Note: Descriptions are shown in the official language in which they were submitted.
CA 02202382 1997-04-1o
WO 96/11954 PCT/CA95/00573
TC OR RE RADIOLABELLED SOMATOSTATIN ANALOGS
BACKGROUND OF THE INVENTION
In the last few years a high incidence of somatostatin
receptors has been demonstrated in a variety of human ~umors, e.g.,
pituitary tumors, neuroendocrine tumors, breast tumors, gastro-
10 enteropancreatic tumors and their metastases. Some of them are smallor slow-growing tumors which are dif~lcult to precisely localize by
conventional diagnosis methods.
In vitro visl.lSlli7~tion of somatostain receptors has been
performed through autoradiography of tumoral tissues using
15 radioioflin~te-l somatostatin analogues, e.g., [125I-Tyrlll somatostatin-
14 (Taylor, J.E. et al., Life Science (1988) 43:421) or [125I-Tyr3] SMS
201-995 also called [125I]204-090 (Reubi, J.C. et al., Br~in Res. (1987)
406:891; Reubi, J.C. etal., J. Clin. Endocr. Metab. (1987) 65:1127;
Reubi, J.C. et al., Cancer Res. (1987) 47:551; Reubi, J.C. et al., Cancer
20 Res. (1987) 47:5758).
Although some somatostatin peptides are useful in
therapeutic or in vivo diagnostic applications, not all radioisotypes
commonly employed in the medical community have been easy to
chelate or label in it.
SUMMARY OF THE INVENTION
According to the invention, synthetic hexapeptides are
chelated with a bifunctional ligand bearing at least one chelating group
for a detectable element, such as technetium or rhenium and
30 furthermore, the chelation can be done at relatively mild conditions.
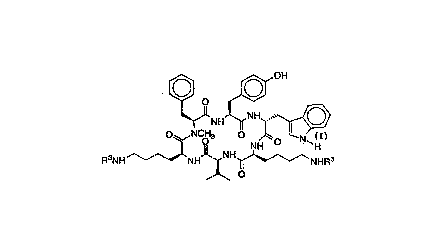
The synthetic hexapeptides useful in this invention are those
disclosed in ~et. Letters, Vol. 32:36, pp 4675-4678 (1991). The best
compound is:
CA 02202382 l997-04-lO
WO 96/11954 PCI~/CA95/00573
[~NH J N~H
oq~NCO3 0 ~0 N
R3NH ~NH~NH~ NHR3
~ O
wherein R3 is hydrogen or an amino protecting group which is selected
from the group consisting of: t-butoxycarbonyl (t-boc), fluorenyl-
methoxycarbonyl (Fmoc), and isonicotinyloxycarbonyl (i-Noc).
S This compound possesses the singular property of being
capable of binding to the a~Lo~liate binding site and also cont~ining a
chelate group capable of attaching radionuclides. Metallic isotopes such
as Tc-m99, In-111, Re-186, or Re-188 are subst~nti~lly better from an
im~gin~ point of view compared to the standard halogen isotopes
(I-123, I-131, I-125) but they are much more difficult to attach to
peptides and proteins.
The synthetic hexapeptides can be chelated with a group of
N3S2 chelates which are a separate invention, (Attorney Docket 19215,
Serial No. 08/322,881, filed October 13, 1994). This group of N3S2
chelates (or lig~n-ls) having ~e following structure:
(CH2)m-N-R2
R~ , ¦ ~ ,R
N N N~
~S ,S~
R1 R1
wherem
~ , ,
CA 02202382 1997-04-lO
WO 96/11954 PCTICA9S/00573
R is hydrogen, loweraLkyl of 1-4 carbon atoms, or loweraLkyl
carboxyl, wherein loweraLkyl is 1-4 carbon atoms;
Rl is hydrogen, a suitable protecting group such as p-
S loweraLkyloxyl (1-4 carbon atoms) benzyl such as p-
methoxylbenzyl; trityl; or Rl and Rl are linked together to
form a bond between the two "S" groups;
and R2 is either a free amino or an isothiocyanato group, which can be
=C=S or --H2;
--C~N=C=S or --C~NH2;
--C (CH2)n~N=C=S or
--C (CH2)n~ NH2;
and n or m are independently an integer from 1-4.
These lig~n(1s are prepared by using either a cyclic
15 approach, when Rl and Rl are linked together to form the disulfide
bond; or an open chain approach when Rl and Rl are either hydrogen
or the protecting group.
The cyclic approach reacts a tris(2-aminoethyl~amine with
the ~L,pr~,iate dithio-dialdehyde in a suitable solvent such as lower
20 aLkanol at reflux. Thereafter, the cyclic amine is optionally reacted on
the secondary amine groups, to provide the desired R groups, and
CA 02202382 1997-04-lo
WO 96/1195~ PCTICA95/00573
subsequently with the a~ro~iate N-hydroxy succinimide ester in
triethylamine and dichloromethane to yield compounds in which R2 is
the amino group; and subsequently optionally reacted with thiophosgene
to yield the corresponding isothiocyanato derivatives.
The open chain approach utilizes a diethylenetri~nine
reaction with first phthalic anhydride; second, p-nitrobenzyl bromide;
and third, 6N HCl at reflux to yield the a~ropLiate N'-(4-nitrobenzyl)-
bis-(2'-pht~ imidoethyl)amine, to which subsequently the pendent
blocking groups are attached, followed by reaction of the amino group
to isothiocyanato if desired.
The N3S2 ligands can then be labelled with the appropriate
radioisotope by reacting in a methanolic solution with the radioisotope
as a glucoheptonate reagent (available commercially) and h~tin~ at 40-
80C for 1-4 hours. Alternatively, the N3S2 ligands can be coupled
with the a~rol)~iate peptide or protein and then radiolabelled using a
.~imil~r procedure.
The N3S2 ligands are therefore useful as radioim~in~
agents after labelling, or as radioph~ c eutical for the treatment of
a~lo~liate tumors. When the ligands are to be first coupled with the
peptide or protein, the isothiocyanate group at R2 is first prepared for
reaction with the amino group of the peptide or protein; and thereafter
chelated with the a~lol,liate radiolabelling agent.
As previously mentioned, the li~n~l~ are coupled with the
desired synthetic hexapeptide, plefel~bly:
Me\ ~ Tyr ~
Phe Trp
I
Lys~ ~Lys
Val
The preferred radioisotope is one of those of rhenium or
technetium, preferably Re-186 or Tc-m99, respectively.
Other radioisotopes can be used which can be any
detectable element. By detectable element is meant any element,
30 preferably a metal ion which exhibits a property detectable in
CA 02202382 1997-04-10
WO 96111954 PCT/CA95/00573
~ therapeutic or in vivo diagnostic techniques, e.g., a metal ion which
emits a detectable radiation or a metal ion which is capable of
influencing NMR relaxation properties.
Suitable detectable metal ions include, for example heavy
5 elements or rate earth ions, e.g., as used in CAT scanning (Computer
axial tomography), par~m~netic ions, e.g., Gd3+, Fe3~, Mn2+ and
Cr2~, fluorescent metal ions, e.g., Eu3+, and radionuclides, e.g.,
~-emitting radionuclides, ,B-emittin~ radionuclides, a-ennitting
- radionuclides, positron-emitting radionuclides, e.g., 68Ga.
The products of this invention are useful either as an
im~ging agent, e.g., visn~ tjon of the particular (peptide) receptor
positive tumors and met~ct~es when complexed with a par~m~netic, a
~-e~ metal ion or a positron-emitting radionuclide9 or as a
radioph~ ceutical for the trea~ment in vivo of (peptide) receptor
15 positive tumors and metastases when complexed with a a- or ,B-
radionuclide, as indicated by standard tests.
The particular radioisotope chosen is relevant to the organ
or system to be radioimaged. For instance~ in the last few years a high
incidence of somatostatin receptors has been demonstrated in a variety
20 of human tumors, e.g., ~iluil~y tumors, central nervous ~,y~lelll tumors,
breast tumors, gastroelltelopancreatic tumors and their metastases.
Some of them are small or slow-growing tumors which are difficult to
precisely locali~e by c~llvelllional diagnosis methods, but in vitro
vis~ tion of somatostatin receptors has been pe.rolmed through
25 autoradiography of tumoral tissues using radioio-lin~te(l somatostatin
analogues.
The products of this inven~ion when used as im~ing agents
may be ~(lmini~tered part;lller~lly~ ~refcr~bly intravenously, e.g., in the
form of injectable solutions or suspensions, preferably in a single
30 injection. The a~pro~ iate dosage will of course vary depending upon,
for example, the precise chelating ligand and the type of detectable
e.lemP.nt used, e.g., the radionuclide. A suitable dose to be injected is in
the range to enable im~ging by photosc~nnin~ procedures known in the
art. It may advantageously be ~-lmini~tered in a dose having a
CA 02202382 1997-04-10
WO 96/11954 PCTICA95/00573
radioactivity of from 0.1 to 50 mCi, preferably 0.1 to 30 mCi, more
efelably 0.1 to 20 mCi. An indicated dosage range may be of from 1
to 200 ,ug product labelled with 0.1 to 50 mCi, preferably 0.1 to 30
mCi, e.g., 3 to 15 mCi, ~-emitting radionuclide, depending on the ~-
S emitting radionuclide used.
The enrichment in the tumorigenic sites with the products
may be followed by the corresponding im~in~ techniques, e.g., using
nuclear medicine im~gin~ instrumentation, for example a scanner, ~-
camera, rotating ~y-camera, each preferably computer assisted; PET-
10 sc~nner (Positron emission tomography); MRI equipment or CATsc~nnin~ equipment.
These products can also be used for in vivo tre~tment of
peptide receptor positive tumors ànd metastases in a subject in need of
such a tre~tm~t which comprises ~Clminietering to said subject a
15 therapeutically effective amount of the product.
Dosages employed in practicing the therapeutic method of
the present invention will of course vary depending, e.g., on the
particular condition to be treated, for example the volume of the tumor,
the particular product employed, for example the half-life of the
20 product in the tumor, and the therapy desired. In general, the dose is
calculated on the basis of radioactivity distribution to each organ and on
observed target uptake. For example, the product may be ~r~mini.ctered
at a daily dosage range having a radioactivity of from 0.1 to 3 mCi/kg
body weight, e.g., 1 to 3 mCi, preferably 1 to 1.5 mCi~g body weight.
25 An indicated daily dosage range is of from 1 to 200 ~g ligand labelled
with 0.1 to 3 mCi/kg body weight, e.g., 0.1 to 1.5/kg body weight oc- or
,I~-emitting radionuclide, conveniently ~-iminietered in divided doses up
to 4 hmes a day.
These products may be ~-lminietered by any conventional
30 route, in particular parenterally, e.g., in the form of injectable solutions
or suspensions. They may also be ~(1minietered advantageously by
infusion, e.g., an infusion of 30 to 60 min. Depen~lin~ on the site of the
tumor, they may be ~(1minietered as close as possible to the tumor site,
e.g., by means of a c~th~-t~-r. The mode of ~-lminietration selected rnay
-
CA 02202382 1997-04-10
WO 96/11954 PCT/CA95100573
- depend on the dissociation rate of the product used and the excretion
rate.
These products may be ~lmini~tered in free folm or in
ph~ ce.utically acceptable form, such as salLts which may be ~re~aled
S in conventional manner and exhibit the same order of activity as the
free compounds.
The products for use in the method of the present invention
may preferably be prepared shortly before the ~tlminictration to a
subject, i.e., the radiolabelling with the desired detectable metal ion,
particularly the desired a-"B- or~y- radionuclide, may be performed
shortly before the ~-lmini.stratiorl.
They are then suitable for im~in~ or treating tumors such
as pituitary, gastroenteropancreatic, central nervous system, breast,
prostatic, ovarian or colonic tumors, small cell lung cancer,
paragangliomas, neuroblastomas, pheochromocytomas, medullary
thyroid carcinomas, myelomas, etc. and metastases thereof, as well as
lymphomas.
According to a further aspect of the invention, there is
20 provided:
i. a ph~rm~ceutical composition comprising the
radiolabelled product of the invention in free or in
ph~ celltically acceptable salt form, together with
one or more ph~rm~ceutically acceptable c~ r.~ or
diluents therefor; or
ii. a ph~ ceutical composition comprising a chel~te-
peptide product according to the invention in free or
in ph~rm~- eutically acceptable salt form, together
with one or more ph~ celltically acceptable
carriers or diluents therefor.
Such compositions may be m~nllf~çtured in col~vel,liona
manner.
CA 02202382 1997-04-10
WO 96/11954 PCT/CA95/00573
A composition according to the invention may also be
presented in separate package with instructions for mixing the chelate-
peptide product with the metal ion and for the ~(lmini~tration of the
resulting radiolabelled product. It may also be presented in twin-pack
S form, that is, as a single package cont~ining separate unit dosages of the
ligand and the detectable metal ion with instructions for mixing them
and for ?~lmini~tration of the product. A diluent or carrier may be
present in the unit dosage forms.
EXPERIMENTAL
Mater~als and methods. Unless otherwise specified, all
reactions were carried out in oven-dried flasks at room temperature
under an argon atmosphere with magnetic stirring. After extraction,
15 organic solvents were dried over MgSO4, filtered, and removed under
reduced pressure on a rotary evaporator. Reagent grade solvents,
starting m~teri~ls and deuterated solvents were purchased from Aldrich
Chemical Co. (Milwaukee,WI) and used without further purification.
lH NMR spectra were obtained on a Bruker Model AM
20 500, AM 400, and AM 300. Samples were dissolved in CDC13,
MeOD4, or DMSO-d6 and chemical shifts were reported as ~ values
with the solvent or tetramethyl~ ne resonance as the internal standard.
The multiplicity is defimed by s(singlet), d(doublet), t(triplet),
q(quartet), and m(multiplet). The relative peak heights of the
25 resonances are reported as integers following the multiplicity. 13C
NMR spectra were recorded on a Bruker AM-300 spectrometer at 75.5
MHz and the degree of substitution of each carbon atom was determin~
by complete decoupling and DEPT composed 135 pulsed sequence
experimPntC. For 13C the carbon and proton signals were assigned by
30 heterocorrelation experim~nt~.
Infrared(IR) spectra (solution cells-CDC13 as solvent) were
recorded on a Perkin Elmer 681 hlrlaled spectrophotometer. Melting
points were lletermine~1 on a Thomas Hoover c~pill~ry meltin~ point
apparatus and are uncolle~ d. Mass spectra(MS) were recorded either
,
CA 02202382 1997-04-10
WO 96/11954 PCT/CA95/00573
- in the CI(methane gas) or FAB mode using Finni~n 4500 single
quadrupole mass spectrometer and were run by Oneida Research
- Services, Inc. (Whitesboro, NY). Flash chromatography was
performed essentially as described in the literab~rel using Merck silica
5 gel 60 (230-400 mesh) as stationary phase with the use of the following
solvents: methanol(M), methylene chloride(C), ammonium
hydroxide(H).
Part 1: Cyclic Approaches to N_S2 Chelates
Synthesis of N3S2-isothiocyanate (11) TPPBI
3,3,13 ,1 3-Tetramethyl- 1 ,2-dithia-5,8, 1 1 -triazacyclotridecan-8-yl-
ethanamine (4)
A round bottom flask was charged with tris~2-aminoethyl)-
amine (O (989 mg, 6.76 mmol), oc,a'-dithiodiiso-butyraldehyde (O
(1,380 mg, 6.69 mmol prepared according to reference 2b) and ethanol
(200 ml). The mix~lre was s~irred at room temp~l~Lure for 1.5 hours
and then refluxed for 3 hours. A~ter cooling the volatile materials were
20 removed in vacuo to give the crude di-imine 3 as a glassy solid. 1H
NMR analysis of the di-imine 3 gave three singlets centered at 7.58 ppm
(17:67:17) co..li....in~ the formation of an imine.
To the crude di-imine 3 in refluxing ethanol (200 mT.) was
added sodium borohydride (1.346 g, 35.58 mmol) in two portions over
25 3.5 hours. The reaction mixtllre was refluxed for a total of 17 hours
and acetone (100 ml) was added to destroy excess reagent. After
cooling the solvent was removed zn vacuo, water was ~d~ , and the
product was extracted with 5% MeOH/CH2CL2. The organic layer was
dried (MgSO4), f;ltered, and concentrate ~ I vacuo to give a yellow oil.
30 Flash chromato~graphy of this oil using 8'- .~% C/15.0% M/2.5~o H gave
amine 4 as a light yellow viscous oil (1.268 g, 59.5% yield).
lH NMR (in CDCl3): ~ 2.83 (t,j=5.9 HZ~2H~N-cH2cH2-NH2)~ 2.72
(s,4H,2N-CH2-C-S), 2.70 (m,4H,2N-CH2CH2-NH), 2.58 (m,4H,2N-
CH2-CH2-NH), 2.50 (t,J=S.9 HZ~2H~N-cH2cH~-NH2)~ 1.73 (br
_
CA 02202382 1997-04-10
WO 96/11954 PCT/CA9S/00573
- 10-
s,4H,4NH), 1.33 ~s,12H,4CH3-C-S) ppm. 13C NMR (in CDC13): 59.2
(t,2NH-CH2-C-S), 56.6 (t,N-CH2CH2-NH2), 54.5 (t,2N-CH2CH2-NH),
50.5 (s,2CH3-C-S), 47.4 (t,2N-CH2CH2-NH), 39.6 (t,N-CH2CH2-NH2),
27.4 (q,4CH3-C-S) ppm. MS (mlz,CI): 321(100, M~+1). IR (CDC13):
5 3350, 2940, 2820, 1455, 1360, 1120 cm-1.
3,3,13,13-Tetramethyl-1,2-dithia-5,8,1 1-triazacyclotridecan-8-yl-(2'-N-
phthaloyl)-ethanamine (5)
A round bottom flask was charged with amine 4 (111.2 mg,
10 0.347 mmol), N-carboethoxyphtl-~limide (108.3 mg, 0.494 mmol), and
dichloromethane (10 mL). The reaction mixture was stirred at room
temperature for 1 hour and the solvent was removed in vacuo. Flash
chromatography of the residue using 90% C/9.5% M/0.5% H gave ~e
p~th~l~te 5 as a yellowish oil (119.2 mg, 76.3%).
lH NMR (in CDC13): ~ 7.86 (m,2H,H2&H5-Ar), 7.71 (m,2H,H3&H4-
Ar), 3.84 (t,j=6.4 Hz,2H,N-CH2CH2-NPhth), 2.81 (t,j_6.4 Hz,2H,N-
CH2CH2-NPhth), 2.69 (m,4H,2N-CH2CH2-NH), 2.66 (m,4H,2N-
CH2CH2-NH), 2.59 (s,4H,2N-CH2-C-S), 1.87 (br m,2H,2NH), 1.23
(s,12H,4CH3-C-S) ppm. 13C NMR (in CDC13): 168.1 (s,2CO), 133.7
20 (d,C3&C4-Ar), 131.9 (s,Cl&C6-Ar), 123.3 (d,C2&C5-Ar), 58.7
(t,2NH-CH2-C-S), 53.9 (t,2N-CH2CH2-NH), 52.8 (t,N-CH2CH2-
NPhTh), 50.2 (s,2CH3-C-S), 47.6 (t,2N-CH2CH2-NH), 35.8 (t,N-
CH2CH2-NPhTh), 27.3 (q,4CH3-C-S) ppm. MS (m/z,CI): 451(100,
M++l). IR (CDC13): 3400, 2950, 2810, 1770, 1705, 1465, 1395 cm~l.
3,3,5,1 1,13,13-Hex~methyl-1,2-dithia-5,8,1 l-tri~7~cyclotridecan-8-yl-
ethanamine (7)
A round bottom flask was charged with phth~l~tç 5 (775.7
mg, 1.72 mmol), formic acid (10 mL, 265 mmol), and formaldehyde
30 (37% wt in water, 15 mL, 200 mmol). The reaction mixt~lre was
refluxed for 20 hours and then allowed to cool and the solvent was
removed in vacuo. A solution of 10% KOH was added to the solid
residue and the compound ex~acted wi~ 5~ MeOH/CH2CL2. The
CA 02202382 1997-04-10
WO 96/11954 PCT/CA95tO0573
- organic solvent was dried (MgSO4), ~lltered, and concentrated in vacuo
to give crude 6 as a viscous oil.
To a round bottom flask was added crude 6, hydrazine
monohydrate (1 mL, 20.6 mmol), and ethanol (40 mL). The reaction
5 mixture was refluxed for 20 hours and then allowed to cool. The
solvent was removed in vacuo, water added, and the residue extracted
with 5% MeOH/CH2C12. The organic solvent was dried (MgSO4),
filtered, and concentrated in vacuo to give a yellowish viscous oil.
Flash chromatography of the crude product using 80% C/19.5%
10 M/1.0% H gave dimethyltetramine 7 as a light yellow oil (375 mg,
62.4% overall yield from 4).
lH NMR (in CDC13): ~ 2.78 (t,j=5.9 Hz,2H,N-CH2CH2 NH2), 2.72
(t,j--5.5 Hz,4H,2N-CH2CH2-NClH3), 2.63 (S~4H~2N-cH2-c-s)~ 2.60
(t,j=5.5 HZ~4H~2N-cH2cH2-NcH3)~ 2.42 (t,j=5.9 HZ,2H,N-CH2CH2-
NH2), 2.36 (s,6H,2NCH3), 1.77 (br s,2H,NH2), 1.28 (s,12H,4CH3-C-S)
ppm. 13C NMR (in CDCl3): 67.2 (t,2N-CH2-C-S), 57.9 (t,2N-
CH2CH2-NCH3), 55.6 (t,N-CH2CH2-NH2), 52.1 (t,2N-CH2CH2-
NCH3), 51.3 (s,2CH3-C-S) 45.1 (q,2NCH3), 39.5 (t,N-C.H2CH2-NH2),
27.4 (q,4CH3-C-S) ppm. MS (-m-~z~cI): 349(:100,M++13. IR (CDC13):
20 2960~ 2800, 1455, 1355, 1310, 1100 cm-1.
4-Amino-N-[2-(3,3,5,11,13,13-hexamethyl-1,2-dithia-5,8,11-
triazacyclo-tridecan-8-yl)ethyllbenzamide (10)
A round bottom flask was charged with the dimethyl-
25 tetla ~ e 7 ~514.9 mg, 1.48 mmol), tBoc-p-aminobenzoyl N-
hyd~oxysuccinimide ester (O (493.8 mg, 1.48 mmol, ~l~aled
accor~ing to le~el~lce 3), triethylamine (0.21 mL, 1.51 mmol), and
dichloromethane (40 mL). The reaction mixtllre was stirred at room
temp~ ure for 12 hours and the solvent was removed i~ vacuo to give
30 crude 2.
To a round bottom flask was added crude 9, dichloro-
methane (10 mL), and trifll~oroacetic acid (10 mL) and the resulting
mixture was stirred at room tempel~lule for 1 hour. The solvent was
removed in vacuo and purification by flash chromatography of the
CA 02202382 1997-04-lo
WO 96/11954 PCT/CA95/00573
residue using 94.5~o C/ 5.0% M/O.5~o H gave the aniline 10 as a
yellowish white solid (345.8 mg, 50.8% overall yield from
dimethyltetramine 7).
lH NMR (in CDCl3): ~ 7.65 (d,j=8.6 Hz,2H,H2&H6-Ar), 6.87 (br
s,lH,NH-CO), 6.67 (d,j=8.6 Hz,2H,H3&H5-Ar), 3.95 (S,2H,NH2), 3.49
(t,j=5.4 HZ~2H~N-cH2cH2-Nco)~ 2.74 (m,4H,2N-CH2CH2-NCH3),
2.65 (m,4H,2N-CH2CH2-NCH3), 2.61 (s,4H,2N-CH2-C-S), 2.53
(t,j=5.4 Hz,2H,N-CH2CH2-NCO), 2.30 (s,6H,2NCH3), 1.29
(s,12H,4CH3-C-S) ppm. 13C NMR (in CDC13): 167.1 (s,NHCO), 149.6
(s,Cl-Ar), 128.6 (d,C3&C5-Ar), 123.9 (s,C4-Ar), 113-9 (d,C2&C6-
Ar), 67.2 (t,N-CH2-C-S), 57.6 (t,2N-CH2CH2-NCH3), 53.7 (t,N-
CH2CH2-NHCO), 53.4 (s,2CH3-C-S), 51.4 (t,2N-CH2CH2-NCH3) 45.0
(q,2NCH3), 37.0 (t,N-CH2CH2-NHCO), 27.2 (q,4CH3-C-S) ppm. MS
(m/z,FAB): 468.2(100, M++l). IR (CDCl3): 3405, 2960, 2800, 1620,
1495, 1280, 1100, 835 cm-l.
4-Isothiocyanato-N-[2-(3,3,5,1 1,13,13-hexamethyl-1,2-dithia-5,8,1 1-
triazacyclotridecan-8-yl)ethyllbenzamide (11) rTPPBIl
To a round bottom flask with the aniline 10 (53.3 mg,
0.114 mmol) and dichloromethane (5 mr.) was added 0.2007 M solution
of thiophosgene (0.60 mL, 0.120 mmol) in dichloromethane. The
heterogeneous reaction mixture was stirred at room tempelaLur~ for 1
hour and ~e solvent was removed in vacuo to give ~e isothiocyanate 11
as a reddish solid (67.8 mg, 116~o).
lH NMR (in DMSO-d6): ~ 8.88 (br s,lH,NH-CO), 7.99 (d,j=8.4
Hz,2H,H2&H6-Ar), 7.54 (d,j=8.4 Hz,2H,H3&H5-Ar), 3.49 (m,2H,N-
CH2CH2-NCO), 3.40 (m~8H~2N-CH2CH2-NCH3)~ 3.01 (s~4H~2N-CH2-
C-S), 2.89 (s,6H,2NCH3), 2.71 (m,2H,N-CH2CH2-NCO), 1.45
(s,12H,4CH3-C-S) ppm. MS (m/z,CI): 510(100, M++l). IR (CDCl3):
3400, 2960, 2100, 1640, 1600, 1545, 1500, 1470, 1300 cm~l.
Preparation of Somatoscan(Fmoc)TPPBI (13)
In a 5 mL Reacti-Vial (Pierce) a solution of Somatoscan-
Fmoc (~) [cyclo(Trp-Lys(Fmoc)-Val-Lys-NMe-Phe-Tyr)] (1.97 mg;
-
CA 02202382 l997-04-lO
WO 96/11954 PCT/CA95/00573
- 1.515 umol), TPPBI (11) (8.4 mg, 16.495 umol), and DMF (400 ,uL)
was stirred while bicarbonate/phosphate buffer (0.2 M, pH 8.2, 100 ,uL,
freshly prepared) was added. The heterogeneous solution was
monitored by HPLC and stirred ~or 4 hours at room temperature. The
5 solvents were removed in vacuo and the residue was partitioned with lN
HCl (300 ~L) and MeOH (100 ~L). Puri~lcation of this solution by
HPLC (Hamilton PRP-l 12-20 ~m preparative column 250 x 21.5 mm)
using a 30% to 100% gradient of acetonitrile: water (con~inin~ 0.1%
TFA) over 40 minutes (flow rate of 12 ml/min) afforded pure
10 Somat,oscan(Fmoc)-TPPBI (13) (Rt= 20.64 min).
MS (Electrospray, Hypermass) 799.4(z=2), Calc. Compound Mass =
1596.8, Meas. Compound Mass = 1597.8.
Part 2: Acyclic Approaches to N_S_ Chelates
Synthesis of Acyclic Dimercaptoanisidine Arylisothiocyanates
Synthesis of open chain N_S2-a~yl isothiocyanate (21)
20 Bis(2'-phth~limidoethyl)amine. (1~)
l~his was prepared according to reference 4.
Phth~lic anhydride (32 g; 0.22 mol) was dissolved in 333
mT. of hot chlorofc)~ and the mixture was filtered to elimin~t~- phth~lic
acid~. A Diethylenetri~mine 14 (7.97 g; 0.077 mol) solution in
2~ chloroform (64 mL) was slowly added (over a period of 50 minlltes) to
the phth~lic anhydride mixtllre m~int~ined at a tempel~lule of 50C.
Tempelatule was raised to 110C after the addition was over. The
reaction mixhlre was then stirred for 48 hours and slowly concentrated.
The conce~ ale solution was then treated with activated charcoal. 31.8
30 g of a yellow solid was recovered after evaporation of the solvent under
redllce-l pressure. The solid was Llitulated successively with ether,
ethanol and then dissolved in methylene chloride. The methylene
chloride solution was washed with 10% sodium carbonate (3 x ~00 mL),
water and salul~led sodium chloride solution. The organic phase was
CA 02202382 1997-04-10
WO 96/11954 PCT/CA95100573
- 14-
dried with magnesium sulfate, filtered and evaporated to dryness under
reduced pressure. A pale yellow solid (14.47 g; 52%) was obtained. A
portion (4.45 g) of that product was purified by flash chromatography
(silica gel) using a mixture of methylene chloride, ethyl acetate and
5 triethyl~mine as elution system (79/20/1). The purification give 2.798 g
of bis(phth~limi~loethyl)amine (~).
* 6.96 g of phthalic acid was recovered.
1H NMR (in CDC13): ~ 7.70 (m,8H,H-Ar(phth)), 3.77 (t,J=6Hz,4H,
-NH(-CH2-CH2-NPhth)2), 2.95 (t~J=6Hz~4H~-NH(-cH2-cH2-Nphth)2)~
1.41 (broad, lH,-NH(-CH2-CH2-NPhth)2) ppm. IR (in CDC13/NaCl):
3460 (N-H,w, sec amine), 2940-2820 (C-H), 1770-1710 (C=O, Phth),
1465, 1425, 1390, 1360, 1185, 1035 cm~l. MS (EI; m/z): 363(0.4,M+),
364(4,M++1), 216(3,M+-Phth), 204(18), 203(100,M+-(Phth-CH2-)),
174(57,Phth-CH2-CH2+), 160(5), 147(6), 130(12) and 56(6).
N'-(4-Nitrobenzyl) bis(2'-phth~limidoethyl)amine (16)
(See Ref. 4) In a 250 mL round bottom flask potassium
hydroxyde (1.6 g; 28 mmol) was dissolved in hot ethanol (100 mL). To
that ethanolic solution Bis(2'-phth~limidoethyl)amine C~) (10.02 g; 28
20 mmol) was added. The solution was magnetically stirred and refluxed
for 2 1/2 hours be~ore p-nitrobenzyl bromide (5.95 g; 28 mmol; 1 eq)
was added. The reaction mixhlre was heated at reflux for 16 additional
hours then filtered hot. The solid obtained previously was washed with
absolute ethanol and dried under vacuum to yield 7.441 g (54%) of a
25 white solid Cl~-nitrobenzyl bisphth~limide). The filtrate was evaporated
under re~-lce~l pressure to give 8.19 g of a yellow solid. That residue
was purified by flash chromatography (silica gel: 400 g) using
methylene chloride-methanol (98/2) system as eluent. The purification
by chromatography produced 3.13 g (23%) of the desired product. The
30 aLkylation reaction yielded 10.571 g of N'-(4-nitrobenzyl) bis(2'-
phth~limidQethyl)amine, (,~
1H NMR (in CDCl3): ~ 7.70 (m,10H,H-Ar(Phth)+o(H)-Ar-NO2), 7.20
(d, J=9Hz,2H, m(H)-Ar-NO2), 3.75 (t, J=6Hz, 4H,-NH
,
CA 02202382 1997-04-10
WO 96/11954 PCT/CA95/00573
(-CH2-CH2-NPhth)2), 3.71 (s, 2H,-N-CH2-Ar-NO2) and 2.80 (t,
J=6H~,4H,-NH(-CH2-CH2-NPh~)2) ppm. M[S (EI; m/z): 498(1,M+),
499(0.6,M++1), 362(l~M+--cH2Ar-No2)~ 339 (32, M++l-(Phth-
CH2~)), 338 (100, M+-(Phth-CH2-)), 324 (2, M+-(Phth-CH2-CH2-)),
174(58,Phth-CH2-CH2+), 173(42), 165(6), 163(8), 1611~6), 160(43),
149(12), 136(24), 130(12), 106(21), 105(12), 104(17), gO(22), 89(18),
78(23), 77(21) and 76(12).
Hydrolysis of N'-(4-nitrobenzyl) bis(2'-phth~limidoethyl)amine
In a 250 mL round bottom flask, provided with a
condenser, N'-(4-nitrobenzyl) bis(2'-phth~limicloethyl)amine (16) (2.80
g; 5.62 mmol) and 6 N hydrochloric acid (lS0 mL) were introduced.
The reaction mixture was stirred and refluxed for 23 hours. The
solution was cooled with an ice bath and filtered. The filtrate was
washed with ether (3 x 100 ml) and dried by vacuum to give a yellow
foam-like m~t~ l (2.17 g). The residue was dissolved in water (10
mL) a~d the pH of that solution was brought basic with 1 N sodium
hydroxide (25 ml). Then the mixture was extracted wilh methylene
chloride (3 x 75 mL). The organic extracts were combined, dried with
m~nesium slllf~t~ filtered and evaporated to dryness to yield 1.347 g
of N'-(4-nitrobenzyl) bis(2'-aminoethyl)amine (17) as a light orange oil
(which turn dark red with time).=
Note: The p-nitrobenzyltri~min~. (11~) is stored for short term
away from light and in an inert atmosphere of argon. For
long term storage it is better to keep that compound as the
- hydrochlorate form~
lH-NMR (in CDC13): o 8.13 (d,J=9Hz,2H,o(H)-Ar-N02), 7.46 (d,
J=9Hz, 2H,m(H)-Ar-N02), 3.65 (s,2H,-N-CH2-Ar-N02), 2.74 (t,J=6Hz,
4H, -N(-cH2-cH2-NH2)2)~ 2.50 (t,J--6Hz,4H,-N(-CH2-CH2-NH2)2) and
1.43 (broad s, 4H, -N(-CH2-CH2-NH2)2) ppm. IR (film): 3370-3290
(N-H,-NH2), 2940-2800(C-H), 1605 (C=C,Ar), lS10 (N=O,Ar), 1450,
1340 (N=O,Ar), l lOS, 1010, 850 (C-N,Ar-NO2) and 730 cm~l.
CA 02202382 1997-04-10
WO 96/11954 PCT/CA9S/00573
- 16-
N'-4-Aminobenzyl-diethylenetriamine (18)
This was prepared according to reference 4.
lH NMR (in CDC13): ~ 7.08 (d,j-8.3 Hz,2H,H3&Hs-Ar), 6.64 (d,j=8.3
Hz,2H,H2&H6-Ar), 3.62 (br s,2H,Ar-NH2), 3.48 (s,2H,N-CH2-Ar),
5 2.74 (t,j=6.0 HZ~4H~2N-cH2cH2-NH2)~ 2.50 (t,j=6.0 Hz,4H,2N-
CH2CH2-NH2), 1.52 (br s,4H,2NH2) ppm.
N,N"-Bis[2-((4-methoxybenzyl)thio)-2-methyl-propionyl]-N'-(4-
aminobenzyl)-diethylenetriamine (20)
To a solution of the ~niline 18 (230 mg, 1.10 mmol, freshly
prepared) in ethanol (20 mL) was added a solution of 2-[(p-methoxy-
benzyl)thio]-2-methylpropionic acid chloride 19 (1.33 g, 5.10 mmol,
prepared according to reference 5) in dichloromethane (10 mL) over 15
minutes. The resulting solution was stirred for 48 hours and the solvent
15 was removed in vacuo, 1 N NaOH was ~-lde(17 and the product was
extracted with CH2C12. The organic layer was washed with water,
dried (MgSO4), filtered, and concentrated in vacuo to give a red oil.
Flash chromatography of this oil using 5~o MeOH/CH2C12 gave the
aniline 20 as a yellow oil (126. mg, 17.5% yield).
20 lH NMR (in CDC13): ~ 7.15 (d,j=8.6 Hz,4H,2H3&H5-Ar-OCH3), 7.07
(t,j=8.2 Hz,2H,H3&Hs-Ar-NH2), 7.07 (m,2H,2NHCO), 6.78 (d,j=8.6
Hz,4H,2H2&H6-Ar-OCH3), 6.58 (d,j=8.2 Hz,2H,H2&H6-Ar-NH2), 3.74
(s,6H,2CH30), 3.72 (br s,2H,NH2), 3.65 (s,4H,2S-CH2-Ar), 3.49
(s,2H,N-CH2-Ar), 3.24 (q,j=5.9 HZ~4H~2N-cH2cH2-NHco)~ 2.55
25 (t,j=6.2 Hz~4H~2N-cH2cH2-NHco)~ 1.50 (s,12H,4CH3-C-S) ppm. 13C
NMR (in CDC13): ~ 174.6 (s,2NCO), 158.9 (s,2Cl-Ar-OCH3), 146.0
(s,Cl-Ar-NH2), 130.2 (d,C3&Cs-Ar-NH2 + 2C3&C5-Ar-OCH3), 129.6
(s,2Cl&C4-Ar-OCH3), 129.1 (s,C4-Ar-NH2), 115.4 (d,C2&C6-Ar-
NH2), 114.3 (d,2C2&C6-Ar-OCH3), 58.3 (t,N-CH2-Ar), 55.5
30 (q,2CH30), 53.0 (t,2N-CH2CH2-NHCO), 50.3 (s,2S-C-CH3), 37.8
(t,2N-CH2CH2-NHCO), 34.4 (t,2S-CH2-Ar), 27.1 (q,4CH3-C-S) ppm.
MS (m/z,FAB): 653.5(100, MH+). IR (CDC13): 3380, 3000, 2930,
2835, 1665, 1510, 1245, 1195, 1175, 1035 cm~l.
CA 02202382 1997-04-10
WO 96111954 PCT/CA95100573
- N,N"-Bis[2-((4-methoxybenzyl)thio)-2-methyl-propionyl]-N'-(4- isothiocyanatobenzyl)-diethylenetriamine (21)
- To a round bottom flask with the ~niline 20 (55.7 mg,
0.0853 mmol) and dichloromethane (5 mL) was added 0.2011 M
5 solution of thiophosgene (0.42 mL, 0.0845 mmol) in dichloromethane.
The heterogeneous reaction mixture was stirred at room temperature
for 1 hour and the solvent was removed in vacuo to give the
isothiocyanate 21 as a brown solid (68.9 mg, 116%).
lH NMR (in CDC13): ~ 7.72 (m,2H,2NHCO), 7.65 (d,j=8.3
10 Hz,2H,H2&H6-Ar-NCS), 7.25 (t,j=8.3 Hz,2H,H3&H5-AI-NCS), 7.17
(d,j=8.5 Hz,4H,2H3&H5-Ar-OCH3), 6.80 (d,j=8.5 Hz~4H~2H2&H6-Ar
OCH3), 4.14 (s,2H,N-CH2-Ar), 3.77 (s,6H,2CH30), 3.70 (s,4H,2S-
CH2 Ar), 3.58 (m,4H,2N-CH2CH2-NHCO), 3.01 (m,4H,2N-CH2CH2-
NHCO), 1.53 (s,12H,4CH3-C-S) ppm. Il~ (CDC13): 33()0, 2930, 2080,
1655, 1605, 1510, 1245, 1170, 1030 cm~l.
Part 3: Syntheci.~ of acyclic dimercapto~ni~i-line aLkyl isothio-
cvanates
20 Synthesis of open chain N3S2-alkylanisidine 26
N,N-Bis(2-aminoethyl)-N'-tert-butyl-oxycarbonyl- 1,2-ethane~ mine
(22)
A solution of tris(2-aminoethyl)amine CO (19.5 g, 133.6
25 mmol) in CH2C12 (300 mL) was cooled to -78C in a dry ice-acetone
bath while di-tert-butyl dicarbonate (14.6 g, 66.9 mmol) in CH2C12
(100 mL) was added slowly over 30 minlltes. The reaction l~ix~ was
slowly allowed to warm up to room temperature and s~ilTed for 18
hours. 1 N NaOH was added and the organic phase was dried (MgSO4),
30 filtered, and concentrated in vacuo to give the amine 22 as a light
yellow oil (9.07 g, 55%).
lH NMR (in CDC13): ~ 5.62 (br m,lH,NH-CO), 3.18 (m~2H,N-
CH2CH2-NCO), 2.98 (m,4H,2N-CH2CH2-NH2), 2.80 (m,4H,2N-
CH2CH2-NH2), 2.57 (m,2H,N-CH2CH2-NHC~0), 2.57 (m,4H,2NH2),
CA 02202382 1997-04-10
WO 96/11954 PCT/CA95/00573
- 18 -
1.44 (s,9H,3CH3-C-O) ppm. MS (m/z,CI): 247 (100, MH+). IR
(CDC13): 3280, 2965, 2815, 1695, 1500, 1165, 905, 730 cm-l.
N,N"-Bis[2-((4-methoxybenzyl)thio)-2-methyl-propionyl]-N'-[2-(N-
5 tert-butoxycarbonyl)aminoethyll-diethylenetriamine (23)
A solution of the tBoc derivative 22 (555.3 mg, 2.25
mmol) in CH2cl2 (25 mL) was cooled to 0C while 2-[(p-methoxy-
benzyl)thio]-2-methylpropionic acid chloride 19 (1.54 g, 6.85 mmol,
prepared according to reference 5) in dichloromethane (10 mL) was
10 added over 5 minutes. The resulting solution was allowed to wa~n to
room temperature and stirred for 12 hours. The solvent was removed
in vacuo, 1 N NaOH was ~ le-l, and the product was extracted with
CH2Cl2. The organic layer was washed with water, dried (MgSO4),
filtered, and concentrated in vacuo to give a yellow oil. Flash
15 chromatography of this oil using 5% MeOH/CH2C12 gave the tBoc
derivative 23 as a yellow oil (938 mg, 60.2% yield).
1H NMR (in CDC13): ~ 7.16 (d,j=8.5 Hz,4H,2H3&H5-Ar-OCH3), 7.07
(t,j=5.2 Hz,2H,2NH-CO), 6.81 (dJ=8.5 Hz,4H,2H2&H6-Ar-OCH3),
4.98 (br s,lH,NH-CO), 3.77 (s,6H,2CH30), 3.63 (s,4H,2S-CH2-Ar),
20 3.17 (m,6H,3N-CH2CH2-NHCO), 2.56 (m~6H~3N-cH2cH2-NHco)7
1.53 (s,12H,4CH3-C-S), 1.42 (s,9H,3CH3-C-O) ppm. MS (mJz,CI): 691
(9, MH+). IR (CDCl3): 3380, 2970, 2930, 2830, 1700, 1650, 1500,
1245, 1170, 1030, 830 cm~l.
25 N,N"-Bis[2-((4-methoxybenzyl)thio)-2-methyl-propionyl]-N'-(2-
aminoethyl)-diethylenetriamine (24)
A solution of 23 (858.3 mg, 1.24 mmol) in 50% TFA in
CH2C12 (30 mL) was stirred at room tempel~ule for 1 hour. The
solvent was removed in vacuo, 1 N NaOH was added, and the product
30 was extracted with CH2cl2. The organic layer was washed with water,
dried (MgSO4), ~lltered, and concenllaled in vacuo to give a yellow oil.
Flash chromatography of this oil using 90.5% C/ 9.5% M/ 0.5~ H gave
the amine 24 as a yellow oil (734 mg, 95.1% yield).
CA 02202382 1997-04-10
WO 96/11954 PCT/CAg5/00573
- 19 -
- lH NMR (in CDC13): ~ 7.25 (m,2H,2NH-CO), 7.16 (d,j=8.5
Hz,4H,2H3&H5-Ar-OCH3), 6.81 (d,j=8.5 Hz~4H~2H2&H6-Ar-ocH3)~
3.77 (s,6H,2CH30), 3.69 (s,4H,2S-CH2-Ar), 3.23 (q,j=6.1 Hz,4H,2N-
CH2CH2-NHCO), 2.72 (t,j=5.9 HZ~2H~N-cH2cH2-NH2)~ 2.54 (m,6H,N-
5 CH2CH2-NH2 ~ 2N-CH2CH2-NH-CO), 1.60 (br s,2H,NH2), 1.52
(s,121H?4CH3-C-S) ppm. 13C NMR (in CDC13): ~ 174.3 (s,2NH-CO),
158.3 (s,2Cl-Ar-OCH3), 129.6 (d~2c2&c6-Ar-ocH3)~ 128.9 (s,2C4-
Ar-OCH3), 113.6 (d,2C3&C5-Ar-OCH3), 54.8 (q,2CH30), 54.2 (t,N-
CH2CH2-NH2), 53.6 (t~2N-cH2cH2-NHco)~ 49.4 (s,2S-C-CH3), 38.5
10 (t,2N-CH2CH2-NHCO), 37.6 (t,N-CH2CH2-NH2), 33.6 ~t,2S-CH2-Ar),
26.4 (q,4CH3-C-S) ppm. MS (rn/z,CI): 591(31, MH+). IR (CDC13):
3770, 2930, 2830, 1655, 1510, 1245, 1170, 1030, 830 c~n~l.
N,N"-Bis[2-((4-methoxybenzyl)thio)-2-methyl-propionyl]-N'-(2-
15 isothiocyanoethyl)-diethylenetriamine (25)
To a round bottom flask co~ lin~ the aniline 24 (28.8
mg, 0.0487 mmol) and dichlorome~ane (10 mL) was added 0.2211 M
solution of thiophosgene (0.22 mL, 0.0509 mmol) in dichloromethane.
The heterogeneous reaction ml~lUlc~ was stirred at room tempel~Lu
20 for 1 hour and the solvent was removed in vacuo. Flash
chromatography of this crude product using 100% EtOAc gave the
isothiocyanate 25 as a clear oil (18.9 mg, 61.3% yield).
lH NMR (in CDC13): ~ 7.17 (d,j=8.5 Hz,4H,2H3&H5-Ar OCH3), 7.07
(m,2H,2NH-CO), 6.82 (d,j=8.5 Hz,4H,2H2&H6-Ar-OCH3), 3.78 (s,6H,
25 2CH30), 3.70 (s,4H,2S-CH2-Ar), 3.49 (t,j=5.9 HZ~2H~N-cH2cH2
NCS), 3.21 (q,j=6.2 HZ~4H~2N-cH2cH2-NHco)~ 2.78 (t,j=5.9
HZ~2H~N-cH2cH2-Ncs)~ 2.58 (t,j=6.4 Hz,4H, 2N-CH2CH2-NHCO),
1.54 (s,12H,4CH3-C-S)ppm. MS (m/z,CI): 633(25, MH~). IR
(CDC13): 3370, 2950, 2920, 2850, 2100, 1720, 1655, 1610, 1510, 1245,
30 1170, 1030, 830 cm~l .
CA 02202382 1997-04-1o
WO 96/11954 PCT/CA95100573
- 20 -
N,N"-Bis[2-((4-methoxybenzyl)thio)-2-methyl-propionyl]-N'-(2-[[[4-
methoxyphenyl)amino]thioxomethyl]aminoethyl]-diethylenetri~mine
(26)
A solution of the isothiocyanate 25 (18.9 mg, 0.02986
5 mmol), triethyl~mine (7.26 mg, 0.07175 mmol), anisidine (6.9 mg,
0.05602 mmol), and CHC13 (15 mL) was refluxed for 3 hours and the
solvent was removed in vacuo. Flash chromatography of this crude
product using 100% EtOAc gave the ~ni~icline derivative 26 as a clear
oil (19.8 mg, 87.6% yield).
lH NMR (in CDC13): o 8.17 (brs,lH,NH-CS-NH-Ar), 7.28 (d,j=8.8
Hz,2H,H2&H6-Ar-N), 7.15 (d,j=8.6 Hz,4H,2H3&H5-Ar-OCH3), 7.06
(brs,lH,NH-CS-NH-Ar), 7.06 (t,j=5.7 Hz,2H,2NH-CO), 6.90 (d,j=8.9
Hz,2H,H3&H5-Ar-N), 6.82 (d,j=8.6 Hz,4H,2H2&H6-Ar-OCH3), 3.79
(s,3H,CH30-Ar-N), 3.77 (s,6H,2CH30-Ar-CH2), 3.67 (s,4H,2S-CH2-
15 Ar), 3.61 (q,j=5.3 HZ~2H~N-cH2cH2-NHcs)~ 3.14 (q,j=6.2 Hz,4H,2N-
CH2CH2-NHCO), 2.69 (t,j=5.6 Hz~2H~N-cH2cH2-NHcs)~ 2.52
(t,j=6.3Hz,4H 2N-CH2CH2-NH-CO), 1.51 (br s,12H,4CH3-C-S) ppm.
13C NMR (in CDC13): o 181.9 (s,N-CS-N), 175.2 (s,2NH-CO), 158.8
(s,3Cl-Ar-OCH3), 130.0 (s,Cl-Ar-N), 130.0 (d,2C2&C6-Ar-OCH3),
20 129.2 (s,2C4-Ar-OCH3), 127.0 (d,C2&C6-Ar-N), 114.5 (d,C3&C5-Ar-
N), 114.2 (d,2C3&C5-Ar-OCH3), 55.4 (q,3CH30), 54.4 (t,2N-
CH2CH2-NHCO), 52.9 (t,N-CH2CH2-NHCS), 50.1 (s,2S-C-CH3), 42.8
(t,N-CH2CH2-NHCS), 38.3 (t,2N-CH2CH2-NHCO), 34.2 (t,2S-CH2-
Ar), 28.3 (q,4CH3-C-S) ppm. MS (m/z,FAB): 756(4.2, M+). IR
25 (CDCl3): 3320, 2960, 1720, 1655, 1510, 1290, 1245, 1030 cm-1.
Part 4: Labelin~ of an N~S2 Chelate
Synthesis of acyclic dimercapto-N.N.N-tris(2-aminoethyl)amine
tion of N,N-dimethyl-2-(3,3,5,11,13,13-hexamethyl-1,2-dithia-
5.8.11-triaza-cyclotridecan-8-yl) ethylamine (27)6
To 2-(3,3,13,1 3-tetramethyl- 1 ,2-dithia-5,8, 1 l-triaza-
cyclotridecan-8-yl) ethy1~mine 4 (202.6 mg, 0.63 mmol), in a 25 mL
-
CA 02202382 1997-04-10
WO 96/11954 PCT/CA95100573
round bottom flask, concentrated formic acid (4 mL) and 37~o
form~lt1ehyde solution (3.7 mL) was added. The mixture was heated
under reflux and stirred for 25 hours. The solution was ~en cooled to
room temperature and extracted three times with ether (SO mL). The
S aqueous phase was rendered basic (~10-11) by ~1flin~ 27% ammonium
hydroxide and extracted with methylene chloride (4 x SO mL). The
organic phase was washed successively with water (SO ~L) and
saturated sodium chloride solution (2 x SO mL), dried with anhydrous
magnesium sulfate, and filtered. The solvent was removed under
vacuum to give 176 mg (74%) of tetramethylated tetr~mine dlisulfide
27 as a yellow oil. The crude product was purified by flash
chromatography (silica gel) using a mixture of me~ylene chlonde,
methanol, and ammonium hydroxide (84.5/lSfO.5) as eluent. Pure N,N-
dimethyl-2-(3,3,5,11,13,13-hexamethyl- 1,2-dithia-5,8,11 -triaza-
cyclotridecan-8-yl) ethyl~mine 27 (108 mg) was recovered from the
purification process.
N,N-dimethyl-2-(3,3,5,11,13,13-hexamethyl- 1,2-dithia-5~8,11 -triaza-
cyclotridecan-8-yl) ethylamine 27
lH NMR (300MHz,in CDCl3): ~ 2.71 (t,J=6Hz,4H,-N-CH2-
CH2-N(CH3)-), 2.64 (s,4H,-N-CH2-C(CH3)2-S-), 2.62 (t,J=6Hz,4H,-N-
CH2-CH2-N(CH3)-), 2.52 (m,2H,-CH2-CH2-N(CH3)2), 2.46 (m,2H,-
CH2-CH2-N(CH3)2), 2.37 (s,6H,-CH2-N(CH3)-CH2-), 2.~5 (s,6H,-CH2-
CH2-N(CH3)2) and 1.28 (S~l2H~-s-c(cH3)2-) ppm. 13C NMR
(75MHz,in CDC13): ~ 74.8, 67.7, 57.6, 57.2, 53.9, 51.7, 51.3, 45.9,
45.1 and 26.9 ppm. MS (EI; m/z): 376(4,M+), 377(2,M++l),
378(0.6,M++2), 318(1,M+-((CH3)2N-CH2-)), 262 (3), 255 (1), 246(1),
187(7), 156(9), 144(8), 133(9), 130(16), 113 (17), 101(27), 99(21),
98(12), 72(32), 71(28), 70(42), 58(100), 56(13), 55(13), 43(31), and
42(38).
CA 02202382 1997-04-1o
WO 96/11954 PCT/CA95/00573
- 22 -
Reduction of N,N-dimethyl-2-(3,3,5,1 1,13,13-hexamethyl-1,2-dithia-
5~8~11-triaza-cyclotridecan-8-yl) ethylamine (27)6
In a 50 mL three-necked flask equipped with a gas inlet
device, a magnetic stirring bar, a stopper and a dry ice condenser 18.9
S mg (0.5 ~mol) of the tetramethylated tetraamine disulfide (~) was
introduced. The condenser was filled with acetone and dry ice, and the
contents of the flask was cooled with a dry ice bath. Liquid ammonia
was then introduced in the flask until about 25 mL was added. The
solution was stirred for several minutes and 36 mg (1.6 mmol) of
10 sodium was added. The mixture was stirred for thirty minutes, and
ammonium chloride (172 mg) was cautiously added. The cooling bath
was then removed to allow the ammonia to evaporate. The white
residue was dissolved with 45 mL of Milli-Q water and concentrated
hydrochloric acid was added to brougth the pH of this solution to 1.
15 The mixture was extracted with ether (3 x 50 mL) and the pH was
raised to about 9 with 37% ammonium hydroxide. The basic solution
was extracted four times with ether (50 mL). The organic phases were
combined, washed sllccessively with water (1 x 30 mL) and brine (2 x
30 mL). The ethereal phase was dried with anhydrous m~gn~cillm
20 sulfate and ~lltered. The solvent was then removed under re~ cerl
pressure to yield 16 mg of a yellow oil. This oil was used as it is to
perform the Tc-m99-technetium labeling study.
Note: The HPLC analysis of this oil has shown that it is a mixl~lre
of two component in a ratio of 2.5/1: the starting m~ri~l
and the reduced compound. The HPLC conditions used for
analysis were as followed: column ~milton PRP-I 10 ,um
analytical, gradient of 10 to 50% of ~etonitrile (with 0.1%
TFA) in 40 minutes. Retention times: tetramethyl
tetr~mine disulfide= 7.73 min; tetramethyl tetr~mine
dithiol= 21.55 min.
,
CA 02202382 1997-04-10
WO 96/11954 PCTICA95/00573
- 23 -
Tc-m99-technetium labeling of a mixture obtained from the reduction
of N,N-dimethyl-2-(3,3,5, 1 1 , 13, 1 3-hexamethyl- 1 ,2-dithia-5,8, 11 -
triaza-cyclotridecan-8-vl) ethvlamine
5 Experiment A:
In a l.S mL plastic conical vial 30 ~L of a methanolic
solution (~2 ~lg/~L) of the mixture obtained from the reduction of N,N-
dimethyl-2-(3,3 ,5,1 1,13,1 3-hexamethyl- 1 ,2-dithia-5,8, 1 1 -tnaza-
cyclotridecan-8-yl) ethylamine, 0.1 mL Tc-m99-technetium
10 glucoheptonate (prepared by the reconstitution of a vial from a
FROSSTIM~GE GLUCOHEPTONATE kit with 1 mL of sodium
per99mtechnetate) were introduced. This solution was stirred for 20
seconds with a vortex mixer and heated at 63C for 2 1/2 hours. The
mixtllre was then analyzed by ITLC@ (Instant Thin Layer
15 Chromatography) ran in acetone and in normal saline and demonstrated
that 90% of the radioactivity was tagged with the chelate. HPLC
analysis* of the labeling mixture showed the presence of three
radioactive products. The first radioactive compound was identi~led as
99mtechnetium glucoheptonate (RT:2.59 min; 19%) while the two
20 others are 99mTc-N3S2 chelated species (RT:20.72 and 23.08 min; 68
and 8% respectively).
Experiment B:
- In a l.S mL plastic conical vial 30 ,uL of a methanolic
25 solution (~2 ~Lg/,uL) of the mixtllre obtained from the reduction of N,N-
lim~thyl-2-(3,3,5,1 1,13,13-hexamethyl-1,2-dithia-5,8,1 l-triaza-
cyclotridecan-8-yl) ethyl~min~, 0.2 mL 99mtech~etium glucoheptonate
(prepared by the reconstitution of a vial from a FROSSTrMAGE
GLUCOHEPTONATE kit with 1 mT of Tc-m99-sodium per techn~ te)
30 were introduced. This solution was stirred for 20 seconds with a vortex
mixer and he~te(1 at 65C for 1 hour. The mixtnre was then analyzed
by ITLC9~ (~n.~t~nt Thin Layer Chromatography) ran in acetone and in
normal saline and demonstrated that 85% of the radioactivity was
tagged with the chelate. HPLC analysis of the labeling mixtnre showed
CA 02202382 1997-04-10
WO 96/11954 PCTICA95/00573
- 24 -
the presence of three radioactive products. The first radioactive
compound was identified as Tc-m99technetium glucoheptonate (RT:2.72
min; 30%) while the two others are 99mTc-N3S2 chelated species
(RT:20.70 and 23.08 min; 56 and 8~o respectively).
s
g[: Gelman SG chromatography paper.
*: The high performance liquid chromatography analysis was
performed with a Waters 625LC instrument equipped with a
Hamilton PRP-I 10 ,um analytical column using a 10 to 50%
acetonitrile (+TFA) gradient in 40 minutes and a flow rate of
1 mL/min.
1. W.C. Still, M. Kahn, and A. Mitra, J. Org. Chem., 43, 2923-
2925 (1978).
2. a) J.J. D'Amico and W.E. Dahl, J. Org. Chem., 40, 1224-1227
(1975) b) K. Masao, T. Ueno, K. Kojima and Y. Morimoto,
(Nippon Shokubai K~ kll Kogyo Co., Ltd.) Jpn. Kokai Tokkyo
Koho JP 63,222,155 [88,222,155].
3. E.A. Bayer, B. Haya and M. Wilchek, Biochem. and Biophys.
Res. Commun., 138, 872-879 (1986).
25 4. M.R.A. Pillai, J.M. Lo, C.S. John and D.E. Troutner, Nucl. Med.
Biol., 19, 791 (1992).
5. Y. Ohmomo, L. Francesconi, M.P. Kung and H.F. Kung, J. Med.
Chem., 35, 157 (1992).
6. M. Apparu, S. Drouillard, J.P. Mathieu, A. DuMoulinet
D'Hardemare, R. Pasqualini, and M. Vidal, Appl. Radiat. Isot., 43
(5), 585-596 (1992).