.
Crowley et al., J. Poly. Sc., Poly. Phys. Ed., 14, 1769
(1976). Crowley et~ al. studied the ionic mobility in a
graft copol~rmer of low density polyethylene (79 wt.~)
and sulfonat:ed polystyrene (21 wt.~) as a function of
ion type, water content and temperature. Sodium,
potassium and silver ions travel along polymer bound
sulfonate groups by exchange with hydronium cations. At
high water contents of 3-6 wt.~ phase separation of ion
clusters in a hydrophobic matrix is likely. The
reported si:Lver ion mobility and mobile ion
concentration is quite high under these conditions (fit =
3.0X10'9 cm2iStatV~-sec, C = 3.3X10-4 mol/cc) . However, in
"dry" films both the mobility and mobile ion
concentration decrE~ase substantially (fit = 1.4X10'4
cmz/StatV-sec, C =- 8.3X10-' mol/cc) . The ion diffusion
constant D can be calculated from the reported ion
mobility us:W g the equation D = (kT~t) /q, where k is
WO 96/39200 PCT/US96/09202
58
Boltzman's constant, T is the absolute temperature, ~. is
ion mobility and q is electron charge. The calculated
ion diffusion constants are 1.21X10-8 cm2/sec and 2.58X10-
8 cm2/sec for a dry and wet (6 wt.% water) silver
counterion loaded film, respectively.
The morphology of such a copolymer would be very
similar to the two material system of the present
invention in that both include partially connected ion
clusters localized at spherulite boundaries within the
hydrophobic layer.
The total amount of hydronium ion that has diffused
across boundary AC (moles/cmz) in time t is represented
by the function Q:
Q(t) /1CA = (Dt/12) - 1/6 - 2 (n)-2 E (-1)nn-zexp(-Dn2n2t/1~)
n=1
Breakthrough of hydronium ion into hydrophilic
layer B will occur at (Dt/12)=0.1 (t=10.4 min, 1=5 mil or
1.27X10-ZCm) and steady state diffusion is reached at
(Dt/lz)=0.45 (t=46.9 min, 1=5 mil). The first two terms
in the above equation dominate after steady state is
reached. Thus under "wet" conditions (6 wt~ water),
Q(t) - 1CA [ (Dt/12) - 1/6] - 5.72x10-5 mole/day-cm2 at 5
mil thickness. The hydronium ion in a 1 cmz area film
and 1.27X10-z cm thickness (1.65X10-5 mole hydronium ion
initially) should be almost completely reacted in the
chlorite layer in 7 hours. In the "dry" film, which is
typical of polyethylene contaminated with ions, Q(t) -
DtCA/1 - 6 . 83X10-8 mole/day-cmz at 5 mil thickness .
Because of the much lower mobile ion concentration, 247
days are required for the hydronium ion to completely
diffuse into the hydrophilic layer B. Thus, a multiple
layered composite providing from about one day to about
247 days of chlorine dioxide release can be formulated
using the two layered composites of the present
invention.
WO 96/39200 ~ ~ 9 6 l 7 8 pCT/CTS96/09202
59
The chlorine dioxide release rate is generally
rapid when chlorine dioxide release is initiated in a
composite containing an intermediate layer because
chlorine decomposition is a function of pH. A minimum
concentration of hydronium ion is transferred before
chlorite decomposition into chlorine dioxide occurs due
to the buffering <~Ction of the hydrophilic layer
containing the chlorite.
The effect of viscosity on reaction rate, the rate
of hydration. of they film required to produce the minimal
amount of free water necessary for catalysis of chlorine
dioxide production, and the changing mobile ion
concentration and diffusion constant supported by the A,
B and C layers can affect hydronium ion transport.
An amount of water must be present in intermediate
layer C for transport of hydronium ion. Water is
transported through a hydrocarbon matrix as single
molecules, except a.t higher water activities where some
tendency to form clusters is noticed. The permeation
rate of water through a 5 mil thick high density
polyethylene film of 1 cm2 face area would be 6.89X10 -6
mole/day/cm2/5mi1 (90~ RH, 38 °C) as reported by Wessling
et al., Encycl.Po~~Ly.Sci.Eng., 17, 510 (1989). This
permeation rate is significantly less than that seen for
polyethylene ionorners which typically contain 3.35x10 -4
mole/cc ionic groups at a minimum (4.08x10-5
mole/day/cm2/5 mil) [Zutty et al., Encycl.Poly.Sci.Tech.,
6, 425 (1967)]. ~('he latter ionic content is suitable
for layers P., B and C, each of which has the potential
to absorb 3.3X10 -4 mole/cc x 10 moles of water
(assuming 10 Hz0/H30' ion) or 4.2X10 -5 mole water/cm2/5
mil (6 wt~ water)., Therefore, 5 mil A and B layers would
require about 1 day to saturate to 6~ water from an
initially dry stage. At most, an additional day would
then be required t:o saturate the intermediate layer C.
WO 96/39200 PCT/US96/09202 I
While the invention is susceptible to various
modifications and alternative forms, specific
embodiments thereof have been shown by way of example in
the drawings and have been described herein in detail.
5 It should be understood, however, that it is not
intended to limit the invention to the particular form
disclosed, but on the contrary, the intention is to
cover all modifications, equivalents and alternatives
falling within the spirit and scope of the invention as
10 defined by the appended claims.