Note: Descriptions are shown in the official language in which they were submitted.
CA 02203155 1997-04-18
WO 96/12727 PCT/FR95/01388
DERIVES HYDROSOLUBLES D'EPIPODOPHYLLOTOXINE,
LEUR PROCEDE DE PREPARATION, LEUR UTILISATION A TITRE
DE MEDICAMENT, ET LEUR UTILISATION DESTINEE AUX
TRAITEMENTS ANTICANCEREUX.
Parmi la classe des épipodophylloïdes, certains composés comme l'étoposide
ou le' téniposide, dérivés de l'épipodophyllotoxine, composés hémi-
synthétiques, issus de lignane naturel, sont utilisés dans la préparation de
médicaments pour traiter de nombreuses formes de cancer. Ils sont
io considérés, actuellement comme des produits majeurs de l'arsenal
thérapeutique.
Parmi les différents cancers traités par ce type de composés on peut citer le
cancer du poumon à petites cellules, les tumeurs embryonnaires,
neuroblastomes, cancer du rein, lymphomes, maladie de Hodgkin, leucémies
is aigües, et méme cancer du sein. L'étoposide est avantageusement utilisé en
association avec d'autres produits anticancéreux et en particulier les dérivés
du Platine comme le Cis Platine.
L' inconvénient important de ce dérivé, et de même pour son dérivé
apparenté le téniposide, est son manque d'hydrosolubilité. Il n'existe pas
2o de formes commercialisées hydrosolubles pour des administrations
intraveineuses. Au contraire la mise en solution est faite actuellement dans
des solvants partiellement non aqueux, nécessite une administration en
perfusion lente et provoque certains effets indésirables voire même toxiques.
Il existe ainsi un besoin de formes hydrosolubles pour des produits dérivés
~s de cette classe de composés, afin d'améliorer l'administration chez la
malade, ainsi que sa biodisponibilité. La présente invention concerne donc
des dérivés de l'étoposide hydrosolubles grâce à la présence de groupements
fonctionnels phosphates ou carboxylates dont les sels d'addition organiques
ou minéraux forment des entités solubles dans l'eau. Cette formulation
~o aqueuse présente l'avantage d'être moins toxique et plus facilement
administrable que les formes commercialisées actuellement.
La préparation de dérivés de l'étoposide a suscité de nombreux travaux et de
nombreux brevets, et en particulier des dérivés diesters 2", 3" et triesters
2",
3", 4' d'étoposide ont été revendiqués dans le brevet FR ~2 699 535-Al.
s Certains de ces dérivés ont m~~ntré une activité égale ou supérieure à
l'étoposide et une moindre toxici;é. Une amélioration supplémentaire est
apportée maintenant grâce à une solubilité aqueuse qui leur confère une
CA 02203155 1997-04-18
WO 96/12727 PCT/FR95/01388
2
facilité d'administration et laisse présager une biodisponibilité accrue par
un
meilleur passage dans les différentes membranes biologiques.
La littérature mentionne des brevets relatifs à des composés apparentés à
l'étoposide cherchant à améliorer l'hydrosolubilité, notamment (US
s 4 904 768, EP 0 369 369-A2, EP 0 196 618-A1, EP 0 320 988, EP
0 415 453-A2).
L'avantage de modules la solubilité aqueuse des composés par des
groupements phosphates a été utilisé dans quelques cas favorablement soit
dans le domaine des anticancéreux WO 8707609, soit dans le domaine des
lo analgésiques (BE 893,563) malgré tout rien ne laisse prévoir que le composé
ainsi obtenu puisse conserver totalement une activité biologique intéressante
en lui-même ainsi que celle du dérivé dont il est issu.
Il a été trouvé que les dérivés phosphates et carboxylates possèdent une
hydrosolubilité permettant l'administration par voie injectable, et de plus
I s manifestent une activité anticancéreuse améliorée par rapport à
l'étoposide.
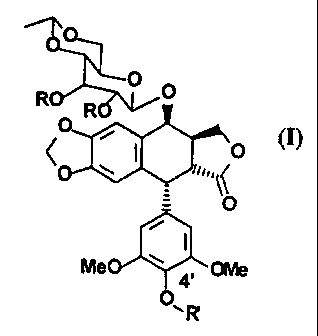
La présente invention concerne donc un composé de formule générale I
0
0
R R
O
I ' ~o
i~"-'~
O
i
f
Me ~ 4, OMe
O
I
2o dans laquelle R' représente soit un atome d'hydrogène, soit un groupement
phosphate monoester, soit un groupement carbamate de type -CO-N (R1R2)
où N (R1R2) représente des groupements aminodiacétiques et une amine
polycyclique comme la 3-amino-quinuclidine, soit un groupement acyle de
type phosphonoacétic H203P-CH2-CO, soit un radical R,
2s R représente un groupe acyle de formule
A-Z-CH2-CO
CA 02203155 2007-02-02
3
où Z représente un atome d'oxygène, de soufre, un groupement 502, un alkylène
linéaire
ou ramifié en C1~, dans ce cas A représente un noyau phényl substitué ou non,
à la
condition que:
- dans le cas où R' = R, c'est-à-dire les dérivés triacylés, A représente un
noyau
aromatique possédant une fonction salifiable, sauf 4-hydroxy-phényle,
- dans le cas où R' ~R, A représente un reste benzyl, naphtyl, hétéroaryl,
phényl substitué
ou non, dans ce cas le phényl pouvant être substitué une, deux, trois, quatre
ou cinq
plusieurs fois quelle que soit sa position sur le noyau aromatique par des
groupes
identiques ou différents choisis parmi les groupes tels que halogènes, F, Cl,
Br, alcoxy
linéaire ou cycliques en CI~, alkyl C,-C6, méthylène dioxy, OCF3, CF3, N02,
CN, OCHZ
Aryl, OH, OP03H2, CHZ P03H2, OCHZC02H, COOH, CH2COOH, COCH3, CHO,
A-Z peut également représenter un groupement OCH2COZH, SOZCHZCOOH, P03H2,
ainsi que leur sel avec des acides ou des bases, minérales ou organiques,
thérapeutiquement acceptables et hydrosolubles,
à l'exception des composés pour lesquels R' = H, et Z représente un atome
d'oxygène ou
un atome de soufre, et
A représente un aryle, choisi parmi les radicaux phényle, phényle-alkyle,
linéaire ou
ramifié en C1-C4, naphtyle, et ces mêmes radicaux substitués par un à trois
substituants
choisis parmi les radicaux alcoxy, linéaire ou ramifié en C1-C4,
éventuellement
perhalogéné avec des atomes de chlore ou de fluor, alkyle linéaire ou ramifié
en C1-C4, et
les atomes d'halogène, en particulier le chlore ou le fluor.
L'invention s'étend à un composé de formule générale I
QO
RO' ~O
RO 'O
".,...
0 0
n~eo~ ~'''~onn~
O~ R'
1
CA 02203155 2007-02-02
3a
dans laquelle R' représente un atome d'hydrogène, soit un groupement phosphate
monoester, soit un groupement carbamate de type -CO-N(R1R2) où N (R,R2)
représente
des groupements aminodiacétiques ou une amine polycyclique, soit un groupement
phosphonoacétique H203P-CH2-CO , soit CO-CH2S02-CH2COOH, soit COCH2-OCH2
COOH, soit un radical R,
R représente un cyclohexyloxyacétyl ou un groupe acyle de formule
A-Z-CH2-CO
où
Z représente un atome d'oxygène, de soufre, un groupement 502, un alkylène
linéaire ou
ramifié en C1_4, A représente un noyau phényl, naphtyl ou benzyl non substitué
ou
substitué jusqu'à substitué cinq fois par des groupes identiques ou différents
choisis parmi
les groupes halogènes, F, Cl, Br, alcoxy linéaire ou cycliques en C1_6, alkyl
C,-C6,
méthylène dioxy, OCF3, CF3, N02, CN, OH, OP03H2, CH2 P03H2, P03H2, OCH2C02H,
COOH, CH2COOH, COCH3, CHO.
De manière avantageuse les composés de formule générale I seront choisis avec
R'
représentant un groupe phosphate monoester (P03H2), carbamate CONR,R2 et NR~R2
représentant un groupement aminodiacétique ou une amino-3 quinuclidine, R'
représentant également un groupe phosphonoacétique et leurs sels.
De préférence, R est choisi parmi les radicaux:
phénoxyacétyl, 3,4-méthylènedioxyphénoxyacétyl, 4-méthoxyphénoxyacétyl, 4-
hydroxyphénoxyacétyl, 4-phosphonooxyphénoxyacétyl, 4-carboxyméthyl-phénoxy
acétyl, 4-carboxyméthoxyphénoxyacétyl, 4-carboxyphénoxyacétyl, 4-
trifluorométhylphénoxyacétyl, 4-trifluorométhoxyphénoxyaétyl, 4-
cholorophénoxyacétyl,
4-nitrophénoxyacétyl, 4-fluorophénoxyacétyl, cyclohexyloxy-
CA 02203155 2005-03-30
WO 96/I2727 PCT/F'R.95101388
4
acétyl, phénylsulfonylacétyl, pentafluorophénoxyacétyl, 2 et 4 formyl-
phénoxyacétyl, 4-cyanophénoxyacétyl.
La prësente invention concerne également les procédés dé préparation d'un
composé de formule I, représentés sur le schéma 1 (voie A), pour lequel on
fait réagir un intermédiaire glycosylé de formule générale II.
'~C~-~~ O H
RO Ra 11
avec un intermédiaire de formule générale III pour fournir un intermédiaire
1 o IV
oH
0
~ j ~o
0
M e ~ atr~e
0
'R4
R ayant les si~nitlcations précédentes. R~ est un groupe protecteur par
p exemple le benzyloxycarbonyl au un restè earbamate. Ce procédé de
prépâration est décrit dans le brevet antérieur FR 2 699 53~ à l'exemple I7.
Pour fournir un composé de formule IV dans lequel R4 est un groupement
protecteur et R dé¿~mi précëdemment. Ce dérivé IV est dëprotégé sur sa
position 4' (R.4), soit par hydrogénol~~se, soit par hydrolyse faiblement
?o basique pour fournir Ie dérivé I (R'=H). II est également possible de
préparer les composés de formule I où R' =R par cette méthode, en utilisant
l'intermédiaire de formule III dans lequel R~ représente un groupe acyl R,
cette méthode est décrite également dans Ie brevet antérieur FR 2 699 53~ à
l'exemple 1. Selon les compatibilités des substituants R du glucosyl, il est
'S possible également de synthétiser les composés de formule I à partir de
l'étoposide lui-méme (voie B).
Dans une première étape l'étoposidé peut étre protégé en position 4' (I~,)
I~ar
un groupement R~.=benzyloxycarbonyl, ou par un groupement quinuclidine
carbamate (R~=CCNH3-quinucliàinyl) obtenu par réaction successive du
CA 02203155 1997-04-18
WO 96/12727 PCT/FIt95/01388
phosgène suivi de l'amino-3-Quinuclidine sur l'étoposide, pour fournir
l' intermédiaire V .
D'une façon générale les intermédiaires V sont acylés par des chlorures
d'acide dérivés des groupes R définis précédemment, (formés par action du
s chlorure d'oxalyl) en présence de pyridine, dans le chlorure de méthylène
à bassé température, sous réserve que les autres fonctions du groupe R
soient inertes dans ces conditions, sinon, les substituants phénoliques,
carboxyliques ou phosphoriques sont protégés sous forme d'éthers ou
d' esters benzyliques respectivement, ce qui permet le déblocage, au stade
~o suivant de la synthèse, par hydrogénolyse (V donnant I R' =H). Les dérivés
pour lesquels R' =R sont préparés à partir de l'étoposide par triacylation sur
les positions 2", 3" et 4' (voie C).
Dans le cas des dérivés fonctionnels R Sensibles aux cnnrtitinnc
d'hydrogénolyse comme par exemple mais non exclusivement, la présence
~ s de Cl ou de N02, le groupement protecteur choisi sur la position 4' peut
être
un dérivé carbamate de type CONH-3-quinuclidinyl ou d'un carbonate ou
d'un ester de faible poids moléculaire, comme les chloroacétates qui peuvent
se cliver ultérieurement entre autres dans des conditions faiblement basiques
comme une solution aqueuse de bicarbonate de sodium à basse température
2o sans influencer la stéréochimie de la lactone trans.
L'étape ultime I (R' =H) donnant I (R'~H) consiste en une phosphorylation
pour former un phosphate monoester du ou des phénols avec POC13 en
présence de pyridine suivi d'une hydrolyse lente en milieu aqueux acide.
L'obtention des dérivés possédant en position 4' un reste carbamate
2s diacétique de type R' - CON (CH2 C02H)2 se préparent par action du
phosgène sur le composé de formule I (R' = H) pour former l'intermédiaire
chlorocarbonate non isolé (R' =COC1) puis en le faisant réagir avec le
diester benzylique de l'acide aminodiacétique suivi d'une hydrogénolyse
pour libérer ensuite les fonctions acides sous forme libre.
~o Les dérivés phosphonoacétiques en position 4' s'obtiennent en faisant
réagir
le phénol libre en cette position avec le chlorure d'acide de l'acide
. diéthylphosphonoacétique (Synthesis 1978, 131) ou l'acide dibenzyl
prosphonoacétique (Tet. Let. 1974, N°9, 711), puis en hydrolysant les
fonctions esters phosphoriques par le bromure de triméthyl silyle, en
~s présence de pyridine dans l'acétonitrile, dans le cas des esters
éthyliques, ou
par hydrogénolyse, dans le cas des esters benzyliques. Les dérivés de
CA 02203155 1997-04-18
. . , ", ,
. , . , , ,
., ". ~, " ~,
6
formule I où R' =R, c'est-à-dire possédant la même substitution acyle sur les
positions 2", 3" et 4' s'obtiennent, par la voie C, par triacylation de
l'étoposide lui-même avec des groupements acyles AZCH2C0 ayant une
fonction carboxylique sur le groupement A défini précédemment, protégée
s par exemple sous forme d' ester benzyl ique, que l' on déprotège par
hydrogénolyse ultérieurement.
Les dérivés carboxyliques, ou les dérivés phosphates ou phosphonates ainsi
obtenus sont salifiés dans l' eau en présence éventuellement d' un cosolvant
organique, par addition de bases organiques ou minérales en proportion
)o stoechiométrique par rapport aux acidités en présence, et par exemple avec
le N-méthylglucamine, la triéthanolamine, la lysine, etc.
Les sels amorphes ou cristallisés sont obtenus par simple lyophylisation.
Schéma 1
Voie A Voie B Voie C
II III Etoposide Etoposide
~O ~O
O O--
~O O
R R O HO HO O
O ~ O
O
nr,,,! ~~ ~ ~ rrr.,,lO
\\O \'O
i i
Me OMe Me OMe
IV OR, V OR,
1) Diacylation
Déprotection 2) Déprotection
I (R'=H)
I (R'-H) Triacylation
R'=R
r ; C~,! çlF~
CA 02203155 2007-02-02
6a
L'invention s'étend également à la préparation d'un composé de formule
générale I, telle
que définie précédemment, caractérisé en ce que l'on fait réagir fétoposide
protégé en
position 4' par un groupe benzyloxycarbonyle ou quinuclidinecarbamate de
formule V
~O
~7
HO
HO
O i C?
~0.,. '...
O
'',., ~'
~''l'~Or ~~rle
4'
V
dans laquelle R4 est tel que défini précédemment, avec un réactif acylant de
type A-Z-
CH2C0, dans lequel A et Z sont tels que définis précédemment, pour conduire
après
hydrogénolyse ou hydrolyse au composé de formule I tel que R' = H.
L'invention s'étend également à l'utilisation d'un composé de formule I de la
présente
invention pour la préparation d'un médicament destiné au traitement
anticancéreux.
CA 02203155 1997-04-18
wo 96ni727
7
PGT/FR95/01388
L~lesure de l'hvdro~colubilitP
La solubilité dans l'eau des sels des dérivés phosphates ou carboxylates par
addition d'amines organiques physiologiquement acceptables comme par
exemple la N-méthylglucamine, la triéthanolamine, la lysine, ou les sels avec
s des cations minéraux comme le sodium, obtenus sous forme lyophilisée ou
par addition extemporanée d'une base sur le composé acide libre, a fourni
les résultats suivants à titre d'exemple.
io
Composés Sel Solubilité % (poids/volume)
exprimé en g pour 100m1 d'eau
Exemple N-mthylglucamine 0,5
1
Exemple N-mthylglucamine
3
Exemple N-mthylglucamine 10
4
Exemple N-mthylglucamine 10
6
Exemple trithanolamine 20
6
Exemple lysine 20
6
Exemple Sodium 1
6
Exemple N-mthylglucamine 10
7
Exemple N-mthylglucamine 10
11
Exemple N-mthylglucamine 10
21
Exemple N-mthylglucamine 10
26
Exemple N-mthylglucamine 0,5
27
Les dérivés ainsi préparés sont stables dans les conditions usuelles de pH
neutre et acide et de température. Les dérivés phosphates sur la position 4'
ont une stabilité chimique pour pouvoir se prêter aux différentes
t s formulations pharmaceutiques.
CA 02203155 1997-04-18
w0 96/12727
8
PCT/FR95/01388
Les molécules ont été testées in vitro en expérimentation biologique et ont
montré leur intérêt en tant qu'agents anticancéreux dans les tests suivants.
La mesure de l'inhibition de l'activité de la topoisomérase II est faite selon
s le protocole décrit dans la littérature : "Nuclear topoisomérase II levels
correlàte with the sensitivity of mammalien cells to intercalating Agents and
Epipodophyllotoxins". LD. HICKSON et coll., J. Biol. Chem. (1988), 263,
17724-1772.
Cette mesure a fourni les~résultats suivants.
to
Composs Test d'inhibition de l'activit de
la
topoisomrase II (ED$p M)
Etoposide 5.6 . 10-5
Etopofos > 10_4
Exemple 1 5.6 . 10-6
Exemple 3 3.2 . 10-7
Exemple 4 1.8 . 10-6
Exemple 6 3.2 . 10-7
Exemple 7 5.6 . 10-5
Exemple 11 5.6 . 10-6
Exemple 21 5.6 . 10-6
Exemple 26 5.6 . 10-6
Exemple 27 7.6 . 10-7
La comparaison de l'étoposide avec son analogue soluble 4'-phosphate
~s étopofos (US-4 904 768) montre une perte d'activité it~ vitro. Ici les
composés de l'invention se trouvent aussi, sinon plus, actifs que l'étoposide.
Les groupements R et R' définis précéderr~ment confèrent aux composés de
l' invention une augmentation d' un facteur 10 à 100 de l' inhibition de
l'activité enzymatique in vitro par rapport à l'étoposide.
CA 02203155 1997-04-18
WO 96/12727 PCTlFR95/01388
9
On peut apprécier, au vu de ces résultats, l'intérêt de composés ayant une
activité anticancéreuse égale ou supérieure à celle de l'étoposide, et une
moindre toxicité, sur différentes formes de cancers comme en particulier le
cancer du poumon à petites cellules, les tumeurs embryonnaires, les
s neuroblastomes, le cancer du rein, les tumeurs pédiatriques, les lymphomes
hodgküniens et non hodgkimiens, les leucémies aigües, les choriocarcinomes
placentaires, les adénocarcinomes mammaires.
Ces dérivés peuvent être utilisés également dans les pathologies induites par
le papilloma virus humain ainsi que l'arthrite rhumatoïde associée ou non à
lo des pathologies cancéreuses.
De plus ces dérivés peuvent être utilisés pour augmenter l'efficacité
thérapeutique des composés inhibiteurs de topoisomérase II et en particulier
le traitement des tumeurs normalement réfractaires à la thérapeutique
usuelle, c'est-à-dire les cancers colorectaux et les mélanomes. De plus on
t s peut apprécier l' intérêt de ces produits présentant, d'une part une
importante
hydrosolubilité qui permet une administration par voie intraveineuse et orale
aisée, et d'autre part une meilleure biodisponibilité que celle de
l'étoposide.
La présente invention concerne également les compositions pharmaceutiques
comprenant au moins un composé de formule générale I selon l' invention et
?o un excipient approprié.
Les compositions pharmaceutiques peuvent ëtre présentées de façon adaptée
pour l'administration par voie injectable ou par voie orale sous forme de
capsule, de gélules, de comprimés à la posologie de 2 à 200 mg/m2 par voie
injectable et de 5 à 400 mg/m2 par 24 h pour la voie orale.
2s A titre d'exemple et de façon non limitative les exemples suivants
décrivent
la préparation des composés de l' invention
R - ~ ~ ~~O , R'= phosph
F~1~1- formule I
4'-déméthyl-4-O-(2,3-bis-phénoxyacétyl-4,6-éthylidène-~3-D-glucosyl)-
~o épipodophyllotoxine-4'-désoxy-4'-phosphate.
A une solution de 4'-déméthyl-4-O-(2,3-bis-phénoxyacétyl-4,6-ethylidène-~i-
D-glucosyl)-épipodophyllotoxine (lg, 1,16 mmole) dans 50 ml de THF
à 10°C sont ajoutés 0,22 ml (2,33 mmoles) de P~~C13 puis 0,~ ml
(3,5 mmoles) de triéthylamine. L'agitation est maintenue 30 mn à cette
~s température. L'hydrolyse est effectuée ensuite par addition. dans le milieu
de
CA 02203155 1997-04-18
WO 96/12727 PCT/FR95/01388
20 ml d'acide chlorhydrique N puis agité une nuit à température ambiante.
Le milieu réactionnel est extrait avec l'acétate d'éthyle pour obtenir le
dérivé
phosphate cristallisant dans l'éther isopropylique avec un rendement
quantitatif.
s Les caractéristiques sont les suivantes
F°C ~ 175°C Anal. C45H45020P : 1,SH20 ; PM = 963,831
C H
Calc. % 56,07 5,02
Tr. % 56,18 4,73
lo Spectre de Masse (FAB) m/e 959 (M++Na)
1H 200MHz RMN CDC13 8 1,30 (3H, d, J = 4,4Hz, Hg~~) ; 2,9 (1H, m,
H3) ; 3,1 (1H, m, H2) ; 5,0 (1H, dd, J = 8,8Hz, H2«) ; 5,3 (1H, dd, J ---
9,2Hz, H3~~) ; 5,5 (1H, s, OCHAO) ; 5,7 (1H, s, OCHBO) ; 6,25 (2H, s,
H2~H6~) ; 6,44 (1H, s, Hg).
Is IR v (KBr) 2941, 1774, 1599, 1487.
Sel de N-méthyl glucamine
Le dérivé phosphate précédent est mis en suspension dans l'eau et on ajoute
2 équivalents de N-méthyl glucamine en solution 0,1 M dans l'eau. La
solution est agitée sous ultrasons et diluée à 200 ml. Après filtration la
2o solution est glacée puis lyophilisée pendant 12 h. Le résidu est alors
repris
dans l'acétone et cristallisé, filtré séché pour fournir 350 mg d'un solide
blanc F ~ 135°C.
Anal. C59H79N2O30P, H20 PM = 1345, 256
C H N
?s Calc. 52,68 6,06 2,08
Tr. 52,69 5,86 1,68
IR v (KBr) 3426, 1772, 1599, 1487.
RMN 1H 200MHz CDC13 8 1,22 (3H, d, J = 4,8Hz, Hg~~) ; 2,3 (6H, s, N
CH3), 2,6-3,0 (2H, m, H2-H3) ; 5,36 (1H, dd, J = 7,8Hz, H3~~) ; 5,74
30 (1H, s, OCHAO) ; 5,97 (1H, s, OCHBO) ; 6,15 (2H, s, H2~-H6~) ; 6,5
(1H, s, Hg) ; 7,10 (1H, s, HS) ; 6,65 (2H, d, J = 8Hz, Ar Ortho) ; 6,8 à
6,96 (2H, d, J = 8Hz Ar ortho et 2H, t, J = 7Hz, Ar para); 7,24 (4H, m,
Ar meta).
Sel de Sodium
~s Le dérivé phosphate précédent est agité en solution dans l'acétone avec une
résine échangeuse d'ion (Dowex 50 x 8 - 100) préparée par élution avec la
CA 02203155 1997-04-18
WO 96/12727 PCT/FR95101388
soude N. Le milieu est allongé avec de l'eau, filtré et concentré. Le résidu
aqueux est lyophillisé pour fournir le di-sel de sodium
F° ~ 190°C Anal C45H43Na2020P3.3H20 PM = 1040,208
C H
s Calc. 51,96 4,81
Tr. 51,56 4,51
Par la même méthode que celle de l'exemple 1 mais en utilisant les
composés intermédiaires de formule I (R' = H) correspondants les nouveaux
dérivés suivants ont été préparés
~o
R = , R'=phosph
Exem~ - formule I
4'-déméthyl-4-O-(2,3-bis-cyclohexyloxyacétyl-4,G-éthylidène-~3-D-
glucosyl)-épipodophyllotoxine-4'-désoxy-4'-phosphate.
Rdt=90%
~s F° ~ 160°CAnal. C45H45~20P~ H2~ PM = 966,920
C H
Calc. 55,89 6,15
Tr. 55,70 6,11
Sel de N-méthylglucamine
20 4'-déméthyl-4-O-(2,3-bis-cyclohexyloxyacétyl-4,G-éthylidène-~-D-
glucosyl)-épipodophyllotoxine-4'-désoxy-4'-phosphate, di-sel de N-
méthylglucamine.
Rdt=50%
F° - 112°C Anal. C59H9, N2030P, 4H20 PM = 1411,420
2s C H N
Calc. 50,21 7,07 1,99
Tr. 50,16 6,60 2,30
R =CF3 ~_~ O O R'=phosph
Ex-elle 3 - formule I
30 4'-déméthyl-4-O-(2,3-bis-(4-trifluorométhoxyphenoxyacétyl)-4,6-
éthylidène-~3-D-glucosyl)-épipodophyllotoxine-4'-désoxy-4'-phosphate, di-
sel de N-méthyl glucamine.
Rdt = 68
o-~
CA 02203155 1997-04-18
WO 96/12727
PCT/FR95/01388
12
F -- 132 ° C Anal . C61 H77N2032F6P, 2, 6H20 PM = 1541, 840
C H N
Calc. 47,52 5,37 1,82
Tr. 47,15 5,51 2,11
R = F ~~ , R'= hos h
\ / P P
s Exe~le 4 - formule I
4'-déméthyl-4-O-(2,3-bis-(4-fluorophénoxyacétyl)-4,6-éthylidène-~3-D-
glucosyl)-épipodophyllotoxine-4'-désoxy-4'-phosphate di-sel de N-méthyl
glucamine.
Rdt=60%
lo F - 130°C Anal. CS~H77N203pF2P, 2,7 H20 PM = 1363,240
C H N
Calc. 50,17 5,88 1,98
Tr. 49,74 5,67 1,92
0
o ~ o~h
R = ~~ , R'=phosph
~ s F~mple 5 - formule I
4'-déméthyl-4-O-(2,3-bis-(3,4-méthylènedioxyphenoxyacétyl)-4,6-
éthylidène-~i-D-glucoxyl)-épipodophyllotoxine-4'-désoxy-4'-phosphate di-
sel de N-méthylglucamine.
Rdt=50%
2o F - 120°C Anal. C61H79N2034P~ H2~ PM = 1433,274
C H IV
Calc. 51,08 5,70 1,95
Tr. 50,68 5,58 1,94
0
R = R'~10-P- /- ~ ~~'1
I
Exemple 6 - formule I
4'-(4-phosphonooxyphénoxyacétyl)-4'-déméthyl-4-O-(2,3-bis-(4-
phosphonooxyphénoxyacétyl)-4,6-éthylidène-~3-D-gIucosyl)=
épipodophyllotoxine.
Le dérivé phénolique de formule I correspondant à R=R' _
~0 4-hydroxyphénoxyacétyl est décrit dans le brevet FR 2 699 535-A1 à
l'exemple 16 préparé par la voie A. lg de ce dérivé (9,6 mmoles) est placé
CA 02203155 1997-04-18
wo 96nzn~
PCT/FR95/01388
13
dans SO ml de THF à -10°C sous atmosphère d'azote et on ajoute 1,2 ml
(8,7 mmoles) de triéthylamine puis goutte à goutte 0,53 ml (5,8 mmoles) de
POC13, l'agitation est maintenue 30 mn. Après filtration du chlorhydrate de
triéthylamine formé, le THF est évaporé. Le résidu est repris dans HCl 1N
s et agité à température ambiante pendant 30 mn. Le précipité blanc est
filtré,
lavé à l'eau et séché sous vide à 60°C pendant une nuit. On obtient 800
mg
de dérivé sous forme de phosphate libre. Rdt = 80 % .
F ~ 160°C Anal. C53H53031P3 PM = 1278,898
Spectre de masse (FAB) ~~ m/e : 1277 (M+-1)
lo IR (KBr) v (cm-1) 3404, 1768, 1603, 1500, 1485, 1203, 1086.
RMN 1H 200Mz (DMSO) 8 : 1,23 (3H, d, J = 4,26Hz Hg~~) ; 3,03 (2H, m,
H2-H3) ; 5,37 (2H, m, H2~~-H3~~) ; 5,77 (1H, s, O-CHA-p) ; 5,97 (1H, s,
O-CHg_O) ; 6,3 (2H, s, H2.-H6~).
La préparation du sel de glucamine se fait de façon similaire à l'exemple 1
~ s mais avec addition de 3 équivalents de N-méthylglucamine. Le composé
s'obtient directement après lyophilisation, avec un rendement de 88
donnant les analyses suivantes
F ~ 155°C Anal. C74 H104N3046P3 PM = 1864,54
C H N
'-o Calc. 47,67 5,62 2,25
Tr. 47,26 5,62 2,38
IR (KBr) v (cm-1) : 3458, 1768, 1604, 1500, 1485, 1199, 1084.
0
R =Ii0-IP- / \ ~ R, = O P.O
Exe~mol -7 - formule I
OH
2s 4'-déméthyl-4-O-(2,3-bis-(4-phosphonooxyphénoxyacétyl)-4,6-éthylidène-
~3-D-glucosyl)-épipodophyllotoxine-4'-désoxy-4'-phosphate.
Par la même suite de réactions que pour l'exemple 6 mais en utilisant le
dérivé de formule I (R=4-hydroxyphénoxyacétyl et R' =H). Décrit dans le
brevet FR 2 699 535-A 1 à l' exemple 20. on obtient le composé avec un
3o rendement de
F ~ 130°C Anal. C66H9g N3O43P3, 6H20 PM = 1822, 503
C H N
Calc. 43,49 6,08 2,31
Tr. 43,30 x,88 2,77
CA 02203155 1997-04-18
WO 96/12727 PCT/FR95/01388
14
IR (KBr) v (cm-1) : 3429, 1763, 1508, 1199, 1084.
_/
R _ R~ ~i203P--~ ~-~ O Ö
Exemple 8 - formule I
4'-(4-phosphonométhylphénoxyacétyl)-4'-déméthyl-4-O-(2,3-bis-(4-
s phosphonométhylphénoxyacétyl)-4,6-ethylidène-~3-D-glucosyl)-épipodo-
phyllotoxine.
1 er stade : 4-benzyloxyphénylméthyldiéthylphosphonate
lg (4,3 10'3 moles) de chlorure de 4-benzyloxybenzyle sont portés 6 h au
reflux avec 0,9 ml (5,15.10-3 moles) de triéthylphosphite. Le milieu
~o réactionnel est filtré sur 150 g de Si02, éluée par un mélange d'heptane
acétate d'éthyle (20-80) pour fournir après évaporation 1,4 g du dérivé
phosphonate (Rdt = 100 % ).
2e stade : 4-hydroxyphénylméthyldiéthylphosphonate
Dans un autoclave, 1,1 g (3,3.10-3 moles) du dérivé benzyloxy du ler stade
ts sont hydrogénés sous une pression d'hydrogène de 7 bars en présence de
200 mg de charbon palladié à 10 % dans 15 ml d'un mélange d'acétate
d'éthyle - éthanol (90-10) à une température de 80°C pendant 12 h sous
agitation. Après filtration du catalyseur, le filtrat est évaporé sous
pression
réduite pour obtenir 800 mg (Rdt 100 % ) du dérivé phénolique.
20 3e stade : Acide diéthyl phosphonométhylphénoxyacétique
A une solution THF (250 ml) de 2,7 g (11 mmoles) du dérivé phénolique
précédent sont ajoutés 1,3 g (26 mmoles) de NaH (50 % dispersion) à
température ambiante, puis 1,8 g (13 mmoles) d'acide bromoacétique sont
introduits et le milieu réactionnel est porté 8 h au reflux. Le milieu
2s réactionnel est versé sur 1 I d'eau glacé et extrait par l'éther éthylique.
Les
phases aqueuses sont acidifiées à pH 1.2 et extraites par l'acétate d'éthyle,
séchées, évaporées pour fournir 3,1 g (Rdt 94 %) du dérivé acétique.
1H 200MHz RMN (CDCI3) 8 8.09 (massif 1H, échangeable) 7,16-7,26 (dd,
2H, J = 8Hz, 2Hz, H aromatiques), 6,85 (2H, d, J = 8Hz, H aromatiques),
0 4,6 (2H, s, OCH2C02H), 4,0 (4H, m, ester phosphonate OCH2), 3,12 (2H,
d, J - 21,7Hz, CH2P), 1,23 (6H, t, J - 7Hz, ester phosphonate
OCH2CH3).
4e stade : condensation de l'acide obtenu au 3e stade sur l'étoposide
A 3 g (10,2 mmoles) d'acide précédemment obtenu au 3e stade en solution
>; dans du chlorure de méthylène ( 15 ml j et 0,2 ml de DMF à 0 ° C
sous azote,
CA 02203155 1997-04-18
WO 96112727 PCTIFR95/01388
ls
sont ajoutés goutte à goutte 1,4 g (11,2 mmoles) de chlorure d'oxalyle, après
un important dégagement de C02 on laisse revenir le milieu réactionnel à
température ordinaire. On refroidit de nouveau à O°C pour introduire
une
solution contenant 1 g (1,7 mmole) d'étoposide, .2 g (25,5 mmoles) de
s pyridine dans du chlorure de méthylène (45 ml) goutte à goutte. En fin
d'addition le milieu est agité encore pendant 4 h avec retour à température
ordinaire. Après évaporation sous pression réduite le milieu réactionnel est
repris par du toluène et évaporé, le résidu est agité avec de l'acétate
d'éthyle
et de l'acide chlorhydrique N. Après extraction, la phase organique est lavée
1 o par une solution glacée de bicarbonate de sodium puis par une solution
saturée de NaCI, décantée, séchée, évaporée, elle fournit une mousse brune
qui est chromatographiée sur Si02 (éluant CH2Cl2 - MeOH-98-2) et donne
après évaporation un résidu solide de 220 mg (Rdt 10 %), Masse (FAB) m/e
1441 (M+).
i s 1 H RMN 200MHz fournit les pics caractéristiques de ces molécules
(CDCl3) 8 7,20 (6H, m, H arom-phosphonate), 6,65-6,93 (6H, d, J =
8,4Hz, H arom. phénoxy) 6,75 (1H, s, HS), 6,47 (1H, s, Hg), 6,24 (2H, s,
H2~ et H6~), 5,87 (1H, s, OCHAO), 5,61 (1H, s, OCHBO), 5,35 (1H, t,
H3~~), 5,05 (1H, t, H2~~), 3,0 (6H, d, J = 2lHz, CH2 Phosphonate).
2o Se stade : Hydrolyse des esters phosphoriques
220 mg (0,16 mmoles) du dérivé triester phosphorique obtenu au 4e stade
sont placés dans CH3CN (50 ml) à 0°C sous azote, on additionne 0,26 ml
de
pyridine (3,2 mmoles) puis goutte à goutte 0,49 g (3,2 mmoles) de bromure
de triméthylsilyle. L'agitation est maintenue 24 h avec retour à température
2s ambiante. On évapore à sec, reprend le milieu par HCl N et le produit
précipite, on filtre le précipité blanc, rince à l'eau jusqu'à neutralité. Le
précipité est solubilisé dans du méthanol, filtré, évaporé. Le résidu est
repris
dans l'eau, cristallisé, pour fournir 120 mg du dérivé phosphorique (Rdt
57%).
~o F° = 190°C Anal C56 H59 028 P3~ SH20 PM = 1301, 41
C H
Calc % 51,68 5,34
Tr % 51,91 4,90
IR v (KBr) 3431, 2922, 1774, 1608, 1512, 1485
_ ~s 1H 200MHz RMN (CD30D) 8 7,15-7,23 (6H, m, H arom. méthyl
phosphorique), 7,04 (1H, s, HS), 6,9 (2H, d, H arorl phénoxy), 6,8 (2H, d,
CA 02203155 1997-04-18
WO 96/12727
16
PCT/FR95/01388
H arom phénoxy), 6,67 (2H, d, H arom. phénoxy), 6,50 (1H, s, Hg), 6,35
(2H, s, H2~, H6~), 5,85 (1H, s, OCHAO), 5,58 (1H, s, OCHBO), 3,05
(2H, d, CH2P), 2,9 (1H, dd, H3), 1,3 (3H, d, Hg~~).
Par la même réaction que pour l'exemple 8 4e stade ou l'exemple 25, on
s prépare (voie C) par triacylation sur l'étoposide les composés suivants à
partir des acides A-Z-CH2-C02H correspondants
_ / \
F- formule I ~ co~~~
4'-(phosphonoacétyl)-4'-déméthyl-4-O-(2,3-bis-phénoxyacétyl-4,6-
t o éthylidène-~3-D-glucosyl)-épipodophyllotoxine.
A une solution de 400 mg (2 mlnoles) d'acide diéthyl phosphonoacétique
dans 5 ml de CH2C12 et 3 gouttes de DMF sous azote à 0°C, sont ajoutés
266 mg (2,1 mmoles) de chlorure d'oxalyle. L'agitation est maintenue 15 mn
à 0°C, puis une solution de 500 mg (0,583 mmoles) de 4'-déméthyl-4-0-
ls (2,3-bis-phénoxyacétyl-4,6-éthylidène-~i-D-glucosyl)-épipodophyllotoxine
dans S ml de CH2C12 et 184 mg (188 ~1, 23 mmoles) de pyridine sont
introduits au milieu réactionnel à 0°C. Le contact est maintenu 2,5 h,
puis le
milieu réactionnel est versé sur HCI N. La phase organique est décantée,
lavée avec une solution de NaCI, séchée, évaporée. Le résidu est cristallisé
?o dans l'éther éthylique pour fournir un précipité blanc (450 mg, Rdt 75 % ).
320 mg (0,31 mmoles) de ce dérivé sont placés sous agitation dans 10 ml
d'acétonitrile en présence de 470 mg (0,4 ml, 0,31 mmoles) de bromure de
triméthyl silyle et 240 mg (0,25 ml, 0,31 mmoles) de pyridine, à
température ambiante pendant 6 h.
2s Après évaporation le résidu est repris dans HCl N pour fournir un solide
blanc qui est filtré, lavé à l'eau, séché. On obtient 180 mg (Rdt 57 %) du
dérivé phosphonique
F° - 140°C Anal C47 H47 021 P, 2H20 (PM = 1014, 996)
C H
Calc % 55,61 5,06
Tr % 55, 87 4, 80
IR v (KBr) 3431, 1774, 1601, 1487
1H 200MHz RMN (CDC13) 8 7,16-7,27 (4H, m, H m. Arom.,, 6,68-6,95
(7H, m, H o.p. Arom, HS), 6,44 (1H, s, Hg), 6,24 (2H, s, H2~-H6~), 5,82
CA 02203155 1997-04-18
WO 96/12727 PC"T/FR95/01388
17
(1H, s, OCHAO), 5,56 (1H, s, OCHBO), 5,32 (1H, dd, H3~~), 5,02 (1H, t,
H2~~), 3,2 (m, 3H, CH2P, H2), 1,32 (d, 3H, J -- 4.4Hz, Hg~~).
,-~ 0
R = C~ / \ O~ R' = C~~ ~O~
Exem ln e 10 - formule I
s 4'-(phosphonoacétyl)-4'-déméthyl-4-O-(2,3-bis-(4-
trifluorométhoxyphénoxyacétyl)-4,6-éthylidène-~i-D-glucosyl)-
épipodophyllotoxine. Sel de N-méthyl glucamine.
Ce dérivé est obtenu par la même méthode que pour l' exemple 9 mais
en partant du dérivé de formule I pour lequel R=4-trifluoro
io méthoxyphénoxyacétyl et R' =H.
Préparation du sel de N-méthyl glucamine : on introduit 810 mg
(0,7 mmoles) du dérivé phosphonique dans l'éthanol (15 ml) et 0,5 ml
d'acétone puis 14,1 ml d'une solution O,1N de N-méthyl glucamine
(1,41 mmoles) dans l'éthanol sont introduits goutte à goutte sous agitation.
is L'agitation est maintenue 1 h. Le milieu réactionnel est évaporé et le
résidu
repris dans l'eau puis filtré (filtre de 0,45 ~c). La solution aqueuse est
lyophylisée, et le résidu est repris dans l'isopropanol, cristallisé, filtré,
séché
pour fournir 740 mg (Rdt 65 % ) de sel de N-méthyl glucamine.
F° = 120°C Anal C63 H79 N2 F6 033 P, 3,SH20
'-o PM = 1600, 47
C H N
Calc % 47,28 5,42 1,75
Tr % 46,95 5,47 2,07
IR v (KBr) 3426, 1774, 1601, 1500, 1487
R= ~ \ O~ R'=CON
2s - formule I
4'-(dicarboxyméthylaminocarbonyl)-4'-déméthyl-4-O-(2,3-bis-(p-
hydroxy-phénoxyacétyl)-4,6-éthylidène-~-D-glucosyl)-
épipodophyllotoxine.
1 er stade : préparation du diesterbenzylique de l'acide aminodiacétique
o A une solution de 10 g (49,6 mmoles) de chlorhydrate de l'ester benzyliqLe
de la glycine dans 200 ml de CH3CN sont ajoutés 13,7 g (99 mmoles) de
K2C03, et 8 ml (49,6 mmoles) de bromoacétate de benzyle goutte à goutte.
Le milieu est agité à température ambiante pendant 2 jours. 200 ml d'eau
CA 02203155 1997-04-18
WO 96/12727 PCT/FR95/01388
18
sont ajoutés et le milieu est acidifié à pH 2-3 par HCl concentré, puis
extrait
par l'acétate d'éthyle, pour obtenir 12 g (Rdt 80 %) du diester utilisé dans
l'étape suivante.
2e stade : préparation du carbamate
s Une solution de phosgène (0,79 ml, 1,5 mmole) dans le toluène à 1,93 M est
introdûite dans 50 ml d'acétonitrile puis refroidie à - 10°C sous
atmosphère
d'azote. On introduit goutte à goutte 820 mg (0,76 mmoles) de 4'-
démetéhyl-4-0-(2,3-bis-(p-benzyloxyphénoxyacétyl)-4,6-éthylidène-~i-D-
glucosyl)-épipodophyllotoxine dans 13 ml d'acétonitrile et 0,24 g de
lo düsopropyléthylamine. Le milieu réactionnel est agité 2 h à -10°C,
puis on y
introduit 0,24 g du diester benzylique de l'acide aminodiacétique, obtenu au
ler stade, dans 6 ml d'acétonitrile à - 5°C. L'agitation est maintenue
6 h. Le
milieu réactionnel est ensuite évaporé puis filtré sur Si02 et élué avec un
gradient de solvant : éther de pétrole - acétate d'éthyle 70.30 puis 60.40 et
~ s enfin 50.50 pour obtenir 700 mg (Rdt 65 % ) du dérivé diester benzylique.
3e stade : Hydro-énolyse des fonctions benzyhques
700 mg du dérivé obtenu au 2e stade sont placés en solution dans un mélange
de 13 ml d'acétate d'éthyle et 3 ml d'éthanol, dans un autoclave sous
atmosphère d'hydrogène en présence de 70 mg de charbon palladié à 10 %.
2o L'agitation est maintenue pendant 24 h puis le milieu réactionnel est
filtré,
évaporé. Le résidu est cristallisé dans l'éther isopropylique pour obtenir
480 mg (Rdt 92 % ) du dérivé diacide.
F° - 150°C Anal C50 H49 NO24 PM = 1047,94
C H N
2s Calc % 57,31 4,71 1,34
Tr % 57,07 4,96 1,13
IR v (KBr) 3433, 1768, 1603, 1512, 1485, 1460, 1236, 1199
1H 200MHz RMN (DMSO) 8 9,03 et 8,98 (2H, 2s, C02H, échangeables),
6,45-6,67 (lOH, m, H5, Hg, ArH), 6,21 (2H, s, H2~, H6~), 6,0 (1H, s,
~o OCHAO), 5,79 (1H, s, OCHBO), 5,32 (2H, m, H2~~ et H3~~), 3,39 (s, N
CH2-C02H), 1,20 (3H, d, Hg~~).
Par la même méthode que l'exemple 11 - 2e stade, mais ~en utilisant les
composés intermédiaires de formule I (R' - H) correspondants, les
nouvea;ix dérivés suivants ont été préparés.
3~
CA 02203155 1997-04-18
WO 96/12727 PC"T/FR95/01388
19
R = O~O ~ R' = CON (CltC00li~
- formule I
4'-(dicarboxyméthylaminocarbonyl)-4'-déméthyl-4-O-(2,3-bis
cyclohexyloxy acétyl-4,6-ethylidène-~i-D-glucosyl)-épipodophyllotoxine.
Rdt = 98
s F° = 170°C Anal C50 H61 N022 PM = 1028,037
'~ R = C~ ~ ~ ~ , R' = CON ( lCOO
- formule I
4'-(dicarboxyméthylaminocarbonyl)-4'-déméthyl-4-O-(2,3-bis(p-trifluoro
méthoxy phénoxy acétyl)-4,6-éthylidène-~i-D-glucosyl)-épipodophyllo
1 o toxine.
Rdt = 87 %
F - 150°C Anal C52 H47 N024 F6 PM = 1183,94
C H N
Calc % 52,75 4,00 1,20
ls Tr % 52,64 4,10 1,30
R = / ~ ~ , R' = CON (CltC00
lïxemnle 14 - formule I
4'-(dicarboxyméthylaminocarbonyl)-4'-déméthyl-4-O-(2,3-bis
phénoxyacétyl-4,6-éthylidène-~3-D-glucosyl)-épipodophyllotoxine.
2o Rdt = 25
F - 170°C Anal CSp H49 N 022, H20 PM = 1033,955
C H N
Calc % 58,08 4,97 1,35
Tr % 58,43 5,05 1,28
?s
R = HOC6-1 ,..~
/ \ O' 'O ,R'_
Exemple 15 - formule I p
4'-déméthyl-4-O-(2,3-bis-(4-carboxyméthoxyphénoxyacétyl)-4,6-
éthylidène-~3-D-glucosyl)-épipodophyllotoxine, sel de N-méthylglucamine.
1 er stade : 4-hydroxy'phénoxyacétate de benwle
CA 02203155 1997-04-18
WO 96/12727 PCT/FR95/01388
A une suspension de 10 g (59 mmoles) d'acide p-hydroxyphénoxyacétique
dans 100 ml de CH2Cl2 à 0°C sous atmosphère d'azote sont ajoutés 0,2 ml
de DMF puis goutte à goutte 9 g de chlorure d'oxalyle (71 mmoles).
L'agitation est maintenue 12 h à température ordinaire, puis 7,7 g
s (71 mmoles) d'alcool benzylique sont introduits et l'agitation est gardée 8
h à
température ambiante. On jette le milieu réactionnel sur une solution
ammoniacale glacée et on extrait par du chlorure de méthylène. La phase
organique est lavée avec HCl N, décantée, séchée, évaporée. Le résidu est
filtré sur 150 g de Si02~et éluée par un mélange heptane - acétate d'éthyle
lo (75-25) pour fournir 3,8 g (Rdt 25 %) d'un solide blanc.
1H 200MHz RMN (CDCl3) b 7,36 (SH, s, Ar), 6,75 (4H, d, ArOH), 5,24
(2H, s, CH2 Ar), 4,61 (2H, s, OCH2C0).
2e stade : acide 4-benzylo~ycarbonylméthoxyphénoxyacétique
3, 8 g du phénol obtenu au 1 er stade sont portés au reflux du THF (200 ml)
i5 en présence de 1,3 g de NaH (dispersion à 60 %) et 2 g d'acide
bromoacétique pendant 48 h. On verse ensuite le milieu réactionnel sur de la
glace et on extrait à l'éther isopropylique puis à l'acétate d'éthyle. La
phase
aqueuse est acidifiée et extraite par du CH2C12 pour 2,8 g (Rdt 60 %) d'un
solide crème.
20 1H 200MHz RMN (CDC13) cS 7,35 (SH, s, Ar), 6,78 (4H, s, Ar0), 5,15
(2H, s, CH2 Ar), 4,55 (2H, s, OCH2 ester), 4,45 (2H, s, OCH2 acide)
3e stade : complage de l'acide du 2e stade avec le 4'-benzyloxy carbonyl
étoposide
A 1,75 g (5,5 mmoles) de l'acide du 2e stade en solution dans 40 ml de
CH2C12 avec 0,2 ml de DMF, on additionne à 0°C sous atmosphère
d'azote
goutte à goutte 770 mg (6,1 mmoles) de chlorure d'oxalyle. L'agitation est
maintenue 2 h à température ambiante. On ajoute alors, après retour à
0°C,
une solution de 1 g (1,38 mmoles) de 4'-benzyloxycarbonyl étoposide et de
1,1 g (1,38 mmoles) de pyridine dans 10 ml de CH2C12. Après 3 h
o d'agitation à température ambiante, le milieu réactionnel est concentré,
repris par l'acétate d'éthyle et lavé à l'eau, puis par une solution glacée de
bicarbonate de sodium, après un nouveau lavage par du HCl N, puis par une
solution saturée de NaCI, la phase organique est décantée, séchée, évaporée
pour fournir 800 mg (Rdt 66 iô) qui sont directement hydrogénolysés au
~s stade suivant (CCM Si02 heptane-.AcOEt 20-80 Rf=0,9)
4e stade : Hydrogénolyse
CA 02203155 1997-04-18
WO 96/12727 PCT/FR95/01388
21
800 mg (0,6 mmoles) de dérivé obtenu au 3e stade sont placés sous
atmosphère d'hydrogène à pression atmosphérique dans 15 ml d'acétate
d' éthyle et 5 ml d' éthanol en présence de 100 mg de charbon palladié, sous
agitation importante pendant une heure. Le catalyseur est filtré et le filtrat
s évaporé. Le résidu est repris dans l'acétone et filtré à nouveau et évaporé
pour fburnir quantitativement (650 mg) le dérivé débenzylé. Ce dérivé est
transformé en sel de N-méthyl glucamine par addition de 2 équivalents d'une
solution 0,1 M de N-méthyl glucamine dans le mélange EtOH, H20-Acétone.
Après 1 h d'agitation, la~ solution est évaporée, reprise par H20, filtrée sur
to filtre 0,45 ~, et lyophylisée on obtient alors 700 mg du dérivé
carboxylique.
F - 130°C Anal C63 Hg2 N2 033 6H2 O PM = 1503,422
C H N
Calc % 50,33 6,30 1,86
Tr % 49,89 5,93 2,19
t s IR v (KBr) 3427, 1772, 1618, 1508, 1425, 1205, 1087
1H 200MHz RMN (DMSO) b 7,04 (1H, s, HS), 6,59-6,70 (8H, m, OArO),
6,47 (1H, s, Hg), 6,13 (2H, s, H2~ et H6~), 5,96 (1H, s, OCHAO), 5,73
(1H, s, OCHBO), 5,33 (2H, m, H2~~ et H3~~), 2,46 (6H, s, NCH3), 1,20
(3H, d, J=4Hz, Hg~~).
2o Par la même suite de réactions de l'exemple 15, mais en utilisant les
réactifs
appropriés les dérivés de formule générale I (R' =H) sont obtenus
R = HOC ~ ~ O~O ~ R
Exemyle 16 - formule I
4'-déméthyl-4-O-(2,3-bis (4-carboxyphénoxyacétyl)-4,6-éthylidène-(3-D-
2s glucosyl)-épipodophyllotoxine
En utilisant l'acide 4-benzyloxycarbonylphénoxyacétique, on obtient selon le
procédé de l'exemple 15 le dérivé carboxylique avec un rendement de 45 %.
F-170°C Anal C47 H44 021, 1,3H20 PM = 968,469
C H
3o Calc % 58,28 4,81
Tr % 58,19 4,85
Sel de N-méthylglucamine
F = 138°C Anal C61 H7g N2 031, SH20 PM = 1425,355
C H N
~s Calc % 51,40 6,22 1,97
CA 02203155 1997-04-18
WO 96/12727
22
Tr % 51,27 5,85 2,19
PC'T'/FR95/01388
R = HOCO / \ O' ''O ~ R~ _
FxemTl7 - formule I
4'-déméthyl-4-O-(2,3-bis-(4-carboxyméthylphénoxyacétyl)-4,6-éthylidène-
s ~i-D-glucosyl) épipodophyllotoxine.
En utilisant l'acide 4-benzyloxycarbonylméthylphénoxyacétique, on obtient
selon l'exemple 15, le dérivé acétique (Rdt 66 %).
F--150°C Anal C49 H4g 021, 2H20 PM = 1008, 932
C H
Calc % 58,33 5,19
Tr % 57, 74 4, 85
Spectrographie de Masse (FAB) m/e 972 (M+)
Par la méthode de l'exemple 15 3e stade uniquement, les dérivés suivants
sont préparés
is
R= / \ ~~ , R~=
- formule I
4'-déméthyl-4-O-(2,3-bis-(phénylsulfonylacétyl)-~3-D-glucosyl)-
épipodophyllotoxine.
Rdt=92%
?o F = 248 ° C Anal . C45 H44 O 19 S2 PM = 976, 790
C H
Calc. % 56,72 4,65
Tr. % 56,40 4,66
F F /
R _- F / \ p~0 , R' __
Exemple 19 - formule I F
2s 4'-déméthyl-4-O-(2,3-bis-(pentafluorophénoxyacétyl)-~3-D-glucosyl)-
épipodophyllotoxine.
Rdt = 75 % Anal. C45 H34 017 F10 PM = 1036,75
C H
Calc. % 52,13 3,30
''° Tr. % 51,88 3,25
CA 02203155 1997-04-18
WO 96/12727
23
PCT/FR95/01388
R =F3C / \ ~~ , R' _
Exemple 20 - formule I
4'-déméthyl-4-O-(2,3-bis-(4-trifluorométhylphénoxyacétyl)-~i-D-glucosyl)-
épipodophyllotoxine.
Rdt = 53 % Anal. C47 H42 O 17 F6 PM = 992, 820
C H
Calc. % 56,86 4,26
Tr. % 56,78 4,22
R = / \ ~~ , R' qohospha
- formule I
i o 4'-déméthyl-4'-désoxy-4'-phosphate-4-O-(2,3-bis-(phénylsulfonylacétyl)-
4,6-éthylidène-~3-D-glucosyl)-épipodophyllotoxine.
Di-sel de N-méthylglucamine
En utilisant la méthode de l' exemple 1, mais avec le composé de l' exemple
18, le dérivé phosphate sous forme de sel de N-méthylglucamine est obtenu.
is Rdt = 60 % Anal. Csg H7g N2 032 PS2; 3,8H20
F~ 148°C
PM = 1492,85
C H N
Calc. % 49,79 5,59 1,97
2o Tr. % 47,47 5,85 1,88
F F
R = F / \ o"p , R' ~hospha
- formule I F
4'-déméthyl-4'-désoxy-4'-phosphate-4-O-(2,3-bis-(2,3,4,5,6-pentafluoro-
phénoxyacétyl)-4,6-éthylidène-~3-D-glucosyl)-épipodophyllotoxine. Di-sel
de N-méthylglucamine.
2s En utilisant la méthode de l'exemple 1 mais avec le composé de l'exemple
19, le dérivé phosphate sous forme de sel de N-méthylglucamine est obtenu.
Rdt = 78
F ~ 140°C Anal. Csg H6g F10 N2 030 P; 3,SH20 PM = 1507,147
C H N
3o Calc. % 45,11 4,88 1,78
Tr. % 45,34 4,67 1,82
CA 02203155 1997-04-18
WO 96/12727 PCT/FR95/01388
24
R =F3C ~ ~ O~ ~ R' = Phosph
- formule I
4'-déméthyl-4'-désoxy-4'-phosphate-4-O-(2,3-bis-(4-trifluoro-
méthylphénoxyacétyl)-4,6-éthylidène-~3-D-glucosyl)-épipodophyllotoxine.
s Di-sel de N-méthylglucamine.
En utilisant la méthode de l'exemple 1 mais avec le composé obtenu à
l'exemple 20, le dérivé.. phosphate sous forme de N-méthylglucamine est
obtenu.
Rdt = 33
io F = 120°C Anal. C61 H77 N2 030 F6 P; 4,15 H20 PM = 1538,170
C H N
Calc. % 47,63 5,59 1,82
Tr. % 47,16 5,12 1,84
R=R'=HO ~ ~ O
~ s F-xemyle 24 - formule I
Voie C
4'-déméthyl-4'-(4-carboxyphénoxyacétyl)-4-O-(2,3-bis-(4-carboxy-
phénoxyacétyl)-4,6-éthylidène-~3-D-glucosyl)-épipodophyllotoxine - sel de
glucamine.
20 1 er stade : Condensation de l'acide 4-benzyloxycarbonylphénoxy acétique
avec l'étoposide
A une solution de 4,9 g (17 mmoles) d'acide 4-benzyloxycarbonyl-
phénoxyacétique dans 100 ml de CH2C12 et 0,2 ml de DMF sont ajoutés à
0°C sous azote, goutte à goutte 2,4 g (18.7 mmoles) de chlorure
d'oxalyle,
2s après 2 h d'agitation à température ambiante, on introduit à cette solution
2 g
(3,4mMoles) d'étoposide en solution dans 15 ml de CH2C12 et 3,2 g
(41 mmoles) de pyridine goutte à goutte à 0°C. Après retour à
température
ambiante en 3 heures, le milieu réactionnel est versé sur HCI N puis extrait
par CH2CI2 et lavé par une solution de NaHC03, et NaCI saturé
3o successivement pour obtenir après évaporation un produit brut qui est
c:~romatographié sur Si02 par élution dans un mélange heptane-AcOEt (60-
4G~. On obtient 1,8 g (Rdt 38 %) de dérivé trisubstitué qui est utilisé
dirFCtement dans l'étape suivante.
CA 02203155 1997-04-18
WO 96/12727 PCT/FR95/01388
2e stade : Hydrogénolyse
1,5 g (1,07 mmoles) du triester benzylique précédent est hydrogénolysé en
présence d'hydrogène à pression atmosphérique dans un mélange EtOH
( 15 ml) AcOEt (60 ml) avec 300 mg de charbon palladié à 10 % sous bonne
s agitation pendant 8 h à température ambiante. Le catalyseur -est filtré, le
filtrat -évaporé est chromatographié sur Si02 et élué dans un mélange
CH2C12-MeOH (96,4) pour obtenir un solide blanc (600 mg Rdt 50 %). Le
sel de glucamine effectué directement, est obtenu en plaçant 3 équivalents
d'une solution aqueuse ~O,1M de N-méthyl glucamine dans un mélange
~o EtOH-H20 (80-20), cette solution est ajoutée à la solution du triacide dans
l'acétone. Un précipité gommeux est obtenu, le milieu est évaporé puis
repris par H20 et filtré à travers un filtre de 0,45 ~,. Le filtrat est alors
lyophilisé pour obtenir le sel de glucamine. Rdt = 50
F -135 ° C Anal C77 H 101 N3 04, 7H2O PM = 1836,103
C H N
Calc % 50,37 6,32 2,29
Tr % 49,99 5,77 2,43
IR v (KBr) 3404, 1772, 1604, 1545, 1385, 1086.
1H 200MHz RMN (DMSO), 7,77-7,85 (6H, m, Ar), 7,1 (1H, s, HS), 6,89,
20 6,79, 6,64 (6H, d, J=8,7Hz, Ar0), 7,08 (1H, s, HS), 6,46 (1H, s, Hg),
6,26 (2H, s, H2~ et H6~), 6,13 (1H, s, OCHAO), 5,95 (1H, s, OCHBO),
2,42 (9H, s, N-CH3), 1,20 (3H, d, J=4,9Hz, Hg~~).
R=R'=HO i \ O
- formule I '----/
4'-déméthyl-4'-carboxyméthylphénoxyacétyl-4-O-(2,3-bis-(4-carboxy-
2s méthylphénoxy acétyl)-4,6-éthylidène-~i-D-glucosyl)-épipodophyllotoxine.
Ce dérivé est obtenu par la même méthode que pour l'exemple 24, mais en
utilisant l'acide benzyloxycarbonylméthylphénoxyacétique. Rdt = 20
F-150°C Anal C59 H56 025 1.4H20 PM = 1190,703
C H
o Calc % 59,63 4,81
Tr % 59,19 4,84
R=R'=HOC i \
~O
Exemple 26 - formule ~/I
CA 02203155 1997-04-18
WO 96/12727 PCT/FR95/01388
26
4'-déméthyl-4'-carboxyméthoxyphénoxyacétyl-4-O-(2,3-bis-(4-carboxy-
méthoxy phénoxyacétyl)-4,6-ethylidène-~3-D-glucosyl) épipodophyllo-
toxine - Sel de N-méthyl glucamine.
Ce dérivé est obtenu par la même méthode que pour l'exemple 24, mais en
s utilisant l'acide benzyloxycarbonylméthoxyphénoxyacétique. Rdt = 84
F -110°C Anal Cgp H107 N3 043, 6.3 H20 PM=1912,036
C H N
Calc % 50,25 6,30 2,20
Tr % 50,13 6,17 2,56
R._ C / \ O~ ~ R,=P~"~2
Exemple 27 - formule I
4'-déméthyl-4'-désoxy-4'-phosphate-4-O-(2,3-bis-(4-
chlorophénoxyacétyl)-4,6-éthylidène-~-D-glucosyl)-épipodophyllotoxine.
Préparation des intermédiaires (voie B)
ts 4'-déméthyl-4'-(3-quinuclidinylaminocarbonyl)-4-O-(2,3-bis-(4-chloro
phénoxy acétyl)-4,6-éthylidène-~3-D-glucosyl)-épipodophyllotoxine.
1 er stade : 4'-(3-quinuclidinylaminocarbonyl) étoposide
formule V R4 = COHN 3-quinuclidinyl
A une solution 1,93M de phosgène dans l'acétonitrile (8,8 ml, 16,9 mmoles)
2o refroidie à 0°C sous azote, est ajoutée goutte à goutte une solution
d'étoposide (5 g, 8,49 mmoles) dans 200 ml de CH3CN et 2,74 g
(21,2 mmoles) de N, N-düsopropyléthylamine en 10 mn, puis introduire
1,07 g (8,49 mmoles) de 3-aminoquinuclidine en solution dans 20 ml de
CH3CN. Le milieu est agité 24 h. Après évaporation, le résidu est
2s chromatographié sur Si02 avec un mélange de solvants : CHC13-MeOH-
NH40H (93-7-0,7) puis (90-10-1). On obtient 1,67g (Rdt 26 %) de produit
carbamate, V homogène en CCM qui est mis aussitôt en réaction dans le 2e
stade.
2e stade : Condensation avec l'acide p-chlorophenoxyacétique
o A une solution de 1,68 g (8,98 mmoles) d'acide p-chlorophénoxyacétique
dans du chloroforme (40 ml) et 0,5 ml de DMF à 0°C sous atmosphère
d'azote, sont introduits 1,25 g (9,88 mmoles) de chlorure d'oxalyle goutte à
goutte. L'agitation est maintenue 1 h. Cette solution est ensuite ajoutée
goutte à goutte à la solution du dérivé de l'étoposide obtenu au ler stade
(1,66 g, 2,24 mmoles) dans ~50 ml de chloroforme et 1,77 g de pyridine à
CA 02203155 1997-04-18
wo 96/12n~
27
PCT'/FR95/01388
0°C. Cette nouvelle solution est agitée 5 h avec retour à la
température
ambiante. Le milieu réactionnel est ensuite versé sur HCl N (100 ml)
décanté, puis lavé par une solution saturée de NaCI, séchée, évaporée pour
fournir une huile qui est chromatographiée sur silice. L'élution par un
s mélange de CHCl3, MeOH, NH40H (95, 5, 0,5) fournit 1,97 g (Rdt 80 %)
du dérivé du titre de l'exemple 26. On en forme le chlorhydrate (par addition
d'une solution d'éther saturé de gaz chlorhydrique à la solution de la base
dans l'acétone. Après agitation pendant lOmn, le précipité obtenu est filtré
lentement, lavé à l'éther et séché (Rdt 50 % ).
to F° --180°C Anal C53 H54 Cl2 N2 018, HCI, 2H20 PM = 1150,422
C H N
Calc % 55,33 5,08 2,43
Tr % 55,74 4,96 2,61
Spectre de masse (FAB) m/e 1077 (M+)
t s 3e stade : hydrolyse
A une solution dans 70 ml d'acétone de 1,08 g du dérivé du 2e stade, on
introduit 30 ml d'une solution saturée de NaHC03. Le milieu est agité
pendant 2 jours, on évapore l'acétone et acidifie le milieu avec HCl
concentré jusqu'à pH 2, puis on extrait par du chlorure de methylène.
?o Une chromatographie sur Si02 (élution CH2C12-MeOH 97-3) fournit le
dérivé du titre de l'exemple 27. Une nouvelle chromatographie sur Si02
(élution éther de pétrole - AcOEt 1-1) fournit après évaporation un résidu qui
cristallise dans l'éther isopropylique (200mg Rdt=21 % ).
F-125°C Anal C45 H42 C12 017 PM = 925,730
Zs C H
Calc % 58,38 4,57
Tr % 58,63 4,69
Spectre de masse (FAB) m/e 924 (M+-1)
IR v (KBr) 3458, 1774, 1618, 1491.
1H 200MHz RMN (CDC13) b 7,15-7,22 (4H, m, Ar), 6,75 (1H, s, HS),
6,77 (2H, d, J=8,8Hz, Ar0), 6,64 (2H, d, J=8,8Hz, Ar0), 6,51 (1H, s,
Hg), 6,22 (2H, s, H2~ et H6~), 5,91 (1H, s, OCHAO); 5,71 (1H, s,
OCHBO), 5,33 (1H, t, J=9Hz, H3~~), 5,02 (1H, t, J=7,8Hz, H2~~), 4,91
(1H, d, J=7,8Hz, Hl ~~), 4,83 (1H, d, ~ =3,2Hz, H4), 4,69 (1H, q, H7~~).
~s 3,1 (1H, dd, H2), 2,9 (1H, m, H3), 1,34 (3H, d, J=SHz, Hg~~).
Préparation du phosphate
CA 02203155 1997-04-18
WO 96/12727 PCT/FR95/01388
28
Ce dérivé est obtenu selon le mode opératoire de l'exemple 1 mais en
utilisant le dérivé obtenu au 3e stade.
Sel de N-méthylglucamine
Rdt=70%
s F--140°C Anal C59 H77 Cl2 N2 030 P, 6H20 PM = 1461,49
- C H N
Calc % 48,49 5,8 1,90
Tr % 48,78 5,55 1,88
R=R'=HOC'Ö~
~ o Exemyle 28 - formule I o
4'-déméthyl-4'-carboxyméthoxyacétyl-4-O-(2,3-bis-carboxyméthoxy-
acétyl-4,6-éthylidène-~3-D-glucosyl) épipodophyllotoxine.
Ce dérivé est préparé selon la méthode de l'exemple 24, ler et 2e stade dans
lequel on utilise l'acide benzyloxycarbonylméthoxy acétique au lieu de
~s l'acide 4-benzyloxycarbonylphénoxyacétique, pour conduire avec des
rendements successifs de 77 % et de 75 % .
F =138 ° C Anal C41 H44 025 ~ H20 PM = 954, 805
C H
Calc % 51,57 4,85
'-o Tr % 51,22 4,72
Spectre de masse (FAB) m/e 959 (M+ +Na)
R = R' = HOC'~SOi~
- formule I o
4'-déméthyl-4'-carboxyméthylsulfonylacétyl-4-O-(2,3-bis-carboxyméthyl-
2s sulfonyl acétyl-4,6-éthylidène-~3-D-glucosyl) épipodophyllotoxine.
Ce dérivé est préparé comme le précédent selon la méthode de l'exemple 24,
ler et 2e stades en utilisant l'acide benzyloxycarbonylméthylsulfonylacétique
avec des rendements successifs de 55 % et 90 % .
F --165 °C Anal C41 H44 028 S3 ~ H20 PM = 1098,98
C H
Calc % 44,81 4,22
Tr % 44,94 4,34
Spectre de masse (FAB) m/e 1103 (M++Na)
Exemple 30 - formule I R = Hococ~e~c~co, R' _
CA 02203155 1997-04-18
WO 96/12727 PCT/FR95/01388
29
4'-déméthyl-4-O-(2,3-bis-carboxyméthoxyacétyl-4,6-éthylidène-~3-D-
glucosyl) épipodophyllotoxine.
Ce dérivé est obtenu par la méthode décrite pour l'exemple 15, 3e et 4e
stades, mais en utilisant l'acide benzyloxycarbonylméthoxyacétique au lieu
s de benzyloxycarbonylméthoxyphénoxyacétique. Les rendements successifs
sont 42 % et 71 % .
F -192 °C Anal C37 H40 021, 0.2 H20 PM = 824,32
C H
Calc % 54,59 5,20
'o Tr % 54,81 5,23
Exem le 1 - formule I R = R' =~o3c~c
4'-déméthyl-4'-Phosphonoacétyl-4-O-(2,3-bis-phosphonoacétyl-4,6-
éthylidène-~i-D-glucosyl) épipodophyllotoxine.
~ s Ce dérivé est obtenu de façon identique à la méthode de l' exemple 24, 1
er et
2e stades, mais en utilisant l'acide dibenzylphosphonoacétique (Tet. Let.
1974, N°9, 711). Les modifications par rapport à l'exemple 24 sont les
suivants : le premier stade se conduit à 0°C avec retour à température
ambiante pendant 18 h ; l'hydrogénolyse au second stade se fait dans le
2o solvant : THF 25 ml et EtOH 50 ml et la réaction est conduite à 0°C
pendant
4h. Les rendements sont successivement 34 % et 83 % .
F-184°C Anal C35 H41 025 P3~ 4H20 PM = 1026, 58
C H H20
Calc % 40,94 4,81 7,02
Tr % 40,95 4,38 7,54
Sprectre de masse (FAB) m/e 955 (M+ + 1)
Exemple 32 - formule I R = ~°spc~4co, R' _
4'-déméthyl-4-O-(2,3-bis-phosphonoacétyl-4,6-éthylidène-~3-D-glucosyl)
~o épipodophyllotoxine.
Ce dérivé est obtenu de façon identique à la méthode de l'exemple 15, 3e et
4e stades, mais en utilisant l'acide dibenzylphosphonoacétique, déjà utilisé
pour l'exemple 31.
Le stade d'hydrogénolyse se fait en utilisant un mélange de solvant : THF-
EtOH (30-70) à 0°C pendant 4h. On obtient alors le dérivé avec un
rendement global de 45 % .
CA 02203155 1997-04-18
WO 96/12727 PCT/FR95/01388
F--168°C Anal C33 H3g 021 P2 PM = 832,606
Spectre de masse (FAB) m/e 833 (M+ + 1).
Les composés suivants selon l'invention ont également été préparés
4'-déméthyl-4'-désoxy, 4'-phosphate-4-O-(2,3-bis(4-cyanophénoxyacétyl)-
s 4,6-éthylidène-~i-D-glucosyl) épipodophyllotoxine. Sel de N-méthyl-
glucamine.
F - 205 °C Anal C47 H43 N2 020 P, 6 H20 = 1 112,93
C H N
Calc % 51,56 5,06 2,56
io Tr % 51,11 4,27 2,31
4'-déméthyl-4'-désoxy, 4'-phosphate-4-O-(2,3-bis(4-nitrophénoxyacétyl)-
4,6-éthylidène-~i-D-glucosyl) épipodophyllotoxine. Sel de N-méthyl-
glucamine.
F--190°C Anal C45 H43 N2 024 P~ 4 H20 = 1 098,88
~s C H
Calc % 49,18 4,68 2,55
Tr % 49,38 4,25 2,46
4'-déméthyl-4'-désoxy, 4'-phosphate-4-O-(2,3-bis(4-méthylphénoxy-
acétyl)-4,6-éthylidène-~i-D-glucosyl) épipodophyllotoxine.
2o F--190°C Anal C47 H4g N2 020 P, 1,25 H20 = 987,378
C H
Calc % 57,12 5,21
Tr % 56,6 5,04
4'-déméthyl-4-O-(2,3-bis(4-phosphonooxyphénoxyacétyl)-4,6-éthylidène-~3
2s -D-glucosyl) épipodophyllotoxine. Sel de N-méthylglucamine.
F--145-150°C Anal Csg Hg0 N2 035 P2, 1,5 H20 = 1 466,240
C H N
Calc % 48,33 5,71 1,91
Tr % 48,46 5,93 2,09
30 4'-déméthyl-4'-désoxy-4'-phosphate-4-O-(2,3-bis(4-hydroxyphénoxy-
acétyl)-4,6-éthylidène-~i-D-glucosyl) épipodophyllotoxine. Sel de N-
méthylglucamine.
F--145°C Anal Csg H7g N2 032 P, H20 = 1 377,26
C H N
3s Calc % 51,45 5,93 2,03
Tr % 51,44 6,17 2,04
FE~IIL#.E ~ECTt~IEF ~HEGLE 91~
ISAIEP