Note: Descriptions are shown in the official language in which they were submitted.
DEMANDES OU BREVETS VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE OU CE BREVET
COMPREND PLUS D'UN TOME.
CECI EST LE TOME ~ DE ~2
NOTE: Pour les tomes additionels, veuiilez contacter le Bureau canadien des
brevets
~z~ 3~~~7
JUMBO APPL1CAT10NS/PATENTS -
THIS SECT10N OF THE APPLlCATIONIPATENT CONTAINS MORE
THAN ONE VOLUME
THIS IS VOLUME OF -
NOTE: For additional volumes-phase contact the Canadian Patent Ofific~
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
RAPID DETECTION AND IDENTIFICATION OF
NUCLEIC ACID VARIANTS AND PATHOGENS
FIELD OF THE INVENTION
The present invention relates to methods and compositions for treating nucleic
acid,
and in particular, methods and compositions for detection and characterization
of nucleic acid
sequences and sequence changes.
BACKGROUND OF THE INVENTION
The detection and characterization of specific nucleic acid sequences and
sequence
changes have been utilized to detect the presence of viral or bacterial
nucleic acid sequences
indicative of an infection, the presence of variants or alleles of mammalian
genes associated
with disease and cancers, and the identification of the source of nucleic
acids found in
forensic samples, as well as in paternity determinations.
I S Various methods are known in the art which may be used to detect and
characterize
specific -nucleic acid sequences and sequence changes. Nonetheless, as nucleic
acid sequence
data of the human genome, as well as the genomes of pathogenic organisms
accumulates, the
demand for fast, reliable, cost-effective and user-friendly tests for specific
sequences continues
to grow. Importantly, these tests must be able to create a detectable signal
from a very low
copy number of the sequence of interest. The following discussion examines
three levels of
nucleic acid detection currently in use: I. Signal Amplification Technology
for detection of
rare sequences; II. Direct Detection Technology for detection of higher copy
number
sequences; and III. Detection of Unknown Sequence Changes for rapid screening
of sequence
changes anywhere within a defined DNA fragment,
I. Signal Amplification Technology Methods For Amplification
The "Polymerase Chain Reaction" (PCR) comprises the first generation of
methods for
nucleic acid amplification. However, several other methods have been developed
that employ
the same basis of specificity, but create signal by different amplification
mechanisms. These
methods include the "Ligase Chain Reaction" (LCR), "Self Sustained Synthetic
Reaction"
(3SR/NASBA), and "Q(3-Replicase" (Q(3).
CA 02203627 1997-04-24
WO 96/15267 PCTIUS95/14673
Polymerise Chain Reaction (PCR)
The polymerise chain reaction (PCR), as described in U.S. Patent Nos. 4,683,19
and
4,683,202 to Mullis and Mullis et al., describe a method for increasing the
concentration of a
segment of target sequence in a mixture of genomic DNA without cloning or
purification.
This technology provides one approach to the problems of low target sequence
concentration.
PCR can be used to directly increase the concentration of the target to an
easily detectable
level. This process for amplifying the target sequence involves introducing a
molar excess of
two oligonucleotide primers which are complementary to their respective
strands of the
double-stranded target sequence to the DNA mixture containing the desired
target sequence.
The mixture is denatured and then allowed to hybridize. Following
hybridization, the primers
are extended with polymerise so as to form complementary strands. The steps of
denaturation, hybridization, and polymerise extension can be repeated as often
as needed, in
order to obtain relatively high concentrations of a segment of the desired
target sequence.
The length of the segment of the desired target sequence is determined by the
relative
positions of the primers with respect to each other, and, therefore, this
length is a controllable
parameter. Because the desired segments of the target sequence become the
dominant
sequences (in terms of concentration) in the mixture, they are said to be "PCR-
amplified."
Lipase Chain Reaction (LCR or LAR)
The lipase chain reaction (LCR; sometimes referred to as "Lipase Amplification
Reaction" (LAR) described by Barany, Proc. Natl. Acid. Sci., 88:189 (1991);
Barany, PCR
Methods and Applic., 1:5 ( 1991 ); and Wu and Wallace, Genomics 4:560 ( 1989)
has
developed into a well-recognized alternative method for amplifying nucleic
acids. In LCR,
four oligonucleotides, two adjacent oligonucleotides which uniquely hybridize
to one strand of
target DNA, and a complementary set of adjacent oligonucleotides, which
hybridize to the
opposite strand are mixed and DNA lipase is added to the mixture. Provided
that there is
complete complementarity at the junction, lipase will covalently link each set
of hybridized
molecules. Importantly, in LCR, two probes are ligated together only when they
base-pair
with sequences in the target sample, without gaps or mismatches. Repeated
cycles of
denaturation, hybridization and ligation amplify a short segment of DNA. LCR
has also been
used in combination with PCR to achieve enhanced detection of single-base
changes. Segev:
PCT Public. No. W090Q1069 A1 (1990). However, because the four
oligonucleotides used in
this assay can pair to form two short ligatable fragments, there is the
potential for the
-2-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95114673
generation of target-independent background signal. The use of LCR for mutant
screening is
limited to the examination of specific nucleic acid positions.
__ Self Sustained Synthetic Reaction (3SR/NASBA)
The self sustained sequence replication reaction (3SR) (Guatelli et al., Proc.
Natl.
Acid. Sci., 87:1874-1878 [1990], with an erratum at Proc. Natl. Acid. Sci.,
87:7797 [1990])
is a transcription-based in vitro amplification system (Kwok et al., Proc.
Natl. Acid. Sci.,
86:1173-1177 [1989]) that can exponentially amplify RNA sequences at a uniform
temperature. The amplified RNA can then be utilized for mutation detection
(Fahy et al.,
PCR Meth. Appl., 1:25-33 [1991]). In this method, an oligonucleotide primer is
used to add
a phage RNA polymerise promoter to the 5" end of the sequence of interest. In
a cocktail of
enzymes and substrates that includes a second primer, reverse transcriptase,
RNase H, RNA
polymerise and ribo-and deoxyribonucleoside triphosphates, the target sequence
undergoes
repeated rounds of transcription, cDNA synthesis and second-strand synthesis
to amplify the
area of interest. The use of 3SR to detect mutations is kinetically limited to
screening small
segments of DNA (e.g., 200-300 base pairs).
Q-Beta (Q~) Replicase
In this method, a probe which recognizes the sequence of interest is attached
to the
replicatable RNA template for Q(3 replicase. A previously identified major
problem with false
positives resulting from the replication of unhybridized probes has been
addressed through use
of a sequence-specific' ligation step. However, available ther.mostable DNA
ligases are not
effective on this RNA substrate, so the ligation must be performed by T4 DNA
lipase at low
temperatures (37°C). This prevents the use of high temperature as a
means of achieving
specificity as in the LCR, the ligation event can be used to detect a mutation
at the junction
site, but not elsewhere.
Table 1 below, lists some of the features desirable for systems useful in
sensitive
nucleic acid diagnostics, and summarizes the abilities of each of the major
amplification
methods (See also, Landgren, Trends in Genetics 9:199 [1993]).
A successful diagnostic method must be very specific. A straight-forward
method of
' controlling the specificity of nucleic acid hybridization is by controlling
the temperature of the
reaction. While the 3SRlNASBA, and Q[3 systems are all able to generate a
large quantity of
signal, one or more of the enzymes involved in each cannot be used at high
temperature ( i. e. ,
- J -
CA 02203627 1997-04-24
WO 96/15267 PG"TlUS95/14673
>55°C). Therefore the reaction temperatures cannot be raised to prevent
non-specific
hybridization of the probes. If probes are shortened in order to make them
melt more easily
at low temperatures, the likelihood of having more than one perfect match in a
complex
genome increases. For these reasons, PCR and LCR currently dominate the
research field in
detection technologies. -
TABLE 1
METHOD:
FEATURE PCR &
3SR
PCR LCR
LCR
NASBA
Qa
Amplifies Target + + + +
Recognition of Independent+ + + + +
Sequences Required
Performed at High Temp.+ +
Operates at Fixed Temp. + +
Exponential Amplification+ + + + +
Generic Signal Generation +
Easily Automatable
The basis of the amplification procedure in the PCR and LCR is the fact that
the
products of one cycle become usable templates in all subsequent cycles,
consequently
doubling the population with each cycle. The final yield of any such doubling
system can be
expressed as: (1+X)° = y, where "X" is the mean efficiency (percent
copied in each cycle),
"n" is the number of cycles, and "y" is the overall efficiency, or yield of
the reaction (Mullis,
PCR Methods Applic., 1:1 [19910. If every copy of a target DNA is utilized as
a template in
every cycle of a polymerise chain reaction, then the mean efficiency is 100%.
If 20 cycles of
PCR are performed, then the yield will be 2'-°, or 1,048.576 copies of
the starting material. If
the reaction conditions reduce the mean efficiency to 85%, then the yield in
those 20 cycles
will be only 1.85'-°, or 220,513 copies of the starting material. In
other words, a PCR running
a
_q._
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
at 85% efficiency will yield only 21% as much final product,, compared to a
reaction running
at 100% efficiency. A reaction that is reduced to 50% mean efficiency will
yield less than
1 % of the possible product.
In practice, routine polymerase chain reactions rarely achieve the theoretical
maximum
yield, and PCRs are usually run for more than 20 cycles to compensate for the
lower yield.
At 50% mean efficiency, it would take 34 cycles to achieve ~.he million-fold
amplification
theoretically possible in 20, and at lower efficiencies, the number of cycles
required becomes
prohibitive. In addition, any background products that amplify with a better
mean efficiency
than the intended target will become the dominant products.
Also, many variables can influence the mean efficiency of PCR, including
target DNA
length and secondary structure, primer length and design, primer and dNTP
concentrations,
and buffer composition, to name but a few. Contamination of the reaction with
exogenous
DNA (e.g., DNA spilled onto lab surfaces) or cross-contamination is also a
major
consideration. Reaction conditions must be carefully optimized for each
different primer pair
and target sequence, and the process can take days, even for an experienced
investigator. The
laboriousness of this process, including numerous technical considerations and
other factors,
presents a significant drawback to using PCR in the clinical setting. Indeed,
PCR has yet to
penetrate the clinical market in a significant way. The same concerns arise
with LCR, as
LCR must also be optimized to use different oligonucleotide sequences for each
target
sequence. In addition, both methods require expensive equipment, capable of
precise
temperature cycling.
Many applications of nucleic acid detection technologies, such as in studies
of allelic
variation, involve not only detection of a specific sequence in a complex
background, but also
the discrimination between sequences with few, or single, nucleotide
differences. One method
for the detection of allele-specific variants by PCR is based upon the fact
that it is difficult
for Tack polymerase to synthesize a DNA strand when there is a mismatch
between the
template strand and the 3' end of the primer. An allele-specific variant may
be detected by
the use of a primer that is perfectly matched with only one of the possible
alleles; the
mismatch to the other allele acts to prevent the extension of the primer,
thereby preventing
the amplification of that sequence. This method has a substantial limitation
in that the base
composition of the mismatch influences the ability to prevent extension across
the mismatch,
and certain mismatches do not prevent extension or have only a minimal effect
(Kwol: et cal.,
Nucl. Acids Res., 18:999 [1990]).)
-5-
CA 02203627 1997-04-24
WO 96/15267 PCTIUS95I14673
A similar 3'-mismatch strategy is used with greater effect to prevent ligation
in the
LCR (Barany, PCR Meth. Applic., 1:5 [1991]). Any mismatch effectively blocla
the action
of the thermostable ligase, but LCR still has the drawback of target-
independent background
ligation products initiating the amplification. Moreover, the combination of
PCR with
A
subsequent LCR to identify the nucleotides at individual positions is also a
clearly
cumbersome proposition for the clinical laboratory.
II. Direct Detection Technology
When a sufficient amount of a nucleic acid to be detected is available, there
are
advantages to detecting that sequence directly, instead of making more copies
of that target,
(e.~T., as in PCR and LCR). Most notably, a method that does not amplify the
signal
exponentially is more amenable to quantitative analysis. Even if the signal is
enhanced by
attaching multiple dyes to a single oligonucleotide, the correlation between
the final signal
intensity and amount of target is direct. Such a system has an additional
advantage that the
products of the reaction will not themselves promote further reaction, so
contamination of lab
surfaces by the products is not as much of a concern. Traditional methods of
direct detection
including Northern and Southern blotting and RNase protection assays usually
require the use
of radioactivity and are not amenable to automation. Recently devised
techniques have sought
to eliminate the use of radioactivity and/or improve the sensitivity in
automatable formats.
Two examples are the "Cycling Probe Reaction" (CPR), and "Branched DNA"
(bDNA).
The cycling probe reaction (CPR) (Duck et al., BioTech., 9:142 [1990]), uses a
long
chimeric oligonucleotide in which a central portion is made of RNA while the
two termini are
made of DNA. Hybridization of the probe to a target DNA and exposure to a
thermostable
RNase H causes the RNA portion to be digested. This destabilizes the remaining
DNA
portions of the duplex, releasing the remainder of the probe from the target
DNA and
allowing another probe molecule to repeat the process. The signal, in the form
of cleaved
probe molecules, accumulates at a linear rate. While the repeating process
increases the
signal, the RNA portion of the oligonucleotide is vulnerable to RNases that
may carried
through sample preparation.
Branched DNA (bDNA), described by Urdea et al., Gene 61:253-264 (1987),
involves
oligonucleotides with branched structures that allow each individual
oligonucleotide to carry '
3~ to 40 labels (e.g., alkaline phosphatase enzymes). While this enhances the
signal from a
hybridization event, signal from non-specific binding is similarly increased.
-6-
CA 02203627 1997-04-24
WO 96115267 PCT/US95/14673
III. Detection Of Unknown Sequence Changes
The demand for tests which allow the detection of spe;ciflc nucleic acid
sequences and
sequence changes is growing rapidly in clinical diagnostics. As nucleic acid
sequence data for
genes from humans and pathogenic organisms accumulates, the demand for fast,
cost-
s effective, and easy-to-use tests for as yet unknown mutations within
specific sequences is
rapidly increasing.
A handful of methods have been devised to scan nucleic acid segments for
mutations.
One option is to determine the entire gene sequence of each lest sample (e.g.,
a bacterial
isolate). For sequences under approximately 600 nucleotides, this may be
accomplished using
amplified material (e.g., PCR reaction products). This avoids the time and
expense associated
with cloning the segment of interest. However, specialized equipment and
highly trained
personnel are required, and the method is too labor-intense and expensive to
be practical and
effective in the clinical setting.
In view of the difficulties associated with sequencing, a given segment of
nucleic acid
may be characterized on several other levels. At the lowest resolution, the
size of the
molecule can be determined by electrophoresis by comparison to a known
standard run on the
same gel. A more detailed picture of the molecule may be achieved by cleavage
with
combinations of restriction enzymes prior to electrophoresis, to allow
construction of an
ordered map. The presence of specific sequences within the fragment can be
detected by
hybridization of a labeled probe, or the precise nucleotide sequence can be
determined by
partial chemical degradation or by primer extension in the presence of chain-
terminating
nucleotide analogs.
For detection of single-base differences between like sequences, the
requirements of
the analysis are often at the highest level of resolution. For cases in which
the position of the
nucleotide in question is known in advance, several methods have been
developed for
examining single base changes without direct sequencing. For example, if a
mutation of
interest happens to fall within a restriction recognition sequence, a change
in the pattern of
digestion can be used as a diagnostic tool (e.g., restriction fragment length
polymorphism
[RFLP] analysis).
Single point mutations have been also detected by the creation or destruction
of
' RFLPs. Mutations are detected and localized by the presence and size of the
RNA fragments
generated by cleavage at the mismatches. Single nucleotide :mismatches in DNA
heteroduplexes are also recognized and cleaved by some chemicals, providing an
alternative
CA 02203627 1997-04-24
WO 96/15267 PC"T/US95/14673
strategy to detect single base substitutions, generically named the "Mismatch
Chemical
Cleavage" (MCC) (Gogos et al. , Nucl. Acids Res., 18:6807-6817 [ 1990]).
However, this
method requires the use of osmium tetroxide and piperidine, two highly noxious
chemicals
which are not suited for use in a clinical laboratory.
RFLP analysis suffers from low sensitivity and requires a large amount of
sample.
F
When RFLP analysis is used for the detection of point mutations, it is, by its
nature, limited
to the detection of only those single base changes which fall within a
restriction sequence of a
known restriction endonuclease. Moreover, the majority of the available
enzymes have 4 to 6
base-pair recognition sequences, and cleave too frequently for many large-
scale DNA
manipulations (Eckstein and Lilley (eds.), Nucleic Acids and Molecular°
Biolo~~, vol. 2,
Springer-Verlag, Heidelberg [1988]). Thus, it is applicable only in a small
fraction of cases,
as most mutations do not fall within such sites.
A handful of rare-cutting restriction enzymes with 8 base-pair specificities
have been
isolated and these are widely used in genetic mapping, but these enzymes are
few in number,
are limited to the recognition of G+C-rich sequences, and cleave at sites that
tend to be highly
clustered (Barlow and Lehrach, Trends Genet., 3:167 [1987]). Recently,
endonucleases
encoded by group I introns have been discovered that might have greater than
12 base-pair
specificity (Penman and Butow, Science 246:1106 [1989]), but again, these are
few in
number.
If the change is not in a recognition sequence, then allele-specific
oligonucleotides
(ASOs), can be designed to hybridize in proximity to the unknown nucleotide,
such that a
primer extension or ligation event can be used as the indicator of a match or
a mis-match.
Hybridization with radioactively labeled allelic specific oligonucleotides
(ASO) also has been
applied to the detection of specific point mutations (Conner et al., Proc.
Natl. Acad. Sci.,
80:278-282 [1983]). The method is based on the differences in the melting
temperature of
short DNA fragments differing by a single nucleotide. Stringent hybridization
and washing
conditions can differentiate between mutant and wild-type alleles. The ASO
approach applied
to PCR products also has been extensively utilized by various researchers to
detect and
characterize point mutations in ras genes (Vogelstein et crl., N. Eng. J.
Med., 319:52-~3?
[1988]; and Farr et al., Proc. Natl. Acad. Sci., 85:1629-1633 [1988]), and
gaplgip oncogenes
(Lyons et al., Science 249:655-659 [1990]). Because of the presence of various
nucleotide
changes in multiple positions, the ASO -method requires the use of many
oligonucleotides to
cover all possible oncogenic mutations.
_g_
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
With either of the techniques described above (i.e., RF LP and ASO), the
precise
location of the suspected mutation must be known in advance: of the test. That
is to say, they
are inapplicable when one needs to detect the presence of a mutation of an
unknown character
and position within a gene or sequence of interest.
Two other methods rely on detecting changes in electrophoretic mobility in
response to
r
minor sequence changes. One of these methods, termed "Denaturing Gradient Gel
Electrophoresis" (DGGE) is based on the observation that slightly different
sequences will
display different patterns of local melting when electrophoretically resolved
on a gradient gel.
In this manner, variants can be distinguished, as differences in melting
properties of
homoduplexes versus heteroduplexes differing in a single nucleotide can detect
the presence
of mutations in the target sequences because of the corresponding changes in
their
electrophoretic mobilities. The fragments to be analyzed, usually PCR
products, are
"clamped" at one end by a long stretch of G-C base pairs (30-80) to allow
complete
denaturation of the sequence of interest without complete dissociation of the
strands. The
attachment of a GC "clamp" to the DNA fragments increases the fraction of
mutations that
can be recognized by DGGE (Abrams et al., Genomics 7:463-475 [1990]).
Attaching a GC
clamp to one primer is critical to ensure that the amplified sequence has a
low dissociation
temperature (Sheffield et al., Proc. Natl. Acad. Sci., 86:232-236 [1989]; and
Lerman and
Silverstein, Meth. Enzymol., 155:482-SO1 [1987]). Modifications of the
technique have been
developed, using temperature gradients (Wartell et al., Nucl. Acids Res.,
18:2699-2701
[1990]), and the method can be also applied to RNA:RNA duplexes (Smith et al.,
Genomics
3:217-223 [1988]).
Limitations on the utility of DGGE include the requirement that the denaturing
conditions must be optimized for each type of DNA to be tested. Furthermore,
the method
requires specialized equipment to prepare the gels and maintain the needed
high temperatures
during electrophoresis. The expense associated with the synthesis of the
clamping tail on one
oligonucleotide for each sequence to be tested is also a major consideration.
In addition. long
running times are required for DGGE. The long running time of DGGE was
shortened in a
modification of DGGE called constant denaturant gel electrophoresis (CDGE)
(Borrensen et
ul., Proc. Natl. Acad. Sci. USA 88:8405 [1991]). CDGE requires that gels be
performed
under different denaturant conditions in order to reach high efficiency for
the detection of
unknown mutations.
-9-
CA 02203627 1997-04-24
WO 96115267 PCTIUS95114673
An technique analogous to DGGE, termed temperature gradient gel
electrophoresis
(TGGE), uses a-thermal gradient rather than a chemical denaturant gradient
(Scholz; et al.,
Hum. Mol. Genet. 2:2155 [1993]). TGGE requires the use of specialized
equipment which
can generate a temperature gradient perpendicularly oriented relative to the
electrical field.
TGGE can detect mutations in relatively small fragments of DNA therefore
scanning of large
gene segments requires the use of multiple PCR products prior to running the
gel.
Another common method, called "Single-Strand Conformation Polymorphism" (SSCP)
was developed by Hayashi, Sekya and colleagues (reviewed by Hayashi, PCR Meth.
Appl..
1:34-38, [1991]) and is based on the observation that single strands of
nucleic acid can take
on characteristic conformations in non-denaturing conditions, and these
conformations
influence electrophoretic mobility. The complementary strands assume
sufficiently different
structures that one strand may be resolved from the other. Changes in
sequences within the
fragment will also change the conformation, consequently altering the mobility
and allowing
this to be used as an assay for sequence variations (Orita, et al., Genomics
5:874-879,
[1989]).
The SSCP process involves denaturing a DNA segment (e.g., a PCR product) that
is
labelled on both strands, followed by slow electrophoretic separation on a non-
denaturing
polyacrylamide gel, so that intra-molecular interactions can form and not be
disturbed during
the run. This technique is extremely sensitive to variations in gel
composition and
temperature. A serious limitation of this method is the relative difficulty
encountered in
comparing data generated in different laboratories, under apparently similar
conditions.
The dideoxy fingerprinting (ddF) is another technique developed to scan genes
for the
presence of unknown mutations (Liu and Sommer, PCR Methods Appli., 4:97
[1994]). The
ddF technique combines components of Sanger dideoxy sequencing with SSCP. A
dideoxy
sequencing reaction is performed using one dideoxy terminator and then the
reaction products
are electrophoresised on nondenaturing polyacrylamide gels to detect
alterations in mobility of
the termination segments as in SSCP analysis. While ddF is an improvement over
SSCP in
terms of increased sensitivity, ddF requires the use of expensive
dideoxynucleotides and this
technique is still limited to the analysis of fragments of the size suitable
for SSCP (i.c.,
fragments of 200-300 bases for optimal detection of mutations).
In addition to the above limitations, all of these methods are limited as to
the size of
the nucleic acid fragment that can be analyzed. For the direct-sequencing
approach.
sequences of greater than 600 base pairs require cloning, with the consequent
delays and
- 10-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
expense of either deletion sub-cloning or primer walking, in order to cover
the entire
fragment. SSCP and DGGE have even more severe size limitations. Because of
reduced
sensitivity to sequence changes, these methods are not considered suitable for
larger
fragments. Although SSCP is reportedly able to detect 90% of single-base
substitutions
y 5 within a 200 base-pair fragment, the detection drops to less than 50% for
400 base pair
fragments. Similarly, the sensitivity of DGGE decreases as the length of the
fragment reaches
500 base-pairs. The ddF technique, as a combination of direct sequencing and
SSCP, is also
limited by the relatively small size of the DNA that can be screened.
Clearly, there remains a need for a method that is less sensitive to size so
that entire
genes, rather than gene fragments, may be analyzed. Such a. tool must also be
robust, so that
data from different labs, generated by researchers of diverse backgrounds and
skills will be
comparable. Ideally, such a method would be compatible with "multiplexing,"
(i.c., the
simultaneous analysis of several molecules or genes in a single reaction or
gel lane, usually
resolved from each other by differential labelling or probing j. Such an
analytical procedure
would facilitate the use of internal standards for subsequent analysis and
data comparison, and
increase the productivity of personnel and equipment. The ideal method would
also be easily
automatable.
SUMMARY OF THE INVENTION
The present invention relates to methods and compositions for treating nucleic
acid,
and in particular, methods and compositions for detection and characterization
of nucleic acid
sequences and sequence changes in human gene sequences and in microbial gene
sequences.
The present invention provides means for cleaving a nucleic acid cleavage
structure in a site-
specific manner. In one embodiment, the means for cleaving is an enzyme
capable of
cleaving cleavage structures on a nucleic acid substrate, forming the basis of
a novel method
of detection of specific nucleic acid sequences. The present invention
contemplates use of the
novel detection method for, among other uses, clinical diagnostic purposes,
including but not
limited to detection and identification of 1 ) mutations in human gene
sequences and 2 )
pathogenic organisms.
In one embodiment, the present invention contemplates a DNA sequence encoding
a
DNA polymerase altered in sequence (i.e., a "mutant" DNA polymerase) relative
to the native
sequence such that it exhibits altered DNA synthetic activity from that of the
native (i.c..
"wild type") DNA polymerase. With regard to the polymera.se, a complete
absence of
CA 02203627 1997-04-24
WO 96/15267 PCT/LTS95/14673
synthesis is not required; it is desired that cleavage reactions occur in the
absence of
polymerise activity at a level where it interferes with the method. It is
preferred that the
encoded DNA polymerise is altered such that it exhibits reduced synthetic
activity from that
of the native DNA polymerise. In this manner, the enzymes of the invention are
nucleases
and are capable of cleaving nucleic acids in a structure-specific manner.
Importantly, the
nucleases of the present invention are capable of cleaving cleavage structures
to create discrete
cleavage products.
The present invention contemplates nucleases from a variety of sources,
including
nucleases that are thermostable. Thermostable nucleases are contemplated as
particularly
useful, as they are capable of operating at temperatures where nucleic acid
hybridization is
extremely specific, allowing for allele-specific detection (including single-
base mismatches).
In one embodiment, the thermostable 5' nucleases are selected from the group
consisting of
altered polymerises derived from the native polymerises of various Thermus
species,
including, but not limited to Thermus aquaticzrs, Thermus.flavus and Thermus
thermophilu.s.
The present invention is not limited to the use of thermostable nucleases. As
demonstrated herein nucleases from mesophilic organisms may also be employed
in the
methods of the invention (e.g., E. coli Exo III, Saccharomyce.s cerevisiae
Radi/RadlO
complex).
The present invention utilizes nucleases in methods for detection and
characterization
of nucleic acid sequences and sequence changes. The present invention relates
to means for
cleaving a nucleic acid cleavage structure in a site-specific manner. Nuclease
activity is used
to screen for known and unknown mutations, including single base changes, in
nucleic acids.
In one embodiment, the present invention contemplates a method for treating
nucleic
acid, comprising: a) providing: i) a cleavage means and ii) nucleic acid
substrate; b) treating
the nucleic acid substrate under conditions such that the substrate forms one
or more cleavage
structures; and c) reacting the cleavage means with the cleavage structures so
that one or more
cleavage products are produced.
In one embodiment, the cleavage means is an enzyme. In a preferred embodiment,
the
cleavage means is a nuclease. In an alternative preferred embodiment, the
nuclease is selected
from the group consisting of the CleavaseTM BN enzyme, Thermus aquaticus DNA
polymerise, Thermus thermophilZrs DNA polymerise, Escherichia coli Exo III.
and the
Saccharomyces cerevisiae Radl/RadlO complex.
-12-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
It is contemplated that the nucleic acid substrate comprise a nucleotide
analog.
including but not limited to the group comprising 7-deaza-dATP, 7-deaza-dGTP
and dUTP.
In one embodiment, the nucleic acid of step is substantially single-stranded.
It is not intended
that the nucleic acid substrate be limited to any particular form, indeed, it
is contemplated that
the nucleic acid substrate is single stranded or double stranded DNA or RNA.
In one embodiment, when a double stranded nucleic acid substrate is employed,
the
treating step (b) comprises rendering the double-stranded nucleic acid
substantially single-
stranded and exposing the single-stranded nucleic acid to conditions such that
the single-
stranded nucleic acid assumes a secondary or characteristic folded structure.
In one preferred
embodiment, the double stranded nucleic acid is rendered substantially single-
stranded by
increased temperature.
In an alternative embodiment, the method of the present invention further
comprises
the step of detecting said one or more cleavage products.
In a preferred embodiment, the nucleic acid substrate comprises an
oligonucleotide
containing human p53 gene sequences. In an alternative embodiment, the nucleic
acid
substrate comprises an oligonucleotide containing microbial gene sequences.
The present invention contemplates further a method for treating nucleic acid,
comprising: a) providing: i) a cleavage means in a solution comprising
manganese and ii) a
nucleic acid substrate; b) treating the nucleic acid substrate with increased
temperature; c)
reducing the temperature under conditions such that the substrate forms one or
more cleavage
structures; d) reacting the cleavage means with the cleavage structures so
that one or more
cleavage products are 'produced: and e) detecting the cleavage products.
Again, the cleavage
means may be an enzyme. As noted above, the cleavage means may be a nuclease.
In an
alternative preferred embodiment. the nuclease is selected from the group
consisting of the
CleavaseTM BN enzyme, Thermus aguaticus DNA polymerase, Thermus thermophilz~s
DNA
polymerase, Escherichia coli Exo III, and the Saccharornyces cerevisiae
Radl/RadlO complex.
It is contemplated that the nucleic acid substrate comprise a nucleotide
analog,
including, but not limitedto, the group comprising 7-deaza-dATP, 7-deaza-dGTP
and dUTP.
In one embodiment, the nucleic acid of step is substantially single-stranded.
It is not intended
that the nucleic acid substrate be limited to any particular form, indeed, it
is contemplated that
the nucleic acid substrate is single stranded or double stranded DNA or RNA.
- 13 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95114673
In a preferred embodiment, the nucleic acid substrate comprises an
oligonucleotide
containing human p53 gene sequences. In an alternative embodiment, the nucleic
acid
substrate comprises an oligonucleotide containing microbial gene sequences.
The present invention contemplates further, a method for detecting mutation in
the
human p53 gene, comprising: a) providing: i) a cleavage means and ii) a
nucleic acid
substrate containing human p53 gene sequences; b) treating the nucleic acid
substrate under
conditions such that the substrate forms one or more cleavage structures; c)
reacting the
cleavage means with the cleavage structures so that one or more cleavage
products are
produced; and d) comparing said cleavage products to the cleavage products
produced by
cleavage of a reference p53 gene sequence.
In a preferred embodiment, the cleavage products produced by cleavage of a
reference
p53 gene sequence are generated by the cleavage of a nucleic acid substrate
containing the
human p53 gene sequences selected from the group consisting of SEQ ID NOS:79-
81, 84-89
and 94-97. Additional p53 mutant sequences are provided herein; SEQ ID N0:79
lists the
I ~ sequence of the wild-type p53 cDNA. Table 2 below provides the identity
and location of
numerous known p53 mutations. Combination of the information in Table 2 with
the
sequence of the wild-type p53 cDNA in SEQ ID N0:79 allows the generation of
the complete
nucleotide sequence for cDNAs corresponding to the numerous p53 mutations
described in
Table 2. In addition, as described fully herein, the method of the invention
permits the
screening of or "scanning" for heretofore uncharacterized mutations within
human gene
sequences, such as the human p53 gene.
The present invention also contemplates a process for creating a record
reference
library of genetic fingerprints characteristic (i.e., diagnostic) of one or
more alleles of the
human p53 gene comprising: a) providing: i) a cleavage means, and ii) nucleic
acid substrate
derived from human p53 gene sequences; b) contacting said nucleic acid
substrate with a
cleavage means under conditions such that said extracted nucleic acid forms
one or more
secondary structures and said cleavage means cleaves said secondary structures
resulting in the
generation of multiple cleavage products; c) separating said multiple cleavage
products; and d)
maintaining a testable record reference of said separated cleavage products. .
By the term "genetic fingerprint" it is meant that changes in the sequence of
the
nucleic acid (e.g., a deletion, insertion or a single point substitution)
alter the structures
formed, thus changing the banding pattern (i.e., the "fingerprint" or "bar
code") to reflect the
difference in the sequence, allowing rapid detection and identification of
variants.
- I4-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
The present invention also contemplates a process for creating a record
reference
library of genetic fingerprints characteristic (i.e., diagnostic) of one or
more alleles of one or
more genes from a eukaryotic organism (e.g., mammals) comprising: a)
providing: i) a
cleavage means; and ii) nucleic acid substrate derived from one or more
alleles of a gene
derived from a eukaryotic organism; b) contacting said nucleic acid substrate
with a cleavage
means under conditions such that said extracted nucleic acid forms one or more
secondary
structures and said cleavage means cleaves said secondary structures resulting
in the
generation of multiple cleavage products; c) separating said multiple cleavage
products; and d)
maintaining a testable record reference of said separated cleavage products.
The present invention also contemplates a method for identifying strains of
microorganisms comprising: a) providing i) a cleavage means; and ii) a nucleic
acid substrate
containing sequences derived from one or more microorgani sm; b) treating said
nucleic acid
substrate under conditions such that said substrate forms one or more cleavage
structures; and
c) reacting said cleavage means with said cleavage structure s so that one or
more cleavage
I S products are produced.
The preferred cleavage means is an enzyme, such as a nuclease. Examples of
enzymes
that can be used with success with the method of the present invention include
(but are not
limited to) the CleavaseTM BN enzyme, Thermus aquaticus DNA polymerase,
Thermus
thermophilus DNA polymerase, Escherichia coli Exo III, and the Saccharomvcc~s
cef°evi.siae
Rad 1 /Rad 10 complex.
It is contemplated that the nucleic acid substrate comprise a nucleotide
analog,
including but not limited to the group comprising 7-deaza-d,ATP, 7-deaza-dGTP
and dUTP.
In one embodiment, the nucleic acid of step is substantially single-stranded.
It is not intended
that the nucleic acid substrate be limited to any particular form, indeed, it
is contemplated that
the nucleic acid substrate is single stranded or double stranded DNA or RNA.
In one embodiment, when a double stranded nucleic acid substrate is employed,
the
treating step (b) comprises rendering the double-stranded nucleic acid
substantially single-
stranded and exposing the single-stranded nucleic acid to conditions such that
the single-
stranded nucleic acid assumes a secondary or characteristic folded structure.
In one preferred
embodiment, the double stranded nucleic acid is rendered substantially single-
stranded by
increased temperature.
In an alternative embodiment, the method of the present invention further
comprises
the step of detecting said one or more cleavage products.
-15-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
It is contemplated that the microorganisms) of the present invention be
selected from
a variety of microorganisms; it is not intended that the present invention be
limited to any
particular type of microorganism. Rather, it is intended that the present
invention will be
used with organisms including, but not limited to, bacteria, fungi, protozoa,
ciliates, and
r
viruses. It is not intended that the microorganisms be limited to a particular
genus, species,
strain, or serotype. Indeed, it is contemplated that the bacteria be selected
from the group
comprising, but not limited to members of the genera Campylobacter,
Escherichia,
Mvcobacterium, Salmonella, Shigella,and Staphylococczrs. In one preferred
embodiment, the
microorganisms) comprise strains of mufti-drug resistant Mycobacterium
tuberculosis. It is
also contemplated that the present invention be used with viruses, including
but not limited to
hepatitis C virus and simian immunodeficiency virus.
Another embodiment of the present invention contemplates a method for
detecting and
identifying strains of microorganisms, comprising the steps of extracting
nucleic acid from a
sample suspected of containing one or more microorganisms and contacting the
extracted
nucleic acid with a cleavage means under conditions such that the extracted
nucleic acid
forms one or more secondary structures, and the cleavage means cleaves the
secondary
structures to produce one or more cleavage products.
In one embodiment, the method further comprises the step of separating said
cleavage
products. In yet another embodiment, the method further comprises the step of
detecting said
cleavage products.
In one preferred embodiment, the present invention further comprises comparing
said
detected cleavage products generated from cleavage of the extracted nucleic
acid isolated from
the sample with separated cleavage products generated by cleavage of nucleic
acids derived
from one or more reference microorganisms. In such a case, the sequence of the
nucleic acids
from one or more reference microorganisms may be related but different (e.g.,
a wild-type
control for a mutant sequence or a known or previously characterized mutant
sequence).
In an alternative preferred embodiment, the present invention further
comprises the
step of isolating a polymorphic locus from said extracted nucleic acid after
the extraction step,
so as to generate a nucleic acid substrate, wherein the substrate is_
contacted with the cleavage
means. In one embodiment, the isolation of a polymorphic locus is accomplished
by nucleic
acid amplification. The invention is limited by the method of nucleic acid
amplification
employed. One method of achieving nucleic acid amplification is the polymerase
chain
reaction. In an alternate embodiment, the nucleic acid amplification is
conducted in the
-16-
CA 02203627 1997-04-24
WO 96/15267 PC"T/US95/14673
presence of a nucleotide analog, including but not limited to the group
comprising 7-deaza-
dATP, 7-deaza-dGTP and dUTP. It is contemplated that the nucleic acid
amplification (e.g.,
PCR) will employ oligonucleotide primers which either 1 ) match consensus gene
sequences
derived from the polymorphic locus (i. e., the primers comprise the same
sequence found on a
strand of nucleic acid derived from the polymorphic locus) o:r 2) are
complementary to
consensus gene sequences derived from said polymorphic locus (i. e., they are
the complement
to a strand of nucleic acid derived from the polymorphic locus). In one
embodiment, the
polymorphic locus comprises a ribosomal RNA gene. In a particularly preferred
embodiment,
the ribosomal RNA gene is a 16S ribosomal RNA gene.
In one embodiment of this method, the cleavage means is an enzyme, such as a
nuclease. In a particularly preferred embodiment, the nuclease is selected
from the group
including, but not limited to CleavaseTM BN, Thermus aquaticus DNA polymerase,
Thermus
thc~rmophilus DNA polymerase, Escherichia coli Exo III, and. the Saccharomyces
cerevisicre
Rad 1 /Rad I 0 complex. It is also contemplated that the enzyme may have a
portion of its
amino acid sequence that is homologous to a portion of the amino acid sequence
of a
thermostable DNA polymerase derived from a eubacterial the:rmophile, the
latter being
selected from the group consisting of Thermus aquaticus, Thermus , flavus and
Thermus
thermophilus.
It is not intended that the nucleic acid substrate be limited to any
particular form,
indeed, it is contemplated that the nucleic acid substrate is single stranded
or double-stranded
RNA or DNA. When a double stranded nucleic acid substrate is employed, the
treating step
of the method may comprise rendering double-stranded nucleic acid
substantially single-
stranded, and exposing the single-stranded nucleic acid to conditions such
that the single-
stranded nucleic acid has secondary structure. In one preferred embodiment,
double-stranded
nucleic acid is rendered substantially single-stranded by increased
temperature.
It is contemplated that the microorganisms) of the present invention be
selected from
a variety of microorganisms; it is not intended that the present invention be
limited to any
particular type of microorganism. Rather, it is intended that the present
invention will be
used with organisms including, but not limited to, bacteria, fungi, protozoa,
ciliates, and
viruses. It is not intended that the microorganisms be limited to a particular
genus, species,
strain, or serotype. Indeed, it is contemplated that the bacteria be selected
from the group
comprising, but not limited to members of the genera Campylobacter°,
Escher-ichicr,
Mycobacterium. Salmonella, Shigella, and Staphylococcus. In one preferred
embodiment, the
-17-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
microorganisms) comprise strains of mufti-drug resistant Mycobacterium
tuberculosis. It is
also contemplated that the present invention be used with viruses, including
but not .limited to
hepatitis C virus and simian immunodeficiency virus.
The present invention also contemplates a process for creating a record
reference
library of genetic fingerprints characteristic (i.e., diagnostic) of one or
more alleles of the ,
various microorganisms, comprising the steps of providing a cleavage means and
nucleic acid
substrate derived from microbial gene sequences; contacting the nucleic acid
substrate with a
cleavage means under conditions such that the extracted nucleic acid forms one
or more
secondary structures and the cleavage means cleaves the secondary structures,
resulting in the
generation of multiple cleavage products; separating the multiple cleavage
products; and
maintaining a testable record reference of the separated cleavage products.
It is not intended that the present invention be limited by the nature of the
microorganism. The detection and identification is application to all
organisms, including
viruses and bacteria. -
The present invention also contemplates a process for creating a record
reference (e.g.,
library) of genetic fingerprints characteristic (i.e., diagnostic) of
pathogenic microorganisms
comprising: a) providing: i) a cleavage means; and ii) a nucleic acid
substrate characteristic of
(e.~T., derived from a polymorphic locus) isolated from a known pathogenic
microorganism; b)
contacting said nucleic acid substrate with a cleavage means under conditions
such that said
extracted nucleic acid forms one or more secondary structures and said
cleavage means
cleaves said secondary structures resulting in the generation of multiple
cleavage products; c)
separating said multiple cleavage products; and d) maintaining a record
reference of said
separated cleavage products.
The present invention also contemplates a nucleic acid treatment kit,-
comprising: a) an
enzyme capable of reacting with cleavage structures so as to generate cleavage
products, and
b) a solution comprising manganese. The enzyme of the kit may be a nuclease.
In a
preferred embodiment the nuclease is elected from the group including, but not
limited to
CleavaseTM BN, Thermus aquaticus DNA polymerase, Thernzus--thermophilus DNA
polymerase, Escherichia coli Exo III, and the Saccharomyces cerevisiae
Radl/RadlO complex.
The present invention contemplates other reagents useful for the treatment of
nucleic acid.
For example, the kit may include reagents for detecting said cleavage
products. Furthermore,
the kit may include reagents for the cleavage reaction including salt
solutions (e.~.. IiCI and
-18-
CA 02203627 1999-07-26
NaCl solutions), manganese chloride solutions, buffer solutions
and solutions which terminate the cleavage reaction.
The methods of the present invention allow for simultaneous
analysis of both strands (e. g., the sense and antisense strands)
and are ideal for high-level multiplexing. The products
produced are amenable to qualitative, quantitative and
positional analysis. The methods may be automated and may be
practiced in solution or in the solid phase (e. g., on a solid
support). The methods are powerful in that they allow for
analysis of longer fragments of nucleic acid than current
methodologies.
More specifically, the present invention provides a
method for treating nucleic acid, comprising: (a) providing:
i) an enzymatic cleavage means comprising a nuclease; and
ii) a nucleic acid substrate; (b) treating said nucleic acid
substrate under conditions such that said substrate forms at
least one cleavage structure; and (c) reacting said cleavage
means with said cleavage structure so that at least one cleavage
product is produced.
The present invention also provides A method for
treating nucleic acid, comprising: (a) providing: i) an
enzymatic cleavage means comprising a nuclease, in a solution
comprising manganese; and ii) a nucleic acid substrate; (b)
treating said nucleic acid substrate with increased temperature;
(c) reducing said temperature under conditions such that said
substrate forms at least one cleavage structure; (d) reacting
said cleavage means with said cleavage structure so that at
least one cleavage product is produced; and (e) detecting said
at least one cleavage product.
The present invention also provides a method for detecting
mutation in the human p53 gene, comprising:(a) providing:
i) an enzymatic cleavage means wherein comprising a nuclease;
and ii) a nucleic acid substrate containing human p53 gene
sequences; (b) treating said nucleic acid substrate under
conditions such that said substrate forms at least one cleavage
19
CA 02203627 1999-07-26
structure; (c) reacting said cleavage means with said cleavage
structure so that at least one cleavage product is produced; and
(d) comparing said cleavage product to the cleavage products
produced by cleavage of a reference p53 gene sequence.
The present invention also provides a method for
identifying strains of microorganisms comprising: (a) providing:
i) an enzymatic cleavage means comprising a nuclease; and ii) a
nucleic acid substrate containing sequences derived from at
least one microorganism; (b) treating said nucleic acid
substrate under conditions such that said substrate forms at
least one cleavage structure; (c) reacting said cleavage means
with said cleavage structure so that at least one cleavage
product is produced; and (d) comparing said cleavage product to
the cleavage products produced by cleavage of a reference
sequence derived from a microorganism.
The present invention also provides a method comprising:
(a) extracting nucleic acid from a sample suspected of
containing at least one microorganism; and (b) contacting said
extracted nucleic acid with an enzymatic cleavage means
comprising a nuclease, under conditions such that said extracted
nucleic acid forms one or more secondary structures, and said
cleavage means cleaves said secondary structures to produce at
least one cleavage product.
The present invention also provides a method comprising:
(a) providing: i) an enzymatic cleavage means comprising a
nuclease; and ii) a nucleic acid target substrate suspected of
containing sequence variation relative to a reference control;
(b) mixing said cleavage means and said substrate under
conditions such that said substrate forms at least one secondary
structure and said cleavage means cleaves said secondary
structure resulting in the generation of multiple cleavage
products; and (c) separating said multiple cleavage products so
as to detect said sequence variation.
The present invention also provides a method comprising:
(a) providing: i) an enzymatic cleavage means comprising a
19a
CA 02203627 1999-07-26
nuclease; and ii) a nucleic acid target substrate suspected of
containing sequence variation relative to a reference control;
(b) mixing said cleavage means and said substrate at an elevated
temperature and under conditions such that said substrate forms
at least one secondary structure and said cleavage means cleaves
said secondary structure resulting in the generation of multiple
cleavage products; and (c) separating said multiple cleavage
products so as to detect said sequence variation.
The present invention also provides a method comprising:
(a) providing: i) a thermostable DNA polymerase altered in amino
acid sequence such that it exhibits reduced DNA synthetic
activity from that of the wild-type DNA polymerase but retains
substantially the same 5' nuclease activity of the wild-type DNA
polymerase; and ii) a nucleic acid target substrate suspected of
containing sequence variation relative to a reference control;
(b) mixing said polymerase and said substrate under conditions
such that said substrate forms at least one secondary structure
and said polymerase cleaves said secondary structure resulting
in the generation of multiple cleavage products; and (c)
separating said multiple cleavage products so as to detect said
sequence variation.
DESCRIPTION OF THE DRAWINGS
Figure 1 is a comparison of the nucleotide structure of the
DNAP genes isolated from Thermus aquaticus (SEQ ID NO:1),
Thermus flavus (SEQ ID N0:2) and Thermus thermophilus (SEQ ID
N0:3); the consensus sequence (SEQ ID N0:7) is shown at the top
of each row.
Figure 2 is a comparison of the amino acid sequence of the
DNAP isolated from Thermus aquaticus (SEQ ID N0:4), Thermus
flavus (SEQ ID N0:5) and Thermus thermophilus (SEQ ID N0:6); the
consensus sequence (SEQ ID N0:8) is shown at the top of each
row.
19b
CA 02203627 1999-07-26
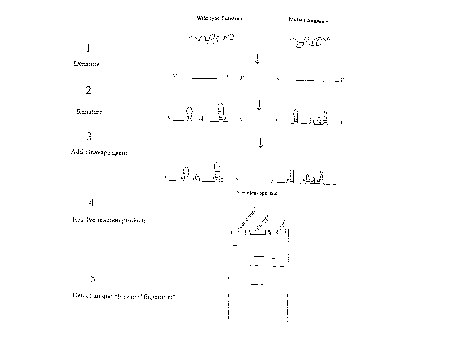
Figure 3 is a schematic showing the CFLPT"" method of
generating a characteristic fingerprint from a nucleic acid
substrate.
Figure 4 depicts the organization of the human p53 gene;
exons are represented by the solid black boxes and are labelled
1-11. Five hot spot regions are shown as a blow-up of the
region spanning exons 5-8; the hot spot regions are labelled A,
A', B, C, and D.
Figure 5 provides a schematic showing the use of a first 2-
step PCR technique for the generation DNA fragments containing
p53 mutations.
Figure 6 provides a schematic showing the use of a second
2-step PCR technique for the generation DNA fragments containing
p53 mutations.
Figure 7 depicts a structure which cannot be amplified
using DNAPTaq.
Figure 8 is an ethidium bromide-stained gel demonstrating
attempts to amplify a bifurcated duplex using either DNAPTaq or
DNAPStf(Stoffel).
Figure 9 is an autoradiogram of a gel analyzing the
cleavage of a bifurcated duplex by DNAPTaq and lack of cleavage
by DNAPStf.
19c
CA 02203627 1997-04-24
WO 96/15267 PC"TlUS95/14673
Figures 10 A-B are a set of autoradiograms of gels analyzing cleavage or lack
of
cleavage upon addition of different reaction components and change of
incubation temperature
during attempts to cleave a bifurcated duplex with DNAPTaq.
Figures 11 A-B are an autoradiogram displaying timed cleavage reactions. with
and
without primer.
Figures 12 A-B are a set of autoradiograms of gels demonstrating attempts to
cleave a
bifurcated duplex (with and without primer) with various DNAPs.
Figures 13A shows the substrates and oligonucleotides used to test the
specific
cleavage of substrate DNAs targeted by pilot oligonucleotides.
Figure 13B shows an autoradiogram of a gel showing the results of cleavage
reactions
using the substrates and oligonucleotides shown Fig. 13A.
Figure 14A shows the substrate and oligonucleotide used to test the specific
cleavage
of a substrate RNA targeted by a pilot oligonucleotide.
Figure 14B shows an autoradiogram of a gel showing the results of a cleavage
reaction
using the substrate and oligonucleotide shown in Fig. 14A. -
Figure 15 is a diagram of vector pTTQlB.
Figure 16A-G are a set of diagrams of wild-type and synthesis-deficient
DNAPTuq
genes. _.
Figure 17 is a diagram of vector pET-3c.
Figure 18A depicts the wild-type Thermus flavus polymerase gene.
Figure 18B depicts a synthesis-deficient Thermus.flavus polymerase gene.
Figures 19A-E depict a set of molecules which are suitable substrates for
cleavage by
the 5' nuclease activity of DNAPs.
Figure 20 is an autoradiogram of a gel showing the results of a cleavage
reaction run
with synthesis-deficient DNAPs.
Figure 21 is an autoradiogram of a PEI chromatogram resolving the products of
an
assay for synthetic activity in synthesis-deficient DNAPTaq clones.
Figure 22A depicts the substrate molecule used to test the ability of
synthesis-deficient
DNAPs to cleave short hairpin structures.
Figure 22B shows an autoradiogram of a gel resolving the products of a
cleavage
reaction run using the substrate shown in Fig. 22A.
Figure 23 provides the complete 206-mer duplex sequence employed as a
substrate for
the 5' nucleases of the present invention
-20-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
Figures 24A and B show the cleavage of linear nucleic acid substrates (based
on the
206-mer of Figure 23) by wild type DNAPs and 5' nucleases isolated from
Thermus~
ayuaticus and Thermus,flavus.
Figure 25A shows the "nibbling" phenomenon detected with the DNAPs of the
present
invention.
Figure 25B shows that the "nibbling" of Figure 25A is 5' nucleolytic cleavage
ald not
phosphatase cleavage.
Figure 26 demonstrates that the "nibbling" phenomenon is duplex dependent.
Figure 27 shows an autoradiograph of a gel resolving the products of cleavage
reactions run in the presence of either MgCh or MnCI,.
Figure 28 shows an autoradiograph of a gel resolving the products of cleavage
reactions run on four similarly sized DNA substrates.
Figure 29 shows an autoradiograph of a gel resolving the products of cleavage
reactions run using a wild-type and two mutant tyrosinase gene substrates.
Figure 30 shows an autoradiograph of a gel resolving the products of cleavage
reactions run using either a wild-type or mutant tyrosinase substrate varying
in length from
157 nucleotides to 1.587 kb.
Figure 31 shows an autoradiograph of a gel resolving the products of cleavage
reactions run in various concentrations of MnCl2.
Figure 32 shows an autoradiograph of a gel resolving the products of cleavage
reactions run in various concentrations of KCI.
Figure 33 shows an autoradiograph of a gel resolving the products of cleavage
reactions run for different lengths of time.
Figure 34 shows an autoradiograph of a gel resolving the products of cleavage
reactions run at different temperatures.
Figure 35 shows an autoradiograph of a gel resolving the products of cleavage
reactions run using different amounts of the enzyme CleavaseTM BN.
Figure 36 shows an autoradiograph of a gel resolving the products of cleavage
reactions run using four different preparations of the DNA substrate.
Figure 37 shows an autoradiograph of a gel resolving the products of cleavage
reactions run on either the sense or antisense strand of four different
tyrosinase gene
substrates.
-21 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
Figure 38 shows an autoradiograph of a gel resolving the products of cleavage
reactions run on a wild-type (3-globin substrate in two different
concentrations of KCl and at
four different temperatures.
Figure 39 shows an autoradiograph of a gel resolving the products of cleavage
reactions run on two different mutant (3-globin substrates in five different
concentrations of
K
KCI.
Figure 40 shows an autoradiograph of a gel resolving the products of cleavage
reactions run on a wild-type and three mutant (3-globin substrates.
Figure 41 shows an autoradiograph of a gel resolving the products of cleavage
reactions run on an RNA substrate.
Figure 42 shows an autoradiograph of a gel resolving the products of cleavage
reactions run using either the enzyme CleavaseTM BN or Taq DNA polymerise as
the 5"
nuclease.
Figure 43 shows an autoradiograph of a gel resolving the products of cleavage
reactions run on a double-stranded DNA substrate to demonstrate multiplexing
of the cleavage
reaction.
Figure 44 shows an autoradiograph of a gel resolving the products of cleavage
reactions run on double-stranded DNA substrates consisting of the 419 and 422
mutant alleles
derived from exon 4 of the human tyrosinase gene in the presence of various
concentrations
of MnCI,.
Figure 45 displays two traces representing two channel signals (JOE and FAM
fluorescent dyes) for cleavage fragments derived from a cleavage reaction
containing two
differently labelled substrates (the wild-type and 422 mutant substrates
derived from exon 4 of
the tyrosinase gene). The thin lines represent the JOE-labelled wild-type
substrate and the
thick lines represent the FAM-labelled 422 mutant substrate. Above the tracing
is an
autoradiograph of a gel resolving the products of cleavage reactions run on
double-stranded
DNA substrates consisting of the wild-type and 422 mutant alleles derived from
exon 4 of the
tyrosinase gene.
Figure 46 depicts the nucleotide sequence of six SIV LTR clones corresponding
to
SEQ ID NOS:63-68. -
Figure 47 shows an autoradiograph of a gel resolving the products of cleavage
reactions run on six different double-stranded SIV LTR substrates which
contained a biotic
label on the 5' end of the (-) strand.
-22-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
Figure 48 shows an autoradiograph of a gel resolving the products of cleavage
reactions run on six different double-stranded SIV -LTR substrates which
contained a biotin
label on the 5' end of the (+) strand.
Figure 49 shows an autoradiograph of a gel resolving the products of single-
stranded
cleavage reactions run in various concentrations of NaCI.
f
Figure 50 shows an autoradiograph of a gel resolving the products of single-
stranded
cleavage reactions run in various concentrations of (NH4)~SO4.
Figure 51 shows an autoradiograph of a gel resolving the products of single-
stranded
cleavage reactions run in increasing concentrations of KCI.
Figure 52 shows an autoradiograph of a gel resolving the products of single-
stranded
cleavage reactions run in two concentrations of KCl for various periods of
time.
Figure 53 shows an autoradiograph of a gel resolving the products of cleavage
reactions run on either the single-stranded or double-stranded form of the
same substrate.
Figure 54 shows an autoradiograph of a gel resolving the products of double-
stranded
I S cleavage reactions run in various concentrations of KCI.
Figure 55 shows an autoradiograph of a gel resolving the products of double-
stranded
cleavage reactions run in various concentrations of NaCI.
Figure 56 shows an autoradiograph of a gel resolving the products of double-
stranded
cleavage reactions run in various concentrations of (NH4)~504.
Figure 57 shows an autoradiograph of a gel resolving the products of double-
stranded
cleavage reactions run for various lengths of time.
Figure 58 shows an autoradiograph of a gel resolving the products of double-
stranded
cleavage reactions run using various amounts of CleavaseTM BN enzyme for
either ~ seconds
or 1 minute.
Figure 59 shows an autoradiograph of a gel resolving the products of double-
stranded
cleavage reactions run at various temperatures.
Figure 60 shows an autoradiograph of a gel resolving the products of double-
stranded
cleavage reactions run using various amounts of CleavaseTM BN enzyme.
Figure 61 A shows an autoradiograph of a gel resolving the products of single-
stranded
cleavage reactions run in buffers having various pHs.
Figure 61 B shows an autoradiograph of a gel resolving the products of single-
stranded
cleavage reactions run in buffers having a pH of either 7.5 or 7.8.
_ 23 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
Figure 62A shows an autoradiograph of a gel resolving the products of double-
stranded cleavage reactions run in buffers having a pH of either 8.2 or 7.2.
Figure 62B shows an autoradiograph of a gel resolving the products of double-
stranded
cleavage reactions run in buffers having a pH of either 7.5 or 7.8.
Figure 63 shows an autoradiograph of a gel resolving the products of single-
stranded
cleavage reactions run in the presence of various amounts of human genomic
DNA.
Figure 64 shows an autoradiograph of a gel resolving the products of single-
stranded
cleavage reactions run using the Tfl DNA polymerase in two different
concentrations of KCI.
Figure 65 shows an autoradiograph of a-gel- resolving the products of single-
stranded
cleavage reactions run using the Tth DNA polymerase in two different
concentrations of KCI.
Figure 66 shows an autoradiograph of a gel resolving the products of single-
stranded
cleavage reactions run using the E. coli Exo III enzyme in two different
concentrations of
KCI.
Figure 67 shows an autoradiograph of a gel resolving the products of single-
stranded
cleavage reactions run on three different tyrosinase gene substrates (SEQ ID
NOS:34, 41 and
42) using either the Tth DNA polymerase, the E. coli Exo III enzyme or
CleavaseTM BN.
Figure 68 is a schematic drawing depicting the location of the ~' and 3"
cleavage sites
on a cleavage structure.
Figure 69 shows an autoradiograph of a gel resolving the products of single-
stranded
cleavage reactions run on three different tyrosinase gene substrates-(SEQ ID
NOS:34, 41 and
42) using either CleavaseTM BN or the Radl/RadlO complex.
Figure 70 shows an autoradiograph of a gel resolving the products of double-
stranded
cleavage reactions run on- a wild-type and two mutant (3-globin substrates.
Figure 71A shows an autoradiograph of a gel resolving the products of single-
stranded
cleavage reactions run on a wild-type and three mutant (3-globin substrates.
Figure 71B shows an autoradiograph of a gel resolving the products of single-
stranded
cleavage reactions run on five mutant (3-globin substrates. -
Figure 72 shows an autoradiograph of a gel resolving the products of double-
stranded
cleavage reactions which varied the order of addition of the reaction
components.
Figure 73 shows an autoradiograph of a gel resolving the products of cleavage
reactions run on a wild-type and two mutant p53 substrates.
Figure 74 shows an autoradiograph of a gel resolving the products of cleavage
4
reactions run on a wild-type and three mutant p53 substrates.
-24-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
Figure 75 shows an autoradiograph of a gel resolving the products of cleavage
reactions run on a wild-type and a mutant p53 substrate where the mutant and
wild-type
substrates are present in various concentrations relative to one another.
Figure 76 provides an alignment of HCV clones 1.1 (SEQ ID N0:108), HCV2.1 (SEQ
ID N0:109), HCV3.1 (SEQ ID NO:110), HCV4.2 (SEQ ID NO:111), HCV6.1 (SEQ ID
N0:112) and HCV7.1 (SEQ ID N0:113).
Figure 77 shows a fluoroimager scan of a gel resolving the products of
cleavage
reactions run on six double-stranded HCV substrates labeled on either the
sense or anti-sense
strand.
Figure 78 shows an autoradiogram of a gel resolving i:he products of cleavage
reactions run on a wild-type and two mutant M. tuberculosis rpoB substrates.
Figure 79A shows a fluoroimager scan of a gel resolving the products of
cleavage
reactions run on a wild-type and two mutant M. tuberculosis rpoB substrates
prepared using
either dTTP or dUTP.
Figure 79B shows a fluoroimager scan of the gel shown in Figure 85A following
a
longer period of electrophoresis.
Figure 80 shows an autoradiogram of a gel resolving 'the products of cleavage
reactions run on a wild-type and three mutant M. tuberculosis katG substrates
labeled on the
sense strand.
Figure 81 shows a fluoroimager scan of a gel resolving the products of
cleavage
reactions run on a wild-type and three mutant M. tuberculosis katG substrates
labeled on the
anti-sense strand.
Figure 82 shows the location of primers along the sequence of the E. coli rrsE
gene
(SEQ ID N0:145).
Figure 83 provides an alignment of the E. coli rrsE (SEQ ID N0:145), Ccrm.
jcejuni~
(SEQ ID N0:146), and Stp.aureus (SEQ ID N0:147) rRNA genes with the location
of
consensus PCR rRNA primers indicated .in bold type.
Figure 84 shows a fluoroimager scan of a gel resolving the products of
cleavage
reactions run on four bacterial 16S rRNA substrates.
Figure 85A shows a fluoroimager scan of a gel resolving the products of
cleavage
reactions run on five bacterial 16S rRNA substrates.
Figure 85B shows bacterial a fluoroimager scan of a gel resolving the products
of
cleavage reactions run on five bacterial 16S rRNA substrate s.
-25-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95I14673
Figure 86 shows bacterial a fluoroimager scan of a gel resolving the products
of
cleavage reactions run on various bacterial 16S i-RNA substrates.-
Figure 87 shows bacterial a fluoroimager scan of a gel resolving the products
of
cleavage reactions run on eight bacterial 16S rRNA substrates.
Figure 88 shows an autoradiogram of a gel resolving the products of cleavage
reactions run on a wild-type and mutant tyrosinase gene substrates prepared
using naturally
occurring deoxynucleotides or deoxynucleotide analogs.
DEFINITIONS
To facilitate understanding of the invention, a number of terms are defined
below.
The term "gene" refers to a DNA sequence that comprises control and coding
sequences necessary for the production of a polypeptide or precursor. The
polypeptide can be
encoded by a full length coding sequence or by any portion of the coding
sequence so long as
the desired enzymatic activity is retained.
The term "wild-type" refers to a gene or gene product which has the
characteristics of
that gene or gene product when isolated from a naturally occurring source. A
wild-type gene
is that which is most frequently observed in a population and is thus
arbitrarily designed the
"normal" or "wild-type" form of the gene. In contrast, the term "modified" or
"mutant" refers
to a gene or gene product which displays modifications in sequence and or
functional
properties (i.c., altered characteristics) when compared to the wild-type gene
or gene product.
It is noted that naturally-occurring mutants can be isolated; these are
identified by the fact that
they have altered characteristics when compared to the wild-type gene or gene
product.
The term "recombinant DNA vector" as used herein refers to DNA sequences
containing a desired coding sequence and appropriate DNA sequences necessary
for the
expression of the operably linked coding sequence in a particular host
organism. DNA
sequences necessary for expression in procaryotes include a promoter,
optionally an operator
sequence, a ribosome binding site and possibly other sequences. Eukaryotic
cells are known
to utilize promoters, polyadenlyation signals and enhancers.
The term "LTR" as used herein refers to the long terminal repeat found at each
end of
a provirus (i.c., the integrated form of a retrovirus). The LTR contains
numerous regulatory
signals including transcriptional control elements,-polyadenylation signals
and sequences
needed for replication and integration of the viral genome. The viral LTR is
divided into
three regions called U3, R and U5.
-26-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
The U3 region contains the enhancer and promoter elements. The US region
contains
the polyadenylation signals. The R (repeat) region separates the U3 and US
regions and
transcribed sequences of the R region appear at both the 5' and 3' ends of the
viral RNA.
i
The term "oligonucleotide" as used herein is defined as a molecule comprised
of two
or more deoxyribonucleotides or ribonucleotides, preferably more than three,
and usually
more than ten. The exact size will depend on many factors, which in turn
depends on the
ultimate function or use of the oligonucleotide. The oligonucleotide may be
generated 11 any
manner, including chemical synthesis, DNA replication, reverse transcription,
or a
combination thereof.
Because mononucleotides are reacted to make oligonucleotides in a manner such
that
the 5' phosphate of one mononucleotide pentose ring is attached to the 3'
oxygen of its
neighbor in one direction via a phosphodiester linkage, an end of an
oligonucleotide is
referred to as the "5' end" if its 5' phosphate is not linked to the 3' oxygen
of a
mononucleotide pentose ring and as the "3' end" if its 3' oxygen is not linked
to a 5'
phosphate of a subsequent mononucleotide pentose ring. As used herein, a
nucleic acid
sequence, even if internal to a larger oligonucleotide, also may be said to
have 5' and 3' ends.
When two different, non-overlapping oligonucleotides anneal to different
regions of
the same linear complementary nucleic acid sequence, and the 3' end of one
oligonucleotide
points towards the 5' end of the other, the former may be called the
"upstream"
oligonucleotide and the latter the "downstream" oligonucleotide.
The term "primer" refers to an oligonucleotide which is capable of acting as a
point of
initiation of synthesis when placed under conditions in which primer extension
is initiated.
An oligonucleotide "primer" may .occur naturally, as in a purified restriction
digest or may be
produced synthetically.
A primer is selected to be "substantially" complementary to a strand of
specific
sequence of the template. A primer must be sufficiently complementary to
hybridize with a
template strand for primer elongation to oc-cur. A primer sequence need not
reflect the exact
sequence of the template. For example, a non-complementary nucleotide fragment
may be
attached to the 5' end of the primer, with the remainder of the primer
sequence being
substantially complementary to the strand. Non-complementary bases or longer
sequences can
be interspersed into the primer, provided that the primer sequence has
sufficient
complementarity with the sequence of the template to hybridize and thereby
form a template
primer complex for synthesis of the extension product of the primer.
-27-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
"Hybridization" methods involve the annealing of a complementary sequence to
the
target nucleic acid (the sequence to be detected). The ability of two polymers
of nucleic acid
containing complementary sequences to find each other and anneal through base
pairing
interaction is a well-recognized phenomenon. The initial observations of the
"hybridization"
process by Marmur and Lane, Proc. Natl. Acad. Sci. USA 46:453 (1960) and Doty
et al.,
Proc. Natl. Acad. Sci. USA 46:461 ( 1960) have been followed by the refinement
of this
process into an essential tool of modern biology. Nonetheless, a number of
problems have
prevented the wide scale use of hybridization as a tool in human diagnostics.
Among the
more formidable problems are: 1 ) the inefficiency of hybridization; 2) the
low concentration
of specific target sequences in a mixture of genomic DNA; and 3) the
hybridization of only
partially complementary probes and targets.
With regard to efficiency, it is experimentally observed that only a fraction
of the
possible number of probe-target complexes are formed in a hybridization
reaction. This is
particularly true with short oligonucleotide probes (less than 100 bases in
length). There are
three fundamental causes: a) hybridization cannot occur because of secondary
and tertiary
structure interactions; b) strands of DNA containing the target sequence have
rehybridized
(reannealed) to their complementary strand; and c) some target molecules are
prevented from
hybridization when they are used in hybridization formats that immobilize the
target nucleic
acids to a solid surface.
Even where the sequence of a probe is completely complementary to the sequence
of
the target, i.c~., the target's primary structure, the target sequence must be
made accessible to
the probe via rearrangements of higher-order structure. These higher-order
structural
rearrangements may concern either the secondary structure or tertiary
structure of the
molecule. Secondary structure is determined by intramolecular bonding. In the
case of DNA
or RNA targets this consists of hybridization within a single, continuous
strand of bases (as
opposed to hybridization between two different -strands). Depending on the
extent and
position of intramolecular bonding, the probe can be displaced from the target
sequence
preventing hybridization.
Solution hybridization of oligonucleotide probes to denatured double-stranded
DNA is
further complicated by the fact that the longer complementary target strands
can renature or
reanneal. Again, hybridized probe is displaced by this process. This results
in a low yield of
hybridization (low "coverage") relative to the starting concentrations of
probe and target.
- 28 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95114673
With regard to low target sequence concentration, the DNA fragment containing
the
target sequence is usually in relatively low abundance in genomic DNA. This
presents great
technical difficulties; most conventional methods that use oligonucleotide
probes lack the
sensitivity necessary to detect hybridization at such low levels.
r 5 One attempt at a solution to the target sequence concentration problem is
the
amplification of the detection signal. Most often this entails placing one or
more labels on an
oligonucleotide probe. In the case of non-radioactive labels, even the highest
affinity reagents
have been found to be unsuitable for the detection of single copy genes in
genomic DNA with
oligonucleotide probes. See Wallace et al., Biochimie 67:755 (1985). In the
case of
radioactive oligonucleotide probes, only extremely high specific activities
are found to show
satisfactory results. See Studencki and Wallace, DNA 3:1 (1984) and Studenclci
et al., Human
Genetics 37:42 (1985).
With regard to complementarity, it is important for some diagnostic
applications to
determine whether the hybridization represents complete or partial
complementarity. For
example, where it is desired to detect simply the presence or absence of
pathogen DNA (such
as from a virus, bacterium, fungi, mycoplasma, protozoan) it is only important
that the
hybridization method ensures hybridization when the relevant sequence is
present; conditions
can be selected where both partially complementary probes and completely
complementary
probes will hybridize. Other diagnostic applications, however, may require
that the
hybridization method distinguish between partial and complete complementarity.
It may be of
interest to detect genetic polymorphisms. For example, human hemoglobin is
composed, in
part, of four polypeptide chains. Two- of these chains are identical chains of
141 amino acids
(alpha chains) and two of these chains are identical chains of 146 amino acids
(beta chains).
The gene encoding the beta chain is known to exhibit polymorphism. The normal
allele
2~ encodes a beta chain having glutamic acid at the sixth position. The mutant
allele encodes a
beta chain having valine at the sixth position. This difference in amino acids
has a profound
(most profound when the individual is homozygous for the mutant allele)
physiological impact
known clinically as sickle cell anemia. It is well known that the genetic
basis of the amino
acid change involves a single base difference between the normal allele DNA
sequence and
the mutant allele DNA sequence.
-29-
CA 02203627 1997-04-24
WO 96/15267 PG"f/LTS95/14673
Unless combined with other techniques (such as restriction enzyme analysis),
methods
that allow for the same level of hybridization in the case of both partial as
well as complete
complementarity are typically unsuited for such applications: the probe will
hybridize to both
the normal and variant target sequence. Hybridization, regardless of the
method used,
requires some degree of complementarity between the sequence being assayed
(the target
sequence) and the fragment of DNA used to perform the test (the probe). (Of
course, one can
obtain binding without any complementarity but this binding is nonspecific and
to be
avoided.)
The complement of a nucleic acid sequence as used herein refers to an
oligonucleotide
which, when aligned with the nucleic acid sequence such that the 5' end of one
sequence is
paired with the 3' end of the other, is in "antiparallel association." Certain
bases not
commonly found in natural nucleic acids may be included in the nucleic acids
of the present
invention and include, for example, inosine and 7-deazaguanine.
Complementarity need not
be perfect; stable duplexes may contain mismatched base pairs or unmatched
bases. Those
1 S skilled in the art of nucleic acid technology can determine duplex
stability empirically
considering a number of variables including, for example, the length of the
oligonucleotide,
base composition and sequence of the oligonucleotide, ionic strength and
incidence of
mismatched base pairs.
Stability of a nucleic acid duplex is measured by the melting temperature, or
"Tn,."
The Tm of a particular nucleic acid duplex under specified conditions is the
temperature at
which on average half of the base pairs have disassociated.
The term "probe" as used herein refers to a labeled oligonucleotide which
forms a
duplex structure with a sequence in another nucleic acid, due to
complementarily of at least
one sequence in the probe with a sequence in the other nucleic acid.
The term "label" as used herein refers to any atom or molecule which can be
used to
provide a detectable (preferably quantifiable) signal, and which can be
attached to a nucleic
acid or protein. Labels may provide signals detectable by fluorescence,
radioactivity,
colorimetry, gravimetry, X-ray diffraction or absorption, magnetism, enzymatic
activity, and
the like. -
The term "cleavage structure" as used herein. refers to a region of a single-
stranded
nucleic acid substrate containing secondary structure, said region being
cleavable by a
cleavage means, including but not limited to an enzyme. The cleavage structure
is a substrate
for specific cleavage by said cleavage means in contrast to a nucleic acid
molecule which is a
-30-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
substrate for non-specific cleavage by agents such as phosphodiesterases which
cleave nucleic
acid molecules without regard to secondary structure (i.e., no folding of the
substrate is
required).
The term "cleavage means" as used herein refers to any means which is capable
of
S cleaving a cleavage structure, including but not limited to enzymes. The
cleavage means may
include native DNAPs having 5' nuclease activity (e.g., Taq DNA polymerise, E.
coli DNA
polymerise I) and, more specifically, modified DNAPs having 5' nuclease but
lacking
synthetic activity. The ability of 5' nucleases to cleave naturally occurring
structures in
nucleic acid templates (structure-specific cleavage) is useful to detect
internal sequence
differences in nucleic acids without prior knowledge of the specific sequence
of the nucleic
acid. In this manner, they are structure-specific enzymes. Structure-specific
enzymes are
enzymes which recognize specific secondary structures in a nucleic molecule
and cleave these
structures. The site of cleavage may be on either the 5' or 3' side of the
cleavage structure;
alternatively the site of cleavage may be between the 5' and 3' side (i. e. ,
within or internal to)
of the cleavage structure. The cleavage means of the invention cleave a
nucleic acid molecule
in response to the formation of cleavage structures; it is not necessary that
the cleavage means
cleave the cleavage structure at any particular location within the cleavage
structure.
The cleavage means is not restricted to enzymes having 5' nuclease activity.
The
cleavage means may include nuclease activity provided from a variety of
sources including
the enzyme CleavaseTM, Taq DNA polymerise, E. coli DNA polymerise I and
eukaryotic
structure-specific endonucleases, marine FEN-1 endonucleases [Harrington and
Liener, ( 1994)
Genes and Develop. 8:1344] and calf thymus 5' to 3' exonuclease [Murante,
R.S., et al.
(1994) J. Biol. Chem. 269:1191]). In addition, enzymes having 3' nuclease
activity such as
members of the family of DNA repair endonucleases (e.g., the Rrpl enzyme from
Drosophila
melcrnogaster, the yeast RAD1/RAD10 complex and E. coli Exo III), are also
suitable
cleavage means for the practice of the methods of the invemion.
The term "cleavage products" as used herein, refers to products generated by
the
reaction of a cleavage means with a cleavage structure (i.e., the treatment of
a cleavage
structure with a cleavage means).
The terms "nucleic acid substrate" and nucleic acid template" are used herein
interchangeably and refer to a nucleic acid molecule which 'when denatured and
allowed to
renature (i. e. , to fold upon itself by the formation of intra-strand
hydrogen bonds), forms at
-31 -
CA 02203627 1997-04-24
WO 96/15267 PG"T/US95114673
least one cleavage structure. The nucleic acid substrate may comprise single-
or double-
stranded DNA or RNA.
The term "substantially single-stranded" when used in reference to a
nucleic~acid
substrate means that the substrate molecule exists primarily as a single
strand of nucleic acid
in contrast to a double-stranded substrate which exists as two strands of
nucleic acid which
are held together by inter-strand base pairing interactions.
Nucleic acids form secondary structures which depend on base-pairing for
stability.
When single strands of nucleic acids (single-stranded DNA, denatured double-
stranded DNA
or RNA) with different sequences, even closely related ones, are allowed to
fold on
themselves, they assume characteristic secondary structures. At "elevated
temperatures" the
duplex regions of the structures are brought to the brink of instability, so
that the effects of
small changes in sequence are maximized, and revealed as alterations in the
cleavage pattern.
In other words, "an elevated temperature" is a temperature at which a given
duplex region of
the folded substrate molecule is near the temperature at which that duplex
melts. An
alteration in the sequence of the substrate will then be likely to cause the
destruction of a
duplex regions) thereby generating a different cleavage pattern when a
cleavage agent which
is dependent upon the recognition of structure is utilized in the reaction.
While not being
limited to any particular theory, it is thought that individual molecules in
the target (i.c., the
substrate) population may each assume only one or a few of the potential
cleavage structures
(i.e., duplexed regions), but when the sample is. analyzed as a whole, a
composite pattern
representing all cleavage sites is detected. Many of the structures recognized
as active
cleavage sites are likely to be only a few base-pairs long and would appear to
be unstable
when elevated temperatures used in the cleavage reaction. Nevertheless;
transient formation
of these structures allows recognition and cleavage of these structures by
said cleavage means.
The formation or disruption of these structures in response to small sequence
changes results
in changes in the patterns of cleavage. Temperatures in the range of 40-
85°C. with the range
of 55-85°C being particularly preferred, are suitable elevated
temperatures for the practice of
the method of the invention.
The term "sequence variation" as used herein refers to differences in
nucleic,acid
sequence between two nucleic acid templates. For example, a wild-type
structural gene and a
mutant form of this wild-type structural gene may vary in sequence by the
presence of single
base substitutions and/or deletions or insertions of one or more nucleotides.
These two forms
of the structural gene are said to vary in sequence from one another. A second
mutant form
-32-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
of the structural gene may exits. This second mutant form is said to vary in
sequence from
both the wild-type gene and the first mutant form of the gene. It is noted,
however, that the
invention does not require that a comparison be made between one or more forms
of a gene
to detect sequence variations. Because the method of the invention generates
.a characteristic
and reproducible pattern of cleavage products for a given nucleic acid
substrate. a
characteristic "fingerprint" may be obtained from any nucleic substrate
without reference to a
wild-type or other control. The invention contemplates the use of the method
for both
"fingerprinting" nucleic acids without reference to a control and
identification of mutant forms
of a substrate nucleic acid by comparison of the mutant form of the substrate
with a wild-type
or known mutant control.
The term "liberating" as used herein refers to the release of a nucleic acid
fragment
from a larger nucleic acid fragment, such as an oligonucleotide, by the action
of a 5" nuclease
such that the released fragment is no longer covalently attached to the
remainder of the
oligonucleotide.
The term "substrate strand" as used herein, means that strand of nucleic acid
in a
cleavage structure in which the cleavage mediated by the 5' nuclease activity
occurs.
The term "template strand" as used herein, means that strand of nucleic acid
in a
cleavage structure which is at least partially complementary to the substrate
strand and which
anneals to the substrate strand to form the cleavage structure.
The term "K"," as used herein refers to the Michaelis-Menten constant for an
enzyme
and is defined as the concentration of the specific substrate ai: which a
given enzyme yields
one-half its maximum velocity in an enzyme catalyzed reaction.
The term "nucleotide analog" as used herein refers to modified or non-
naturally
occurring nucleotides such as 7-deaza purines (i. e., 7-deaza-dATP and 7-deaza-
dGTP).
Nucleotide analogs include base analogs and comprise modified forms of
deoxyribonucleotides as well as ribonucleotides. As used herein the term
"nucleotide analog"
when used in reference to substrates present in a nucleic acid amplification
mixture (e.g., a
PCR mixture) refers to the use of nucleotides other than dAT'P, dGTP, dCTP and
dTTP; thus,
the use of dUTP (a naturally occurring dNTP) in a PCR would comprise the use
of a
nucleotide analog in the PCR. A PCR product generated using dUTP. 7-deaza-
dATP, 7-
deaza-dGTP or any other nucleotide analog in the reaction mixture is said to
contain
nucleotide analogs.
_»_
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
"Oligonucleotide primers matching or complementary to a gene sequence" refers
to
oligonucleotide primers capable of facilitating the template-dependent
synthesis of single or
double-stranded nucleic acids. Oligonucleotide primers matching or
complementary to a gene
sequence may be used in PCRs, RT-PCRs and the like.
A "consensus gene sequence" refers to a gene sequence which is derived by
comparison of two or more gene sequences arid which describes the nucleotides
most often
present in a given segment of the genes; the consensus sequence is the
canonical sequence.
The term "polymorphic locus" is a locus present in a population which shows
variation
between members of the population (i. e., the most common allele has a
frequency of less than
0.95). In contrast, a "monomorphic locus" is a genetic locus at little or no
variations seen
between members of the population (generally taken to be a locus at which the
most common
allele exceeds a frequency of 0.9~ in the gene pool of the population).
The term "microorganism" as used herein means an organism too small to be
observed
with the unaided eye and includes, but is not limited to bacteria, virus,
protozoans, fungi, and
ciliates.
The term "microbial gene sequences" refers to gene sequences derived from a
microorganism. -
The term "sequences derived from one or more microorganisms" refers to nucleic
acid
sequences extracted from one or a mixture of more than one microorganism. The
extracted
sequences may be subjected to further treatment, such as nucleic acid
amplification (e.g.,
polymerase chain reaction) prior to treatment to form and subsequently cleave
cleavage
structures comprising the microbial nucleic acid sequences.
The term "bacteria" refers to any bacterial species including eubacterial and
archaebacterial species.
The term "virus" refers to obligate, ultramicroscopic, intracellular parasites
incapable
of autonomous replication (i.e., replication requires the use of the host
cell's machinery).
The term "mufti-drug resistant" or multiple-drug resistant" refers to a
microor~~anism
which is resistant to more than one of the antibiotics or antimicrobial agents
used in the
treatment of said microorganism.
The term "CFLPTM (CleavaseTM Fragment Length Polymorphism) analysis" as used
herein refers to analysis, often by electrophoresis, of the products of a
reaction in which
strands of nucleic acid are i) denatured; ii) cooled, or otherwise allowed to
form intra-strand
secondary structures and iii) cleaved with a cleavage agent which recognizes
and cleaves the
-34-
CA 02203627 1997-04-24
WO 96!15267 PCT/US95/14673
nucleic acid having intra-strand secondary structure in response to said
structures, thereby
creating a collection of fragments (i. e., cleavage products) that are
characteristic of the nucleic
acid substrate. Nucleic acid substrates which differ in sequence from a
control or reference
nucleic acid substrate will, when analyzed by this method, show an altered
representation of
the fragments within the pool of cleavage products, indicating the presence of
said differences
,
in sequence between the substrates.
DESCRIPTION OF THE INVENTION
The present invention relates to methods and compositions for treating nucleic
acid,
and in particular, methods and compositions for detection and characterization
of nucleic acid
sequences and sequence changes.
The present invention relates to means for cleaving a nucleic acid cleavage
structure in
a site-specific manner. In particular, the present invention relates to a
cleaving enzyme
having 5' nuclease activity without interfering nucleic acid synthetic
ability.
1 ~ This invention provides 5' nucleases derived from thermostable DNA
polymerises
which exhibit altered DNA synthetic activity from that of native thermostable
DNA
polymerises. The 5' nuclease activity of the polymerise is retained while the
synthetic
activity is reduced or absent. Such 5' nucleases are capable of catalyzing the
structure-
specific cleavage of nucleic acids in the absence of interfering synthetic
activity. The lack of
synthetic activity during a cleavage reaction results in nucleic; acid
cleavage products of
uniform size.
The novel properties of the polymerises of the invention form the basis of a
method of
detecting specific nucleic acid sequences. This method relies upon the
amplification of the
detection molecule rather than upon the amplification of the target sequence
itself as do
existing methods of detecting specific target sequences.
DNA polymerises (DNAPs), such as those isolated from E. coli or from
thermophilic
bacteria of the genus Thermus, are enzymes that synthesize n.ew DNA strands.
Several of the
known DNAPs contain associated nuclease activities in addition to the
synthetic activity of the
enzyme.
Some DNAPs are known to remove nucleotides from the 5' and 3' ends of DNA
chains jKornberg, DNA Replication, W.H. Freeman and Co., San Francisco, pp.
127-139
( 1980)]. These nuclease activities are usually referred to as .5' exonuclease
and 3'
exonuclease activities, respectively. For example, the 5' exonuclease activity
located in the
5_
CA 02203627 1997-04-24
WO 96/15267 PCT/US95114673
N-terminal domain of several DNAPs participates in the removal of RNA primers
during
lagging strand synthesis during DNA replication and the removal of damaged
nucleotides
during repair. Some DNAPs, such as the E. coli DNA polymerase (DNAPEc 1 ).
also have a
3' exonuclease activity responsible for proof reading during DNA synthesis
(Kornberg,
supra).
A DNAP isolated from Thermus aquaticus, termed Taq DNA polymerase (DNAPTaq),
has a 5' exonuclease activity, but lacks a functional 3' exonucleolytic domain
[Tindall and
Kunkell, Biochem. 27:6008 (1988)]. Derivatives of DNAPEcI and DNAPTaq,
respectively
called the Klenow and Stoffel fragments, lack 5' exonuclease domains as a
result of
enzymatic or genetic manipulations [Brutlag et al., Biocherrz. Biophys. Re.s~.
Commun. 37:982
( 1969); Erlich et al., Science 252:1643 ( 1991 ); Setlow and Kornberg, J.
Biol. ChenZ. 247:232
(1972)].
The 5" exonuclease activity of DNAPTaq was reported to require concurrent
synthesis
[Gelfand; PCR Technology - Principles and Applications,for- DNA Amplifrcatior7
(H.A. Erlich,
Ed.), Stockton Press, New York, p. 19 (1989)]. Although mononucleotides
predominate
among the digestion products of the 5' exonucleases of DNAPTaq and DNAPEc 1,
short
oligonucleotides (_< 12 nucleotides) can also be observed implying that these
so-called 5'
exonucleases can function endonucleolytically [Setlow, .supoa; Holland et al.,
Pf~oc. Natl.
Acac~ Sci. USA 88:7276 (1991)].
In WO 92/06200, Gelfand et al. show that the preferred. substrate of the,5'
exonuclease activity of the thermostable DNA polymerases is displaced single-
stranded DNA.
Hydrolysis of the phosphodiester bond occurs between the displaced single-
stranded DNA and
the double-helical DNA with the preferred exonuclease cleavage site being a
phosphodiester
bond in the double helical region. Thus, the 5' exonuclease activity usually
associated with
DNAPs is a structure-dependent single-stranded endonuclease and is more
properly referred to
as a 5' nuclease. Exonucleases are enzymes which cleave nucleotide molecules
from the ends
of the nucleic acid molecule. Endonucleases, on the other hand, are enzymes
which cleave
the nucleic acid molecule at internal rather than terminal sites.- The
nuclease activity
associated with some thermostable DNA polymerases cleaves endonucleolytically
but this
cleavage requires contact with the 5' end of the molecule being cleaved.
Therefore, these
nucleases are referred to as 5' nucleases.
When a 5' nuclease activity is associated with a eubacterial Type A DNA
polymerase.
it is found in the one-third N-terminal region of the protein as an
independent functional
-36-
CA 02203627 1997-04-24
WO 96/15267 PC"T/US95/14673
domain. The C-terminal two-thirds of the molecule constitute the
polymerization domain
which is responsible for the synthesis of DNA. Some Type A DNA polymerases
also have a
3' exonuclease activity associated with the two-third. C-terminal region of
the molecule.
The 5' exonuclease activity and the polymerization activity of DNAPs have been
separated by proteolytic cleavage or genetic manipulation of the polymerase
molecule. To
r
date thermostable DNAPs have been modified to remove or reduce the amount of
5' nuclease
activity while leaving the polymerase activity intact.
The Klenow or large proteolytic cleavage fragment of DNAPEc 1 contains the
polymerase and 3' exonuclease activity but lacks the 5' nuclease activity. The
Stoffel
fragment of DNAPTaq (DNAPStf) lacks the 5' nuclease activity due to a genetic
manipulation which deleted the N-terminal 289 amino acids of the polymerase
molecule
[Erlich et al., Science 252:1643 ( 1991 )]. WO 92/06200 describes a
thermostable DNAP with
an altered level of 5' to 3' exonuclease. U.S. Patent No. 5,108,892 describes
a Thermu.s
czquaticus DNAP without a 5' to 3' exonuclease. However, the art of molecular
biology lacks
a thermostable DNA polymerase with a lessened amount of synthetic activity.
The present invention provides 5' nucleases derived from thermostable Type A
DNA
polymerases that retain 5' nuclease activity but have reduced or absent
synthetic activity. The
ability to uncouple the synthetic activity of the enzyme from the 5' nuclease
activity proves
that the 5' nuclease activity does not require concurrent DNA synthesis as was
previously
reported (Gelfand, PCR Technology, supra).
The description of the invention is divided into: I. Generation of 5'
Nucleases
Derived From Thermostable DNA Polymerases; II. CleavaseTM Fragment Length
Polymorphism for the Detection of Secondary Structure; III. Detection of
Mutations in the
p53 Tumor Suppressor Gene Using the CFLPTM Method andTV.~etection
and_Tdentificat,'_on
of Pathogens Using the CFLPTM Method.
I. Generation Of 5' Nucleases From Thermostable DNA Polymerases
The methods of the present invention employ 5' nucleases for the detection of
specific
nucleic acid sequences. The 5' nuclease may be derived from a thermostable DNA
polymerase; however, the methods of the invention are not limited to the use
of a 5" nuclease,
any cleavage agent capable of generating a unique (i. e.. characteristic)
pattern of cleavage
products from a substrate nucleic acid may be employed. When a 5' nuclease is
to be
- 37 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95114673
employed, the 5' nuclease may be derived from a thermostable DNA polymerase as
described
below.
The genes encoding Type A DNA polymerases share about 85°l°
homology to each
other on the DNA sequence level. Preferred examples of thermostable
polymerases include
those isolated from Thermus aquaticus, Thermus flavus, and Thermus
thermophilus. However,-
other thermostable Type A polymerases which have 5' nuclease activity are also
suitable.
Figures 1 and 2 compare the nucleotide and amino acid sequences of the three
above
mentioned polymerases. In Figures 1 and 2, the consensus or majority sequence
derived from
a comparison of the nucleotide (Fig. 1 ) or amino acid (Fig. 2) sequence of
the three
thermostable DNA polymerases is shown on the top line. A dot appears in the
sequences of
each of these three polymerases whenever an amino acid residue in a given
sequence is
identical to that contained in the consensus amino acid sequence. Dashes are
used to
introduce gaps in order to maximize alignment between the displayed sequences.
When no
consensus nucleotide or amino acid is present at a given position, an "X" is
placed in the
consensus sequence. SEQ ID NOS:1-3 display the nucleotide sequences and SEQ ID
NOS:4-
6 display the amino acid sequences of the three wild-type polymerases. SEQ ID
NO:l
corresponds to the nucleic acid sequence of the wild type Thermus aquatica~s
DNA
polymerase gene isolated from the YT-1 strain [Lawyer et al., .I. Bivl. Chem.
264:6427
(1989)]. SEQ ID N0:2 corresponds to the nucleic acid sequence of the wild type
Thel"n22fs
.flavus DNA polymerase gene [Akhmetzjanov and Vahhitov, Nucl. Acids Res.
20:5839 (1992)].
SEQ ID N0:3 corresponds to the nucleic acid sequence of the wild type Thermu.s
thermophilus DNA polymerase gene jGelfand et al.. WO 91/09950 (1991)]. SEQ ID
NOS:7-
8 depict the consensus nucleotide and amino acid sequences, respectively for
the above three
DNAPs (also shown on the top row in Figs. 1 and 2).
The 5' nucleases o f the invention derived from thermostable polymerases have
reduced
synthetic ability, but retain substantially the same 5' exonuclease activity
as the native DNA
polymerase. The term "substantially the same 5' nuclease activity" as used
herein means that
the 5' nuclease activity of the modified enzyme retains the ability to
function as a structure-
dependent single-stranded endonuclease but not necessarily at the same rate of
cleavage as
- compared to the unmodified enzyme. Type A DNA polymerases may also be
modified so as
to produce an enzyme which has increases 5' nuclease activity while having a
reduced level
of synthetic activity. Modified enzymes having reduced synthetic activity and
increased 5'
nuclease activity are also envisioned by the present invention.
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
By the term "reduced synthetic activity" as used herein it is meant that the
modified
enzyme has less than the level of synthetic activity found in the unmodified
or "native"
enzyme. The modified enzyme may have no synthetic activity remaining or may
have that
level of synthetic activity that will not interfere with the use of the
modified enzyme in the
detection assay described below. The 5' nucleases of the present invention are
advantageous
in situations where the cleavage activity of the polymerise is desired, but
the synthetic ability
is not (such as in the detection assay of the invention).
As noted above, it is not intended that the invention be limited by the nature
of the
alteration necessary to render the polymerise synthesis deficient. The present
invention
contemplates a variety of methods, including but not limited to: 1)
proteolysis; '?)
recombinant constructs (including mutants); and 3) physical and/or chemical
modification
and/or inhibition.
1. Proteolysis
Thermostable DNA polymerises having a reduced level of synthetic activity are
produced by physically cleaving the unmodified enzyme with proteolytic enzymes
to produce
fragments of the enzyme that are deficient in synthetic activity but retain 5'
nuclease activity.
Following proteolytic digestion, the resulting fragments are separated by
standard
chromatographic techniques and assayed for the ability to synthesize DNA and
to act as a 5'
nuclease. The assays to determine synthetic activity and 5' nuclease activity
are described
below.
2. Recombinant Constructs
The examples below describe a preferred method for creating a construct
encoding a
5' nuclease derived from a thermostable DNA polymerise. As the Type A DNA
polymerises
are similar in DNA sequence; the cloning strategies employed for the Thermus
aquaticu.s and
.flavus polymerises are applicable to other thermostable Type A polymerises.
In general. a
thermostable DNA polymerise is cloned by isolating genomic DNA using molecular
biological methods from a bacteria containing a thermostable Type A DNA
polymerise. This
genomic DNA is exposed to primers which are capable of amplifying the
polymerise gene bS~
PCR. __
-39-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
This amplified polymerise sequence is then subjected to standard deletion
processes to
delete the polymerise portion of the gene. Suitable deletion processes are
described below in
the examples.
. The example below discusses the strategy used to determine which portions of
the
DNAPTaq polymerise domain could be removed without eliminating the 5' nuclease
activity.
Deletion of amino acids from the protein can be done either by deletion of the
encoding
genetic material, or by introduction of a translational stop codon by mutation
or frame shift.
In addition, proteolytic treatment of the protein molecule can be performed to
remove
segments of the protein.
In the examples below, specific alterations of the Taq gene were: a deletion
between
nucleotides 1601 and 2502 (the end of the coding region), a 4 nucleotide
insertion at position
2043, and deletions between nucleotides 1614 and 1848 and between nucleotides
875 and
1778 (numbering is as in SEQ ID NO:1 ). These modified sequences are described
below in
the examples and at SEQ ID NOS:9-12.
Those skilled in the art understand that single base pair changes can be
innocuous in
terms of enzyme structure -and function. Similarly, small additions and
deletions can be
present without substantially changing the exonuclease or polymerise function
of these
enzymes.
Other deletions are also suitable to create the 5' nucleases of the present
invention. It
is preferable that the deletion decrease the polymerise activity of the 5'
nucleases to a level at
which synthetic activity will not interfere with the use of the 5' nuclease in
the detection
assay of the invention. Most preferably, the synthetic ability is absent.
Modified polymerises
are tested for the presence of synthetic and 5' nuclease activity as in assays
described below.
Thoughtful consideration of these assays allows for the screening of candidate
enzymes whose
structure is heretofore as- yet unknown. In other words, construct "X" can be
evaluated
according to the protocol described below to determine whether it is a member
of the genus
of 5' nucleases of the present invention as defined functionally, rather than
structurally.
In the example below, the PCR product of the amplified Thermtrs aquaticus
genomic
DNA did not have the identical nucleotide structure of the native genomic DNA
and did not
have the same synthetic ability of the original clone. Base pair changes which
result due to
the infidelity of DNAPTaq during PCR amplification of a polymerise gene are
also a method
by which the synthetic ability of a polymerise gene may be inactivated. The
examples below
and Figs. 4A and SA indicate regions in the native Thermos ayuaticZrs
and.flcnurs DNA
-40-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
polymerises likely to be important for synthetic ability. There are other base
pair changes
and substitutions that will likely also inactivate the polymerise.
It is not necessary, however, that one start out the process of producing a 5'
nuclease
from a DNA polymerise with such a mutated amplified product. This is the
.method by
which the examples below were performed to generate the synthesis-deficient
DNAPTay
mutants, but it is understood by those skilled in the art that a wild-type DNA
polymerise
sequence may be used as the starting material for the introduction of
deletions, insertion and
substitutions to produce a 5' nuclease. For example, to generate the synthesis-
deficient
DNAPTfI mutant, the primers listed in SEQ ID NOS:13-14 were used to amplify
the wild
type DNA polymerise gene from Thermus flavus strain AT-62. The amplified
polymerise
gene was then subjected to restriction enzyme digestion to delete a large
portion of the
domain encoding the synthetic activity.
The present invention contemplates that the nucleic acid construct of the
present
invention be capable of expression in a suitable host. Those in the art know
methods for
attaching various promoters and 3' sequences to a gene structure to achieve
efficient
expression. The examples below disclose two suitable vectors and six suitable
vector
constructs. Of course, there are other promoter/vector combinations that would
be suitable. It
is not necessary that a host organism be used for the expression of the
nucleic acid constructs
of the invention. For example, expression of the protein encoded by a nucleic
acid construct
may be achieved through the use of a cell-free in vitro trap
scription/translation system. An
example of such a cell-free system is the commercially available TnTTM Coupled
Reticulocyte
Lysate System (Prorriega Corporation, Madison, WI).
Once a suitable nucleic acid construct has been made°_, the 5' nuclease
may be
produced from the construct. The examples below and standard molecular
biological
teachings enable one to manipulate the construct by different suitable
methods.
Once the 5' nuclease has been expressed, the polymerise is tested for both
synthetic
and nuclease activity as described below.
3. Physical And/Or Chemical Modification And/Or Inhibition
The synthetic activity of a thermostable DNA polymerise may be reduced by
chemical
and/or physical means. In one embodiment, the cleavage reaction catalyzed by
the 5'
nuclease-activity of the polymerise is run under conditions which
preferentially inhibit the
synthetic activity of the polymerise. The level of synthetic activity need
only be reduced to
-41 -
CA 02203627 1999-07-26
that level of activity which does not interfere with cleavage reactions
requiring no significant
synthetic activity.
As shown in the examples below. concentrations of M~_-- greater than ~ mNl
inhibit
the polymerization activity of the native DNAPTa~I. The ability of the ~'
nuclease to function
under conditions where synthetic activity is inhibited is tested by running
the assays for
synthetic and 5' nuclease activity. described below, in the presence of a
range of M~~-
concentrations (~ to 10 mM). The effect of a given concentration of M~_~~ is
determined by
quantitation of the amount of synthesis and cleavage in the test reaction as
compared to the
standard reaction for each assay.
The inhibitory effect of other ions, polyamines. denaturants. such as urea.
formamide.
dimethvlsulfoxide. glycerol and non-ionic detergents (Triton X-100*and Tween-
?0). nucleic
acid binding chemicals such as. actinomvcin D, ethidium bromide and psoralens.
are tested b~~
their addition to the standard reaction buffers for the synthesis and ~'
nuclease assays. Those
compounds havin~_ a preferential inhibitory effect on the synthetic activity
of a thermostable
I ~ polymerase are then used to create reaction conditions under which ~'
nuclease activim
(cleava~_e) is retained while synthetic activity is reduced or eliminated.
Physical means may be used to preferentially inhibit the synthetic actiyim of
a
polymerase. For example. the synthetic activity of thermostable polvmerases is
destrwed by
etposure of the polymerase to extreme heat (typically 96 to 100°C) for
extended periods of
time ~~reater than or equal to ?0 minutes). While these are minor differences
with respect to
the specific heat tolerance for each of the enzymes. these are readily
determined. Polymerases
are treated with heat for various periods of time and the effect of the heat
treatment upon the
synthetic and ~' nuclease activities is determined.
II. CleavaseT"' Fragment Length Polymorphism For The Detection Of
Secondan~ Structure
Nucleic acids assume secondary structures which depend on base-pairin~~ for
stabilim.
When single strands of nucleic acids (single-stranded DNA. denatured DNA or
R'vA) with
different sequences. even closely related ones. are allowed to fold on
themselves. they assume
characteristic secondary structures. These differences in structures account
for the ability of
sin'le strand conformation polymorphism (SSCP) analysis to distinguish between
DNA
fragments having closely related sequences.
* Trade-mark
_ :1~ _
CA 02203627 1997-04-24
WO 96115267 PCT/US95/14673
The 5' nuclease domains of certain DNA polymerases are specific endonucleases
that
recognize and cleave nucleic acids at specific structures rather than in a
sequence-specific
manner (as do restriction endonucleases). The isolated nuclease domain of
DNAPTaq
described herein (termed the enzyme CleavaseTM) recognizes the end of a duplex
that has non-
base paired strands at the ends. The strand with the ~' end is cleaved at the
junction between
the single strand and the duplex.
Figure 3 depicts a wild-type substrate and a mutant substrate wherein the
mutant
substrate differs from the wild-type by a single base change (A to G as
indicated). According
to the method of the present invention, substrate structures form when nucleic
acids are
denatured and allowed to fold on themselves (See Figure 3, steps 1 and 2). The
step of
denaturation may be achieved by treating the nucleic acid with heat, low (<3)
or high pH
(>10), the use of low salt concentrations, the absence of cations, chemicals
(e.g., urea,
formamide) or proteins (e.g., helicases). Folding or renaturation of the
nucleic acid is
achieved by lowering of the temperature, addition of salt, neutralization of
the pH, withdrawal
of the chemicals or proteins.
The manner in which the substrate folds is dependent. upon the sequence of the
substrate. The 5' nucleases of the invention cleave the structures (See Figure
3, step 3). The
end points of the resulting fragments reflect the locations of the cleavage
sites. The cleavage
itself is dependent upon the formation of a particular structure, not upon a
particular sequence -
at the cleavage site.
When the 5' nucleases of the invention cleave a nucleic acid substrate, a
collection of
cleavage products or fragments is generated. These fragments constitute a
characteristic
fingerprint of the nucleic acid which can be detected (e.g., by
electrophoresis on a gel (see-
step 4)J. Changes in the sequence of a nucleic acid (e.g., single point
mutation between a
wild-type and mutant gene) alter the pattern of cleavage structures formed.
When the 5'
nucleases of the invention cleave the structures formed by a wild-type and an
altered or
mutant form of the substrate, the distribution of the cleavage fragments
generated will differ
between the two substrates reflecting the difference in the sequence of the
two substrates (See
Figure 3, step 5): -
The CleavaseTM enzyme generates a unique pattern of cleavage products for a
substrate
nucleic acid. Digestion with the CleavaseTM enzyme can be used to detect
single base
changes in DNA molecules of great length (e.g., 1.6 kb in length) to produce a
characteristic
pattern of cleavage products. The method of the invention is termed
"CleavaseTM Fragment
- 43 -
CA 02203627 1999-07-26
Length Polymorphism" (CFLPTM). However. it is noted that the invention is not
limited to
the use of the enzyme CleavaseT'': suitable enzymatic cleavage activity may be
provided from
a variety of sources including the CleavaseT"' enzyme. Taq DNA polymerase. F.
coli DNA
polymerase 1 and eukaryotic structure-specific endonucleases (~~.~.. the yeast
R4D? protein
and RAD 1 /R.AD 10 complex [Harrington. J.J. and Liener ( 1994) Genes and
Develop. 8:1 s44].
murine FEN-1 endonucleases (Harrington and Liener. .supra) and calf thymus ~'
to 3'
exonuclease [Murante, R.S.. et al. (1994) J. Biol. Chem. 269:1191]). Indeed
actual
experimental data is provided herein which demonstrates that numerous enzymes
may be used
to generate a unique pattern of cleavage products for a substrate nucleic
acid. Enzymes which
are shown herein to be suitable for use in the CFLPT"' method include the
CleayaseT"' BN
enzyme. Tuy DNA polymerase. T~h DNA polvmerase. Tn DNA polvmerase. E. roll Exo
Ill.
and the yeast Radl/RadlO complex.
The invention demonstrates that numerous enzymes may be suitable for use in
the
CFLPT" method includine enzymes which have been characterized in the
literature a bein~_ ~'
1 ~ exonucleases. In order to test whether an enzyme is suitable for use as a
cleava~_e means in
the CFLPT'" method (i.c~.. capable of generating a unique pattern of cleavage
products for a
substrate nucleic acid). the following steps are taken. Careful consideration
of the steps
described below allows the evaluation of any enzyme ("enzyme X") for use in
the CFLPT~'
method.
?0 .An initial CFLPTM reaction is prepared using a previously characterized
substrate
nucleic acid [for example the 1~7 nucleotide fragment of exon 4 of the human
tyrosinase ~=ene
(SEQ ID N0:34)]. The substrate nucleic acid (approximately 100 fmoles: the
nucleic acid
template may contain a ~' end or other label to permit easy detection of the
cleaya~Te
products) is placed into a thin wall microcentrifu~e tube in a solution which
comprises
'_'s reaction conditions reported to be optimal for the characterized activity
of the enzyme ( i. ~-. .
enzyme X). For example. if the enzyme X is a DNA polymerase. the initial
reaction
conditions would utilize a buffer which has been reported to be optimal for
the polymerization
activity of the polymerase. If enzyme X is not a polymerase. or if no specific
components are
reported to be needed for activity. the initial reaction may be assembled by
placin'= the
s0 substrate nucleic acid in a solution comprising IX CFLPTM buffer (10 mM
MOPS. 0.0~°a
Tween-?0. 0.0~% Nonidet P-40~. pH 7.2 to 8.2. 0.2 to 1.0 mM MnCI,.
The substrate nucleic acid is denatured by heating the sample tube to
9~°C for
seconds and then the reaction is cooled to a temperature suitable for the
enzyme being tested
*Trade-mark
_4:~_
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
(e.g., if a thermostable polymerase is being tested the cleavage reaction may
proceed at
elevated temperatures such as 72°C, if a mesophilic enzyme is being
tested the tube is cooled
to 37°C for the cleavage reaction). Following denaturation and cooling
to the target
temperature, the cleavage reaction is initiated by the addition of a solution
comprising 1 to
200 units of the enzyme to be tested (i. e. , enzyme X; the enzyme may be
diluted into 1 X
CFLPTM buffer, pH 8.2 if desired).
Following the addition of the enzyme X solution, thc: cleavage reaction is
allowed to
proceed at the target temperature for 2 to 5 minutes. The cleavage reaction is
then terminated
[this may be accomplished by the addition of a stop solution (95% formamide,
10 mM
EDTA, 0.05% bromophenol blue, 0.05% xylene cyanol)] and the cleavage products
are
resolved and detected using any suitable method (e.g., electrophoresis on a
denaturing
polyacrylamide gel followed by transfer to a solid support and nonisotopic
detection). The
cleavage pattern generated is examined by the criteria described below for the
CFLPTM
optimization test.
1 S An enzyme is suitable for use in the CFLPTM method if it is capable of
generating a
unique (Z.L'., characteristic) pattern of cleavage products from a substrate
nucleic acid; this
cleavage must be shown to be dependent upon the presence of the enzyme.
Additionally, an
enzyme must be able to reproducibly generate the same cleavage pattern when a
given
substrate is cleaved under the same reaction conditions. To test for
reproducibility, the
enzyme to be evaluated is used in at least two separate cleavage reactions run
on different
occasions using the same reaction conditions. If substantially the same
cleavage pattern is
obtained on both occasions, the enzyme is capable of reproducibly generating a
cleavage
pattern and is therefore suitable for use in the CFLPTM method.
When enzymes derived from mesophilic organisms are to be tested in the CFLPTM
reaction they may be initially tested at 37°C. However it may be
desirable to use theses
enzymes at higher temperatures in the cleavage reaction. The ability to cleave
nucleic acid
substrates over a range of temperatures is desirable when the cleavage
reaction is being used
to detect sequence variation (i.e., mutation) between different substrates.
Strong secondary
structures that may dominate the cleavage pattern are less likely to be
destabilized by single-
base changes and may therefore interfere with mutation detf:ction. Elevated
temperatures can
then be used to bring these persistent structures to the brink of instability,
so that the effects
of small changes in sequence are maximized and revealed as alterations in the
cleavage
pattern. Mesophilic enzymes may be used at temperatures greater than
37°C under certain
- 4~ -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
conditions known to the art. These conditions include the use of high (i.e.,
10-30%)
concentrations of glycerol in the reaction conditions. Furthermore. it is
noted that while an
enzyme may be isolated from a mesophilic organism this fact alone does not
mean that the
enzyme may not demonstrate thermostability; therefore when testing the
suitability of a
mesophilic enzyme in the CFLPTM reaction, the reaction should be run at
37°C and at higher
temperatures. Alternatively, mild denaturants can be used to destabilize the
nucleic acid
substrate at a lower temperature (e.g., 1-10% formamide, 1-10% DMSO and 1-10%
glycerol
have been used in enzymatic reactions to mimic thermal destabilization).
Nucleic acid substrates that may be analyzed using a cleavage means, such as a
5'
nuclease, include many types of both RNA and DNA. Such nucleic acid substrates
may all
be obtained using standard molecular biological techniques. For example,
substrates may be
isolated from a tissue sample, tissue culture cells, bacteria or viruses, may
be transcribed in
vitro from a DNA template, or may be chemically synthesized. Furthermore,
substrates may
be isolated from an organism, either as genomic material or as a plasmid or
similar
extrachromosomal DNA, or it may be a fragment of such material generated by
treatment
with a restriction endonuclease or other cleavage agents or it may be
synthetic.
Substrates may also be produced by amplification using the PCR. When the
substrate
is to be a single-stranded substrate molecule, the substrate may be produced
using the PCR
with preferential amplification of one strand (asymmetric PCR). Single-
stranded substrates
may also be conveniently generated in other ways. For example, a double-
stranded molecule
containing a biotin label at the end of one of the two strands may be bound to
a solid support
(e.g., a magnetic bead) linked to a streptavidin moiety. The biotin-labeled
strand is
selectively captured by binding to the streptavidin-bead complex. It is noted
that the
subsequent cleavage reaction may be performed using substrate attached to the
solid support,
as the enzyme CleavaseTM can cleave the substrate while it is bound to the
bead. A single-
stranded substrate may also be produced from a double-stranded molecule by
digestion of one
strand with exonuclease.
The nucleic acids of interest may contain a label to aid in their detection
following the
cleavage reaction. The label may be a radioisotope (e.g., a ''P or ''"S-
labeled nucleotide)
placed at either the 5' or -3' end of the nucleic acid or alternatively the
label may be
distributed throughout the nucleic acid (i.e., an internally labeled
substrate). The label may be
a nonisotopic detectable moiety, such as a fluorophore which can be detected
directly. or a
reactive group which permits specific recognition by a secondary agent. For
example.
-46-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
biotinylated nucleic acids may be detected by probing with a streptavidin
molecule which is
coupled to an indicator (e.g., alkaline phosphatase or a fluorophore), or a
hapten such as
digoxigenin may be detected using a specific antibody coupled to a similar
indicator.
Alternatively, unlabeled nucleic acid may be cleaved and vi sualized by
staining (e.g., ethidium
r 5 bromide staining or silver staining) or by hybridization using a labeled
probe. In a preferred
embodiment, the substrate nucleic acid is labeled at the 5' end with a biotin
molecule and is
detected using avidin or streptavidin coupled to alkaline phosphatase. In
another preferred
embodiment the substrate nucleic acid is labeled at the 5' end with a
fluorescein molecule and
is detected using an anti-fluorescein antibody-alkaline phosphatase conjugate.
The cleavage patterns are essentially partial digests of the substrate in the
reaction.
When the substrate is labelled at one end (e.g., with biotin), all detectable
fragments share a
common end. Many of the structures recognized as active cleavage sites are
likely to be only
a few base-pairs long and would appear to be unstable at the elevated
temperatures used in
the CleavaseTM reaction. The formation or disruption of these structures in
response to small
1 ~ sequence changes results in changes in the patterns of cleavage.
The products of the cleavage reaction are a collection of fragments generated
by
structure specific cleavage of the input nucleic acid. Nucleic acids which
differ in size may be
analyzed and resolved by a number of methods including electrophoresis,
chromatography,
fluorescence polarization, mass spectrometry and chip hybridization. The
invention is
illustrated using electrophoretic separation. However, it is rkoted that the
resolution of the
cleavage products is not limited to electrophoresis. Electrophoresis is chosen
to illustrate the
method of the invention because electrophoresis is widely practiced in the art
and is easily
accessible to the average practitioner.
If abundant quantities of DNA are available for the analysis, it may be
advantageous
to use direct fluorescence to detect the cleavage fragments, raising the
possibility of analyzing
several samples in the same tube and on the same gel. This "multiplexing"
would permit
automated comparisons of closely related substrates such as wild-type and
mutant forms of a
gene. _ _
The CFLPTM reaction is useful to rapidly screen for differences between
similar
nucleic acid molecules. To optimize the CFLPTM reaction for any desired
nucleic acid system
(e.g.. a wild-type nucleic acid and one or more mutant forms of the wild-type
nucleic acid). it
is most convenient to use a single substrate from the test system (for
example, the wild-type
substrate) to determine the best CFLPTM reaction conditions. A single suitable
condition is
-47-
CA 02203627 1997-04-24
WO 96/15267 PC"T/US95/14673
chosen for doing the comparison CFLPTM reactions on the other molecules of
interest. For
example, a cleavage reaction may be optimized for a wild-type sequence and
mutant
sequences may subsequently be cleaved under the same conditions for comparison
with the
wild-type pattern. The objective of the CFLPTM optimization test is the
identification of a set
of conditions which allow the test molecule to form an assortment (i.e., a
population) of intra-
strand structures that are sufficiently stable such that treatment with a
structure-specific
cleavage agent such as the CleavaseTM enzyme or DNAPTaq will yield a signature
array of
cleavage products, yet are sufficiently unstable that minor or single-base
changes within the
test molecule are likely to result in a noticeable change in the array of
cleavage products.
The following discussion illustrates the optimization of the CFLPTM method for
use
with a single-stranded substrate.
A panel of reaction conditions with varying salt concentration and temperature
is first
performed to identify an optimal set of conditions for the single-stranded
CFLPTM. "Optimal
CFLPTM" is defined for this test case as the set of conditions that yields the
most widely
spaced set of bands after electrophoretic separation, with the most even
signal intensity
between the bands.
Two elements of the cleavage reaction that significantly affect the stability
of the
nucleic acid structures are the temperature at which the cleavage reaction is
performed and the
concentration of salt in the reaction solution. Likewise, other factors
affecting nucleic acid
structures, such as, formamide, urea or extremes in pH may be used. The
initial test typically
will comprise reactions performed at four temperatures (50°C,
~5°C, 60°C and 65°C) in three
different salt concentrations (0 mM, 25 mM and 50 mM) for a total of twelve
individual
reactions. It is not intended that the present invention be limited by the
salt utilized. The salt
utilized may be chosen from potassium chloride, sodium chloride, etc. with
potassium chloride
being a preferred salt.
For each salt concentration to be tested, 30 pl of a master mix containing a
DNA
substrate, buffer and salt is prepared. When the substrate is DNA, suitable
buffers include 3-
jN-Morpholino]propanesulfonic acid (MOPS), pH 6.5 to 9.0, with pH 7.2 to 8.4
being
particularly preferred and other "Good" biological buffers such as
tris[Hydroxymethyl]aminomethane (Tris) or N,N-bis[2-Hydroxyethyl]glycine
(Bicine). pH G.5
to 9.0, with pH 7.5 to 8.4 being particularly preferred. When the nucleic acid
substrate is
RNA. the pH of the buffer is reduced to the range of 6.0 to 8.5, with pH 6.0
to 7.0 being
particularly preferred. When manganese is to used as the divalent cation in
the reaction, the
-48-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
use of Tris buffers is not preferred. Manganese tends to precipitate as
manganous oxide in
Tris if the divalent cation is exposed to the buffer for prolonged periods
(such as in
incubations of greater than 5 minutes or in the storage of a stock buffer).
When manganese is
to be used as the divalent cation, a preferred buffer is the MOPS buffer.
For reactions containing no salt (the "0 mM KCI" mix), the mix includes enough
detectable DNA for 5 digests (e.g., approximately 500 fmolf;s of 5'
biotinylated DNA or
approximately 100 fmoles of 3'-P-S' end labeled DNA) in 30 p,l of 1 X CFLPTM
buffer ( 10 mM
MOPS, pH 7.2 to 8.2) with 1.7 mM MnCh or MgClz (the final concentration of the
divalent
cation will be 1 mM). Other concentrations of the divalent cation may be used
if appropriate
for the cleavage agent chosen (e.g., E. coli DNA polymerase I is commonly used
in a buffer
containing 5 mM MgCI,). The "25 mM KCl" mix includes 41.5 mM KCI in addition
to the
above components; the "50 mM KCl" mix includes 83.3 mM KCl in addition to the
above
components.
The mixes are distributed into labeled reaction tubes (0.2 ml, 0.5 ml or 1.5
ml
"Eppendorf' style microcentrifuge tubes) in 6 ~l aliquots, overlaid with light
mineral oil or a
similar barrier, and stored on ice until use. Sixty microliters of an enzyme
dilution cocktail is
assembled, comprising a 5' nuclease at a suitable concentration in 1X CFLPTM
buffer without
MnCI,. Preferred 5' nucleases and concentrations are 25 to 100 ng of the
CleavaseTMBN
enzyme. with 25 ng being particularly preferred or 5 units of Taq DNA
polymerase (or
another eubacterial Pol A-type DNA polymerase). Suitable amounts of a similar
structure-
specific cleavage agent in 1X CFLPTM buffer without MnCI., may also be
utilized.
If a strong (i.e., stable) secondary structure is formed by the substrates, a
single
nucleotide change is unlikely to significantly alter that structure, or the
cleavage pattern it
produces. Elevated temperatures can be used to bring structures to the brink
of instability. so
that the effects of small changes in sequence are maximized, and revealed as
alterations in the
cleavage pattern within the target substrate, thus allowing the cleavage
reaction to occur at
that point. Consequently, it is often desirable to run the reaction at an
elevated temperature
(i.e., above 50°C).
Preferably, reactions are performed at 50°C, 55°C, 60°C
and 65°C. For each
temperature to be tested, a trio of tubes at each of the three KCI
concentrations are brought to
95°C for 5 seconds. then cooled to the selected temperature. The
reactions are then started
immediately by the addition of 4 ~l of the enzyme cocktail. A duplicate trio
of tubes may be
included (these tubes receiving 4 p,l of 1X CFLPTM buffer without enzyme or
MnCh), to
-49-
CA 02203627 1997-04-24
WO 96/15267 PG"T/US95/14673
assess the nucleic acid stability in these reaction conditions. All reactions
proceed for 5
minutes, and are stopped by the addition of 8 ~l of 95% formamide with 20 mM
EDTA and
0.05% xylene cyanol and 0.05% bromophenol blue.. Reactions may be assembled
and stored
on ice if necessary. Completed reactions are stored on ice until all reactions
_in the series have
been performed.
x
Samples are heated to 72°C for 2 minutes and 3 to 7 p.l of each
reaction is resolved by
electrophoresis through a suitable gel, such as 6 to 10% polyacrylamide ( 19:1
cross-link), with
7M urea, in a buffer of 45 mM Tris-Borate, pH 8.3, 1.4 mM EDTA for nucleic
acids up to
approximately 1.5 kb, or native or denaturing agarose gels for larger
molecules. The nucleic
acids may be visualized as described above, by staining, autoradiography (for
radioisotopes)
or by transfer to a nylon or other membrane support with subsequent
hybridization and/or
nonisotopic detection. The patterns generated are examined by the criteria
described above
and a reaction condition is chosen for the performance of the variant
comparison CFLPsTM.
A "no enzyme" control allows the assessment of the stability of the nucleic
acid
substrate under particular reaction conditions. In this instance, the
substrate is placed in a
tube containing all reaction components except the enzyme and treated the same
as the
enzyme-containing reactions. Other control reactions may be run. A wild-type
substrate may
be cleaved each time a new mutant substrate is tested. Alternatively, a
previously
characterized mutant may be run in parallel with a substrate suspected of
containing a
different mutation. Previously characterized substrates allow for the
comparison of the
cleavage pattern produced by the new test substrate with a known cleavage
pattern. In this
manner, alterations in the new test substrate may be identified.
When the CFLPTM pattern generated by cleavage of a single-stranded substrate
contains an overly strong (i.e., intense) band, this indicates the presence of
a very stable
structure. The preferred method for redistributing the signal is to alter the
reaction conditions
to increase structure stability (e.g., lower the temperature of the cleavage
reaction, raise the
monovalent salt concentration); this allows other less stable structures to
compete more
effectively for cleavage.
When the CFLPTM reaction is to optimized for the cleavage a double-stranded
substrate
the following steps are taken. The cleavage of double-stranded DNA substrates
up to 2.000
base pairs may be optimized in this manner.
The double-stranded substrate is prepared such that it contains a single end-
label using
any of the methods known to the art. The molar amount of DNA used in the
optimization
-50-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
reactions is the same as that use for the optimization of reactions utilizing
single-stranded
substrates. The most notable differences between the optimization of the
CFLPTM reaction for
single- versus double-stranded substrates is that the double-stranded
substrate is denatured in
distilled water without buffer, the concentration of MnCI, in the reaction is
reduced to 0.2
mM, the KCl (or other monovalent salt) is omitted, and the enzyme
concentration is reduced
t
to 10 to 25 ng per reaction. In contrast to the optimization of the single-
stranded CFLPTM
reaction (described above) where the variation of the monovalent salt (e.g.,
KCl)
concentration is a critical controlling factor, in the optimization of the
double-stranded
CFLPTM reaction the range of temperature is the more critical controlling
factor for
optimization of the reaction. When optimizing the double-stranded CFLPTM
reaction a
reaction tube containing the substrate and other components described below is
set up to allow
performance of the reaction at each of the following temperatures:
40°C, 45°C, 50°C, 55°C,
60°C,-65°C, 70°C, and 75°C.
For each temperature to be tested, a mixture comprising the single end
labelled double-
stranded DNA substrate and distilled water in a volume of 15 ~,l is prepared
and placed into a
thin walled microcentrifuge tube. This mixture may be overlaid with light
mineral oil or
liquid wax (this overlay is not generally required but may provide more
consistent results with
some double-stranded DNA substrates).
A 2 mM solution of MnCh is prepared. For each CFLPTM reaction, 5 ~l of a
diluted
enzyme solution is prepared comprising 2 ~.1 of 1 OX CFLPTM buffer ( 100 mM
MOPS, pH 7.2
to 8.2, 0.5% Tween-20, 0.5% Nonidet P-40), 2 Pl of 2 mM MnCI., and 25 ng of
CleavaseTM
BN enzyme and distilled water to yield a final volume of 5 ~.1.
The DNA mixture is heated to 95°C for 10 to 30 seconds and then
individual tubes are
cooled to the reaction temperatures to be tested (e.g.; 40°C,
45°C, 50°C, 55°C, 60°C, 65°C,
70°C, and 75°C). The cleavage reaction is started by adding 5 ql
of the dilute enzyme
solution to each tube at the target reaction temperature. The reaction is
incubated at the target
temperature for ~ minutes and the reaction is terminated (e.g., by the
addition of 16 q.l of stop
solution comprising 95% formamide with 10 mM EDTA and 0.05% xylene cyanol and
0.05%
bromophenol blue).
Samples are heated to 72°C for 1 to 2 minutes and 3 to 7 pl of each
reaction is
resolved by electrophoresis through a suitable gel, such as 6 to 10%
polyacrylamide ( 19:1
cross-lil~l:), with 7M urea, in a buffer of 4~ mM Tris-Borate, pH 8.3. 1.4 mM
EDTA for
nucleic acids up to approximately 1.5 kb, or native or denaturing agarose gels
for larger
-51-
CA 02203627 1997-04-24
WO 96115267 PCT/US95114673
molecules. The nucleic acids may be visualized as described above, by
staining.
autoradiography (for radioisotopes) or by transfer to a nylon or other
membrane support with
subsequent hybridization and/or nonisotopic detection. The patterns generated
are examined
by the criteria described above and a reaction condition is chosen for the
performance of the
double-stranded CFLPTM. Control reactions may be run as described above to
assess nucleic
acid stability or to create patterns fox reference.
When performing double-stranded CFLPTM reactions the MnCI, concentration
preferably will not exceed 0.25 mM. If the end label on the double-stranded
DNA substrate
disappears (i.e., loses its 5' end label as judged by a loss of signal upon
detection of the
cleavage products), the concentration of MnCI, may be reduced to 0.1 mM. Any
EDTA
present in the DNA storage buffer will reduce the amount of free Mn" in the
reaction, so
double-stranded DNA should be dissolved in water or Tris-HCI with a EDTA
concentration of
0.1 mM or less.
When the nucleic acid substrate is labelled at one end (e.g., with biotin or
''-P) all
detectable fragments share a common end. For short DNA substrates (less than
2~0
nucleotides) the concentration of the enzyme (e.g., CleavaseTM BN) and the
length of the
incubation have minimal influence on the distribution of signal intensity,
indicating that the
cleavage patterns are not partial digests of a single structure assumed by the
nucleic acid
substrate, but rather are relatively complete digests of a collection of
stable structures formed
by the substrate. With longer DNA substrates (greater than 250 nucleotides)
there is a greater
chance of having multiple cleavage sites on each structure, giving apparent
overdigestion as
indicated by the absence of any residual full-length materials. For these DNA
substrates, the
enzyme concentration may be lowered in the cleavage reaction (for example, if
~0 ng of the
CleavaseTM BN enzyme were used initially and overdigestion was apparent, the
concentration
of enzyme may be reduced to 25, 10 or 1 ng per reaction). Alternatively, a
combination of
Mn'-~ and Mg'+ can be used in CFLPTM buffer, to attenuate the rate of
cleavage. When 0.2
mM MnCh is used in a CFLPTM reaction, as described above (with either a single-
or double
stranded nucleic acid substrate), the use of 1 mM Mg'~' in addition to the
Mn'~ slows down
the rate of cleavage, in the case of the 1059 by amplicon seen in Figure 30,
the rate of
cleavage is reduced approximately three-fold (in the Mn'-T/Mg'~ mixture as
compared to Mn'--
alone). If overdigestion is observed when the substrate is incubated at the
reaction
temperature for 2 to 5 minutes in the presence of 0.2 to 1.0 mM Mn~~, the 0.2
mM
Mn'~'/1mM Mg'- mixture may be used in conjunction with a reaction time of ~ to
20 minutes.
-52-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
Cleavage products produced by cleavage of either single-or double-stranded
substrates
which contain a biotin label may be detected using the following nonisotopic
detection
method. The following description is exemplary only: the art knows alternative
methods for
the detection of biotin-labelled products. After electrophoresis of the
reaction products, the
gel plates are separated allowing the gel to remain flat on one plate. A
positively charged
nylon membrane (preferred membranes include Nytran~Plus, 0.2 or 0.45 mm-pore
size,
Schleicher and Schuell, Keene, NH), cut to size and pre-wetted in O.SX TBE (45
mM tris-
Borate, pH 8.3, 1.4 mM EDTA), is laid on top of the exposc;d gel. All air
bubbles trapped
between the gel and the membrane are removed (e.g., by rolling a 10 ml pipet
firmly across
the membrane). Two pieces of 3MM filter paper (Whatman) are then placed on top
of the
membrane, the other glass plate is replaced, and the sandwich is clamped with
binder clips or
pressed with books or weights. The transfer is allowed to proceed 2 hours to
overnight (the
signal increases with longer transfer).
After transfer, the membrane is carefully peeled from the gel and allowed to
air dry.
Distilled water from a squeeze bottle can be used to loosen any gel that
sticks to the
membrane. After complete drying, the membrane is agitated for 30 minutes in
1.2X
Sequenase Images Blocking Buffer (United States Biochemical, Cleveland, OH;
avoid any
precipitates in the blocking buffer by decanting or filtering); 0.3 ml of the
buffer is used per
cm'- of membrane (e.g., 30 mls for a lOcm x lOcm blot). A streptavidin-
alkaline phosphatase
conjugate (SAAP, United Stated Biochemical) is added at a 1:4000 dilution
directly to the
blocking solution (avoid spotting directly on membrane), and agitated for 15
minutes. The
membrane is rinsed briefly with dH~O and then washed 3 times (5 minutes of
shaking
per/wash) in 1X SAAP buffer (100 mM Tris-HCI, pH 10; 50 mM NaCI) with 0.1%
sodium
dodecyl sulfate (SDS), using 0.5 ml buffer/cm' of membrane, with brief water
rinses between
each wash. The membrane is then washed twice in 1X SAAP buffer (no SDS) with 1
mM
MgCI,, drained thoroughly, and placed in a plastic heat-sealable bag. Using a
sterile pipet tip,
0.05 ml/cm' of CDP-StarTM (Tropix, Bedford, MA) is added to the bag and
distributed over
the entire membrane for 5 minutes. The bag is drained of all excess liquid and
air bubbles,
sealed, and the membrane is exposed to X-ray film (e.g., Kadak XRP) for 30 W
mutes.
Exposure times are adjusted as necessary for resolution and clarity.
To date, every nucleic acid substrate tested in the CFLPTM system has produced
a
reproducible pattern of fragments. The sensitivity and specificity of the
cleavage reaction
make this method of analysis very suitable for the rapid screening of
mutations in cancer
-53-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95114673
diagnostics, tissue typing, genetic identity, bacterial and- viral typing,
polymorphism analysis,
structure analysis, mutant screening in genetic crosses, etc. It could also be
applied to
enhanced RNA analysis, high level multiplexing and .extension to longer
fragments. One
distinct benefit of using the CleavaseTM reaction to characterize nucleic
acids is that the
pattern of cleavage products constitutes a characteristic fingerprint, so a
potential mutant can
be compared to previously characterized mutants without sequencing. Also, the
place in the
fragment pattern where a change is observed gives a good indication of the
position of the
mutation. But it is noted that the mutation need not be at the precise site of
cleavage, but
only in an area that affects the stability of the structure.
III. Detection of Mutations in the p53 Tumor Suppressor Gene Using the
CFLPTM Method
Tumor -suppressor genes control cellular proliferation and a variety of other
processes
important for tissue homeostasis. One of the most extensively studied of
these. the p53 gene,
1 ~ encodes a regulator of the cell cycle machinery that can suppress the
growth of cancer cells as
well as inhibit cell transformation (Levine, Annu. Rev. Biochem. 62:623
[1993)). Tumor
suppressor mutations that alter or obliterate normal p53 function are common.
Mutations in the p53 tumor suppressor gene are found in about half of all
cases of
human cancer making alterations in the p53 gene the most common cancer-related
genetic
20 change known at the gene level. In the wild-type or non-mutated form, the
p53 gene encodes
a 53-kD nuclear phosphoprotein, comprising 393 amino acids, which is involved
in the
control of cellular proliferation. Mutations in the p53 gene are generally
(greater than 90%)
missense mutations which cause a change in the identity of an amino acid
rather than
nonsense mutations which cause inactivation of the protein. It has been
postulated that the
2~ high frequency of p53 mutation seen in human tumors is due to the fact that
the missense
mutations cause both a loss of tumor suppressor function and a gain of
oncogenic function
jLane, D.P. and Benchimol, S., Genes Dev. 4:1 (1990)).
The gene encoding the p53 protein is large, .spanning 20,000 base pairs, and
is
divided into 11 exons (see Figure 4). The ability to scan the large p~3 gene
for the presence
30 of mutations has important clinical applications. In several major human
cancers the presence
of a tumor p53 mutation is associated with a poor prognosis. p53 mutation has
been shown
to be an independent marker of reduced survival in lymph node-negative breast
cancers. a
finding that may assist clinicians in reaching decisions regarding more
aggressive therapeutic
-54-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
treatment. Also, Lowe and co-workers have demonstrated tUat the vulnerability
of tumor cells
to radiation or chemotherapy is greatly reduced by mutations which abolish p~3-
dependent
apoptosis [Lowe et al., Cell 74:957 (1995)].
Regions of the p53 gene from approximately 10,000- tumors have been sequenced
in
the last 4 to 5 years, resulting in characterization of over 3,700 mutations
of which
approximately 1,200 represent independent p53 mutations (i.e., point
mutations, insertion or
deletions). A database has been compiled and deposited wish the European
Molecular
Biology Laboratory (EMBL) Data Library and is available in electronic form
[Hollstein, M. et
al. (1994) Nucleic Acids Res. 22:3551 and Cariello, N.F. et al. (1994) Nucleic
Acids Res.
22:3549]. Iri addition, an IBM PC compatible software package to analyze the
information in
the database has been developed. [Cariello et al., Nucl. Acids Res. 22:3551
(1994)]. The
point mutations in the database were identified by DNA sequencing of PCR-
amplified
products. In most cases, preliminary screening for mutations by SSCP or DGGE
was
performed.
Analysis of the p53 mutations shows that the p53 gene contains 5 hot spot
regions
(HSR) most frequently mutated in human tumors that show a tight correlation
between
domains of the protein that are evolutionary highly conserved (ECDs) and seem
to be
specifically involved in the transformation process (see Figure 4; the height
of the bar
represent the relative percentage of total mutations associated with the five
HSRs). The five
HSRs are confined to exons 5 to 8 and account for over 85~% of the mutations
detected.
However, because these studies generally confined their analysis to PCR
amplifications and
sequencing of regions located between exons 5 to 8, it should be kept in mind
that mutations
outside this region are underrepresented. As 10% to 15% of the mutations lie
outside this
region, a clinically effective p53 gene DNA diagnostic should be able to cost-
effectively scan
for life-threatening mutations scattered across the entire gene.
The following table lists a number of the known p53 mutations.
-55-
CA 02203627 1997-04-24
WO 96!15267 PC"TIUS95114673
TABLE 2
Human p53 Gene Mutations
CODON NO. WILD-TYPE MUTANT EVENT TUMOR TYPE
36 CCG - CCA GOAT Lung '
49 GAT CAT GC~CG CML
53 TGG TGT GC~TA CML
60 CCA TCA GOAT CML
68 GAG TAG GC-ETA SCLC
110 CGT TGT GC-SAT Hepatoca
113 TTC TGT Double M NSCLC
128 CCT CCG T-~G Breast
128 TCT C-~T Breast
129 GCC GAC GC-ETA Neurofibrosa
130 CTC CTG GC-aCG MDS
132 AAG AAC GC-~CG Colorectal
ca
132 CAG AT~CG Breast ca
132 AAT GC~TA Lung (NSCLC)
ca
132 CAG AT~CG Pancreatic
ca
132 AGG AT-~GC CML
133 ATG TTG AT~TA Colorectal
ca
133 AAG AT~TA Burkitt lymphoma
134 TTT TTA AT-ETA Lung (SCLC)
ca
135 TGC TAC GOAT Colorectal
ca
135 TCC GC-~CG AML
135 TAC GC-SAT Lung (NSCLC)
ca
135 TGG GC-~CG MDS
136 CAA GAG Double M Breast ca
138 GCC GTC GC-SAT Rhabdomyosa
138 GGC . GC-~CG _ Lung (SCLC)
_ ca
140 ACC TAC AT~TA CML
141 TGC TAC GC-SAT Colorectal
ca
141 TAC GOAT Bladder ca
143 GTG GCG AT~GC Colorectal
ca
143 TTG GC-ETA Lung (NSCLC)
ca
144 CAG TAG GOAT Esophageal
ca
-56-
CA 02203627 1997-04-24
WO 96/15267 PG"T/US95/14673
144 CCG AT-~CG Burkitt lymphoma
151 CCC CAT Double M Leiomyosa
151 CAC ~ GC-ETA Lung (SCLC)
ca
151 TCC GC-SAT Glioblastoma
151 TCC GC-SAT Lung (NSCLC)
ca
152 CCG- CTG GC--SAT Leiomvosa
152 TCG GC-SAT Breast ca
154 GGC GTC GC~TA Esophageal
ca
154 GTC GC-ETA. LunQ
(NSCLC) ca
154 GTC GC~TA Lung (NSCLC)
ca
154 GTC GC~TA Lung (NSCLC)
ca
15G CGC CCC GC-~CG Rhabdomyosa
I SG CCC GC~CG Osteosa
156 CGT GC-SAT Lung (NSCLC)
ca
15G CCC GC-~CCi Lun
(NSCLC) ca
I 57 GTC TTC GC-ETA Hepatoca
157 TTC GC-ETA Lung (SCLC)
ca
157 TTC GC-ETA Lung (NSCLC)
ca
157 TTC GC-ETA Breast ca
157 TTC GC~TA Lung (SCLC)
ca
157 TTC GC~TA Bladder ca
158 CGC CGT GC-SAT Neurofibrosa
158 CAC GC-~A'l.' Burkitt lymphoma
159 GCC GTC GC-SAT Lung (NSCLC)
ca
159 CCC GC->CG Lung (NSCLC)
ca
163 TAC TGC AT~GC Breast ca
163 CAC AT~GC Burkitt lymphoma
164 AAG CAG AT-~CG Breast ca
171 GAG TAG GC~TA Lung (SCLC)
ca
172 GTT TTT ~ GC~TA Burkitt lymphoma
173 GTG TTG GC->TA Lung (NSCLC)
ca'
173 TTG GC-ETA Lung (NSCLC)
ca
17 3 GGG AT~CG Burkitt lymphoma
173 GTA GOAT Gastric ca-
3J 175 CGC CAC GC-SAT Colorectal
ad
175 CAC GC-SAT Colorectal
ad
175 ~ [ CAC I GC-SAT Colorectal
ad
-57-
CA 02203627 1997-04-24
WO 96115267 PCTIUS95/14673
175 CAC GC-SAT Colorectal
ca
175 CAC GC-aAT Colorectal
ca
175 CAC GOAT T-ALL
175 CAC GC-SAT Brain tumor
175 CAC GC-SAT Colorectal
ca
175 CAC GOAT Colorectal
ca
175 CAC GC-SAT Leiomyosa
175 CAC GOAT Esophageal
ca
175 CAC GOAT Glioblastoma
175 CAC GOAT Colorectal
ca
175 CAC GOAT T-ALL
175 CAC GC-SAT Breast ca
175 CTC GC->TA Breast ca
175 AGC GC~TA Hepatoca
175 CAC GC-SAT B-ALL
175 CAC GOAT B-ALL
175 CAC GOAT Burkitt lymphoma
175 CAC GC-SAT Burkitt lymphoma
175 CAC GOAT Burkitt lymphoma
175 CAC GOAT Burkitt lymphoma
175 CAC GC-SAT Gastric ca
176 TGC TTC GC~TA Lung (NSCLC)
ca
176 TTC GC-ETA Esophageal
ca
176 TTC GC-ETA Lung (NSCLC)
ca
176 TAC GC-SAT Burkitt lymphoma
177 CCC CGC GC-~CG PTLC
179 CAT TAT GOAT Neurofibrosa
179 CAG AT->CG Lung (SCLC)
ca
179 CTT AT-ETA Esophageal
ca
179 GAT ~ GC~CG Breast ca
179 CTT AT-ETA Cholangiosa
179 CTT AT~TA Cholanaiosa
181 CGC CAC GOAT Li-Fraumeni
sdm
187 GGT TGT GC~TA Breast ca
192 CAG TAG GOAT Esophageal
ca
193 CAT CGT AT~GC Lung (SCLC)
ca
193 TAT GOAT Esophageal
ca
-58-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
193 CGT AT->GC AML
194 CTT TTT GOAT Breast ca
194 CGT AT~CG Lung (SCLC)
ca
194 CGT AT~CG Esophageal
ca
194 CGT AT->CG Esophageal
r ca
194 CGT AT~CG B-CLL
196 CGA TGA GC-SAT Colorectal
ca
196 TGA GOAT T-ALL
196 TGA GOAT T-cell lymphoma
196 TGA GOAT Lung (SCLC)
ca
196 TGA GC-SAT Bladder ca
198 GAA TAA GC-ETA Lung (SCLC)
ca
198 TAA GC~TA Lung (SCLC)
ca
202 CGT CTT GC~TA CML
204 GAG GGG AT~GC CML
205 TAT TGT AT->GC B-ALL
205 TGT AT-~GC B-CLL
205 TTT AT-ETA Gastric ca
211 ACT GCT AT->GC Colorectal
ca
213 CGA TGA GC-SAT Colorectal
ca
213 CAA GOAT B-cell lymphoma
213 CAA GOAT Burkitt lymphoma
213 CGG AT->GC Lung (SCLC)
ca
213 CGG AT-~GC Esophageal
ca
213 TGA GC~A'r Lung (NSCLC)
ca
213 CGG AT-~GC Lung (NSCLC)
ca
213 TGA GC-SAT Burkitt lymphoma
213 TGA GC-->A'T Burkitt lymphoma
215 AGT GGT AT~GC Colorectal
ca
21G GTG ATG ~ GC-SAT Brain tumor
216 GAG AT-ETA Burkitt lymphoma
216 TTG GC~TA Gastric ca
216 ATG GC->AT Ovarian ca
220 TAT TGT AT-~GC Colorectal
ca
229 TGT TGA AT~TA Lung (SCLC)
ca
232 ATC AGC AT-~CG B-CLL
234 TAC CAC AT~GC B-cell lymphoma
-59-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
234 CAC AT-~GC Burkitt lymphoma
234 TGC AT--~GC Burkitt lymphoma
236 TAC TGC AT-~GC Burkitt lymphoma
237 ATG AGG AT~CG T-ALL
237 ATA GOAT Lung (SCLC)
ca
237 ATA GC-SAT AML
237 ATA GC-SAT Breast ca
237 ATA GC->AT Burkitt lymphoma
237 ATA GOAT Richter's sdm
238 TGT TTT GC-ETA Larynx ca
23g TAT GOAT Burkitt lymphoma
23 g TAT GOAT C M L
239 AAC AGC AT~GC Colorectal
ca
239 AGC AT~GC Colorectal
ca
239 AGC AT-~GC Burkitt lymphoma
239 AGC AT~GC CML
239 AGC AT-~GC CML
239 AGC AT~GC B-CLL
241 TCC TTC GOAT Colorectal
ca
2U 241 TGC GC-~CG Colorectal
ca
241 TGC GC-~CG Bladder ca
242 TGC TCC GC~CG Lung (SCLC)
ca
242 TTC GC-ETA Breast ca
242 TCC GC-~CG MDS
242 TAC GC-SAT Ependymoma
244 GGC TGC GC-ETA T-ALL
244 TGC GC->TA Esphageal ca
244 TGC GC-ETA Lung (SCLC)
ca
244 AGC GC-SAT Hepatoca
245 GGC GTC ~ GC-ETA Esophageal
ca
245 TGC GC~TA Li-Fraumeni
sdm
245 AGC GC-SAT Leyomyosa
245 GAC GOAT Li-Fraumeni
sdm
245 AGC GOAT Esophageal
ca
245 GCC GC-~CG - Bladder ca
245 GAC GOAT Breast ca
245 GAC GOAT Li-Fraumeni
sdm
-60-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
24~ GGC TGC GC-ETA Li-Fraumeni
sdm
245 GTC GC~TA Cervical ca
246 ATG GTG AT~GC AML
246 ATC GC-~CG Lung (NSCLC)
ca
246 GTG AT~GC Hepatoca
246 GTG AT~GC Bladder ca
247 AAC ATC AT-ETA Lung (NSCLC)
ca
248 CGG TGG GC-SAT Colorectal
ad
248 TGG GC-SAT Colorectal
ca
248 CAG GOAT Colorectal
ca
248 CAG GC->AT Colorectal
ca
248 CAG GC-SAT T-ALL
248 CAG GC-SAT Esophageal
ca
248 TGG GC-~A'T Li-Fraumeni
sdm
248 TGG GC~A'i' Li-Fraumeni
sdm
248 TGG GC~A'1' Colorectal
ca
248 TGG GC-~A'r Colorectal
ca
248 TGG GC-SAT Rhabdomyosa
248 CTG GC~TA Esophageal
ca
248 TGG GOAT Lun
(NSCLC) ca
248 CAG GC-~A'T Lung (SCLC)
ca
248 CTG GC-ETA Lung (SCLC)
ca
248 CAG GC-~A'T T-ALL
248 TGG GC-SAT Lun
5 (NSCLC) ca
248 CTG GC-ETA Lung (SCLC)
ca
248 TGG GC-SAT Colorectal
ca
248 CAG GC->A'T Bladder ca
248 CAG GC-SAT MDS
248 TGG GC-SAT Burkitt lymphoma
248 CAG ~ GOAT Breast ca
248 CAG GOAT B-CLL
248 CAG GC-aAT Burkitt lymphoma
248 TGG GC-SAT Burkitt lymphoma
248 CAG GC->AT Burkitt lymphoma
248 TGG GC-SAT Burkitt lymphoma
248 CAG GOAT Gastric ca
248 TGG GC-SAT Lung (SCLC)
ca
-61 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
248 CAG GOAT Breast ca
248 CAG GC-SAT CML
248 TGG GC-SAT Li-Fraumeni
sdm
248 CAG GC-SAT Li-Fraumeni
sdm
S 248 TGG GG~AT Colorectal
ca
249 AGG AGT GC-ETA Hepatoca
249 AGT GC~TA Hepatoca
249 AGT GC~TA Hepatoca
249 AGC GC-~CG Hepatoca
249 AGT GC~TA Hepatoca
249 AGT GC-ETA Hepatoca
249 AGT GC~TA Hepatoca
249 AGT GC~TA Hepatoca
249 AGT GC-ETA Hepatoca
249 AGT GC->TA Hepatoca
249 AGT GC-ETA Hepatoca
249 AGT GC~TA Esophageal
ca
249 AGC GC-~CG Breast ca
249 AGT GC-ETA Lung (NSCLC)
ca
249 AGT GC~TA Hepatoca
250 CCC CTC GOAT Burkitt lymphoma
251 ATC AGC AT~CG Gastric ca
252 CTC CCC AT-~GC Li-Fraumeni
sdm
252 CTC CCC AT-~GC Li-Fraumeni
sdm
254 ATC GAC Double M Burkitt lymphoma
254 AAC AT-ETA Breast ca
256 ACA GCA AT~GC T-ALL
258 GAA AAA GOAT Li-Fraumeni
sdm
25g AAA GOAT Burkitt lymphoma
258 AAA ~ GC-SAT Li-Fraumeni
sdm
259 GAC GGC AT~GC T-ALL
260 TCC GCC AT~CG T-ALL
266 GGA GTA GC iTA Lung (NSCLC)
ca
26G GTA GC-aTA Lun~~ (NSCLC)
ca
266 GTA GC-ETA Breast ca
267 CGG CCG GC-~CG Luna (SCLC)
ca
270 TTT TGT AT~CG Esophageal
ca
- 62 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
270 TGT AT-~CG T-ALL
272 GTG ATG GOAT' Brain tumor
272 CTG ~ GC-~CG Lung (SCLC)
ca
i
272 ATG GOAT Hepatoca
272 ATG GC->AT AML
273 CGT TGT GC-SAT' Colorectal
ad
273 TGT GOAT' Brain tumor
273 CAT GC-SAT Breast ca
273 CAT GC-SAT Colorectal
ca
273 TGT GOAT' Lung (NSCLC)
ca
273 CTT GC-ETA Lung (SCLC)
ca
273 CAT GC-SAT Colorectal
ca
273 CAT GOAT Colorectal
ca
273 CAT GC-SAT Colorectal
ca
273 CAT GC-SAT Lun
b (NSCLC)
ca
273 CCT GC~CG Lung (NSCLC)
ca
273 CTT GC~TA Lung (NSCLC)
ca
273 CTT GC-ETA Lun
(NSCLC) ca
273 CAT GC-aAT Thyroid ca
273 CAT GOAT Lung (SCLC)
ca
273 TGT GC-SAT B-cell lymphoma
273 TGT GC-SAT B-ALL
273 TGT GC-SAT Burkitt lymphoma
273 TGT GOAT Burkitt lymphoma
273 CAT GC-SAT Li-Fraumeni
sdm
273 TGT GOAT Cervical ca
273 TGT GOAT AML
273 CAT GOAT B~CLL
273 CTT GC-ETA B-CLL
274 GTT GAT ~ AT-ETA Erythroleukemia
276 GCC CCC GC-~CG B-ALL
276 GAC GC~TA Hepatoca
277 TGT TTT GC~TA Lung (SCLC)
ca
278 CCT TCT GOAT Esophageal
ca
278 CTT GOAT Esophageal
ca
278 GCT GC-~CG Breast ca
278 TCT GC-SAT LunQ (SCLC)
ca
-63-
CA 02203627 1997-04-24
WO 96/15267 PC"TlUS95114673
278 CGT GC-~CG Ovarian ca
280 AGA AAA GOAT Esophageal
ca
280 AAA ~ GOAT Breast ca
281 GAC GGC AT~GC Colorectal
ca
281 GGC AT~GC Breast ca
281 GAC GAG GC-~CG Richter's sdm
281 TAC GC~TA B-CLL
282 CGG TGG GOAT Colorectal
ad
282 TGG GC-SAT Colorectal
ca
282 CGG TGG GC-SAT Rhabdomyosa
2g2 GGG GC~CG Lung (NSCLC)
ca
282 CCG GC-~CG Breast ca
2g2 TGG GC-SAT Bladder ca
2g2 TGG GC-SAT AML
282 CTG GC~TA Breast ca
2g2 TGG GOAT B-ALL
2g2 TGG GC-SAT Burkitt lymphoma
282 TGG GOAT Richter's sdm
2g2 TGG GC-SAT Ovarian ca
282 TGG GC-SAT Li-Fraumeni
sdm
283 CGC TGC GC-SAT Colorectal
ca
283 CCC GC-~CG Lung (NSCLC)
ca
285 GAG AAG GC->AT Breast ca
28G GAA AAA GC-SAT Colorectal
ca
28G GGA AT-~GC Lung (SCLC)
ca
28G - GCA AT-~CG Li-Fraumeni
sdm
287 GAG TAG GC-ETA Burkitt lymphoma
293 GGG TGG GC-ETA Glioblastoma
298 GAG TAG GC~TA Bladder ca
302 GGG GGT ~ GC~TA Lung (SCLC)
ca
305 AAG TAG AT~TA Esophageal
ca
305 TAG AT~TA Esophageal
ca
307 GCA ACA GC-SAT Breast ca
309 CCC TCC GOAT Colorectal
ca
334 GG GTG GC~TA Lung (SCLC)
G ca
342 CGA TGA GC-SAT Luna (SCLC)
ca
-64-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95I14673
DELETIONS/INSERTIONS
CODON EVENT TUMOR TYPE
137 del 7 Gastric ca
143 del 1 Gastric ca
152 del 13 Colorectal ad
167 del 1 Breast ca
168 del 31 He atoca
175 del 18 Breast ca
190 del 3 nu 1 ALL
201 del 1 Breast ca
206 del 1 Burkitt I m homa
206 del 1 Burkitt 1 m homa
214 del I B-ALL
236 del 27 Bladder ca
239 del 1 Lung (NSCLC) ca
262 del 1 Astroc toms
262 del 24 Gastric ca
262 del 24 Lun~ (NSCLC) ca
263 del 1 Eso haaeal ca
264 del 1 AML
286 del 8 He atoca
293 del 1 Lun~ (NSCLC) ca
307 del 1 Li-Fraumeni sdm
381 del 1 He atoca
Exon 5 del 15 B-ALL
152 ins 1 B-CLL
239 ins 1 Waldenstrom sdm
252 ins 4 Gastric ca
256 ins 1 AML
275 ins I B-CLL
301 ins 1 MDS
307 ins 1 ~ Glioblastoma
Exon 8 ins 25 HCL
-65-
CA 02203627 1997-04-24
WO 96115267 PC"T/US95/14673
SPLICE MUTATIONS
INTRON SITE EVENT TUMOR TYPE
Intron 3 Accept GC->CG Lung (SCLC) ca
Intron 4 Donor GC-ETA Lung (SCLC) ca
Intron 4 Donor GOAT T-BALL
Intron 5 Donor GOAT CML
Intron 6 Donor AT~CG Lung (SCLC) ca
Intron 6 Accept AT->TA Lung (SCLC) ca
Intron 6 Accept AT-ETA Lung (NSCLC) ca
Intron 7 Donor GC~TA Lung (NSCLC) ca
Intron 7 Accept GC-~CG Lung (SCLC) ca
Intron 7 Accept CG~AT AML
Intron 7 Donor GC~TA Lung (SCLC) ca
1 J Intron 9 Donor GC-ETA Lung (SCLC) ca
A. CFLPTM Analysis of p53 Mutations in Clinical Samples
To permit the identification of mutations in the p53 gene from clinical
samples,
nucleic acid comprising p53 gene sequences are prepared. The nucleic acid may
comprise
genomic DNA, RNA or cDNA forms of the p53 gene. Nucleic acid may be extracted
from a
variety of clinical samples [fresh or frozen tissue, suspensions of cells
(c.g.. blood), cerebral
spinal fluid, sputum, etc.] using a variety of standard techniques or
commercially available
kits. For example, kits which allow the isolation of RNA or DNA from tissue
samples are
available from Qiagen, Inc. (Chatsworth, CA) and Stratagene (LaJolla, CA),
respectively.
Total RNA may be isolated from tissues and tumors by a number of methods known
to those
skilled in the art and commercial kits are available to facilitate the
isolation. For example, the
RNeasy~ kit (Qiagen Inc., Chatsworth, CA) provides protocol, reagents and
plasticware to
permit the isolation of total RNA from tissues, cultured cells or bacteria,
with no modification
to the manufacturer's instructions, in approximately 2U minutes. Should it be
desirable. in the
case of eukaryotic RNA isolates, to further enrich for messenger RNAs, the
polyadenylated
RNAs in the mixture may be specifically isolated by binding to an oligo-
deoxythymidine
matrix, through the use of a kit such as the Oligotex~ kit (Qiagen).
Comparable isolation ,
kits for both of these steps are available through a number of commercial
suppliers.
In addition, RNA may be extracted from samples, including biopsy specimens. ,
conveniently by lysing the homogenized tissue in a buffer containing 0.22 M
NaCI, 0.7~ 111M
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
MgCI,, 0.1 M Tris-HCI, pH 8.0, 12.5 mM EDTA, 0.25% NP40, 1% SDS, 0.5 mM DTT,
500
u/ml placental RNAse inhibitor and 200 p.g/ml Proteinase K. Following
incubation at 37°C
for 30 min, the RNA is extracted with phenol:chloraform (1:1) and the RNA is
recovered by
ethanol precipitation.
Since the majority of p53 mutations are found within exons 5-8, it is
convenient as a
first analysis to examine a PCR fragment spanning this region. PCR fragments
spanning
exons 5- 8 may be amplified from clinical samples using the technique of RT-
PCR (reverse
transcription-PCR); kits which permit the user to start with tissue and
produce a PCR product
are available from Perkin Elmer (Norwalk, CT) and Stratagene (LaJolla, CA).
The RT-PCR
technique generates a single-stranded cDNA corresponding to a chosen segment
of the coding
region of a gene by using reverse transcription of RNA; the single-stranded
cDNA is then
used as template in the PCR. In the case of the p53 gene, an approximately 600
by fragment
spanning exons 5-8 is generated using primers located in the coding region
immediately
adjacent to exons 5 and 8 in the RT-PCR. The PCR amplified segment is then
subjected to
the CFLP reaction and the reaction products are analyzed as described above in
section VIII.
Fragments suitable for CFLP analysis may also be generated by PCR
amplification of
genomic DNA. DNA is extracted from a sample and primers corresponding to
sequences
present in introns 4 and 8 are used to amplify a segment of the p53 gene
spanning exons 5-8
which includes introns 5-7 (an approximately 2 kb fragment). If it is
desirable to use smaller
fragments of DNA in the CFLP reaction, primers may be chosen to amplify
smaller ( 1 lcb or
less) segments of genomic DNA or alternatively a large PCR. fragment may be
divided into
two or more smaller fragments using restriction enzymes.
In order to facilitate the identification of p53 mutations in the clinical
setting, a library
containing the CFLP pattern produced by previously characterized mutations may
be
provided. Comparison of the pattern generated using nucleic acid derived from
a clinical
sample with the patterns produced by cleavage of known and. characterized p53
mutations will
allow the rapid identification of the specific p53 mutation present in the
patient's tissue. The
comparison of CFLP patterns from clinical samples .to the patterns present in
the library may
be accomplished by a variety of means. The simplest and least expensive
comparison involves
visual comparison. Given the large number of unique mutations known at the p53
locus,
visual (i.e., manual) comparison may be too time-consuming. especially when
large numbers
of clinical isolates are to be screened. Therefore the CFLP patterns or "bar
codes" may be
provided in an electronic format for ease and efficiency in camparison.
Electronic entry may
-67-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
comprise storage of scans of gels containing the CFLP products of the
reference p53
mutations (using for example, the GeneReader and Gel Doctor Fluorescence Gel
documentation system (BioRad. Hercules, CA) or the ImageMaster (Pharmacia
Biotech,
Piscataway, NJ). Alternatively, as the detection of cleavage patterns may be
automated using
DNA sequencing instrumentation (see Example 18), the banding pattern may be
stored as the
signal collected from the appropriate channels during an automated run
[examples of
instrumentation suitable for such analysis and data collection include
fluorescence-based gel
imagers such as fluoroimagers produced by Molecular Dynamics and Hitachi or by
real-time
electrophoresis detection systems such as the ABI 377 or Pharmacia ALF DNA
Sequencer].
B. Generation of a Library of Characterized p53 Mutations
The generation of a library ofcharacterized mutations will enable clinical
samples to
be rapidly and directly screened for the presence of the most common p53
mutations.
Comparison of CFLP patterns generated from clinical samples to the p53 bar
code library will
establish both the presence of a mutation in the p53 gene and its precise
identity without the
necessity of costly and time consuming DNA sequence analysis.
The p53 bar code library is generated using reverse genetics. Engineering of
p53
mutations ensures the identity and purity of each of the mutations as each
engineered mutation
is confirmed by DNA sequencing. The individual p53 mutations in p53 bar code
library are
generated using the 2-step "recombinant PCR" technique [Higuchi, R. (1991) In
Ehrlich, H.A.
(Ed.). PCR Technology: Principles and Applications for DNA Amplification,
Stoclcton Press,
New Yorlc, pp. 61-70 and Nelson, R.M. and Long, G.L. (1989) Analytical
Biochem.
180:147]. Figure 5 provides a schematic representation of one method of a 2-
step
recombinant PCR technique that may be used for the generation of p53
mutations.
The template for the PCR amplifications is the entire human p53 cDNA gene. In
the
first of the two PCRs (designated "PCR 1" in Fig. 5), an oligonucleotide
containing the
engineered mutation ("oligo A" in Fig. 5) and an oligonucleotide containing a
5' arm of
approximately 20 non-complementary bases ("oligo B") are used.to amplify a
relatively small
region of the target DNA (100-200 bp). The resulting amplification product
will contain the
mutation at its extreme 5' end and a foreign sequence at its 3' end. The 3'
sequence is
designed to include a unique restriction site (e.g., EcoRI) to aid in the
directional cloning of
the final amplification fragment (important for purposes of sequencing and
archiving the DNA
containing the mutation). The product generated in the upstream or first PCR
may be gel
-68-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
purified if desired prior to the use of this first PCR product in the second
PCR; however gel
purification is not required once it is established that this fragment is the
only species
amplified in the PCR.
The small PCR fragment containing the engineered mutation is then used to
direct a
second round of PCR (PCR 2). In PCR 2, the target DNA is a larger fragment
(approximately 1 kb) of the same subcloned region of the p53 cDNA. Because the
sequence
at the 3' end of the small PCR fragment is not complementary to any of the
sequences present
in the target DNA, only that strand in which the mismatch is at the extreme 5'
end is
amplified in PCR 2 (a 3' non-templated arm cannot be extended in PCR).
Amplification is
accomplished by the addition of a primer complementary to a region of the
target DNA
upstream of the locus of the engineered mutation ("oligo C") and by the
addition of a primer
complementary to the 5' noncomplementary sequence of the small product of PCR
1 ("oligo
D"). By directing amplification from the noncomplementary sequence, this
procedure results
in the specific amplification of only those sequences containing the mutation.
In order to
facilitate cloning of these PCR products into a standard vector, a second
unique restriction site
can be engineered into oligo C (e.g., HindIII).
The use of this 2-step PCR approach requires that only one primer be
synthesized for
each mutant to be generated after the initial set-up of the system (i.e.,
oligo A). Oligos B, C
and D can be used for all mutations generated within a given region. Because
oligos C and D
are designed to include different and unique restriction sites, subsequent
directional cloning of
these PCR products into plasmid vectors (such as pUC 19) is greatly
simplified. Selective
amplification of only 'those sequences that include the desired mutational
change simplifies
identification of mutation-containing clones as only verification of the
sequence of insert
containing plasmids is required. Once the sequence of the insert has been
verified, each
mutation-containing clone may be maintained indefinitely as a bacterial master
stoclc. In
addition, DNA stocks of each mutant can be maintained in the form of large
scale PCR
preparations. This permits distribution of either bacteria harboring plasmids
containing a
given mutation or a PCR preparation to be distributed as individual controls
in kits containing
reagents for the scanning of p53 mutations in clinical samples or as part of a
supplemental
master p53 mutation library control kit.
An alternative 2-step recombinant PCR is diagrammed in Figure 6, and described
in
Example 30. In this method two mutagenic oligonucleotides, one for each
strand, are
synthesized. These oligonucleotides are substantially complementary to each
other but are
-69-
CA 02203627 1997-04-24
WO 96/15267 PCTlI1595/14673
opposite in orientation.. That is, one is positioned to allow amplification of
an "upstream"
region of the DNA, with the mutation incorporated into the 3' proximal region
of the,.upper,
or sense strand, while the other is positioned to allow amplification of a
"downstream"
segment with the intended mutation incorporated into the 5' proximal region of
the upper, or
sense strand. These two double stranded products share the sequence provided
by these
mutagenic oligonucleotides. When purified, combined, denatured and annealed,
those strands
that anneal with recessed 3' ends can be extended or filled in by the action
of DNA
polymerase, thus recreating a full length molecules with the mutation in the
central region.
This recombinant can be amplified by the use of the "outer" primer pair,those
used to make
the 5' end of the "upstream" and the 3' end of the "downstream" intermediate
fragments.
While extra care must be taken with this method (in comparison with the method
described above) because the outer primers can amplify both the recombinant
and the un-
modified sequence, this method does allow rapid recombinant PCR to be
performed using
existing end primers, and without the introduction of foreign sequences. In
summary, this
I S method is often used if only a few recombinations are to be performed.
When large volumes
of mutagenic PCRs are to be performed, the first described method is
preferable as the first
method requires a single oligo be synthesized for each mutagenesis and only
recombinants are
amplified. -
An important feature of kits designed for the identification of p53 mutations
in clinical
samples is the inclusion of the specific primers to be used for generating PCR
fragments to be
analyzed for CFLP. While DNA fragments from 100 to over 1500 by can be
reproducibly
and accurately analyzed for the presence of sequence polymorphisms by this
technique, the
precise patterns generated from different length fragments of the same input
DNA sequence
will of course vary. Not only are patterns shifted relative to one another
depending on the
length of the input DNA, but in some cases, more long range interactions
between distant
regions of long DNA fragments may result in the generation of additional
cleavage products
not seen with shorter input DNA products. For this reason, exact matches with
the bar code
library will be assured through the use of primers designed to amplify the
same size fragment
from the clinical samples as was used to generate a given version of the p53
bar code library.
_70_
CA 02203627 1997-04-24
WO 96/15267 PC"T/US95/14673
C. Detection of Unique CFLPTM Patterns for p53 Mutations
The simplest and most direct method of analyzing the DNA fragments produced in
the
CFLPTM reaction is by gel electrophoresis. Because electrophoresis is widely
practiced and
easily accessible, initial efforts have been aimed at generating a database in
this familiar
format. It should, however, be noted that resolution of DNA fragments
generated by CFLPTM
analysis is not limited to electrophoretic methods. Mass spectrometry,
chromatography,
fluorescence polarization, and chip hybridization are all approaches that are
currently being
refined and developed in a number of research laboratories. Once generated,
the CFLPTM
database is easily adapted to analysis by any of these methods.
There are several possible alternatives available for detection of CFLP
patterns. A
critical user benefit of CFLP analysis is that the results are not dependent
on the chosen
method of DNA detection. DNA fragments may be labeled with a radioisotope
(e.g., a ''-P or
3sS_labeled nucleotide) placed at either the S' or 3' end of the nucleic acid
or alternatively the
label may be distributed throughout the nucleic acid (i.e., an internally
labeled substrate). The
label may be a nonisotopic detectable moiety, such as a fluorophore which can
be detected
directly, or a reactive group which permits specific recognition by a
secondary agent. CFLP
patterns have been detected by immunostaining, biotin-avidin interactions,
autoradiography
and direct fluorescence imaging. Since radiation use is in rapid decline in
clinical settings
and since both immunostaining and biotin-avidin based detection schemes
require time-
consuming transfer of DNA onto an expensive membrane support, fluorescence-
based
detection methods may be preferred. It is important to note, however, that any
of the above
methods may be used to generate CFLP bar codes to be input into the database.
In addition to their being a direct, non-isotopic means of detecting CFLP
patterns.
fluorescence-based schemes offer a noteworthy additional advantage in clinical
applications.
CFLP allows the analysis of several samples in the same tube and in the same
lane on a gel.
This "multiplexing" permits rapid and automated comparison of a large number
of samples in
a fraction of the time and for a lower cost than can be realized through
individual analysis of
each sample. This approach opens the door to several alternative applications.
A researcher
could decide to double, triple or quadruple (up to 4 dyes have been
demonstrated to be
detectable and compatible in a single lane in commercially available DNA
sequencing
instrumentation such as the ABI 373/377) the number of samples run on a given
gel.
Alternatively, the analyst may include a normal p53 gene sample in each tube.
and each gel
lane, along with a differentially labeled size standard, as a internal
standard to verify both the
-71 -
CA 02203627 1997-04-24
WO 96115267 PCTIUS95I14673
presence and the exact locations) of a pattern differences) between the normal
p53 gene and
putative mutants.
VI. Detection and Identification of Pathogens Using the CFLPTM Method
A. Detection and Identification of Hepatitis C Virus
Hepatitis C virus (HCV) infection is the predominant cause of post-transfusion
non-A,
non-B (NANB) .hepatitis around the world. In addition, HCV is the major
etiologic agent of
hepatocellular carcinoma (HCC) and chronic liver disease world wide. HCV
infection is
transmitted primarily to blood transfusion recipients and intravenous drug
users although
maternal transmission to offspring and transmission to recipients of organ
transplants have
been reported.
The genome of the positive-stranded RNA hepatitis C virus comprises several
regions
including 5' and 3' noncoding regions (i. e., 5' and 3' untranslated regions)
and a polyprotein
coding region which encodes the core protein (C), two envelope glycoproteins
(E1 and
E2/NS 1 ) and six nonstructural glycoproteins (NS2-NSSb). Molecular biological
analysis of
the small (9.4 kb) RNA genome has showed that some regions of the genome are
very highly
conserved between isolates, while other regions are fairly rapidly changeable.
The 5'
noncoding region (NCR) is the most highly conserved region in the HCV. These
analyses
have allowed these viruses to be divided into six basic genotype groups, and
then further
classified into over a dozen sub-types [the nomenclature and division of HCV
genotypes is
evolving; see Altamirano et al., J. Infect. Dis. 171:1034 (1995) for a recent
classification
scheme]. These viral groups are associated with different geographical areas,
and accurate
identification of the agent in outbreaks is important in monitoring the
disease. While only
Group 1 HCV has been observed in the United States, multiple HCV genotypes
have been
observed in both Europe arid Japan.
The ability to determine the genotype of viral isolates also allows
comparisons of the
clinical outcomes from infection by the different types of HCV, and from
infection by
multiple types in a single individual. HCV type has.also been associated with
differential
efficacy of treatment with interferon, with Group 1 infected individuals-
showing little
response [Kanai et al., Lancet 339:1543 (1992) and Yoshioka et al., Hepatologv
16:293
(1992)]. Pre-screening of infected individuals for the viral type will allow
the clinician to
make a more accurate diagnosis, and to avoid costly but fruitless drug
treatment.
-72-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
Existing methods for determining the genotype of HC:V isolates include PCR
amplification of segments of the HCV genome coupled with either DNA sequencing
or
hybridization to HCV-specific probes, RFLP analysis of PCF, amplified HCV DNA
anything
else?. All of these methods suffer from the limitations discussed above (i.
e., DNA sequencing
is too labor-intensive and expensive to be practical in clinical laboratory
settings; RFLP
analysis suffers from low sensitivity).
Universal and genotype specific primers have been dfaigned for the
amplification of
HCV sequences from RNA extracted from plasma or serum [Okamoto et al. J. Gen.
Virol.
73:673 (1992);Yoshioka et al., Hepatology 16:293 (1992) and Altamirano c~t
al.. supra].
These primers can be used to generate PCR products which serve as substrates
in the CFLPTM
assay of the present invention. As shown herein CFLPTM analysis provides a
rapid and
accurate method of typing HCV isolates. CFLPTM analysis of HCV substrates
allows a
distinction to be made between the major genotypes and subtypes of HCV thus
providing
improved methods for the genotyping of HCV isolates.
B. Detection and Identification of Multi-Drug Resistant M. tuberculosis
In the past decade there has been a tremendous resurgence in the incidence of
tuberculosis in this country and throughout the world. In thc~ United States,
the incidence of
tuberculosis has risen steadily during past decade, accounting for 2000 deaths
annually, with
as many as 10 million Americans infected with the disease. The situation is
critical in New
York City, where the incidence has more than doubled in the past decade,
accounting for 14%
of all new cases in the United States in 1990 [Frieden et al., New Engl. J.
Med. 328:521
(1993)].
The crisis in New York City is particularly dire because a significant
proportion (as
many as one-third) of the recent cases are resistant to one or more
antituberculosis drugs
[Frieden et al, supra and Hughes, Scrip Magazine May (1994)]. Multi-drug
resistant
tuberculosis (MDR-TB) is an iatrogenic disease that arises from incomplete
treatment of a
primary infection [Jacobs, Jr., Clin. Infect. Dis. 19:1 (1994)]. MDR-TB
appears to pose an
especially serious risk to the immunocompromised, who are more likely to be
infected with
MDR-TB strains than are otherwise healthy individuals [Jacobs, Jr., supra].
The mortality
rate of MDR-TB in immunocompromised individuals is alarmingly high, often
exceeding
90%, compared to a mortality rate of <50% in otherwise uncompromised
individuals
[Donnabella et al.. Am. J. Respir. Dis. 11:639 (1994)].
- 73 -
CA 02203627 1997-04-24
WO 96!15267 PCT/US95t14673
From a clinical standpoint, tuberculosis has always been difficult to diagnose
because
of the extremely long generation time of Mycobacterium tuberculosis as well as
the
environmental prevalence of other, faster growing mycobacterial species. The
doubling. time
of M. tuberculosis is 20-24 hours, and growth by conventional methods
typically requires 4 to '
6 weeks to positively identify M. tuberculosis [Jacobs, Jr. et al., Science
260:819 (1993) and
Shinnick and Jones in Tuberculosis: Pathogenesis, Protection and Control,
Bloom, ed.,
American Society of Microbiology, Washington, D.C. (1994), pp. 517-530]. It
can take an
additional 3 to 6 weeks to diagnose the drug susceptibility of a given strain
[Shinnick and
Jones, supra]. Needless to say, the health risks to the infected individual,
as well as to the
public, during a protracted period in which the patient may or may not be
symptomatic, but is
almost certainly contagious, are considerable. Once a drug resistance profile
has been
elucidated and a diagnosis made, treatment of a single patient can cost up to
$250,000 and
require 24 months.
The recent explosion int he incidence of the disease, together with the dire
risks posed
by MDR strains, have combined to spur a burst of research activity and
commercial
development of procedures and products aimed at accelerating the detection of
M. tubes°culosis
as well the elucidation of drug resistance profiles of M. tuberculosis
clinical isolates. A
number of these methods are devoted primarily to the task of determining
whether a given
strain is M. tuberculosis or a mycobacterial species other than tuberculosis.
Both culture
based methods and nucleic-acid based methods have been developed that allow M.
tuberculosis to be positively identified more rapidly than by classical
methods: detection times
have been reduced from greater than 6 weeks to as little as two weeks (culture-
based
111ethOdS) or two days (nucleic acid-based methods). While culture-based
methods are
currently in wide-spread use in clinical laboratories, a number of rapid
nucleic acid-based
methods that can be applied directly to clinical samples are under
development. For all of the
techniques described below, it is necessary to first "decontaminate" the
clinical samples, such
as sputum (usually done by pretreatment with N-acetyl L-cysteine and NaOH) to
reduce
contamination by~ non-mycobacterial species [Shinnick and Jones, supra.]
The polymerise chain reaction (PCR) has been applied to the detection of M.
tzcherculosis and can be used to detect its presence directly from clinical
specimens within one
to two days. The more sensitive techniques rely on a two-step procedure: the
first step is the
PCR amplification itself, the second is an analytical step such as
hybridization of the
_~4_
CA 02203627 1997-04-24
WO 96!15267 PCT/US95/14673
amplicori to a M. tuberculosis-specific oligonucleotide probe, or analysis by
RFLP or DNA
sequencing [Shinnick and Jones, supra].
The Amplified M. tuberculosis Direct Test (AMTDT; Gen-Probe) relies on
Transcription Mediated Amplification jTMA; essentially a self sustained
sequence reaction
(3SR) amplification] to amplify target rRNA sequences directly from clinical
specimens.
Once the rRNA has been amplified, it is then detected by a dye-labeled assay
such as the
PACE2. This assay is highly subject to inhibition by substances present in
clinical samples.
The Cycling Probe Reaction (CPR; ID Biomedical). This technique, which is
under
development as a diagnostic tool for detecting the presence of M.
tuberculosis, measures the
accumulation of signal probe molecules. The signal amplification is
accomplished by
hybridizing tripartite DNA-RNA-DNA probes to target nucleic acids, such as M.
tuberculosis-specific sequences. Upon the addition of RNAse H, the RNA portion
of the
chimeric probe is degraded, releasing the DNA portions, which accumulate
linearly over time
to indicate that the target sequence is present [Yule, Bio/Technology 12:1335
(1994)]. The
need to use of RNA probes is a drawback, particularly for use in crude
clinical samples,
where RNase contamination is often rampant.
The above nucleic acid-based detection and differentiation methods offer a
clear time
savings over the more traditional, culture-based methods. While they are
beginning to enter
the clinical setting, their usefulness in the routine diagnosis of M.
tuberculosis is still in
question, in large part because of problems with associated with cross-
contamination and
low-sensitivity relative to culture-based methods. In addition, many of these
procedures are
limited to analysis of respiratory specimens [Yule, Bio/Technology 12:1335
(1994)].
ii) Determination of the antibiotic resistance profile of M. tuberculosis
a) Culture-based methods: Once a positi~re identification of M.
tuberczclosis has been made, it is necessary to characterize the extent and
nature of the strain's
resistance to antibiotics. The traditional method used to determine antibiotic
resistance is the
direct proportion agar dilution method, in which dilutions of culture are
plated on media
containing antibiotics and on control media without antibiotics. This method
typically adds an
additional 2-6 weeks to the time required for diagnosis and characterization
of an unknown
clinical sample [Jacobs, Jr., supra].
The Luciferase Reporter Mycobacteriophage (LRM) assay was first described in
1993
[Jacobs, Jr. et al., Science 260:819 (1993)]. In this assay, a
mycobacteriophage containing a
cloned copy of the luciferase gene is used to infect mycobacterial cultures.
In the presence of
_7j_
CA 02203627 1997-04-24
WO 96115267 PG"TlUS95/14673
luciferin and ATP, the expressed luciferase produces photons, easily
distinguishable by eye or
by a luminometer, allowing a precise determination of the extent of
mycobacterial growth in
the presence of antibiotics. Once sufficient culture has been obtained
(usually 10-14 days
post-inoculation), the assay can be completed in 2 days. This method suffers
from the fact
that the LRM are not specific for M. tuberculosis: they also infect M.
smegmatis and M.
bovis (e.g., BCG), thereby complicating the interpretation of positive
results. Discrimination
between the two species must be accomplished by growth on specialized media
which does
not support the growth of M. tuberculosis (e.g., NAP media). This confirmation
requires
another 2 to 4 days.
The above culture-based methods for determining antibiotic resistance will
continue to
play a role in assessing the effectiveness of putative new anti-mycobacterial
agents and those
drugs for which a genetic target has not yet been identified. However, recent
success in
elucidating the molecular basis for resistance to a number of anti-
mycobacterial agents,
including many of the front-line drugs, has made possible the use of much
faster, more
accurate and more informative DNA polymorphism-based assays-
b) DNA-based methods: Genetic loci involved in resistance to isoniazid,
rifampin, streptomycin, fluoroquinolones, and ethionamide have been identified
[Jacobs, Jr.,
supra; Heym et al., Lancet 344:293 (1994) and Morris et al., J. Infect. Dis.
171:954 (1995)].
A combination of isoniazid (inh) and rifampin (rif) along with pyrazinamide
and ethambutol
or streptomycin, is routinely used as the first line of attack against
confirmed cases of M.
tuberculosis [Banerjee et al., Science 263:227 (1994)]. Consequently,
resistance to one or
more of these drugs can have disastrous implications for short course
chemotherapy treatment.
The increasing incidence of such resistant strains necessitates the
development- of rapid assays
to detect them and thereby reduce the expense and community health hazards of
pursuing
ineffective, and possibly detrimental, treatments. The identification of some
of the genetic
loci involved in drug resistance has facilitated the adoption of mutation
detection technologies
for rapid screening of nucleotide changes that result. in drug resistance. The
availability of
amplification procedures such as PCR and SDA, which have been successful in
replicating
large amounts of target DNA directly from clinical specimens, makes DNA-based
approaches
to antibiotic profiling far more rapid than conventional, culture-based
methods.
The most widely employed techniques in the genetic identification of mutations
leading to drug resistance are DNA sequencing, Restriction Fragment Length
Polymorphism
(RFLP). PCR-Single Stranded Conformational Polymorphism (PCR-SSCP), and
-76-
CA 02203627 1997-04-24
WO 96/15267 PCT/iJS95/14673
PCR-dideoxyfingerprinting (PCR-ddF). All of these techniques have drawbacks as
discussed
above. None of them offers a rapid, reproducible means of precisely and
uniquely identifyinn
individual alleles.
In contrast the CFLPTM method of the present invention provides an approach
that
relies on structure specific cleavage to generate distinct collections of DNA
fragments. This
i
method is highly sensitive (>98%) in its ability to detect sequence
polymorphisms, and
requires a fraction of the time, skill and expense of the techniques described
above.
The application of the CFLPTM method to the detection of MDR-TB is illustrated
herein using segments of DNA amplified from the rpoB and katG genes. Other
genes
associated with MDR-TB, including but not limited to those involved in
conferring resistance
to isoniazid (inhA), streptomycin (rpsL and rrs), and fluoroquinoline (gvrA),
are equally well
suited to the CFLPTM assay.
C. Detection and Identification of Bacterial Pathogens
Identification and typing of bacterial pathogens is critical in the clinical
management
of infectious diseases. Precise identity of a microbe is used not only to
differentiate a disease
state from a healthy state, but is also fundamental to determining whether and
which
antibiotics or other antimicrobial therapies are most suitable for treatment.
Traditional
methods of pathogen typing have used a variety of phenotypic features,
including growth
characteristics, color, cell or colony morphology, antibiotic susceptibility,
staining, smell and
reactivity with specific antibodies to identify bacteria. All of these methods
require culture
of the suspected pathogen, which suffers from a number of serious
shortcomings, including
high material and labor costs, danger of worker exposure, false positives due
to mishandling
and false negatives due to low numbers of viable cells or due to the
fastidious culture
requirements of many pathogens. In addition, culture methods require a
relatively long time
to achieve diagnosis, and because of the potentially life-threatening nature
of such infections,
antimicrobial therapy is often started before the results can be obtained. In
many cases the
pathogens are very similar to the organisms that make up the normal flora, and
may be
indistinguishable from the innocuous strains by the methods cited above. In
these cases,
determination of the presence of the pathogenic strain may require the higher
resolution
afforded by more recently developed molecular typing methods.
A number of methods of examining the genetic material from organisms of
interest
have been developed. One way of performing this type of analysis is by
hybridization of
species-specific nucleic acid probes to the DNA or RNA from the organism to be
tested. This
_77_
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
may be done by immobilizing the denatured nucleic acid to be tested on a
membrane support,
and probing with labeled nucleic acids that will bind only in the presence of
the DNA or
RNA from the pathogen. In this way, pathogens can be identified. Organisms can
be further
differentiated by using the RFLP method described above, in which the genomic
DNA is
digested with one or more restriction enzymes before electrophoretic
separation and transfer to
a nitrocellulose or nylon membrane support. Probing with the species-specific
nucleic acid
probes will reveal a banding pattern that, if it shows variation between
isolates, can be used
as a reproducible way of discriminating between strains. However, these
methods are
susceptible to the drawbacks outlined above: hybridization-based assays are
time-consuming
and may give false or misleading results if the stringency of the
hybridization is not well
controlled, and RFLP identification is dependent on the presence of suitable
restriction sites
in the DNA to be analyzed.
To address these concerns about hybridization and RFLP as diagnostic tools,
several
methods of molecular analysis based on polymerase chain reaction (PCR)
amplification have
gained popularity. In one well-accepted method, called PCR fingerprinting, the
size of a
fragment generated by PCR is used as an identifier. In this type of assay. the
primers are
targeted to regions containing variable numbers of tandem repeated sequences
(referred to as
VNTRs an eukaryotes). The number of repeats, and thus the length of the PCR
alnplicon, can
be characteristic of a given pathogen, and co-amplification of several of
these loci in a single
reaction can create specific and reproducible fingerprints, allowing
discrimination between
closely related species. -
In some cases where organisms are very closely related, however, the target of
the
amplification does not display a size difference, and the amplified segment
must be further
probed to achieve more precise identification. This may be done on a solid
support, in a
fashion analogous to the whole-genome hybridization described above,- but this
has the same
problem with variable stringency as that assay. Alternatively, the interior of
the PCR
fragment may be used as a template for a sequence-specific ligation event. As
outlined
above for the LCR, in this method, single stranded probes to be Iigated are
positioned along
the sequence of interest on either side of an identifying polymorphism, so
that the success or
failure of the ligation will indicate the presence or absence of a specific
nucleotide sequence
at that site. With either hybridization or ligation methods of PCR product
analysis,
knowledge of the precise sequence in the area of probe binding must be
obtained in advance,
_78_
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
and differences outside the probe binding area are not detected. These methods
are poorly
suited to the examination and typing of new isolates that have not been fully
characterized.
In the methods of the present invention, primers that recognize conserved
regions of
bacterial ribosomal RNA genes allow amplification of segments of these genes
that include
sites of variation. The variations in ribosomal gene sequences have become an
accepted
method not only of differentiating between similar organisms on a DNA sequence
level, but
their consistent rate of change allows these sequences to be used to evaluate
the evolutionary
relatedness of organisms. That is to say, the more similar the nucleic acid is
at the sequence
level, the more closely related the organisms in discussion are considered to
be. [Woese,
Bacterial Evolution. Microbiological Reviews, vol 51, No. 2. 1987]. The
present invention
allows the amplification products derived from these sequences to be used to
create highly
individual barcodes (i. e., cleavage patterns), allowing the detection of
sequence
polymorphisms without prior knowledge of the site, character or even the
presence of said
polymorphisms. With appropriate selection of primers, amplification can be
made to be
either all-inclusive (e.g., using the most highly conserved ribosomal
sequences) to allow
comparison of distantly related organisms, or the primers can be chosen to be
very specific
for a given genus, to allow examination at the species and subspecies level.
While the
examination of ribosomal genes is extremely useful in these characterizations,
the use of the
CFLPTM method in bacterial typing is not limited to these genes. Other genes,
including but
not limited to those associated with specific growth characteristics, (e.g.,
carbon source
preference, antibiotic resistance, resistance to methycillin or antigen
production), or with
particular cell morphologies (such as pilus formation) are equally well suited
to the CFLPTM
assay.
D. Extraction of Nucleic Acids From Clinical Samples
To provide nucleic acid substrates for use in the detection and identification
of
microorganisms in clinical samples using the CFLPTM assay, nucleic acid is
extracted from the
sample. The nucleic acid may be extracted from a variety of clinical samples
[fresh or frozen
tissue, suspensions of cells (e.g., blood), cerebral spinal fluid, sputum,
urine, etc.] using a
variety of standard techniques or commercially available kits. For example,
kits which allow
the isolation of RNA or DNA from tissue samples are available from Qiagen,
Inc.
(Chatsworth, CA) and Stratagene (LaJolla, CA). For example, the QIAamp Blood
kits permit
the isolation of DNA from blood (fresh, frozen or dried) as well as bone
marrow, body fluids
-79-
CA 02203627 1997-04-24
WO 96115267 PC"T/US95/14673
or cell suspensions. QIAamp tissue kits permit the isolation of DNA from
tissues such as
muscles, organs and tumors. , _ _ .
It has been found that crude extracts from relatively homogenous specimens
(such as
blood, bacterial colonies, viral plaques, or cerebral spinal fluid) are better
suited to severing as
templates for the amplification of unique PCR products than are more composite
specimens
(such as urine, sputum or feces;) [Shibata in PCR: The Polymerase Chaifz
Reaction, Mullis et
al., eds., Birkhauser, Boston (1994), pp. 47-54]. Samples which contain
relatively few copies
of the material to be amplified (i. e., the target nucleic acid), such as
cerebral spinal fluid, can
be added directly to a PCR. Blood samples have posed a special problem in PCRs
due to the
inhibitory properties of red blood cells. The red blood cells must be removed
prior to the use
of blood in a PCR; there are both classical and commercially available methods
for this
purpose [e.g., QIAamp Blood kits, passage through a Chelex 100 column
(BioRad), etc.].
Extraction of nucleic acid from sputum, the specimen of choice for the direct
detection of M.
tuberculosis, requires prior decontamination to kill or inhibit the growth of
other bacterial
species. This decontamination is typically accomplished by treatment of the
sample with N-
acetyl L-cysteine and NaOH (Shinnick and Jones, supra). This decontamination
process is
necessary only when the sputum specimen is to be cultured prior to analysis.
-80-
CA 02203627 1997-04-24
WO 96115267 PCT/US95/14673
EXPERIMENTAL
The following examples serve to illustrate certain preferred embodiments and
aspects
of the present invention and are not to be construed as limiting the scope
thereof.
In the disclosure which follows, the following abbreviations apply:°C
(degrees
Centigrade); g (gravitational field); vol (volume); w/v (weight to volume);
v/v (volume to
volume); BSA (bovine serum albumin); CTAB (cetyltrimethylammonium bromide);
HPLC
(high pressure liquid chromatography); DNA (deoxyribonucleic acid); IVS
(intervening
sequence); p (plasmid); p,l (microliters); ml (milliliters); ~.g (micrograms);
pmoles
(picomoles); mg (milligrams); MOPS (3-jN-Morpholino]propanesulfonic acid); M
(molar);
mM (milliMolar); ~M (microMolar); nm (nanometers); kdal (kilodaltons); OD
(optical
density); EDTA (ethylene diamine tetra-acetic acid); FITC (fluorescein
isothiocyanate); SDS
(sodium dodecyl sulfate); NaP04 (sodium phosphate); Tris (tris(hydroxymethyl)-
aminomethane); PMSF (phenylmethylsulfonylfluoride); TBE (Tris-Borate-EDTA,
i.c., Tris
buffer tifrated with boric acid rather than HCl and containing EDTA) ; PBS
(phosphate
buffered saline); PPBS (phosphate buffered saline containing 1 mM PMSF); PAGE
(polyacrylamide gel electrophoresis); Tween (polyoxyethylene-sorbitan);
Boehringer
Mannheim (Boehringer Mannheim, Indianapolis, IN); Dynal (Dynal A.S., Oslo,
Norway);
Epicentre (Epicentre Technologies, Madison, WI); National Biosciences
(National Biosciences,
Plymouth, MN); New England Biolabs (New England Biolabs, Beverly, MA); Novagen
(Novagen, Inc., Madison, WI); Perkin Elmer (Perkin Elmer, Norwalk, CT);
Promega Corp.
(Promega Corp., Madison, WI); RJ Research (RJ Research, Inc., Watertown, MA);
Stratagene
(Stratagene Cloning Systems, La Jolla, CA); USB (U.S. Biochemical, Cleveland,
OH).
EXAMPLE 1
Characteristics Of Native Thermostable DNA Polvmerases
A. 5' Nuclease Activity Of DNAPTaq
During the polymerase chain reaction (PCR) [Saiki et al., Science 239:487
(1988);
Mullis and Faloona, Methods in Enzymology 155:335 (1987)], DNAPTaq is able to
amplify
many, but not all, DNA sequences. One sequence that cannot be amplified using
DNAPTay
is shown in Figure 7 (Hairpin structure is SEQ ID NO:15, PRIMERS are SEQ ID
NOS:16-
-81-
CA 02203627 1997-04-24
WO 96115267 PCT/US95ii4673
17.) This DNA sequence has the distinguishing characteristic of being able to
fold on itself to
form a hairpin with two single-stranded arms, which correspond to the primers
used in PCR.
To test whether this failure to amplify is due .to the 5' nuclease activity of
the enzyme,
we compared the abilities of DNAPTaq and DNAPStf to amplify this DNA sequence
during
30 cycles of PCR. Synthetic oligonucleotides were obtained from The
Biotechnology Center
at the University of Wisconsin-Madison. The DNAPTaq and DNAPStf were from
Perkin
Elmer (i.e., AmpliTaq DNA polymerase and the Stoffel fragment of Amplitaq DNA
poly-
merase). The substrate DNA comprised the hairpin structure shown in Figure 7
cloned in a
double-stranded form into pUCl9. The primers used in the amplification are
listed as SEQ
ID NOS:16-17.Primer SEQ ID N0:17 is shown annealed to the 3' arm of the
hairpin struc-
ture in Fig. 7. Primer SEQ ID N0:16 is shown as the first 20 nucleotides in
bold on the 5'
arm of the hairpin in Fig. 7.
Polymerase chain reactions comprised 1 ng of supercoiled plasmid target DNA, 5
pmoles of each primer, 40 ~,M each dNTP, and 2.5 units of DNAPTaq or DNAPStf,
in a 50
~.l solution of 10 mM Tris Cl pH 8.3. The DNAPTaq reactions included 50 mM KCl
and 1.5
mM MgCI,. The temperature profile-was 95°C for 30 sec., 55°C for
1-min. and 72°C for 1
min., through 30 cycles. Ten percent of each reaction was analyzed by gel
electrophoresis
through 6% polyacrylamide (cross-linked 29:1) in a buffer of 45 mM Tris
Borate, pH 8.3, 1.4
mM EDTA (O:SX-TBE).
The results are shown in Figure 8. The expected product was made by DNAPStf
(indicated simply as "S") but not by DNAPTaq (indicated as "T"). We conclude
that the 5'
nuclease-activity of DNAPTaq is responsible for the lack of amplification of
this DNA se-
quence. - - - . _ . _ _
To test whether the 5' unpaired nucleotides in the substrate region of this
structured
DNA are removed by DNAPTaq, the fate of the end-labeled 5' arm during four
cycles of
PCR was compared using the same two polymerases (Figure 9). The hairpin
templates, such
as the one described in Figure 6, were made using DNAPStf and a 3'P-5"-end-
labeled primer.
The 5'-end of the DNA was released as a few large fragments by DNAPTarI but
not by
DNAPSt~ The sizes of these fragments (based on their mobilities) show that
they contain
most or all of the unpaired 5' arm of the DNA. Thus, cleavage occurs at or
near the base of
the bifurcated duplex. These released fragments terminate with 3' OH groups.
as evidenced
by direct sequence analysis, and the abilities of the fragments to be extended
by terminal
deoxynucleotidyl transferase.
_g2_
CA 02203627 1997-04-24
WO 96115267 PC"TlUS95/14673
Figures 10-12 show the results of experiments designed to characterize the
cleavage
reaction catalyzed by DNAPTag. Unless otherwise specified, the cleavage
reactions com-
prised 0.01 pmoles of heat-denatured, end-labeled hairpin DNA (with the
unlabeled comple-
mentary strand also present), 1 pmole primer (complementary to- the 3' arm)
and 0.5 units of
DNAPTaq (estimated to be 0.026 pmoles) in a total volume of 10 pl of 10 mM
Tris-Cl, pH
8.5, 50 mM KCl and 1.5 mM MgCI,. As indicated, some reactions had different
concentra-
tions of KCI, and the precise times and temperatures used in each experiment
are indicated in
the individual figures. The reactions that included a primer used the one
shown in Figure 6
(SEQ ID N0:17). In some instances, the primer was extended to the junction
site by
providing polymerase and selected nucleotides.
Reactions were initiated at the final reaction temperature by the addition of
either the
MgCI, or enzyme. Reactions were stopped at their incubation temperatures by
the addition of
8 p.l of 95% formamide containing 20 mM EDTA and 0.05% marker dyes (stop
solution).
The T", calculations listed were made using the OligoTM primer analysis
software from
National Biosciences, Inc. These were determined using 0.2 i ~M as the DNA
concentration,
at either 1 ~ or 65 mM total salt (the 1.5 mM MgCI, in all reactions was given
the value of 15
mM salt for these calculations).
Figure 10 is an autoradiogram containing the results of a set of experiments
and
conditions on the cleavage site. Figure l0A is a determination of reaction
components that
enable cleavage. Incubation of 5'-end-labeled hairpin DNA was for 30 minutes
at 55°C, with
the indicated components. The products were resolved by denaturing
polyacrylamide gel
electrophoresis and the lengths of the products, in nucleotide s, are
indicated. Figure 1 OB .
describes the effect of temperature on the site of cleavage in the absence of
added primer.
Reactions were incubated in the absence of KCl for 10 minul:es at the
indicated temperatures.
The lengths of the products, in nucleotides, are indicated.
Surprisingly, cleavage by DNAPTag requires neither a primer nor dNTPs (.see
Fig.
l0A). Thus, the 5' nuclease activity can be uncoupled from polymerization.
Nuclease
activity requires magnesium ions, though manganese ions can be substituted.
albeit with
potential changes in specificity and activity. Neither zinc nor calcium ions
support the
cleavage reaction. The reaction occurs over a broad temperature range. from
2~°C to 85°C.
with the rate of cleavage increasing at higher temperatures.
Still referring to Figure 10, the primer is not elongated in the absence of
added
dNTPs. However, the primer influences both the site and th~° rate of
cleavage of the hairpin.
- g3 -
CA 02203627 1997-04-24
WO 96/15267 PG"T/U595I14673
The change in the site of cleavage (Fig. l0A) apparently results from
disruption of a short
duplex formed between the arms of the DNA substrate. In the absence of primer,
the
sequences indicated by underlining in Figure 7 could. pair, forming an
extended duplex.
Cleavage at the end of the extended duplex would release the 11 nucleotide
fragment seen on
the Fig. 1OA lanes with no added primer. Addition of excess primer (Fig. 1OA,
lanes 3 and
4) or incubation at an elevated temperature (Fig. lOB) disrupts the short
extension of the
duplex and results in a longer 5' arm and, hence, longer cleavage products.
The location of the 3' end of the primer can influence the precise site of
cleavage.
Electrophoretic analysis revealed that in the absence of primer (Fig. lOB),
cleavage occurs at
the end of the substrate duplex (either the extended or shortened form,
depending on the
temperature) between the first and second base pairs. When the primer extends
up to the base
of the duplex, cleavage also occurs one nucleotide into the duplex. However,
when a gap of
four or six nucleotides exists between the 3' end of the primer and the
substrate duplex, the
cleavage site is shifted four to six nucleotides in the 5' direction.
Fig. 11 describes the kinetics of cleavage in the presence (Fig. 11 A) or
absence (Fig.
I lB) of a primer oligonucleotide. The reactions were run at 55°C with
either 50 mM KCl
(Fig. 11A) or 20 mM KCl-(Fig. 11B). The reaction products were resolved by
denaturing
polyacrylamide gel electrophoresis and the lengths of the products, in
nucleotides, are
indicated. "M", indicating a marker, is a 5' end-labeled 19-nt
oligonucleotide. Under these
salt conditions, Figs. 11A and 11B indicate that the reaction appears to be
about twenty times
faster in the presence of primer than in the absence of primer. This effect on
the efficiency
may be attributable to' proper alignment and stabilization of the enzyme on
the substrate.
The relative influence of-primer on cleavage rates becomes much greater when
both
reactions are run in 50 mM KCI. In the presence of primer, the rate of
cleavage increases
with KCl concentration, up to about 50 mM. However, inhibition of this
reaction in the
presence of primer is apparent at 100 mM and is complete at 150 mM KCI. In
contrast, in
the absence of primer the rate is enhanced by concentration of KCl up to 20
mM, but it is
reduced at concentrations above 30 mM. At 50 mM KCI, the reaction is almost
completely
inhibited. The inhibition of cleavage by KCl in the absence of primer is
affected by
temperature, being more pronounced at lower temperatures.
Recognition of the 5' end of the arm to be cut appears to be an important
feature of '
substrate recognition. Substrates that lack a free 5' end, such as circular M
13 DNA, cannot
be cleaved under any conditions tested. Even with substrates having defined 5'
arms. the rate
- 84 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
of cleavage by DNAPTaq is influenced by the length of the arm. In the presence
of primer
and 50 mM KCI, cleavage of a 5' extension that is 27 nucleotides long is
essentially complete
within 2 minutes at 55°C. In contrast, cleavages of molecules with 5'
arms of 84 and 188
nucleotides are only about 90% and 40% complete after 20 minutes. Incubation
at higher
temperatures reduces the inhibitory effects of long extensions indicating that
secondary
structure in the 5' arm or a heat-labile structure in the enzyme may inhibit
the reaction. A
mixing experiment, run under conditions of substrate excess, shows that the
molecules with
long arms do not preferentially tie up the available enzyme in non-productive
complexes.
These results may indicate that the 5' nuclease domain gains access to the
cleavage site at the
end of the bifurcated duplex by moving down the 5' arm fram one end to the
other. Longer
5' arms would be expected to have more adventitious secondary structures
(particularly when
KCl concentrations are high), which would be likely to impede this movement.
Cleavage does not appear to be inhibited by long 3' arms of either the
substrate strand
target molecule or pilot nucleic acid, at least up to 2 kilobases. At the
other extreme, 3' arms
of the pilot nucleic acid as short as one nucleotide can support cleavage in a
primer-
independent reaction, albeit inefficiently. Fully paired oligonucleotides do
not elicit cleavage
of DNA templates during primer extension.
The ability of DNAPTaq to cleave molecules even when the complementary strand
contains only one unpaired 3' nucleotide may be useful in optimizing allele-
specific PCR.
PCR primers that have unpaired 3' ends could act as pilot oligonucleotides to
direct selective
cleavage of unwanted templates during preincubation of potential template-
primer complexes
with DNAPTaq in the absence of nucleoside triphosphates.
S. 5' Nuclease Activities Of Other DNAPs
To determine whether other 5' nucleases in other DNAPs would be suitable for
the
present invention, an array of enzymes, several of which were reported in the
literature to be
free of apparent 5' nuclease activity, were examined. The ability of these
other enzymes to
cleave nucleic acids in a structure-specific manner was tested using the
hairpin substrate
shown in Fig. 7 under conditions reported to be optimal for synthesis by each
enzyme.
DNAPEcI and DNAP Klenow were obtained from Promega Corporation; the DNAP of
Pyrococcus _ furious ["Pfu", Bargseid et al., Strategies 4:34 ( 1991 )] was
from Stratagene; the
DNAP of Thermococcus litoralis ["Tli", Vent(exo-), Perler et al., Proc. Natl.
Acad. Sci. USA
89:5577 (1992)] was from New England Biolabs; the DNAF of ThermZrs,flavus
["Tfl",
-85-
CA 02203627 1997-04-24
WO 96115267 PCTIUS95/14673
Kaledin et al., Biokhimiya 46:1576 (1981)] was from Epicentre Technologies;
and the DNAP
of Thermus thermophilus ["Tth", Carballeira et crl., Biotechniques 9:276
(1990): Myers et al..
Biochem. 30:7661 (1991)] was from U.S. Biochemicals.
0.5 units of each DNA polymerase was assayed in a 20 ~.l reaction, using
either the
buffers supplied by the manufacturers for the primer-dependent reactions, or
10 mM Tris Cl,
pH 8.5, 1.5 mM MgCl2, and 20 mM KCI. Reaction mixtures were at held
72°C before the
addition of enzyme.
Figure 12 is an autoradiogram recording the results of these tests. Figure 12A
demonstrates reactions of endonucleases of DNAPs of several thermophilic
bacteria. The
reactions were incubated at 55°C for 10 minutes in the presence of
primer or at 72°C for 30
minutes in the absence of primer, and the products were resolved by denaturing
polyacrylamide gel electrophoresis. The lengths of the products, in
nucleotides, are indicated.
Figure 12B demonstrates endonucleolytic cleavage by the 5' nuclease of
DNAPEcI. The
DNAPEcl and DNAP Klenow reactions were incubated for 5 minutes at 37°C.
Note the light
1 ~ band of cleavage products of 25 and 11 nucleotides in the DNAPEcI lanes
(made in the
presence and absence of primer, respectively). Figure 12B also demonstrates
DNAPTaq
reactions in the presence (+) or absence (-) of primer. These reactions were
run in 50 mM
and 20 mM KCI, respectively, and were incubated at 55°C for 10 minutes.
Referring to Figure 12A, DNAPs from the eubacteria Thermus -thermophilus and
Thermzrs , flavus cleave the substrate at the same place as DNAPTaq, both in
the presence and
absence of primer. In contrast, DNAPs from the archaebacteria Pyrococcus ,
furioszts and
Thermococcus litoralis are unable to cleave the substrates
endonucleolytically. The DNAPs
from Pyrococcus,furious and Thermococcus litoralis share little sequence
homology with
eubacterial enzymes (Ito et al. , Nucl. Acids Res. 19:4045 ( 1991 ); Mathur et
al. , Nucl. Acids.
Res. 19:6952 (1991); see also Perler et al.). Referring to Figure 12B, DNAPEcI
also cleaves
the substrate, but the resulting cleavage products are difficult to detect
unless the 3'
exonuclease is inhibited. The amino acid sequences of the 5' nuclease domains
of DNAPEcI
and DNAPTaq are about 38% homologous (Gelfand, supra).
The 5' nuclease domain of DNAPTaq also shares about 19% homology with the ~~
exonuclease encoded by gene 6 of bacteriophage T7 [Dune et al., J. Mol. Biol.
166:477
(1983)]. This nuclease, which is not covalently attached to a DNAP
polymerization domain.
is also able to cleave DNA endonucleolytically, at a site similar or identical
to the site that is
cut by the 5' nucleases described above, in the absence of added primers
-86-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
C. Transcleavage
The ability of a 5' nuclease to bedirected to cleave efficiently at any
specific sequence
was demonstrated in the following experiment. A partially complementary
oligonucleotide
termed a "pilot oligonucleotide" was hybridized to sequences at the desired
point of cleavage.
The non-complementary part of the pilot oligonucleotide provided a structure
analogous to the
3' arm of the template (see Figure 7), whereas the 5' region of the substrate
strand became
the 5' arm. A primer was provided by designing the 3' region of the pilot so
that it would
fold on itself creating a short hairpin with a stabilizing tetra-loop [Antao
et al., Nucl. Acids
Res. 19:5901 (1991)]. Two pilot oligonucleotides are shown in Figure 13A.
Oligonucleotides
19-12 (SEQ ID N0:18) and 30-12 (SEQ ID N0:19) are 31 or 42 or nucleotides
long,
respectively. However, oligonucleotides 19-12 ($EQ ID NO:18) and 34-19 (SEQ ID
N0:19)
have only 19 and 30 nucleotides, respectively, that are complementary to
different sequences
in the substrate strand. The pilot oligonucleotides are calculated to melt off
their
complements at about 50°C (19-12) and about 75°C (30-12). Both
pilots have 12 nucleotides
at their 3' ends, which act as 3' arms with base-paired primers attached.
To demonstrate that cleavage could be directed by a pilot oligonucleotide, we
incubated a single-stranded target DNA with DNAPTaq in the presence of two
potential pilot
oligonucleotides. The transcleavage reactions, where the target and pilot
nucleic acids are not
covalently linked, includes 0.01 pmoles of single end-labeled substrate DNA, 1
unit of
DNAPTaq and 5 pmoles of pilot oligonucleotide in a volume of 20 p.l of the
same buffers.
These components were combined during a one minute incubation at 95°C,
to denature the
PCR-generated double-stranded substrate DNA, and the temperatures of the
reactions were
then reduced to their final incubation temperatures. Oligonucleotides 30-12
and 19-12 can
hybridize to regions of the substrate DNAs that are 85 and 27 nucleotides from
the 5' end of
the targeted strand.
Figure 23 shows the complete 206-mer sequence (SEQ ID N0:26). The 206-mer was
generated by PCR . The M13/pUC 24-mer reverse sequencing (-48) primer and the
M13/pUC
sequencing (-47) primer from New England Biolabs (catalogue nos. 1233 and 1224
respectively) were used (50 pmoles each) with the pGEM3z(f+) plasmid vector
(Promega
Corp.) as template (10 ng) containing the target sequences. The conditions for
PCR were as
' follows: 50 ~.M of each dNTP and 2.5 units of Taq DNA p~olymerase in 100 ~l
of 1X PCR
Buffer (20 mM Tris-Cl, pH 8.3, 1.5 mM MgCI,, 50 mM K~CI with 0.05% Tween-20
and
0.05% NP-40). Reactions were cycled 35 times through 95°C for 45
seconds, 63°C for 4~
_87_
CA 02203627 1997-04-24
WO 96/15267 PC'T/US95/14673
seconds, then 72°C for 75 seconds. After cycling, reactions were
finished off with an
incubation at 72°C for 5 minutes. The resulting fragment was purified
by electrophoresis
through a 6% polyacrylamide gel (29:1 cross link) in a buffer of O.SX TBE (45
mM Tris-
Borate, pH 8.3, 1.4 mM EDTA), visualized by ethidium bromide staining or
autoradiography,
excised from the gel, eluted by passive diffusion, and concentrated by ethanol
precipitation.
Cleavage of the substrate DNA occurred in the presence of the pilot
oligonucleotide
I9-12 at 50°C (Figure 13B, lanes 1 and 7) but not at 75°C (lanes
4 and 10). In the presence
of oligonucleotide 30-12 cleavage was observed at both temperatures. Cleavage
did not occur
in the absence of added oligonucleotides (lanes 3, 6 and 12) or at about
80°C even though at
50°C adventitious structures in the substrate allowed primer-
independent cleavage in the
absence of KCl (Figure 13B, lane 9). A non-specific oligonucleotide with no
complementarity to the substrate DNA did not direct cleavage at SO°C,
either in the absence
or presence of 50 mM KCl (lanes 13 and 14). Thus, the specificity of the
cleavage reactions
can be controlled by the extent of complementarity to the substrate and by the
conditions of
incubation.
D. Cleavage Of RNA -
An shortened RNA version of the sequence used in the transcleavage experiments
discussed above was tested for its ability to serve as a substrate in the
reaction. The RNA is
cleaved at the expected place, in a reaction that is dependent upon the
presence of the pilot
oligonucleotide. The RNA substrate, made- by T7 RNA- polymerase in the
presence of [a.-
3'P]UTP, corresponds to a truncated version of the DNA substrate used in
Figure I3B.
Reaction conditions were similar to those in used for the DNA substrates
described above,
with 50 mM KCI; incubation was for 40 minutes at 55°C. The pilot
oligonucleotide used is
termed 30-0 (SEQ ID N0:20) and is shown in Figure 14A.
The results of the cleavage reaction is shown in Figure 14B. The reaction was
run
either in the presence or absence of DNAPTaq or pilot oligonucleotide as
indicated in Figure
14B.
Strikingly, in the case of RNA cleavage, a 3' arm is not required for the
pilot
oligonucleotide. It is very unlikely that this cleavage is due to previously
described RNaseH,
-which would be expected to cut the RNA in several places along the 30 base-
pair long RNA-
DNA duplex. The 5' nuclease of DNAPTaq is a'structure-specific RNaseH that
cleaves the
RNA at a single site near the 5' end of the heteroduplexed region.
_88_
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
It is surprising that an oligonucleotide lacking a 3' arm is able to act as a
pilot in
directing efficient cleavage of an RNA target because such ol.igonucleotides
are unable to
direct efficient cleavage of DNA targets using native,DNAPs. However, some 5'
nucleases of
the present invention (for example, clones E, F and G shown in Figure 16) can
cleave DNA
in the absence of a 3' arm. In other words, a non-extendable cleavage
structure is not
required for specific cleavage with some 5' nucleases of the present invention
derived from
thermostable DNA polymerases.
We tested whether cleavage of an RNA template by L>NAPTczq in the presence of
a
fully complementary primer could help explain why DNAPTag is unable to extend
a DNA
oligonucleotide on an RNA template, in a reaction resembling; that of reverse
transcriptase.
Another thermophilic DNAP, DNAPTth, is able to use RNA as a template, but only
in the
presence of Mn++, so we predicted that this enzyme would not cleave RNA in the
presence of
this cation. Accordingly, we incubated an RNA molecule with an appropriate
pilot
oligonucleotide in the presence of DNAPTaq or DNAPTth, in buffer containing
either MgT'
or Mn'*. As expected, both enzymes cleaved the RNA in the presence of Mg++.
However,
DNAPTczq, but not DNAPTth, degraded the RNA in the presence of Mn++. We
conclude that
the 5" nuclease activities of many DNAPs may contribute to their inability to
use RNA as
templates:
EXAMPLE 2
Generation Of 5' Nucleases From Thermostable DNA Polymerases
Thermostable DNA polymerases were generated which have reduced synthetic
activity,
an activity that is an undesirable side-reaction during DNA cleavage in the
detection assay of
the invention, yet have maintained thermostable nuclease activity. The result
is a
thermostable polymerase which cleaves nucleic acids DNA with extreme
specificity.
Type A DNA polymerases from eubacteria of the genus Then-mus share extensive
protein
sequence identity (90% in the polymerization domain, using the Lipman-Pearson
method in
the DNA analysis software from DNAStar, WI) and behave similarly in both
polymerization
and nuclease assays. Therefore, we have used the genes for t:he DNA polymerase
of Tlzez°m2zs
"' aquaticus (DNAPTaq) and Thermus flavus (DNAPTfl) as representatives of this
class.
Polymerase genes from other eubacterial organisms, such as ThermZCS
thermophilus, Thez°mzzs~
sp.. Thermotoga maritima, Thermosipho af>"icanzc.s and Bacillzzs
stearothermophilus are equally
-89-
CA 02203627 1997-04-24
WO 96/15267 PC"T/US95t14673
suitable. The DNA polymerases from these thermophilic organisms are capable of
surviving
and performing at elevated _ temperatures, and can thus be used in reactions
in which
temperature is used as a selection against non-specific hybridization of
nucleic acid strands.
The restriction sites used for deletion mutagenesis, described below, were
chosen for
convenience. Different sites situated with similar convenience are available
in the Thermiss
thermophilus gene and can be used to make similar constructs with other Type A
polymerase
genes from related organisms.
A. Creation Of 5' Nuclease Constructs
1. Modified DNAPTaq Genes
The first step was to place a modified gene for the Taq DNA polymerise on a
plasmid
under control of an inducible promoter. The modified Taq polymerise gene was
isolated as
follows: The Taq DNA polymerise gene was amplified by polymerise chain
reaction from
genomic DNA from Thermus aquaticus, strain YT-1 (Lawyer et al., supra), using
as primers
the oligonucleotides described in SEQ ID NOS:13-14. The resulting fragment of
DNA has a
recognition sequence for the restriction endonuclease EcoRI at the 5' end of
the coding
sequence and a BgIII sequence at the 3' end. Cleavage with BgIII leaves a 5'
overhang or
"sticlcy end" that is compatible with the end generated by BamHI. The PCR-
amplified DNA
was digested with EcoRI and BamHI. The 2512 by fragment containing the coding
region for
the polymerise gene was gel purified and then ligated into a plasmid which
contains an
inducible promoter.
In one embodiment of the invention, the pTTQ 18 vector, which contains the
hybrid
trp-lac (tic) promoter, was used jM.J.R. Stark, Gene 5:255 (1987) and shown in
Figure 15.
The tic promoter is under the control of the E. coli lac repressor. Repression
allows the
synthesis of the gene product to be suppressed until the desired level of
bacterial growth has
been achieved, at which point repression is removed by addition of a specific
inducer,
isopropyl-b-D-thiogalactopyranoside (IPTG). Such a system allows the
expression of foreign
proteins that may slow or prevent growth of transformants.
Bacterial promoters, such as tic, may not be adequately suppressed when they
are
present on a multiple copy plasmid. If a highly toxic protein is placed under
control of such
a promoter, the small amount of expression leaking through can be harmful to
the bacteria.
In another embodiment of the invention, another option for repressing
synthesis of a cloned
gene product was used. The non-bacterial promoter, from bacteriophage T7,
found in the
plasmid vector series pET-3 was used to express the cloned mutant Taq
polymerise genes
-90-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95I14673
[Figure 15; Studier and Moffatt, J. Mol. Biol. 189:113 (1986)].- This promoter
initiates
transcription only by T7 RNA polymerise. In a suitable strain, such as BL21
(DE3)pLYS. the
gene for this RNA polymerise is carried on the bacterial genome under control
of the lac
operator. This arrangement has the advantage that expression of the multiple
copy gene (on
the plasmid) is completely dependent on the expression of T7 RNA polymerise,
which is
easily suppressed because it is present in a single copy.
For ligation into the pTTQl8 vector (Figure 15), the PCR product DNA
containing the
Taq polymerise coding region (mutTaq, clone 4B, SEQ ID NO:21 ) was digested
with EcoRI
and BgIII and this fragment was ligated under standard "sticky end" conditions
[Sambrook et
al. Molecular Cloning, Cold Spring Harbor Laboratory Press, Cold Spring
Harbor, pp. 1.63-
1.69 ( 1989)] into the EcoRI and BamHI sites of the plasmid vector pTTQ 18.
Expression of
this construct yields a translational fusion product in which the first two
residues of the native
protein (Met-Arg) are replaced by three from the vector (Met-Asn-Ser), but the
remainder of
the natural protein would not change. The construct was transformed into the
JM109 strain of
E. coli and the transformants were plated under incompletely repressing
conditions that do not
permit growth of bacteria expressing the native protein. These plating
conditions allow the
isolation of genes containing pre-existing mutations, such as those that
result from the
infidelity of Taq polymerise during the amplification process.
Using this amplification/selection protocol, we isolated a clone (depicted in
Figure 16B) containing a mutated Taq polymerise gene (mutTirq, clone 4B). The
mutant was
first detected by its phenotype, in which temperature-stable 5' nuclease
activity in a crude cell
extract was normal, but polymerization activity was almost absent
(approximately less than
1 % of wild type Taq polymerise activity).
DNA sequence analysis of the recombinant gene showed that it had changes in
the
polymerise domain resulting in two amino acid substitutions: an A to G change
at nucleotide
position 1394 causes a Glu to Gly change at amino acid position 465 (numbered
according to
the natural nucleic and amino acid sequences, SEQ Il7 NOS:1 and 4) and another
A to G
change at nucleotide position 2260 causes a Gln to Arg change at amino acid
position 7~4.
Because the Gln to Gly mutation is at a nonconserved position and because the
Glu to Ark
mutation alters an amino acid that is conserved in virtually all of the known
Type A
polymerises, this latter mutation is most likely the one responsible for
curtailing the synthesis
activity of this protein. The nucleotide sequence for the clone 4B construct
(Figure 16B) is
-91 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
given in SEQ ID N0:21. The corresponding amino acid sequence encoded by the
nucleotide
sequence of SEQ ID N0:21 is listed in SEQ ID N0:72. ,
Subsequent derivatives of DNAPTaq constructs were made from the mutTcrg gene,
thus, they all bear these amino acid substitutions in addition to their other
alterations, unless
these particular regions were deleted. These mutated sites are indicated by
black boxes at
these locations in the diagrams in Figure 16. In Figure 16, the designation
"3' Exo" is used
to indicate the location of the 3' exonuclease activity associated with Type A
polymerases
which is not present in DNAPTaq. All constructs except the genes shown in
Figures 16E, F
and G were made in the pTTQ 18 vector.
The cloning vector used for the genes in Figures 16E and F was from the
commercially available pET-3 series, described above. Though this vector
series has only a
BamHI site for cloning downstream of the T7 promoter, the series contains
variants that allow
cloning into any of the three reading frames. For cloning of the PCR product
described
above, the variant called pET-3c was used (Figure 17). The vector was digested
with BcrrnHl,
dephosphorylated with calf intestinal phosphatase, and the sticky ends were
filled in using the
Klenow fragment of DNAPEc 1 and dNTPs. The gene for the mutant Tack DNAP shown
in
Figure 16B (mutTaq, clone 4B) was released from pTTQlB by digestion with EcoRI
and .ScrlI,
and the "sticky ends" were filled in as was done with the vector. The fragment
was ligated to
the vector under standard blunt-end conditions (Sambrook et crl., Moleculcrr~
Cloning, supra),
the construct was transformed into the BL21(DE3)pLYS strain of E. coli, and
isolates were
screened to identify those that were ligated with the gene in the proper
orientation relative to
the promoter. This construction yields another translational fusion product,
in which the first
two amino acids of DNAPTaq (Met-Arg) are replaced by 13 from the vector plus
two from
the PCR primer (Met-Ala-Ser-Met-Thr-Gly-Gly-Gln-Gln-Met-Gly-Arg-Ile-Asn-Ser)
(SEQ ID
N0:23).
Our goal was to generate enzymes that lacked the ability to synthesize DNA.
but
retained the ability to cleave nucleic acids with a 5' nuclease activity. The
act of primed,
templated synthesis of DNA is actually a coordinated series of events, so it
is possible to
disable DNA synthesis by disrupting one event while not affecting the others.
These steps
include, but are not limited to, primer recognition and binding, dNTP binding
and catalysis of
the inter-nucleotide phosphodiester bond. Some of the amino acids in the
polymerization '
domain of DNAPEcI have been linked to these functions, but the precise
mechanisms are as
yet poorly defined.
-92-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95I14673
One way of destroying the polymerizing ability of a DMA polymerise is to
delete all
or part of the gene segment that encodes that domain for the protein, or to
otherwise render
the gene incapable of making a complete polymerization domain. Individual
mutant enzymes
may differ from each other in stability and solubility both inside and outside
cells. For
instance, in contrast to the 5' nuclease domain of DNAPEcI, which can be
released in an
active form from the polymerization domain by gentle proteolysis [Setlow and
Kornberg, J.
Biol. ChenZ. 247:232 (1972)], the Thermus nuclease domain, when treated
similarly, becomes
less soluble and the cleavage activity is often lost.
Using the mutant gene shown in Figure 16B as starting material, several
deletion
constructs were created. All cloning technologies were standard (Sambrook et
al., sup~cr) and
are summarized briefly, as follows:
Figure 16C: The mutTaq construct-was digested with .PstI, which cuts once
within the
polymerise coding region, as indicated, and cuts immediately downstream of the
gene in the
multiple cloning site of the vector. After release of the fragment between
these two sites, the
vector was re-ligated, creating an 894-nucleotide deletion, and bringing into
frame a stop
codon 40 nucleotides downstream of the junction. The nucleotide sequence of
this 5'
nuclease (clone 4C) is given in SEQ ID N0:9. The corresponding amino acid
sequence
encoded by the nucleotide sequence of SEQ ID N0:9 is listed in SEQ ID N0:73.
Figure 16D: The mutTaq construct was digested with .NheI, which cuts once in
the
gene at position 2047. The resulting four-nucleotide 5' overhanging ends were
filled in, as
described above, and the blunt ends were re-ligated. The resulting four-
nucleotide insertion
changes the reading frame and causes termination of translation ten amino
acids downstream
of the mutation. The nucleotide sequence of this 5' nuclease (clone 4D) is
given in SEQ ID
NO:IO. The corresponding amino acid sequence encoded by the nucleotide
sequence of SEQ
ID NO:10 is listed in SEQ ID N0:74.
Figure 16E: The entire mutTaq gene was cut from pTTQl8 using EcoRI and SaII
and
cloned into pET-3c, as described above. This clone was digested with BstXI and
XcmI, at
unique sites that are situated as shown in Figure 16E. The DNA was treated
with the Klenow
fragment of DNAPEcl and dNTPs, which resulted in the 3' overhangs of both
sites being
trimmed to blunt ends. These blunt ends were ligated together, resulting in an
out-of frame
deletion of 1540 nucleotides. An in-frame termination codon occurs 18 triplets
past the
junction site. The nucleotide sequence of this 5' nuclease (clone 4E) is given
in SEQ ID
NO:11 [The corresponding amino acid sequence encoded by the nucleotide
sequence of SEQ
-93-
CA 02203627 1997-04-24
WO 96!15267 PG"T/US95114673
ID NO:l 1 is listed in SEQ ID N0:75]., with the appropriate leader sequence
given in SEQ ID
N0:24 (The corresponding amino acid sequence encoded by the nucleotide
sequence of SEQ
ID N0:24 is listed in SEQ ID N0:76. It is also referred to as the CleavaseTM
BX enzyme.
Figure 16F: The entire mutTaq gene was cut from pTTQ 18 using EcoRI and SaII
and
cloned into pET-3c, as described above. This clone was digested with BstXI and
BamHI, at
unique sites that are situated as shown in the diagram. The DNA was treated
with the
Klenow fragment of DNAPEcl and dNTPs, which resulted in the 3' overhang of the
BstXI
site being trimmed to a blunt end, while the 5' overhang of the BamHI site was
filled in to
make a blunt end. These ends were ligated together, resulting in an in-frame
deletion of 903
nucleotides. The nucleotide sequence of the 5' nuclease (clone 4F) is given in
SEQ ID
N0:12. It is also referred to as the CleavaseTM BB enzyme. The corresponding
amino acid
sequence encoded by the nucleotide sequence of SEQ ID NO:1? is listed in SEQ
ID N0:77.
Figure 16G: This polymerase is a variant of that shown in Figure 16E. It was
cloned
in the plasmid vector pET=21 (Novagen). The non-bacterial promoter from
bacteriophage T7,
found in this vector, initiates transcription only by T7 RNA polymerase. Sec
Studier and
Moffatt, sups°a. In a suitable strain, such as (DES)pLYS, the gene for
this RNA polymerase
is carried on the bacterial genome under control of the lac operator. This
arrangement has the
advantage that expression of the multiple copy gene (on the plasmid) is
completely dependent
on the expression of T7 RNA polymerase, which is easily suppressed because it
is present in
a single copy. Because the expression of these mutant genes is under this
tightly controlled
promoter, potential problems of toxicity of the expressed proteins to the host
cells are less of
a concern.
The pET-21 vector also features a "His-Tag", a stretch of six consecutive
histidine
residues that are added on the carboxy terminus of the expressed proteins. The
resulting
proteins can then be purified in a single step by metal chelation
chromatography, using a
commercially available (Novagen) column resin with immobilized NiT' ions. The
2.5 ml
columns are reusable, and can bind up to 20 mg of the target protein under
native or
denaturing (guanidine-HCl or urea) conditions. -
E. coli (DES)pLYS cells are transformed with the constructs described above
using
standard transformation techniques, and used to inoculate a standard growth
medium (e.g.,
Luria-Bertani broth). Production of T7 RNA polymerase is induced during log
phase growth '
by addition of IPTG and incubated for a further 12 to 17 hours. Aliquots of
culture are
removed both before and after induction and the proteins are examined by SDS-
PAGE.
-94-
CA 02203627 1999-07-26
Staining with Coomassie Blue allows visualization of the foreign proteins if
they account for
about 3-5% of the cellular protein and do not co-migrate with any of the major
host protein
bands. Proteins that co-migrate with major host proteins must be expressed as
more than 10°~0
of the total protein to be seen at this stage of analysis.
Some mutant proteins are sequestered by the cells into inclusion bodies.
.These are
granules that form in the cytoplasm when bacteria are made to express high
levels of a .
forei'n protein. and they can be purified from a crude lysate. and analyzed by
SDS-PAGE to
determine their protein content. If the cloned protein is found in the
inclusion bodies. it must
be released to assay the cleavage and polymerise activities. Different methods
of
solubilization may be appropriate for different proteins, and a variety of
methods are known.
.See e.g.. Builder & Ogez. U.S. Patent No. 4,~ 11.502 ( 1980: Olson. U.S.
Patent No.
4.~ 18.26 ( I 98~): Olson & Pai. U.S. Patent No. 4.511.03 ( 1985): Jones of
ul.. U.S. Patent
No. 4.~ l 2.922 ( 198 ) .
The solubilized protein is then purified on the Ni~ column as described above.
I ~ followin~_ the manufacturers instructions (Novagen). The washed proteins
are eluted from the
column by a combination of imidazole competitor ( 1 Ml and high salt (0.~ M
NaCI ). and
dialyzed to exchange the buffer and to allow denatured proteins to refold.
Typical recoveries
result in approximately 20 ~g of specific protein per ml of startin~~ culture.
The DNAP
mutant is rei~erred to as the CleavaseTM BN enzyme and the sequence is given
in SEQ ID
2() NO:'_'~. The corresponding amino acid sequence encoded by the nucleotide
sequence of SEQ
ID N0:2~ is listed in SEQ ID N0:78.
2. Modified DNAPTfl Gene
The DNA polymerise gene of Thermus.navu.s was isolated from rlTO "T
.flcn~tr.v" AT-G'_'
strain obtained from the American Type Tissue Collection (ATCC 33923 ). This
strain has a
different restriction map then does the T. flavus strain used to generate the
sequence published
by Akhmetzianov and Vakhitov. supra. The published sequence is listed as SEQ
ID N0:2.
No sequence data has been published for the DNA polymerise gene from the AT-62
strain of
T. llantr.v.
Genomic DNA from T. ,flavus was amplified using the same primers used to
amplify
30 the T. aquaticu.s DNA polymerise gene (SEQ ID NOS:13-14). The approximately
200 base
pair PCR fragment was digested with EcoR1 and BamHI. The over-hanging ends
were made
blunt with the Klenow fragment of DNAPEcl and dNTPs. The resulting
approximately 1800
base pair fragment containing the coding region for the N-terminus was ligated
into pET-3c.
-9~-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
as described above. This construct, clone SB, is depicted in Figure 18B. The
wild type T.
flavus DNA polymerase gene is depicted in Figure 18A. In Figure 18, the
designation " 3'
Exo" is used to indicate the location of the 3' exonuclease activity
associated with Type A
polymerases which is not present in DNAPTfI. The SB clone has the same leader
amino acids '
as do the DNAPTaq clones 4E and F which were cloned into pET-3c; it is not
known
precisely where translation termination occurs, but the vector has a strong
transcription
termination signal immediately downstream of the cloning site.
S. Growth And Induction Of Transformed Cells
Bacterial cells were transformed with the constructs described above using
standard
transformation techniques and used to inoculate 2 mls of a standard growth
medium (e.g.,
Luria-Bertani broth). The resulting cultures were incubated as appropriate for
the particular
strain used, and induced if required for a particular expression system. For
all of the
constructs depicted in Figures 16 and 18, the cultures were grown to an
optical density (at
600nm wavelength) of 0.5 OD.
To induce expression of the cloned genes, the cultures were brought to a final
concentration of 0.4 mM IPTG and the incubations were continued for 12 to 17
hours. 50 q l
aliquots of each culture were removed both before and after induction and were
combined
with 20 p,l of a standard gel loading buffer for sodium dodecyl sulfate-
polyacrylamide gel
electrophoresis (SDS-PAGE). Subsequent staining with Coomassie Blue (Sambrook
et al.,
supra) allows visualization of the foreign proteins if they account for about
3-5% of the
cellular protein and do not co-migrate with any of the major E. coli protein
bands. Proteins
that do co-migrate with a major host protein must be expressed as- more than
10% of the total
protein to be seen at this stage of analysis.
C. Heat Lysis And Fractionation
Expressed thermostable proteins, i.e., the 5' nucleases, were isolated by
heating crude
bacterial cell extracts to cause denaturation and precipitation of the less
stable E. coli proteins.
The precipitated E. coli proteins were then, along with other cell debris,
removed by
centrifugation. 1.7 mls of the culture were pelleted by microcentrifugation at
12.000 to
14,000 rpm for 30 to 60 seconds. After removal of the supernatant, the cells
were
resuspended in 400 ~l of buffer A (50 mM Tris-HC1, pH 7.9, 50 mM dextrose, 1
mM
EDTA), re-centrifuged, then resuspended in 80 ql of buffer A with 4 mg/ml
lysozyme. The '
cells were incubated at room temperature for 15 minutes, then combined with 80
~1 of buffer
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
B (10 mM Tris-HC1, pH 7.9, 50 mM KCI, 1 mM EDTA, 1 mM PMSF. 0.5% Tween-20,
0.5% Nonidet-P40).
This mixture was incubated at 75°C for 1 hour to denature and
precipitate the~host
proteins. This cell extract was centrifuged at 14,000 rpm for 15 minutes at
4°C, and the
supernatant was transferred to a fresh tube. An aliquot of 0.5 to 1 ~.l of
this supernatant was
used directly in each test reaction, and the protein content of the extract
was determined by
subjecting 7 ~,l to electrophoretic analysis, as above. The native recombinant
Taq DNA
polymerase [Englke, Anal. Biochem 191:396 (1990)], and the double point
mutation protein
shown in Figure 16B are both soluble and active at this point.
The foreign protein may not be detected after the heat treatments due to
sequestration
of the foreign protein by the cells into inclusion bodies. These are granules
that form in the
cytoplasm when bacteria are made to express high levels of a foreign protein,
and they can be
purified from a crude lysate, and analyzed SDS PAGE to determine their protein
content.
Many methods have been described in the literature, and one approach is
described below.
D. Isolation And Solubilization Of Inclusion Bodies
A small culture was grown and induced as described above. A 1.7 ml aliquot was
pelleted by brief centrifugation, and the bacterial cells were resuspended in
100 ~.l of Lysis
buffer (50 mM Tris-HCl, pH 8.0, 1 mM EDTA, 100 mM NaCI). 2.5 ~l of 20 mM PMSF
were added for a final concentration of 0.5 mM, and lysozyme was added to a
concentration
of 1.0 mg/ml. The cells were incubated at room temperature for 20 minutes,
deoxycholic acid
was added to 1 mg/ml ( 1 ~.1 of 100 mg/ml solution), and the mixture was
further incubated at
37°C for about 15 minutes or until viscous. DNAse I was added to 10
~.g/ml and the mixture
was incubated at room temperature for about 30 minutes or until it was no
longer viscous.
From this mixture the inclusion bodies were collected by centrifugation at
14,000 rpm
2~ for 1 ~ minutes at 4°C, and the supernatant was discarded. The
pellet was resuspended in 100
~.1 of lysis buffer with lOmM EDTA (pH 8.0) and 0.5% Triton X-100. After ~
minutes at
room temperature, the inclusion bodies were pelleted as before, and the
supernatant was saved
for later analysis. The inclusion bodies were resuspended in 50 ~.1 of
distilled water, and ~ ~l
was combined with SDS gel loading buffer (which dissolves the inclusion
bodies) and
analyzed electrophoretically, along with an aliquot of the supernatant.
' If the cloned protein is found in the inclusion bodies, it may be released
to assay the
cleavage and polymerase activities and the method of solubilization must be
compatible with
the particular activity. Different methods of solubilization may be
appropriate for different
-97-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95114673
proteins, and a variety of methods are discussed in Molecula~° Cloning
(Sambrook et al.,
supra). The following is an adaptation we have used for several of our
isolates.
20 p,l of the inclusion body-water suspension were pelleted by centrifugation
at 14,000
rpm for 4 minutes at room temperature, and the supernatant was discarded. To
further wash '
the inclusion bodies, the pellet was resuspended in 20 p.l of lysis buffer
with 2M urea, and ,
incubated at room temperature for one hour. The washed inclusion bodies were
then
resuspended in 2 p.l of lysis buffer with 8M urea; the solution clarified
visibly as the inclusion
bodies dissolved. Undissolved debris was removed by centrifugation at 14,000
rpm for 4
minutes at room temperature, and the extract supernatant was transferred to a
fresh tube.
I0 To reduce the urea concentration, the extract was diluted into KH,P04. A
fresh tube
was prepared containing 180 ~1 of 50 mM KH~P04, pH 9.5, 1 mM EDTA and 50 mM
NaCI.
A 2 q.l aliquot of the extract was added and vortexed briefly to mix. This
step was repeated
until all of the extract had been added for a total of 10 additions. The
mixture was allowed
to sit at room temperature for 15 minutes, during which time some precipitate
often forms.
Precipitates were removed by centrifugation at 14,000 rpm, for 15 minutes at
room
temperature, and the supernatant was transferred to a fresh tube. To the 200
ql of protein in
the KH,P04 solution, 140-200 ~l of saturated (NH4),SO4 were added, so that the
resulting
mixture was about 41 % to 50% saturated (NH4)~504. The mixture was chilled on
ice for 30
minutes to allow the protein to precipitate, and the protein was then
collected by
centrifugation at 14,000 rpln, for 4 minutes at room temperature. The
supernatant was
discarded, and the pellet was dissolved in 20 p.l Buffer C (20 mM HEPES, pH
7.9, 1 mM
EDTA, 0.5% PMSF, 25 mM KCl and 0.5 % each of Tween-20 and Nonidet P 40). The
protein solution was centrifuged again for 4 minutes to pellet insoluble
materials, and the
supernatant was removed to a fresh tube. The protein contents of extracts
prepared in this
manner were visualized by resolving 1-4 pl by SDS-PAGE; 0.5 to 1 pl of extract
was tested
in the cleavage and polymerization assays as described.
E. Protein Analysis For Presence Of Nuclease And Synthetic Activity
The 5' nucleases described above and shown.in Figures 16 and 18 were analyzed
by
the following methods.
3p 1. Structure Specific Nuclease Assay
A candidate modified polymerase is tested for S' nuclease activity by
examining its
ability to catalyze structure-specific cleavages. By the term "cleavage
structure" as used
-98-
CA 02203627 1997-04-24
WO 96/15267 PC"T/US95/14673
herein, is meant a nucleic acid structure which-is a substrate for cleavage by
the 5' nuclease
activity of a DNAP.
The polymerase is exposed to test complexes that have the structures shown in
Figure
19. Testing for 5' nuclease activity involves three reactions: 1 ) a primer-
directed cleavage
(Figure 19B) is performed because it is relatively insensitive i:o variations
in the salt
concentration of the reaction and can, therefore, be performed in whatever
solute conditions
the modified enzyme requires for activity; this is generally the same
conditions preferred by
unmodified polymerases; 2) a similar primer-directed cleavage is performed in
a buffer which
permits primer-independent cleavage, i.e., a low salt buffer, to demonstrate
that the enzyme is
viable under these conditions; and 3) a primer-independent cleavage (Figure
19A) is
performed in the same low salt buffer.
The bifurcated duplex is formed between a substrate strand and a template
strand as
shown in Figure 19. By the term "substrate strand" as used herein, is meant
that strand of
nucleic acid in which the cleavage mediated by the 5' nuclease activity
occurs. The substrate
1 ~ strand is always depicted as the top strand in the bifurcated complex
which serves as a
substrate for 5' nuclease cleavage (Figure 19). By the term "template strand"
as used herein.
is meant the strand of nucleic acid which is at least partially complementary
to the substrate
strand and which anneals to the substrate strand to form the cleavage
structure. The template
strand is always depicted as the bottom strand of the bifurcated cleavage
structure (Figure 19).
If a primer (a short oligonucleotide of 19 to 30 nucleotides in. length) is
added to the
complex. as when primer-dependent cleavage is to be tested, it is designed to
anneal to the 3'
arm of the template strand (Figure 19B). Such a primer would be extended along
the
template strand if the polymerase used in the reaction has synthetic activity.
The cleavage structure may be made as a single hairpin molecule, with the 3'
end of
the target and the 5' end of the pilot joined as a loop as shown in Figure
19E. A primer
oligonucleotide complementary to the 3' arm is also required for these tests
so that the
enzyme's sensitivity to the presence of a primer may be tested.
Nucleic acids to be used to form test cleavage structures can be chemically
synthesized, or can be generated by standard recombinant DNA techniques. By
the latter
method, the hairpin portion of the molecule can be created by inserting into a
cloning vector
duplicate copies of a short DNA segment, adjacent to each other but in
opposing orientation.
The double-stranded fragment encompassing this inverted repeat, and including
enough
flanking sequence to give short (about 20 nucleotides) unpaired 5' and 3'
arms, can then be
-99-
CA 02203627 1997-04-24
WO 96/15267 PC"TIUS95/14673
released from the vector by restriction enzyme digestion, or by PCR performed
with an
enzyme lacking a 5' exonuclease (e.g., the Stoffel fragment of AmplitaqTM DNA
polymerase,
VentTM DNA polymerase). .
The test DNA can be labeled on either end, or internally, with either a
radioisotope, or '
with a non-isotopic tag. Whether the hairpin DNA is a synthetic single strand
or a cloned
double strand, the DNA is heated prior to use to melt all duplexes. When
cooled on ice, the
structure depicted in Figure 19E is formed, and is stable for sufficient time
to perform these
assays.
To test for primer-directed cleavage (Reaction 1 ), a detectable quantity of
the test
molecule (typically 1-100 fmol of 3'P-labeled hairpin molecule) and a 10 to
100-fold molar
excess of primer are placed in a buffer known to be compatible with the test
enzyme. For
Reaction 2, where primer-directed cleavage is performed under condition which
allow primer-
independent cleavage, the same quantities of molecules are placed in a
solution that is the
same as the buffer used in Reaction 1 regarding pH, enzyme stabilizers (e.g.,
bovine serum
albumin, nonionic detergents, gelatin) and reducing agents (e.g.,
dithiothreitol, 2-
mercaptoethanol) but that replaces any monovalent cation salt with 20 mM KCI;
20 mM hCl
is the demonstrated optimum for primer-independent cleavage. Buffers for
enzymes, such as
DNAPEc 1, that usually operate in the absence of salt are not supplemented to
achieve this
concentration. To test for primer-independent cleavage (Reaction 3) the same
quantity of the
test molecule, but no primer, are combined under the same buffer conditions
used for
Reaction 2.
All three test reactions are then exposed to enough of the enzyme that the
molar ratio
of enzyme to test complex is approximately 1:1. The reactions are incubated at
a range of
temperatures up to, but not exceeding, the temperature allowed by either the
enzyme stability
or the complex stability, whichever is lower, up to 80°C fox enzymes
from thermophiles, for a
time sufficient to allow cleavage ( 10 to 60 minutes). The products of
Reactions 1, 2 and 3
are resolved by denaturing polyacrylamide gel electrophoresis, and visualized
by
autoradiography or by a comparable method appropriate to the labeling system
used.
Additional labeling systems include chemiluminescence detection, silver or
other stains,
blotting and probing and the like. The presence of cleavage products is
indicated by the
presence of molecules which migrate at a lower molecular weight than does the
uncleaved test "
structure. These cleavage products indicate that the candidate polymerase has
structure-
specific 5' nuclease activity.
- 100 -
CA 02203627 1997-04-24
WO 96/15267 PCTlUS95/14673
. To determine whether a modified DNA polymerise has substantially the same 5'
nuclease activity as that of the native DNA polymerise, the results of the
above-described
tests axe compared with the results obtained from these tests performed with
the native DNA
polymerise. By "substantially the same 5' nuclease activity" we mean that the
modified
y 5 polymerise and the native polymerise will both cleave test molecules in
the same manner. It
is not necessary that the modified polymerise cleave at the same rate as the
native DNA
polymerise.
Some enzymes or enzyme preparations may have other associated or contaminating
activities that may be functional under the cleavage conditions described
above and that may
interfere with 5' nuclease detection. Reaction conditions can be modified in
consideration of
these other activities, to avoid destruction of the substrate, or other
masking of the 5' nuclease
cleavage and its products. For example, the DNA polymerise I of E. coli (Pol
I), in addition
to its polymerise and 5' nuclease activities, has a 3' exonuclease that can
degrade DNA in a
3' to 5' direction. Consequently, when the molecule in Figure 19E is exposed
to this
polymerise under the conditions described above, the 3' exonuclease quickly
removes the
unpaired 3' arm, destroying the bifurcated structure required of a substrate
for the 5'
exonuclease cleavage and no cleavage is detected. The true ability of Pol I to
cleave the
structure can be revealed if the 3' exonuclease is inhibited by a change of
conditions (e.g.,
pH), mutation, or by addition of a competitor for the activity. Addition of
500 pmoles of a
single-stranded competitor oligonucleotide, unrelated to the Figure 19E
structure, to the
cleavage reaction with Pol I effectively inhibits the digestion of the 3' arm
of the Figure 19E
structure without interfering with the 5' exonuclease release of the 5' arm.
The concentration
of the competitor is not critical, but should be high enough to occupy the 3'
exonuclease for
the duration of the reaction.
Similar destruction of the test molecule may be caused by contaminants in the
candidate polymerise preparation. Several sets of the structure specific
nuclease reactions
may be performed to determine the purity of the candidate nuclease and to find
the window
between under and over exposure of the test molecule to the polymerise
preparation being
investigated.
The above described modified polymerises were tested for 5' nuclease activity
as
follows: Reaction 1 was performed in a buffer of 10 mM Tr:is-Cl, pH 8.5 at
20°C, l.~ mM
MgCI, and 50 mM KCl and in Reaction 2, the KCl concentration was reduced to 20
mM. In
Reactions 1 and 2, 10 fmoles of the test substrate molecule shown in Figure
16E were
- 101 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95114673
combined with 1 pmole of the indicated primer and 0.5 to 1.0 pl of extract
containing the
modified polymerise (prepared as described above). This mixture was then
incubated for 10
minutes at 55°C. For all of the mutant polymerises tested these
conditions were sufficient to
give complete cleavage. When the molecule shown in Figure 19E was labeled at
the S' end,
the released 5' fragment, 25 nucleotides long, was conveniently resolved on a
20%
polyacrylamide gel ( 19:1 cross-linked) with 7 M urea in a buffer of O.SX TBE.
Clones 4C-F
and SB exhibited structure-specific cleavage comparable to that of the
unmodified DNA
polymerise. Additionally, clones 4E, 4F and 4G have the added ability to
cleave DNA in the
absence of a 3' arm as discussed above. Representative cleavage reactions are
shown in
Figure 20.
For the reactions shown in Figure 20, the mutant polymerise clones 4E (Taq
mutant)
and SB (Tfl mutant) were examined for their ability to cleave the hairpin
substrate molecule
shown in Figure 19E. The substrate molecule was labeled at the 5' terminus
with 3'P. Ten
fmoles of heat-denatured, end-labeled substrate DNA and 0.5 units of DNAPTuq
(lane 1 ) or
1 ~ 0.5 p.l of 4e or Sb extract (Figure 20, lanes 2-7, extract was prepared as
described above)
were mixed together in a buffer containing 10 mM Tris-CI, pH 8.5, 50 mM KCl
and l.~ mM
MgCI,. The final reaction volume was 10 p.l~ Reactions shown in lanes 4 and 7
contain in
addition 50 p.M of each dNTP. Reactions shown in lanes 3, 4, 6 and 7 contain
0.2 pM of the
primer oligonucleotide (complementary to the 3' arm of the substrate and shown
in Figure
19E). Reactions were incubated at 55° C for 4 minutes. Reactions were
stopped by the
addition of 8 ~.1 of stop solution per 10 ~l reaction volume. Samples were
then applied to
12% denaturing acrylamide gels. Following electrophoresis, the gels were
autoradiographed.
Figure 20 shows that clones 4E and SB exhibit cleavage activity similar to
that of the native
DNAPTaq. Note that some cleavage occurs in these reactions in the absence of
the primer.
When long hairpin structure, such as the one used here (Figure 19E), are used
in cleavage
reactions performed in buffers containing 50 mM KCl a low level of primer-
independent
cleavage is seen. Higher concentrations of KCl suppress, -but do not
eliminate, this primer-
independent cleavage under these conditions.
2. Assay For Synthetic Activity
The ability of the modified enzyme or proteolytic fragments is assayed by
adding the
modified enzyme to an assay system in which a primer is annealed to a template
and DNA '
synthesis is catalyzed by the added enzyme. Many standard laboratory
techniques employ
- 102 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
such an assay. For example, nick translation and enzymatic sequencing involve
extension of a
primer along a DNA template by a polymerase molecule.
In a preferred assay for determining the synthetic activity of a modified
enzyme an
oligonucleotide primer is annealed to a single-stranded DNA template, e.g.,
bacteriophage
M13 DNA, and the primer/template duplex is incubated in the presence of the
modified
polymerase in question, deoxynucleoside triphosphates (dNTPs) and the buffer
and salts
known to be appropriate for the unmodified or native enzyme. Detection of
either primer
extension (by denaturing gel electrophoresis) or dNTP incorporation (by acid
precipitation or
chromatography) is indicative of an active polymerase. A label, either
isotopic or non-
isotopic, is preferably included on either the primer or as a dNTP to
facilitate detection of
polymerization products. Synthetic activity is quantified as the amount of
free nucleotide
incorporated into the growing DNA chain and is expressed as amount
incorporated per unit of
time under specific reaction conditions.
Representative results of an assay for synthetic activity is shown in Figure
21. The
synthetic activity of the mutant DNAPTaq clones 4B-F was tested as follows: A
master
mixture of the following buffer was made: 1.2X PCR buffer, 50 p.M each of
dGTP, dATP
and dTTP, 5 ~M dCTP and 0.125 ~M a.-3'-P-dCTP at 600 Ci/mmol. Before adjusting
this
mixture to its final volume, it was divided into two equal aliquots. One
received distilled
water up to a volume of 50 p.l to give the concentrations above. The other
received 5 yg of
single-stranded Ml3mpl8 DNA (approximately 2.5 pmol or 0.05 ~M final
concentration) and
250 pmol of M13 sequencing primer (5 pM final concentration) and distilled
water to a final
volume of 50 pl. Each cocktail was warmed to 75°C for 5 minutes and
then cooled to room
temperature. This allowed the primers to anneal to the DNA in the DNA-
containing mixtures.
For each assay, 4 ~,l of the cocktail with the DNA wa s combined with 1 p.l of
the
mutant polymerise, prepared as described, or 1 unit of DNAPTaq (Perkin Elmer)
in 1 EGl of
dH~O. A "no DNA" control was done in the presence of the DNAPTaq (Figure 21,
lane 1 ),
and a "no enzyme" control was done using water in place of the enzyme (lane
2). Each
reaction was mixed, then incubated at room temperature (approx. 22°C)
for 5 minutes, then at
55°C for 2 minutes, then at 72°C for 2 minutes. This step
incubation was done to detect
polymerization in any mutants that might have optimal temperatures lower than
72°C. After
the final incubation, the tubes were spun briefly to collect an5~ condensation
and were placed
on ice. One ~.l of each reaction was spotted at an origin 1.5 cm from the
bottom edge of a
polyethyleneimine (PEI) cellulose thin layer chromatography plate and allowed
to dry. The
-103-
CA 02203627 1997-04-24
WO 96115267 PCT/US95/14673
chromatography plate was run in 0.75 M NaH,P04, pH 3.5, until the buffer front
had run
approximately 9 cm from the origin. The plate was dried, wrapped in plastic
wrap. marked
with luminescent ink, and exposed to X-ray film. Incorporation was detected as
counts that
stuck where originally spotted, while the unincorporated nucleotides were
carried by the salt
solution from the origin.
Comparison of the locations of the counts with the two control lanes confirmed
the
lack of polymerization activity in the mutant preparations. Among the modified
DNAPTac~
clones, only clone 4B retains any residual synthetic activity as shown in
Figure 21.
- E~MPLE 3
5' Nucleases Derived From Thermostable DNA
Polymerases Can Cleave Short Hairpin Structures With Specificity
The ability of the 5' nucleases to cleave hairpin structures to generate a
cleaved
hairpin structure suitable as a detection molecule was examined. The structure
and sequence
of the hairpin test molecule is shown in Figure 22A (SEQ ID NO:15). The
oligonucleotide
(the primer in Figure 22A, SEQ ID N0:22) is- shown annealed to its
complementary sequence
on the 3' arm of the hairpin test molecule. The hairpin test molecule was
single-end labeled
with 3'P using a labeled T7 promoter primer in a polymerase chain reaction.
The label is
present on the 5' arm of the hairpin test molecule and is represented by the
star in Figure
22A.
The cleavage reaction was performed by adding 10 fmoles of heat-denatured. end-
labeled hairpin test molecule, 0.2 p.M of the primer oligonucleotide
(complementary to the 3'
arm of the hairpin), 50 ~M of each dNTP and 0.5 units of DNAPTaq (Perkin
Elmer) or 0.5
pl of extract containing a 5'nuclease (prepared as described above) in a total
volume of 10 ~l
in a buffer containing 10 W M Tris-Cl, pH 8.5, 50 mM KCl and 1.5 mM MgCI,.
Reactions
shown in lanes 3, 5 and 7 were run in the absence of dNTPs.
Reactions were incubated at 55° C for 4 minutes. Reactions were stopped
at 55° C by
the addition of 8 ~1 of stop solution per 10 ~l reaction volume. Samples were
not heated
before loading onto denaturing polyacrylamide gels (10% polyacrylamide, 19:1
crosslinking.
and 7 M urea, in a buffer of 1X TBE [89 mM Tris-borate, pH 8.3, 2.8 mM EDTA]).
The
samples were not heated to allow for the resolution of single-stranded and re-
duplexed
uncleaved hairpin molecules.
- 104 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
Figure 22B shows that altered polymerises lacking any detectable synthetic
activity
cleave a hairpin structure when an oligonucleotide is annealed to the single-
stranded 3' arm of
the hairpin to yield a single species of cleaved product (Figure 22B, lanes 3
and 4). 5'
- nucleases, such as clone 4D, shown in lanes 3 and 4, produce a single
cleaved product even in
the presence of dNTPs. 5' nucleases which retain a residual amount of
synthetic activity (less
y
than 1 % of wild type activity) produce multiple cleavage products as the
polymerise can
extend the oligonucleotide annealed to the 3' arm of the hairpin thereby
moving the site of
cleavage (clone 4B, lanes 5 and 6). Native DNATaq produces even more species
of cleavage
products than do mutant polymerises retaining residual synthetic activity and
additionally
converts the hairpin structure to a double-stranded form in the presence of
dNTPs due to the
high level of synthetic activity in the native polymerise (Figure 22B, lane 8)
EXAMPLE 4
Cleavage Of Linear Nucleic Acid Substrates
1S
From the above, it should be clear that native (i.e., "wild type")
thermostable DNA
polymerises are capable of cleaving hairpin structures in a specific manner
and that this
discovery can be applied with success to a detection assay. In this example,
the mutant
DNAPs of the present invention are tested against three different cleavage
structures shown in
Figure 24A. Structure 1 in Figure 24A is simply single stranded 206-mer (the
preparation
and sequence information for which was discussed above). Structures 2 and 3
are duplexes;
structure 2 is the same hairpin structure as shown in Figure 13A (bottom),
while structure 3
has the hairpin portion of structure 2 removed.
The cleavage reactions comprised 0.01 pmoles of the resulting substrate DNA,
and 1
pmole of pilot oligonucleotide in a total volume of 10 p.l of 10 mM Tris-Cl,
pH 8.3, 100 mM
KCI, 1 mM MgCh. Reactions were incubated for 30 minutes at 55°C, and
stopped by the
addition of 8 q.l of stop solution. Samples were heated to 75°C for 2
minutes immediately
before electrophoresis through a 10% polyacrylamide gel (19:1 cross lil~l:),
with 7M urea. in a
buffer of O.SX TBE.
The results were visualized by autoradiography and are shown in Figure 24B
with the
° enzymes indicated as follows: I is native Taq DNAP; II is native Tfl
DNAP; III is the
CleavaseTM BX enzyme shown in Figure 16E; IV is the Cle<~vaseTM BB enzyme
shown in
Figure 16F; V is the mutant shown in Figure 18B; and VI is the CleavaseTM BN
enzyme
-105-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
shown in Figure 16G. Structure 2 was used to "normalize" the comparison. For
example. it
was found that it took 50 ng of Taq DNAP and 300 ng of the CleavaseTM BN
enzyme to give
similar amounts of cleavage of Structure 2 in thirty (30) minutes. .-Under
these conditions
native TayDNAP is unable to cleave Structure 3 to any significant degree.
Native Tfl DNAP '
cleaves Structure 3 in a manner that creates multiple products.
By contrast, all of the mutants tested cleave the linear duplex of Structure
3. This
finding indicates that this characteristic of the mutant DNA polymerises is
consistent of
thermostable polymerises across thermophilic species.
- - EXAMPLE 5
5' Exonucleolvtic Cleavage ("Nibbling"1 By Thermostable DNAPs
It has been found that thermostable DNAPs, including those of the present
invention,
have a true 5' exonuclease capable of nibbling the 5' end of a linear duplex
nucleic acid
structures. In this example, the 206 base pair DNA duplex substrate is again
employed (see
above). In this case, it was produced by the use of one 3'P-labeled primer and
one unlabeled
primer in a polymerise chain reaction. The cleavage reactions comprised 0.01
pmoles of
heat-denatured, end-labeled substrate DNA (with the unlabeled strand also
present), 5 pmoles
of pilot oligonucleotide (see pilot oligos in Figure 13A) and 0.5 units of
DNAPTaq or 0.5 ~.1
of the CleavaseTM BB enzyme in the E. coli extract (see above), in a total
volume of 10 ~.l of
10 mM Tris~Cl, pH 8.5, 50 mM KCI, 1.5 mM MgCl2.
Reactions were initiated at 65°C by the addition of pre-warmed enzyme,
then shifted to
the final incubation temperature for 30 minutes. The results are shown in
Figure 25A.
Samples in lanes 1-4 are the results with native Taq DNAP, while lanes 5-8
shown the results
with the CleavaseTM BB enzyme. The reactions for lanes 1, 2, 5, and 6 were
performed at
65°C and reactions for lanes 3, 4, 7, and 8 were performed at
50°C and all were stopped at
temperature by the addition of 8 ~1 of 95% formamide with 20 mM EDTA and 0.05%
marker
dyes. Samples were heated to 75°C for 2 minutes immediately before
electrophoresis through
a 10% acrylamide gel (19:1 cross-linked), with 7 M urea, in a buffer of 0.5X
TBE. The
expected product in reactions l, 2, 5, and 6 is 85 nucleotides long; in
reactions 3 and 7, the
expected product is 27 nucleotides long. Reactions 4 and 8 were performed
without pilot. and °
should remain at 206 nucleotides. The faint band seen at 24 nucleotides is
residual end-
labeled primer from the PCR.
d
- 106 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
The surprising result is that the CleavaseTM BB enzyme under these conditions
causes
all of the label to appear in a very small species, suggesting the possibility
that the enzyme
completely hydrolyzed the substrate. To determine the composition of the
fastest-migrating
band seen in lanes 5-8 (reactions performed with the deletion mutant), samples
of the 206
~ base pair duplex were treated with either T7 gene 6 exonucle;~se (USB) or
with calf intestine
alkaline phosphatase (Promega), according to manufacturers" instructions, to
produce either
labeled mononucleotide (lane a of Figure 25B) or free ''-P-labeled inorganic
phosphate (lane b
of Figure 25B), respectively. These products, along with the products seen in
lane 7 of panel
A were resolved by brief electrophoresis through a 20% acrylamide gel (19:1
cross-link), with
7 M urea, in a buffer of O.SX TBE. The CleavaseTM BB enzyme is thus capable of
converting the substrate to mononucleotides.
EXAMPLE 6
_ Nibblin I~ s Duplex Dependent
The nibbling by the enzyme CleavaseTM BB is duplex dependent. In this example,
internally labeled, single strands of the 206-mer were produced by 15 cycles
of primer
extension incorporating a.-3'-P labeled dCTP combined with all four unlabeled
dNTPs, using
an unlabeled 206-by fragment as a template. Single and double stranded
products were
resolved by electrophoresis through a non-denaturing 6% polyacrylamide gel
(29:1 cross-link)
in a buffer of O.SX TBE, visualized by autoradiography, excised from the gel,
eluted by
passive diffusion, and concentrated by ethanol precipitation.
The cleavage reactions comprised 0.04 pmoles of substrate DNA, and 2 p.l of
the
enzyme Cleavase BB (in an E. coli extract as described above) in a total
volume of 40 l:~l of
10 mM Tris~Cl, pH 8.5, 50 mM KCI, 1.5 mM MgCI,. Reactions were initiated by
the
addition of pre-warmed enzyme; 10 ~l aliquots were removed at 5, 10, 20, and
30 minutes,
and transferred to prepared tubes containing 8 ~,1 of 95% forrnamide with 30
mM EDTA and
0.05% marker dyes. Samples were heated to 75°C for 2 minutes
immediately before
electrophoresis through a 10% acrylamide gel (19:1 cross-linked), with 7 M
urea, in a buffer
of O.SX TBE. Results were visualized by autoradiography as shown in Figure 26.
Clearly,
the cleavage by the CleavaseTM BB enzyme depends on a duplex structure; no
cleavage of the
single strand structure is detected whereas cleavage of the 206-mer duplex is
complete.
- 107 -
CA 02203627 1997-04-24
WO 96115267 PCT/US95/14673
EXAMPLE 7
Purification Of CleavaseTM Enzymes
As noted above, expressed thermostable proteins, i.c., the 5' nucleases. were
isolated
by crude bacterial cell extracts. The precipitated E. coli proteins were then,
along with other
cell debris, removed by centrifugation. In this example, cells expressing the
CleavaseTM BN
clone were cultured and collected (500 grams). For each gram (wet weight) of
E. coli, 3 ml
of lysis buffer (SO mM Tris-HCI, pH 8.0, 1 mM EDTA, 100 pM NaCI) was added.
The cells
were lysed with 200 ~,g/ml lysozyme at room temperature for 20 minutes.
Thereafter
deoxycholic acid was added to make a 0.2% final concentration and the mixture
was
incubated 15 minutes at room temperature.
The lysate was sonicated for approximately 6-8 minutes at 0°C. The
precipitate was
removed by centrifugation (39,OOOg for 20 minutes). Polyethyleneimine was
added (0.5%)
to the supernatant and the mixture was incubated on ice for 15 minutes.
The mixture was centrifuged (S,OOOg for 15 minutes) and the supernatant was
retained.
This was heated for 30 minutes at 60°C and then centrifuged again
(S,OOOg for 15 minutes)
and the supernatant was again retained.
The supernatant was precipitated with 35% ammonium sulfate at 4°C for
15 minutes.
The mixture was then centrifuged (S,OOOg for 15 minutes) and the supernatant
was removed.
The precipitate was then dissolved in 0.25 M KCI, 20 mM Tris, pH 7.6, 0.2%
Tween and 0.1
EDTA) and then dialyzed against Binding Buffer (8X Binding Buffer comprises:
40 mM
imidazole, 4 M NaCI, 160 mM Tris-HCI, pH 7.9).
The solubilized protein was then purified on the Ni++ column (Novagen). The
Binding
Buffer was allowed to drain to the top of the column bed and the column was
then loaded
with the prepared extract. A flow rate of about 10 column volumes per hour is
optimal for
efficient purification. If the flow rate is too fast, more impurities will
contaminate the eluted
fraction.
The column was washed with 25 ml ( 10 volumes) of 1 X Binding Buffer and then
washed with I S ml (6 volumes) of 1X Wash Buffer (8X Wash Buffer comprises:
480mM
imidazole, 4 M NaCI, 160 mM Tris-HCI, pH 7.9). The bound protein was eluted
with 1 ~ ml
(6 volumes) of 1X Elute Buffer (4X Elute Buffer comprises: 4 mM imidazole, 2 M
NaCI, 80
mM Tris-HCI, pH 7.9). Protein is then reprecipitated with 35% Ammonium Sulfate
as
above. The precipitate was then dissolved and dialyzed against: 20 mM Tris,
100 mM KC 1,
- 108 -
CA 02203627 1997-04-24
WO 96115267 PCT/US95/14673
1 mM EDTA). The solution was brought up to 0.1 % each of Tween 20 and NP-40
and
stored at 4°C.
EXAMPLE 8
5' Nucleases Cut Nucleic Acid Substrates At
Naturally Occurring Areas Of Secondary Structure
The ability of a 5' nuclease to recognize and cleave nucleic acid substrates
at naturally
occurring areas of secondary structure in the absence of a pilot
oligonucleotide (i.e., primer
independent cleavage) was shown in Example 1 C (Figure 13, lane 9). When
DNAPTaq was
incubated at 50°C in the presence of a 206 by DNA substrate (single end
labeled, double
stranded template) in a buffer containing 10 mM Tris-HCI, pH 8.5 and 1.5 mM
MgCI,,
adventitious (i.e., naturally occurring) structures in the DNA substrate were
cleaved by the 5'
nuclease activity of the enzyme. This cleavage generated three prominent
fragments (Figure
13, lane 9); this cleavage pattern provides a "fingerprint" of t:he DNA
template.
The ability of 5' nucleases to cleave naturally occurring structures in
nucleic acid
templates (structure-specific cleavage) is useful to detect internal sequence
differences in
nucleic acids without prior knowledge of the specific sequence of the nucleic
acid. To
develop a general method to scan nucleic acids for mutations [e.g., single
base changes (point
mutations), small insertions or deletions, etc.] using 5' nucleases, the
following series of
experiments were performed.
A. The Substitution Of MnCl2 For MgCl2 In The Cleavage Reaction Produces
Enhanced Cleavage Patterns
The effect of substituting of Mn'+ in place of Mg'- upon the cleavage pattern
created
by 5' nuclease activity on a double-stranded DNA substrate was examined. A 157
by
fragment derived from exon 4 of either the wild-type (SEQ ID N0:27) or the
mutant G419R
(SEQ ID N0:28) tyrosinase gene was prepared by PCR as follows.
The primer pair 5' biotin-CACCGTCCTCTT~AAGAAG 3' (SEQ ID N0:29) and 5'
fluorescein-CTGAATCTTGTAGATAGCTA 3' (SEQ ID N0:30) was used to prime the
PCRs. Tlie synthetic primers were obtained from Promega; the primers were
labeled on the
5' end with biotin or fluorescein during synthesis.
The target DNA for the generation of the 157 by fragment of mutant G419R
(Ding,
R.A., et al., (1991) Mol. Biol. Med. 8:19; here after referred to as the 419
mutant) was a 339
by PCR product (SEQ ID N0:31) generated using genomic DNA homozygous for the
419
- 109 -
CA 02203627 1997-04-24
WO 96115267 PCT/LTS95/14673
mutation. Genomic DNA was isolated using standard techniques from peripheral
blood
leukocytes isolated from patients. This 339 by PCR product was prepared as
follows. '
The symmetric PCR reaction comprised 10 ng .of genomic DNA from the 419
mutant,
100 pmoles of the primer 5' biotin-GCCTTATTTTACTTTAAAAAT-3' (SEQ ID N0:32), '
100 pmoles of the primer 5' fluorescein-TAAAGTTTTGTGTTATCTCA-3' (SEQ ID
N0:33), ,
and 50 p.M of each dNTP in 1X PCR buffer. The primers of SEQ ID NOS:32 and 33
were
obtained from Integrated DNA Technologies, Coralville, IA. A tube containing
45 yl of the
' above mixture was overlaid with two drops of light mineral oil and the tube
was heated to
95°C for 1 min. Tay polymerase was then added as 1.25 units of enzyme
in 5 p,l of 1X PCR
buffer. The tube was heated-to 94°C -for 40 sec, cooled to 55°C
for 50 sec, heated to 72°C
for 70 sec for 29 repetitions with a 5 min incubation at 72°C after the
last repetition.
The PCR products were gel purified as follows. The products were resolved by
electrophoresis through a 6% polyacrylamide gel (29:1 cross-link) in a buffer
containing O.SX
TBE. The DNA was visualized by ethidium bromide staining and the 339 by
fragment was
excised from the gel. The DNA was eluted from the gel slice by passive
diffusion overnight
into a solution containing 0.5 M NH40Ac, 0.1% SDS and 0.1 M EDTA. The DNA was
then
precipitated with ethanol in the presence of 4 ~.g of glycogen carrier. The
DNA was pelleted
and resuspended in 40 ~,l of TE (10 mM Tris-Cl, pH 8.0, 0.1 mM EDTA).
To generate the 157 by fragment from the 419 mutant, the purified 339 by 419
PCR
fragment was used as the target in an asymmetric PCR. The asymmetric PCR
comprised 100
pmoles of the biotinylated primer of SEQ ID N0:32, 1 pmole of the
fluoresceinated primer of
SEQ ID N0:33, 50 p.M of each dNTP, in 1X PCR buffer. A tube containing 45 ~.t
of the
above mixture was overlaid with two drops of light mineral oil and the tube
was heated to
95°C for 5 sec and then cooled to 70°C. Taq polymerase was then
added as 1.25 units of
enzyme in 5 p.l of 1X PCR buffer. The tube was heated to 95°C for 45
sec. cooled to 50°C
for 45 sec, heated to 72°C -for 1 min 15 sec for 30 repetitions with a
5 min incubation at
72°C -after the last repetition.
The asymmetric PCR products were gel purified as follows. The products were
resolved by electrophoresis through a 6% polyacrylamide gel (29:1 cross-link)
in a buffer
containing O.SX TBE. The DNA was visualized by ethidium bromide staining; the
double-
stranded DNA was differentiated from the single-stranded DNA due to the
mobility shift
commonly seen with single-stranded DNA produced from asymmetric PCR (in an
asymmetric
PCR both single-stranded and double-stranded products are produced; -typically
the single-
- 110 -
CA 02203627 1997-04-24
WO 96!15267 PCT/US95/14673
stranded product will have a slower speed of migration through the gel and
will appear closer
to the origin than will the double-stranded product). The double-stranded 1~7
by substrate
corresponding to the 419 mutant (SEQ ID N0:28) was excised from the gel.
The 157 by wild-type fragment was generated by asymmetric PCR as described
above
for the 419 mutant with the exception that the target DNA was 10 ng of
supercoiled pcTYR-
NlTyr plasmid DNA. The pcTYR-NlTyr plasmid contains the entire wild-type
tyrosinase
cDNA [Geibel, L.B., et al. (1991) Genomics 9:435].
Following the asymmetric PCRs, the reaction products were resolved on an
acrylamide
gel and the double-stranded fragments of interest were excisf:d, eluted and
precipitated as
described above. The precipitated 157 by wild-type (SEQ ID N0:27) and 419
mutant (SEQ
ID N0:28) fragments were resuspended in 40 ~l of TE.
Cleavage reactions comprised 100 fmoles of the resulting double-stranded
substrate
DNAs (the substrates contain a biotin moiety at the 5' end of the sense
strand) in a total
volume of 10 ~1 of 10 mM MOPS, pH 8.2; 1 mM divalent ration (either MgCI, or
MnCI~)
and 1 unit of DNAPTaq. The reactions were overlaid with a drop of light
mineral oil.
Reactions were heated to 95°C for 5 seconds to denature the substrate
and then the tubes were
quickly cooled to 65°C (this step allows the DNA assume its unique
secondary structure by
allowing the formation of intra-strand hydrogen bonds between complimentary
bases). The
reaction can be performed in either a thermocycler (MJ Research, Watertown,
MA)
programmed to heat to 95°C for 5 seconds then drop the temperature
immediately to 65°C or
alternatively the tubes can be placed manually in a heat block set at
95°C and then transferred
to a second heat block set at 65°C.
The reaction was incubated at 65°C for 10 minutes and was stopped by
the addition of
8 p.l of stop buffer. Samples were heated to 72°C for 2 minutes and 5
p.l of each reaction
were resolved by electrophoresis through a 10% polyacrylamide gel ( 19:1 cross-
link), with 7
M urea, in a buffer containing O.SX TBE.
After electrophoresis, the gel plates were separated allowing the gel to
remain flat on
one plate. A 0.2 p.m-pore positively-charged nylon membrane (Schleicher and
Schuell,
Keene, NH), pre-wetted in O.SX TBE, was laid on top of the exposed acrylamide
gel. All air
bubbles trapped between the gel and the membrane were removed. Two pieces of
3MM filter
' paper (Whatman) were then placed on top of the membrane, the other glass
plate was
replaced, and the sandwich was clamped with binder clips. Transfer was allowed
to proceed
overnight. After transfer, the membrane was carefully peeled from the gel and
allowed to air
- 111 -
CA 02203627 1997-04-24
WO 96/15267 PG"TIUS95/14673
dry. After complete drying, the membrane was washed in 1.2X Sequenase Images
Blocking
Buffer (United States Biochemical) fox 30 minutes. Three tenths of a ml of the
buffer was
used per cm' of membrane. A streptavidin-alkaline pliosphatase conjugate
(SAAP, United
States Biochemical) was added to a 1:4000 dilution directly to the blocking
solution, and '
agitated for 15 minutes. The membrane was rinsed briefly with HBO and then
washed 3 times
(5 minutes/wash) in 1X SAAP buffer (100 mM Tris-HCL, pH 10; 50 mM NaCI) with
0.1%
sodium dodecyl sulfate (SDS) using 0.5 ml buffer/cm'- of the buffer, with
brief HBO rinses
between each wash. Similarly, for fluorescein-labeled DNA, anti-fluorescein
fragment
(Boehringer Mannheim Biochemicals, Indianapolis, IN) at a 1:20.000 final
dilution maybe
added followed by three washes (5 min/wash) in 1X SAAP buffer containing 0.1%
SDS and
0.025% Tween 20. The membrane was then washed once in 1X SAAP buffer without
SDS,
drained thoroughly and placed in a plastic heat-sealable bag. Using a sterile
pipet tip, 0.05
ml/cm'- of CDP-Star'"' (Tropix, Bedford, MA) was added to-the bag and
distributed over the
entire membrane for 5 minutes. The bag was drained of all excess liquid and
air bubbles.
The membrane was then exposed to X-ray film (Kodax XRP) for an initial 30
minutes.
Exposure times were adjusted as necessary for resolution and clarity. The
results are shown
in Figure 27. - -
In Figure 27, the lane marked "M" contains molecular weight markers. The
marker
fragments were generated by digestion of pUCl9 with HaeIII followed by the
addition of
biotinylated dideoxynucleotides (Boehringer Mannheim, Indianapolis, IN) to the
cut ends
using terminal transferase (Promega). Lanes 1, 3 and 5 contain the reaction
products from the
incubation of the wild type 157 nucleotide substrate in the absence of the
DNAPTaq enzyme
(lane 1), in the presence of MgCh and enzyme (lane 3) or in the-presence of
MnCh and
enzyme (lane 5). Lanes 2, 4 and 6 contains the reaction products from the
incubation of the
?5 157 nucleotide substrate derived from the 419 mutant in the absence of
enzyme (lane 2), in
the presence of MgChand enzyme (lane 4) or in the presence of MnCI, and enzyme
(lane 6).
Figure 27 demonstrates that the use of MnCh rather than MgCI, in the cleavage
reaction results in the production of an enhanced cleavage pattern. It is
desirable that the
cleavage products are of different sizes so that the products do not all
cluster at one end of
the gel. The ability to spread the cleavage products out over the entire
length of the gel
makes it more likely that alterations in cleavage products between the wild
type and mutant '
substrates will be identified. Figure 27 shows that when Mg'~' is used as the
divalent canon,
the majority of the cleavage products cluster together in the upper portion of
the gel. In
- 112 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95114673
contrast when Mn'+ is used as the divalent cation, the substrate assumes
structures which,
when cleaved, generate products of widely differing mobilities. These results
show that Mn'-
is the preferred divalent cation far the cleavage reaction.
B. 5' Nuclease Cleavage Of Different But Similarly Sized DNAs. Generates
Unique Cleavage Fragments
The ability of 5' nuclease to generate a cleavage pattern or "fingerprint"
which is
unique to a given piece of DNA was shown by incubating four similarly sized
DNA
substrates with the Cleavase'~"' BN enzyme. The four DNA substrates used were
a 157
nucleotide fragment from the sense (or coding) strand of exon 4 of the wild-
type tyrosinase
gene (SEQ ID N0:34); a 157 nucleotide fragment from the anti-sense (or non-
coding) strand
of exon 4 of the wild-type tyrosinase gene (SEQ ID N0:35); a 165 nucleotide
DNA fragment
derived from pGEM3Zf(+) (SEQ ID N0:36) and a 206 nucleotide DNA fragment
derived
from pGEM3Zf(+) (SEQ ID N0:37). The DNA substrates contained either a biotin
or
fluorescein label at their 5' or 3' ends. The substrates were made as follows.
To produce the sense and anti-sense single-stranded substrates corresponding
to exon 4
of the wild-type tyrosinase gene, a double-stranded DNA fragment, 157
nucleotides in length
(SEQ ID N0:27), was generated using symmetric PCR. The target for the
symmetric PCR
was genomic DNA containing the wild-type tyrosinase gene. The symmetric PCR
comprised
50-100 ng of genomic wild-type DNA, 25 pmoles each of primers SEQ ID NOS:42
and 43,
50 pM each dNTP and 1.25 units of Taq polymerase in 50 pl of 1X PCR buffer.
The
reaction mixture was overlaid with two drops of light mineral oil and the tube
was heated to
94°C for 30 sec, cooled to 50°C for 1 min, heated to 72°C
for 2 min for 30 repetitions. The
double-stranded PCR product was gel purified, precipitated and resuspended in
40 p.l of TE
buffer as described above in a).
The single-stranded sense and anti-sense 157 nucleotide DNA fragments were
generated using the above 157 by wild-type DNA fragment (SEQ ID N0:27) in two
asymmetric PCR reactions. The sense strand fragment was generated using 5 p.l
of the above
purified 157 by fragment (SEQ ID N0:27) as the target in an asymmetric PCR.
The reaction
mixtures for the asymmetric PCR were as above for the syrmnetric PCR with the
exception
that 100 pmoles of the biotin-labeled sense primer (SEQ ID N0:29) and 1 pmole
of the
fluorescein-labeled anti-sense primer (SEQ ID N0:30) was used to prime the
reaction. The
anti-sense fragment was generated using 5 pl of the above purified 157 by
fragment as the
- 113 -
CA 02203627 1997-04-24
WO 96115267 PCT/US95114673
target in an asymmetric PCR. The reaction conditions for the asymmetric PCR
were as above
for the symmetric PCR with the exception that 1 pmole of the sense primer (SEQ
ID N0:29)
and 100 pmoles of the anti-sense primer (SEQ ID N0:30) was used to prime the
reaction.
The reaction conditions for the asymmetric PCR were 95°C for 45
sec,.50°C for 45
sec, 72°C for 1 min and 15 sec for 30 repetitions with a 5 min
incubation at 72°C after the
last repetition. The reaction products were visualized. extracted and
collected as described
above with the single stranded DNA being identified by a shift in mobility
when compared to
a double stranded DNA control.
The single-stranded 165 nucleotide fragment from pGEM3Zf(+) (SEQ ID N0:36) was
generated by asymmetric PCR. The PCR comprised 50 pmoles of 5' biotin-
AGCGGATAACAATTTCACACAGGA-3' (SEQ ID N0:38: Promega) and 1 pmole of 5'-
CACGGATCCTAATACGACTCACTATAGGG-3' (SEQ ID NO:39; Integrated DNA
Technologies. Coralville, IA), 50 ~M each dNTP, in 1 X PCR buffer. Forty-five
microliters
of this reaction mixture was overlaid with two drops of light mineral oil and
the tube was
heated to 95°C for 5 sec and then cooled to 70°C. Taq polymerase
was then added at 1.25
units in 5 ~l of 1X PCR buffer. The tubes were heated to 95°C fox 45
sec, cooled to 50°C
for 45 sec, heated to 72°C for 1 min 15 sec for 30 repetitions with a 5
min incubation at
72°C after the last repetition. The reaction products were visualized,
extracted and collected
as described above with the 164 nucleotide DNA fragment being identified by a
shift in
mobility when compared to a double stranded DNA control.
The 206 nucleotide DNA fragment (SEQ ID N0:37) was prepared by asymmetric
PCR, performed as described above, using 1 pmole of a double-stranded 206 by
PCR product
(generated as described in Example 1C), and 50 pmoles of the primer 5'-
CGCCAGGGTTTTCCCAGTCACGAC-3' (SEQ ID N0:40). The tubes were heated to
95°C
for 45 sec, cooled to 63°C for 45 sec, heated to 72°C for I min
I S sec for I S repetitions with
a 5 min incubation at 72°C after the last repetition. The reaction
products were visualized.
extracted and collected as described above with the 206_ nucleotide DNA
fragment being
identified by a shift in mobility when compared to a double stranded DNA
control. The
precipitated DNA was resuspended in 70 p.l of TE buffer. _
Twenty-five microliters of the above product was biotinylated on the 3' end
using 10-
20 units of terminal deoxynucleotidyl transferase (TdT) (Promega) in a 50 ~l
reaction. The
reaction comprised 0.5 nmoles of biotin-16-ddUTP(Boehringer Mannheim) and 1X
TdT
buffer (500 mM -cacoodylate buffer, pH 6.8, 5 mM CoCI,, 0.5 mM DTT and 500
~.g/ml °
- 114 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
BSA). The tubes were incubated at 37°C for 15 min followed by ethanol
precipitation in the
presence of 4 ~g of glycogen. The DNA was ethanol precipitated a second time
and then
resuspended in 25 q.l of TE. - . '
The cleavage reactions were carried out in a final volume of 10 q,l of 10 mM
MOPS,
pH 8.2, with 1 mM MnCh using approximately 100 fmoles of substrate DNA and 250
ng of
the enzyme CleavaseTT'' BN. Parallel reactions lacking the enzyme CleavaseT"'
BN (no
enzyme control) were set up as above with the exception that one third as much
DNA
template was used (approximately 33 fmoles of each template) to balance the
signal on the
autoradiograph.
Each substrate DNA was placed in a 200 ~l thin wall microcentrifuge tube
(BioRad,
Hercules, CA) in 5 ~1 of 10 mM MOPS, pH 8.2, with 2 mM MnCh. The solution was
overlaid with one drop of light mineral oil. Tubes were brought to 95°C
for ~ seconds to
denature the substrates and then the tubes were quickly cooled to 65°C.
Cleavage reactions were started immediately by the addition of a diluted
enzyme
mixture comprising 1 pl of the enzyme CleavaseTM BN [250 ng/q.l in 1X dilution
buffer
(0.5% NP40, 0.5% Tween20, 20 mM Tris-HCI, pH 8.0, 50 mM KCI, 10 mg/ml BSA)] in
5
~l of 10 mM MOPS, pH 8.2, without MnCh. The enzyme solution was at room
temperature
before addition to the cleavage reaction. After 5 minutes at 65°C, the
reactions were stopped
by the addition of 8 p.l of stop buffer. Samples were heated t:o 72°C
for 2 minutes and 5 p.l
of each reaction were resolved by electrophoresis through a I 0%
polyacrylamide gel ( 19:1
cross-link), with 7 M urea, in a buffer containing O.SX TBE.
After electrophoresis, the gel plates were separated and overlaid with a 0.45
q.m-pore
positively charged nylon membrane (United States Biochemical). The DNA was
transferred
to the membrane and the membrane was dried, washed in 1.2X Sequenase Images
Blocking
Buffer, treated with 1 X SAAP buffer as described above. The signal was
developed using
Lumiphos-530 (United States Biochemical) or Quantum Yield Chemiluminescent
Substrate
(Promega) in place of the CDP-Star~'~"''; the membrane was then exposed to X-
ray film as
described above. The resulting autoradiograph is shown in Figure 28.
Figure 28 shows the results of incubation of the four substrates described
above in the
presence or absence of the CleavaseT"'' BN enzyme. Four sets of reactions are
shown. Set
one contains the reaction products from the incubation of the 157 nucleotide
sense strand
fragment of the tyrosinase gene (SEQ ID N0:34) in the absence or presence of
the
CleavaseTM BN enzyme. Set two contains the reaction products from the
incubation of the
- 11~ -
CA 02203627 1997-04-24
WO 96115267 PCT/US95/14673
157 nucleotide anti-sense strand fragment of the tyrosinase gene (SEQ ID
N0:35) in the
absence or presence of the CleavaseT"~' BN enzyme. Set three contains the
reaction products
from the incubation of the 165 base bottom strand fragment of the plasmid
pGEM3Zf(+)
(SEQ ID N0:36) in the absence or presence of the CleavaseT"' BN enzyme. Set
four contains '
the reaction products from the incubation of the 206 base top strand fragment
of the plasmid
pGEM3Zf(+) (SEQ ID N0:37) in the absence or presence of the CleavaseTM BN
enzyme.
Lanes marked "M" contain biotin-labeled molecular weight markers prepared as
described
above; the sizes of the marker fragments are indicated in Figure 28. In the
absence of the
CleavaseT"'' BN enzyme, no cleavage of the substrates is observed. In the
presence of the
CleavaseTM BN enzyme, each substrate is cleaved generating a unique set of
cleavage
products. When these cleavage products are resolved on a polyacrylamide gel, a
unique
pattern or fingerprint is seen for each substrate DNA. Thus, although the four
substrates are
similar in size ( 157 to 206 bases), the CleavaseT"' BN enzyme generates a
unique collection of
cleavage products from each substrate. These unique cleavage patterns result
from the
1 ~ characteristic conformation each substrate DNA assumes.
The present invention contemplates the ability to generate a unique cleavage
pattern for
two or more DNA substrates of the same size as part of a method for the
detection of genetic
mutations. This method compares a normal (or wild type or non-mutated)
substrate with a
substrate from a patient suspected of having a mutation in that substrate. The
two substrates
would be of the same length and the cleavage reaction would be used to probe
the patient
DNA substrate for conformational changes relative to the pattern seen in the
wild type control
substrate. -
EXAMPLE 9
Cleavage Directed By The CleavaseT"' BN Enzyme
Can Detect Single Base Changes In DNA Substrates
The ability of the CleavaseT"'' BN enzyme to cleave DNA substrates of the same
size
but which contain single base changes between the substrates is herein
demonstrated. The
human tyrosinase gene was chosen as a model system because numerous single
point.
mutations have been identified in exon 4 of this gene [Spritz, R.A. (1994)
Human Molecular
Genetics 3:1469]. Mutation of the tyrosinase gene leads to oculocutaneous
albinism in
humans.
- 116 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
Three single-stranded substrate DNAs were prepared; the substrates contain a
biotin
label at their 5' end. The wild type substrate comprises the 157 nucleotide
fragment from the
sense strand of the human tyrosinase gene [(SEQ ID N0:34); Geibel, L.B., et
al. ( 1991 )
Genomics 9:435]. Two mutation-containing substrates were used. The 419
substrate (SEQ
ID N0:41 ) is derived from the tyrosinase mutant G419R which contains a
glycine (GGA) to
arginine (AGA) substitution; this mutant differs from the wild-type exon 4
fragment by a
single base change at nucleotide 2675 [King, R.A., et al. ( 1991 ) Mol. Biol.
Med. 8:19]. The
422 substrate (SEQ ID N0:42) is derived from the tyrosinasc~ mutant R422Q
which contains
an arginine (CGG) to glutamine (CAG) substitution; this mutant differs from
the wild type
exon 4 fragment by a single base change at nucleotide 2685 [Giebel, L.B., et
al. (1991) J.
Clin. Invest. 87:1119].
Single-stranded DNA containing a biotin label at the 5" end was generated for
each
substrate using asymmetric PCR as described in Example 8a with the exception
that the
single-stranded PCR products were recovered from the gel rather than the
double-stranded
products.
The following primer pair was used to amplify each DNA (the 419 and 422
mutations
are located internally to the exon 4 fragment amplified by the primer pair
thus the same
primer pair can be used to amplify the wild type and two mutant templates).
The primer
listed as SEQ ID N0:29 sense primer) contains a biotin label at the 5' end and
was used in a
100-fold excess over the anti-sense primer of SEQ ID N0:30.
To generate-the single stranded substrates the following templates were used.
Ten ng
of supercoiled plasmid DNA was used as the target to generate the wild-type
(plasmid
pcTYR-NlTyr) or 422 mutant (plasmid pcTYR-A422) 157 nucleotide fragments. Five
microliters of the gel purified 339 by PCR fragment (SEQ ID N0:31 ) derived
from genomic
DNA homozygous for the 419 mutation (described in Example 8a) was used as the
target to
generate the 157 nucleotide 419 mutant fragment (SEQ ID N0:41).
For each target DNA, the asymmetric PCR comprised 100 pmoles of SEQ ID N0:29
and I pmole of SEQ ID N0:30, and 50 ~.M each dNTP in 1X PCR buffer. The
reaction
mixture (45 p.l) was overlaid with two drops of light mineral oil and the
tubes were heated to
95°C for 5 sec then cooled to 70°C. Tag polymerise was then
added as 1.25 units of enzyme
in 5 ~1 of 1X PCR buffer. The tubes were heated to 95°C for 45 sec.
cooled to 50°C for 4~
sec, heated to 72°C for 1 min 15 sec for 30 repetitions with a 5 min
incubation at 72°C after
- 117 -
CA 02203627 1997-04-24
WO 96/15267 PC"T/US95/14673
the last repetition. The single stranded PCR products were gel purified,
precipitated and
resuspended in 40 ~,l of TE buffer as described above. .
Cleavage reactions were performed as descibed in Example 8b. The samples were
heated to -72°C for 2 minutes and 7 p.l of each reaction were resolved
by electrophoresis
through a 10% polyacrylamide gel (19:1 cross-link), with 7 M urea, in a buffer
containing
O.SX TBE. After electrophoresis, the DNA was transferred to a nylon membrane
and
processed with SAAP and CDPStar as described in Example 8a. The resulting
autoradiograph
is shown in Figure 29.
In Figure 29, lanes marked "M" contain molecular weight markers prepared as
described in Example 8. Lanes 1-3 contain the no enzyme control for the wild
type (SEQ ID
N0:34), the 419 mutant (SEQ ID N0:41) and the 422 mutant (SEQ ID N0:42)
substrates,
respectively. Lane 4 contains the cleavage products from the wild type
template. Lane 5
contains the cleavage products from the 419 mutant. Lane 6 contains the
cleavage products
from the 422 mutant.
Figure 29 shows that a similar, but distinctly different, pattern of cleavage
products is
generated by digestion of the three template DNAs with the CleavaseTM BN
enzyme. Note
that in the digest of mutant 419, the bands below about 40 nucleotides are
absent. when
compared to wild-type, while in the digest of mutant 422 several new bands
appear in the 53
nucleotide range.
Although the three template DNAs differed in only one of the 157 nucleotides,
a
unique pattern of cleavage-fragments was generated for each. Thus a single
base change in a
157 nucleotide fragment gives rise to different secondary structures which are
recognized by
the CleavaseTM enzyme.
-__ EXAMPLE 10
Single Base Changes In Large DNA
Fragments Are Detected By The Enzyme CleavaseTM BN
The previous example demonstrated that the 5' nuclease activity of the
CleavaseTM BN
enzyme could be used to detect single point mutations within a 157 nucleotide
DNA
fragment. The ability of the CleavaseT"' BN enzyme to detect single point
mutations within ,
larger DNA fragments is herein demonstrated.
Increasingly larger fragments derived from the 422 tyrosinase mutant was
compared to
the same size fragments derived from the wild-type tyrosinase gene. Four sets
of single-
- 118 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
stranded substrates were utilized: 1 ) a 157 nucleotide template derived from
the sense strand
of exon 4 from the wild-type (SEQ ID N0:34) and 422 mutant (SEQ ID N0:42), 2)
a 378
nucleotide fragment containing exons 4 and 5 from the wild-type (SEQ ID N0:43)
and 422
mutant (SEQ ID N0:44), 3) a 1.059 kb fragment containing exons 1-4 from the
wild-type
(SEQ ID N0:45) and 422 mutant (SEQ ID N0:46) and 4) a 1.587 kb fragment
containing
exons 1-5 from the wild-type (SEQ ID N0:47) and 422 mutant (SEQ ID N0:48). The
only
difference between the wild type and 422 mutant templates is the G to A change
in exon 4
regardless of the length of the template used. The G to A point mutation is
located 27, 27,
929 and 1237 nucleotides from the labeled ends of the 157 base, 378 base,
1.059 kb and 1.6
lcb substrate DNAs, respectively.
A) Preparation Of The Substrate DNA
A cDNA clone containing either the wild-type [pcTYR-NlTyr, Bouchard, B., et
crl.
( 1989) J. Exp. Med. 169:2029] or 422 mutant [pcTYR-A422, Giebel, L.B., et al.
( 1991 )
87:1119] tyrosinase gene was utilized as the target DNA in PCRs to generate
the above
substrate DNAs. The primer pair consisting of SEQ ID NOS:42 and 43 were used
to generate
a double stranded 157 by DNA fragment from either the mutant of wild-type cDNA
clone.
The primer pair consisting of SEQ ID N0:29 and SEQ ID N0:49 was used to
generate a
double stranded 378 by DNA fragment from either the wild-type or mutant cDNA
clone. The
primer pair consisting of SEQ ID NO:50 and SEQ ID N0:3G was used to generate a
double
stranded 1.059 kbp DNA fragment from either the wild-type or mutant cDNA
clone. The
primer pair consisting of SEQ ID NO:51 and SEQ ID N0:49 was used to generate a
double
stranded 1.587 kbp DNA fragment from either the wild-type or mutant cDNA
clone. In each
case the sense strand primer contained a biotin label at the 5' end.
The PCR reactions were carried out as follows. One to two ng of plasmid DNA
from
the wild-type or 422 mutant was used as the target DNA in a 100 p.l reaction
containing 50
~,M of each dNTP, 1 p,M of each primer in a given primer pair, in 1 X PCR
buffer. Tubes
containing the above mixture were overlaid with three drops of light mineral
oil and the tubes
were heated to 94°C for 1 min, then cooled to 70°C.. Taq
polymerase was then added as 2.~
units of enzyme in 5 p,l of 1X PCR buffer. The tube was heated to 93°C
for 45 sec, cooled
to 52°C for 2 min, heated to 72°C for 1 min 45 sec for 35
repetitions, with a 5 min
incubation at 72°C after the last repetition.
Following the PCR, excess primers were removed using a QIA Quick-Spin PCR
Purification kit (Qiagen, Inc. Chatsworth, CA) following the manufacturer's
instructions; the
- 119 -
CA 02203627 1997-04-24
WO 96/15267 PG"TlUS95/14673
DNA was eluted in 50 p.l of TE. The sense strand of each of the double-
stranded fragments
from the wild-type and 422 mutant gene were isolated as follows. Streptavidin-
coated
paramagnetic beads (Dynal M280 beads) [0.5 mg in 50 p.l; pre-washed in 2X bind
and wash
(B&W) buffer (2 M NaCI, 10 mM Tris-HCI, pH 7.5, 1 mM EDTA, 0.1 % Tween 20))
were
added to each purified PCR product. The samples were incubated at room
temperature for 15 ,
minutes with occasional shaking. The beads were removed from the supernatant
by exposing
the tube to a magnetic plate and the supernatant was discarded. The bead-DNA
complexes
were washed twice in 2X B&W buffer. One hundred microliters of 0.1 M NaOH were
added
to the beads and the samples were incubated at room temperature for 15 minutes
(for the 157,
378 by DNAs); for DNA fragments larger than 1 kb, the beads were incubated at
47°C for 30
minutes. After incubation, the beads were washed twice with 2X B&W buffer.
Finally, the
bead-ssDNA complexes were resuspended in 50 p.l 2X B&W buffer and stored at
4°C.
B) Cleavage Reaction Conditions
The cleavage reactions were performed directly on the single-stranded DNA-bead
complexes. 5 to 10 ~,l of DNA-bead complex (about 100 fmoles of DNA) were
placed in a
200 pl microcentrifuge tube and washed once with 10 p.l of sterile HBO. 7.5
microliters of 10-
mM -MOPS, pH 8.2, with 1.3 mM MnCI, (to yield a final concentration of 1 mM)
was then
added to each tube. The reaction tubes were prewarmed to 65°C for 2
minutes and cleavage
was initiated by the addition of 2.5 ~.1 of the enzyme Cleavase-'~"'' BN ( 10-
50 ng in 1 X dilution
buffer). The reaction was carried out at 65°C for 5 min.
Immediately after this 5 min incubation, the beads were allowed to settle to
the bottom
of the tube and the supernatant was removed and discarded. Ten to forty
microliters of stop
buffer was then added to the beads and the sample was incubated at 90°C
for 5-10 minutes.
The formamide/EDTA solution releases the biotinylated DNA from the beads. The
beads
were allowed to settle to the bottom of the tube. The supernatant containing
the cleavage
products was collected. Two to eight microliters of the supernatant solution
loaded onto 6%
polyacrylamide gel (19:1 cross-link), with 7 M urea, in a buffer containing
O.SX TBE.
After electrophoresis, the DNA was transferred to a nylon membrane and
processed
with SAAP and CDPStar as described in Example 8a. The resulting autoradiograph
is shown
in Figure 30.
In Figure 30, lanes marked "M" contain molecular weight- markers prepared as '
described in Example 8. Lanes l, 3, ~ and 7 contain cleavage products using
the 157, 378.
1056 or 1587 nucleotide sense strand fragment from the wild-type tyrosinase
gene.
- 120 -
CA 02203627 1997-04-24
WO 96115267 ~ PCT/US95/14673
respectively. Lanes 2. 4, 6 and 8 contain cleavage products using the 157,
378, 1056 or 1587
nucleotide sense strand fragment from the 422 mutant tyrosinase gene,
respectively.
As shown in Figure 30, the clear pattern of cleavages seen between the wild
type and
' 422 mutant was not obscured when the single base change was located in
longer DNA
fragments. Thus, thecleavage reaction of the invention can be used to scan
large fragments
of DNA for mutations. Fragments greater than about 500 by in length cannot be
scanned
using existing methodologies such as SSCP or DGGE analysis.
EXAMPLE 11
The Cleavase~'~'' Reaction Is Insensitive
To Large Changes In Reaction Conditions
The results shown above demonstrated that the CleavaseTM BN enzyme can be used
to
probe DNA templates in a structure-specific but sequence independent manner.
These results
demonstrated that the Cleavase~'~'' BN enzyme could be used as an efficient
way to recognize
conformational changes in nucleic acids caused by sequence variations. This
suggested that
the 5' nuclease activity of the CleavaseT"' BN enzyme could be used to develop
a method to
scan nucleic acid templates for sequence alterations relative to a wild-type
template. The
experiments below showed that this was the case. Furthermore it is
demonstrated below that
the method of the invention is relatively insensitive to large changes in
conditions thereby
making the method suitable for practice in clinical laboratories.
First, the effect of varying the concentration of MnCh on the cleavage
reaction was
determined. Second, the effect of different amounts of salt (KCl) on the
cleavage pattern was
examined. Third, a time course was performed to investigate when complete
cleavage was
obtained. Fourth, a temperature titration was performed to determine the
effect of
temperature variations on the cleavage pattern. Next, the enzyme was titrated
to determine
the effect of a 50-fold variation in enzyme concentration on the cleavage
reaction. The
results of these experiments showed that the Cleavase'~"' reaction is
remarkably robust to large
changes in conditions.
. 30 A) MnClz Titration
To determine the sensitivity of the cleavage reaction 1:o fluctuations in the
concentration of MnCI,. a single template was incubated in the presence of a
fixed amount of
the CleavaseTM BN enzyme (250 ng) in a buffer containing 10 mM MOPS, pH 8.2,
and
various amount of MnCI,. The cleavage reaction was performed as follows. One
hundred
- 121 -
CA 02203627 1997-04-24
WO 96/15267 PG"T/US95I14673
fmoles of the 157 nucleotide sense strand- fragment of the tyrosinase gene
(SEQ ID N0:42;
prepared by asymmetric PCR .as described in Example 9) was placed i11 a 200 ul
thin wall
microcentrifuge tube (BioRad) in 5 ~.l of 10 mM MOPS, pH 8.2, with 0, 2, 4. 8,
12 or 20
mM MnCh (to yield a final concentration of either 0, 1, 2, 4, 6, 8 or 10 -mM
MnCh). A tube
containing 100 fmoles -template DNA in 5 q.l of 10 mM MOPS, pH 8.2 with 10
MnCI, was y
prepared and served as the no enzyme (or uncut) control. Each reaction mixture
was overlaid
with a drop of light mineral oil. The tubes were heated to 95°C for 5
sec and then cooled to
GS°C. .. _
Cleavage reactions were initiated and terminated as descibed in Example 8b.
After the
addition of stop solution, the samples were heated to 72°C for 2
minutes and 8 ~.1 of each
reaction were resolved by electrophoresis through a 10% polyacrylamide gel
(19:1 cross-link).
with 7 M urea, in a buffer containing O.SX TBE. After electrophoresis, the DNA
was
transferred to a nylon membrane and processed with SAAP and CDPStar as
described in
Example 8a. The resulting autoradiograph is shown in Figure 31.
In Figure 31, lanes marked "M" contain molecular.weight markers. Lane I
contains
the no enzyme control and shows the migration of the uncleaved template DNA.
Lanes 2
through 8 contain reaction products-incubated in the presence of the enzyme
CleavaseT"'~ BN
in a buffer containing 10, 8, 6, 4, 2, 1, or 0 mM MnCI,, respectively.
Figure 31 shows that no cleavage occurs in the absence of divalent canons
(lane 8, 0
mM MnCI,). Efficient production of cleavage fragments was promoted by the
inclusion of
MnCI,. The most distinct pattern of cleavage seen at 1 mM MnCh (lane 7), but
little change
in the pattern was seen when the Mn'-T concentration varied from 1 to 4 mM;
High
concentrations of MnCh tend to suppress the cleavage reaction (concentrations
above 6 mMj.
These results show that the cleavage reaction requires a divalent cation but
that changes in the
amount of divalent cation present have little effect upon the cleavage
pattern.
B) Effect Of Salt Concentration On The Cleavage Reaction
To determine the effect of salt concentration upon the cleavage reaction, a
single
template was incubated in the presence of a fixed amount of the CleavaseTM BN
enzyme (250
ng) -in a buffer containing 10 mM MOPS, pH 8.2, 1mM MnCI, and various amounts
of IhCI.
.One hundred fmoles of the 157 base fragment derived from the sense strand of
exon 4
of the tyrosinase gene (SEQ ID N0:34; prepared as described in Example 8a) was
placed in a
200 yl thin wall microcentrifuge tube (BioRad) in a buffer containing 10 mM
MOPS, pH 8.?
- 122 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
and I mM MnCh. KCl was added to give a final concentration of either 0, 10,
20, 30, 40, or
50 mM KCI; the final reaction volume was 10 pl.
A tube containing 10 mM MOPS, pH 8.2, I mM MnCh, 33 fmoles template DNA and
50 mM KCl was prepared and served as the no enzyme (or uncut) control. Each
reaction
S mixture was overlaid with a drop of light mineral oil. The tubes were heated
to 95°C for 5
seconds and then cooled to 65°C.
Cleavage reactions were initiated and terminated as descibed in Example 8b.
After the
addition of stop solution, the samples were heated to 72°C for 2
minutes and 8 ~.l of each
reaction were resolved by electrophoresis through a 10% polyacrylamide gel
(19:1 cross-link),
with 7 M urea, in a buffer containing O.SX TBE. After electrophoresis, the DNA
was
transferred to a nylon membrane and processed with SAAP and Lumiphos-530
(United States
Biochemical) or Quantum Yield Chemiluminescent Substrate (Promega Corp.,
Madison WI)
and exposed to X-ray film as described in Example 8b. The resulting
autoradiograph is
shown in Figure 32.
I S In Figure 32 , lanes marked "M" contain molecular weight markers. Lane I
contains
the no enzyme control and shows the migration of the uncleaved template DNA.
Lanes 2
through 7 contain reaction products incubated in the presence of the
CleavaseT"' BN enzyme
in a buffer containing 50, 40, 30, 20, 10 or 0 mM KCI, respectively.
The results shown in Figure 32 show that the CleavaseT"' reaction is
relatively
insensitive to variations in salt concentration. The same cleavage pattern was
obtained when
the 157 nucleotide tyrosinase DNA template (SEQ ID N0:34) was incubated with
the
CleavaseT"' enzyme regardless of whether the KCI concentration varied from 0
to 50 mM.
C) Time Course Of The Cleavage Reaction
To determine how quickly the cleavage reaction is completed, a single template
was
incubated in the presence of a fixed amount of the CleavaseT"'' BN enzyme for
various lengths
of time. A master mix comprising 20 ~.l of a solution containing 10 mM MOPS,
pH 8.2, ~
mM MnCI,, and 400 fmoles of the 157 base fragment derived from the sense
strand of exon 4
of the tyrosinase gene [(SEQ ID N0:34); prepared as described in Example 8b]
was made.
Five microliter aliquots were placed in 200 p.l thin wall microcentrifuge tube
(BioRad) for
each time -point examined. A no enzyme control tube was run; this reaction
contained 33
fmoles of the template DNA in 10 mM MOPS, pH 8.2 with 1 mM MnCI, (in a final
reaction
volume of 10 pl). The solutions were overlaid with one drop of light mineral
oil. The tubes
-123-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
were brought to 95°C for 5 seconds to denature the templates and then
the tubes were cooled
to 65°C.
Cleavage reactions were started by the addition of a diluted enzyme mixture as
described in Example 8b. At the indicated time points, the reactions were
stopped
immediately by the addition of 8 p.l of stop solution. The no enzyme control
was incubated
at 65°C, for 10 minutes and treated in the same manner as the other
reactions by the addition
of 8 ~.1 of stop buffer. Samples were heated to 72°C for 2 minutes and
5 ~.l of each reaction
were resolved by electrophoresis through a 10% polyacrylamide gel (19:1 cross-
link), with 7
M urea, in a buffer containing O.SX TBE. After electrophoresis, the DNA was
transferred to
a nylon membrane and processed with SAAP, then reacted with and Lumiphos-530
(United
States Biochemical) or Quantum Yield Chemiluminescent Substrate (Promega
Corp., Madison
WI) and exposed to X-ray film as described in Example 8b. The resulting
autoradiograph is
shown in Figure 33.
In Figure 33, lanes marked "M" contain molecular weight markers prepared as
described in Example 8. Lane 1 contains the no enzyme control incubated for 10
minutes.
Lanes 2-5 contain the cleavage products from reactions incubated for 0.1, 1, 5
or 10 minutes
at 65°C. Figure 36 shows that the cleavage reaction mediated by the
Cleavase~'~'' BN enzyme
is very rapid. Cleavage is already apparent at less than 6 seconds (<0.1 min)
and is complete
within one minute. These results also show that the same pattern of cleavage
is produced
whether the reaction is run for I or 10 minutes.
D) Temperature Titration Of The Cleavase Reaction
To determine the effect of temperature variation on the cleavage pattern, the
157 base
fragment derived from the sense strand of exon 4 of the tyrosinase gene (SEQ
ID N0:34) was
incubated in the presence of a fixed amount of the CleavaseTM BN enzyme for 5
minutes at
various temperatures. One hundred fmoles of substrate DNA (prepared as
described in
Example 8b) was placed in a 200 pl thin wall microcentrifuge tube (BioRad) in
5 ~.l of 10
mM _MOPS, pH 8.2 with 2 mM MnCI,. .Two "no enzyme" test control tubes were set-
up as
above with the exception that these reactions contained 33 fmoles of substrate
DNA in 10 yl
of the above buffer with 1 mM MnCh. The solution was overlaid with one drop of
light
mineral oil. Tubes were brought to 95°C for 5 seconds to denature the
templates and then the
tubes were cooled to the desired temperature.
Cleavage reactions were started immediately by the addition of a diluted
enzyme
mixture as described in Example 8b. The tubes placed at either 55°,
60°, 65°, 70°, 7~° or
- 124 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
80°C. After 5 minutes at a given temperature, the reactions were
stopped by the addition of 8
yl of stop buffer.
Samples were heated to 72°C for 2 minutes and 5 ql of each reaction
were resolved by
electrophoresis through a 10% polyacrylamide gel (19:1 cross-link), with 7 M
urea, in a
s 5 buffer containing O.SX TBE. After electrophoresis, the DNA was transferred
to a nylon
membrane and processed with SAAP, then reacted with and Lumiphos-530 (United
States
Biochemical) or Quantum Yield Chemiluminescent Substrate (Promega Corp.,
Madison WI)
and exposed to X-ray film as described in Example 8b. The resulting
autoradiograph is
shown in Figure 34.
In Figure 34, the lanes marker "M" contain molecular weight markers prepared
as
described in Example 8. Lanes 1 and 2 contain no enzyme controls incubated at
55°C and
80°C, respectively. Lanes 3-8 contain the cleavage products from the
CleavaseT"1 enzyme-
containing reactions incubated at 55°C, 60°C, 65°C,
70°C, 75°C or 80°C, respectively.
Figure 34 shows that the CleavaseTM reaction can be performed over a wide
range of
temperatures. The pattern of cleavages changed progressively in response to
the temperature
of incubation, in the range of 55°C to 75°C. Some bands were
evident only upon incubation
at higher temperatures. Presumably some structures responsible for cleavage at
the
intermediate temperatures were not favored at the lower temperatures. As
expected, cleavages
became progressively less abundant in the high end of the temperature range
tested as
structures were melted out. At 80°C cleavage was inhibited completely
presumably due to
complete denaturation of the template.
These results show that the cleavage reaction can be performed over a wide
range of
temperatures. The ability to run the cleavage reaction at elevated
temperatures is important. If
a strong (i.e., stable) secondary structure is assumed by the templates, a
single nucleotide
change is unlikely to significantly alter that structure, or the cleavage
pattern it produces.
Elevated temperatures can be used to bring structures to the brink of
instability, so that the
effects of small changes in sequence are maximized, and revealed as
alterations in the
cleavage pattern.within the target template, thus allowing the cleavage
reaction to occur at that
point.
E) Titration Of The CleavaseT"' BN Enzvme
The effect of varying the concentration of the CleavaseTM BN enzyme in the
cleavage
reaction was examined. One hundred fmoles of the 157 base fragment derived
from the sense
strand of exon 4 of the tyrosinase gene (SEQ ID N0:34) was placed in 4
microcentrifuge
_ 12$ _
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
tubes in 5 q.l of 10 mM MOPS, pH 8.2 with 2 mM MnCh. A no enzyme control tube
was
run; this reaction contained 33 fmoles of substrate DNA in 10 q.l of 10 mM
MOPS, pH 8.2
containing 1 mM MnCI,. The solutions were overlaid with one drop of light
mineral oil.
The tubes were brought to 95°C for 5 seconds to denature the templates
and then the tubes
were cooled to 65°C.
Cleavage reactions were started immediately by the addition of a diluted
enzyme
mixture comprising 1 ~.l of the CleavaseTT'' BN enzyme in 1X dilution buffer
such that 10, 50,
100 or 250 ng of enzyme was in the tubes in 5 p,l of 10 mM MOPS, pH 8.2
without MnCh.
After 5 minutes at 65°C, the reactions were stopped by the addition of
8 ql of stop buffer.
The samples were heated to 72°C for 2 minutes and 7 p.l of each
reaction were resolved by
electrophoresis through a 10% polyacrylamide gel (19:1 cross-link), with 7 M
urea, in a
buffer containing O.SX TBE.
After electrophoresis, the DNA was transferred to a nylon membrane and
processed
with SAAP, then reacted with and Lumiphos-530 (United States Biochemical) or
Quantum
Yield Chemiluminescent Substrate (Promega Corp., Madison WI) and exposed to X-
ray film
as described in Example 8b. The resulting autoradiograph is shown in Figure
3~.
The lanes marked "M" in Figure 35 contain molecular weight markers. Lane 1
contains the no enzyme control and shows the migration of the uncut substrate.
Lanes 2-~
contain reaction products from reactions containing 10, 50, 100 or 250 ng of
the CleavaseT"''
BN enzyme , respectively.
These results show that the same cleavage pattern was obtained using the 157
nucleotide tyrosinase DNA substrate regardless of whether the amount of enzyme
used in the
reaction varied over a 25-fold range. Thus, the method is ideally suited for
practice in
clinical laboratories where reactions conditions are not as controlled as in
research
laboratories.
F) Consistent Cleavage Patterns Are Obtained Using Different DNA
Preparations
To demonstrate that the same cleavage pattern is consistently obtain from a
given
substrate, several different preparations of the 157 base fragment derived
from the sense
strand of exon 4 of the tyrosinase gene (SEQ ID N0:34) were generated. The
substrate. was
generated as described in Example 8b. Three independent PCR reactions
performed on '
separate days were conducted. One of these PCR samples was split into two and
one aliquot
- 126 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
was gel-purified on the day of generation while the other aliquot was stored
at 4°C overnight
and then gel-purified the next dav.
Cleavage reactions were performed as described in Example 8b. Samples were run
on
' an acrylamide gel and processed as described in Example 8b. The resulting
autoradiograph is
shown in Figure 36.
In Figure 36, the lanes marked "M" contain biotinylated molecular weight
markers.
Set 1 contains the products from a cleavage reaction performed in the absence
(-) or presence
(+) of enzyme on preparation no. 1. Set 2 contains the products from a
cleavage reaction
performed in the absence (-) or presence (+) of enzyme on I>reparation no. 2.
Set 3 contains
the products from a cleavage reaction performed in the absence (-) or presence
(+) of enzyme
on preparation no. 3. Preparation no. 3 was derived from preparation 2 and is
identical except
that preparation no. 3 was gel-purified one day after preparation no. 2. Set 4
contains the
products from a cleavage reaction performed in the absence (-) or presence (+)
of enzyme on
preparation no. 4. The same pattern of cleavage products is generated from
these
independently prepared substrate samples.
These results show that independently produced prep~~rations of the 157
nucleotide
DNA fragment gave identical cleavage patterns. Thus, the CleavaseTM reaction
is not effected
by minor differences present between substrate preparations.
EXAMPLE 12
Point Mutations Are Detected Using Either The
Sense Or Anti-Sense Strand Of The Tyrosinase Gene
The ability of the Cleavase'~"'~ enzyme to create a unique pattern of cleavage
products
(i.e., a fingerprint) using either the sense (coding) or anti-sense (non-
coding) strand of a gene
fragment was examined.
Single stranded DNA substrates corresponding to either the sense (SEQ ID
N0:34) or
anti-sense strand (SEQ ID N0:35) of the 157 nucleptide fragment derived from
the wild-type
tyrosinase gene were prepared using asymmetric PCR as described in Example 8a.
The sense
strand wild-type substrate contains a biotin label at the 5' end; the anti-
sense strand contains a
fluorescein label at the 5' end.
A single stranded DNA substrate corresponding to thf: sense strand of the 157
nucleotide fragment derived from the 419 mutant tyrosinase gene (SEQ ID N0:41
) was
_ 127 _
CA 02203627 1997-04-24
WO 96/15267 PCT/US95114673
prepared using asymmetric PCR as described in Example 9. The, sense strand 419
mutant
substrate contains a biotin label at the 5' end.
A single stranded DNA substrate corresponding to the anti-sense strand of the
157
nucleotide fragment derived from the 419 mutant tyrosinase gene (SEQ ID N0:52)
was
prepared using asymmetric PCR as described in Example 9, with the exception
that 100
pmoles of the fluorescein-labeled anti-sense primer (SEQ ID N0:30) and 1 pmole
of the
biotin-labelled sense primer (SEQ ID N0:29) were used. The resulting anti-
sense strand 419
mutant substrate contains a fluorescein label at the 5' end.
A single stranded DNA substrate corresponding to the sense strand of the 157
nucleotide fragment derived from the 422 mutant tyrosinase gene (SEQ ID N0:42)
was
prepared using asymmetric PCR as described in Example 9. The sense strand 422
mutant
substrate contains a biotin label at the 5' end.
A single stranded DNA substrate corresponding to the anti-sense strand of the
157
nucleotide fragment derived from the 422 mutant tyrosinase gene (SEQ ID N0:53)
was
prepared using asymmetric PCR as described in Example 9 with the exception
that 100
pmoles of the fluorescein-labeled anti-sense primer (SEQ ID N0:30) and 1 pmole
of the
biotin-labelled sense primer (SEQ ID N0:29) were used. The resulting anti-
sense strand 422
mutant substrate contains a fluorescein label at the 5' end.
Following asymmetric PCR, the single stranded PCR products were gel purified,
precipitated and resuspended in 40 p,l of TE buffer as described in Example 8.
Cleavage reactions were performed as described in Example 9, and were resolved
by
electrophoresis as described in Example 8a. After electrophoresis, the DNA was
transferred
to a nylon membrane and processed with SAAP conjugate and antifluorescein
antibody (Fab)-
allcaline phosphatase conjugate, and visualized using CDPStar as described in
Example 8a.
The resulting autoradiograph is shown in Figure 37.
In Figure 37, lanes marked "M" contain biotinylated molecular weight markers
prepared as described in Example 8. Lanes 1-6 contain biotinylated sense
strand substrates
from the wild-type, 419 and 422 mutant 157 nucleotide fragments. Lanes 1-3
contain no
enzyme controls for the wild-type, 419 and 422 mutant fragments, respectively.
Lanes 4-6
contain the reactionproducts from the incubation of the sense strand of the
wild-type, 419
and 422 mutant fragments with theCleavase~'T'' BN enzyme , respectively. Lanes
7-12 contain '
fluoresceinated anti-sense strand substrates from the wild-type, 419 and 422
mutant 157
z
nucleotide fragments. Lanes 1-3 contain "no enzyme" controls for the wild-
type, 419 and 422
- 128 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
mutant fragments, respectively. Lanes 4-6 contain the reaction products from
the incubation
of the anti-sense strand of the wild-type, 419 and 422 mutant fragments with
the CleavaseTh'
BN , respectively. '
As expected, distinct but unique patterns of cleavage products are generated
for the
wild-type, 419 and 422 mutant fragments when either the sense or anti-sense
fragment is
V
utilized. The ability to use either the sense or anti-sense strand of a gene
as the substrate is
advantageous because under a given set of reaction conditions one of the two
strands may
produce a more desirable banding pattern (i. e., the cleavage products are
spread out over the
length of the gel rather than clustering at either end), or may have a
mutation more favorably
placed to create a significant structural shift. This could be more important
in the analysis of
long DNA substrates which contain mutations closer to one end or the other.
Additionally,
detection on both strands serves as a confirmation of a sequence change.
EXAMPLE 13
Detection Of Mutations In The Human
Beta-Globin Gene Using The Enzyme CleavaseT"'
The results shown in Examples 8-12 showed that the CleavaseTM reaction could
be
used to detect single base changes in fragments of the tyrosi:nase gene
ranging from 157
nucleotides to 1.6 kb. To demonstrate that the CleavaseT"' reaction is
generally applicable for
the detection of mutations, a second model system was examined.
The human (3-globin gene is known to be mutated in a number of
hemoglobinopathies
such as sickle cell anemia and [3-thalassemia. These disorders generally
involve small ( 1 to 4)
nucleotide changes in the DNA sequence of the wild type (3-globin gene [Orkin,
S.H. and
Kazazian, H.H., Jr. (1984) Annu. Rev. Genet. 18:131 and Collins, F.S. and
Weissman, S.M.
( 1984) Prog. Nucleic Acid Res. Mol. Biol. 31:315]. At least 47 different
mutations in the [3-
globin gene have been identified which give rise to a [i-thala.ssemia:
Three [3-globin mutants were compared to the wild type (3-globin gene [Lawn,
R.M., et
al. ( 198-0) Cell 21:647] using the CleavaseT"' reaction. Mutant 1 contains a
nonsense mutation
in codon 39; the wild-type sequence at codon 39 is CAG; the mutant 1 sequence
at this codon
is TAG [Orkin, S.H. and Goff, S.C. (1981) J. Biol. Chem. 256:9782]. Mutant 2
contains a T
to A substitution in codon 24 which results in improper splicing of the
primary transcript
[Goldsmith, M.E., et al. (1983) Proc. Natl. Acad. Sci. USA 80:2318]. Mutant 3
contains a
deletion of two A residues in codon 8 which results in a shift in the reading
frame; mutant 3
- 129 -
CA 02203627 1999-07-26
also contains a silent C to T substitution in codon 9 [Orkin. S.H. and Goff.
S.C. ( 1981 ) ,1.
Biol. Chem. 26:9782].
A) Preparation Of Wiid Type And Mutant -Globin Gene Substrates
Single stranded substrate DNA was prepared from the above wild type and mutant
[3-
globin genes as follows. Bacteria harboring the appropriate plasmids were
streaked onto
antibiotic plates and grown overnight at 37°C (bacteria with the wild-
type plasmid and the
plasmid containing the mutant 3, were grown on tetracycline plates: bacteria
with the plasmids
containin= the mutant 1 and mutant 2 sequences were grown on ampicillin
plates). Colonies
from the plates were then used to isolate plasmid DNAs using the Wizard
Minipreps DNA
Purification System (Promega Corp., Madison, WI). The colonies were
resuspended in ?00 pl
of "Cell Resuspension Buffer" from the kit. The DNA was extracted according to
the
manufacturers protocol. Final yields of approximately ?.3 pg of each plasmid
were obtained.
A 5 36 (wild-type. mutants 1 and ?) or X34 (mutant 3) nucleotide fragment was
amplified from each of the above plasmids in polvmerase chain reactions
comprisin~T ~ n~~ ~i~
1 ~ plasmid DNA. ?3 pmoles each of 3~-biotinylated KM?9 primer (SEQ ID NO:>.~)
and ~'-
fiuorescein labeled RS4? primer (SEQ ID NO:>j). 50 ~M each dNTP and 1.'_'~
units of lcrc~
DNA Polvmerase in sU ul of 1X PCR buffer. The reactions were overlaid with '_'
drops of~
light mineral oiI and were heated to 93°C for 30 seconds. cooled to
»°C for s0 seconds.
heated to 7'?°C for 60 seconds, for 3~ repetitions in a thermocvcler
(MJ Research. Vvatert~wn.
?0 MA). The products of these reactions were purified from the residual dNTPs
and primers by
use of a Vl~'izard'~P~R Cleanup kit (Promega Corp.. Madison. WI). leaving the
duplex D'~.a in
50 l.tl of TE.
To generate single stranded copies of these DNAs. the PCRs described above
were
repeated using= 1 pl of the duplex PCR DNA as template. and omitting the
RS4'_' primer. The
products of this asymmetric PCR were loaded directly on a 6% polyacrylamide
gel (?9:1
cross-link) in a buffer of O.~X TBE. alongside an aliquot of the original PCR
DNA to identify
the location of the double-strand DNA product. After electrophoretic
separation. the DN.as
were visualized by staining with ethidium bromide and the single stranded
DNAs. identified
by altered mobility when compared to the duplex DNAs. were excised and eluted
from the «el
30 slices by passive diffusion overnight into a solution comprising 0.5 M
NH,OAc. U.1 °r SDS '
and 0.? mM EDTA. The products were collected by ethanol precipitation and
dissolved in -IO
pl of TE.
* Trade-mark
- 1 s0 -
CA 02203627 1997-04-24
WO 96/15267 PG"T/US951i4673
The sequence of the 536 nucleotide fragment from the wild-type (3-globin gene
is
listed in SEQ ID N0:56. The sequence of the 534 nucleotide fragment from
mutant 3 is
listed in SEQ ID N0:57. The sequence of the 536 nucleotide fragment from
mutant 1 is
listed in SEQ ID N0:58. The sequence of the 536 nucleotide fragment from
mutant 2 is
listed in SEQ ID N0:59.
B) Optimization Of The Cleavage Reaction Using The Wild-Type Beta-Globin
Substrate
The optimal conditions (salt concentration, temperature) which produce an
array of
cleavage products having widely differing mobilities from the (3-globin
substrate were
determined. Conditions which produce a cleavage pattern having the broadest
spread array
with the most uniform intensity between the bands were determined (the
production of such
an array of bands aids in the detection of differences seen between a wild-
type and mutant
substrate). This experiment involved running the cleavage reaction on the wild
type ~3-globin
substrate (SEQ ID N0:56) at several different temperatures in the presence of
either no KC1
or 50 mM KCI.
For each KCL concentration to be tested, 30 q.l of a master mix containing
DNA,
CFLPT"1 buffer and salts was prepared. For the "0 mM KCl" reactions, the mix
included
approximately 500 fmoles of single-stranded, 5' biotinylated 536-mer PCR DNA
from
plasmid pHBG 1 in 30 p.l of 10 mM MOPS, pH 8.2, with 1.7 mM MnC 1 ~ (for 1 mM
in the
final reaction); the "50 mM KCl" mix included 83.3 mM KC 1 in addition to the
above
components. The mixes were distributed into labeled reaction tubes in 6 q,l
aliquots, and
stored on ice until use. An enzyme dilution cocktail included 450 ng of the
enzyme
CleavaseT"' BN in 10 mM MOPS, pH 8.2 without MnCl.,.
Cleavage reactions were performed at 60°C, 65°C, 70°C
and 75°C. For each
temperature to be tested, a pair of tubes with and without KC 1 were brought
to 95 ° C for 5
seconds, then cooled to the selected temperature. The reactions were then
started immediately
by the addition of 4 p.l of the enzyme cocktail. In the 75°C test, a
duplicate pair of tubes was
included, and these tubes received 4 ~.1 of 10 mM MOPS, pH 8.2 without MnCl~
in place of
the enzyme addition. No oil overlay was used. All reactions proceeded for 5
minutes. and
were stopped by the addition of 8 q.l of stop buffer. Completed and yet-to-be-
started
reactions were stored on ice until all reactions had been performed. Samples
were heated to
72°C for 2 minutes and 5 ~1 of each reaction was resolved by
electrophoresis through a 6%
polyacrylamide gel (19:1 cross-link), with 7 M urea, in a buffer of O.SX TBE.
After
- 131 -
CA 02203627 1999-07-26
electrophoresis. the gel plates were separated allowing the gel to remain flat
on one plate. A
0.? mm-pore positively-charged nylon membrane (NYTRAIv':~Schleicher and
Schuell. Keene.
NH). pre-wetted in H,O. was laid on top of the exposed gel. All air bubbles
were removed.
Two pieces of 3MM filter paper (Whatman) were then placed on top of the
membrane. the
other glass plate was replaced. and the sandwich was clamped with binder
clips. Transfer was
allowed to proceed overnight. After transfer. the membrane was carefully
peeled from the gel
and allowed to air dry. After complete drying the membrane was washed in l.'?X
Sequenase
Images Blocking Buffer (United States Biochemical) using 0.3 ml of buffer/cmv
of membrane.
The wash was performed for 30 minutes. A streptavidin-alkaline phosphatase
conjugate
(SAAP. United States Biochemical) was added to a 1:4000 dilution directly to
the blockin«
solution. and agitated for 1 ~ minutes. The membrane was rinsed briefly with
H,O and then
washed three times for ~ minutes per wash using 0.~ ml/cm= of IX SA.4P buffer
( 100 m~~1
Tris-HCI. pH 10. ~0 mM NaCI) with O.l% sodium dodecvl sulfate (SDS). The
membrane
was rinsed brief7v with H,0 bemveen each wash. The membrane was then washed
once in 1 X
1~ SA.~P''1 mi\~t MgCI= without SDS. drained thoroughl~ and placed in a
plastic heat-sealable
ba~_. Using a sterile pipet. ~ mls of either CSPDT''' or CDP-StarT"' (Tropix.
Bedford. '\-La i
chemiluminescent substrates for alkaline phosphatase were added to the bag and
distributed
over the entire membrane for ?-3 minutes. The CSPDT"'-treated membranes were
incubated at
37°C for 30 minutes before an initial exposure to XRP X-ray filth
(Kodak) for 60 minutes.
?0 CDP-StarT"'-treated membranes did not require preincubation. and initial
exposures were fur
10 minutes. Exposure times were adjusted as necessary for resolution and
clarity. The results
are shown in Figure 38.
In Fib=ure 38 he lane marked "M" contains molecular weight markers. Lanes 1-
contain reaction products from reactions run in the absence of KCI. Lane 1
contains the a
reaction run without enzyme at 7~°C. Lanes 2-~ contain reaction
products from reactions run
at 60°C. 6~'C. 70°C and 75°C. respectively. Lanes 6-10
contain reaction products from
reactions run in the presence of ~0 mM KC1. Lane 6 contains the a reaction run
without
enzyme at 7~°C. Lanes 7-10 contain reaction products from reactions run
at 60°C. 6~'C.
70°C and 7>°C. respectively.
O) In general. a preferred pattern of cleavage products was produced when the
reaction
included ~0 mM KCI. As seen in Lanes 7-10, the reaction products are more
widely spaced
in the ~0 m:~i KCL-containing reactions at every temperature tested as
compared to the
reactions run in the absence of KCL (lanes ?-~: more of the cleava~~e products
are found
*Trade-mark
- I;~ _
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
clustered at the top of the gel near the uncut substrate). As seen in Lane 7
of Figure 41,
cleavage reactions performed in 50 mM KCl at 60.°C produced a pattern
of cleavage products
in which the products are maximally spread out, particularly in the upper
portion of the gel,
when compared to other reaction condition patterns.
From the results obtained in this experiment, the optimal cleavage conditions
for the
536 nucleotide sense strand fragment derived from the wild-type (3-globin gene
(SEQ ID
N0:56) were determined to be 10 mM MOPS, pH 8.2 containing 1 mM MnCh and 50 mM
KCl at 60°C.
C) Optimization Of The Cleavage Reaction Using Two Mutant Beta-Globin
Substrates
From the results obtained above in a) and b), 60°C was chosen as the
optimum
temperature for the cleavage reaction when a (3-globin substrate was to be
used. When the
wild-type substrate was utilized, running the cleavage reaction in the
presence of 50 mM KCl
generate the optimal pattern of cleavage products. The effect of varying the
concentration of
KCl upon the cleavage pattern generated when both wild-type and mutant (3-
globin substrates
were utilized was next examined to determine the optimal salt concentration to
allow a
comparison between the wild-type and mutant (3-globin substrates.
Single stranded substrates, 536 nucleotides in length, corresponding to mutant
I (SEQ
ID N0:58) and mutant 2 (SEQ ID N0:59) mutations were prepared as described
above in a).
These two mutants each differ from the wild-type sequence by 1 nucleotide;
they differ from
each other by 2 nucleotides.
For each substrate tested, 39 p.l of a master mix containing DNA,
CFLPT°~ buffer and
MnCI, was prepared. These mixes each included approximately 500_ fmoles of
single-
stranded, 5' biotinylated 536 nucleotide substrate DNA, 39 p.l of 10 mM MOPS.
pH 8.2
2~ containing 1.54 mM MnCI? (giving a final concentration of 1 mM MnCI,). The
mixes were
distributed into labeled reaction tubes in 6.5 p.l aliquots. Each aliquot then
received 0.5 ~.1 of
200 mM KCl for each 10 mIvl final KCl concentration (e.g., 2.0 p.l added to
the 40 mM
reaction tube) and all volumes were brought to 9 ~.l,with dH~O. No oil overlay
was used.
The reactions were brought to 95°C for 5 seconds, then cooled to
65°C. The reactions were
then started immediately by the addition of 50 ng of the enzyme CleavaseTM BN
in 1 pl of
enzyme dilution buffer (20 mM Tris-HCI, pH 8.0, 50 mM KCI, 0.5% NP40, 0.5%
Tween 20,
10 mg/~1 BSA). All reactions proceeded for 5 minutes, and were stopped by the
addition of
8 ~l of stop buffer. Samples were heated to 72°C for 2 minutes and 5 ~1
of each reaction
- 133 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
was resolved by electrophoresis through a 6% polyacrylamide gel ( I 9:1 cross-
link), with 7 M
urea, in a buffer of O.SX TBE.
After electrophoresis, the gel plates were separated and overlaid with a nylon
membrane as described above. The DNA was transferred to the membrane and the
membrane
was treated as described above in b) and then exposed to X-ray-film. The
resulting
autoradiograph is shown in Figure 39
In Figure 39, the lane marked "M" contains molecular weight markers. Lanes 1,
3, 5,
7, 9 and 11 contain reaction products from cleavage reactions using the mutant
1 substrate in
the presence of 0,- 10, 20, 30, 40 or 50 mM KCI, respectively. Lanes 2, 4, 6,
8, 10 and 12
contain reaction products from cleavage reactions using the mutant 2 substrate
in the presence
of 0, 10, 20, 30, 40 or 50 mM KCI, respectively.
Figure 39 shows that while the pattern of cleavage products- generated from
each
mutant changes as the KCl concentration is increased, distinct patterns are
generated from
each mutant and differences in banding patterns are seen between the two
mutants at every
1~ concentration of KCl tested. In the mid-salt ranges (10 to 20 mM KCl), the
larger cleavage
bands disappear and smaller molecular weight bands appear (lanes 3-6). At
higher salt
concentrations (30 to 50 mM KCl), the larger molecular weight cleavage bands
reappear with
the cominant loss of the low molecular weight bands (lanes 7-12). Reaction
conditions
comprising the use of 50 mM KCl were chosen as optimal- from the results shown
in Figure
39. Clear differences in the intensities of a band appearing about 200
nucleotides (see arrow
in Figure 39) is seen between the two mutant substrates under these reaction
conditions.
D) The Enzyme CleavaseTnt Generates Unique Cleavage Products From Wild-
Type And Mutant Beta-Globin Substrates
From the experiments performed above, the optimal reaction conditions when the
wild-
type or mutant (3-globin substrates were determined to be the use of 50 mM KCl
and a
temperature of 60°C. These conditions were then used to allow the
comparison-of the
cleavage patterns generated when the wild-type substrate (SEQ ID N0:56) was
compared to
the mutant 1 (SEQ ID N0:58), mutant 2 (SEQ ID N0:59) and mutant 3 (SEQ ID
N0:57)
substrates.
Single-stranded substrate DNA, 534 or 536 nucleotides in length,- was prepared
for the
wild-type, mutant 1, mutant 2 and mutant 3 [3-globin genes as described above
in a). '
Cleavage reactions were performed as follows. Reaction tubes were assembled
which
contained approximately 100 fmoles of each DNA substrate in 9 ~.1 of 1.10 mM
MOPS, pH '
- 134 -
CA 02203627 1997-04-24
WO 96115267 PG"T/US95/14673
8.2 ( 1 X final concentration) with 1.1 mM MnCh ( 1 mM final concentration)
and 55.6 mM
KCl (50 mM final concentration). A "no enzyme" or uncut control was set up for
each
substrate DNA. The uncut controls contained one third as much DNA (
approximately 33
fmoles) as did the enzyme-containing reactions to balance thf; signal seen on
the
autoradiograph.
The tubes were heated to 95°C for 5 sec, cooled to 60°C and the
reactions were started
immediately by the addition of 1 ~l of the enzyme CleavaseT"'' BN (50 ng per
~l in 1X
dilution buffer). The uncut controls received 1 ~,l of 1 X dilution buffer.
Reactions proceeded for 5 min and then were stopped by the addition of 8 ~.l
of stop
buffer. The samples were heated to 72°C for 2 min and 5 ~.l of each
reaction was resolved
by electrophoresis through a 6% polyacrylamide gel (19:1 cross-link), with 7 M
urea, in a
buffer of 0.5X TBE.
After electrophoresis, the gel plates were separated and overlaid with a nylon
membrane as described above. The DNA was transferred to the membrane and the
membrane
was treated as described above in b) and then exposed to X-ray film. The
resulting
autoradiograph is shown in Figure 40.
In Figure 40, two panels are shown. The first panel shows the reaction
products from
the above cleavage reactions; the uncut controls are shown in lanes 1-4 and
the cleavage
products are shown in lanes 5-6. The second panel is a magnification of lanes
5-8 to better
shown the different banding patterns seen between the substrate DNAs in the
upper portion of
the gel.
In Figure 40, the lanes marked "M" contain biotinylal:ed molecular weight
markers
prepared as described in Example 8. Lanes 1-4 contain the uncut controls for
the wild-type,
mutant l, mutant 2 and mutant 3 (3-globin substrates, respectively. Lanes 5-8
contain the
cleavage products from the wild-type, mutant 1, mutant 2 anal mutant 3
substrates,
respectively.
From the results shown in Figure 40, the CleavaseT"'' BN enzyme generates a
unidue
pattern of cleavage products from each (3-globin substrate tested. Differences
in banding
patterns are seen between the wild-type and each mutant; dii:ferent banding
patterns are seen
for each mutant allowing not only a discrimination of the mutant from the wild-
type but also
a discrimination of each mutant from the others.
-135-
CA 02203627 1997-04-24
WO 96115267 PCTlUS95/14673
The results shown here for the (3-globin gene and above for the tyrosinase
gene
demonstrate that the Cleavase~ reaction provides a powerful ne~~ tool for the
detection of
mutated genes.
EXAMPLE 14
Treatment Of-RNA Substrates Generates Unique Cleavage Patterns
The present invention contemplates 5' nuclease cleavage of single- or double-
stranded
DNA substrates to generate a unique pattern of bands characteristic of a given
substrate. The
ability of the 5' nuclease activity of the enzyme CleavaseTT' BN to utilize
RNA as the
substrate nucleic acid was next demonstrated. This experiment showed that RNA
can be
utilized as a substrate for the generation of a cleavage pattern using
appropriate conditions
(Lowering of the pH to 6.5 from 8.2 to reduce manganese-mediated degradation
of the RNA
substrate). The experiment was performed as follows.
An RNA transcript internally labelled with biotin was produced to serve as the
substrate. The plasmid pGEM3Zf (Promega) was digested with EcoRI. EcoRI cuts
the
plasmid once generating a linear template. An RNA transcript 64 nucleotides in
length (SEQ
ID NO:GO) was generated by SP6 transcription of the linearized template using
a Riboprobe
Gemini System kit from Promega, Corp.; the manufacturer's directions were
followed with
the exception that 25% of the UTP in the reaction was replaced with biotin-UTP
(Boehringer
Mannheim) to produce an internally labelled transcript. Following the
transcription reaction
(1 hour at 37°C), the DNA template was removed by treatment with RQ1
RNase-free DNAse
(from the Riboprobe kit and used according to the manufacturer's instructions)
and the RNA
was collected and purified by precipitating the sample twice in the presence
of 2 M NH40Ac
and ethanol. The resulting RNA pellet was rinsed with 70% ethanol, air dried
and
resuspended in 40 ~.1 of 10 mM Tris-HCI, pH 8.0 and 1 mM EDTA.
Cleavage reactions contained 1 ~,1 of the above RNA substrate and 50 ng of the
enzyme
Cleavase~'~'"' BN in 10 ~.l of 1X RNA-CFLP~ buffer (10 mM MOPSz pH G.3) and 1
mM of
either MgCI, or MnCh. The reactions were assembled with, all the components
except, the
enzyme and were warmed to 45°C for 30 sec. Reactions were started by
the addition of 50
ng of the enzyme CleavaseT"' BN in 1 ~l of dilution buffer (20 mM Tris-HCI, pH
8.0, ~0 mM '
ICCI, 0.5% NP40, 0.5% Tween 20, 10 ~.g/ml BSA). Reactions proceeded for 10 min
and
were stopped by the addition of 8 ~.1 of stop buffer. The samples were heated
to 72°C for ? '
- 13G -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95114673
minutes and 5 ~.l of each reaction were resolved by electrophoresis through a
10%
polyacrylamide gel ( 19:1 cross-link), with 7 M urea, in a buffer containing
O.~X TBE.
After electrophoresis, the gel plates were separated and overlaid with a nylon
membrane as described in Example 8b. The DNA was transferred to the membrane
and the
membrane was dried, washed in 1.2X Sequenase Images Blacking Buffer, treated
with 1X
SAAP buffer and reacted with Lumiphos-530 (United States Biochemical) or
Quantum Yield
Chemiluminescent Substrate (Promega) and exposed to X-ray film as described in
Example
8b. The resulting autoradiograph is shown in Figure 41.
In Figure 41 , lanes marked "M" contain molecular weight markers. Lane 1
contains
the no enzyme control and shows the migration of the uncut substrate. Lanes 2
and 3 contain
reaction products from the incubation of the RNA substrate in a buffer
containing MgCI, in
the presence or absence of the Cleavase~'~"' BN enzyme , respectively. Lanes
4~ and 5 contain
reaction products from the incubation of the RNA substrate in a buffer
containing MnCI, in
the presence or absence of the CleavaseT'''' BN enzyme , respectively. A
pattern of cleavage
I S products is seen when the enzyme is incubated with the RNA substrate in
the presence of
MnCI,, (lane 5).
These results show that the Cleavase~'~"'' enzyme can be used to probe RNA
substrates
for changes in sequence (i. e., point mutations, deletions, substitutions).
This capability
enables the examination of genes which have very large introns (e.g., greater
than 10 kb)
interrupting the coding sequences. The spliced RNA transcript represents a
simpler target for
the scanning for mutations. In addition, the structural (i, e., folding)
information gained by
cleavage of the RNA would be useful in targeting of hybridization or ribozyme
probes to
unstructured regions of RNAs. Furthermore, because the cleavage reaction
occurs so quickly,
the Cleavase'~"'' enzyme can be used to study various types of RNA folding and
the kinetics
with which this folding occurs.
EXAMPLE 15
The 5' Nuclease Activity From Both the CleavaseT"' 13N Enzyme Taq Polymerase
Generates Unickue Cleavage Patterns Using Double-Stranded DNA Substrates
The ability of both the enzyme CleavaseT"' BN and Taq polymerase to generate
cleavage patterns on single-stranded DNA templates was examined. The
substrates utilized in
this experiment were the 378 nucleotide fragment from either the wild-type
(SEQ ID N0:43)
- 137 -
CA 02203627 1997-04-24
WO 96115267 PCT/US95/14673
or 422 mutant (SEQ ID N0:44) tyrosinase gene. These single-stranded substrates
were
generated as described in Example 10a.
Cleavage reactions were performed as described in Example lOb with the
exception
that half of the reactions were cut with the enzyme CleavaseT"'' BN as
described and a parallel
set of reaction was cut with Taq polymerase. The Taq polymerase reactions
contained 1.25 ;
units of Taq polymerase in 10 mM MOPS, pH 8.2. The reaction products were
resolved by
electrophoresis and the autoradiograph was generated as described in Example
lOb. The
autoradiograph is shown in Figure 42.
In Figure 42, lanes marked "M" contain biotinylated molecular weight markers.
Lanes
1 and 2 contain the wild-type or 422 mutant substrate cleaved with the
CleavaseT"'' BN
enzyme, respectively. Lanes 3 and 4 contain the wild-type or 422 mutant
substrate cleaved
with Taq polymerase, respectively.
Figure 42 shows that both the Cleavase~'~"'' BNenzyme and Tag polymerase
generate a
characteristic set of cleavage bands for each substrate allowing the
differentiation of the wild-
type and 422 mutant substrates. The two enzyme produce similar but distinct
arrays of bands
for each template.
These results show that the 5' nuclease of both the CleavaseT"'' BN enzyme and
Taq
polymerase are useful for practicing the cleavage reaction of the invention.
Cleavage with
Taq polymerase would find application when substrates are generated using the
PCR and no
intervening purification step is employed other than the removal of excess
nucleotides using
alkaline phosphatase
EXAMPLE 16
Multiplex Cleavage Reactions
The above Examples show that the cleavage reaction can be used to generate a
characteristic set of cleavage products from single-stranded DNA and RNA
substrates. The
ability of the cleavage reaction to utilize double-stranded DNA templates was
examined. For
many applications, it would be ideal to run the cleavage reaction directly
upon a double-
stranded PCR product without the need to isolate a single-stranded substrate
from the initial
PCR. Additionally it would be advantageous to be able to analyze multiple
substrates in the
same reaction tube ("multiplex" reactions).
1J8 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
Cleavage reactions were performed using a double-stranded template which was
carried
a 5' biotin label on the sense-strand and a 5' fluorescein label on the anti-
sense strand. The
double-stranded substrate was denatured prior to cleavage. 7.'he double-
stranded substrate was
cleaved using Taq polymerase. Taq polymerase was used in this experiment
because it has a
w 5 weaker duplex-dependent 5' to 3' exonuclease activity than does the enzyme
CleavaseT"' BN
and thus Taq polymerase does not remove the 5' end label from the re-natured
DNA duplexes
as efficiently as does the enzyme Cleavase~'~"'' BN; therefore less signal is
lost in the reaction.
The substrate utilized was a 157 by fragment derived from either the wild-type
(SEQ
ID N0:34), 419 mutant (SEQ ID N0:41) or 422 mutant (SEQ ID N0:42) of the
tyrosinase
gene. The wild-type fragment was generated as described in Example 8a, the 419
mutant
fragment was generated as described in Example 8a and the 422 mutant fragment
was
generated as described in Example 9 using PCR. The sense strand primer (SEQ ID
N0:29)
contains a 5' biotin label and the anti-sense primer (SEQ ID N0:30) contains a
5' fluorescein
label resulting in the generation of a double-stranded PCR product label on
each strand with a
I S different label. The PCR products were gel purified as described in
Example 8a.
The cleavage reactions were performed as follows. Reaction tubes were
assembled
with approximately 100 fmoles of the double-stranded DNA substrates in 5 ~.l
of 10 mM
MOPS, pH 8.2, 1 mM MnCI,. The solutions were overlaid with a drop of mineral
oil. The
tubes were heated to 95°C for 30 sec and 1 unit of Taq polymerase
(Promega) was added.
Uncut controls contained 33 fmoles of double-stranded DNA substrates in 5 pl
of 10 mM
MOPS, pH 8.2, 1 mM MnCl2. The reactions were cooled to 65°C and
incubated at this
temperature for 15 minutes. The reactions were stopped by the addition of 8
~,l of stop
buffer. The samples were heated to 72°C for 2 min and 5 yl of reaction
were resolved by
electrophoresis through a 10% polyacrylamide gel ( 19:1 cross-link), with 7 M
urea in a buffer
containing O.SX TBE. The entire set of reactions was loaded in duplicate on
the gel such that
duplicate nylon membranes containing the full set of reactions were created.
After transfer to
a nylon membrane (performed as described in Example 8a), the membrane was cut
in half:
one half was probed using a streptavidin-alkaline phosphatase conjugate to
visualize the
biotinylated sense-strand products (as described in Example 8a). The other
half of the
membrane was probed with an anti-fluorescein antibody-alkaline phosphatase
conjugate to
visualize the fluorescein-labelled anti-sense strand products (as described in
Example ~). The
blots were visualized using the chemiluminescent procedures described in
Examples 8a and ~
- 139 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
for biotin-labeled or fluorescein-labeled DNA, respectively. The
autoradiographs are shown
side-by side in Figure 43.
In Figure 43, the lane labeled "Ml" contains biotinylated molecular weight
markers
prepared as described in Example 8a. The lane labeled "M2" contains molecular
weight '
markers generated by digestion of pUCl9 with MspI, followed by Klenow
treatment to fill-in
the ends. The blunt ends were then labeled with fluoresceinated
dideoxynucleotides
(Boehringer Mannheim) using terminal transferase (Promega). Lanes Ml and 1-6
were
developed using the protocol for biotinylated DNA. Lanes 7-12 and M2 were
developed
using the protocol for fluorescein-labeled DNA. Note that in all lanes both
strands of the
substrate are present; only one strand is visualized in a given development
protocol.
In Figure 43, lanes 1-3 and 7-9 contain the "no enzyme" or uncut controls
using the
wild-type, 419 or 422 mutant substrates, respectively. Lanes 4-6 and 10-12
contain cleavage
products from the wild-type, 419 or 422 mutant substrates, respectively.
Unique patterns of
cleavage products are seen for each strand of each of the three substrates
examined. Thus, a
single reaction allowed the -generation of a unique fingerprint from either
the sense or anti-
sense strand of each of the three tyrosinase fragments tested.
The results shown in Figure 43 demonstrate that a cleavage pattern can be
generated
from a double-stranded DNA fragment by denaturing the fragment before
performing the
cleavage reaction. Note that in Figure 43 the cleavage patterns were generated
in the course
of a single round of heating to denature and cooling to cleave and that much
of the substrate
remains in an uncut form. This reaction would be amenable to performing
multiple cycles of
denaturation and cleavage in a thermocycler. Such cycling conditions would
increase the
signal intensity seen for the cleavage products. Substrates generated by the
PCR performed in
the standard PCR buffer (50 mM KCI, 10 mM Tris-Cl, pH 8.3, 1.5 mM MgCI,, 0.01
gelatin) can be treated to remove remaining dNTPs (e.g., addition of alkaline
phosphatase)
and to provide Mn'+. Under these conditions the cleavase reaction can be
performed on both
strands of one or more products generated in that PCR. Such a protocol reduces
sample
preparation to a minimum resulting in a savings of both time and expense.
The above example also demonstrates that two distinct substrates can be
analyzed in a
single reaction thereby allowing the "multiplexing" of the cleavage reaction.
- 140 -
CA 02203627 1997-04-24
WO 96/15267 PG"T/US95/14673
EXAMPLE 17
Optimization Of Manganese Ion Concentration
For Cleavage Of Double Stranded DNA Substrates
As discussed above, it may be desirable to run the cleavage reaction on double-
stranded DNA substrates such restriction fragments or segments generated by
balanced or
symmetric PCR. The effect of varying the concentration of Mn'-+ in cleavage
reactions using
double-stranded DNA substrates was investigated. The results shown below
demonstrate that
the optimal concentration of Mn'-+ is lower when a double-stranded DNA
substrate is
employed in the cleavage reaction as compared to single-stranded DNA
substrates.
Two double-stranded (ds) DNA substrates, 157 by in length, derived from the
tyrosinase mutants 419 (SEQ ID N0:27) and 422 (SEQ ID 1'J0:71 ) were prepared
by PCR
amplification of the exon 4 region of human tyrosinase gene as described above
in Example
16. The sense strand of the 419 and 422 tyrosinase mutant substrates contained
a biotin-
1 ~ labeled at the 5' end following the PCR. The PCR products were gel
purified as described in
Example 8a.
The cleavage reactions were performed as follows. Reaction tubes were
assembled
with approximately 40 fmoles of the ds DNA substrates in 10 ~,l of water. The
tubes were
brought to 95°C for 10 seconds in a PTC-100TT' Programmable Thermal
Controller (M,T
Research, Inc.) to denature the DNA. The cleavage reactions were started by
the addition of
10 ~l of 2X CFLPT"' buffer (pH 8.2) containing 1 p.l of the enzyme
Cleavase~'~"' BN (25 ng in
1 X dilution buffer) and different concentrations of MnCh such that the final
concentration of
MnCh in reaction mixture (20 ~,1 final volume) was either 0,.5 mM, 0.2~ mM,
0.15 mM, 0.1
mM, 0.05 mM and 0 mM. After mixing, the samples were immediately cooled to
65°C and
incubated at this temperature for 5 minutes. The reactions were terminated by
placing the
samples on ice and adding 10 ~1 of stop buffer. The samplers were heated to
85°C for 30 sec
and 10 p.l of each reaction were resolved by electrophoresis through a 10%
polyacrylamide
gel ( 19:1 cross-link), with 7 M urea in a buffer containing O.SX TBE.
After electrophoresis, the gel plates were separated and overlaid with a nylon
membrane as described in Example 8a. The DNA was transferred to the membrane
and the
membrane was dried, washed in 1X Sequenase Images Blocking Buffer (USB),
treated with
1 X SAAP buffer and reacted with CDP-Star'"'' (Tropix) and exposed to X-ray
film as
described in Example 8a. The resulting autoradiograph is shown in Figure 44.
-141-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
In Figure 44, the lane marked "M" contains molecular weight markers prepared
as
described in Example 8. Lanes 1-6 contain the cleavage products generated by
cleavage of
the 419 mutant and lanes 7-12 contain the cleavage products generated by
cleavage ~of the 422
mutant. The reaction products generated by cleavage of the ds DNA substrates
in 10 mM '
MOPS, pH 8.2 containing 0.5 mM (lanes 1,7); 0.25 mM (lanes 2,8); 0.15 mM
(lanes 3,9); 0.1
mM (lanes 4,10); 0.05 mM (lanes S,11 ) and 0 mM MnCh (lanes 6,12) are shown.
The results shown in Figure 44 show no cleavage is seen in the absence of
divalent
cations as is also the case for cleavage of ss DNA substrates [see Example 11
(a) and Figure
31 ]. Optimal cleavage (i. e., production of the most distinct pattern of
cleavage fragments) of
ds DNA substrates was seen in the presence of 0.25 mM MnCh. This optimum is
considerably lower than that obtained using ss DNA substrates [Example 11 and
Figure 31
show that cleavage of ss DNA substrates was optimal in 1 mM MnCh.]. Figure 44
shows
that the efficiency of cleavage of ds DNA substrates decreases as the
concentration of MnCh
is lowered; this effect is likely due to the lower efficiency of the enzyme in
decreasing
concentrations of MnCh.
Figure 44 shows that the cleavage pattern for dsDNA substrates apparently
disappears
when high concentrations of MnCh (0.5 mM, lanes 1 and 7) are-employed in the
cleavage
reaction. This result is in contrast to the results obtained when cleavage
reactions are
performed on single-stranded DNA (ssDNA) substrates. Example 11 (a) showed
that efficient
cleavage of ss DNA substrates were obtained in 1 mM MnCI, and little change in
the
cleavage pattern was seen when the Mn'+ concentration varied from 1 to 4 mM.
The loss of the signal seen when ds DNA substrates are cleaved in buffers
containing
high concentrations of MnCh may be explained as follows. The presence of high
concentrations of divalent ions promotes the reannealling of the DNA strands
of the ds
substrate during the course of the cleavage reaction. The enzyme CleavaseT"'
BN can nibble
ds DNA substrates from the 5' end (i.e., the enzyme removes short DNA
fragments from the
5' end in an exonucleolytic manner; see Example 5). This nibbling results in
the apparent
removal of the label from the substrate DNA (as the DNA contains a 5' end
label). Very
short DNA fragments which contain the 5' end label are not visualized as they
run out of the
gel or are not efficiently transferred to the membrane.
- 142 -
CA 02203627 1997-04-24
WO 96115267 PC"T/US95/14673
EXAMPLE 18
Detection of Cleavage Patterns Can Be Automated
The ability to detect the characteristic genetic fingerprint of a nucleic acid
substrate
generated by the cleavage reaction using fluorescently labelled substrates in
conjunction with
automated DNA sequencing instrumentation would facilitate the use of the
CFLPT"' method in
both clinical and research applications. This example demonstrates that
differently labelled
isolates (two different dyes) can be cleaved in a single reaction tube and can
be detected and
analyzed using automated DNA sequencing instrumentation.
Double-stranded DNA substrates, which contained either the N-hydroxy
succinimidyl
ester JOE-NHS (JOE) or FAM-NHS (FAM) on the sense-strand, were generated using
the
PCR and primers labelled with fluorescent dyes. The anti-sense strand
contained a biotin
label. The substrates utilized in this experiment were the 157 by fragments
from the wild-
type (SEQ ID N0:27) and 422 mutant (SEQ ID N0:71 ) of exon 4 of the tyrosinase
gene.
The wild-type and 422 mutant tyrosinase gene substrates were amplified from
cDNA
plasmid clones containing either the wild-type [pcTYR-N 1 Tyr, Bouchard, B.,
et al. ( 1989) J.
Exp. Med. 169:2029] or the 422 mutant [pcTYR-A422, Giebel, L.B., et al. ( 1991
) 87:1119]
forms of the tyrosinase gene. Each double-stranded substrate was amplified and
the 5' ends
labelled with either a biotin moiety or a fluorescent dye by using the
following primer pairs in
the PCR. The anti-sense primer of SEQ ID N0:30 containing a 5'-biotin moiety
was
obtained from Integrated DNA Technologies, Inc. (IDT, Coralville, IA). The
biotinylated
anti-sense primer was used to prime the synthesis of both the wild-type and
422 mutant
substrates. The sense primer of SEQ ID N0:29 labelled with JOE was used to
prime
synthesis of the wild-type tyrosinase gene. The sense primer of SEQ ID N0:29
labelled with
FAM was used to prime synthesis of the 422 mutant tyrosinase gene. The JOE and
FAM-
labelled primers were obtained from Genset (Paris, France).
The PCR reactions were carried out as follows. One to two nanograms of plasmid
DNA from the wild-type or 422 mutant were used as, the target DNA in a 100 p.l
reaction
containing 50 q.M of each dNTP, and 1 ~.M of each primer in a given primer
pair, in 1 X PCR
buffer. Tubes containing the above mixture were overlaid with 70 ~1 Chill Out
14TM liquid
wax (MJ Research, Watertown, MA). The tubes were heated to 95°C for 1
min and then
cooled to 70°C. Taq DNA polymerase (Perkin-Elmer) was then added as 2.5
units of enzyme
in 5 q,l of 1X PCR buffer. The tubes were heated to 95°C for 45 sec,
cooled to 50°C for 45
-143-
CA 02203627 1997-04-24
WO 96115267 PC"T/US95/14673
sec, heated to 72°C for 1 min and 15 sec for 35 repetitions. Following
the last repetition, the
tubes were incubated at 72°C for 5 min.
The PCR products were gel purified as follows. The products were resolved by
electrophoresis through a 6% polyacrylamide gel (29:1 cross-link) in a buffer
containing O.SX
TBE. The DNA was visualized by ethidium bromide staining and the 157 by
fragments were ,
excised from the gel. The DNA was eluted from the gel slices by passive
diffusion overnight
into a solution containing 0.5 M NH40Ac, 0.1 % SDS and 0.1 M EDTA. The DNA was
then
precipitated with ethanol in the presence of 4 p.g of glycogen carrier. The
DNA was pelleted
and resuspended in 30 p.l of H,O.
The cleavage reactions were performed as follows. Approximately 100 fmoles of
each
double-stranded DNA substrate (1-3 p.l of each gel purified DNA) in a total
volume of 6 yl in
HBO was placed in a 500 ~,l thin wall microcentrifuge tube (Perkin-Elrner) _
The tube was
heated to 95°C for 10 seconds to denature the substrates and then the
tube was quickly cooled
to 50°C (this step allows the DNA to assume its unique secondary
structure by allowing the
formation of intra-strand hydrogen bonds between complimentary bases). The
cleavage
reaction was started by adding 2 p,l of 50 mM MOPS (pH 7.2), 1 p.l of 1 mM
MnCh and 1
p.l of CleavaseT"' BN (50 ng/p.l). The cleavage reaction was performed in a
thermocycler
(Perkin-Elmer DNA Thermal Cycler 480, Norwalk, CT) programmed to heat to
95°C for 10
seconds and then cooled immediately to 50°C. The reaction was then
incubated at 50°C for 5
minutes and stopped by the addition of 1 pl of 1.0 mM EDTA.
Following the cleavage reaction, the sample was resolved by gel
electrophoresis using
an ABI 373A DNA Sequericer (Foster City, CA). Prior to loading, the sample was
denatured
by adding 5 ~l of a solution containing 95% formamide and 10 mM EDTA and
heating to
90°C for 2 minutes. Fivemicroliters of the sample was resolved by
electrophoresis through a
6% polyacrylamide gel ( 19:1 cross-link), with 6 M urea, in 1 X TBE buffer (89
mM Tris-
Borate, pH 8.3, 2 mM EDTA). The gel was run at 30 watts for 14 hours. Signals
from four
wavelength channels were collected using the Applied Biosystem Data Collection
program on
a Macintosh computer. The raw data was analyzed with the BaseFinder program
[Giddings.
M., et al. (1993) Nucl. Acids Res. 21:4530 which corrects for the fluorescent
spectrum
overlap in the four channel signals and mobility shifts caused by the use of
different dye
labels.
The results are shown in Figure 45. Figure 45 shows two traces representing
the two
channel signals for the wild-type and mutant samples. The wild-type sample.
which was
- 144 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
labeled with JOE dye, is shown by the thin lines. The mutant sample (R422Q).
which was
labeled with FAM dye, is shown by the thick lines. For comparison, a
photograph of a high
resolution polyacrylamide gel ( 10% gel with 19:1 crosslink) containing the
resolved cleavage
- products is shown above the traces (the top lane contains cleavage fragments
produced by
r 5 clevage of the wild-type substrate; the bottom lane contains cleavage
fragments produced by
clevage of the R422Q mutant substrate). The cleavage products shown in the
gel, which
contain biotin labels at the 5' end of the sense strand, were l;enerated,
transferred to a nylon
membrane and visualized as described in Example 8a. Arrows point from selected
bands seen
upon cleavage of the 422 mutant substrate_to the corresponding peaks in the
trace generated
by the automated DNA sequencer (the arrows are labelled 1 through 7 beginning
with the
left-hand side of Figure 45).
Comparison of the two traces shows that differences i.n the cleavage patterns
generated
from the cleavage of the wild-type and 422 mutant substrates in the same
reaction are
detected using automated DNA sequencing instrumentation. For example, cleavage
of the 422
substrate generates a cleavage product of approximately 56 nucleotides which
is not seen
when the wild-type substrate is cleaved. This 56 nucleotide product is seen as
the peak
depicted by arrow 6 in Figure 45. Figure 45 shows that not only are new
cleavage products
generated by cleavage of the mutant substrate, but that the cleavage of
certain structures is
enhanced in the mutant substrate as compared to the wild-type substrate
(compare the
intensity of the peaks corresponding to arrows 2-5 in the wild-type and mutant
traces). In
addition, certain cleavage products are shared between the two substrates and
serve as
reference markers (see arrows l and 7).
The above results show that automated DNA sequencing instrumentation can be
used
to detect the characteristic genetic fingerprint of a nucleic acid substrate
generated by the
cleavage reaction. The results also demonstrate that the cleavage reaction can
be run as a
multiplex reaction. In this experiment both the wild-type and the mutant ds
DNA substrates
were cleaved in the same reaction (i. e., a multiplex reaction) and then were
resolved on the
same electrophoretic run using an automated DNA sequencer.
- 145 -
CA 02203627 1997-04-24
WO 96115267 PCT/US95114673
EXAMPLE 19
Identification of Viral Strains Using the CleavaseT"'' Reaction
The above examples demonstrate that the CleavaseT"'' reaction could be used to
detect
single base changes in fragments of varying size from the human ~3-globin and
tyrosinase
genes. These examples showed the utility of the Cleavase~ reaction for the
detection and
characterization of mutations in the human population. The ability of the
Cleavase~"' reaction
to detect sequence variations characteristic of different strains of a virus
was next examined.
The simian immunodeficiency virus (SIV) infection of monkeys is a widely used
animal model for the transmission of human immunodeficiency virus type-1 (HIV)
in humans.
Biological isolates of SIV contain multiple virus strains. When a monkey is
infected with a
biological isolate of SIV, unique subsets of the virus stock are recovered
from the infected
animals (specific strains are also able to infect tissue culture cells).
Different genotypes of the
virus are isolated from infected animals depending on the route of infection
[Trivedi, P. c~t crl.
Journal of Virology 68:7649 (1994)]. The SIV long terminal repeat (LTR)
contains sequences
which vary between the different viral strains and can be used as a marker for
the
identification of the viral genotype.
In order to develop a rapid method for the identification of viral strains) in
a sample
(c.g., a clinical isolate), the CleavaseTM reaction was used to characterized
SIV genotypes
isolated after infection of cultured cells in vitro or after infection of
rhesus monkeys by either
intravenous or intrarectal inoculation with uncloned biological SIV stocla .
Six clones
generated from viral DNA isolated following in vitro infection of the CEMx174
cell line
(L.CEM/251/12 clone), after intravenous inoculation of monkeys (L100.8-1
clone), after
intrarectal low-dose inoculation of monkeys (L46.16-10 and L46.16-12 clones)
and after
intrarectal high-dose inoculation of monkeys (L19.16-3 and L36.8-3 clones)
were obtained
from C. David Pauza (Wisconsin Primate Research Center, Madison, WI). These
clones were
generated as described by Trivedi, P. et al. Journal of Virology 68:7649
(1994). These
plasmid clones contained viral LTR sequences and were utilized to generate
double-stranded
DNA (ds DNA) substrates for the cleavage reaction.
A) Preparation Of The Substrate DNA
The six SIV plasmids were utilized as templates in PCRs in order to generate
dsDNA
substrates for the cleavage reaction. The primer pair utilized spans the U3-R
boundary in the
- 146 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
SIV LTR and amplifies an approximately 350 by fragment. This portion of the
SIV LTR
contains recognition sequences for transcription factors (including Sp 1 and
NF-kB) as well as
the TATA box for transcription initiation and is polymorphic in different
viral strains
- [Trivedi, P. et al., supra].
The primer pair consisting of SEQ ID NOS:74 and 75 was used to amplify the SIV
LTR clones in the PCR. SEQ ID N0:61 primes synthesis of the (+) strand of the
SIV LTR
and comprises 5'-GGCTGACAAGAAGGAAACTC-3'. SEQ ID N0:62 primes synthesis of
the (-) strand of the SIV LTR and comprises 5'-CCAGGCGGCG GCTAGGAGAGATGGG-
3'. To visualize the cleavage pattern generated by cleavage of the (+) strand
of the LTR, the
PCR was performed using the primer consisting of SEQ ID N0:61 containing a
biotin label at
5' end and unlabeled primer consisting of SEQ ID N0:62. To visualize the
cleavage pattern
generated by clevage of the (-) strand of the viral LTR, the PCR was performed
using the
primer pair consisting of SEQ ID N0:62 containing a biotin label at the 5' end
and unlabeled
primer SEQ ID N0:61.
The PCR reactions were carried out as follows. Ten to twenty nanograms of
plasmid
DNA from each of the above 6 SIV LTR clones was used as the target DNA in
separate 100
~,1 reactions containing 60 pM of each dNTP, 0.2 p.M of each primer in a given
primer pair,
10 mM Tris-Cl, pH 9.0 (at 25°C), 2 mM MgCh, 50 mM K<:1, with 0.1%
Triton X-100.
Tubes containing the above mixture were overlaid with two drops of light
mineral oil and the
tubes were heated to 96°C for 3 min and Taq DNA polymerase (Perkin-
Elmer) was then
added as 2.5 units of enzyme in 0.5 ~.l of 20 mM Tris-HCI, pH 8.0, 100 mM KCI,
0.1 mM
EDTA, 1 mM DTT, 50% glycerol and 0.5% Tween 20 and 0.5% Nonidet P-4~0. The
tubes
were heated to 96°C for 45 seconds, cooled to 60°C for 45
seconds, heated to 72°C for 1
minute for 35 repetitions. Following the PCR, the reaction mixture was
separated from the
mineral oil and 5 p,l of SM NaCI, 4 pl of 10 mg/ml glycogen and 250 ~,l of
100% ethanol
were added to the aqueous PCR samples. After incubation at -20°C for 1
hour, the DNA was
pelleted by centrifugation in a Marathon Micro A centrifuge (Fisher
Scientific) at maximum
speed for 5 minutes and resuspended in 40 q,l of 10 mM Tris-HCI, pH 8.0, 0.1
mM EDTA
TE.
The PCR products were gel purified as follows. The DNA was mixed with 0.5
' volume of loading buffer (95% formamide, ~mM EDTA, 0.02% bromphenol blue,
0.02%
xylene cyanol) and heated to 75°C for 2 minutes. The products were
resolved by
r
electrophoresis through a 6% polyacrylamide denaturing gel. ( 19:1 cross-link)
in a buffer
- 147 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
containing 7M urea, O.SX TBE. The DNA was visualized by ethidium bromide
staining and
the product bands were excised from the gel. The DNA was eluted from the gel
slices by
passive diffusion overnight into a solution containing 0.5 M NH40Ac, 0.1 % SDS
and 0.1 M
EDTA. The DNA was then precipitated with ethanol in the presence of 4 p.g of
tRNA
carrier. The DNA was pelleted and resuspended in 50 ~.l of 0.2 M NaCI, 10 mM
Tris-HCI,
pH8.0, 0.1 mM EDTA. The DNA was precipitated with ethanol and resuspended in
50 E~l of
TE. The final DNA concentration was estimated to be 40 fmole/~1 for each
double-stranded
SIV LTR PCR product.
B) DNA Sequence Analysis Of The SIV LTR PCR Products
The DNA sequence of the six PCR fragments generated in section a) above was
determined using the fmol~'~"'' DNA Sequencing System (Promega) according to
the
manufacturer's instructions. For each set of the sequencing reactions 0.2
pmoles of the PCR
product and 2 pmoles of one of the two 5'-biotinylated PCR primers SEQ ID
NOS:74 and 75
was used (i.e., both strands of the PCR fragments were sequenced). Following
the sequencing
reactions, the sequencing products were resolved by electrophoresis. After
electrophoresis, the
DNA bands were visualized following transfer to a nylon membrane as described
in Example
17 with the following modification. A solution containing 0.2% Blocking
reagent
(Boehringer-Mannheim) and 0.2% SDS in TBS buffer ( 100-mM Tris-HCI, pH7.4; 68
mM
NaCI) was used in place of the 1X Sequenase Images Blocking Buffer (USB).
The sequence of the 351 by fragment derived from the L100.8-1 LTR clone is
listed in
SEQ ID N0:63. The sequence of the 340 by fragment from the L46.16-10 LTR clone
is
listed in SEQ ID N0:64. The sequence of the 340 by fragment derived from the
L46.16-12
LTR clone is listed in SEQ ID N0:65. The sequence of the 351 by fragment from
the
L19.16-3 LTR clone is listed in SEQ ID N0:66. The sequence of the 351 by
fragment
derived from the LCEM/251/12 LTR clone is listed in SEQ ID N0:67. The sequence
of the
351 by fragment derived from the L36.8-3 LTR clone is listed in SEQ-ID N0:68.
Analysis of sequenced LTR fragments shows that they have multiple
substitutions and
a deletion relative to the L100.8-1 LTR sequence (SEQ ID N0:63); the L100.8-1
LTR
sequence was chosen as a reference to permit comparisons between the six LTR
clones. For
the ease of discussion, the first or 5'-terminal nucleotide of the (+) strand
of L 100.8-1 LTR
sequence (SEQ ID N0:63) is defined as number 1 and the last or 3'-terminal
nucleotide of
this sequence is defined as number 351.
- 148 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95114673
Figure 46 displays the nucleotide sequence of the six LTR clones. The
reference
clone, L.100.8-1 (SEQ ID N0:63), is shown on the top line. Sequences appearing
in bold
type represent sequence changes relative to the sequence of clone L.100.8-1
(SEQ ID N0:63).
The sequences outlined by the brackets in Figure 46 represent palindromic
sequences which
,; 5 can form a very stable hairpin structure having a stem of 14 base pairs
(12/14 bases in the
stem are complementary) and a loop of 7 nucleotides in the reference clone
L.100.8-1 (SEQ
ID N0:63). This hairpin structure is present in all six LTR clones although
the sequence of
the stem and loop structures varies between the clones.
In comparison with L100.8-1 sequence (SEQ ID N0:63), the L46.16-10 sequence
(SEQ ID N0:64) has seven substitutions and one 11 nucleotide deletion
corresponding to
nucleotides 65-75 of SEQ ID N0:63. The substitutions are: C to T in position
28 (C28T),
C57T, G90A, C97T, G238A, G242A and G313A. The L46.16-12 sequence (SEQ ID
N0:65)
has seven substitutions and one 11 nucleotide deletion corresponding to
nucleotides 65-75 of
SEQ ID N0:63. The substitutions are: C28T, C57T, G90A, C97T, A103G, G242A and
G313A. L 19.16-3 sequence (SEQ ID N0:66) has two substitutions: A94C and
A317T.
LCEM/251/12 sequence (SEQ ID N0:67) has seven substitutions: G26A, G72A, C97T,
G258A, A281C, G313A and C316T. L36.8-3 sequence (SEQ ID N0:68) has six
substitutions: G60A, G172A, G207A, G221A, T256C and C316T.
C) Cleavage Reaction Conditions And CFLPT"° Analysis Of The (-)
Strand Of
The SIV LTR
Double-stranded substrates corresponding to the SIV LTR sequences listed in
SEQ ID
NOS:62-6$ were labelled on the (-) strand using the PCR and the primer pair
corresponding
to SEQ ID NOS:61 and 62. The primer of SEQ ID N0:62 [the (-) strand
primer]contained a
biotin label at the 5' end. The PCR was performed and the reaction products
were isolated as
described in section a).
The cleavage reactions were performed as follows. Reaction tubes were
assembled
with approximately 60 fmoles of the ds DNA substrates in 6 q.l of water. The
following
reagents were added to the DNA: 2 ql of SX CFLPT"' buffer (pH 7.2) containing
150 mM
KCl (to yield a final concentration of 30 mM KCl) and 1 ql of the CleavaseT"'
BN enzyme
(25 ng in 1X dilution buffer). A reaction tube containing the above components
with the
exception that 1 p.l of HBO was added in place of the Cleava.seTM BN enzyme
was prepared
and run as the uncut or no enzyme control. The tubes were brought
to95°C for 10 seconds
in a PTC-100T"'' Programmable Thermal Controller (MJ Rese;arch, Inc.) to
denature the DNA.
- 149 -
CA 02203627 1997-04-24
WO 96/15267 PCTIUS95/14673
Following the denaturation step, the tubes were immediately cooled to
40°C. The cleavage
reaction was immediately started by the addition of 1 ~.l of 2 mM MnCI, (to
achieve a final
concentration of 0.2 mM). The tubes were incubated at 40°C for 5
minutes. The reactions
were terminated by adding 6 ~.1 of stop buffer. The samples were-heated to
85°C for 30 sec
and 5 ~.l of each reaction were resolved by electrophoresis through a 12%
polyacrylamide gel
( 19:1 cross-link), with 7 M urea in a buffer containing O.SX TBE.
After electrophoresis, the gel plates were separated and overlaid with a nylon
membrane as described in Example 8a. The DNA was transferred to the membrane
and the
membrane was dried, washed in 0.2% Blocking reagent (Boehringer Mannheim);
0.2°I° SDS
in 100 mM Tris-HCI, pH 7.43 68 mM NaCI, treated with 1X SAAP buffer and
reacted with
CDP-StarTn'' (Tropix) and exposed to X-ray film as described in Example 8a.
The resulting
autoradiograph is shown in Figure 47.
Figure 47 shows the cleavage patterns which correspond to the cleavage of the
(-)
strand of the double-stranded LTR substrates. In Figure 47, the lane marked
"M" contains
molecular weight markers (prepared as described in Example 8). Lanes 1-6
contain the
cleavage products generated by cleavage of the L100.8-l, L46.16-10, L46.16-12,
L19.16-3,
LCEM/251/12 and L36.8-3 LTR PCR fragments, respectively. Lanes 7-12 contain
the uncut
controls of each of the 6 LTR substrates in the same order as that described
for Lanes 1-6.
The results shown in Figure 47 show that the cleavage or CFLP~'~'~'' pattern
for each
LTR substrate contains multiple bands which range in size from approximately
350
nucleotides (the uncut substrate) to less than 24 nucleotides. The bands
located below about
100 nucleotides in length show differences between the six clones which
reflect nucleotide
changes characteristic of the-different- SIV LTR isolates. Examination of the
CFLPT''~ patterns
revealed that the reaction detected five unique cleavage patterns among the
six SIV LTR
isolates. From the DNA sequence data, it was known that all six LTR clones
were unique.
However, the CFLPT"'' pattern appeared to be identical for the clones shown in
lanes 2 and 3.
The CFLP~ patterns generated by cleavage of the (-) strand from all six
substrates
contain a strong band which corresponds to a fragment of approximately 100
nucleotides in
length. This band corresponds to cleavage of all six LTR substrates at the
long palindromic
sequence located 97 nucleotides from the 5' end of the (-) strand (see the
bracketed region in
Figure 46). This palindromic sequence- forms a very stable hairpin structure
in single-stranded
DNA and provides an optimal substrate for the Cleavase~ BN enzyme. Cleavage of
this
hairpin structure is predicted to generate a fragment of approximately 100
nucleotides.
- 150 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
The LTR substrates, L46.16-10 (SEQ ID N0:64) and. L46.16-12 (SEQ ID N0:65),
shown in lanes 2 and 3 were generated from the same animal using the same
route of
infection [Trivedi, P. et al., supra]. These substrates. have identical
sequences in the region
corresponding to the detectable cleavage sites (i. e., below 100 nucleotides)
with the exception
S of a single base; the L46.16-10 clone (SEQ ID N0:64) contains a G to A
change at position
239 (G239A) relative to the reference sequence listed in SEQ ID N0:63 .
Examination of the
DNA sequence of these two clones reveals that this substitution is located in
the loop region
of a strong hairpin structure (see the palindromic region bracketed in Figure
46). Because the
single base difference between these two sequences is located in the loop
region of the hairpin
structure, it may not change DNA secondary structure of the two substrates
sufficiently to
generate different CFLP~'~"'' patterns under the conditions utilized here. It
may be possible to
detect this single base difference between these two clones by varying the
reaction conditions
in a way that destabilizes the strong hairpin structure.
The results shown in Figure 47 demonstrate that the CFLP-'~"'' reaction can be
used to
detect the majority (5/6 or 83%) of the sequence variations present in the six
SIV LTR clones
studied. In addition, Figure 47 demonstrates that the CFLPTM reaction is a
sensitive means for
probing the secondary structure of single strands of nucleic acids.
D) Cleavage Reaction Conditions And CFLPT"' Analysis Of The (+) Strand Of
The SIV LTR
Double-stranded substrates corresponding to the SIV :LTR sequences listed in
SEQ ID
NOS:76-81 were labelled on the (+) strand using the PCR and the primer pair
corresponding
to SEQ ID NO: 74 and 75. The primer of SEQ ID N0:61 [the (+) strand
primer]contained a
biotin label at the 5' end. The PCR was performed and the reaction products
were isolated as
described in section a). The cleavage reactions, electrophoresis and DNA
visualization were
performed as described above in section c). The resulting autoradiograph is
shown in Figure
48.
Figure 48 shows the resulting pattern corresponding to the cleavage products
of the (+)
strand of six SIV LTR fragments. The lane marked."M" contains molecular weight
markers
(prepared as described in Example 8). Lanes 1-6 contain the cleavage products
generated by
cleavage of the L100.8-1, L46.16-10, L46.16-12, L19.16-3, LCEM/251/12 and
L36.8-3 LTR
PCR fragments, respectively. Lanes 7-12 contain the uncut controls of each of
the 6 LTR
substrates in the same order as that described for Lanes 1-6.
-151-
CA 02203627 1997-04-24
WO 96/15267 PC"TlUS95l14673
As was shown for cleavage of the (-) strand of the LTR clones, the CFLP~'T''
pattern for
each (+) strand of the SIV LTR substrates contains unique features that
characterize the
majority of specific nucleotide substitutions. For example, deletion of 11
nucleotides can be
easiiy detected for L46.16-10 (SEQ ID N0:64) and L46.16-12 (SEQ ID NO:65)
(Figure 48, '
lanes 2 and 3). This deletion removes one of the three SpI binding sites and
is a change ,
characteristic of the genotype of SIV which predominates in animals which are
infected using
low-doses of virus stock via intrarectal inoculation [Trivedi, P. et al.,
supra]. The CFLP-'~"''
pattern generated by cleavage of the (+) strand of the substrates derived from
clones L46.16-
and L46.16-12 again were identical under these reaction conditions.
10 The results shown above demonstrate that the CFLP'~"'' reaction can be used
as a means
to rapidly identify different strains (i. e., genotypes) of virus. The ability
to rapidly identify
the particular strain of virus or other pathogenic organism in a sample is of
clinical
importance. The above results show that the CFLP~ reaction can be used to
provide a fast
method of strain or species identification.
- EXAMPLE 20
The Effects Of Alterations In Salt Conditions In
Cleavage Reactions Using A Single-Stranded DNA Substrate
: _ _ - r
In Example 11 it was shown that the Cleavase~'~"'' reaction is insensitive to
large
changes in reactions conditions when a single-stranded DNA is-employed as the
substrate.
Example 11 showed that the cleavage reaction can be performed using a range of
salt
concentrations (0 to 50 mM KCl) in conjunction with single-stranded
substrates. In this
example, the effect of substituting other salts in place of KCI was examined
in cleavage
reactions using single-stranded DNA substrates.
A) Effect Of Substituting NaCI For KCl In Cleavage Reactions Using A
Single-Stranded Template
To determine the effect of substituting NaCI in place of KCI upon the cleavage
pattern
created by 5' nuclease activity on a single-stranded DNA substrate, the
following experiment
was performed. A single template was incubated in the presence of a fixed
amount of the
Cleavase~'~"' BN enzyme (50 ng) in a buffer containing 10 mM MOPS, pH 8.2, 1mM
MnCh
and various amounts of NaCI.
- 1~2 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
Approximately 100 fmoles of the 157 nucleotide fragment derived from the sense
strand of exon 4 of the tyrosinase gene (SEQ ID NO 47; prepared as described
in Example
8b) were placed in a 500 p.l thin wall microcentrifuge tubes (Perkin Elmer,
Norwalk, CT) in 1
X CFLP-'~"' buffer, pH 8.2 and 1.33 mM MnCI, (to yield a final concentration
of 1 mM
MnCI,) in a volume of 15 p,l. NaCI was added to yield a final concentration of
0, 10, 20, 30,
40, 50, 75 or 100 mM. The final reaction volume was 20 yl.
A tube containing 10 mM MOPS, pH 8.2, 1 mM MnCI, and 100 fmoles substrate
DNA was prepared and served as the no salt, no enzyme control (sterile
distilled water was
substituted for Cleavase~'~"'' BN and all reaction components were added prior
to denaturation at
95°C).
The tubes were heated to 95°C for 20 seconds and then rapidly cooled to
65°C. The
cleavage reaction was started immediately by the addition of 5 p,l of a
diluted enzyme mixture
comprising 1 pl of CleavaseT"'' BN [50 ng/~1 in 1 X dilution buffer (0.5%
NP40, 0.5% Tween
20, 20 mM Tris-HCI, pH 8.0, 50 mM KCI, 10 ~g/ml BSA)J in 10 mM MOPS, pH 8.2,
without MnCI,.
After 5 minutes at 65°C, reactions were stopped by the addition of 16
pl of stop buffer
(95% formamide, 10 mM EDTA, 0.05% bromophenol blue, 0.05% xylene cyanol).
Samples
were heated to 72°C for 2 minutes and 7 ~1 of each reaction were
resolved by electrophoresis
through a 10% polyacrylamide gel (19:1 cross-link), with 7M urea, in a buffer
containing
O.SX TBE, as described in Example 8a.
After electrophoresis, the gel plates were separated and overlaid with a nylon
membrane, as described in Example 8a. The DNA was transferred to the membrane
and the
membrane was dried, blocked in 1 X I-Block (Tropix, Bedford, MA), conjugated
with
streptavidin-alkaline phosphatase (United States Biochemical), washed, reacted
with CDP-
StarT'"' (Tropix, Bedford, MA) as described in Example 8a with the exception
that 0.01 ml
CDP-Star"' was added per cm'- of membrane. The membrane was exposed to X-ray
film as
described in Example 8a. The results are shown in Figure 49.
In Figure 49, the lane marked "M" contains molecular weight markers as
described in
Example 8a. Lane 1 contains the no salt, no enzyme control and shows the
mobility of the
uncleaved template DNA. Lanes 2 through 9 contain reaction products incubated
in the
presence of CleavaseT"' BN enzyme in a buffer containing 0, 10, 20, _ 30, 40,
~ 0 75 or I 00
mM NaCI, respectively.
-153-
CA 02203627 1997-04-24
WO 96/15267 PG"T/US95114673
The results shown in Figure 49 demonstrate that the substitution of NaCI in
place of
KCl has little or no effect upon the cleavage pattern generated using the 157
nucleotide
tyrosinase DNA substrate (SEQ ID N0:34). Essentially the same dependence of
the cleavage
pattern on salt concentration was observed using this single-stranded DNA
substrate when
either KCl (See example 13b, Figure 32) or NaCI (Figure 49) was employed in
the cleavage
reaction.
B) Effect Of Substituting (NIi4)ZS04 For KCl In Cleavage Reactions Using A
Single-Stranded Template _
In an approach similar to that described in above in section a), the effect of
substituting (NH4)~504 in place of KCl upon the-cleavage pattern created by 5'
nuclease
activity on a single-stranded DNA substrate was tested. Cleavage reactions
were set up
exactly as described in section a) with the exception that variable amounts of
(NH4)~SOa were
used in place of the NaCI.
Approximately 100 fmoles of the 157 nucleotide fragment derived from the sense
strand of exon 4 of the tyrosinase gene (SEQ ID NO 47; prepared as described
in Example
8a) were placed in 500 p.l thin wall microcentrifuge tubes (Perkin Elmer.
Norwalk, CT) in 1
X CFLPT"'' buffer, pH 8.2 and 1.33 mM MnCI, (to yield a final-concentration of
1 mM) in a
volume of 15 p,l. (NH4),504 was added to yield a final concentration of 0, 10,
20, 30, 40, 50,
75 or 100 mM. The final reaction volume was 20 pl.
A tube containing 10 mM MOPS, pH 8.2, 1 mM MnCl2 and 100 fmoles substrate
DNA was prepared and served as the no salt, no enzyme control (sterile
distilled water was
substituted for Cleavase~ BN and all reaction components were added prior to
denaturation at
95°C). _
The tubes were heated to 95°C for 20 seconds and then rapidly cooled to
65°C. The
cleavage reaction was started immediately by the addition of 5 pl of a diluted
enzyme mixture
comprising 1 p.l of CleavaseT'~' BN [50 ng/ml in 1 X dilution buffer (0.5%
NP40, 0.5%
Tween 20, 20 mM Tris-HCI, pH 8.0, 50 mM KCI, 10 mg/pl BSA)~ in 10 mM MOPS, pH
8.2, without MnCh.
After 5 minutes at 65°C, reactions were stopped by the addition of 16
pl of stop
buffer. Samples were heated to 72°C for 2 minutes and 7 p.l of each
reaction were resolved
by electrophoresis through a 10% polyacrylamide gel ( 19:1 cross-link), with
7M urea, in a
buffer containing O.SX TBE, as described in,Example 8a.
- 154 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95114673
. After electrophoresis, the DNA was transferred to a membrane and the
detected as
described in section a) above. The resulting autoradiograph is shown in Figure
50.
In Figure 50, the lane marked "M" contains molecular weight markers as
described in
example 10a. Lane 1 contains the no enzyme control and shows the mobility of
the
uncleaved template DNA. Lanes 2 through 9 contain reaction products incubated
in the
presence of Cleavase~'~"'' BN enzyme in a buffer containing 0, 10, 20, 30, 40,
50 75 or 100
mM (NH4)~SO4, respectively.
The results shown in Figure 50 demonstrate that the cleavage reaction is
severely
inhibited by the presence of (NH4)~SO4. The reaction is completely inhibited
by as little as 20
mM (NH4)~504; the extent of the cleavage reaction in 10 mM (NH4)~504 is
comparable to
that obtained in 50 mM KCl or NaCI and is significantly reduced relative that
obtained at 0
mM (NH4),SO4. The pattern of cleavage obtained at 10 mM (NH4)~SO4, however, is
identical
to that observed when the 157 nucleotide template (SEQ ID N0:34) incubated in
the absence
of (NH4)~504 or in KCl or NaCI. This indicates that the choice of salt
included in the
cleavase reaction has no effect on the nature of the sites recognized as
substrates by the
CleavaseT"'' BN enzyme (i.e., the inhibitory effect seen is duc: the effect of
(NH4)~504 upon
enzyme activity not upon the formation of the cleavage structures).
C) Effect of Increasing KCl Concentration on the Cleavage of Single-Stranded
Substrates
The effect of increasing the concentration of KCl in cleavage reactions using
a single-
stranded DNA substrate was examined by performing the cleavage reaction in
concentrations
of KCl which varied from 0 to 100 mM. The cleavage reactions were performed as
described
in section a) with the exception that KCl was added to yield final
concentrations of 0, 2~. 50,
75 or 100 mM and 200 fmoles of the substrate were used in the reaction;
additionally the
substrate DNA was denatured by incubation at 95°C for 5 seconds.
Following the cleavage reaction, the samples were ele:ctrophoresed,
transferred to a
membrane and detected as described in section a) above. The resulting
autoradiograph is
shown in Figure 51.
In Figure 51, the lanes marked "M" contains molecular weight markers as
described in
Example 8a. Lane 1 is the no enzyme control; Lanes 2-7 contain reactions
carried out in the
presence of 0, 25, 50, 75, 100 or 100 mM KCl (the 100 mlVl sample was repeated
twice).
respectively.
-155-
CA 02203627 1997-04-24
WO 96!15267 PCT/US95/14673
The results shown in Figure 51 demonstrate that the extent of cleavage in the
cleavage
reaction decreased as a function of increasing KCl concentration (although
residual cleavage
was detectable at 100 mM KCl). Furthermore, the pattern of fragments generated
by cleavage
of single-stranded substrates with Cleavase~ BN is unaffected by the
concentration of KC1
present in the reactions.
D) Effect Of High KCl Concentrations On Cleavage Reactions Using A Single-
Stranded Substrate
The ability of the Cleavase~ reaction to be carried out at relatively high
concentrations of KCl was tested by performing the cleavage reaction in the
presence of
variable concentrations of KCl in excess of 100 mM. The reactions were
performed using the
157 nucleotide fragment from exon 4 of the tyrosinase gene (SEQ ID N0:34) as
described
above in section c), with the exception that KCl was added to yield a final
concentration of 0;
100, 150, 200. 250 or 300 mM.
Following the cleavage reaction, the samples were electrophoresed, transferred
to a
membrane and detected as described in section a) above. The results (data not
shown)
indicated that the cleavage reaction was severely inhibited by KCl-
concentrations in excess of
100 mM. Some residual cleavage did, however, persist at these elevated salt
concentrations,
up to and including 300 mM KCI.
E) Effect Of KCl Concentration On The Stability Of The Cleavage Pattern
During Extended Incubation Periods
The results presented above demonstrate that the Cleavase~ reaction is
inhibited by
elevated concentrations (i.e., above 50 mM) of either KCl or NaCI. To
determine whether
this iWibition would effectively result in the stabilization of the cleavage
pattern after
extended reaction times (i.e., due to inhibition of enzyme activity),
reactions were examined at
varying extended time points at both 0 and 50 mM KCI.
Approximately 100 fmoles of the 157 nucleotide fragment derived from the sense
strand of exon 4 of the tyrosinase gene (SEQ ID NO 47; prepared as described
in example
l0a) were placed in 200 p,l thin wall microcentrifuge tubes (BioRad, Richmond,
CA) in 1 X
CFLP~'~''' buffer, pH 8.2, 1.33 mM MnCh (to yield a final concentration of I
mM) and KC1 to
yield a final concentration of 0 or 50 mM KCI. The final reaction volume was
20 p.l.
Control reactions which lacked enzyme were set up in parallel for each time
point
examined; these no enzyme controls were prepared as described above with the
exception that
- I56 -
CA 02203627 1997-04-24
WO 96115267 PCT/US95/14673
sterile distilled water was substituted for Cleavase~'~"'' BN and all reaction
components were
added prior to denaturation at 95°C.
The tubes were heated to 95°C for 20 seconds and then rapidly cooled to
65°C. The
cleavage reaction was started immediately by the addition of 5 ~l of a
diluted, enzyme mixture
comprising 1 ~1 of Cleavase~ BN [50 ng/ml in 1 X dilution buffer (0.5% NP40,
0.5%
Tween 20,-20 mM Tris-HCI, pH 8.0, 50 mM KCI, 10 ~g/ml BSA)] in 10 mM MOPS, pH
8.2, without MnCl2. Twenty microliters of Chill Out 14T"'' (MJ Research,
Watertown, MA)
were added to each tube after the addition of the enzyme. 'the reactions were
allowed to
proceed at 65°C for 5 min, 30 min, 1 hour, 2 hours, 4 hours and 17
hours.
At the desired time point, the reactions were stopped by the addition of 16
p.l of stop
buffer. Samples were heated to 72°C for 2 minutes and 7 ~1 of each
reaction were resolved
by electrophoresis through a 10% polyacrylamide gel ( 19:1 cross-link), with
7M urea, in a
buffer containing 0.5X TBE, as described in Example 8a.
After electrophoresis, the DNA was transferred to a membrane and the detected
as
described in section a) above. The resulting autoradiograph is shown in Figure
52.
In Figure 52, the lane marked "M" contains molecular weight markers as
described in
example 10a. Lanes 1-10 contain products from reactions carried out in the
absence of KCI;
lanes 11-20 contain products from reactions carried out in the presence of 50
mM KCI.
Lanes l, 3, 5, 7, and 9 contain no enzyme controls incubated for 5 minutes, 30
minutes, 1
hour, 2 hours, 4 hours and 17 hours, respectively. Lanes 2, 4, 6, 8, ald 10
contain the
reaction products from reactions incubated at 65°C for 5 minutes, 30
minutes, 1 hour, 2 hours,
4 hours and 17 hours, respectively. Lanes 11, 13, 15, 17, and 19 contain no
enzyme controls
incubated in 50 mM KCl for 5 minutes, 30 minutes, 1 hour, 2 hours, 4 hours and
17 hours,
respectively. Lanes 12, 14, 16, 18 and 20 contain reaction products from
CFLP'~"'' reactions
incubated in 50 mM KCl at 65°C for 5 minutes, 30 minutes, 1 hour, 2
hours, 4 hours and 17
hours, respectively.
The results indicated that cleavage was retarded in the presence of 50 mM KCl
which
resulted in a significant stabilization of the cleavage pattern (i.e., the
cleavage pattern
remained the same over time because the rate of cleavage was dramatically
slowed and thus
the larger cleavage fragments are not further cleaved to produce smaller
fragments). Note
that at the extended incubation times, the reactions carried out in the
absence of KCl were
significantly overdigested; after 1 hour at 65°C, essentially no large
fragments remain, and
there is substantial accumulation of small cleavage products. In contrast, the
reactions carried
- 157 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
out at 50 mM KCI were essentially static between 30 minutes and 4 hours;
overdigestion was
only apparent at the longest time point and was not as extensive as that
observed in the
absence of KCI. .
EXAMPLE 21
Comparison Of The Patterns Of Cleavage Generated By
Cleava e-Of Sin le-Stranded And Double-Stranded DNA Substrates
In CleavaseT"'' BN-mediated primer-independent cleavage of double-stranded DNA
substrates, the two strands of DNA are separated in a denaturation step prior
to the addition of
enzyme. Therefore, the patterns generated by cleaving double-stranded
templates should be
identical to those generated by cleaving single-stranded template. This
assumption was
verified by the experiment described below.
The single-stranded substrate comprising the 157 nucleotide fragment derived
from the
sense strand of exon 4 of the tyrosinase gene (SEQ ID N0:34) was prepared as
described in
example lOb with the following modification. After gel purification and
precipitation in the
presence of glycogen carrier, the PCR products were resuspended in TE (lOmM
Tris-CI. pH
8.0, 1 mM EDTA) and then reprecipitated with 2 M NH40Ac and 2.5 volumes of
ethanol.
The DNA was then resuspended in 400 ~l of TE.
Approximately 50 or 100 fmoles of the single-stranded 157 nucleotide fragment
(SEQ
ID NO: 47) were placed in a 200 pl centrifuge tube (BioRad, Richmond, CA) in 1
X CFLP~
buffer, pH 8.2 and 1.33 mM MnClz (final concentration was 1 mM MnCh) in a
volume of 15
~1. The final reaction volume was 20 ~.1. A 20 ~l no salt, no enzyme control
was set up in
parallel; this reaction contained sterile distilled water in place of the
Cleavase~ BN enzyme
and all reaction components were added prior to denaturation at 95°C.
The reaction tubes were heated to 95°C for 5 seconds and then rapidly
cooled to 65°C.
The cleavage reactions were started immediately by the addition of 5 p.l of a
diluted enzyme
mixture comprising 1 ~,l of Cleavase'~ BN [50 ng/~.l in 1 X dilution buffer
(0.5% NP40,
0.5% Tween 20, 20 mM Tris-HCI, pH 8.0, 50 mM KCI, 10 ~.g/ml BSA)] in 10 mM
MOPS,
pH 8.2, without MnCh. After 5 minutes at 65°C, reactions were stopped
by the addition of ,
16 ~.I of stop buffer.
A double stranded form of the 157 nucleotide substrate was cleaved with
CleavaseT"'' -
BN in the same experiment. This double-stranded substrate (SEQ ID N0:27) was
generated
- 158 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
as described in Example 8b with the following modification s. After gel
purification and
precipitation in the presence of glycogen carrier, the PCR products were
resuspended in TE
( 1 OmM Tris-Cl, pH 8.0, 1 mM EDTA) and then reprecipitated with 2 M NH40Ac
and 2.5
' volumes of ethanol. The DNA was then resuspended in 400 ~.1 of TE.
w 5 Approximately 33 or 66 fmoles of the double-stranded 157 by fragment (SEQ
ID
N0:27) were placed in a 200 p,l thin walled microcentrifuge tube (BioRad,
Richmond, CA).
Sterile distilled water was added to a volume of 15 pl.
The reaction tubes were heated to 95°C for 5 seconds and then rapidly
cooled to 65°C.
The cleavage reactions were started immediately by the addition of 5 p.l of a
diluted enzyme
mixture comprising 10 mM MOPS, pH 8.2, 0.8 mIVI MnCh (to yield a final
concentration of
10 mM MOPS, pH 8.2 and 0.2 mM MnCI,) and 0.5 ~.l of CleavaseTM BN [50 ng/p.l
in 1 X
dilution buffer (0.5% NP40, 0.5% Tween 20, 20 mM Tris-HCI, pH 8.0, 50 mM KCI,
10
~,g/ml BSA)]. A 20 pl no salt, no enzyme double-stranded substrate control was
set up in
parallel; this reaction contained sterile distilled water in place of the
CleavaseTM BN enzyme.
After 5 minutes at 65°C, the reactions were stopped by the addition of
16 pl of stop
buffer. The samples were then heated to 72°C for 2 minutes and the
reaction products were
resolved by electrophoresis through a 10% polyacrylamide gel ( 19:1 cross-
link), with 7M
urea, in a buffer containing O.SX TBE, as described in Example 8a.
After electrophoresis, the gel plates were separated and overlaid with a nylon
membrane, as described in example 10a. The DNA was transferred to the membrane
and the
membrane was dried, blocked in 1 X I-Block (Tropix, Bedford, MA), conjugated
with
streptavidin-alkaline phosphatase (United States Biochemical), washed, reacted
with CDP-Star
(Tropix, Bedford, MA), and exposed to X-ray film as described in Example 20a.
The
resulting autoradiograph is shown in Figure 53.
In Figure 53, lanes 1-3 contain reaction products derived from reactions
containing the
single-stranded substrate; lanes 4-7 contain reaction products derived from
reactions
containing the double-stranded substrate. Lanes 1 and 3 contain 7.0 p.l of the
reaction products
derived from the cleavage reactions which contained either 50 or 100 fmoles of
the single-
stranded substrate, respectively. Lane 2 contains 7.0 yl of the uncut single-
stranded substrate
control reaction. Lanes 4 and 6 contain 7.0 ~.l of the uncut double-stranded
control reactions
which contained either 33 or 66 fmoles of the substrate, respectively. Lanes 5
and 7 contain
7.0 p.l of the reaction products derived from cleavage reactions which
contained either 33 or
66 fmoles of the double-stranded substrate, respectively. Note that the uncut
double-stranded
- 159 -
CA 02203627 1997-04-24
WO 96/15267 PG"TILTS95/14673
control shows a doublet underneath the prominent band containing the 157 by
substrate; it is
believed that this doublet represents alternative structures which migrate
with an altered
mobility rather than degradation products. This doublet does not appear in
experiments
performed using double-stranded DNA purified from a denaturing gel (See
Example 22)
Comparison of the cleavage patterns generated by cleavage of either the single-
stranded or double-stranded substrate shows that identical patterns are
generated.
EXAMPLE 22
The Cleavase"~'' Reaction Using A Double Stranded DNA
Template Is Sensitive to Large Changes In Reaction Conditions
The results presented in Example 11 demonstrated that the Cleavase~l reaction
is
relatively insensitive to significant changes in numerous reaction conditions
including, the
concentration of MnCh and KCI, temperature, the incubation period, the amount
of
CleavaseT"' BN enzyme added and DNA preparation. The results shown in Example
11
demonstrated that when the Cleavase~ reaction is performed using a single-
stranded
substrate, the reaction is remarkably robust to large changes in conditions.
The experiments
shown below show that the cleavage of double-stranded substrates is restricted
to a somewhat
narrower range of reaction conditions.
A) Generation Of The Double-Stranded 157 by Fragment Of Exon 4 Of The
Tyrosinase Gene
The following experiments examine the effect of changes in reaction conditions
when
double-stranded DNA templates are used in the Cleavase~'~"'' reaction. The
double-stranded
substrate utilized was the157 by fragment of the wild type tyrosinase gene
(SEQ ID N0:27).
This 157 by fragment was generated using symmetric PCR as described in Example
8b.
Briefly, approximately 75 fmoles of double-stranded substrate DNA were
incubated with ~0
pmoles of the primer 5' biotin-GCCTTATTTTACTTTAAAAAT-3' (SEQ ID N0:32), 50
pmoles of the primer 5' fluorescein-TAAAGTTTTGTGTTATCTCA-3' (SEQ ID N0:33).
and
50 mM of each dNTP in 1X PCR buffer. Tubes containing 95 p.l of the above
mixture were
heated to 95°G for 5 seconds and cooled to 70°C. Five
microliters of enzyme mix containin;~
1.25 units of Taq DNA polymerase in 1X PCR buffer were then added. Each tube
was
overlaid with 50 ~l of Chill Out 14~ (MJ Research, Watertown, MA).
- 160 -
CA 02203627 1997-04-24
WO 96115267 PCT/US95/14673
The tubes were heated to 95°C for 4~ seconds, cooled to 50°C for
45 seconds, heated
to 72°C for 75 seconds for 30 repetitions with a ~ minute incubation at
72°C after the last
repetition. The reactions were then ethanol precipitated to rf:duce the volume
to be gel
purified. NaCI was added to a final concentration of 400 mM and glycogen (in
distilled
water) was added to a final concentration of 200 p.g/ml. Two and one-half
volumes of 100%
ethanol were added to each tube, and the tubes were chilled to -70°C
for two and one-half
hours. The DNA was pelleted and resuspended in one-fifth the original volume
of sterile
distilled water.
The PCR products were gel purified as follows. An equal volume of stop buffer
was
added to each tube and the tubes were heated to 72°C for 2 minutes. The
products were
resolved by electrophoresis through a 6 % denaturing polyacrylamide gel ( I
9:1 cross-link) and
7 M urea in a buffer containing 45 mM Tris-Borate, pH 8.3 and 1.4 mM EDTA. The
DNA
was visualized by ethidium bromide staining and the 157 by fragment was
excised from the
gel. The DNA was eluted from the gel slice by passive diffusion as described
in Example 8a
with the exception that diffusion was allowed to occur over 'two days at room
temperature.
The DNA was then precipitated with ethanol in the presence of 200 mM NaCI and
no added
carrier molecules. The DNA was pelleted and resuspended in 150 pl TE.
S) Effect Of KCl Concentration On The Double-Stranded Cleavage Reaction
To determine the effect of salt concentration upon thf: cleavage reaction when
a
double-stranded substrate was utilized, a single substrate was incubated in
the presence of a
fixed amount of the enzyme Cleavase'~"' BN (25 ng) in a buffer containing 10
mM MOPS, pH
7.5, 0.2 mM MnCI, and varying concentrations of KCl from 0 to 100 mM.
Approximately 100 fmoles of the 157 by fragment derived from the exon 4 of the
tyrosinase gene (SEQ ID N0:27; prepared as described above in section a) were
placed in 200
q.l thin wall microcentrifuge tubes (BioRad, Richmond, CA) in sterile
distilled water in a
volume of 6.25 ~l (the final reaction volume was 10 pl). The tubes were heated
to 95°C for
15 seconds and then rapidly cooled to 45°C. The cleavage reactions were
started by the
addition of 3.75 p.l of an enzyme mix containing 2.7 X CFLP~ buffer, pH 7.5
(to yield a
final concentration of 1 X), 0.531 mM MnCh (to yield a final concentration of
0.2 mM), 0.5
yl CleavaseT"'' BN [50 ng/p,l in 1 X dilution buffer (0.5% NP40, 0.5% Tween
20, 20 mM
Tris-HCI, pH 8.0, 50 mM KCI, 10 p,g/ml BSA)], and KCl to yield a final
concentration of 0,
2.5, 5. 10, 15, 20, 25, 30, 50 or 100 mM. The final reaction volume was 10 ~1.
The enzyme
solution was brought to room temperature before addition to the cleavage
reaction. No
-161-
CA 02203627 1997-04-24
WO 96/15267 PCTIUS95/14673
enzyme (i. e., uncut) controls were set up in parallel at either 0 or 100 mM
KCI, with the
difference that sterile distilled water was substituted for the CleavaseTM BN.
After 5 minutes at 45°C, the reactions were stopped by the addition of
8 ~.l of stop
buffer. Samples were heated to 72°C for 2 minutes and 4 ~.I of each
reaction were resolved "
by electrophoresis through a 10% polyacrylamide gel ( 19:1 cross-link), with 7
M urea, in a
buffer containing O.SX TBE.
After electrophoresis, the gel plates were separated and overlaid with a nylon
membrane, as described in Example 8b. The DNA was transferred to the membrane
and the
membrane was dried. washed in 1X I-Block Blocking Buffer, washed and exposed
to X-ray
film as described in Example 20a, except that the distilled water washes were
omitted. The
resulting autoradiograph is shown in Figure 54.
In Figure 54, the lane marked "M" contains molecular weight markers. Lane 1
contains the uncut control in 0 mM KCl and shows the mobility of the uncleaved
template
DNA. Lanes 2 through 11 contain reaction products generated by incubation of
the substrate
in the presence of CleavaseT"'' BN enzyme in a buffer containing 0, 2.5, 5,
10, 15, 20, 25, 30,
50, or 100 mM KCI, respectively. Lane 12 contains the uncut control incubated
in a buffer
containing 100 mM KCI.
The results shown in Figure 54 demonstrate that the Cleavase-"~' reaction
carried out on
double-stranded DNA template was sensitive to variations in salt
concentration. Essentially
no cleavage was detected in reactions containing- greater than 30 mM KCI. The
same
cleavage pattern was obtained when the 157 by tyrosinase DNA substrate (SEQ ID
N0:27)
was incubated with the CleavaseT"'' BN enzyme regardless of whether the
concentration of
KCl was varied from 0 to 30 mM.
C) Effect Of NaCI On The Double-Stranded Cleavage Reaction
The effect of substituting NaCI in place of KCI upon the cleavage pattern
created by 5'
nuclease activity on a double-stranded DNA substrate was examined.
Approximately 100
fmoles of the 157 by fragment derived from exon 4 of the tyrosinase gene (SEQ
ID NO 40;
prepared as described in Example 22a) were placed .in 200 ~.l thin wall
microcentrifuge tubes
(BioRad, Richmond, CA) in sterile distilled water in a volume of 6.25 ~1 and
were heated to
95°C for 15 seconds. The tubes were cooled to 45°C. The cleavage-
reactionyvas started by
the addition of 3.75 ~.l of an enzyme mix containing 2.7 X CFLPT"' buffer, pH
7.5 (to yield a '
final concentration of 1 X), 0.53 mM MnCI, (to yield a final concentration of
0.2 mM). 0.5
~.1 Cleavase'~"'' BN [50 ng/~I in 1 X dilution buffer (0.5% NP40, 0.5% Tween
20. 20 mM '
- 162 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
Tris-HCI, pH 8.0, 50 mM KCI, 10 ~.g/ml BSA)], and NaCI to yield a final
concentration of 0,
2.5, 5, 10, 15, 20, 25, 30, 50 or 100 mM. The final reaction volume was 10 ~1.
No enzyme
(i. e., uncut) controls were set up in parallel at either.0 or 100 mM NaCI,
with the difference
that sterile distilled water was substituted for the CleavaseT"' BN.
The reactions were incubated at 45°C for 5 minutes and were stopped by
the addition
of 8 ~l of stop buffer. Samples were heated to 72°C for 2 minutes and 4
~1 of each reaction
were resolved by electrophoresis through a 10% polyacrylamide gel ( 19:1 cross-
link), with 7
M urea, in a buffer containing O.SX TBE.
After electrophoresis, the gel plates were separated and overlaid with a nylon
membrane, as described in Example 8b. The DNA was transferred to the membrane
and the
membrane was dried, washed in 1X I-Block Blocking Buffer, washed and exposed
to X-ray
film as described in Example 20a with the exception that the distilled water -
washes were
omitted. The resulting autoradiograph is shown in Figure 55.
In Figure 55 , the lane marked "M" contains molecular weight markers. Lane 1
contains the no enzyme control incubated in a buffer containing 0 mM NaCI and
shows the
mobility of the uncleaved template DNA. Lanes 2 through 11 contain reaction
products
generated by cleavage of the 157 by substrate (SEQ ID N0:27) with the
CleavaseTM BN
enzyme in a buffer containing 0, 2.5, 5, 10, 15, 20, 25, 30, 50, or 100 mM
NaCI,
respectively. Lane 12 contains the no enzyme control incubated in a buffer
containing 100
mM NaCI.
The results shown in Figure 55 demonstrate that the CleavaseT"'' reaction
carried out on
a double-stranded DNA template was sensitive to variations in NaCI
concentration.
Essentially no cleavage was detected above 20 mM NaCI. The same cleavage
pattern was
obtained when the 157 by tyrosinase DNA template (SEQ ID N0:27) was incubated
with the
CleavaseTM BN enzyme regardless of whether the NaCI concentration was varied
from 0 to 20
mM.
D) Effect Of Substituting (NH4)ZSO4 For KCI In Cleavage Of Double-Stranded
Template
In an approach similar to that described in Example 20b, the ability of
(NH4),SO~, to
substitute for KCl in the cleavage reaction when double-stranded substrates
were utilized was
tested. Cleavage reactions were set up exactly as described in Examples 22b
and c with the
exception that variable amounts of (NH4)~504 were substituted for the KCl or
NaCI.
-163-
CA 02203627 1997-04-24
WO 96115267 PCT/US95/14673
Approximately 100 fmoles of the 157 by fragment derived exon 4 of the
tyrosinase
gene (SEQ ID NO 40; prepared as described above in section a) were placed in
200 ~l thin
wall microcentrifuge tubes (BioRad, Richmond, CA) , in sterile distilled water
in a volume of
6.25 p.l and were heated to 95°C for 15 seconds. The tubes were cooled
to 45°C.
Cleavage reactions were started by the addition of 3.75 p.l of an enzyme mix ,
containing 2.7 X CFLPT"'' buffer, pH 7.5 (to yield a final concentration of 1
X), 0.53 mM
MnCI, (to yield a final concentration of 0.2 mM MnCI,), 0.5 ~,l CleavaseT"" BN
[50 ng/p,l in
1 X dilution buffer (0.5% NP40, 0.5% Tween 20, 20 mM Tris-HCI, pH 8.0, 50 mM
KCI, 10
Elg/ml BSA)], and (NH4)~SO4 to yield a final concentration of 0, 2.5, 5, 10,
15, 20, 25, 30, 50
or 100 mM. The final reaction volume was 10 p.l. No enzyme (i. e., uncut)
controls were set
up in parallel at either 0 or 100 mM (NH4).,SO4, with the difference that
sterile distilled water
was substituted for the Cleavase'M BN.
The reactions were incubated at 45°C for 5 minutes and were stopped by
the addition
of 8 yl of stop buffer. Samples were heated to 72°C for 2 minutes and 4
~l of each reaction
1 S were resolved by electrophoresis through a 10% polyacrylamide gel ( 19:1
cross-link), with 7
M urea, in a buffer containing O.SX TBE.
After electrophoresis, the gel plates were separated and overlaid with a nylon
membrane, as described in Example 8b. The DNA was transferred to the membrane
and the
membrane was dried, washed in 1X I-Block Blocking Buffer, washed and exposed
to X-ray
film as described in Example 20a, except that the distilled water washes were
omitted. The
resulting autoradiograph is shown in Figure 56.
In Figure 56, the lane marked "M" contains molecular weight markers. Lane 1
contains the no enzyme control incubated in a buffer containing 0 mM
(NH~,)~S04 and shows
the migration of the uncleaved substrate DNA. Lanes 2 through 11 contain
reaction products
generated by incubation of the substrate in the presence of Cleavase~'~"' BN
enzyme in a buffer
containing 0, 2.5, 5, 10, 15, 20, 25, 30, 50, or 100 mM (NH4)~504,
respectively. Lane 12
contains the no enzyme control incubated in a buffer containing 100 mM
(NH4),SO~.
The results shown in Figure 56 demonstrate that the Cleavase'~''' reaction was
severely
inhibited by the presence of (NH4)~SO~. The reaction was completely inhibited
by as little as
15 mM -(NH4),504; the extent of the cleavage reaction in 5 mM (NH4),SO~ v~ras
comparable to
that ~ obtained in 20 mM KCl and was significantly reduced relative to that
obtained in 0 mM
(NH~),S04. The pattern of cleavage- obtained using 5 mM (NHQ),504, however,
was identical
to that observed when the 157 by substrate was incubated in the absence of
(NH~),SO~ or in
- 164 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
KCl or NaCI, indicating that the choice of salt included in the CleavaseT"'
reaction has no
effect on the nature of the sites recognized by the enzyme.
E) Time Course Of The Double-Stranded Cleavage Reaction
. To determine how quickly the double-stranded cleavage reaction is completed,
a single
substrate was incubated in the presence of a fixed amount of CleavaseTM BN
enzyme for
various lengths of time. Approximately 100 fmoles of the double-stranded 157
by fragment
of exon 4 of the tyrosinase gene (SEQ ID NO 40; prepared as described above in
Example
22a) were placed in sterile distilled water in 200 ~.l thin walled centrifuge
tubes (BioRad,
Richmond, CA) in a volume of 6.25 p.l. The tubes were heated to 95°C
for 15 seconds, as
described in section b), and cooled to 45°C.
Cleavage reactions were started by the addition of 3.'75 ~.1 of an enzyme mix
containing 2.7 X CFLPTM buffer, pH 7.5 (to yield a final concentration of 1
X), 0.53 mM
MnCI, (to yield a final concentration of 0.2 mM MnCI,), 0.5~,l CleavaseT"' BN
[50 ng/yl in
1 X dilution buffer (0.5% NP40, 0.5% Tween 20, 20 mM Tris-HCI, pH 8.0, 50 mM
KC1. 10
~.g/ml BSA)]. The final reaction volume was 10 ~l. No enzyme (i.e., uncut]
controls were
set up in parallel and stopped after either 5 minutes or 120 minutes, with the
difference that
sterile distilled water was substituted for the CleavaseT"' BN enzyme.
The cleavage reactions were stopped by the addition of 8 ~,l of stop buffer at
the
following times: 5 seconds, l, 2, 5, 10, 15, 20, 30, 60 or 120 minutes.
Samples were heated
to 72°C for 2 minutes and 4 p.l of each reaction were resolved by
electrophoresis through a
10% polyacrylamide gel (19:1 cross-link), with 7 M urea, in. a buffer
containing O.SX TBE.
After electrophoresis, the gel plates were separated and overlaid with a nylon
membrane, as described in Example 8b. The DNA was transferred to the membrane
and the
membrane was dried, washed in 1X I-Block Blocking Buffer, washed and exposed
to X-ray
film as described in Example 20a with the exception that the distilled water
washes were
omitted. The resulting autoradiograph is shown in Figure 57.
In Figure 57, lane 1 contains the no enzyme control after a ~ minute
incubation at
45°C and shows the mobility of the uncleaved template DNA. Lanes 2-10
contain cleavage
fragments derived from reactions incubated in the presence of the CleavaseT"'
BN enzyme for
305 sec, l, 2, 5, 10, 15, 20, 30, 60 (i hr), or 120 minutes (2 hr),
respectively. Lane 11 contains
the no enzyme control after a 120 minute incubation at 45°C.
Figure 57 shows that the cleavage of a double-stranded DNA template mediated
by the
CleavaseT"'' BN enzyme was rapid. A full cleavage pattern was apparent and
essentially
-165-
CA 02203627 1997-04-24
WO 96115267 PCT/US95114673
complete within one minute. Unlike the example of cleavage of a single-
stranded DNA
template (Example 11 c), very little cleavage is detectable after 5 seconds.
This reaction
contained one-tenth the amount of enzyme used in the reaction described in
Example 11 c. In
addition, whereas incubation of single-stranded cleavage reactions for
extended periods "
generated a pattern of increasingly truncated fragments (Example 20e),
extended incubation of ,
the double-stranded cleavage reaction resulted in a complete loss of signal
(Figure 57, lane
10); this result is probably due to nibbling by the enzyme of the 5' biotin
moiety from the
reannealed strands. It is important to note that these results show that the
same pattern of
cleavage was produced for cleavage of double-stranded DNA, as for single-
stranded, whether
the reaction is run for 1 or 30 minutes. That is, the full representation of
the cleavage
products (i.e., bands) is seen over a 30-fold difference in time of
incubation; thus the double-
stranded CFLP~ reaction need not be strictly controlled in terms of incubation
time.
The results shown in Figure 58 contain short time courses of cleavage
reactions
performed at a variety of enzyme concentrations. Approximately 100 fmoles of
the double
stranded 157 by fragment of exon 4 of the tyrosinase gene (SEQ ID N0:27) were
placed in
sterile distilled water in 200 p.l thin walled centrifuge tubes (BioRad,
Richmond, CA) in a
volume of 6.25 p.l. The tubes were heated to 95°C for 15 seconds, as
described in Example
22b, and cooled to 45°C. Cleavage reactions were started by the
addition of 3.75 yl of an
enzyme mix containing 2.7X CFLP~ buffer, pH 7.5 (to yield a final
concentration of 1 X),
0.53 mM MnCI, (to yield a final concentration of 0:2 mM MnCh), 0.5 pl
CleavaseT"'' BN [at
either 50, 100, 200, or S00-rig/p.l in 1 X dilution buffer (0.5% NP40, 0.5%
Tween 20, 20 mM
Tris-HCI, pH 8Ø 50 mM KCh 10 p.g/ml BSA) to yield a final amount of enzyme
of 25, 50,
100, or 250 ng]. The final- reaction volume was 10 p,l. A no enzyme control
was set up in
parallel, with the difference that sterile distilled water was substituted for
the CleavaseT"' BN
enzyme, and stopped after 1 minute at 45°C.
The cleavage reactions were stopped by the addition of 8 ~1 of stop buffer
after either
5 seconds or 1 minute. Samples were heated to 72°C for 2 minutes and 4
~l of each reaction
were resolved by electrophoresis through a 10% polyacrylamide gel (19:1 cross-
link), with 7
M urea, in a buffer containing O.SX TBE.
After electrophoresis, the gel plates were separated and overlaid with a nylon
membrane, as described in Example 8b. The DNA was transferred to the membrane
and the '
membrane was dried, washed in 1 X I-Block Blocking Buffer, washed and exposed
to X-ray
- 166 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
film as described in Example 20a, except that the distilled water washes were
omitted. The
resulting autoradiograph is shown in Figure 58.
In Figure 58, lane "M" contains molecular weight markers as described in
Example 8a.
Lane 1 contains the no enzyme control. Lanes 2 and 3 each contain reaction
products
S generated by incubation of the substrate in the presence of 2 5 ng of the
CleavaseT"' BN
enzyme; the reaction in lane 2 was stopped after 5 seconds, that in lane 3,
after 1 minute.
Lanes 4 and 5 contain reaction products generated by cleavage of the substrate
in the presence
of 50 ng of the CleavaseT"'' BN enzyme; the reaction in lane 4 was stopped
after 5 seconds,
that in lane 5, after 1 minute. Lanes 6 and 7 contain reactian products
generated by cleavage
of the substrate in the presence of 100 ng of Cleavase~'~"'' BN enzyme; the
reaction in lane 6
was stopped after 5 seconds, that in lane 7, after 1 minute. The reactions
shown in lanes 8
and 9 each contain 250 ng of the Cleavase~'~'' BN enzyme; that in lane 8 was
stopped after 5
seconds, that in lane 9, after 1 minute.
The results presented in Figure 58 indicate that the rate of cleavage of
double-stranded
DNA increased with increasing enzyme concentration. Note that as the
concentration of
enzyme was increased, there was a corresponding reduction in the amount of
uncut DNA that
remained after 1 minute. As was demonstrated below, in Figure 60, the
concentration of
enzyme included in the cleavage reaction had no effect on the cleavage pattern
generated.
Comparison of the 250 ng reaction (shown in Figure 58, lanes 8 and 9) to the
short time point
digestion described in Example 11 c, indicates that the amount of enzyme
rather than the
double-stranded or single-stranded nature of the substrate controls the extent
of cleavage in
the very early time points.
F) Temperature Titration Of The Double-Stranded Cleavage Reaction
To determine the effect of temperature variation on the cleavage pattern, the
157 by
fragment of exon 4 of the tyrosinase gene (SEQ ID N0:27) was incubated in the
presence of
a fixed amount of CleavaseT"'' BN enzyme for 5 minutes at various
temperatures.
Approximately 100 fmoles of substrate DNA (prepared as described in Example
22a) were
placed in sterile distilled water in 200 p.l thin walled centrifuge tubes
(BioRad, Richmond.
CA) in a volume of 6.25 p.l. The tubes were heated to 95°C for 15
seconds and cooled to
either 37, 40, 45, 50, 55, 60, 65, 60, 75, or 80°C. _ Cleavage
reactions were started by the
addition of 3.75 p.l of an enzyme mix containing 2.7 X CFLPT"' buffer, pH 7.5
(to yield a
final concentration of 1 X), 0.53 mM IVInCh (to yield a final concentration of
0.2 mM
MnCI,), 0.5 p.l Cleavase~ BN [50 ng/p.l in 1 X dilution buffer (0.5% NP40,
0.5% Tween 20,
- 167 -
CA 02203627 1997-04-24
WO 96/15267 PG"T/US95/14673
20 mM Tris-HCI, pH 8.0, 50 mM KCI, 10 ~.g/ml BSA)]. The enzyme mix was kept on
ice
throughout the duration of the experiment, but individual aliquots of the
enzyme mix were
allowed to come to room temperature before being added to the reactions. A
second reaction
was run at 37°C at the end of the experiment to control for any loss of
enzyme activity that
may have occurred during the course of the experiment. No enzyme controls were
set up in a
parallel and incubated at either 37°C or 80°C, with the
difference that sterile distilled water
was substituted for the Cleavase'~'' BN. The reactions were stopped by the
addition of 8 ~1 of
stop buffer.
Samples were heated to 72°C for 2 minutes and 5 pl of each reaction
were resolved by
I 0 electrophoresis through a 10% polyacrylamide gel ( 19:1 cross-link), with
7 M urea, in a
buffer containing O.SX TBE.
After electrophoresis, the gel plates were separated and overlaid with a nylon
membrane as described in Example 8b. The DNA was transferred to the membrane
and the
membrane was dried, washed in 1X I-Block Blocking Buffer, washed and exposed
to X-ray
film as described in Example 20a, except that the distilled water washes were
omitted. The
resulting autoradiograph is shown in Figure 59.
In Figure 59, the lane marked "M" contains molecular weight markers, prepared
as
described in Example 8a. Lane 1 contains the no enzyme control after a 5
minute incubation
at 37°C. Lanes 2 and 3 contain reactions incubated at 37°C, run
at the beginning and end of
the experiment, respectively. Lanes 4-13 contain reactions incubated at 40,
45, 50, 55, 60,
65, 70, 75, or 80°C [there are two 80°C samples; the first was
not covered with Chill Out
14T"'' (MJ Research, Watertown, MA), the second was overlaid with 20 ~,l Chill
Out 14~'''
after the addition of the enzyme mix], respectively. Lane 14 contains a no
enzyme control
incubated at 80°C for 5 minutes.
Figure 59 shows that cleavage of double-stranded DNA substrates proceeded most
effectively at lower temperatures. The distribution of signal and pattern of
cleavage changed
smoothly in response to the temperature of incubation over the range of
37°C to 60°C. Some
cleavage products were evident only upon incubation at higher temperatures,
whereas others
were far more predominant at lower temperatures. Presumably, certain
structures that are
substrates for the CleavaseT"'' BN enzyme at one end of the temperature range
are not favored ,
at the other. As expected, the production of cleavage fragments became
progressively less '
abundant in the high end of the temperature range as the cleavage structures
were melted out.
Above 70°C, the cleavage products were restricted to small fragments,
presumably due to
-168-
CA 02203627 1997-04-24
WO 96115267 PCT/US95/14673
extensive denaturation of the substrate. When longer DNAs (350 to 1000
nucleotides) are
used, it has been found that useful patterns of cleavage are l;enerated up to
75°C.
These results show that the cleavage reaction can be performed over a fairly
.wide
range of temperatures using a double-stranded DNA substrate. As in the case of
the single-
r 5 stranded cleavage reaction, the ability to cleave double-stranded DNA over
a range of
temperatures is important. Strong secondary structures that may dominate the
cleavage
pattern are not likely to be destabilized by single-base changes and may
therefore interfere
with mutation detection. Elevated temperatures can then be used to bring these
persistent
structures to the brink of instability, so that the effects of small changes
in sequence are
maximized and revealed as alterations in the cleavage pattern. This also
demonstrates that
within the useful temperature range, small changes in the reaction temperature
due to heating
block drift or similar device variations will not cause radical changes in the
cleavage pattern.
g) Titration Of The CleavaseT"' BN Enzyme In Double-Stranded Cleavage
Reactions
The effect of varying the concentration of the CleavaseTM BN enzyme in the
double-
stranded cleavage reaction was examined. Approximately 100 fmoles of the 157
by fragment
of exon 4 of the tyrosinase gene (SEQ ID N0:27; prepared as described in
Example 22a)
were placed in sterile distilled water in 200 p.l thin walled centrifuge tubes
(BioRad,
Richmond, CA) in a total volume of 6.25 q.l. These tubes were heated to
95°C for 15
seconds and then rapidly cooled to 45°C.
Cleavage reactions were started immediately by the addition of 3.75 ~1 of a
diluted
enzyme mix containing 2.7 X CFLPT"'' buffer, pH 7.5 (to yield a final
concentration of 1 X),
0.53 mM MnCI, (to yield a final concentration of 0.2 mM MnCh), 0.5 pl
CleavaseT"' BN [2,
10, 20, 50, 100, 200, 500 ng/~l in 1 X dilution buffer (0.5% NP40, 0.5% Tween
20, 20 mM
Tris-HCI, pH 8Ø 50 mM KCI, 10 ~,g/ml BSA) such that 1, 5, 10, 25, 50, 100 or
250 ng of
enzyme was added to the reactions]. No enzyme controls were set up in
parallel, with the
difference that 1X dilution buffer was substituted for the CleavaseT"' BN.
After 5 minutes at
45 ° C, the reactions were stopped by the addition of 8 q.l of stop
buffer. The samples were
heated to 72°C for 2 minutes and 4 ~l of each reaction were resolved by
electrophoresis
through a 10% polyacrylamide gel (19:1 cross-link), with 7 M urea, in a buffer
containing
0.5X TBE.
After electrophoresis, the gel plates were separated and overlaid with a nylon
membrane as described in Example 8b. The DNA was tran sferred to the membrane
and the
- 169 -
CA 02203627 1997-04-24
WO 96/15267 PCTIUS95/14673
membrane was dried, washed in 1X I-Block Blocking Buffer, washed and exposed
to X-ray
film as described in Example 20a, except that the distilled water washes were
omitted. The
resulting autoradiograph is shown in Figure 60.
The lane marked "M" in Figure 60 contains molecular weight markers. Lane 1
contains the no enzyme control and shows the migration of the uncut substrate.
Lanes 2-8
contain cleavage products derived from reactions containing 1, 5, 10, 25, 50,
100 or 250 ng of
the Cleavase~'' BN enzyme, respectively.
These results show that the same cleavage pattern was obtained using the 157
by
tyrosinase DNA substrate (SEQ ID N0:27) regardless of whether the amount of
enzyme used
in the reaction varied over a 50-fold range. Thus, the double-stranded
cleavage reaction is
ideally suited for practice in clinical laboratories where reaction conditions
are not as
controlled as in research laboratories. Note, however, that there is a
distinct optimum for
cleavage at intermediate enzyme concentrations for a double-stranded template,
in marked
contrast to what was observed on single-stranded substrates (Example 11 e).
The progressive
loss of signal in the double-stranded reactions at increasing concentrations
of CleavaseTM BN
is likely due to the nibbling of the 5' biotin label off the end of the
reannealed double-
stranded template. -
E~MPLE 23
Determination Of The pH Optimum For Single
Stranded And Double-Stranded Cleavage Reactions
In order to establish optimal pH conditions for the two types of primer-
independent
cleavage reactions (i. e., single-stranded and double-stranded cleavage
reactions), the
CleavaseT"'' reaction buffer was prepared at a range of different pHs.
A) Establishment Of A pH Optimum For The Single-Stranded Cleavage
Reaction
The effect of varying the pH of the CleavaseT"'' reaction (i.e., CFLPTM)
buffer upon the
cleavage of single-stranded substrates was examined. Several 10 X buffer
solutions were
made with 0.5 M MOPS at pH 6.3, 7.2, 7.5, 7.8, 8.0 and 8.2 by titrating a 1 M
solution of
MOPS at pH 6.3 with 6 N NaOH. The volume was then adjusted to yield a 0.5 M
solution at
each pH.
- 170 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
Approximately 100 fmoles of a single-stranded substrate prepared from the
sense
strand of exon 4 of the tyrosinase gene (SEQ ID N0:34; prepared as described
in Example
8a), were placed in 200 ~l thin walled centrifuge tubes (BioRad, Richmond, CA)
in 15 pl of
1 X CFLPT"' buffer, at varying pH, and 1.33 mM MnCh (to yield a final
concentration of 1
mM). The final reaction volume was 20 ~.1. The reaction mixes were heated to
95°C for 5
seconds and rapidly cooled to 65°C. The reactions were started by the
addition of 5 ~,1 of
diluted enzyme mix containing 1 pl of Cleavase"''' BN [50 ng/~.l in 1 X
dilution buffer (0.5%
NP40, 0.5% Tween 20, 20 mM Tris-HCI, pH 8.0, 50 mM KCI, 10 p.g/ml BSA)] in 1 X
CFLPT"' buffer (without MnCh), again at the appropriate pH. A 20 p.l no salt,
no enzyme
control was set up in parallel and incubated at 65°C for each of the
indicated pHs, with the
difference that sterile distilled water was substituted for CleavaseT"'' BN
and all reaction
components were added prior to denaturation. Reactions were stopped by the
addition of 16
p.l of stop buffer after 5 minutes.
Samples were heated to 72°C for 2 minutes and 7 ~,l of each reaction
were resolved by
electrophoresis through a 10% polyacrylamide gel (19:1 cross-link), with 7M
urea, in a buffer
containing O.SX TBE, as described in Example 8a.
After electrophoresis, the gel plates were separated and overlaid with a nylon
membrane, as described in Example 8a. The DNA was transferred to the membrane
and the
membrane was dried, blocked in 1 X I-Block (Tropix, Bedford, MA), conjugated
with
streptavidin-alkaline phosphatase (United States Biochemical), washed, reacted
with CDP-
StarT"'' (Tropix, Bedford, MA), and exposed to X-ray film as described in
Example 20a,
except that the distilled water washes were omitted. The results are presented
in Figure 61.
In Figure 61, panels A and B contain reactions which used single-stranded DNA
substrates. In panel A, 5 pairs of reactions are presented. In each case, the
first lane of the
pair is the no enzyme control and the second is the single-stranded cleavage
reaction. Lanes 1
and 2 depict reaction products obtained using a reaction buffer at pH 6.3;
lanes 3 and 4, at pH
7.2; lanes 5 and 6, pH 7.8; lanes 7 and 8, pH 8.0; lanes 9 and 10, at pH 8.2.
Panel B
contains the results of a separate experiment comparing cleavage reactions
performed using a
reaction buffer at pH 7.5 (lanes 1 and 2, uncut and cut, respectively) and at
pH 7.8 (lanes 3
and 4, uncut and cut, respectively).
°
The results shown in Figure 61, panels A and B, indicate that the cleavage of
the
single-stranded DNA template was sensitive to relatively small changes in pH.
There v~~as a
pH optimum for the reaction between pH 7.5 and 8Ø Because the pK~ of MOPS is
7.2. the
-171-
CA 02203627 1997-04-24
WO 96/15267 PC"T/US95/14673
pH closest to that value which supported cleavage, pH 7.5, was determined to
be optimal for
the single-stranded cleavage-reaction.
B) Establishment Of A pH Optimum For The Double-Stranded Cleavage
Reaction
The effect of varying the pH of the CleavaseT"'' reaction (i. e., CFLPT"'')
buffer upon the ,
cleavage of double-stranded substrates was examined. Several 10 X buffer
solutions were
made with 0.5 M MOPS at pH 7.2, 7.5, 7.8, and 8.0, as described above in
section a).
Approximately 100 fmoles of the double-stranded 157 by fragment of exon 4 of
the tyrosinase
gene (SEQ ID N0:27; prepared as described in Example 8) were placed in 200 pl
thin walled
centrifuge tubes (BioRad, Richmond, CA) in a total volume of 6.25 p.l. The
tubes were
heated to 95°C for 15 seconds and cooled to 45°C. The clevage
reactions were started by the
addition of 3.75 ~I of diluted enzyme mix containing 2.7 X CFLP~'~"'' buffer,
pH 7.5 (to yield
a final concentration of 1 X), 0.53 mM MnCh (to yield a final concentration of
0.2 mM
MnCI,), 0.5 ~1 of CleavaseT"'' BN [50 ng/p.l in 1 X dilution buffer (0.5%
NP40, 0.5% Tween
20, 20 mM Tris-HCI, pH 8.0, 50 mM KCI, 10 p.g/ml BSA)].
The cleavage reactions were incubated for 5 minutes and then were terminated
by the
addition of 8 p,l of stop buffer.
Samples were heated to 72°C for 2 minutes and 4 p.l of each reaction
were resolved by
electrophoresis through a 10% polyacrylamide gel (19:1 cross-link), with 7M
urea, in a buffer
containing O.SX TBE.
After electrophoresis, the gel plates were separated and overlaid with a nylon
membrane, as described in Example 8b. The DNA was transferred to the membrane
and the
membrane was dried, washed in 1X I-Block Blocking Buffer, washed and exposed
to X-ray
film as described in Example 20a, except that the distilled water washes were
omitted. The
resulting autoradiographs are shown in Figure 62, panels A and B.
In Figure 62, panel A, lanes 1 and 2 contain cleavage products from reactions
run in a
buffer at pH 8.2 (lane 1 contains the cleavage reaction; lane 2 is the uncut
control). Lanes 3
and 4 contain cleavage products from reactions run in a buffer at pH 7.2 (lane
3 contains the
cleavage reaction; lane 4 is the uncut control). In panel B, lanes 1 and 2
contain cleavage
products from reactions run in a buffer at pH 7.5 (lane 1 is the uncut
control; lane 2 contains
the cleavage reaction). Lanes 3 and 4 contain cleavage products from reactions
run in a
buffer at pH 7.8 (lane 3 contains the uncut control; lane 4 contains the
cleavage reaction).
-172_
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
. The results in Figure 62, panels A and B, demonstrate that the cleavage of
double-
stranded DNA was not sensitive to changes in pH over the range of buffer
conditions tested.
Because the cleavage of single-stranded DNA, however, was sensitive to changes
in~ pH, the
buffer conditions that were determined to be optimal for the single-stranded
cleavage reaction
a 5 were chosen for subsequent double-stranded cleavage experiments.
EXAMPLE 24
The Presence Of Competitor DNA Does Not Alter The Cleavage Pattern
The effect of the presence of competitor (i. e., non-labelled substrate) DNA
upon the
cleavage reaction was examined. The cleavage reaction was run using the 157
nucleotide
fragment from the sense strand of the human tyrosinase gent: (SEQ ID N0:34)
and human
genomic DNA. The results shown below demonstrate that the presence of non-
substrate DNA
has no effect on the CFLPT"'' pattern obtained in the cleavage reaction.
A) Preparation Of The Substrate DNA And The Cleavage Reactions
The 157 nucleotide single-stranded wild type tyrosinase substrate (SEQ ID
N0:34)
containing a biotin label on the 5' end was prepared as described in Example
9. Human
genomic DNA (Promega) present at 235 p.g/ml in Tris-HCI, pH 8.0; I mM EDTA was
ethanol precipitated and resuspended in Tris-HCI, pH 8.0; 0.1 mM EDTA to final
concentration 400 ~g/ml. This DNA was used as a competitor in standard CFLPTM
single-
stranded reactions (described in Example 9). Tyrosinase DNA substrate (SEQ ID
N0:34) and
human genomic DNA were mixed in H,O in final volume of 6 p,l. Samples were
heated at
95°C for 10 seconds to denature the DNA, cooled to the target
temperature of 65°C, and
mixture of 2 ~,I SX CFLPT"' buffer, pH 7.5, 1 ~1 10 mM MnCh and 1 p,l (2~ ng)
the enzyme
Cleavase~'~"'' BN in dilution buffer was added. After 5 minutes at
65°C, 6 ~.1 of stop buffer
was added to terminate reaction and 5 pl of each sample was separated on a 10%
denaturing
polyacrylamide gel. Membrane transfer and DNA visualization were performed as
described
in Example 19.
B) The Presence Of Genomic DNA Does Not Alter The CFLPT"' Pattern
Figure 63 shows the resulting pattern corresponding to the cleavage products
of the
sense strand of the wild type tyrosinase substrate (SEQ ID N0:34) in the
presence of 0 yg/ml
(lane 2), 20 pg/ml (lane 3), 40 p.g/ml (lane 4), 80 ~g/ml (lane 5), 120 ~,g/ml
(lane 6) and 200
-173-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95114673
q.g/ml (lane 7) unlabeled human genomic DNA. Lane 1 shows an uncut control in
the
absence of the enzyme Cleavase~ BN and lane marked "M" contains the molecular
weight
markers prepared as described in Example 8.
Figure 63 shows that the presence of genomic DNA in the cleavage reaction did
not
change either the position or the relative intensity of the product bands
produced. Increasing
the amount of nonspecific DNA in the reaction did, however, decrease the
efficiency of the
cleavage reaction and reduced the overall intensity of the pattern. These
results can be
explained by the binding of the Cleavase~'~"' BN enzyme to the nonspecific DNA
which has the
effect of decreasing the effective enzyme concentration in the reaction. This
effect became
significant when the concentration of genomic DNA in the reaction was equal to
or greater
than 120 ~g/ml jFigure 63 , lanes 6 (120 p.g/ml) and 7 (200 ~.g/ml)]. Under
these
conditions, the genomic DNA was present at more than a 20,000-fold-excess
relative to the
specific substrate DNA; nonetheless the CFLP~ pattern could still be
recognized under these
conditions. The observed stability of the CFLPT"' pattern in the presence of
genomic DNA
1 S ruled out the possibility that nonspecific DNA could significantly change
the structure of the
substrate DNA or alter the interaction of the Cleavase~'~"' BN enzyme with the
substrate.
EXAMPLE 25
The CFLP~ Reaction Can Be Practiced Using A Variety of Enzymes
The above Examples demonstrated the ability of the CleavaseTM BN enzyme, a 5'
nuclease derived from Taq DNA polymerase, to generate a characteristic set of
cleavage
fragments from a nucleic acid substrate. The following experiments demonstrate
that a
number of other enzymes can be used to generate a set of cleavage products
which are -
characteristic of a given nucleic acid. These enzymes are not limited to the
class of enzymes
characterized as 5' nucleases.
A) Cleavage Patterns Generated by Other DNA Polymerases From The Genus
Tl:ermus
To determine whether ~' nuclease activity associated with DNA polymerases
(DNAPs) '
other than Tad DNAP could generate a distinct cleavage pattern from_ nucleic
acid substrates,
DNAPs from two species-of Thermus were examined. The DNAP of Thermus,flavzr.s
["Tfl",
- 174 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95t14673
Kaledin et al., Biokhimiya 46:1576 (1981); obtained from Promega Corp.,
Madison, WI] and
the DNAP of Thermus thermophilus ["Tth", Carballeira et al., Biotechniques
9:276 (1990);
Myers et al., Biochem. 30:7661 (1991); obtained from U.S. Biochemicals,
Cleveland, OH]
were examined for their ability to generate suitable cleavage patterns (i.e.,
patterns which can
. 5 be used to characterize a given nucleic acid substrate).
The ability of these other enzymes to cleave nucleic acids in a structure-
specific
manner was tested using the single-stranded 157 nucleotide fragment of the
sense strand of
exon 4 of the tyrosinase gene (SEQ ID N0:34) under conditions reported to be
optimal for
the synthesis of DNA by each enzyme.
Approximately 100 fmoles of the 157 nucleotide fragment derived from the sense
strand of exon 4 of the tyrosinase gene (SEQ ID NO 47; prepared as described
in example
l0a) were placed in 200 p,l thin wall microcentrifuge tubes (BioRad, Richmond,
CA) in 1 X
CFLPT"' buffer, pH 8.2 and 1.33 mM MnCh (to yield a final concentration of 1
mM) and KCl
to yield a final concentration of either 0 or 50 mM. Final reaction volumes
were 20 ~.1.
Samples were heated to 95°C for 5 seconds and then cooled to
65°C. A 20 p.l no salt, no
enzyme control was set up in parallel, with the differences that sterile
distilled water was
substituted for the CleavaseT"'' BN enzyme and all reaction components were
added prior to
denaturation at 95 ° C.
The cleavage reactions were started by the addition of 5 pl of a diluted
enzyme mix
containing either 1.25 units or 5 units of the indicated enzyme (see below) in
1 X CFLPTM
buffer, pH 8.2. After 5 minutes, reactions were stopped by the addition of 16
p,l of stop
buffer.
Samples were heated to 72°C for 2 minutes and 7 ~l (in the case of the
samples
digested with Tfl) or 5 p.l (in the case of the samples digested with Tth)
were electrophoresed
through a 10% polyacrylamide gel (19:1 cross-link), with 71VI urea, in a
buffer containing
O.SX TBE, as described in Example 8a.
After electrophoresis, the gel plates were separated and overlaid with a nylon
membrane, as described in Example 8a. The DNA was transferred to the membrane
and the
membrane was dried, blocked in 1 X I-Block (Tropix, Bedford, MA), conjugated
with
streptavidin-alkaline phosphatase (United States Biochemical, Cleveland, OH),
washed. reacted
with CDP-StarTT'1 (Tropix, Bedford, MA), and exposed to X-ray film as
described in Example
20a, except that the distilled water washes were omitted. The results are
presented in Figures
64 and 65.
-17~-
CA 02203627 1997-04-24
WO 96/15267 PG"T/US95/14673
In Figure 64, lane 1 contains the no enzyme control and indicates the
migration of the
uncut DNA. Lanes 2-5 contain cleavage products derived from reactions
incubated with Tfl
DNAP: The reactions represented in lane 2 and 3 each contained 5 units of Tfl
DNAP; the
sample in lane 2 was incubated in a reaction buffer containing 0 mM KCI, while
the sample
in lane 3 was incubated in a reaction buffer containing 50 mM KCI. The
reactions in lanes 4
and 5 each contained 1.25 units of T,fl DNAP; the sample in lane 4 was
incubated in a
reaction buffer containing 0 mM KCI; that in lane 5 was incubated in a
reaction buffer
containing 50 mM KCI.
In Figure 65, lanes 1 and 2 each contain cleavage products derived from
reactions
incubated with 1.25 units of Tth DNAP. The sample in lane 1 was incubated in a
reaction
buffer containing 0 mM KCI; that in lane 2 was incubated in a reaction buffer
containing 50
mM KCI. Lanes 3 and 4 contain cleavage products derived from reactions
incubated with S
units of Ttla DNAP. The sample shown in lane 3 was incubated in a reaction
buffer
containing 0 mM KCI; that in lane 4 was incubated in a reaction buffer
containing 50 mM
KCI.
Figures 64 and 65 demonstrates that both Tth DNAP and Tfl DNAP display
structure
specific endonuclease activity similar in nature to that seen in the
Cleavase~"' BN enzyme. A
comparison of the results shown in Figures 64 and 65 showed that the Tth DNAP
was more
efficient at generating a cleavage pattern under the reaction conditions
tested. Comparison of
the cleavage patterns generated by Tth DNAP with those generated by the
Cleavase'~'~' BN
enzyme the indicates that essentially the same structures are recognized by
these two enzymes
[compare Figure 66, lane 2 (Cleavase"~'' BN) with Figure 65 (Tth DNAP)].
B) Enzymes Characterized As 3' Nucleases Can be Used To Generate Distinct
Cleavage Patterns
To determine whether enzymes possessing 3' nucleolytic activity could also
generate a
distinct cleavage pattern, enzymes other than DNAPs (which possess 5' nuclease
activity)
were tested in the cleavage reaction. Exonuclease III from Escher-ichia coli
(E. coli Exo III)
was tested in a cleavage reaction using the 157 nucleotide fragment prepared
from the sense
strand of exon 4 of the tyrosinase gen (SEQ ID N0:34). As a comparison, a
reaction
containing this substrate (SEQ ID N0:34) and the CleavaseT"' BN enzyme was
also prepared.
Approximately 100 fmoles of the 157 nucleotide fragment prepared from the
sense
strand of exon 4 of the tyrosinase gene (SEQ ID N0:34; prepared as described
in Example
8a) were placed in 200 p.l thin wall microcentrifuge tubes (BioRad, Richmond,
CA) in 1 X
- 176 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
CFLP~'~'' buffer, pH 8.2 and 1.33 mM MnCl2 (to yield a final concentration of
1 mM) and KCl
to yield a final concentration of either 0 or 50 mM in a volume of 15 ul.
Final reaction
volumes were 20 ~.1.
The samples were heated to 95°C for 5 seconds and then rapidly cooled
to 37°C. A 20
p.l no salt, no enzyme control was set up in parallel, with the differences
that sterile distilled
water was substituted for the Cleavase'~"'' BN enzyme and all reaction
components were added
prior to denaturation at 95°C.
A reaction tube containing 100 fmoles of the 157 nucleotide fragment (SEQ ID
N0:34) and 50 ng of the Cleavase~'~"'' BN enzyme in a buffer containing 0 mM
KCl was
prepared and treated as described in Example 21 (i.e., denatured by incubation
at 95°C for 5
seconds followed by cooling to 65°C and the addition of the; enzyme and
incubation at 65°C
for 5 minutes).
The cleavage reactions were started by the addition of 5 ~l of a diluted
enzyme mix
containing either 1.25 units or 200 units of Exo III (United States
Biochemical. Cleveland,
OH) in 1 X CFLP~'~"'' buffer, pH 8.2 (without MnCl2) were added to the 15 ~l
reactions, and
the reactions were incubated for 5 minutes. After 5 minutes at 37°C,
the reactions were
stopped by the addition of 16 ~,l of stop buffer.
The samples were heated to 72°C for 2 minutes and 5 ~.I were
electrophoresed through
a 10% polyacrylamide gel (19:1 cross-link), with 7M urea, in a buffer
containing 0.5X TBE,
as described in Example 8a.
After electrophoresis, the gel plates were separated and overlaid with a nylon
membrane, as described in example 10a. The DNA was transferred to the membrane
and the
membrane was dried, blocked in 1 X I-Block (Tropix, Bedford, MA), conjugated
with
streptavidin-alkaline phosphatase (United States Biochemical, Cleveland, OH),
washed, reacted
with CDP-Star'"'' (Tropix, Bedford, MA), and exposed to X-ray film as
described in Example
20a, except that the distilled water washes were omitted. The results are
presented in Figure
Lane 1 in Figure 66 contains the no enzyme control and indicates the mobility
of the
uncut DNA. Lane 2 contains cleavage fragments generated by incubation of the
substrate
with the CleavaseT"'' BN enzyme and provides a comparison of the patterns
generated by the
two different enzymes. Lanes 3-6 contain cleavage fragments generated by
incubation of the
substrate with Exo III. Lanes 3 and 4 each contain reaction products generated
in reactions
which contained 200 units of Exo III; the reaction in lane 3 was run in a
buffer containing 0
- 177 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
mM KCI, that in lane 4 was run in a buffer containing 50 mM KCI. Lanes 5 and 6
each
contain reaction products generated in reactions which contained 1-.25 units
of Exo III: the
reaction in lane 5 was run in a buffer containing 0 mM KCI, that in lane 6 was
run in a
buffer containing 50 mM KCI.
The results presented in Figure 66 demonstrate that Exo III generated a
distinct
cleavage pattern when incubated with a single-stranded DNA substrate. The
pattern generated
by Exo III was entirely distinct from that generated by the CleavaseT"'' BN
enzyme. The
results shown in Figure 66 also show that significant differences in the
cleavage pattern
generated by Exo III were observed depending on the concentrations of both the
enzyme and
KCl included in the reactions. -
C) Ability Of Alternative Enzymes To Identify Single Base Changes
In sections and a) and b) above it was shown that enzymes other than the
Cleavase~'~"'
BN enzyme could generate a distinct pattern of cleavage fragments when
incubated in the
presence of a nucleic acid substrate. Because both Tth DNAP and E. coli Exo
III generated
distinct cleavage patterns on single-stranded DNA, the ability of these
enzymes to detect
single base changes present in DNA substrates of the same size was examined.
As in
Example 9, the human tyrosinase gene was chosen as a model system because
numerous
single point mutations have been identified in exon 4 of this gene.
Three single-stranded substrate DNAs were prepared; all three substrates
contained a
biotin label at their 5' end. The wild type substrate comprises the 157
nucleotide fragment
from the sense strand of the human tyrosinase gene (SEQ ID N0:34). Two
mutation-
containing substrates were used. The 419 substrate (SEQ ID N0:41 ) and the 422
substrate
(SEQ ID N0:42), both of which are described in Example 9. Single-stranded DNA
containing a biotin label at the 5' end was generated for each substrate using
asymmetric PCR
as described in Example 8a with the exception that the single-stranded PCR
products were
recovered from the gel rather than the double-stranded products.
Cleavage reactions were performed as follows. Each substrate DNA
(approximately
100 fmoles) was placed in a 200 ~.l thin wall microcentrifuge tubes (BioRad.
Richmond, CA)
in 5 ~1 of 10 mM MOPS, pH 8.2, with 1.33 mM MnCI, (to yield a final
concentration of 1
mM). A no enzyme control was set up with the wild type DNA fragment in
parallel and
incubated at 65°C for each of the indicated time points, with the
differences that sterile
distilled water was substituted for the CleavaseT"'' BN enzyme and all
reaction components
were added prior to denaturation at 95°C. The reaction tubes were
brought to 95°C for ~
- 178 -
CA 02203627 1997-04-24
WO 96115267 PCT/US95/14673
seconds to denature the substrates and then the tubes were ~u~Ckl~ cZSoZed fo
~5°C~or the
reactions containing Tth DNAP and 37°C for the reactions containing Exo
III.
Cleavage reactions were started immediately by the addition of a diluted
enzyme
mixture containing 1.25 units of the enzyme either Tth DNAP or Exo III in 5
p,l of 10 mM
MOPS, pZI 8.2 without MnCh. The enzyme solution was brought to room
temperature before
addition to the cleavage reaction. After 5 minutes at 65°C, the
reactions were stopped by the
addition of 8 p.l of stop buffer. The samples were heated to 72°C for 2
minutes and 7 ~l of
each reaction were resolved by electrophoresis through a 10~% polyacrylamide
gel ( 19:1 cross-
link), with 7 M urea, in a buffer containing O.SX TBE.
After electrophoresis, the gel plates were separated and overlaid with a nylon
membrane, as described in example 10a. The DNA was transferred to the membrane
and the
membrane was dried, blocked in 1 X I-Block (Tropix, Bedford, MA), conjugated
with
streptavidin-alkaline phosphatase (United States Biochemical), washed, reacted
with CDP-
Star'~"' (Tropix, Bedford, MA), and exposed to X-ray film as described in
Example 20a with
the exception that the distilled water washes were omitted. 'The results are
presented in
Figure 67. _
In Figure 67, lanes 1-3 contain cleavage fragments generated by incubation of
either
the wild-type, mutant 419 and mutant 422 alleles of the tyrosinase gene,
respectively, with
Tth DNAP. Lanes 4-6 contain cleavage fragments generated by incubation of
either the wild
type, mutant 419 and mutant 422 substrates, respectively, with Exo III in a
buffer containing
0 mM KCI. Lanes 7-9 contain cleavage fragments generates( by incubation of
either the wild
type, mutant 419 and mutant 422 substrates, respectively, in<;ubated with Exo
III in a buffer
containing SO mM KCI. Lane 10 contains cleavage fragments generated by
incubation of the
wild type DNA substrate with the Cleavase~'~"'' BN enzyme in a buffer
containing 0 mM KCI:
this reaction provides a comparison of the patterns generated by the three
different enzymes
(i. e.. the CleavaseT"' BN enzyme, -Tth DNAP and Exo III). Lane 11 contains
the no enzyme
control with the wild type DNA substrate incubated in the presence of 50 mM
KC1.
The results shown in Figure 67 demonstrate that both Tth DNAP and Exo III were
able to detect single base changes in a single-stranded DNA substrate relative
to a wild-type
DNA substrate. The patterns generated by Tth DNAP were comparable to those
generated by
the CleavaseT~"'' BN enzyme for all three DNA substrates (See Figure 29 for a
comparison of
the pattern generated by the CleavaseT"' BN enzyme).
- 179 -
CA 02203627 1997-04-24
WO 96115267 PCTlUS95114673
The patterns generated by Exo III v~~ere entirely distinct from those
generated by
enzymes derived from the genus Thermus (i.e., the CleavaseT~"' BN enzyme and
Tth DNAP).
Furthermore, the pattern produced by cleavage of the DNA substrates by Exo III
were distinct
depending on which concentration of KCl was employed in the reaction (Figure
67). A -
distinct pattern change was evident for the 419 mutant at both KCI
concentrations. As shown
in Figure 67, at 0 mM KCI, a band appears in_the 40 nucleotide range in the
419 mutant (lane
5); at 50 mM KCI, the 419 mutant contains an additional band in the 70
nucleotide range
(lane 8). Pattern changes were not discernable for the 422 mutant (relative to
the wild-type)
in the Exo III digestions; this difference in the ability of the E. coli Exo
III enzyme to detect
single base changes could relate to the relative positions- of the changes
with respect to
secondary structures that act as substrates for the structure specific
cleavage reaction, and the
position of the label (5' or 3' end) relative to the preferred cleavage site
(5' or 3'), Figure 68.
D) The Drosophila RrpI Enzyme Can Be Used to Generate Cleavage Patterns
Another protein in the Exo III family of DNA repair endonucleases, RrpI from
Drosophila melanogaster (Nugent, M, Huang, S.-M., and Sander, M.
Biochemistyy~, 199 3: 32,
pp. 11445-11452), was tested for its ability to generate a distinct cleavage
pattern on a single-
stranded DNA template. Because its characteristics in the cleavage assay were
unknown, this
enzyme was tested under a variety of buffer conditions. Varying amounts of
this enzyme (1
ng or 30 ng) were incubated with approximately 100 fmoles of the 157
nucleotide fragment
of the sense strand of exon 4 of the tyrosinase gene (SEQ ID NO: 47) in either
1 mM MnC 1
or 5 mM MgC1? and either 1 X CFLPT"'' buffer, pH 8.2 or 1 X CFLP~'~"' buffer,
pH 7.8, with
10 mM NaCI. Samples were heated to 95°C and begun by the addition of a
diluted enzyme
mix containing either 1 or 30 ng of RrpI in 1 X CFLPT"'' buffer. Reactions
were carried out
at 30°C for either 5 or 30 minutes. The results (data not shown)
indicated that this enzyme
2~ generates a weak, but distinct cleavage pattern on a single-stranded DNA
template.
E) The Radl/RadlO Complex Can Be Used To Generate Cleavage Patterns
The Radl-RadlO endonuclease (Radl/10) from S. cerevisiae is a specific 3"
endonuclease which participates in nucleotide excision repair in yeast. This
enzyme is a
heterodimer consisting of two proteins, Radl and RadlO. Radl and RadlO alone
do not have
enzymatic activity. Radl/10 recognizes structures comprising a bifurcated DNA
duplex and
cleaves the single-stranded 3' arm at the end of the duplex [Bardwell, A.J et
al. ( 1994)
Science 265:2082]. In this sense Radl/10 shares the same substrate specificity
as does the
CleavaseT"'' BN enzyme. However, the cleavage products produced by Radl/10 and
the
- 180 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
CleavaseT"' BN enzyme differ as the Radl/10 cleaves on the 3' single-stranded
arm of the
duplex while the CleavaseT"'' BN enzyme cuts on the 5' single-stranded arm.
Figure 68 provides a schematic drawing depicting the site of cleavage by these
two
enzymes on a bifurcated DNA duplex (formed by the hairpin structure shown). In
Figure 68,
. 5 the hairpin structure at the top shows the site of cleavage by a 5'
nuclease (e.g., the enzyme
Cleavase'~"'' BN enzyme). The hairpin structure shown at the bottom of Figure
68 shows the
site of cleavage by an enzyme which cleaves at the 3' single-stranded arm
(e.g., Radl/10).
Enzymes which cleave on the 5' single-stranded arm are referred to as
CleavaseTM 5'
enzymes; enzymes which cleave on the 3' single-stranded arm are referred to as
CleavaseTM 3'
enzymes.
In order to determine whether the Radl/10 protein is able to detect single
base changes
in DNA substrates, the cleavage patterns created by cleavage of DNA substrates
by the
Radl/10 and CleavaseT"'' BN enzymes were compared. In this comparison the
following
substrates were used. The 157 nucleotide fragment from the wild type (SEQ ID
N0:34). the
419 mutant (SEQ ID N0:41 ) and the 422 mutant (SEQ ID N0:42) .alleles derived
from the
sense strand of exon 4 of the human tyrosinase gene was generated containing a
biotin label
at the 5' end as described in Example 9.
The Radl and RadlO proteins were generously provided by Dr. Errol C. Friedberg
(The University of Texas Southwestern Medical Center, Dallas). The Radl/10
complex was
prepared by mixing Radl and RadlO proteins in 1X dilution buffer (0.5% NP40,
0.5% Tween
20, 20 mM Tris-HCI, pH 8.0, 50 mM KCI, 10 pg/ml BSA) to achieve a final
concentration of
0.1 mM of each protein.
Cleavage reactions using the Radl/10 endonuclease were performed as follows.
The
substrate DNA and 15 ng (0.1 pmole) of Radl/10 complex in 1 ~.1 of 1X dilution
buffer were
mixed on ice in 10 ~.l of 10 mM MOPS, pH 7.8, 1 mM MnCI,. The reaction was
then
incubated at 37°C for 5 minutes. The cleavage reaction was stopped by
addition of G ~.1 of
stop buffer.
Cleavage reactions using the CleavaseT"'' BN enzyme were done exactly as
described
above for the Radl/10 cleavages with the exception that 10 ng of the
CleavaseT"' BN enzyme
was added and the incubation at 37°C was performed for 3 minutes. Uncut
or no enzyme
controls were run for each substrate DNA and were prepared as described for
the reactions
containing enzyme with the exception that sterile water was added in place of
the enzyme
(data not shown).
- 181 -
CA 02203627 1997-04-24
WO 96/15267 PG"TlUS95/14673
The cleavage products (3 ~.1 each) were separated by electrophoresis through a
10%
denaturing polyacrylamide gel, transferred to a membrane and visualized as
described in
Example 19. The resulting autoradiograph is shown in Figure 69.
Figure 69 shows the resulting patterns corresponding to the cleavage products
of the
sense strand of the wild type tyrosinase substrate (SEQ ID N0:34) (lanes 1 and
4), the 419
mutant (SEQ ID N0:41) (lanes 2 and 5) and the 422 mutant (SEQ ID N0:42) (lanes
3 and
6). Lanes 1-3 show the cleavage pattern created by incubation of the three
substrate DNAs
with the Cleavase~ BN enzyme and lanes 4-6 show cleavage patterns created by
incubation
of the three substrate DNAs with the Radl/10 enzyme. Lanes marked "M" contain
molecular
weight markers prepared as described in Example 8.
The results shown in Figure 69 demonstrate that the Radl/10 enzyme was able to
produce distinctive cleavage patterns from the substrate DNAs (lanes 4-6); the
average
product length produced by cleavage of the substrate was longer than that
produced by the
CleavaseT'~' BN enzyme. Importantly, the results shown in Figure 69
demonstrate that the
single base substitutions found in the mutant tyrosinase substrates resulted
in the production of
specific changes in the otherwise similar cleavage patterns of tyrosinase
substrates (compare
lanes 5 and 6 with lane 4). Note that in the digestion of the mutant 419
substrate with
Radl/10, the bands below about 40 nucleotides have lower intensity and one
band is absent,
when compared to wild-type, while in the digest of the mutant 422 substrate
several new
bands appear in the range of 42-80 nucleotides. . Since both enzymes were
tested using the
same reaction conditions, these results show that Radl/10 was able to detect
the same
differences in DNA secondary structure that were recognized by the CleavaseT"'
BN enzyme.
Radl/10 generates a different cleavage pattern relative to that produced by
the CleavaseT"' BN
enzyme, since cleavage takes place at the 3' end of DNA hairpins producing
inherently longer
fragments when the substrate contains- a 5' end label. _ .
EXAMPLE 26
Detection Of Mutations In The Human (3-globin
Gene Using Double-Stranded DNA Substrates
The results shown in Example 13 demonstrated that single base changes in
fragments
of the (3-globin gene can be detected by cleavage of single-stranded DNA
substrates with the .
- 182 -
CA 02203627 1997-04-24
WO 96/15267 PC"T/US95/14673
CleavaseT"' BN enzyme. In this example it is shown that rrautations in the (3-
globin gene can
be detected by cleavage of double-stranded DNA substrates using the
CleavaseT"' BN enzyme.
Double-stranded substrate DNA comprising 536 by fragments derived from the
wild-
' type (3-globin gene (SEQ ID N0:56), mutant 1 (SEQ ID N0:58) and mutant .2
(SEQ ID
N0:59) were generated containing a 5' biotin label on the sense strand using
the PCR. PCR
amplification of these substrates was done as described in Example 13a. Gel
purification and
isolation of double-stranded fragments was performed as described in Example
19a.
The cleavage reactions were performed as described in Example 19c. Briefly, 2
~l of
stock DNA (80 ng) in TE was mixed with 3 ~,l H,O and denatured at 95°C
for 20 seconds.
The denatured DNA was cooled to 70°C and a mixture consisting of 2 ~.1
of SX CFLP~'~"''
buffer pH 7.5, 2 ~.l of 2 mM MnCI, and 1 pl (25 ng) of the enzyme CleavaseT"'
BN in
dilution buffer was added to start the cleavage reaction. The cleavage
reactions were stopped
after 1 minute by the addition of 6 ~,l of stop buffer. Control uncut
reactions were performed
as described above with the exception that of 1 ~.l of HBO vras used in place
of 1 ~l of the
1 S CleavaseT"'' BN enzyme. The cleavage products (5 ~.1 each) were separated
by electrophoresis
through a 6% denaturing polyacrylamide gel, transferred to a membrane and
visualized as
described in Example 19. The resulting autoradiograph is shown in Figure 70.
Figure 70 shows the cleavage patterns which correspond to the cleavage of the
sense
strand of the wild type (3-globin 536 by fragment (lane 4), mutant 1 fragment
(lane 5) and
mutant 2 fragment (lane 6). Lanes 1-3 show the uncut controls for wild-type,
mutant 1 and
mutant 2 substrates, respectively. The lane marked "M" contains biotinylated
molecular
weight markers prepared as described in Example 8.
As shown in Figure 70, the base substitution present in mutant 1 results in a
reduction
in the intensity of a band which migrates close to the uncut DNA (lane 5),
when compared to
wild-type cleavage pattern. The base substitution present in mutant 2 results
in the
disappearance of the band present in the region just above major product band
(approximately
174 nucleotides), when compared to the wild-type cleavage pattern.
For the double-stranded cleavage reactions described above, different reaction
conditions were used than those employed for the cleavage of the single-
stranded (3-globin
DNA substrates described in Example 13. The conditions employed for the
cleavage of the
double-stranded substrates used a lower MnCI, concentration, no KCl was added,
a higher
temperature and shorter time course relative to the conditions used in Example
13. Although
the cleavage patterns generated by cleavage of the double-stranded and single-
stranded ~3-
-183-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
globin DNA were slightly different, the positions of the pattern changes for
mutants 1 and
are similar to those demonstrated in Example 13, and it was possible to detect
the base
substitutions in both double-stranded cases. These results show that the
subtle changes in
DNA secondary structure caused by single base substitutions in larger DNA
substrates can be '
detected by the Cleavase.'~'' BN enzyme whether a single- or double-stranded
form of the t
DNA substrate is employed.
EXAMPLE 27
Identification_Of Mutations In The Human (3-globin Gene CFLPT"''
Patterns Of Unknowns By Comparison To An Existing Library of Patterns
The results shown in Examples 13 demonstrated that the CleavaseT"'' BN enzyme
generates a unique pattern of cleavage products from each (3-globin substrate
tested.
Differences in banding patterns were seen between the wild-type and each
mutant; different
banding patterns were seen for each mutant allowing not only a discrimination
of the mutants
from the wild-type but also a discrimination of each mutant from the others.
To demonstrate
that the products of the Cleavase~ reaction can be compared to previously
characterized
mutants for purposes of identification and classification, a second set of (3-
globin mutants
were characterized and the CFLP~'~"' patterns, by comparison to the set,
analyzed in Example
13, were used to determine if the mutants in the second set were the same as
any in the first
set, or were unique to the second set. Although these isolates have all been
described
previously (specific references are cited for of these isolates at the end of
this example). the
experiment was performed "blind", with the samples identified only by a
number.
Five (3-globin mutants were compared to the CFLPT"'' patterns from the first
set: the
wild type [3-globin gene (SEQ ID N0:56) or mutant 1 (SEQ ID N0:58), mutant 2
(SEQ ID
N0:59)or mutant 3 (SEQ ID N0:57). Plasmids for containing these 5 new isolates
were
grown and purified, and single-stranded substrate DNA, 534 or 536 nucleotides
in length. was
prepared for each of the 5 (3-globin genes as described above in Example 13a.
Cleavage
reactions were performed and reaction products were resolved as described in
Example 13; the
resulting autoradiograph is shown in Figure 71.
In Figure 71, two panels are shown. Panel A shows the reaction products from
the ~-
globin isolates described in Example 13 (and as seen in Figure 40). Panel B
shows the
- 184 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
reaction products of the five additional isolates, numbered 4, 5, 6, 7 and 8.
The lanes marked
"M" contain biotinylated molecular weight markers prepared as described in
Example 8.
By comparison to the CFLP~ patterns shown in Panel A, the isolates shown in
Panel
B can be characterized. It can be seen that the banding pattern of isolate 4
(Panel B, lanel) is
the same as was seen for the wild-type (3-globin substrate shown in Panel A
(lane 1 ); isolate 8
(Panel B, lane 5) is comparable to the previously characterized mutant 3
(Panel A, lane 4);
isolate number 6 (Panel B, lane3) has changes in two areas of the pattern and
appears to have
features of both isolates 2 (Panel A, lane 3)and 3 (Panel A, lane 4); isolates
5 and 7 (Panel B,
lanes 2 and 4, respectively) appear to be identical, and they show a pattern
not seen in panel
A.
To confirm the relationships between the different isolates, the identities of
the
mutations were then determined by primer extension sequencing using
the,finoleT"' DNA
Sequencing System (Promega Corp., Madison, WI) using the PCR primers [5'-
biotinylated
KM29 primer (SEQ ID N0:54) and 5'-biotinylated RS42 primer (SEQ ID NO:55)],
according
to the manufacturer's protocol. The sequencing reactions were visualized by
the same
procedures used for the (3-globin CFLPT"'' reactions, as described in Example
13b.
The two isolates that matched members of the original set by CFLPTM pattern
analysis
matched by sequence also. Isolate 4 is identical to the wild type sequence
(SEQ ID N0:56);
isolate 8 is a duplicate of mutant 3 (SEQ ID N0:57).
Isolate 6 appears by CFLP-'~"'' pattern to have changes similar to both mutant
2 and
mutant 3 of the original set. The sequence of mutant 6 (SEQ ID N0:69) reveals
that it shares
a one base change with mutant 3, a silent C to T substitution in codon 3.
Mutant 6 also has a
G-to-A substitution in codon 26, only 4 bases downstream of that found in
mutant 2 (SEQ ID
N0:59). This mutation has been shown to enhance a cryptic splice site causing
a fraction of
the mRNA to encode a nonfunctional protein [Orkin, S.H., et al. (1982) Nature,
300:768]. It
is worthy of note that while mutant 6 and mutant 2 both showed alteration in
the band that
migrates at about 200 nucleotides (e.g., the band is missing or weak in mutant
2 but appears
to be split into 3 weak bands in mutant 6) these changes are not of identical
appearance.
These CFLPT"' changes, caused by mutations four nucleotides apart, are
distinguishable from
each other.
The last two isolates, 5 and 7, had the same sequence (SEQ ID N0:70), and
revealed a
single base substitution within the first intron, at IVS position 110. This
mutation is
associated with abnormal splicing leading to premature termination of
translation of the (3-
- 18~ -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95114673
globin protein [R.A. Spritz et al. (1981) Proc. Natl. Aead. Sci. USA,
78:2455]. It is worthy
of note that the band that disappears in the CFLP~ patterns for these mutants
(at ,
approximately 260 nucleotides, as compared to the size markers) is between the
indicative
bands in the mutant 1 (at approximately 400 nucleotides) and mutant 2 (at
approximately 200
nucleotides) CFLPT"'' patterns, and the actual mutation (at nucleotide 334
from the labeled 5'
end) is between those of mutants l and 2, at nucleotides 380 and 207,
respectively. Thus, the
CFLPT"' analysis not only indicated the presence of a change, but also gave
positional
information as well.
From the results shown in Figure 71, the unique pattern of cleavage products
generated
by the CleavaseT"'' BN enzyme from each of the first four (wild type plus
three variants) (3-
globin substrates tested was used as reference to characterize additional (3-
globin isolates. The
banding patterns show an overall "familial" similarity, with subtle
differences (c~.g., missing or
shifted bands) associated with each particular variant. Differences in banding
patterns were
seen between the wild-type and each mutant; different banding patterns were
seen for each
mutant allowing not only a discrimination of the mutant from the wild-type but
also a
discrimination of each mutant from the others.
EXAMPLE 28
Effect Of The Order Of Addition Of The Reaction
Components On The Double-Stranded Cleavage Pattern
The cleavage reaction using a double-stranded DNA substrate can be considered
a two-
step process. The first step is the denaturation of the DNA substrate and the
second step is
the initiation of the cleavage reaction at the target temperature. As it is
possible that the
resulting cleavage pattern may differ depending on the conditions present
during denaturation
(e.g., whether the DNA is denatured in water or in a buffer) as well as on the
conditions of
reaction initiation (e.g.. whether the cleavage reaction is started by the
addition of enzyme or
MnCh) the following experiment was performed.
To study the effect of the addition of the reaction components on the
resulting
cleavage pattern, all possible mixing combinations for 4 reaction components
(i.e.. DNA, '
CFLP~' buffer, MnCh and the Cleavase~"~'' BN enzyme) were varied. A single DNA
substrate
was used which comprised the 536 by fragment derived from the wild-type (3-
globin gene
- 186 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
(SEQ ID N0:56). The substrate DNA contained a biotin label at the 5' end of
the sense
strand and was prepared as described in Example 26.
The substrate was cut in 8 different cleavage reactions which employed
different
- combinations for the addition of the reaction components at the denaturing
and initiation
steps. These reactions are described below.
Figure 72 shows the resulting patterns generated by c;leavage of the sense
strand of the
wild-type (3-globin 536-by substrate (SEQ ID NO:56). In lane 1, the substrate
DNA (40
fmoles of DNA in 1 ~1 of TE mixed with 5 ~.1 Hz0) was denatured at 95°C
for 10 seconds,
cooled to 55°C and the reaction was started by the addition of a
mixture containing 2 yl of
SX CFLPT"' buffer with 150 mM KCI, 1 ~,l of 2 mM MnCl7 and 1 1.~I (50 ng) of
the
CleavaseT"' BN enzyme. In lane 2, the DNA was denatured in the presence of 2
p.l of SX
CFLPTM buffer and reaction was started at 55°C by the addition of 1 ~.I
MnCI, and I ~.l (50
ng) of the CleavaseT"' BN enzyme. In lane 3, the DNA was. denatured in the
presence of
MnCh and the reaction was started with addition of the buffer and the enzyme.
In lane 4, the
denaturation mixture included the substrate DNA and the enzyme and the
reaction was started
with addition of the buffer and MnCI,. In lane 5, the substrate DNA was
denatured in the
presence of CFLPTM buffer and MnCh and then the enzyme was added at
55°C. In lane 6, the
substrate DNA was denatured in the presence of CFLPT"' buffer and the enzyme
and then
MnCI= was added at 55°C. Lane 7 shows the uncut control. In lane 8, the
DNA was
denatured in the presence of the enzyme and MnCh and then the buffer was added
at ~5°C.
In lane 9, the substrate DNA was denatured in the presence of the enzyme, MnCh
and the
CFLPT"' buffer and then the mixture was incubated at 55°C :for 5
minutes. The lane marked
"M" contains biotinylated molecular weight markers prepared as described in
Example 8.
In all cases reaction was stopped by addition of 6 ~l of stop buffer. The
reaction
products (5 ~1 each) were resolved by electrophoresis through a 10% denaturing
polyacrylamide gel and the DNA was transferred to a membrane and visualized as
described
in Example 19. The resulting autoradiograph is shown in Figure 72.
The results shown in Figure 72 demonstrate that most of denaturation-
initiation
protocols employed generated identical cleavage patterns with the exception of
the reaction
shown in lane 3. In the reaction shown in lane 3, the DNA was denatured in the
presence of
MnCh and in the absence of CFLP~''' buffer. In the cases where the enzyme and
MnCI, were
added before the denaturation step (lanes 8,9) no labeled material was
detected. In these
cases the label was released in a form of short DNA fragments which were
produced as a
- 187 -
CA 02203627 1997-04-24
WO 96115267 PCT/US95114673
result of nibbling (i.e., the exonucleolytic removal) of the label from the 5'
end of the double-
stranded DNA template.
The results shown in Figure 72 demonstrate that the order of addition of the
reaction
components has little effect upon the cleavage pattern produced with the
exception that 1 ) the
DNA should not be denatured in the presence of MnCI., but in the absence of
any buffering
solution and 2) the CleavaseT'~'' BN enzyme and MnCI, should not be added
together to the
DNA prior to the denaturation step. Under these two exceptional conditions,
the 5' label was
removed from the 5' end of the substrate by the enzyme resulting in a loss of
the signal.
EXAMPLE 29
Detection Of Mutations In Human p53 Gene By
CleavaseT"' Fragment Length Polymorphism (CFLPTMI Analysis
The results shown in preceding examples demonstrated that the CFLPT~' reaction
could
detect single base changes in fragments of varying size from the human (3-
globin and
tyrosinase genes and that the CFLPT"'' -reaction could be used to identify
different strains of
virus. The ability of the CleavaseT"' reaction to-detect single base changes
in the human
tumor suppressor gene p53 was next examined. Mutation of the human p53 gene is
the most
common cancer-related genetic change; mutations in the p53 gene are found in
about half of
all cases of human cancer.
The ability of the Cleavase~"~' BN enzyme to cleave DNA fragments derived from
the
human p53 gene and to detect single base changes in fragments-of the same size
was
examined. Plamsids containing cDNA clones containing either wild type or
mutant p53
sequences were used to generate templates for analysis in the CFLPT"~
reaction. The p53 gene
is quite large, spanning 20,000 base pairs and is divided into 11 exons. The
use of a template
derived from a cDNA allows for maximization of the amount of protein-encoding
sequence
that can be examined in a DNA fragment of a given size.
The nucleotide sequence of the coding region of the wild type human p~3 cDNA
gene
_ is listed in SEQ ID N0:79. The nucleotide sequence of the_coding region of
the mutant 14 3
human p53 cDNA gene is listed in SEQ ID N0:80. The nucleotide sequence of the
coding
region of the -mutant 249 (silent) human p53 cDNA gene is listed in SEQ ID
N0:81. A 601
nucleotide fragment spanning exons-5 through 8 was generated from each of
these three p53
cDNAs as follows.
-188-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
A) Preparation Of The Substrate DNA
Six double stranded substrate DNAs were prepared for analysis in the CFLP~'T'~
reaction. The substrates contained a biotin label at either their 5' or 3'
end. The wild type
substrate comprises a 601 nucleotide fragment spanning exons 5 through 8 of
the cDNA
sequence of the human p53 gene (SEQ ID N0:79) [Baker, S. J. et al., Science (
1990)
249:912]. Two mutation containing substrates were used. The mutant 143
substrate (SEQ
ID:93) is derived from a p53 mutant V 143A which contains a valine (GTG) to
alanine (GCG)
substitution; this mutation differs from the wild type p53 ea;on 5-8 fragment
by a single
nucleotide change [Baker, S. J. et al., Science (1990) 249:912]. The mutant
249 (silent)
substrate is derived from a p53 mutant which contains a single base change at
amino acid
249. from AGG to AGA (SEQ ID N0:81 ). This single base change does not result
in a
corresponding amino acid change and is therefore referred to as a silent
mutation.
The 601 by double stranded PCR fragments were generated as follows. The primer
pair 5'-TCTGGGCTTCTTGCATTCTG (SEQ ID N0:82) and 5'-
GTTGGGCAGTGCTCGCTTAG (SEQ ID N0:83) were used to prime the PCRs. The
synthetic primers were obtained from Integrated DNA Technologies (Coralville,
IA). The
primers were biotinylated on their 5' ends with the Oligonucleotide
Biotinylation Kit
purchased from USB-Amersham (Cleveland, OH) according to the manufacturers'
protocols.
When the sense strand was to be analyzed in the CFLPT"' reaction, the primer
listed in SEQ
ID N0:82 was labeled at the 5' end with the biotin. When the anti-sense strand
was to be
analyzed in the CFLP~ reaction, the primer listed in SEQ ID N0:83 was labeled
at the 5'
end with the biotin.
The target DNA used in the PCR for the generation of the 601 by fragment
derived
from the wild type p~3 cDNA was the plasmid CMV-p53-SN3 [Baker, S. J. et crl.,
.supr°a];
this plasmid contains the coding region listed in SEQ ID N0:79. The target for
the
generation of the 601 by fragment derived from the mutant 143 was the plasmid
CMV-p53-
SCX3 [Baker, S. J. et al., supra]; this plasmid contains the coding region
listed in SEQ ID
N0:80. REF). The target for the generation of the 601 by fragment derived from
mutant 249
(silent) was the plasmid LTR 273 His jChen, P.-L. et al., Science (1990)
250:1576]; this
plasmid contains the coding region listed in SEQ ID N0:81. DNA was prepared
from
i
bacteria harboring each plasmid (plasmid DNA was isolated using standard
techniques). The
601 by PCR products were prepared as follows.
- 189 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
The symmetric PCR reactions contained 50 ng of plasmid DNA, 50 pmoles primer
5"-
TCTGGGCTTCTTGCATTCTG (SEQ ID:95), 50 pmoles of primer 5'-
GTTGGGCAGTGCTCGCTTAG (SEQ ID:96), and .50 pM each dNTP in 95 p.l of 1X PCR
buffer. The reaction mixtures were overlaid with 50 ~.l ChillOutT"'' (MJ
Research, Watertown,
MA) and the tubes were heated to 95°C for 2.5 min. Tag DNA polymerase
(Promega Corp.,
Madison, WI) was then added as 1.25 units of enzyme in 5 ~.l of 1X PCR buffer.
The tubes
were then heated to 95°C for 45 seconds, cooled to 55°C for 45
seconds and heated to 72°C
for 75 seconds for 34 cycles with a 5 min incubation at 72°C after the
last cycle.
The PCR products were gel purified as follows. The products were precipitated
by the
addition of NaCI to a final concentration of 0.4M, 20 p.g glycogen carrier and
500 pl ethanol.
The DNA was pelleted by centrifugation and the PCR products were resuspended
in 25 or 50
q.l sterile distilled water to which was added an equal volume of a solution
containing 95%
formamide, 20 mM EDTA and 0.05% each xylene cyanol and bromophenol blue. The
tubes
were then heated to 85°C for 2 min and the reaction products were
resolved by
electrophoresis through a 6% polyacrylimide gel ( 19:1 cross-link) containing
7 M urea in a
buffer containing 0.5X TBE. The DNA was visualized by ethidium bromide
staining and the
601 by fragments were excised from the gel slices by passive diffusion
overnight into a
solution containing 0.5 M NH40Ac, 0.1 % SDS and 0.1 % EDTA. The DNA was then
precipitated with ethanol in the presence of 4 ~.g of glycogen carrier. The
DNA was pelleted,
resuspended in sterile distilled water and reprecipitated by the addition of
NaCI to a final
aqueous concentration of 0.2 M and 80% ethanol. After the second
precipitation, the DNA
was pelleted and resuspended in 30 ~.l sterile distilled water or TE.
The nucleotide sequence of these 601 by templates are listed in SEQ ID NOS:84-
89.
The sense strand of the 601 nucleotide wild type fragment is listed in SEQ ID
N0:84. The
anti-sense strand of the 601 nucleotide wild type fragment is listed in SEQ ID
N0:85. The
sense strand of the 601 nucleotide mutant 143 fragment is listed in SEQ ID
N0:86. The anti-
sense strand of the 601 nucleotide mutant 143 fragment is listed in SEQ ID
N0:87. The
sense strand of the 601 nucleotide mutant 249 (silent) fragment is listed in
SEQ ID N0:88.
The anti-sense strand of the 601 nucleotide mutant 249 (silent) fragment is
listed in SEQ ID
N0:89.
- 190 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
B) Cleavage Reaction Conditions
Cleavage reactions comprised approximately 100 fmoles of the resulting double
stranded substrate DNAs (the substrates contained a. biotin moiety at the ~'
end of the sense
or antisense strand) in a total volume of 5 ~,l of sterile distilled water.
The reactions were
heated to 95°C for 15 seconds to denature the substrates and then
quickly cooled to 50°C (this
step allows the DNA to assume its unique secondary structure by allowing the
formation of
intra-strand hydrogen bonds between complimentary bases).
The reactions were performed in either a thermocycler (MJ Research. Watertown,
MA)
programmed to heat to 95°C for 15 seconds and then cooled immediately
to 50° C.
Once the tubes were cooled to the reaction temperatwe of 50°C, the
following
components were added: 5 ~.1 of a diluted enzyme mix containing 0.2 pl of
CleavaseT"'' BN
[SOng/p.l 1 X CleavaseT"'t Dilution Buffer (0.5% NP40, 0.5°ro Tween 20,
20 mM Tris-Cl, pH
8.0, 50 mM KCl, 10 ~.g/ml BSA)]; 1 p,l of 10 X CFLP~'~"'' reaction buffer (100
mM MOPS,
pH 7.~. 0.5% NP 40, 0.5% Tween 20), and 1 pl of 2mM IVInCI,.
A no enzyme control ( 10 ~.1) was set up in parallel for each PCR fragment
examined;
this control differed from the above reaction mixture only in that sterile
distilled water was
substituted for Cleavase'~"~ BN enzyme. Reactions were stopped after 3 minutes
by the
addition of 8 p.l of stop buffer.
The samples were then heated to 85°C for 2 minutes and 4 p.l of each
reaction mixture
were resolved by electrophoresis through a 6% polyacrylimide gel (19:1 cross-
link), with 7M
urea, in-a buffer containing O.SX TBE.
After electrophoresis, the gel plates were separated allowing the gel to
remain flat on
one plate. -A 0.2 ~.m-pore positively charged nylon membrane (Schleicher and
Schuell.
Keene. NH), pre-wetted with O.SX TBE, was laid on top of the exposed
acrylamide gel. All
air bubbles trapped between the gel and the membrane were removed. Two pieces
of 3MM
filter paper (Whatman) were then placed on top of the membrane, the other
glass plate was
replaced, and the sandwich was clamped with binder clips. Transfer was allowed
to proceed
overnight. After transfer, the membrane was carefully peelE;d from the gel and
washed in 1 X
Sequencase Images Blocking Buffer (United States Biocherr~ical) for two 15
minute intervals
with gentle agitation. Three tenths of a ml of the buffer was used per cm' of
membrane. A
streptavidin-alkaline phosphatase conjugate (SAAP, United States Biochemical,
Cleveland,
OH) was added to a 1:3000 dilution directly to the blocking solution. and
agitated for 15
minutes. The membrane was washed 3 times (~ min/wash) in 1 X SAAP buffer ( 1
OOmM
-191-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
Tris-HCI, pH 10; 50 mM NaCI) with 0.1% SDS, using 0.5 mls/cm'' of membrane.
The
membrane was then washed twice in 1 X SAAP buffer without SDS, but containing
1 mM
MgCh, drained thoroughly and placed in a heat sealable bag. Using a sterile
pipet ,tip, 0.0~
ml/cm'- of CDP-StarT"'' (Tropix. Bedford, MA) was added to the bag and
distributed over the
membrane for 5 minutes. The bag was drained of all excess liquid and air
bubbles. The
membrane was then exposed to X-ray film (Kodak XRP) for an initial 30 minute
exposure.
Exposure times were adjusted as necessary for resolution and clarity. The
result
autoradiograph is shown in Figure 73.
In Figure 73, the lane marked M contains biotinylated molecular weight
markers. The
marker fragments were purchased from Amersham (Arlington Heights, IL). - Lanes
1-4
contain the reaction products from the incubation of double stranded DNA
substrates in the
absence of the CleavaseT"' BN enzyme (i.e., uncut controls). Lane I contains
the wild type
fragment labeled on the sense strand of the 601 by PCR fragment. Lane 2
contains the
mutant 143 fragment labeled on the sense strand of the 601 by PCR fragment.
Lane 3
contains the wild type fragment labeled on the antisense strand of the PCR
product. Lane 4
contains the fragment encoding the silent mutation at amino acid 249 labeled
on the antisense
strand of the PCR product. Lanes 5-8 contain the reaction products from the
incubation of
the 601 by double stranded substrates with the CleavaseT"'' BN enzyme. Lane 5
contains
products generated using the wild type fragment labeled on the sense strand;
lane 6 contains
products generated using the mutant 143 labeled on the sense strand. Lanes 7
and 8 contain
products generated using the wild type and mutant 249 (silent) substrates,
respectively, labeled
on the anti-sense strand.
The results shown in Figure 73 demonstrate that a similar, but distinctly
different,
pattern of cleavage products was generated by the digestion of wild type and
mutant-
containing templates by the CleavaseT"'' BN enzyme. Comparison of lanes 5 and
6 reveals a
difference in the band pattern in the 100 nucleotide range. Specifically, the
strong band
present in the wild type (at around 100 nucleotides) was missing in the V 143A
mutant while
two bands immediately below this strong band were prominent in the mutant and
not evident
in the wild type. In the 200 nucleotide range, a pronounced doublet seen in
the wild type is
missing from the mutant, which instead contained a strong single band
migrating slightly
a
faster than the wild type doublet. Similarly, comparison of lanes 7 and 8
revealed differences
between the pattern generated from cleavage of the anti-sense strand of the
wild type
fragment and the mutant 249 (silent) fragment. In the 100 nucleotide range,
the wild type -
- 192 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
fragment exhibited a strong doublet whereas tl lupper band of this doublet was
missing in the
mutant 249 (silent) fragment. In addition, two prominent bands present in the
wild type
pattern in the 150-180 by range were completely absent from the mutant 249
(silent) cleavage
products. -
~ 5 Although each mutant fragment analyzed in Figure 73 differs from the wild
type by
only one of the 601 nucleotides, a unique pattern of cleavage: fragments was
generated for
each. Furthermore, at least one pattern change occurred in each mutant in the
immediate
vicinity (i. e., within 10-20 nucleotides) of the DNA sequence change. This
experiment
demonstrates that CFLPT"'' is capable of distinguishing the presence of single
base changes in
PCR fragments containing exons 5 through 8 of the p53 gene.
EXAMPLE 30
Detection Of Genetically Engineered
Mutations In PCR Fragments Of The Human p53 Gene
The ability of the Cleavase~'~'' BN enzyme to detect single base changes
genetically
engineered into PCR fragments containing exons 5 through 8 of the human p53
gene was
analyzed. The single base changes introduced were 1 ) a change from arginine
(AGG) to
serine (AGT) at amino acid 249 (termed the R249S mutation) and 2) a change
from arginine
(CGT) to histidine (CAT) at amino acid 273 (termed the R273H mutation). Both
of these
mutations have been found in human tumors and have been identified as
mutational hot spots
[Hollstein et al., Science 253:49 (1991)]. The R249S mutation is strongly
correlated with
exposure to aflatoxin B and/or infection with hepatitis B virus [Caron de
Fromental and
Soussi, Genes, Chromosomes and Cancer (1992) pp. 1-15]. The R273H mutation
arises as a
result of a transition at a CpG dinucleotide. Such transitions account for
approximately one-
third of the known p53 mutations and are characteristic of a variety of tumor
types [Caron de
Fromental and Soussi, supra; Hollstein et al., supra].
Plasmids containing the R249S and R273H mutations were engineered according to
a
variation of a protocol described by R. Higuchi [in PCR Technolog3e Principles
and
~ Applications,for DNA Amplification, H. A. Ehrlich, Ed.(1989) Stockton Press,
NY, pp. 61-
70]. This methodology allows the generation of collection of plasmids
containing DNA
sequences corresponding to known p53 mutations. The availability of this
collection allows
-193-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
the generation of p53 "bar code" library which contains the CFLPT"'' patterns
generated by
cleavage of the p53 mutants- using the CleavaseTM enzymes. ,
A) Construction of a 601 by PCR fragment Containing the R249S Mutation
To generate a 601 by fragment containing the R249S mutation, a 2-step
recombinant
PCR was performed (see Figure 6 for a schematic representation of the 2-step
recombinant
PCR). In the first or "upstream" PCR, oligonucleotides 5'-TCTGGGCTTCTTGCATTCTG
(SEQ ID N0:82) and 5'-GAGGATGGGACTCCGGTTCATG (SEQ ID N0:90) were used to
amplify a 427 by fragment containing the G to T base change resulting in the
R249S
mutation; the sequence of the 427 by fragment is listed in SEQ ID N0:98. In
the second or
"downstream" PCR, oligonucleotide 5'-CATGAACCGGAGTCCCATCCTCAC (SEQ ID
N0:91 ) and 5'-GTTGGGCAGTGCTCGCTTAG (SEQ ID N0:83) were used to amplify a 196
by fragment containing the same base change on the complementary strand; the
sequence of
the 196 by fragment is listed in SEQ ID N0:99. __
For each PCR, 10 ng of a cDNA clone encoding the wild type p53 gene (coding
region listed in SEQ ID N0:79) were used as the template in a 50 p.l PCR
reaction. In the
case of the upstream fragment, 10 ng of template were added to a tube
containing 5
picomoles of the oligonucleotide 5'-TCTGGGCTTCTTGCATT CTG (SEQ ID N0:82), 5
pmoles of the oligonucleotide 5'-GAGGATGGGACTCC GGTTCATG (SEQ ID N0:90), and
50 q.M each dNTP, in 45 p.l of 1X PCR buffer. For the downstream fragment, 10
ng of the
wild type template, plasmid CMV-p53-SN3 (Example 29) were added to 5 picomoles
of the
oligonucleotide 5'-CATGAACCGGAGTCCCATCCTCAC (SEQ ID N0:91 ) and 5 picomoles
of the oligonucleotide 5'-GTTGGGCAGTGCTCGCTTAG (SEQ-ID N0:83), and 50 ~.M each
dNTP in 1 X PCR buffer.
Tubes containing 45 p.l of the above mixtures for each template to be
amplified were
overlaid with 50 p.l ChillOut~' (MJ Research, Watertown, MA) and the tubes
were heated to
95°C for 2.5 min and then cooled to 70°C. Taq DNA polymerase
(Promega) was then added
as 1.23 units of enzyme in 5 ~.l of 1X PCR buffer. The tubes were then heated
to 95°C for
45 seconds, cooled to 55°C for 45 seconds and heated to 72°C for
75 seconds for 24 cycles
with a 5 min incubation at 72°C after the last cycle.
The PCR products were gel purified as follows. Ten microliters of each PCR
product
were mixed with 10 p.l of stop buffer. The tubes were then heated to
85°C for 2 min and the '
reaction products were resolved by electrophoresis through a 6% polyacrylimide
gel ( 19:1
cross-link) containing 7 M urea in a buffer containing O.SX TBE (the-
polyacrylimide solutions '
- 194 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
used were freshly prepared). The DNA was visualized by eahidium bromide
staining and the
fragment was excised from the gel slice by passive diffusion overnight into a
solution
containing 0.5 M NH40Ac, 0.1 % SDS and 0.1 % EDTA at 37° C.
' Ten microliters of each eluted PCR product were combined to serve as the
recombinant template to prime a second round of PCR. To this template, 10
picomoles of 5'-
' biotin exon 8 primer (SEQ ID N0:83), 10 pmoles of 5'-exon 5 primer (SEQ ID
N0:82). and
50 p.M each dNTP in 1X PCR buffer were added. Tubes containing 90 pl of the
above
mixtures for each template to be amplified were overlaid with 50 q.l
ChillOutT"' (MJ Research,
Watertown, MA) and the tubes were heated to 95°C for 2.5 min and then
cooled to 70°C.
Tuy DNA polymerise (Promega) was then added as 2.5 units of enzyme in 5 ~,1 of
1 X PCR
buffer. The tubes were then heated to 95°C for 45 seconds, cooled to
47°C to allow the two
template molecules to anneal, then heated to 72° C to allow extension
of the primers by Tcrg
DNA polymerise. Following this initial cycle of denaturation, annealing and
extension, 25
cycles in which the reactions were heated to 95°C for 45 seconds,
cooled to 55°C for 45
l~ seconds, and then heated to 72°C for 1 minute were carried out,
followed by a ~ min
extension at 72° C. The fragments were then ethanol precipitated and
gel purified as
described in Example 29.
B) Construction of a 601 by PCR Fragment Containing the R273H Mutation
To generate a 601 by fragment containing the R273H mutation, a 2-step
recombinant
PCR was performed using the procedure described in section a) was used to
simultaneously
amplify PCR fragments encoding a single base change from arginine (CGT) to
histidine
(CAT) at amino acid 273. In the first or "upstream" PCR, oligonucleotide 5'-
TCTGGGC
TTCTTGCATTCTG-3' (SEQ ID N0:82) and 5'-GCACAAACATGCACCTCAAAGCT-3'
(SEQ ID N0:92) were used to generate the 498 by fragment whose sequence is
listed in SEQ
ID NO:100. In the second or "downstream" PCR, oligonucleotide 5'-CAGCTTTG
AGGTGCATGTTTGT-3' (SEQ ID N0:93) was paired with oligonucleotide 5'-GTTGGG
CAGTGCTCGCTTAG-3' (SEQ ID N0:83) to generate a 127 nucleotide fragment whose
sequence is listed in SEQ ID NO:101. The DNA fragments were electrophoresed.
eluted,
combined and used to prime a second round of PCR as described in section a) to
generate a
601 by PCR product containing the R273H mutation.
..
C) Sequence Analysis of the 601 Nucleotide PCR Fragments
The recombinant 601-by PCR products generated through this two step PCR
procedure
were gel purified as described in Example 29. The PCR products were sequenced
using the
-19~-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95114673
fmol~ DNA Sequencing System (Promega) in conjunction with oligonucleotide 5'-
biotin-
GTTGGGCAGTGCTCGCTTAG (SEQ ID N0:83) according to manufacturers' standard
protocols to verify the presence of the engineered mutations.
The nucleotide sequence corresponding to the sense strand of the 601
nucleotide
R249S mutant fragment is listed in SEQ ID N0:94. The anti-sense strand of the
601
nucleotide R249S mutant fragment is listed in SEQ ID N0:95. The sense -strand
of the 601
nucleotide R273H mutant fragment is listed in SEQ ID N0:96. The anti-sense
strand of the
601 nucleotide R273H mutant fragment is listed in SEQ ID N0:97.
D) Cleavage Reactions
In order to generate ample quantities-of DNA for subsequent CFLP~'~"''
analysis, the 601
by fragments containing either the R249S or the R273H mutation were used as
templates in
an additional round of PCR. Approximately 2 fmoles of each 601 by fragment
were added to
pmoles of the primers corresponding to SEQ ID NOS:82 and 83 (SEQ ID N0:83
contained a biotin on the 5' end), 50 ~.M each dNTP, 20 mM Tris-HCI, pH 8.3.
1.5 mM
15 MgCI,, 50 mM KCI, 0.05% Tween 20 and 0.05% NP40. Tubes containing 90 pl of
the
above mixture were assembled for each template to be amplified; the tubes were
overlaid with
50 p.l ChillOutT''~ (MJ Research, Watertown, MA) and the tubes were heated to
95°C for 2.5
min and then cooled to 70°C. Taq DNA polymerise (Promega) was then
added as 2.5 units
of enzyme in 5 p.l of 1X PCR buffer. The tubes were then heated to 95°C
for 45 seconds,
20 cooled to 47°C to allow the two template molecules to anneal, then
heated to 72° C to allow
extension of the primers by Taq DNA polymerise. Following this initial cycle
of
denaturation, annealing and extension, 25 cycles in which the reactions were
heated to 95°C
for 45 seconds, cooled to 55°C for 45 seconds, and then heated to
72°C for 1 minute were
carried out. followed by a 5 min extension at 72° C. The fragments were
then ethanol
precipitated and gel purified as described in Example 29. The gel purified
fragments were
then used in CFLP~' reactions as follows.
Cleavage reactions comprised approximately 100 fmoles of the resulting double
stranded substrate DNAs (the substrates contained a biotin moiety at either
the 5" end of the
sense or anti-sense strand) in a total volume of 5 p.l (sterile distilled
water was used to bring
the volume to 5 ~1). The reactions were heated to 95°C for 15 seconds
to denature the
substrates and then quickly cooled to 50°C (this step allows the DNA to
assume its unique
secondary structure by allowing the formation of intra-strand hydrogen bonds
between
complimentary bases). The reaction were performed in either a thermocycler (MJ
Research,
- 196 -
CA 02203627 1997-04-24
WO 96115267 PCT/US95/14673
Watertown, MA) programmed to heat to 95°C for 15 seconds and then cool
immediately to
50° C or the tubes were placed manually in a heat block set at
95°C and then transferred to a
second heat block set at 50°C.
' Once the tubes were cooled to the reaction temperature of 50°C, 5 ~1
of a diluted
enzyme mix containing 0.2 ~,l of Cleavase'~"" BN enzyme [50 ng/p.l 1 X
CleavaseTM Dilution
Buffer (0.5% NP40, 0.5% Tween 20, 20 mM Tris-Cl, pH 8.0, 50 mM KCI, 10 ~.g/ml
BSA)],
1 ~l of 10 X CFLP~ reaction buffer (100 mM MOPS, pH 7.5, 0.5% NP 40, 0.5%
Tween
20), and 1 ~1 of 2 mM MnCh. A 10 ~l no enzyme control was set up in parallel
for each
PCR fragment examined in which sterile distilled water was substituted for the
Cleavase rM BN
enzyme. After 2 minutes at 50°C, the reactions were stopped by the
addition of 8 ~1 of stop
buffer . -
The samples were heated to 85°C for 2 minutes and 7 ~1 of each
reaction were
resolved by electrophoresis through a 10% polyacrylimide gel ( 19:1 cross-
link), with 7M urea,
in a buffer containing O.SX TBE.
After electrophoresis, the gel plates were separated allowing the gel to
remain flat on
one plate. A 0.2 ~.m-pore positively charged nylon membrane (Schleicher and
Schuell,
Keene, NH), pre-wetted with O.SX TBE, was laid on top of the exposed
acrylamide gel. All
air bubbles trapped between the gel and the membrane were removed. Two pieces
of 3MM
filter paper (Whatman) were then placed on top of the membrane, the other
glass plate was
replaced, and the sandwich was clamped with binder clips. Transfer was allowed
to proceed
overnight. After transfer, the membrane was carefully peeled from the gel and
washed in 1 X
Sequencase Images Blocking Buffer (United States Biochemical) for two 1 ~
minute intervals
with gentle agitation. Three tenths of a ml of the buffer was used per cm? of
membrane. A
streptavidin-alkaline phosphatase conjugate (SAAP, United States Biochemical,
Cleveland,
OH) was added to a 1:3000 dilution directly to the blocking solution, and
agitated for 15
minutes. The membrane was washed 3 times (5 min/wash) in 1 X SAAP buffer ( 100
mM
Tris-HCI, pH 10; 50 mM NaCI) with 0.1% SDS, using 0.5 .mls/cm'- of membrane.
The
membrane was then washed twice in 1 X SAAP buffer witlxout SDS, but containing
1mM
MgCI,, drained thoroughly and placed in a heat sealable bag. Using a sterile
pipet tip, 0.0~
. 30 ml/cm' of CDP-StarT"' (Tropix, Bedford, MA) was added to the bag and
distribu~:.d over the
membrane for S minutes. The bag was drained of all excess liquid and air
bubbles. The
membrane was then exposed to X-ray film (Kodak XRP) far an initial 30 minute
exposure.
- 197 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
Exposure times were adjusted as necessary for resolution and clarity. The
results are shown
in Figure 74.
In Figure 74, the lane marked M contains biotinylated molecular weight
markers.
The marker fragments were purchased from Amersham (Arlington Heights, IL) and
include '
bands corresponding to lengths of 50, 100, 200, 300, 400, 500, 700, and 1000
nucleotides. ,
Lanes 1-4 contain the reaction products from the incubation of double stranded
DNA
substrates labeled on the antisense strand in the absence of the Cleavase~'~''
BN enzyme. Lane
1 contains the reaction products from the wild type fragment (SEQ ID N0:85):
lane 2
contains the reaction products from the engineered R249S mutation (SEQ ID
N0:95); lane 3
contains the reaction products from the 249 (silent) mutation (SEQ ID N0:89);
lane 4
contains the reaction products from the engineered R273H mutation (SEQ ID
N0:97).
Lanes 5-8 contain the cleavage products generated from the sense strand each
of these
templates when incubated in the presence of the CleavaseT"' BN enzyme. Lane ~
contains the
cleavage products from the wild type fragment (SEQ ID N0:84);-lane 6 contains
the cleavage
products from the R249S fragment (SEQ ID N0:94); lane 7 contains the cleavage
products
from the 249 (silent) mutant fragment (SEQ ID N0:88); lane 8 contains the
cleavage products
from the 8273 S fragment (SEQ ID N0:96).
The results shown in Figure 74 demonstrate that similar, but distinctly
different,
patterns of cleavage were generated from each of these templates containing
single-base
changes. Lane 6 shows the attenuation of bands in the 150-180 nucleotide
range, as well as,
the loss of a band in the 100 nucleotide range when compared to the wild-type
pattern shown
in lane 5. In addition, lane 6 shows a new band appearing in the 140
nucleotide range, and
increased intensity in the top band of a doublet at about 120 nucleotides.
Examination of the
silent 249 mutant (lane 7) which differs from wild-type at the same nucleotide
position as
R249S (lane 6), revealed pattern differences relative to both the wild type
(lane 5) as well as
to the R249S (lane 6) mutation. Specifically, comparison to lane 5 shows an
attenuation of
bands in the 150-180 nucleotide range as well as the loss of a band in the 100
nucleotide
range, as was seen in lane 6. However, the sample, in lane 7 does not exhibit
the additional
band in the 140 nucleotide range, nor the increased intensity in the top band
of the doublet in
the 120 nucleotide range seen in lane 6. This result demonstrates that the
CFLPT"1 technique
is capable of distinguishing between changes to a different base at the same
nucleotide "
position.
- 198 -
CA 02203627 1997-04-24
WO 96115267 PCT/US95/14673
Examination of the reaction products in lane 8 reveals the loss of a band in
the 100
nucleotide range in the R273S fragment when compared to the wild-type pattern
in lane 5.
This CFLPT"' pattern is distinct from those in lanes 6 and 7, however, in that
it does not show
attenuation of bands in the 150-180 nucleotide range; in thi:> region of the
gel this pattern is
essentially indistinguishable from that generated from the wild type fragment.
The above results demonstrate that CFLP~ can be used to detect clinically
significant
mutations in the human p53. Further, these results indicate that the CFLPTM
technique is
sufficiently sensitive to distinguish different base changes at the same
position from one
another, as well as from wild type. In addition these results show that the 2-
PCR technique
can be used to generate a collection of PCR fragments containing known p53
mutations; such
a collection allows the generation of a p53 bar code library containing the
CFLPTM patterns
generated by different p53 mutations.
EXAMPLE 31
Detection Of The Presence Of Wild
Type And Mutant Sequences In Mixed Samples
The ability of the CFLP~ reaction to detect the presence of different alleles
of the
same sized PCR fragments in a mixed sample, such as might be found in
heterozygous or
otherwise heterogenous tissue, samples was examined.
PCR products_containing a biotin label on the sense strand were produced and
purified
as described in Example 29 for the wild type p53 (SEQ ID N0:84) and mutant 143
(SEQ ID
N0:86) 601-by fragments. Aliquots of these samples were diluted to a final
concentration of
approximately 12.5 fmols/~,1 and mixed in different proportions to give a
spectrum of ratios of
wild type to mutant DNA. Four microliters of the diluted DNA samples, for an
approximate
total of 50 fmols of DNA in each sample, mixed in various combinations, were
placed in
microfuge tubes and heated to 95 °C for 15 seconds. The tubes were
rapidly cooled to 50°C
and 6 q.l of a diluted enzyme mix containing 0.2 ~.l of the Cleavase'~'' BN
enzyme [SOn~~/q l
1 X CleavaseT"' Dilution Buffer (0.5% NP40, 0.5% Tween 20, 20 mM Tris-Cl, pH
8.0, 50 mM
KCI, 10 q.g/ml BSA)~ , 1 ~l of 10 X CFLPT"'' reaction buffer (100mM MOPS, pH
7.~, 0.5%
NP 40, 0.5% Tween 20), and 1 q,l of 2mM MnCI,. A 10 yl no enzyme control was
set up in
parallel for each PCR fragment examined, with the difference that sterile
distilled water was
substituted for the CleavaseT'~' BN enzyme. After 1.5 minutes at 50°C,
the reactions were
- 199 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95114673
stopped by the addition of 8 ~l of stop buffer. In addition, 4 ~I of wild type
only as well as
4 ~.l of V 143A only were analyzed by the same method for comparison to the
mixed samples.
Samples were heated to 85°C for 2 minutes and 7 p.l of each reaction
were resolved .
by electrophoresis through a 10% polyacrylimide gel ( 19:1 cross-link), with
7M urea, in a '
buffer containing O.SX TBE.
After electrophoresis, the gel plates were separated allowing the gel to
remain flat on
one plate. A 0.2 ~.m-pore positively charged nylon membrane (Schleicher and
Schuell,
Keene, NH), pre-wetted with O.SX TBE, was laid on top of the exposed
acrylamide gel. All
air bubbles trapped between the gel and the membrane were removed. Two pieces
of 3MM
filter paper (Whatman) were then placed on top of the membrane, the other
glass plate was
replaced, and the sandwich was clamped with binder clips. Transfer was allowed
to proceed
overnight. After transfer, the membrane was carefully peeled from the gel and
washed in 1 X
Sequencase Images Blocking Buffer (United States Biochemical) for two I S
minute intervals
with gentle agitation. Three tenths of a ml of the buffer was used per cm' of
membrane. A
streptavidin-alkaline phosphatase conjugate (SAAP, United States Biochemical,
Cleveland,
OH) was added to a 1:300 dilution directly to the blocking solution, and
agitated for 15
minutes. The membrane was washed 3 times (5 min/wash) in 1 X SAAP buffer ( 1
OOmM
Tris-HCI, pH 10; 50 mM NaCI) with 0.1 % SDS, using 0.5 mls/cm' of membrane.
The
membrane was then washed twice in 1 X SAAP buffer without SDS, but containing
1mM
MgCI,, drained thoroughly and placed in a heat sealable bag.- Using a sterile
pipet tip, 0.05
ml/cm' of CDP-Star (Tropix, Bedford, MA) was added to the bag and distributed
over the
membrane for 5 minutes. = The bag was drained of all excess liquid and air
bubbles. The
membrane was then exposed to X-ray film (Kodak XRP) for an initial 30 minute
exposure.
Exposure times were adjusted as necessary for resolution and clarity. The
resulting
autoradiograph is shown in Figure 75.
In Figure 75, the lane marked .M contains biotinylated molecular weight
markers
obtained from Amersham (Arlington Heights, IL) and include bands corresponding
to lengths
of 50, 100, 200, 300, 400s 500, 700, and 1000 nucleotides. Lanes 1 and 2
contain the
reaction products from the no enzyme controls for the wild type and V 143A
mutant
fragments, respectively. Lane 3 contains cleavage products from the sample
containing the
wild type fragment only. Lane 4 contains cleavage products from wild type and
mutant
fragmentsmixed in a 1:1 ratio. Lane ~ contains cleavage products from a
reaction containing
a 1: 2 ratio of wild type to mutant fragment. Lane 6 contains reaction
products present in a '
- 200 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
ratio of wild type to mutant of 1:9. Lane 7 contains cleavage products from a
sample
containing V 143A mutant DNA only. Lane 8 contains cleavage products mixed at
a ratio of
_, wild type to mutant of 2:1. Lane 9 contains cleavage products mixed at a
ratio of wild type
to mutant 4:1. Lane 10 contains cleavage products mixed a ratio of wild type
to mutant to
9:1.
t
The results shown in Figure 75 demonstrate that the presence of different
alleles can
be detected in a mixed sample. Comparison of lanes 4-6 anal lanes 8-10 with
either lane 3 or
lane 7 demonstrates that the lanes containing mixed reactions exhibit distinct
differences from
either sample alone. Specifically, in the 100 nucleotide region, there is a
doublet in the wild
type sample that shifts in the mutant (see discussion of Figure 74 in Example
29). All three
of these bands are present in the mixed samples (lanes 4-6 and lanes 8-10)
whereas only one
or the other pair is detectable in lanes 3 and 7.
EXAMPLE 32
Detection and Identification of Hepatitis C Virus
Genotypes By Cleavase.'~"'' Fragment Length Polymorphism Analysis
Hepatitis C virus (HCV) infection is the predominant cause of post-transfusion
non-A,
non-B (NANB) hepatitis around the world. In addition, HCV is the major
etiologic agent of
hepatocellular carcinoma (HCC) and chronic liver disease world wide. Molecular
biological
analysis of the small (9.4 kb) RNA genome has showed that some regions of the
genome are
very highly conserved between isolates, while other regions are subject to
fairly rapid
mutation. These analyses have allowed these viruses to be divided into six
basic genotype
groups, and then further classified into several sub-types [Altamirano et al.,
J. Infect. Dis.
171:1034 (1995)]. These viral groups are associated with different
geographical areas, and
and accurate identification of the agent in outbreaks is important in
monitoring the disease.
While only genotype 1 HCV has been observed in the United States, multiple HCV
genotypes
have been observed in both Europe and Japan. HCV genotype has also been
associated with
differential efficacy of treatment with interferon, with Group 1 infected
individuals showing
little response. The ability to identify the genotype of HCV present in an
infected individual
allows comparisons of the clinical outcomes from infection by the different
types of HCV,
and from infection by multiple types in a single individual. Pre-screening of
infected
- 201 -
CA 02203627 1997-04-24
WO 96115267 PCTlUS95/14673
individuals for the viral type will allow the clinician to make a more
accurate diagnosis, and
to avoid costly but fruitless drug treatment.
In order to develop a rapid and accurate method of typing HCV present in
infected
individuals, the ability of the Cleavase~ reaction to detect and distinguish
between the major '
genotypes and subtypes of HCV was examined. Plasmids containing DNA derived
from the
conserved 5' untranslated region of six different HCV RNA isolates were used
to generate
templates for analysis in the CFLPTT' reaction. The HCV sequences contained
within these
six plasmids represent genotypes 1 (four sub-types represented; 1 a, 1 b, I c
and Ol c), 2 and 3.
The nomenclature of the HCV genotypes used is that of Simmonds et al. [as
described in
Altamirano et al., supra].
A) Generation of Plasmids Containing HCV Sequences
Six DNA fragments derived from HCV were generated by RT-PCR using RNA
extracted from serum samples of blood donors; these PCR fragments were a gift
of Dr. M.
Altamirano (University of British Columbia, Vancouver). These PCR fragments
represent
HCV sequences derived from HCV genotypes 1 a, 1 b, 1 c, D 1 c, 2c and 3 a.
The RNA extraction, reverse transcription and PCR were performed using
standard
techniques [Altamirano et al., J. Infect. Dis. 171:1034 (1995)]. Briefly, RNA
was extracted
from 100 p,l of serum using guanidine isothiocyanate, sodium lauryl sarkosate
and phenol-
chloroform [Inchauspe et al., Hepatology 14:595 (1991)]. Reverse transcription
was
performed according to the manufacturer's instructions using a GeneAmp rTh
reverse
transcriptase RNA PCR kit (Perkin-Elmer) in the presence of an external
antisense primer.
HCV342. The sequence of the HCV342 primer is 5'-GGTTTTTCTTTGAGGTTTAG-3'
(SEQ ID N0:102). Following termination of the RT reaction, the sense primer
HCV7 [5'-
GCGACACTCCACCATAGAT-3' (SEQ ID N0:103)] and magnesium were added and a first
PCR was performed. Aliquots of the first PCR products were used in the second
(nested)
PCR in the presence of primers HCV46 [5'-CTGTCTTCACGCAGAAAGC-3' (SEQ ID
N0:104)] and HCV308 [5'-GCACGGTCTACGAGACCTC-3' (SEQ ID NO:105)]. The PCRs
produced a 281 by product which corresponds to a.conserved 5' noncoding region
(NCR)
region of HCV between-positions -284 and -4 of the HCV genome [Altramirano eI
crl., .1.
Infect. Dis. 171:1034 (1995)].
.
The six 281 by PCR fragments were used directly for cloning or they were
subjected '
to an additional amplification step using a 50 ~,1 PCR comprising
approximately 100 finoles
of DNA, the HCV46 and HCV308 primers at 0.1 ~.M, 100 pM of all four dNTPs and
2.~ '
-202-
CA 02203627 1997-04-24
WO 96115267 PCT/US95/14673
°-
units of Taq polymerise in a buffer containing 10 mM Tris-HCI, pH"~.~, 50 mM
KCI, 1.5
mM MgCI, and 0.1% Tween 20. The PCRS were cycled 25~ times at 96°C for
45 sec., 55°C
for 45 sec. and 72°C for 1 min. Two microliters,of.either the,original
DNA samples or the
= reamplified PCR products were used for cloning in the linear pT7Blue T-
vector (Novagen,
w 5 Madison,WI) according to manufacturer protocol. After the PCR products
were ligated to the
pT7Blue T-vector; the ligation reaction mixture was used to transform
competent JM 109 cells
(Promega). Clones containing the pT7Blue T-vector with an insert were selected
by the
presence of colonies having a white color on LB plates containing 40 ~.g/ml X-
Gal, 40 l~g/ml
IPTG and 50 ~g/ml ampicillin. Four colonies for each PCR sample were picked
and grown
overnight in 2 ml LB media containing 50 ~.g/ml carbenicillin. Plasmid DNA was
isolated
using the following alkaline miniprep protocol. Cells from 1.5 ml of the
overnight culture
were collected by centrifugation for 2 min. in a microcentrifuge ( 14K rpm),
the supernatant
was discarded and the cell pellet was resuspended in 50 ~,l TE buffer with 10
~.g/ml RNAse
A (Pharmacia). One hundred microliters of a solution containing 0.2N NaOH, 1%
SDS was
added and the cells were lysed for 2 min. The lysate was gently mixed with 100
~,1 of 1.32
M potassium acetate, pH 4.8, and the mixture was centifugated for 4 min. in a
microcentrifuge ( 14K rpm); the pellet comprising cell debris was discarded.
Plasmid DNA
was precipitated from the supernatant with 200 ~.l ethanol and pelleted by
centrifugation a
microcentrifuge (14K rpm). The DNA pellet was air dried forl5 min. and was
then
redissolved in 50 ~.l TE.
To analyze the cloned HCV inserts, 1 ~.1 of plasmid DNA (approximately 10 to
100
ng) reamplified in a 50 p.l PCR using the HCV46 and HCV308 primers as
described above
with the exception that 30 cycles of amplification were employed. The PCR
products were
separated by electrophoresis on a 6% non-denaturing acrylamide gel (29:1 cross
linked) in
O.SX TBE buffer; clones that gave rise to a 281 by PCR product were selected
for further
analysis.
For sequencing purposes, plasmid DNA from selected clones was PEG purified as
follows. To 50 ~1 of plasmid DNA in TE buffer (approximately 10-100 ng/yl), 2~
~l of SM
NaCI and 10 ~I 20% PEG (M.W.8,000; Fisher) was added, mixed well, and the
mixture was
incubated on ice for 1 hour. The mixture was then centrifuged for 5 min in a
table-top
microcentrifuge (at 14K rpm), the pellet was removed and an additional 15 ~.l
of 20% PEG
was added to the supernatant. After incubation for 1 hour on ice, a second
pellet was
collected by centrifugation, the supernatant was discarded, and the pellet was
redissolved in
- 203 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
20 p,l H,O. Two microliters of PEG-purified plasmid DNA (approximately 100 ng)
was used
in cycle-sequencing reactions using the_fmol~' DNA Sequencing System (Promega.
Madison.
WI) according to manufacturer protocol, in conjunction with the HCV46 or
HCV308 primers.
The HCV46 or HCV308 primers were biotinylated at the 5' end using
Oligonucleotide Biotin '
Labeling kit (Amersham, Arlington Heights, IL) prior to use in the sequencing
reactions.
Sequencing reactions were separated on 10% denaturing acrylamide gel.
transferred on nylon
membrane and visualized as described in Example 19.
Alternatively, DNA sequencing was done using either the Blue-Tl [5'-GATCTAC
TAGTCATATGGAT-3' (SEQ ID N0:106)] and Blue-T2 [5'-TCGGTACCCG
GGGATCCGAT-3' (SEQ ID N0:107)] primers labeled at the 5' end with tetra chloro
fluorescein (TET) dye (Integrated DNA Technologies). In this case, the
sequencing reactions
were separated on a 10% denaturing acrylamide gel and the products were
visualized using a
FMBIO-100 Image Analyzer (Hitachi). The six HCV clones were termed HCV1.1,
HCV2.1,
HCV3.1, HCV4.2, HCV6.1and HCV7.1; the double-stranded DNA sequence of these
clones
are listed in SEQ ID NOS:108-113, respectively. The sequence of the sense
strand for each
of the six HCV clones is shown as the top line in SEQ ID NOS:108-113. The
sequence of
the anti-sense strand for HCV clones HCVl.I, HCV2.1, HCV3.1, HCV4.2, HCV6.1
and
HCV7.1 is listed in SEQ ID NOS:114-118, respectively.
The DNA sequences of each of the six HCV clones are aligned in Figure 76. In
Figure 76, nucleotides which represent variations between the six HCV clones
are-indicated
by bold type and underlining; dashes are used to indicate gaps introduced to
maximize
alignment between the sequences (necessary due to the insertion found in clone
HCV4.2).
This alignment shows that these six HCV clones represent six different HCV
genotypes.
HCVI.I represents a genotype lc HCV; HCV2.1 represents a genotype la HCV;
HCV3.1
represents a genotype lb HCV; HCV4.2 represents a genotype lc HCV; HCV6.1
represents a
genotype 2c HCV and HCV7.1 represents a genotype 3a HCV. For one sample,
HCV4.2. an
insertion of an "G" nucleotide was found at position 146 (relative to the
protypical HCV;
Altamirano et al., supra), since no insertion or deletions in the HCV NCR have
been
previously reported, a second independent clone derived from the PCR products
corresponding
to HCV4 was sequenced. This second HCV4 clone was found to have the same
sequence as
that shown for HCV4.2 in Figure 76. '
- 204 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
B) Preparation of HCV Substrates
Six double stranded substrate DNA were prepared for analysis in the CFLP~'~'1
reaction.
The substrates were labelled at the 5' end of either the sense or the anti-
sense strand by the
use of labeled primers in the PCR to permit CFLP~'~"'' analysis of each strand
of the HCV
a 5 DNA substrates.
r
To prepare PCR products for CFLP~'~"'' analysis, the HCV46 and HCV308 primers
were
5' end labeled with TMR dye using the ONLYT"' BODIPY"~ TMR Oligonucleotide
Phosphate
Labeling Kit (Molecular Probes, Inc., Eugene, OR) according to manufacturer
protocol. All
six HCV 281 by NCR sequences were PCR amplified using 10 ng of template and 30
cycles
of amplification as described above in section a).
For sense strand analysis, the PCR was conducted using the HCV46 primer (SEQ
ID
N0:104) labeled with TMR and unlabeled HCV308 primer (SEQ ID NO:105). For
antisense
analysis, the PCR was conducted using unlabeled HCV46 primer (SEQ ID N0:104)
and
HCV308 primer (SEQ ID NO:105) labeled with TMR. The: PCR products were
purified by
I S electrophoresis on a 6% denaturing acrylamide gel and eluted overnight as
described above in
Example 19. The gel-purified DNA substrates were redissolved in 20 pl HBO at
an
approximate concentration of 100 fmoles/~1.
C) Cleavage Reaction Conditions
Cleavage reactions comprised 1 ~,1 of TMR-labeled I'CR products (approximately
100
fmoles of the double-stranded substrates) in a total volume of 10 ~.I 10 mM
MOPS, pH 7.5;
with 0.5% each Tween 20 and NP-40 and 10 ng Cleavase~ BN enzyme. All
components
except the MnCh were assembled in a volume of 8 ~1. The: reactions were heated
to 95°C for
15 seconds to denature the substrates and then quickly cooled to 55°C.
The reaction were
performed in either a thermocycler (MJ Research, Waterto~~n, MA) programmed to
heat to
95°C for 15 seconds and then cool immediately to 55° C or the
tubes were placed manually in
a heat block set at 95°C and then transferred to a second heat block
set at 55°C.
Once the tubes were cooled to the reaction temperature of 55°C, the
cleavage reaction
was started by the addition of 2 p.l of 1 mM MnCI,. After 2 minutes at
55°C, the reactions
were stopped by the addition of 5 ~I of a solution containing 95% formamide.
10 mM EDTA
and 0.02% methyl violet.
i
Five microliters of each reaction mixture were heated at 85°C for 2
min, and where
than resolved by electrophoresis through a 12% denaturing polyacrylamide gel (
19:1 cross
link) with 7M urea in a buffer of O.SX TBE. The gels were run at 33 watts for
1.5 hours.
- 205 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
The labeled reaction products were visualized using the FMBIO-100 Image
Analyzer
(Hitachi), with the resulting fluoroimager scan shown in Figure 77.
In Figure 77, the CFLPTMpatterns produced by cleavage of the six HCV samples
labeled on the sense strand are shown in lanes 1-G; the CFLPT~'' patterns
produced by cleavage '
of the six HCV samples labeled on the anti-sense strand are shown in lanes 7-
12. The
position of molecular weight markers is indicated on the left-hand side of the
fluoroimager
scan by the large arrowheads; the size of the markers is indicated in
nucleotides.
The experiment presented in Figure 77 demonstrates the ability of CFLPT"' to
differentiate six distinct hepatitis C viral subtypes. The six samples in the
left hand side of
the panel (lanes 1-G) were labeled on the 5' end of the sense strand; the six
on the right (lanes
7-12), on the 5' end of the antisense strand. The first four samples in each
set all contain
samples amplified from HCV type 1. Subtypes a, b, and c are represented, as is
a single base
deletion of type 1 c (i. e., Q 1 c). Analysis of either strand points out
numerous similarities as
well as several distinctive differences between the subtypes. Most notable
among the
similarities on the sense strand are prominent bands marked A, B and C.
Specifically,
whereas bands B and C are evident in the patterns generated from both subtypes
I a and 1 b
(and are, in fact, more prominent in subtype 1 b than in 1 a), they are barely
visible in subtype
1 c. Band A, though present in all 4 of these samples, is more prominent in
the patterns
generated from subtypes 1 c and 1 a. Differences between subtypes 2c and 3 a
vs. all of the
subtype 1 samples, are evident in the region between 50 and 100 nt (compare
bands D and E)
on the sense strand and between the 80 and 150 nt on the antisense strand
(compare bands F-
J). Viral type 2 gives rise to the most significantly altered CFLPTM pattern,
while type 3
appears to be similar to type 1; these relationships appear to be consistent
with the relative
number of sequence differences between the different isolates.
The results shown in Figure 77 demonstrate that the CFLPT"'' method provides a
simple
and rapid method to determine the genotype of HCV strains. This method will
facilitate the
diagnosis of HCV infection, permit appropriate treatment of HCV-infected
patients, and aid in
the monitoring of HCV outbreaks.
P
- 20G -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
EXAMPLE 33
Detection of Mutations Associated With
Antiobiotic Resistance in Mvcobacterium tuberculosis '
In the past decade there has been a tremendous resurgence in the incidence of
' tuberculosis in this country and throughout the world. Worldwide, the number
of new cases
reported annually is forecast to increase from 7.5 million in 1990 to 10.2
million by the year
2000. An alarming feature of this resurgence in tuberculosis is the increasing
numbers of
patients presenting with strains of M. tuberculosis which are resistant to one
or more
antituberculosis drugs [i.e., mufti-drug resistant tuberculosis (MDR-TB)].
Resistance to either or both of the antibiotics rifampin (rift and isoniazid
(inh) is the
standard by which M. tuberculosis strains are judged to be mufti-drug
resistant. Both because
of their potent bactericidal activities and because acquisition. of primary
resistance to these
drugs is rare (the spontaneous mutation rate of resistance to rifampin is
approximately 10-h
and to isoniazid, 10-8 to 10-9), until very recently, these two antibiotics
were among the most
powerful front-line drugs used to combat the advance and spread of
tuberculosis. However
surveys of tuberculosis patients in the U.S. reveal that as many as one-third
were infected
with strains resistant to one or more antituberculosis drugs; greater than 25%
of the M.
tuberculosis cultures isolated were resistant to isoniazid and 19% were
resistant to both
isoniazid and rifampin [Frieden et al., New Eng. J. Med. 328:521 (1993)].
As discussed above (Description of the Invention), resistance to rifampin is
associated
with mutation of the rpoB gene in M. tuberculosis. While the exact mechanism
of resistance
to isoniazid is not clear, the majority (as many as 80%) of inh' mutations
occur in the katG
and inhA genes of M. tuberculosis. To investigate whether CFLP'~"'' could be
used to detect
mutations in the genes involved in MDR-TB, DNA fragments were amplified from
the yoB
and katG genes of M. tuberculosis. DNA fragments derived from wild-type (i.
e., antibiotic-
sensitive) or mutant (i.e., antibiotic-resistant) strains of M. tuberculosis
were subjected to
CFLPTM analysis.
A) CFLPTn' Analysis of Mutations in the RpoB Gene of M. tuberculosis
i) Generation of Plasmids Containing RpoB Geue Sequences
Genomic DNA isolated from wild-type M. tuberculosis or M. tuberculosis strains
containing mutations in the rpoB gene associated with rifarnpin resistance
were obtained from
Dr. T. Schinnick (Centers for Disease Control and Prevention. Atlanta, GA).
The rifampin
resistant strain #13 (91-3083) contains a tyrosine residue at codon 451 of the
rpoB gene in
- 207 -
CA 02203627 1997-04-24
WO 96/15267 PCT/U595/14673
place of the histidine residue found in the wild-type strain (i. e., H451 Y);
this mutation is is
present in 28% of rifampin resistant TB isolates. The H451 Y mutation is
hereinafter referred
to as mutant 1. The rifampin resistant strain #56 (91-2763) contains a Ieucine
residue at
codon 456 of the rpoB gene in place of the serine residue found in_the wild-
type strain (i.e., '
S456L); this mutation is present in 52% of rifampin resistant TB isolates. The
S456L
mutation is hereinafter referred to as mutant 2.
A 620 by region of the TB rpoB gene was amplified using the PCR from DNA
derived from the wild-type and mutant 1 and mutant 2 strains. The primers used
to amplify
the rpoB gene sequences v~ere PoIB-SA [S'-ATCAACATCCGGCCGGTGGT-3' (SEQ ID
N0:120] and PoIB-SB [5'-GGGGCCTCGCTACGGACCAG-3' (SEQ ID N0:121 )]; these
PCR primers amplify a 620 by region of the rpoB gene which spans both the H451
Y and
S456L mutations [Miller et al., Antimicrob. Agents Chemother., 38:805 (1994)].
The PCRs
were conducted in a final reaction volume of 50 q.I containing the PoIB-SA and
PoIB-SB
primers at 1 ~.M, 1X PCR buffer and 60 ~M of all four dNTPs. The reaction
mixture was
heated at 95°C for 3 min.- Amplification was started by the addition of
2.5 units of Tcrcf
polymerase and was continued for 35 cycles at 95°C for 1 min,
60°C for 1 min and 72°C for
2 min.
To clone the PCR-amplified fragments, 1 ~.I of each PCR product was used for
ligation in the linear pT7Blue T-vector (Novagen, Madison,WI). The ligation
products were
used to transform competent JM109 cells and clones containing pT7Blue T-vector
with an
insert were selected by white color on LB plates containing 40 ~.g/ml X-Gal,
40 ~g/ml IPTG
and 50 ~.g/ml ampicillin. For each PCR sample (i.e., wild-type and mutants 1
and 2), five
independent colonies were picked and grown overnight in 2 ml of LB media
containing 50
qg/ml carbenicillin. Plasmid DNA was isolated using the alkaline miniprep
protocol
described above in Example 32.
To analyze the cloned fragments, 1 ~.l of plasmid DNA from each clone was
amplified
by PCR using 50 ~.1 reaction containing. the PoIB-SA and PoIB-SB primers at 1
q.M. 1X PCR
buffer, 60 q.M of all 4 dNTPs and 2.5 units of-Taq.polymerase. The PCRs were
cycled 35
times at 95°C forl min, 60°C for 1 min and 72°C for 2
min. The PCR products were
separated by electrophoresis on a 6% native polyacrylamide gel in O.SX TBE
buffer and
clones that gave rise to a 620 by fragment were selected for further analysis.
For sequencing purposes, plasmid DNA from selected clones was PEG-purified as
described in Example 32. Two microliters of PEG-purified plasmid DNA
(approximately 100 '
- 208 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95114673
ng) was used for cycle-sequencing with fmol~ kit (Promega, Madison, WI) in
conjunction
with the PoIB-SA and PoIB-SB primers containing a biotin moiety at the ~' end.
Biotinylation of the primers was performed using an Oligonucleotide Biotin
Labeling kit
(Amersham). Sequencing reactions were separated in a 8% denaturing
polyacrylamide gel,
transferred to a nylon membrane and visualized as described above in Example
19. The DNA
sequences of the 620 by rpoB gene fragment derived from l:he wild-type, mutant
1 and mutant
2 strains are listed in SEQ ID NOS:122-124. The sequence of the sense strand
for each of
the three TB strains is shown as the top line in SEQ ID NOS:122-124. The
sequence of the
anti-sense strand for the wild-type, mutant 1 and mutant 2 TB strains is
listed in SEQ ID
NOS:125-127, respectively.
ii) Preparation of M. tuberculosis rpoB Gene Substrates
In order to generate substrates for use in CFLPT"'' reactions, the cloned 620
by
fragment derived from the wild type and mutants 1 and 2 rpoB gene were
amplified using the
PCR. The PCRs were conducted using one primer of the primer pair labeled at
the 5' end so
that the resulting PCR product would permit the analysis of either the sense
or anti-sense
strand of the rpoB gene fragments. In order to generate substrates labelled on
the anti-sense
strand, ten nanograms of plasmid DNA from the sequenced clones was used as the
template in
50 Ld reactions containing 1 ~.M of each the PoIB-SA primer (unlabelled) and
PoIB-SB primer
biotinylated at the S' end using Oligonucleotide Biotin Labeling kit
(Amersham), 1X PCR
buffer, 60 q.M of all 4 dNTPs and 2.5 units of Taq polymerase. The reactions
were cycled 35
times at 95°C for 1 min, 60°C for 1 min and 72°C for 2
min. The resulting 620 by PCR
products containing a biotin-labeled antisense strand were gel-purified as
described in
Example 19. The purified fragments were dissolved in 20 pl HBO.
To generate substrates labelled on the sense strand of the 620 by fragment of
~poB
gene fragments (wild-type and mutants 1 and 2), the PCRs were conducted using
1 ~.M each
PoIB-SA primer 5' end labeled with TMR dye using ONLY~'~'~' BODIPY~" TMR
Oligonucleotide Phosphate Labeling Kit (Molecular Probes, Inc., Eugene, OR)
and unlabeled
° PoIB-SB primer. The PCR reactions also contained 1X PCR buffer, 60
q.M of all 4 dNTPs, 5
units -of Taq polymerase and 10 ng of plasmid DNA from the sequenced clones as
a template
" 30 in a final volume of 100 ~,1. The reactions were cycled for a total of 35
cycles comprising
95°C for 1 min, 60°C for 1 min and 72°C for 2 min.
In addition to the above PCR conditions, the PCR reactions were also conducted
using
dUTP in place of dTTP to generate uridine-containing PCR fragments. Uridine-
containing
- 209 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
PCR fragments have become the standard type of PCR fragment analyzed in
clinical
laboratories. In order to demonstrate that uridine-containing PCRfragments can
be used to
produce distinct CFLP~'' patterns from substrates which vary by a single base
pair change
within a 620 by fragment, rpoB gene fragments containing a 5' TMR label on the
sense
strand and uridine in place of thymidine were generated as follows. Uridine-
containing 620
by fragments (wild-type and mutants l and 2) were amplified according to the
PCR protocol
described above for the generation of fragments labelled at the 5' end of the
sense strand with
TMR -with the exception that 2.5 mM MgCI, was used in place- of 1.5 mM MgCh
and 100
p.M dATP, 100 p.M dCTP, 100 p.M dGTP and 200 p.M dUTP were used in place of
the
mixture containing 60 p.M each of all 4 dNTPs (i.e., dATP, dCTP, dGTP and
dTTP).
The 620 by PCR products containing a TMR-labeled sense strand (either uridine-
or
thymidine-containing) were purified in 6% denaturing gel as described above,
eluted
overnight, precipitated with ethanol and redissolved in 20 p.l HBO as
described in Example 19,
for a concentration of approximately 15 fmoles/~l.
iii) Cleavage Reaction Conditions -
Cleavage reaction conditions for analysis of the 620 by rpoB fragments
containing a
biotin-labelled antisense strand were as follows. Six microliters of biotin
labeled PCR product
were combined with 1 p,l of lOX CFLP~'~"'' buffer (100 mM MOPS, pH 7.5, 0.5%
each Tween
and NP-40) and 25 ng of the Cleavase~ BN enzyme. Prior to the initiation of
the
20 cleavage reaction, the DNA mixtures were denatured by incubation at
95°C for 10 sec. The
reactions were then cooled to 60°C and reaction was started by the
addition of 1 p,l of 2 mM
MnCI,. The cleavage reactions were conducted at 60°C for 2 min.
Cleavage reactions were
stopped after 2 min. by adding 5 p.l of stop buffer. Six microliters of each
sample were
resolved by electrophoresis on a 6% denaturing polyacrylamide gel and labeled
fragments
were visualized as described in Example 19. The resulting autoradiogram is
shown in Figure
78.
In Figure 78, the lane marked "M" contains biotinylated molecular weight
markers
obtained from Amersham (Arlington Heights, IL) and include bands corresponding
to lengths
of 200, -300, 400, 500 nucleotides. The size of the markers and of the
uncleaved yoB
substrates (620) is indicated on the left-hand side of the autoradiograph
using large ,
arrowheads. Lanes 1-3 contain the reaction products generated by the cleavage
of the mutant
l, wild-type and mutant 2 substrates labelled on the anti-sense strand,
respectively. The
-210-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95114673
distance of the point mutation (relative to the wild-type sequence) from the
5' end label was
511 nucleotides for the mutant 1 substrate and 49~ nucleotides for the mutant
2 substrate.
- The results shown in Figure 78 demonstrate that similar, but distinctly
different
patterns of cleavage were generated from the each of the rpoB substrates
labelled on the anti-
sense strand. In comparison with the cleavage pattern generated by the wild-
type substrate,
' the pattern generated by cleavage of the mutant 1 substrate shows a
disappearance of Band A.
A comparison of the pattern generated by cleavage of the wild-type and mutant
2 substrates
shows that the mutant 2 substrate has a significant reduction of intensity of
Band B. Thus,
the two mutants can be distinguished from the wild-type and from each other.
Cleavage reaction conditions for analysis of the 620 by rpoB fragments
containing a
TMR-labelled sense strand were as follows. Four microliters of TMR-labeled PCR
product
were cleaved as described above. Cleavage reactions were stopped after 2 min.
by adding 5
p.l 95% formamide, 10 mM EDTA and 0.02% of methyl violet (Sigma).
The reactions were heated to 85°C for 2 min. and five microliters of
each reaction
mixture were resolved by electrophoresis through a 12% denaturing
polyacrylamide gel ( 19:1
cross link) with 7M urea in a buffer containing O.SX TBE. The gel was run at
33W (watts)
for 1.5 hours. The labeled reaction products were visualized using the FMBIO-
100 Image
Analyzer (Hitachi) with the resulting fluoroimager scan shown in Figure 79,
Panel A. the gel
was then electrophoresed for another 1 hour, and the second scan is shown in
Panel B.
In Figure 79, two panels, A and B, are shown. Panel B represents a scan of the
same
gel shown in Panel A following a longer period of electrophoresis than that
shown in Panel
A. Thus, Panel B serves to spread out the banding pattern seen in the upper
portion of Panel
A (lines connecting Panels A and B show the region of expansion). In Figure
79, Panels A
and B, lanes 1-4 contain the reaction products produced by cleavage of
thymidine-containing
substrates having a TMR-label on the sense strand derived from the mutant 1,
wild-type.
mutant 2 and a mixture of the wild-type and mutant 2 substrates, respectively.
Lane 5 of
Panels A and B contains the 157 by fragment derived from exon 4 of the
tyrosinase gene
(SEQ ID N0:27) labeled with TET as a marker. Lanes 6-9 of Panels A and B
contain the
reaction products produced by cleavage of uridine-containing substrates having
a TMR-label
on the sense strand derived from the mutant 1, wild-type, mutant 2 and a
mixture of the wild-
type and mutant 2 substrates. respectively. Mixtures of the wild-type and
mutant 2 substrates
(lanes 4 and 9) were generated by mixing together 5 ~.l of each substrate
after the cleavage
reaction; 6 ~1 of the mixture was then loaded on the gel. The distance of the
point mutation
- 211 -
CA 02203627 1997-04-24
WO 96/15267 pC"T~JS95/14673
(relative to the wild-type sequence) from the 5' end label was 100 nucleotides
for the mutant
1 substrate and 116 nucleotides for the mutant 2 substrate.
The results shown in Figure 79 demonstrate that similar, but distinctly
different
patterns of cleavage were generated from the each of the rpoB substrates
labelled on the sense '
strand. The left hand set of each panel contains CFLPTM patterns generated
from PCR .
products containing dNTPs, while the right hand side contains CFLPTM patterns
generated
from PCR products in which dUTP was substituted for dTTP. Comparison of the
CFLPTM
patterns generated from dNTP-containing amplicons of mutant 1 and wild-type
reveals a
marked reduction in intensity of a band approximately 80 nt from the labeled
~' end (band
A), in the vicinity of the sequence change in this mutant (100 by from the
labeled 5' end). In
addition, a band migrating at approximately 200-250 nt from the labeled 5' end
(band B) is
missing in mutant 1. In contrast, comparison of the patterns generated from
wild-type and
mutant 2 reveals the loss of a band 120 nt from the labeled 5' end (band C).
Furthermore,
examination of the region of the gel corresponding to 120 nt shows,
particularly in Panel B,
that band D is shifted downward in mutant 2 relative to wild-type. In Panel B,
another band,
migrating just above band D (labeled band D') also appears to be shifted
downward in mutant
2 relative to wild-type. Lane 4 of each panel, in which aliquots from the wild-
type and
mutant CFLP reactions were mixed prior to electrophoresis demonstrates that
this shift (in
band D') in mutant 2 is real and not due to an electrophoresis artifact.
Examination of the CFLPTM patterns generated from the dUTP-containing
amplicons
demonstrates that the ability to distinguish these mutants from one another,
as well as from
the wt, is not adversely affected by substitution of dUTP for dTTP and may, in
fact, be
enhanced. In this example, both mutants 1 and 2 are more readily distinguished
from the w~t
when the patterns are generated from amplicons containing dUTP than dTTP. In
the right-
hand portion of panel A, comparison of the lanes containing mutant 1 and wt
reveals several
distinctive differences between the two amplicons, while others are new and
unanticipated.
Specifically, band A is reduced in intensity in the mutant, as compared to the
wt, in much the
same way that it is in the left-hand portion of this panel. A band migrating
at approximately -
110 nt (band E) appears to be missing from the mutant, as does a band at
approximately 250
nt (compare to band B in the left-hand portion of the gel). In addition, the
strong band
labeled F, while not noticeably different in the three samples containing
dTTP, is much '
stronger in the wt pattern generated from dUTP-containing amplicons than it is
in the
mutants. Comparison of the patterns- generated from wt and mutant 2 also
reveals a number '
- 212 -
CA 02203627 1997-04-24
WO 96/15267
PGT/US95/146_73
of pronounced differences. Most notably, a band migrating at approximately 60
nt appears in
mutant 2 (band G), as does a complex of 2 new bands migrating at approximately
150 nt
(band H). Interestingly, while some of the elements that make each of these
patterns distinct
from one another are different if dUTP is substituted for dTTP in the PCR
amplification, the
vast majority of the cleavage fragments are identical in the two experiments.
This result
suggests that substitution of dUTP results in subtle alterations in the single-
stranded DNA
substrate which may be the result of altered stability of secondary structures
or an altered
affinity of the CleavaseT"' enzyme for secondary structures containing
modified nucleotides.
These differences in CleavaseTM-based recognition of secondary structures in
DNA fragments
containing dUTP provides an unexpected benefit of using; this nucleotide
substitution.
B) CFLPT"' Analysis of Mutations in the KrntG Gene of M. tuberculosis
Generation of Plasmids Containing Kate Gene Sequences
Genomic DNA isolated from wild-type M. tuberculosis or M. tuberculosis strains
containing mutations in the katG gene associated with isoniazid resistance
were obtained from
Dr. J. Uhl (Mayo Clinic, Rochester, MN). These four strains are termed wild-
type, S315T,
R463L and S315T;R463L [Cockerill, III et al, J. Infect. Dis. 171:240 (1995).
Strain S315T
contains a G to C mutation in codon 315 of the wild-type katG gene. Strain
R463L contains
a G to T mutation in codon 463 of the wild-type gene and strain S315T;R463L
contains both
the G to C mutation in codon 315 and the G to T mutation in codon 463.
A 620 by region of the M. tuberculosis katG gene was amplified using the PCR
from
DNA derived from the above four strains. The primers used to amplify the katG
gene
sequences were Kat~04 [5'-AGCTCGTATGGCACCGGAAC-3' (SEQ ID N0:128) and
Kate 1523 [5'-TTGACCTCCCACCCGACTTG-3' (SEQ ID N0:129)]; these primers amplify
a 620 by region of katG gene which spans both the S315T and R463L mutations.
The PCRs
were conducted in a final reaction volume of 100 ~,l and contained the KatG904
and
KatG1523 primers at 0.5 ~.M, 1X PCR buffer, 60 pM of all 4 dNTPs. The reaction
mixtures
were heated at 95°C for 3 min, then amplification was started with
addition of ~ units of Taq
polymerase and continued for 35 cycles at 95°C for 1 min, 60°C
for 1 min and 72°C for 2
min.
To clone the PCR-amplified katG fragments, 1 p.l of each PCR product was used
for
ligation into the linear pT7Blue T-vector (Novagen, Madison,WI). The ligation
products were
used to transform competent JM109 cells and clones containing pT7Blue T-vector
with an
insert were selected by white color on LB plates containing 40 p,g/ml X-Gal.
40 ~g/ml IPTG
- 213 -
CA 02203627 1997-04-24
WO 96/15267 PGT/US95/14673
and 50 p.g/ml ampicillin. For each of the four PCR samples, four colonies were
picked and
grown overnight in 2 ml LB media containing 50 p,g/ml carbenicillin. Plasmid
DNA was
isolated using the alkaline miniprep protocol described in Example 32.
To analyze the cloned katv fragments, 1 p.l of plasmid DNA from each clone was
S amplified by PCR using 100 p.l reactions containing the KatG904 and Kate
1523 primers at ,
0.5 p.M IX PCR buffer, 60 pM of all 4 dNTPs and 5 units of Tag polymerase. The
PCRs
were cycled 35 times at 95°C for 1 min, 60°C for 1 min and
72°C for 2 min. PCR products
were separated by electrophoresis on a 6% native polyacrylamide gel in O.SX
TBE buffer and
clones that gave rise to a 620 by fragment were selected for further analysis.
For sequencing purposes, plasmid DNA from selected clones was PEG-purified
according to the protocol described in Example 32. Two microliters of plasmid
DNA
(approximately 100ng) was used for cycle-sequencing with fmolR kit (Promega,
Madison. WI)
in conjunction with the KatG904 and KatG1523 primers containing a biotin
moiety at the 5'
end. Biotinylation of the primers was performed using an Oligonucleotide
Biotin Labeling kit
I S (Amersham). Sequencing reactions were separated in a 8% denaturing
polyacrylamide gel.
transferred to a nylon membrane and visualized as described above in Example
19. The DNA
sequences of the 620 by katG gene fragments from the wild-type and mutant
strains S315T.
R463L and S315T;R463L are listed in SEQ ID NOS:130-133, respectively. The
sequence of
the sense strand for each of the four katG gene fragments is shown as the top
line in SEQ ID
NOS:130-133, respectively. The sequence of the anti-sense strand of the 620 by
katG gene
fragments from the wild-type and mutant strains S315T, R463L and S315T;R463L
is listed in
SEQ ID NOS:134-137, respectively.
ii) Preparation of M. tuberculosis Kate Gene Substrates
In order to generate substrates for use in CFLPT"'' reactions, the cloned 620
by
fragments derived from the wild-type and S315T, R463L and S315T;R463L M.
tuherculosi.s
strains were amplified using the PCR. The PCRs were conducted in a final
reaction volume
of 100 p.l and contained 0.5 p.M each KatG904 and Kate 1523 primers, I X PCR
buffer. 60
mM of all 4 dNTPs, 5 units of Taq polymerase and 10 ng of plasmid DNA from the
sequenced clones as a template. The reactions were cycled 35 times at
95°C for 1 min. 60'C
for 1 min and 72°C for 2 min. '
To obtain 620 by PCR fragments of the katG gene having a biotin label on the
sense
strand. and unlabeled KatG1523 primer (SEQ ID N0:129) and S"-biotinylated
KatG904
primer (SEQ ID N0:128) was used in the PCR; biotinylation was achieved using
the
- 214 -
CA 02203627 1997-04-24
WO 96/1267
PCT/US95/14673
Oligonucleotide Biotin Labeling kit (Amersham). To produce the same fragments
having the
TMR label on the antisense strand, unlabeled KatG904 (SEQ ID N0:128) and TMR-
labeled
KatG1523 (SEQ ID N0:129) primers were used in the PCR. Amplified PCR products
were
purified on a 6% denaturing gel, eluted overnight, precipitated with ethanol
and redissolved in
s.
50 p,l H,O as described in Example 19.
iii) Cleavage Reaction Conditions
The cleavage reaction conditions for analysis of Iu~tG substrates labelled on
the sense
strand were as follows. Five microliters of biotin labeled PCR product were
combined with 1
p,l of lOX CFLPTM buffer (100 mM MOPS, pH 7.5, 0.5°ro each Tween 20 and
NP-40) and 25
ng Cleavase~'~"'' BN enzyme. Prior to the initiation of the cleavage reaction,
the DNA
mixtures were denatured by incubation at 95°C for 10 sec. The reactions
were then cooled to
50°C and the reaction was started by the addition of 1 p,l of 2 mM
MnCh. The cleavage
reactions were incubated for 2 min. at 50°C and were stopped by adding
5 p.l of stop buffer.
Four and one-half microliters of each sample were run on a 10% denaturing
polyacrylamide
1 S gel and labeled fragments were visualized following transfer to a nylon
membrane as
described in Example I9. The resulting autoradiogram is shown in Figure 80.
In Figure 80, lanes marked "M" contain biotinylated molecular weight markers
obtained from Amersham (Arlington Heights, IL) and include bands corresponding
to lengths
of S0, I00, 200, 300. 400, 500, 700, and 1000 nucleotides; the size of the
markers is indicated
by the use of large arrowheads. Lanes 1-4 contain the reaction products
obtained by
incubating the R463L. R463L;S315T, S315T and wild-type katG substrates in the
presence of
Cleavase'~"'' BN enzyme, respectively. The mutation distance from the 5' end
label is 485
nucleotides for the R463L mutation and 41 nucleotides for the S315T mutation
when the label
is present on the sense strand.
The results shown in Figure 80 demonstrate that similar, but distinctly
different
patterns of cleavage were generated from the wild-type and S315L mutant (seen
in both the
S3I ST and S3 I5;R463L substrates) katG substrates labelled on the sense
strand. Comparison
' of the CFLP'~"'' pattern for wild-type fragment (lane 4) shows that the
S315T mutation ( seen in
both mutants R463L:S315T and S315T; lanes 2 and 3) results in disappearance of
Band B
which is located around 40 nucleotides from the end label i.n the wild-type
substrate. The
disappearance of Band B correlates very well with the distance of S315T
mutation from the
5' end (41 nucleotides from the 5' end label on the sense strand). Subsequent
experiments
' have demonstrated that the R463L mutant can be distinguished from wild-type
by a mobiliti~
- 21~ -
CA 02203627 1997-04-24
WO 96/15267 PGT/US95/14673
shift in a band migrating at approximately 500 nt from the S' end label on the
sense strand
(the band shifts downward in the R463L mutant), but is difficult to resolve in
many gels
systems.
The cleavage reaction conditions for analysis of katG substrates labeled on
the anti-
s sense strand were as described for the sense strand. Four and one-half
microliters of each
sample were run on a 10% denaturing polyacrylamide gel and labeled fragments
were
visualized using the Hitachi FMBIO-100 fluoroimager as described in Example
33(a)(iii).
The resulting scan is shown in Figure 81.
In Figure 81, lanes marked "M" contain plasmid pUCl9 DNA digested with MspI
and
3' end labeled with fluorescein ddUTP using terminal deoxynucleotidyl
transferase as
described in Example 8. This marker includes bands corresponding to lengths
off 10/111,
147, 190, 242, 331, 404, 489 and 501 bp. Additional marker bands of 26, 34.
and 67 by are
not visible in this figure; the size of the markers is indicated by the use of
large arrowheads.
Lanes 1-4 contain the reaction products obtained by incubating the R436L.
S31~T:R463L.
S315T, and wild-type katG substrates in the presence of CleavaseT"'' BN
enzyme, respectively.
The location of the single base mutation from the 5' end label is 136
nucleotides for the
R463L mutation and 580 nucleotides for the S315T mutation when the label is
present on the
anti-sense strand.
The results shown in Figure 81 demonstrate that wild-type can be distinguished
from
mutants containing the R463L substitution on the anti-sense strand. Comparison
of the lanes
containing the S315T;R463L double mutant or the R463L mutant by itself
demonstrates that
the R463L mutation is associated with the presence of a strong band migrating
at
approximately 130 nt (band A). This result, taken with that presented in
Figure 80.
demonstrates that all three of these mutants can be distinguished from one
another. as well as
from wild type, by CFLP~'~"'' analysis.
The CFLP~'~"'' technology offers cost benefits by reducing gel electrophoresis
processing
time from 12-18 hours down to 5 to 10 minutes. Adapting the readout to mufti-
lane
Fluorescence Image Detectors allows for an expanded volume of work by allowing
simultaneous processing of up to 48 reactions. The consequent decrease in
turnaround time in
performing the analyses reduces the turnaround time of reporting patient
results from days to
hours, or, as in the case of MDR-TB patients, from weeks to hours. Early
detection of MDR-
TB can save thousands of dollars per patient by reducing the expense of
extended stays in
isolation wards, spent while testing various antibiotic treatments for
efficacy.
-216-
CA 02203627 1997-04-24
WO 96115267
PCT/US95/14673
EXAMPLE 34
Rapid Identification of Bacterial Strains by(''Fr pT" ~awsis
' The results shown above demonstrated that CFLPT"~ analysis can be used to
detect the
Y Y
presence of wild-type and drug-resistant mutations of M. tuberculosis by
examining portions
of gene associated with drug resistance (e.g., rpoB and katG). In order to
examine whether
the CFLP~'~"' analysis could be used as a method of detecting and identifying
a wide variety of -.
microorganisms, CFLP'~"' analysis was conducted using substrates derived from
bacterial 16S
rRNA genes.
Bacterial 16S rRNA genes vary throughout the phylogenetic tree; these genes do
contain segments which are conserved at the species, genus or kingdom level.
These features
have been exploited to generate primers containing consensus sequences which
flank regions
of variability. These primers have been used to amplify segments of bacterial
16S rRNA
genes which are then characterized by either Southern blot hybridization
[Greisen et al.. .I.
Clip. Microbiol. 32:335 (1994)] or SSCP analysis [Widjojoatmondjo et al., J.
Clin. Microhiol.
32:3002 (1994)]. These types of analysis, while faster than traditional
culturing methods, are
at best limited to the differentiation of species within a particular genus
and higher bacterial
taxons. However, it is often desirable to differentiate between different
strains of the same
species. For example, a given species may contain subspecies which comprise
harmless as
well as pathogenic organisms. In order to develop a technique which would
allow the
differentiation between species and/or subspecies, CFLPT"' analysis was
applied to segments
derived from bacterial 16S rRNA genes.
A) Bacterial Strains
Table 3 below lists the bacterial strains used in this study. These strains
were derived
from the ATCC strains listed below with the exception of .Desulfurococcus
amylolyticus Strain
Z-533 which was derived from a deposit obtained from the Deutsche Sammlung von
Mikroorganismen (DSM).
f
-217-
CA 02203627 1997-04-24
WO 96/15267 PGT/US95/14673
TABLE 3
ORGANISM STRAIN NO. CHARACTERISTICS
E. coli ATCC 11303 Strain B -
E. coli ATCC 14948 Derived from E. coli strain
K-12
E. coli Serotype 01~7:H7ATCC 43895 Produces Shiga-like toxins ,
I and II
Campylobacter jejuni ATCC 33291 Isolated from human stool
subsp. jejuni
Shigella dysenteriae ATCC 29027 Isolated from human stool
Serotype 2
Salmonella choleraesui.sATCC 6539 Used for germicide testing
subsp. choleraesuis
Serotype typhi
Staphylococcus aureusMethicillin-
subsp. aureus ATCC resistant
33591
S. aureus subsp. aureusATCC 33592 Gentamicin- and methicillin-resistant
S. aureus subsp. aureusATCC 13565 Produces enterotoxin A and
large
amounts of beta-hemolysin
Staphylococcus hominisATCC 29885 Methicillin control for MIC
testing
Staphylococcus warneriATCC 17917 Used for soap germicide testing
Desulfurococcus STRAIN 3822 hermophilic archaebacterium
T
amylolyticus
The strains listed in Table 3 represent pathogenic microorganisms with the
exception
of E. coli strains B and K-12 and Desulfurococcus amylolyticus.
Desulfurococcus
amylolyticus was included in this study to determine whether the consensus
primers, whose
design was based upon known rRNA gene sequences, could also be used to amplify
rRNA
gene fragments sequences from archeabacterial species whose rRNA gene
sequences have not
been reported. The strains listed in Table 3 were selected to provide
representatives from
several different genera (e.~, Escherichia, Shigella, Salmonella,
Campylobacter. etc.) as well
as to provide several representatives of different species (or subspecies)
within a given genus.
For example, three different strains of E. coli were chosen so that the
consistency (or lack
thereof) of the CFLP~ banding pattern generated by cleavage of an rRNA gene
substrate
could be examined between species within a given genus. In addition, E. coli
Serotype
0157:H7 was examined as this strain has been implicated in hemorrhagic colitis
outbreaks. It "_
was of interest to examine whether the CFLP~ pattern observed from clevage of
a rRNA
gene substrate from E. coli strains B or K-12 differed from that produced by
cleavage of a _
rRNA gene substrate from E. coli Serotype 0157:H7.
-218-
CA 02203627 1997-04-24
WO 96/15267
PGT/US95/14673
Table 4 below describes the phylogenic relationship between the strains used
in this example.
.. TABLE 4
Phylogenetic Position of Strains from Prokaryotic Small SubUnit rRNA Taxonomic
List'
1 ARCHAEA
1.2 CRENARCHAEOTA '
1.2.1 CRENARCHAEOTA-GROUP-I
Desul furococcus amylolyticus
2 BACTERIA
2.13 PURPLE-BACTERIA
2.13.3 GAMMA-SUBDIVISION
2.13.3.15 ENTERICS AND RELATIVES
2.13.3.15.2ESCHERICHIA-SALMONELL A ASSEMBLAGE
Escherichia coli Strain B
Escherichia coli Strain K-12-derived
Escherichia coli Serotype 0157: H7
Shigella dysenteriae Seroty.pe 2
Salmonella choleraesuis subsp. choleraesuis
Serotype typhi
2.13.5 EPSILON-SUBDIVISION
2.13.5.2 CAMPYLOBACTER AND RELATIVES
Campylobacter.jejuni subsp..jejuni
2.15 GRAM-POSITIVE PHYLUM
2.15.5 BACILLUS-LACTOBACILLUS-STREPTOCOCCUS
SUBDIVISION
2.15.5.10 STAPHYLOCOCCUS GROUP
2.15.5.10.2STAPHYLOCOCCUS SUBGROUP
Staphylococcus aureus subsp. aureus ATCC
33591
Staphylococcus aureus subsp. aureus ATCC
33592
Staphylococcus aureus subsp. aureus ATCC
13565
Staphylococcus hominis
Staphylococcus warneri
' Data derived from the Ribosomal Database Project; available on the Internet
at
http://rdp.life.uiuc.edu/index.html; Maidak et al. Nucleic Acids Res.. 22:348
( 1994).
r
- 219 -
CA 02203627 1997-04-24
WO 96/15267 PCTlUS95/14673
B) Growth of Microorganisms
In order to minimize handling of the pathogenic strains, the microorganisms
were
grown on slant cultures or on plates rather than in liquid culture.
i) Growth of Escherichia, Shigella, Salmonella, and Staphvlococcus species
All strains were derived from the ATCC strains listed above in Table 3 as
follows. A ~,
loopful of a culture previously frozen in Trypticase Soy Broth and 15%
glycerol (Remel
Corp., Lenexa, KS. Cat. 06-5024) was subcultured onto a trypticase soy agar
slant (Remel,
Cat. 06-4860). The cultures were incubated overnight at 37°C.
ii) Growth of Campylobacter species
A loopful of a culture previously frozen in Trypticase Soy Broth and 15%
glycerol
(Remel Corp., Lenexa, KS. Cat. 06-5024) was subcultured onto Campylobacter
Agar
supplemented with 10% sheep blood, amphotericin B, cephalothin, trimethoprim.
vancomycin,
and polymyxin B (BBL, Cat. 21727). Inoculated plates were sealed in Campy
microaerophilic pouches (BBL, Cat. 4360656) and incubated at 42°C for 3
days.
C) Extraction of Genomic DNA from Microorganisms
For each bacterial sample, 300 p.l of TE buffer and 300 ~I
phenol:chloroform:isoamyl
alcohol (25:24:1 ) were placed in a 1.5 ml microfuge tube. This combination is
referred to as
the extraction buffer. A loopful (approximately 0.1 ml) of the desired
bacterial strain was
removed from a slant culture or plate and combined with the extraction buffer
in a 1.5 ml
microfuge tube and the contents were vortexed for two minutes. The extracted
DNA present
in the aqueous phase was processed for further purification as described
below.
Samples of E. coli and C. jejuni strains were ethanol precipitated and
dissolved in 50
~.l TE buffer. The samples were then treated with 0.5 p.g RNase A at
37°C for 30 min.
DNA was precipitated with ethanol, collected by centrifugation and dissolved
in 200 p.l 10
mM Tris-HCl (pH 8.0 at 25°C).
Samples of Shigella, Salmonella, and Staphylococcus strains were concentrated
using a
Microcon T"' 30 filter (Amicon) to 50 p.l and then transferred to TE buffer
using MicrospinT"'
v
S-200 HR gel filtration columns (Pharmacia Biotech). The samples were then
treated with
0.5 p.g RNase A at 37°C for 80 min. DNA was precipitated with ethanol,
collected by
centrifugation and dissolved in 200 pl 10 mM Tris-HCl (pH 8.0 at 25°C).
Genomic DNA of E. coli Strain B (ATCC 11303) was obtained from Pharmacia
Biotech (Piscataway, NJ; Cat. 27-4566-O1, Lot 411456601 ). The DNA was
dissolved in 10
mM Tris-HCl (pH 8.0 at 25°C).
- 220 -
CA 02203627 1997-04-24
WO 96/15267
PCTJUS95/14673
Genomic DNA from Desulfurococcus amylolyticus Strain Z-533 (DSM 3822) was
isolated and purified using the standard technique of cesium chloride
centrifugation. [Bonch-
Osmolovskaya, et al., Microbiology (Engl. Transl. of Milcrobiologiya) 57: 78
(1988)].
The concentration of the genomic DNA preparations was determined by measuring
the
0 5 OD~~~, of the preparations.
D) Design of Primer for the Amplification of 16s rRNA Genes of Bacterial
Species _
Primers and probes have been reported which allow the amplification or
detection of
16S rRNA sequences from a wide variety of bacterial strains. These
oligonucleotide primers
or probes represent consensus sequences derived from a comparison of the 16s
rRNA ene
g
sequences from a variety of eubacterial species. For example, oligonucleotide
primers suitable
for either PCR amplification or dot blot hybridization of bacterial rRNA gene
sequences have
been reported [e.g., PCT Publication WO 90/15157; Widjojoatmodjo et al., J.
Clin. Microbiol.
32:3002 (1994)]. Typically the conserved primer sequences are designed to
flank
nonconserved regions of the 16s rRNA gene with species-:specific sequences.
A number of previously published consensus primers derived from 16S rRNA gene
sequences were examined for the ability to produce substrates for use in
CFLPT"' reactions.
Primers 1638, 1659 and 1743 were described in PCT Publication WO 90/15157.
Primer
ER10 was described in Widjojoatmodjo et al., supra. Primers SB-1, SB-3 and SB-
4 represent
new primers (i. e., not previously published). The primers used in this
example are listed in
Table 5 below.
- 221 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
TABLE 5
Primers for PCR Amplification of 16S rRNA Genes
PRIMER SEQ ID NO: SEQUENCE
1638 138 5'-AGAGTTTGATCCTGGCTCAG-3'
ER10 139 5'-GGCGGACGGGTGAGTAA-3'
1659 140 5'-CTGCTGCCTCCCGTAGGAGT-3'
SB-4 141 5'-ATGACGTCAAGTCATCATGGCCCTTACGA-3'
1743 142 5'-GTACAAGGCCCGGGAACGTATTCACCG-3'
SB-I 143 5'-GCAACGAGCGCAACCC-3'
SB-3 144 5'-ATGACGTCAAGTCATCATGGCCCTTA -3'
The oligonucleotide primers were obtained from Integrated DNA Technologies,
Inc.
The oligonucleotides were dissolved in 10 mM Tris-HCl (pH 8 at 25°C) at
a concentration of
20 ~.M. Two sets of primers were synthesized; one set having an OH group at
the 5' end
(i.c., unlabelled primers) and the other set having the fluorescent dye TET
(tetrachlorinated
analog of 6-carboxyfluorescein, Applied Biosystems) at the 5' end (i. c., TET-
labelled
primers). TET-labelled primers are indicated by the use of "TET" as a suffix
to the primer
name (for example, TET-1638 indicates the 1638 primer having a 5' TET label).
The location of each of the primers listed in Table 5 is shown along the
sequence of
the E. coli rrsE gene (encodes a 16S rRNA) in Figure 82. In Figure 82 the
primer sequences
are shown in bold type and underlining is used to indicate complete identity
between primer
sequences and E. coli rrsE gene sequences. The sequence of the E. coli rrsE
gene is listed in
SEQ ID N0:145. As shown in Figure 82, the 1638, ER10, SB-1, SB-3, SB-4 primers
2~ correspond to sequences present on the sense strand of the 16S rRNA gene.
The 1659. 174 3
primers correspond to sequences present on the anti-sense strand of the 16S
rRNA gene. .
Figure 83 provides an alignment of the E. coli rrsE gene (SEQ ID N0:145). the
Cam.jejun5 gene (a rRNA gene from C. jejuni) (SEQ ID N0:146) and the
Stp.aureus gene (a
rRNA gene from S. aureus) (SEQ ID N0:147). The location of the 1638. ER10, 169
'
(shown as the complement of 1659), SB-1, SB-3, SB-4 and 1743 (shown as the
complement
of 1743) primers is indicated by the bold type. Gaps (dashes) are introduced
to maximize '
alignment between the rRNA genes.
- 222 -
CA 02203627 1997-04-24
WO 96/15267
PGT/US95/14673 _
In procaryotes the ribosomal RNA genes are present in 2 to 10 copies, with an
average
of 7 copies in Escherichia strains. Any PCR amplification produces a mixed
population of
these genes and is in essence a "multiplex" PCR from that strain. The CFLP
represents a
~Y
composite pattern from the slightly varied rRNA genes within that organism so
no one
S particular rRNA sequence is directly responsible for the entire "bar code."
In some cases
these minor variations (between rRNA genes; see, for example, minor variations
between the
E. cnli rRNA gens in Figure 82) cause shifts in the minor (lower signal) bands
in the CFLPTM~~
pattern, allowing discrimination between very closely related organisms. More
dramatic
sequence variations, found in most or all copies of these genes, are seen when
more distantly
related organisms are compared (see, for example, the extensive variations
between the E.
coli, C. jejuni and S. aureus. rRNA genes in Figure 83) and these larger
differences are
reflected in the CFLP patterns as more dramatic pattern changes. Despite the
variable nature
of these genes, the amplification by PCR can be performed between conserved
regions of the
rRNA genes, so prior knowledge of the entire collection of rRNA sequences for
any microbe
I S of interest is not required.
Three primers (TET-1638, TET-ER10, and TET-SB-4) were used for making the ~'
end fluorescently labeled fragments of the sense strand of 16S rRNA genes; two
other primers
(TET-1659 and TET-1743) were used for making labeled fragments of the
antisense strands.
The predicted size of PCR products produced by amplification of 16s rRNA gene
sequences from a variety of bacterial genera using the indicated primer pairs
is shown in
Table 6. In Table 6, the size of the predicted PCR product is based upon the
known sequence
of the 16S rRNA gene in the indicated species. The following abbreviations are
used in
Table 6: Dco (Desulfurocnccus); E.co (E. coli), Cam (Carr~pylobacter) and Stp
(Staphylococus). The location of the PCR product relative to the sequence of
the E. coli r ~ sE
gene (see Figure 82) is given in the last column.
r
- 273 _
CA 02203627 1997-04-24
WO 96/15267 PCT/U595/14673
TABLE 6
Combinations of Primers for PCR Amplification of 1 GS rRNA Sequences
w
Anti-
Primer Sense Sense Labeled Size Position
Pair Primer Primer Strand (bp)
Dco E.co Cam Stp (E.co)
A TET-1638 1659 sense 350 348 347 8-357
B TET-1638 1743 sense 1388 1365 1397 8-1395
C TET-ERIO 1659 sense 254 254 263 104-357
1
D TET-ER10 1743 sense 1278 1292 1271 1303 104-1395
E 1638 TET-1659 antisense 350 348 347 8-357
F ERIO TET-1659 antisense 254 254 263 104-357
G TET-SB-4 1743 sense 208 208 1188-
1395
H TET-1743 1638 antisense 1388 1365 1397 8-1395
1 TET-1743 ER10 antisense1278 1292 1271 1303 104-1395
J SB-4 TET-1743 antisense 208 208 1188-
1395
K SB-I TET-1743 antisense305 297 29G 296 1099-
1395
L SB-3 TET-1743 antisense 208 208 208 1188-
1395
E) PCR Amplification of 16S rRNA Gene Sequences
The ability of each primer pair listed in Table 6 to amplify 1 GS rRNA gene
sequences
from each bacterial strain listed in Table 3 was examined. It is well known
that commercial
preparations of recombinant Taq DNA polymerase contain various amount of E.
coli I GS
rRNA gene sequences. In order to minimize amplification of contaminating E.
coli 16S
rRNA sequences during the amplification of bacterial DNA samples. AmpliTaq DNA
polymerase, LD (Low DNA) (Perkin Elmer) was used in the PCRs. This preparation
of Tuq
DNA polymerase is tested by the manufacturer to verify that less than or equal
to 10 copies
of bacterial 1GS ribosomal RNA gene sequences are present in a standard 2.5
unit aliquot of
enzyme.
-224-
CA 02203627 1997-04-24
wo 9snsis7
PCT'/US95/14673
Each primer pair (Table 6) was tested in PCRs. The PCR reactions contained 10
mM
Tris-HCl (pH 8.3 at 25°C), 50 mM KCI, 1.5 mM MgCh, 0.001% w/v gelatin.
60 pM each of
dGTP, dATP, dTTP, and dCTP, 1 pM each of one S'-TET labeled and one unlabeled
' primers, 2.5 units AmpliTaq DNA polymerise, LD. The reactions were conducted
in a final
volume of 50 p,l using AmpliTaq DNA polymerise, LD, Lot E0332, or 100 pl
volume using
AmpliTaq DNA polymerise, LD, Lot D0008. The amount of genomic DNA added varied
from 6 to 900 ng per PCR. Control reactions which contained no input bacterial
genomic
DNA were also run to examine the amount of 16S rRNA product produced due to
contaminants in the AmpliTaq DNA polymerise, LD preparations.
PCR reactions were performed on PTC-100'"'' Programmable Thermal Controller
(MJ
Research, Inc.). Two sets of cycling conditions were utilized. The first set
of conditions
comprised 30 cycles of 95°C for 30 sec; 60°C for 1 min;
72°C for 30 sec; after the last cycle
the tubes were cooled to 4°C. The second set of conditions comprised 30
cycles of of 95° for
30 sec; 60°C for 1 min; 72°C for 90 sec; after the last cycle
the tubes were cooled to 4°C.
Thus, the difference between the two cycling conditions i s the length of time
the reactions are
held at the elongation temperature (72°C). These two elongation times
were tested because
the predicted size of the 16S rRNA targets varied from 208 to 1388 by
depending on the
primer pair used in the amplification.
As a rule of thumb, when the target to be amplified is less than 500 by in
length, a 30
sec elongation step is used; when the target is about 500-1000 by in length,
an elongation step
of 30 to 60 sec is used; when the target is greater than 1 kb in length, the
elongation is
conducted for approximately 1 min per 1 kb length. While the first set of PCR
conditions (30
sec elongation step) worked with the longer amplicons, the yield was lower
than that obtained
when the second set of PCR conditions (90 sec elongation) was used.
Following the thermal cycling, 400 ~I of formamidf: containing 1 mM EDTA was
added to each sample and the samples were concentrated to a volume of 40 pl in
a Microcon
30. The samples (40 p,l) were loaded on a denaturing 6% polyacrylamide gel (7
M urea,
O.SX TBE running buffer), that was prewarmed to 50-55°C prior to the
loading of the
samples. The samples were run at 20 W for 20 min (200-350 by fragments) or 40
min (more
than 1 kb fragments). The gels were scanned using a Fluorescent Method Bio
Image
Analyzer Model 100 (FMBIO-100, Hitachi) with a 585 or 505 nm filter.
The results of these PCRs showed that each primer pair (Table 6) tested
successfully
amplified a fragment of the expected size. Thus the primer pairs shown in
Table 6 are
- 225 _
CA 02203627 1997-04-24
WO 96/15267 PGT/US95/14673
suitable for the amplification of end labeled DNA fragments using genomic DNA
from
variety of prokaryotes including archaea, gram-positive and gram-negative
bacteria, different
species of the same genus and different strains of the same species. These
PCRs also
demonstrated that, although the amount of genomic DNA present in the PCR
varied from
strain to strain, the yield of the amplified product was always many-fold
higher than the trace
yield of product from the E. coli genomic DNA present in AmpliTaq DNA
polymerase, LD,
seen in the reactions which contained no input bacterial genomic DNA.
F) Preparation of 16S rRNA Gene Substrates
To generate labelled PCR products corresponding to bacterial 16S rRNA
sequences for
use in CFLPT"'' reactions, the following primer pairs were used in PCRs.
1. The SB-1/TET-1743 pair was used to amplify an approximately 297 by
fragment from genomic DNA derived from Desulfurococcus amylolyticus (DSM
3822), E.
coli Strain K-12 (ATCC 14948), S. aureus subsp. aureus (ATCC 33591) and S.
aureus subsp.
ccacreus (ATCC 33592). The resulting PCR product contains a 5' TET-label on
the antisense
strand.
2. The TET-SB-4/1743 pair was used to amplify an approximately 208 by
fragment from genomic DNA derived from E. coli Stain B (ATCC 11303), E. coli
Strain K-
12 (ATCC 14948), E. coli Serotype 0157: H7 (ATCC 43890, Shigella dysenteriac~
Serotype
2 (ATCC 29027), and Salmonella choleraesuis subsp. choleraesuis Serotype typhi
(ATCC
6539). The resulting PCR product contains a 5' TET-label on the sense strand.
3. The 1638/TET-1659 pair was used to amplify an approximately 350 by
fragment from genomic DNA derived from E. coli Stain B (ATCC 11303), E. coli
Strain K-
12 (ATCC 14948), E. coli Serotype 0157: H7 (ATCC 43895), Shigella dusenteriae
Serotype
2 (ATCC 29027), and Salmonella choleraesuis subsp. choleraesui.s Serotype
typhi (ATCC
6539). The resulting PCR product contains a 5' TET-label on the antisense
strand.
4. The TET-ER10/1743 pair was used to amplify an approximately 1292 by
fragment from genomic DNA derived from E. coli Strain K-12 (ATCC 14948) and
Campylnbacter jejuni subsp. jejuni (ATCC 33291). The resulting PCR product
contains a ~'
TET-label on the sense strand.
5. The 16381T'ET-1659 pair was used to amplify an approximately 350 by o
fragment from genomic DNA derived from E. coliSerotype 0157: H7 (ATCC 43895).
Salmonella choleraesuis subsp. clzoleraesuis Serotype typhi (ATCC 6539),
Shigella
dysenteriae Serotype 2 (ATCC 29027), S. aureus subsp. aureus (ATCC 33591 ). S.
crzrrcu.s
-226-
CA 02203627 1997-04-24
WO 96/15267
PCTlUS95/14673
subsp. aureus (ATCC 33592), S. aureus subsp. aureus (ATCC 13565), S. hominis
(ATCC
29885), and S. warneri (ATCC 17917).
The PCRs were conducted as described in section (e) above. Two separate PCR
sr
reactions were performed using 0.2 pg of genomic DNA derived from
Camylobacter.jejurri
subsp. jejuni (ATCC 33291) and the TET-ER10/1743 primer pair. One reaction was
' conducted in a final volume of 50 ~.l and used an extension step of 30 sec
at 72°C during
thermal cycling. The second reaction was conducted in a final volume of 100 pl
and used an
extension step of 90 sec at 72°C. The yield of PCR product produced in
the second reaction
was 76% higher (as compared to first reaction). Following the amplification
reaction, the
samples were processed for electrophoresis on denaturing polyacrylamide gels
as described in
section (e) above. After electrophoresis, the desired bands were cut from the
gel and eluted
by placing the gel slice into 0.4 ml of a solution containing 0.5 M ammonium
acetate, 0.1
mM EDTA and 0.1 % SDS. The mixture was then incubated at 55°C for 2 h
and then at
37°C for 12 h. The samples were concentrated to 25 ~l using a Microcon
30 (Amicon) and
transferred into water using S-200 microspin columns (Pharmacia).
G) Cleavage Reaction Conditions
Cleavage reactions were conducted in a final volume of 10 ~.1 volume
containing
approximately 0.2 to 1 pmole (as indicated below) 5' TET'-labeled DNA
substrate, 10 ng
CleavaseT'''' BN enzyme (Third Wave Technologies), 1X CFLP buffer and 0.2 mM
MnCI,.
The reactions were first assembled as a 9 p,l mixture lacking MnCh; this
mixture was heated
to 95°C for 10 sec and then cooled down to the desired incubation
temperature (45°C, 50°C or
65°C). Optimal reaction temperature for each substrate was chosen based
on even distribution
of bands, and the presence of some undigested material to indicate
representation of molecules
all the way up to full length. Selected optimal temperatures for each
substrate are indicated in
the description of Figures 84-87 below.
The cleavage reaction was started by the addition of 1 ~.1 of 2 mM MnCI,.
Following incubation at the desired temperature for 2 min, the reaction was
stopped by the
addition of 10 ~,l of a solution containing 95% formamide, 5 mM EDTA, 5%
glycerol and
0.02% methyl violet. Uncut or "no enzyme" controls were set up for each
substrate as
described above with the exception that H,O was used in place of the
CleavaseT"' BN enzyme.
Samples (approximately 4 to 8 ~l) were run on 6 to 12% denaturing
polyacrylamide gels
( 19:1 cross link) with 7 M urea in a buffer containing 45 mM Tris Borate, pH
8.3, 1.4 mM
EDTA at I S to 20 W for 9 minutes (specific gel percentages are indicated
below in the
-227-
CA 02203627 1997-04-24
WO 96/15267 PGTILTS95/14673
descriptions of Figures 84-87). The gels were then scanned using a FMBIO-100
(Hitachi)
with the 585 nm filter.
The resulting fluoroimager scans are shown in Figures 84-87. In Figure 84. the
cleavage products generated by cleavage of an approximately 297 by 16S rRNA
substrate
generated using the SB-1/TET-1743 pair and genomic DNA derived from
Desulfurococcus
amylolyticus (DSM 3822), E. coli Strain K-12 (ATCC 14948), S. aureus
subsp.'aureus
(ATCC 33591) and S. aureus subsp. aureus (ATCC 33592) is shown. Lanes 1-4
contain the
products generated by incubation of the substrate derived from Desu~rococczrs
amylolyticus
(DSM 3822), E. coli Strain K-12 (ATCC 14948), S. aureus subsp. aureus (ATCC
33591 ) and
S. aureus subsp. aureus (ATCC 33592) in the absence of Cleavase~'~"'' BN
enzyme,
respectively. Lanes 5-8 contain the products generated by incubation of the
substrate derived
from Desulfurococcus amylolyticus (DSM 3822), E. coli Strain K-12 (ATCC
14948), S.
aureus subsp. aureus (ATCC 33591) and S. aureus subsp. aureus (ATCC 33592) in
the
presence of CleavaseT"'' BN enzyme, respectively. The CFLPT"'' reactions were
performed
using approximately 1 pmole of each PCR product and the cleavage reactions
were incubated
at 50°C for 2 min. The cleavage products were resolved by
electrophoresis on an 8%
polyacrylamide gel, as described above.
The results shown in Figure 84 demonstrate that distinct CFLPT"'' patterns are
obtained
using the Desulfurococcus amylolyticus (DSM 3822), E.. coli Strain K-12 (ATCC
14948) and
S. aureus subsp. aureus substrates. The same CFLPT"'' pattern was generated by
cleavage of
the two S. aureus subsp. aureus substrates (lanes 7 and 8); these two S.
aurezrs subsp. azrreus
strains (ATCC 33591 and 33592) are considered different subspecies based upon
differences
in sensitivities to the antibiotics methicillin and gentamicin. Resistant or
sensitivity to these
antibiotics is not associated with mutation in the 16S rRNA gene; therefore it
was not
expected that different CFLPT"'' patterns would be observed using a 16S rRNA
substrate.
The results shown in Figure 84 show that the SB-1/TET-1743 pair can be used to
generate substrates for CFLPT"' analysis which allow the identification and
discrimination of
Desulfurococcus amylolyticus (DSM 3822), E. coli Strain K-12 (ATCC 14948) and
.S: aureus -
subsp. aureus. -
In Figure 85, Panel A shows the reaction products generated by cleavage of an
approximately 208 by 16S rRNA substrate generated using the TET-SB-4/1743 pair
and
genomic DNA derived from E. c_oli Stain B (ATCC 11303). E. coli Strain K-12
(ATCC
14948), E. coli Serotype 0157: H7 (ATCC 43890, Shigella dyserrteriae Serotype
2 (ATCC
- 228 -
CA 02203627 1997-04-24
WO 96/15267
PCT'/US95l14673
29027), and Salmonella choleraesuis subsp. choleraesuis Serotype typhi (ATCC
6539). The
TET-SB-4/1743 pair amplifies a portion of the 165 rRNA gene located in the 3'
region of the
gene (see Figure 82).
h_
The CFLPTM reactions shown in Figure 85, Panel A were performed using
s 5 approximately 0.7 pmole of each PCR product and the cleavage reactions
were incubated at
50°C for 2 min. The cleavage products were resolved by electrophoresis
on an 8% denaturing
polyacrylamide gel, as described for Figure 84.
In Figure 85, Panel B shows the reaction products generated by cleavage of an
approximately 350 by 165 rRNA substrate generated using; the 1638/TET-1659
pair and
genomic DNA derived from E. coli Stain B (ATCC 11303), E coli Strain K-12
(ATCC
14948), E. coli Serotype 0157: H7 (ATCC 43895), Shigella dysenteriae Serotype
2 (ATCC
29027), and Salmonella choleraesuis subsp. choleraesuis Serotype typhi (ATCC
6539). The
1638/TET-1659 pair amplifies a portion of the 165 rRNA gene located in the 5'
region of the
gene (see Figure 82).
The CFLPT"' reactions shown in Figure 85, Panel B were performed using
approximately 1 pmole of each PCR product and the cleavage reactions were
incubated at
45°C. The cleavage products were resolved by electrophoresis on an 8%
polyacrylamide gel.
The lanes marked "M" in Figure 85, Panels A and B contain plasmid pUC 19 DNA
digested with MspI and 3' end labeled with fluorescein ddLJTP using terminal
deoxynucleotidyl transferase as described in Example 8. Tllis marker includes
bands
corresponding to lengths of 26, 34, 67, 110/111, 147, 190, 242 and 331 bp.
Additional
marker bands of 404, 489 and 501 by are not visible in this figure. In Panel
A. lanes 1-5
contain the uncut (i.e., no enzyme) controls and lanes 6-10 contain the
cleavage products
generated by the incubation of substrates derived from E. coli Stain B (ATCC
11303), E coli
2~ Strain K-12 (ATCC 14948), E coli Serotype 0157: H7 (ATCC 43895), Shigella
dysenteriae
Serotype 2 (ATCC 2902'7), and Salmonella choleraesuis subsp. choleraesuis
Serotype typhi
(ATCC 6539), respectively. In Panel B, lanes 1-5 contain the uncut (i.e., no
enzyme) controls
'' and lanes 6-10 contain the cleavage products generated by the incubation of
substrates derived
from E. coli Stain K-12 (ATCC 14948), E. coli Strain B (ATCC 11303), E coli
Serotype
0157: H7 (ATCC 43895), Shigella dysenteriae Serotype 2 (.ATCC 29027), and
Salmonella
choleraesuis subsp. choleraesuis Serotype typhi (ATCC 6539), respectively.
The lower molecular weight materials seen in the "uncut" lanes has been found
to be
due to degradation of the gel-purified material after storage for several days
in dH20. This
- 229 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US9St14673
degradation may be due to environmental nucleases that are active when EDTA is
not present
in the storage solution (i. e., the necessary metal ions may be present in
trace amounts). This
degradation is effectively suppressed by inclusion of tRNA in the storage
solution (see
Example 19). The degradation seen in these uncut controls (Pane B, lanes 1-5)
does not
effect the CFLPTM results.
The results shown in Figure 85 demonstrate that some regions of the 16S rRNA
genes
are more variable than others, and that analysis of these regions are
particularly useful when
comparing very closely related organisms. For example, substrates generated by
the
1638/TET-1659 pair (which amplifies a portion of the 16S rRNA gene located in
the 5'
region of the gene) can be used to generate CFLPT"' patterns which distinguish
not only
between the DNA derived from the genera of Escherichia, Shigella, and
Salmonella (Panel B,
lanes 6-10), but which also creates distinct cleavage patterns from the DNA
derived from the
three strains of E. coli tested (i.e., strains B, K-12 and 0157: H7) (Panel B
lanes 6-8).
In contrast, no substantial difference in CFLPT"'' patterns was observed
between the
strains of the Escherichia-Salmonella assemblage for DNA fragments produced
using the
TET-SB-4/1743 pair which generates an approximately 208 by fragment located
near the 3'
end of 16S rRNA genes (Panel A, lanes 6-10). This contrast in the degree of
variation
between the 5' and 3' regions of the 16S rRNA genes is consistent with the
results reported
by Widjojoatmondjo et al., supra, in which the comparisons between strains of
the
Escherichia-Salmonella assemblage were made by SSCP analysis.
Since each organism has multiple copies of the 16S rRNA gene, and these co-
amplify
in each PCR, it was important to show that the products of different
amplifications from the
same organism produced the same cleavage pattern. In Figure 86, the cleavage
products
generated by cleavage of an approximately 1292 by 16S rRNA substrate generated
using the
TET-ER10/1743 pair in two separate PCR reactions from Campylobacter jejuni
subsp. .)ejtll~l
(ATCC 33291 ) are shown in lanes 2 and 3. For comparison, the same region
amplified from
E. coli Strain K-12 (ATCC 14948) is shown in lane 1. The CFLP~ reactions were
performed using approximately 60 fmole of each PCR product and the cleavage
reactions
were incubated at 50°C for 2 min. Reactions were stopped by the
addition of 95%
formamide, 5 mM EDTA, 5% glycerol and 0.02% methyl violet. The cleavage
products were =
resolved by electrophoresis on a 6% denaturing polyacrylamide gel as described
above.
The results shown in Figure 86 demonstrate that very different CFLPT~''
patterns were
generated using substrates from Gamma (Escherichia. lane 1 ) and Epsilon
(Campvlobacter.
-230-
CA 02203627 1997-04-24
WO 96/15267 PCTILTS95/14673
lanes 2 and 3) subdivisions of Purple bacteria, but that the same CFLPTM
pattern was observed
between the products of separate PCR reactions on the same genomic DNA (lanes
2 and 3).
In Figure 87, the cleavage products generated by cleavage of an approximately
350 by
16S rRNA substrate generated using the 1638/TET-1659 pair and genomic DNA
derived from
3 5 E. coli Serotype 0157: H7 (ATCC 43895), S. choleraesuis subsp.
choleraesuis Serotype typhi
' (ATCC 6539), Shigella dysenteriae Serotype 2 (ATCC 29027), S. aureus subsp.
aureus
(ATCC 33591 ), S. aureus subsp. aureus (ATCC 33592), ..f. aureus subsp. aureus
(ATCC
13565), S. hominis (ATCC 29885), and S. warneri (ATCC'. 17917) are shown in
lanes I-8,
respectively. The CFLPT"' reactions were performed as described above, using
approximately
IO 200 fmol of each PCR product; the cleavage reactions were incubated at
65°C for 2 min. The
cleavage products were resolved by electrophoresis on a 10% denaturing
polyacrylamide gel
as described above.
The results shown in Figure 87 demonstrate that very different CFLPTM patterns
were
produced using DNA derived from strains representing Purple bacteria (lanes I-
3) and the
I S Gram-positive phylum (lanes 4-8). A substantial difference between CFLPT"'
patterns was
detected between the genera Escherichia (lane 1), Salmonella (lane 2), and
Shigella (lane 3).
Additionally, a substantial difference between the C:FLPT"' patterns was
detected
between species of Staphylococcus aureus (lanes 4-6), hominis (lane 7), and
warneri (lane 8).
No substantial difference between CFLPTM patterns was observed between the
three strains of
20 Staphylococcus aureus subsp. aureus ATCC 33591 (lane 4), ATCC 33592 (lane
5), and
ATCC 13565 (lane 6). These S. aureus isolates differ in reported antibiotic
resistance, but are
so closely related that the rRNA genes do not yet show divergence by CFLPT"'
analysis.
The above results demonstrate that CFLPTM analysis can be used to discriminate
between bacterial genera as well as between different species and subspecies
(depending on
25 the region of the 16S rRNA gene used as the substrate). A comparison of the
CFLPTM
patterns generated within the same or similar genera (e.g., S'almonella,
Shigella and E. coli)
shows an overall similarity in the banding pattern with differences revealed
as changes in a
'' small subset of the bands. When the comparison is made across different
genera (e.g.,
between E coli and S. aureus) a more striking change in barcode pattern is
evident indicatin~~
30 that CFLPT"' patterns may not only be used to detect differences between
organisms. but the
degree to which the patterns change may be used to assess the degree of
evolutionary
divergence between organisms.
- 231 -
CA 02203627 1997-04-24
WO 96/15267 PC"T/US95/14673
Substrates for CFLPT"'' analysis were produced by PCR amplification using
different
sets of primers. Some primer pairs (sets) are reported to be universal for all
procaryotic
organisms; other primer pairs have been observed to be specific for
representatives of lower ,
taxons (See, PCT Publication WO 90/15157). Except for the primer sequences, no
knowledge of the DNA sequence of the rRNA gene from any specific organisms) is
required
d
for amplification and CFLP~ analysis of bacterial 16S rRNA genes.
Distinct CFLP'~"'' patterns were observed between representatives of archeaea
and
eubacteria, different phyla of eubacteria, different phyla within eubacteria,
different
subdivisions of the same phylum, different genera of the same assemblage,
different species of
the same genus and different strains of the same species. Distinct signatures
in CFLP~'~"''
patterns were found that allowed discrimination of pathogenic isolates,
including those
associated with food poisoning, from innocuous members of the normal flora.
While the PCR products generated using genomic DNA from different organisms
with
the same set of primers are indistinguishable by their mobility during gel
electrophoresis (on
non-gradient polyacrylamide gels), the CleavaseT"'' BN enzyme cleaves these
PCR products
into shorter fragments thereby generating a characteristic set of cleavage
products (i.e., a
distinct CFLP~ signature). The pattern of cleavage products generated is
reproducible; DNA
substrates generated in independent PCRs from the same organism using a given
primer pair
yield the same pattern of cleavage products.
CFLP"'' patterns can be generated using large DNA fragments (e.g., at least
about 1.6
kb) and thus could cover the entire length of the bacterial 16S rRNA gene.
CFLP~'~"'' can also
be used in conjunction with shorter DNA fragments (about 200 bp) which are
located at
different positions throughout the 16S rRNA gene.
EXAMPLE 35
CFLP'~"'' Analysis of Substrates Containing Nucleotide Analogs
The effect of using various nucleotide analogs to generate substrates for
CFLPT"'
reactions was examined. As discussed below, nucleotide analogs are used in
PCRs for several
reasons; therefore, the ability to analyze the modified products of PCRs
(i.e., nucleotide
analog-containing PCR products) by CFLP~ analysis was investigated. The 7-
deaza purine
analogs (7-deaza-dATP and 7-deaza-dGTP) serve to destabilize regions of
secondary structure
by weakening the intrastrand stacking of multiple adjacent purines. This
effect can allow
_ '73'7 _
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
amplification of nucleic acids that, with the use of natural dNTPs, are
resistant to
amplification because of strong secondary structure [McConlogue et al..
Nucleic Acids Res.
16:20 (1988)].
' Similarly, the analog dUTP is often used to replace dTTP, but for different
reasons.
z 5 dUTP-containing DNA (this nomenclature is shorthand for PCR products
generated using
' dUTP; the actual PCR product will contain dUMP) can be destroyed by the
enzymatic activity
of uracil DNA glycosylase (UDG) while dTTP-containing DNA is untouched. When
PCR
products are produced containing dUMP in place of dTMP, UDG can be used in all
subsequent reactions to eliminate false positive results due to carry-over
from the earlier
PCRs, without preventing amplification from the normal I)NA of interest. This
method is
widely used in clinical laboratories for performing PCR and thus this method
would be used
by most clinical laboratories using PCR in conjunction with CFLPT"' for
pathogen typing.
Thus, the ability of the CFLP"~' reaction to suitably cleave dUTP-containing
DNA fragments
(i.e., produce strong reproducible band patterns) was examined.
For these comparisons, substrates corresponding to a 157 by fragment derived
from
exon of of the wild-type and R422Q mutant of the human tryosinase gene were
generated by
PCR amplification using either 1) the standard mixture of dNTPs (i.e., dATP,
dCTP, dGTP
and dTTP); 2) dUTP in place of dTTP; 3) 7-deaza-dGTP (d'GTP) in place of dGTP;
and 4)
7-deaza-dATP (d'ATP) in place of dATP. These substrates were then incubated
with
Cleavase~'~"'' BN enzyme and the effect the presence of the various nucleotide
analogs on the
cleavage pattern was examined.
B) Preparation of Substrates Containing Nucleotide Analogs
A 157 by fragment of the human tyrosinase gene (exon 4) was amplified in PCRs
using the following pair: 5' CACCGTCCTCTTCAAGAAG 3' (SEQ ID N0:29) and 5"
biotin-CTGAATCTTGTAGATAGCTA 3' (SEQ ID N0:30). Plasmids containing cDNA
derived from the wild-type or R422Q mutant of the tyrosinase gene were used as
template
(see Example 8 for a description of these plasmids). The resulting double-
stranded PCR
products contain the 5' biotin label on the anti-sense strand such that
sequence detected in the
CFLPT~'' reaction is SEQ ID N0:35 (wild-type anti-sense strand) or SEQ ID
N0:53 (R422Q
mutant anti-sense strand). All PCRs were conducted in a final volume of 100
q.l. dATP.
h
dCTP, dGTP, dTTP and dUTP were obtained from Perkin Elmer; d'ATP and d'GTP
were
obtained from Pharmacia. Taq DNA polymerase was obtained from Promega. The PCR
' mixtures were assembled as shown below in Table 7.
- 7jj
CA 02203627 1997-04-24
WO 96/15267 PC'~'/US95114673
TABLE 7
Reaction Components [Stock]Aliquot [Final]
Plasmid cDNA 4 ng/~.l1 ~.l 40 pg
PCR Buffer' 1OX 10 ~l 1X ,
Unlabelled primer 100 0.25 ~,l 25 pmole
pM
Labeled primer 100 0.25 ~.1 25 pmole
~,M
dATP 10 mM 1 ~.l 100 ~.m
dCTP 10 -mM 1 ~.l I 00 ~.m
dGTP 10 mM 1 ~.l 100 ~.m
dTTP 10 mM 1 g.l 100 ~,m
d'ATP'- 5 mM 2 ~l 100 ~,m
d'GTP 3 5 mM 2 ~I 100 ~m
dUTP 4 20 mM 4 ~.l 800 ~m
Taq polymerise 5 units0.5 ~.l 2.5 units
/~l
dH20 to 100
~,l
1X concentration contains 20 mM Tris-HCI, pH 8.5; 1.5 mM MgCh; 50 mM KCI; 0.5%
Tween 20; and 0.5% NP-40.
'- d'ATP completely substituted for dATP in the PCR.
3 d'GTP completely substituted for dGTP in the PCR.
4 dUTP completely substituted for dTTP in the PCR. Other nucleotides were
present at a final
concentration of 200 Vim. In this reaction, the PCR buffer used was the lOX
buffer (500 mM
KCI, 100 mM Tris-Cl, pH 9.0, 1.0% Triton X-100) provided by Promega. 25 mM
MgCh
was added separately to a final concentration of 2.5 mM.
Wild-type and the mutant R422Q substrates were amplified using the natural and
substituted nucleotide analogs listed above. For reactions containing the
natural dNTPs,
d'ATP and d'GTP, all reaction components were added together. Reactions
containing dUTP
were initially assembled without the polymerise (see below).
The assembled reactions were placed in a thermocycler (MJ Research, Watertown,
MA) that was preheated to 95°C. The tubes were allowed to incubate for
one minute at 95°C
- 234 -
CA 02203627 1997-04-24
WO 96/15267 PGT/US95/14673
before amplification. The program was then set at 94°C for 30 minutes.
50°C for one minute.
72°C degrees for two minutes for 34 cycles with a final 72°C
incubation for 5 minutes.
Reactions containing dUTP were performed with a "hot start." All components
except
the polymerase were mixed, heated to 95°C for 1 minute, then cooled to
72°C. Taq
z 5 polymerase (2.5 units) was then added in 10 pl of 1X PCR buffer for a
final volume of 100
" l.~.l.
At the end of the amplification, the PCR products were made 0.3M Na04Ac, with
the
exception of reactions containing dUTP; the dUTP-containing reactions were
brought to 2M
NH40Ac; all were then precipitated by the addition of 2.5 volumes (total
aqueous volumes) of
absolute ethanol. The DNA pellets were collected by centrifugation and then
dried under
vacuum. The pellets were resuspended in 10 p,l of TE and 10 pl of STOP buffer
(20 p.l
TIOE0.1 and 16 p.l of STOP for the dUTP-containing reactions). The tubes were
then heated
to 85°C for 2 minutes and the mixtures were resolved by electrophoresis
through 10% (6%
for dUTP) denaturing acrylamide gel (19:1 cross link) with 7M urea in a buffer
of O.SX TBE.
I S The PCR products corresponding to the 157 by substrate derived from the
wild-type
and R422Q~ mutant were gel purified as described in Example 19. The gel-
purified DNAs
were resuspended in T10E0.1 buffer using the following volumes: 40 pl for
fragments
containing only dNTPs; 40 p.l for fragments containing d'ATP; 25 p,l for
fragments containing
d'GTP and 25 p,l for fragments containing dUTP.
B) Cleavage Reaction Conditions
The gel purified 157 by tyrosinase substrates containing natural
deoxynucleotides and
nucleotide analogs were analyzed in cleavage reactions as i:ollows. Final
reaction mixtures
comprised 1 ~1 of the resuspended gel-purified DNA [see section (a) above] and
25 ng
CleavaseT"'' BN in 10 mM MOPS, pH 7.5 with 0.2 mM MllCl,, and 0.05% each Tween
20
and NP-40 in a volume of 20 p.l. No enzyme controls were assembled in which
distilled
water replaced the CleavaseT"'' BN enzyme. The substrate DNAs were distributed
into
reaction tubes and brought to a volume of 15 p.l with H,O. The remaining
reaction
r components were mixed in a volume of 5 pl (i. e., at a 4X concentration).
The DNAs were
heated for 15 sec. at 95°C to denature the DNA. The cleavage reactions
were initiated by the
addition of 5 p.l of the enzyme/buffer mixture (the 4X concentrate). The
cleavage reactions
were incubated at 45°C for three minutes, and the reactions were
terminated by the addition
of 16 ~1 of Stop solution (described in section a). Seven microliters of each
sample was
heated to 85°C for two minutes prior to loading onto a 10% denaturing
acrylamide gel ( 19:1
_ 23j _
CA 02203627 1997-04-24
WO 96115267 PCT/US95114673
cross link), with 7M urea in a buffer of 45 mM Tris Borate pH 8.3, 1.4 mM
EDTA.. The gel
was run at a constant 800 V until the bromophenol blue had migrated the length
of the gel.
Following electrophoresis, the biotinylated fragments were detected as
described in
Example 8 with the exception that 4 p,l of the SAAP conjugate was added to 100
p.l of USB
s
blocking buffer (1:25,000 dilution). After washing, 5 p,ls of CDP-Star~'~''
was used as the
chemiluminescent substrate. The resulting autoradiogram is shown in Figure 88.
In Figure 88, the lanes marked "M" contain biotinylated molecular weight
markers -
obtained from Amersham (Arlington Heights, IL) and include bands corresponding
to lengths
of 50, 100 and 200 nucleotides (size indicated by use of numbers and large
arrowheads).
Lanes 1-8 contain reaction products obtained by incubation of the substrates
in the absence of
CleavaseT"' BN enzyme (i.e., no enzyme or uncut controls). Lanes 9-16 contain
reaction
products obtained by incubation of the substrates in the presence of
CleavaseT"'' BN enzyme.
Lanes 1, 3, 5, 7, 9, 11, 13 and 15 contain the wild-type substrate; lanes 2,
4, G, 8, 10, I?, 14
and I 6 contain the R422Q mutant substrate. The products shown in lanes 1, 2.
9 and 10 were
generated from substrates generated using dNTPs in the PCRs. The products
shown in lanes
3, 4, 11 and 12 were generated from substrates generated using dUTP in place
of dTTP in the
PCRs. The products shown in lanes S, 6, 13 and 14 were generated from
substrates generated
using d'GTP in place of dGTP in the PCRs. The products shown in lanes 7, 8, I
S and 16
were generated from substrates generated using d'ATP in place of dATP in the
PCRs. It can
be seen from this example that modified DNA fragments are suitable for
cleavage in CFLP
reactions. Though the banding pattern is substantially different with these
substitutions, the
wild-type and R422Q mutant DNAs are readily distinguishable in all cases.
While not limiting the invention to any particular theory, the changes in
banding
patterns observed when nucleotide analogs are utilized can be attributed to
two sources. In all
cases, but particularly in reference to the 7-deaza purines, the use of
nucleotide analogs may
substantially change the nature and stability of the intrastrand folded
structures formed during
the cleavage reaction. As a consequence, the locations of the cleavage sites
would naturall~~
shift. In addition, the substitution of the modified nucleotides may change
the affinity of the
cleavage enzyme for the folded cleavage structure, either strengthening or
weakening cleavage
at a particular site. -
Examination of the variations seen between the wild-type and R422Q mutant when
_
different analogs are used also shows that the use of these substituants'can
enhance the
contrast between the variants. For example, with regard to the cleavage
products of the two
-236-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
substrate DNAs (generated using dUTP or dTTP) in the region just above the 50
by marker:
one significant band that reduces in intensity between the wild-type and the
mutant is more
dramatically reduced in the dU-containing samples.
The results shown in Figure 88 demonstrate that nucleotide analogs may be used
for
the generation of CFLP~'~"'' substrates. The substrates derived from the wild-
type or R422Q
' mutant of the tyrosinase gene which contain nucleotide analogs produce
distinct cleavage
patterns which allow the discrimination and identification of the mutant and
wild-type alleles.
This example demonstrates that even with 100% substitution with either 7-deaza-
GTP
for dGTP or 7-deaza-ATP for dATP, robust CFLP patterns are generated, although
the precise
sites of clevage are different in the dNTP-containing and i'-deaza-dNTP
containing substrates.
The above results also demonstrated that single base changes present within
DNA fragments
containing nucleotide analogs still influence the folded structure
sufficiently to cause cleavage
pattern changes similar to those seen when DNA fragments lacking nucleotide
analogs are
analyzed using the CFLPT"'' assay.
From the above it is clear that the invention provides reagents and methods to
permit
the rapid screening of nucleic acid sequences for variations. These methods
allow the
identification of viral and bacterial pathogens as well as permit the
detection of mutations
associated with gene sequences (e.g., mutations associated with multiple drug
resistance in M.
tuberculosis or mutations associated with human disease). These methods
provide improved
means for the identification and characterization of pathogens.
-237-
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
SEQUENCE LISTING
(1) GENERAL
INFORMATION:
(i) APPLICANT: DAHLBERG, JAMES E.
LYAMICHEV, VICTOR I.
S
BROW, MARY ANN D.
OLDENBURG, MARY C.
HEISLER, LAURA M. ,
FORS, LANCE
OLIVE, DAVID M. '
IO (ii) TITLE OF INVENTION: RAPID DETECTION AND IDENTIFICATION OF
NUCLEIC ACID VARIANCE AND PATHOGENS
(iii) NUMBER OF SEQUENCES: 147
IS (iv) CORRESPONDENCE ADDRESS:
(A) ADDRESSEE: MEDLEN & CARROLL
(B) STREET: 220 MONTGOMERY STREET, SUITE 2200
(C) CITY: SAN FRANCISCO
(D) STATE: CALIFORNIA
ZO (E) COUNTRY: UNITED STATES OF AMERICA
(F) ZIP: 94104
(v) COMPUTER READABLE FORM:
(A) MEDIUM TYPE: Floppy disk
(B) COMPUTER: IBM PC compatible
,~S (C) OPERATING SYSTEM: PC-DOS/MS-DOS
(D) SOFTWARE: PatentIn Release #1.0, Version #1.25
(vi) CURRENT APPLICATION DATA:
(A) APPLICATION NUMBER: US
(B) FILING DATE:
(C) CLASSIFICATION:
3O (vii) PRIOR APPLICATION DATA:
(A) APPLICATION NUMBER: US 08/520,946
(B) FILING DATE: 30-AUG-1995
(vii) PRIOR APPLICATION DATA:
3S (A) APPLICATION NUMBER: US 08/484,956
(B) FILING DATE: 07-JUN-1995
(vii) PRIOR APPLICATION DATA:
(A) APPLICATION NUMBER: US 08/402
601
,
(B) FILING DATE: 09-MAR-1995 _
(vii) PRIOR APPLICATION DATA:
(A) APPLICATION NUMBER: US 08/337,164
(B) FILING DATE: 09-NOV-1994
(vii) PRIOR APPLICATION DATA:
(A) APPLICATION NUMBER: US 08/254,359
(B) FILING DATE: 06-JUN-1994
4S (vii) PRIOR APPLICATION DATA: ,
(A) APPLICATION NUMBER: US 08/073,384
(B) FILING DATE: 04-JUN-1993
(vii) PRIOR APPLICATION DATA:
SO (A) APPLICATION NUMBER: US 07/986,330 ;
(B) FILING DATE: 12-DEC-1992
(viii) ATTORNEY/AGENT INFORMATION:
(A) NAME: CARROLL, PETER G.
(B) REGISTRATION NUMBER: 32,837 .,
(C) REFERENCE/DOCKET NUMBER: FORS-02000
-
CA 02203627 1997-04-24
WO 96/15267
PCT/US95/14673
(ix) TELECOMMUNICATION INFORMATION:
(A) TELEPHONE: (415) 705-8410
(B) TELEFAX: (415) 397-8338
(2) INFORMATION FOR SEQ ID NO:1:
S (ij SEQUENCE CHARACTERISTICS:
(A) LENGTH: 2506 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: double
(D) TOPOLOGY: linear
10- ( i t )- ;v~LEGuLE TYPE : DNA ( genomi c )
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:1:
ATGAGGGGGA TGCTGCCCCT CTTTGAGCCC AAGGGCCGGG TCC7.'CCTGGT 60
GGACGGCCAC 120
IS CACCTGGCCT ACCGCACCTT CCACGCCCTG AAGGGCCTCA CCAC'CAGCCG 180
GGGGGAGCCG 240
GTGCAGGCGG TCTACGGCTT CGCCAAGAGC CTCCTCAAGG CCCTCAAGGA 300
GGACGGGGAC 360
GCGGTGATCG TGGTCTTTGA CGCCAAGGCC CCCTCCTTCC GCCA.CGAGGC 420
CTACGGGGGG 480
ZS TACAAGGCGG GCCGGGCCCC CACGCCGGAG GACTTTCCCC GGCAACTCGC 540
CCTCATCAAG 600
GAGCTGGTGG ACCTCCTGGG GCTGGCGCGC CTCGAGGTCC CGGGCTACGA 660
GGCGGACGAC 720
GTCCTGGCCA GCCTGGCCAA GAAGGCGGAA AAGGAGGGCT ACGAGGTCCG 7gp
CATCCTCACC 840
3O GCCGACAAAG ~CCTI'T~C'rCA GCTCCTTfiC~ ~ACCcicATCC ACGTCCTCCA900
CCCCGAGGGG 960
TACCTCATCA CCCCGGCCTG GCTTTGGGAA AAGTACGGCC TGAGGCCCGA 1020
CCAGTGGGCC 1080
GACTACCGGG CCCTGACCGG GGACGAGTCC GACAACCTTC CCGGGGTCAA 1140
GGGCATCGGG 1200
GAGAAGACGG CGAGGAAGCT TCTGGAGGAG TGGGGGAGCC TGGAAGCCCT 1260
CCTCAAGAAC 1320
CTGGACCGGC TGAAGCCCGC CATCCGGGAG AAGATCCTGG CCCACATGGA
CGATCTGAAG
CTCTCCTGGG ACCTGGCCAA GGTGCGCACC GACCTGCCCC TGGAG~GTGGA
CTTCGCCAAA
AGGCGGGAGC CCGACCGGGA GAGGCTTAGG GCCTTTCTGG AGAGGCTTGA
GTTTGGCAGC
CTCCTCCACG AGTTCGGCCT TCTGGAAAGC CCCAAGGCCC TGGAGGAGGC
CCCCTGGCCC
CCGCCGGAAG GGGCCTTCGT GGGCTTTGTG CTTTCCCGCA AGGAGCCCAT
GTGGGCCGAT
CTTCTGGCCC TGGCCGCCGC CAGGGGGGGC CGGGTCCACC GGGCCCCCGA
GCCTTATAAA
GCCCTCAGGG ACCTGAAGGA GGCGCGGGGG CTTCTCGCCA AAGACCTGAG
CGTTCTGGCC
CTGAGGGAAG GCCTTGGCCT CCCGCCCGGC GACGACCCCA TGCTCCTCGC
CTACCTCCTG
GACCCTTCCA ACACCACCCC CGAGGGGGTG GCCCGGCGCT ACGGCGGGGA
GTGGACGGAG
GAGGCGGGGG AGCGGGCCGC CCTTTCCGAG AGGCTCTTCG CCAACCTGTG
GGGGAGGCTT
GAGGGGGAGG AGAGGCTCCT TTGGCTTTAC CGGGAGGTGG AGAGGCCCCT
TTCCGCTGTC
CTGGCCCACA TGGAGGCCAC GGGGGTGCGC CTGGACGTGG CCTATCTCAG 1380
3S GGCCTTGTCC 1440
CTGGAGGTGG CCGAGGAGAT CGCCCGCCTC GAGGCCGAGG TCTTCCGCCT
GGCCGGCCAC
CCCTTCAACC TCAACTCCCG GGACCAGCTG GAAAGGGTCC TCTTTGACGA GCTAGGGCTT 1500
CCCGCCATCG GCAAGACGGA GAAGACCGGC AAGCGCTCCA CCAGCGCCGC CGTCCTGGAG 1560
GCCCTCCGCG AGGCCCACCC CATCGTGGAG AAGATCCTGC AGTACC'.GGGA GCTCACCAAG 1620
_ ~3g
CA 02203627 1997-04-24
WO 96/15267 PCT/US95114673
CTGAAGAGCA CCTACATTGACCCCTTGCCG GACCTCATCC 1680
ACCCCAGGAC GGGCCGCCTC
CACACCCGCT TCAACCAGACGGCCACGGCC ACGGGCAGGCTAAGTAGCTC 1740
CGATCCCAAC
CTCCAGAACA TCCCCGTCCGCACCCCGCTT GGGCAGAGGA 1800 ,,
TCCGCCGGGC CTTCATCGCC
GAGGAGGGGT GGCTATTGGTGGCCCTGGAC TATAGCCAGA 1860
TAGAGCTCAG GGTGCTGGCC
S CACCTCTCCG GCGACGAGAACCTGATCCGG GTCTTCCAGGAGGGGCGGGA 1920 '
CATCCACACG
GAGACCGCCA GCTGGATGTTCGGCGTCCCC CGGGAGGCCGTGGACCCCCTGATGCGCCGG1980
GCGGCCAAGA CCATCAACTTCGGGGTCCTC TACGGCATGTCGGCCCACCGCCTCTCCCAG2040 -
GAGCTAGCCA TCCCTTACGAGGAGGCCCAG GCCTTCATTGAGCGCTACTTTCAGAGCTTC2100
CCCAAGGTGC GGGCCTGGATTGAGAAGACC CTGGAGGAGGGCAGGAGGCGGGGGTACGTG2160
IO GAGACCCTCT TCGGCCGCCGCCGCTACGTG CCAGACCTAGAGGCCCGGGTGAAGAGCGTG2220
CGGGAGGCGG CCGAGCGCATGGCCTTCAAC ATGCCCGTCCAGGGCACCGCCGCCGACCTC2280
ATGAAGCTGG CTATGGTGAAGCTCTTCCCC AGGCTGGAGGAAATGGGGGCCAGGATGCTC2340
CTTCAGGTCC ACGACGAGCTGGTCCTCGAG GCCCCAAAAGAGAGGGCGGAGGCCGTGGCC2400
CGGCTGGCCA AGGAGGTCATGGAGGGGGTG TATCCCCTGGCCGTGCCCCTGGAGGTGGAG2460
IS GTGGGGATAG GGGAGGACTGGCTCTCCGCC AAGGAGTGATACCACC 2506
(2) INFORMATION
FOR SEQ
ID N0:2:
(i) SEQUENCE
CHARACTERISTICS:
(A) LENGTH: 2496 base pairs
(B) ~'~E:
nucleic
acid
20 (C) STRANDEDNESS:
double
(D) TOPOLOGY:
linear
(ii) MOLECULE
TYPE: DNA
(genomic)
(xi) SEQUENCE
DESCRIPTION:
SEQ ID
N0:2:
ATGGCGATGC TTCCCCTCTTTGAGCCCAAA GGCCGCGTGCTCCTGGTGGACGGCCACCAC60
2J CTGGCCTACC GCACCTTCTTTGCCCTCAAG GGCCTCACCACCAGCCGCGGCGAACCCGTT120
CAGGCGGTCT ACGGCTTCGCCAAAAGCCTC CTCAAGGCCCTGAAGGAGGACGGGGACGTG180
GTGGTGGTGG TCTTTGACGCCAAGGCCCCC TCCTTCCGCCACGAGGCCTACGAGGCCTAC240
AAGGCGGGCC GGGCCCCCACCCCGGAGGAC TTTCCCCGGCAGCTGGCCCTCATCAAGGAG300
TTGGTGGACC TCCTAGGCCTTGTGCGGCTG GAGGTTCCCGGCTTTGAGGCGGACGACGTG360
3O CTGGCCACCC TGGCCAAGCGGGCGGAAAAG GAGGGGTACGAGGTGCGCATCCTCACTGCC420
GACCGCGACC TCTACCAGCTCCTTTCGGAG CGCATCGCCATCCTCCACCCTGAGGGGTAC480 '.
CTGATCACCC CGGCGTGGCTTTACGAGAAG TACGGCCTGCGCCCGGAGCAGTGGGTGGAC540
TACCGGGCCC TGGCGGGGGACCCCTCGGAT AACATCCCCGGGGTGAAGGGCATCGGGGAG600
AAGACCGCCC AGAGGCTCATCCGCGAGTGG GGGAGCCTGGAAAACCTCTTCCAGCACCTG660
~J GACCAGGTGA AGCCCTCCTTGCGGGAGAAG CTCCAGGCGGGCATGGAGGCCCTGGCCCTT720
TCCCGGAAGC TTTCCCAGGTGCACACTGAC CTGCCCCTGGAGGTGGACTTCGGGAGGCGC780
CGCACACCCA ACCTGGAGGGTCTGCGGGCT TTTTTGGAGCGGTTGGAGTTTGGAAGCCTC840
- 240 -
CA 02203627 1997-04-24
WO 96/15267
PGT/US95/14673
CTCCACGAGT TCGGCCTCCT GGAGGGGCCG AAGGCGGCAG AGGAGGCCCC
'" CTGGCCCCCT 900
CCGGAAGGGG CTTTTTTGGG CTTTTCCTTT TCCCGTCCCG AGCCCATGTG
S GGCCGAGCTT 960
CTGGCCCTGG CTGGGGCGTG GGAGGGGCGC CTCCATCGGG CACAAGACCC
CCTTAGGGGC 1020
CTGAGGGACC TTAAGGGGGT GCGGGGAATC CTGGCCAAGG ACCTGGCGGT
lO TTTGGCCCTG 1080
CGGGAGGGCC TGGACCTCTT CCCAGAGGAC GACCCCATGC TCCTGGCCTA
CCTTCTGGAC 1140
CCCTCCAACA CCACCCCTGA GGGGGTGGCC CGGCGTTACG GGGGGGAGTG
IS GACGGAGGAT 1200
GCGGGGGAGA GGGCCCTCCT GGCCGAGCGC CTCTTCCAGA CCC7.'AAAGGA
GCGCCTTAAG 1260
GGAGAAGAAC GCCTGCTTTG GCTTTACGAG GAGGTGGAGA AGCC'GCTTTC
CCGGGTGTTG 1320 -
2O GCCCGGATGG AGGCCACGGG GGTCCGGCTG GACGTGGCCT ACCTCCAGGC
CCTCTCCCTG 1380
GAGGTGGAGG CGGAGGTGCG CCAGCTGGAG GAGGAGGTCT TCCGCCTGGC
CGGCCACCCC 1440
TTCAACCTCA ACTCCCGCGA CCAGCTGGAG CGGGTGCTCT TTGACGAGCT
2S GGGCCTGCCT 1500
GCCATCGGCA AGACGGAGAA GACGGGGAAA CGCTCCACCA GCGCTGCCGT
GCTGGAGGCC 1560
CTGCGAGAGG CCCACCCCAT CGTGGACCGC ATCCTGCAGT ACCGGGAGCT
CACCAAGCTC 1620
AAGAACACCT ACATAGACCC CCTGCCCGCC CTGGTCCACC CCAAGACCGG
CCGGCTCCAC 1680
ACCCGCTTCA ACCAGACGGC CACCGCCACG GGCAGGCTTT CCAGCTCCGA
CCCCAACCTG 1740
CAGAACATCC CCGTGCGCAC CCCTCTGGGC CAGCGCATCC GCCGAGCCTT
CGTGGCCGAG 1800
GAGGGCTGGG TGCTGGTGGT CTTGGACTAC AGCCAGATTG AGCT7.'CGGGT
CCTGGCCCAC 1860
CTCTCCGGGG ACGAGAACCT GATCCGGGTC TTTCAGGAGG GGAGGGACAT
CCACACCCAG 1920
ACCGCCAGCT GGATGTTCGG CGTTTCCCCC GAAGGGGTAG ACCCTCTGAT
GCGCCGGGCG 1980
GCCAAGACCA TCAACTTCGG GGTGCTCTAC GGCATGTCCG CCCACCGCCT
CTCCGGGGAG 2040
CTTTCCATCC CCTACGAGGA GGCGGTGGCC TTCATTGAGC GCTACTTCCA
GAGCTACCCC 2100
AAGGTGCGGG CCTGGATTGA GGGGACCCTC GAGGAGGGCC GCCGGCGGGG
GTATGTGGAG 2160
ACCCTCTTCG GCCGCCGGCG CTATGTGCCC GACCTCAACG CCCGGGTGAA
GAGCGTGCGC 2220
GAGGCGGCGG AGCGCATGGC CTTCAACATG CCGGTCCAGG GCACCGCCGC
CGACCTCATG 2280
AAGCTGGCCA TGGTGCGGCT TTTCCCCCGG CTTCAGGAAC TGGGGGCGAG
GATGCTTTTG 2340
CAGGTGCACG ACGAGCTGGT CCTCGAGGCC CCCAAGGACC GGGCGGAGAG
GGTAGCCGCT 2400
TTGGCCAAGG AGGTCATGGA GGGGGTCTGG CCCCTGCAGG TGCCCCTGGA
GGTGGAGGTG 2460
GGCCTGGGGG AGGACTGGCT CTCCGCCAAG GAGTAG
2496
(2) INFORMATION FOR SEQ ID N0:3:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 2504 base pairs
- (B) TYPE: nucleic acid
(C) STRANDEDNESS: double
(D) TOPOLOGY: linear
~S (ii) MOLECULE TYPE: DNA (genomic)
- 241 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
(xi) S EQUENCE CRIPTION:EQ ID
DES S N0:3:
ATGGAGGCGATGCTTCCGCTCTTTGAACCC GGACGGCCAC 60
AAAGGCCGGG
TCCTCCTGGT
CACCTGGCCTACCGCACCTTCTTCGCCCTGAAGGGCCTCA GGGCGAACCG 120
CCACGAGCCG r
GTGCAGGCGGTCTACGGCTTCGCCAAGAGCCTCCTCAAGGCCCTGAAGGAGGACGGGTAC 180
S AAGGCCGTCTTCGTGGTCTTTGACGCCAAGGCCCCCTCCTTCCGCCACGAGGCCTACGAG 240
GCCTACAAGGCGGGGAGGGCCCCGACCCCCGAGGACTTCCCCCGGCAGCTCGCCCTCATC 300
AAGGAGCTGGTGGACCTCCTGGGGTTTACCCGCCTCGAGGTCCCCGGCTACGAGGCGGAC 360
GACGTTCTCGCCACCCTGGCCAAGAAGGCGGAAAAGGAGGGGTACGAGGTGCGCATCCTC 420
ACCGCCGACCGCGACCTCTACCAACTCGTCTCCGACCGCGTCGCCGTCCTCCACCCCGAG 480
IO GGCCACCTCATCACCCCGGAGTGGCTTTGGGAGAAGTACGGCCTCAGGCCGGAGCAGTGG 540
GTGGACTTCCGCGCCCTCGTGGGGGACCCCTCCGACAACCTCCCCGGGGTCAAGGGCATC 600
GGGGAGAAGACCGCCCTCAAGCTCCTCAAGGAGTGGGGAAGCCTGGAAAACCTCCTCAAG 660
AACCTGGACCGGGTAAAGCCAGAAAACGTCCGGGAGAAGATCAAGGCCCACCTGGAAGAC 720
CTCAGGCTCTCCTTGGAGCTCTCCCGGGTGCGCACCGACCTCCCCCTGGAGGTGGACCTC 780
IS GCCCAGGGGCGGGAGCCCGACCGGGAGGGGCTTAGGGCCTTCCTGGAGAGGCTGGAGTTC 840
GGCAGCCTCCTCCACGAGTTCGGCCTCCTGGAGGCCCCCGCCCCCCTGGAGGAGGCCCCC 900
TGGCCCCCGCCGGAAGGGGCCTTCGTGGGCTTCGTCCTCTCCCGCCCCGAGCCCATGTGG 960
GCGGAGCTTAAAGCCCTGGCCGCCTGCAGGGACGGCCGGGTGCACCGGGCAGCAGACCCC 1020
TTGGCGGGGCTAAAGGACCTCAAGGAGGTCCGGGGCCTCCTCGCCAAGGACCTCGCCGTC 1080
2O TTGGCCTCGAGGGAGGGGCTAGACCTCGTGCCCGGGGACGACCCCATGCTCCTCGCCTAC 1140
CTCCTGGACCCCTCCAACACCACCCCCGAGGGGGTGGCGCGGCGCTACGGGGGGGAGTGG 1200
ACGGAGGACGCCGCCCACCGGGCCCTCCTCTCGGAGAGGCTCCATCGGAACCTCCTTAAG 1260
CGCCTCGAGGGGGAGGAGAAGCTCCTTTGGCTCTACCACGAGGTGGAAAAGCCCCTCTCC 1320
CGGGTCCTGGCCCACATGGAGGCCACCGGGGTACGGCTGGACGTGGCCTACCTTCAGGCC 1380
2S CTTTCCCTGGAGCTTGCGGAGGAGATCCGCCGCCTCGAGGAGGAGGTCTTCCGCTTGGCG 1440
GGCCACCCCTTCAACCTCAACTCCCGGGACCAGCTGGAAAGGGTGCTCTTTGACGAGCTT 1500
AGGCTTCCCGCCTTGGGGAAGACGCAAAAGACAGGCAAGCGCTCCACCAGCGCCGCGGTG 1560
CTGGAGGCCCTACGGGAGGCCCACCCCATCGTGGAGAAGATCCTCCAGCACCGGGAGCTC 1620
ACCAAGCTCAAGAACACCTACGTGGACCCCCTCCCAAGCCTCGTCCACCCGAGGACGGGC 1680
3O CGCCTCCACACCCGCTTCAACCAGACGGCCACGGCCACGGGGAGGCTTAGTAGCTCCGAC 1740
CCCAACCTGCAGAACATCCCCGTCCGCACCCCCTTGGGCCAGAGGATCCGCCGGGCCTTC 1800
GTGGCCGAGGCGGGTTGGGCGTTGGTGGCCCTGGACTATAGCCAGATAGAGCTCCGCGTC 1860
CTCGCCCACCTCTCCGGGGACGAAAACCTGATCAGGGTCTTCCAGGAGGGGAAGGACATC 1920
CACACCCAGACCGCAAGCTGGATGTTCGGCGTCCCCCCGGAGGCCGTGGACCCCCTGATG 1980
3S CGCCGGGCGGCCAAGACGGTGAACTTCGGCGTCCTCTACGGCATGTCCGCCCATAGGCTC 2040
- 242 -
CA 02203627 1997-04-24
WO 96/15267 PCT/US95/14673
TCCCAGGAGC TTGCCATCCC CTACGAGGAG GCGGTGGCCT TTATAGAGGCTACTTCCAAA 2100
GCTTCCCCAA GGTGCGGGCC TGGATAGAAA AGACCCTGGA GGA.GGGGAGGAAGCGGGGCT 2160
ACGTGGAAAC CCTCTTCGGA AGAAGGCGCT ACGTGCCCGA CCTCAACGCCCGGGTGAAGA 2220
'" GCGTCAGGGA GGCCGCGGAG CGCATGGCCT TCAACATGCCCGTCCAGGGCACCGCCGCCG 2280
S ACCTCATGAA GCTCGCCATG GTGAAGCTCT TCCCCCGCCT CCGGGAGATGGGGGCCCGCA 2340
TGCTCCTCCA GGTCCACGAC GAGCTCCTCC TGGAGGCCCC CCAAGCGCGGGCCGAGGAGG 2400
TGGCGGCTTT GGCCAAGGAG GCCATGGAGA AGGCCTATCC CCTCGCCGTGCCCCTGGAGG 2460 '
TGGAGGTGGG GATGGGGGAG GACTGGCTTT CCGCCAAGGG TTAG 2504
(2) INFORMATION FOR SEQ ID N0:4:
IO (i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 832 amino acids
(B) TYPE: amino acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
IS (ii) MOLECULE TYPE: protein
(xi) SEQUENCE DESCRIPTION: SEQ ID N0:4:
Met Arg Gly Met Leu Pro Leu Phe Glu Pro Ly~~ Gly
1 5 10 Arg Val
Leu Leu
15
Val Asp Gly His His Leu Ala Tyr Arg Thr Phe His
ZO 20 25 Ala Leu
Lys Gly
30
Leu Thr Thr Ser Arg Gly Glu Pro Val Gln Ala. Val
35 40 Tyr Gly
Phe Ala
45
Lys Ser Leu Leu Lys Ala Leu Lys Glu Asp Gly Asp
50 55 Ala Val
Ile Val
60
ZS Val Phe Asp Ala Lys Ala Pro Ser Phe Arg His Glu Tyr Gly
65 70 Ala Gly
75 80
Tyr Lys Ala Gly Arg Ala Pro Thr Pro Glu Asp Phe Arg Gln
85 90 Pro Leu
95
Ala Leu Ile Lys Glu Leu Val Asp Leu Leu Gly Leu Arg Leu
100 105 Ala Glu
110
Val Pro Gly Tyr Glu Ala Asp Asp Val Leu Ala Ser Ala Lys
115 120 Leu Lys
125
Ala Glu Lys Glu Gly Tyr Glu Val Arg Ile Leu Thr Asp Lys
130 135 Ala Asp
140
3S Leu Tyr Gln Leu Leu Ser Asp Arg Ile His Val Leu Pro Glu
145 150 His Gly
_ 155
160
Tyr Leu Ile Thr Pro Ala Trp Leu Trp Glu Lys Tyr Leu Arg
165 170 Gly Pro
175
Asp Gln Trp Ala Asp Tyr Arg Ala Leu Thr Gly Asp Ser Asp
4'O 180 185 Glu Asn
190
Leu Pro Gly Val Lys Gly Ile Gly Glu Lys Thr Ala Lys Leu
195 200 Arg Leu
205
Glu Glu Trp Gly Ser Leu Glu Ala Leu Leu Lys Asn Asp Arg
210 215 Leu Leu
220
CA 02203627 1997-04-24
WO 96/15267 PGT/US95/14673
Lys Pro Ala Ile Arg Glu Lys Ile Leu Ala His Met Asp Asp Leu Lys
225 230 235
240
Leu Ser Trp Asp Leu Ala Lys Val Arg Thr Asp Leu Pro Leu Glu Val
245 250 255
Asp Phe Ala Lys Arg Arg Glu Pro Asp Arg Glu Arg Leu Arg Ala Phe
260 265 270
Leu Glu Arg Leu Glu Phe Gly Ser Leu Leu His Glu Phe Gly Leu Leu
275 280 285
Glu Ser Pro Lys Ala Leu Glu Glu Ala Pro Trp Pro Pro Pro Glu Gly
290 295 300
Ala Phe Val Gly Phe Val Leu Ser Arg Lys Glu Pro Met Trp Ala Asp
305 310 315
320
Leu Leu Ala Leu Ala Ala Ala Arg Gly Gly Arg Val His Arg Ala Pro
325 330 335
15 Glu Pro Tyr Lys Ala Leu Arg Asp Leu Lys Glu Ala Arg Gly Leu Leu
340 345 350
Ala Lys Asp Leu Ser Val Leu Ala Leu Arg Glu Gly Leu Gly Leu Pro
355 360 365
Pro Gly AspAspProMet LeuLeu AlaTyrLeu Leu Asn
2~ 370 375 Asp
Pro
Ser
380
Thr Thr ProGluGlyVal AlaArg ArgTyrGly GlyGluTrp Glu
385 390 395 Thr
400
Glu Ala GlyGluArgAla AlaLeu SerGluArg LeuPheAlaAsn Leu
405 410 415
2$ Trp Gly ArgLeuGluGly GluGlu ArgLeuLeu TrpLeuTyrArg Glu
420 425 0430
Val Glu ArgProLeuSer AlaVal LeuAlaHis MetGluAlaThr Gly
435 - 440 445
Val Arg LeuAspValAla TyrLeu ArgAlaLeu SerLeuGluVal Ala
450 - 455 460
Glu Glu IleAlaArgLeu GluAla GluValPhe ArgLeuAlaGly His
465 470 475
480
Pro Phe AsnLeuAsnSer ArgAsp GlnLeuGlu ArgValLeuPhe Asp
485 490 495
Glu Leu GlyLeuProAla IleGly LysThrGlu LysThrGlyLys Arg
500 505 510
Ser Thr SerAlaAlaVal LeuGlu AlaLeuArg GluAlaHisPro Ile
515 520 525
Val Glu LysIleLeuGln TyrArg GluLeuThr LysLeuLysSer Thr
40 530 535 540
Tyr Ile AspProLeuPro AspLeu IleHisPro ArgThrGlyArg Leu
545 550 555
560
His Thr ArgPheAsnGln ThrAla ThrAlaThr GlyArgLeuSer Ser
565 570 575
4S Ser Asp Pro LeuGln Ile ProVal ThrProLeuGly
Asn Asn 585Arg 590Gln
580
- 244 -
CA 02203627 1997-04-24
WO 96/15267
PCTJUS95/14673
Arg Ile Arg Glu Gl
Arg Tr
Ala L
Phe
Ile
Ala
Glu
y
595 600 p
eu Leu Val Ala
605
Leu Asp Tyr
Ser
Gln
Ile
Glu
Leu
Arg
Val
Leu
Ala
Hi
L
s
610 615 eu Ser Gly
620
.. ~ Asp Glu Asn Phe
Leu Gln
Ile Glu
Arg Gly
Val Ar
As
Il
g
625 630 p
e His Thr
635
640
' Glu Thr AlaSerTrpMet Gly Pro Ar
Phe Val Gl
Al
g
s 645 u
a Val Asp Pro
650
655
1~ Leu Me't ArgArgAlaAla Thr Asn Phe: Gl
Lys Ile Val L
y
660 eu Tyr Gly
665
670
Met Ser AlaHisArgLeu Gln Leu Ala
Ser Glu Il
.
675 e Pro Tyr Glu Glu
680
685
Ala 69n AlaPheIleGlu Tyr Gln Ser Ph
Arg Phe P
0 e
695 ro Lys Val Arg
700
15 Ala Trp IleGluLysThr Glu Gly Ar
Leu Glu Ar
A
g
705 710 g
rg Gly Tyr Val
715 720
Glu Thr LeuPheGlyArg Arg Val Pro As
Arg Tyr Leu Gl
p
725 u Ala Arg
730
735
Val Lys SerValArgGlu Ala Arg Met Ala Ph
Ala Glu
e Asn Met Pro
740
745
750
Val Gln GlyThrA1aAla Leu Lys Leu Al
Asp Met M
a
755 et Val Lys Leu
760
765
Phe Pro Arg LeuGluGlu Gly Arg Met Le
Met Ala L
u
770 775 eu Gln Val His
780
25 Asp Glu Leu ValLeuGlu Pro Glu Ar
Ala Lys Ala Gl
g
785 790 u Ala Val Ala
795 800
Arg Leu Ala LysGluVal Val Tyr Pro Leu Al
Met
Glu
Gly
a Val Pro
805
810
815
Leu Glu Val Glu Gly Asp Trp Leu Ser Al
Val Ile L
Gly
Glu
a
- 820 825 ys Glu
830
(2) INFORMATION FOR SEQ ID N0:5:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 831 amino acids
(B) TYPE: amino acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: protein
(xi) SEQUENCE DESCRIPTION: SEQ ID N0:5:
Met Ala Met Leu Pro Leu Phe Glu Pro Lys Gly Arg Val Leu Leu Val
1 5 10 15
Asp Gly His His Leu Ala Tyr Arg Thr Phe Phe Ala Leu Lys Gly Leu
20 25 30
Thr Thr Ser Arg Gly Glu Pro Val Gln Ala Val Tyr Gly Phe Ala Lys
35 40 45
4S Ser Leu Leu Lys Ala Leu Lys Glu Asp Gly Asp Val Val Val Val Val
50 55 60
- 24~ -
CA 02203627 1997-04-24
WO 96/15267 PGT/US95/14673
Phe Asp Ala Lys Ala Pro Ser Phe Arg His Glu Ala Tyr Glu Ala Tyr
65 70 75 80
Lys Ala Gly Arg Ala Pro Thr Pro Glu Asp Phe Pro Arg Gln Leu Ala
85 90 95
$ Leu Ile Lys Glu Leu Val Asp Leu Leu Gly Leu ValArg Leu Glu Val
100 105 110
Pro Gly Phe G1-a Ala Asp Asp Val Leu Ala Thr Leu Ala Lys Arg Ala
115 120 125
Glu Lys Glu Gly Tyr Glu Val Arg Ile Leu Thr Ala Asp Arg Asp Leu
130 135 140
Tyr Gln Leu Leu Ser Glu Arg Ile Ala Ile Leu His Pro Glu Gly Tyr
145 150 155
160
Leu Ile Thr Pro Ala Trp Leu Tyr Glu Lys Tyr Gly Leu Arg Pro Glu
165 170 175
l$ Gln Trp Val Asp Tyr Arg Ala Leu Ala Gly Asp Pro Ser Asp Asn Ile
180 185 190
Pro Gly Val Lys Gly Ile Gly Glu Lys Thr Ala Gln Arg Leu Ile Arg
195 200 205
Glu TrpGly SerLeu GluAsnLeu PheGlnHisLeu AspGlnVal L
ys
210 215 220
Pro SerLeu ArgGlu LysLeuGln AlaGlyMetGlu AlaLeuAla Leu
225 230 235
240
Ser ArgLys LeuSer GlnValHis ThrAspLeuPro LeuGluVal Asp
245 250 255
2$ Phe GlyArg ArgArg ThrProAsn LeuGluGlyLeu ArgAlaPhe Leu
260 265 270
Glu ArgLeu GluPhe GlySerLeu LeuHisGluPhe GlyLeuLeu Glu
275 280 285
Gly ProLys AlaAla GluGluAla ProTrpProPro ProGluGly Ala
290 295 300
Phe Leu Gly Phe Ser Phe Ser Arg Pro Glu Pro Met Trp Ala Glu Leu
305 310 315
320
Le~u Ala Leu Ala Gly Ala Trp Glu Gly Arg Leu His Arg Ala Gln Asp
325 330 335
3$ Pro Leu Arg Gly-Leu Arg Asp Leu Lys Gly Val Arg Gly Ile Leu Ala
340 345 350
Lys Asp Leu Ala Val Leu Ala Leu Arg Glu Gly Leu Asp Leu Phe Pro
355 360 365
Glu Asp Asp Pro Met Leu Leu Ala Tyr Leu Leu AspJPro~Ser Asn Thr
370 375 380
Thr Pro Glu Gly Val Ala Arg Arg Tyr Gly Gly Glu Trp Thr Glu Asp
385 -- 390 395
400
Ala Gly Glu Arg Ala Leu Leu Ala Glu Arg Leu Phe Gln Thr Leu Lys
405 410 415
4$ Glu Arg Leu Lys Gly Glu Glu Arg Leu Leu Trp Leu Tyr Glu Glu Val
420 425 430
- 24G -
CA 02203627 1997-04-24
WO 96/15267 _PCTlUS95/14673
Glu Lys Pro Leu Ser Arg Val Leu Ala Arg Met Glu ~~'~
435 440 445
Arg Leu Asp Val Ala Tyr Leu Gln Ala Leu Ser Leu Glu Val Glu Ala
450 455 460
'_ S Glu Val Arg Gln Leu Glu Glu Glu Val Phe Arg Leu Ala Gly His Pro
465 470 47-'°.
480
Phe Asn Leu Asn Ser Arg Asp Gln Leu Glu Arg Val Leu Phe Asp Glu
485 490 495
Leu Gly Leu Pro Ala Ile Gly Lys Thr Glu Lys Thr Gly Lys Arg Ser
500 505 510
Thr Ser Ala Ala Val Leu Glu Ala Leu Arg Glu Ala His Pro Ile Val
515 520 525
Asp Arg Ile Leu Gln Tyr Arg Glu Leu Thr Lys Leu Lys Asn Thr Tyr
530 535 540
15 Ile Asp Pro Leu Pro Ala Leu Val His Pro Lys Thr Gly Arg Leu His
545 550 555
560
Thr Arg Phe Asn Gln Thr Ala Thr Ala Thr Gly Arg Leu Ser Ser Ser
565 570 575
Asp ProAsn LeuGln AsnIlePro ValArgThr ProLeuGly GlnArg
580 585 590
Ile ArgArg AlaPhe ValAlaGlu GluGlyTrp ValLeuVal ValLeu
595 600 605
Asp TyrSer GlnIle GluLeuArg ValLeuAla HisLeuSer GlyAsp
610 615 620
25 Glu AsnLeu IleArg ValPheGln GluGlyArg AspIleHis ThrGln
625 630 635
640
Thr AlaSer TrpMet PheGlyVal SerProGlu GlyValAsp ProLeu
645 650 655
Met ArgArg AlaAla LysThrIle AsnPheGly ValLeuTyr GlyMet
660 665
670
Ser AlaHis ArgLeu SerGlyGlu LeuSerIle ProTyrGlu GluAla
675 680 685
Val AlaPhe IleGlu ArgTyrPhe GlnSerTyr ProLysVal ArgAla
690 695 700
35 Trp IleGlu GlyThr LeuGluGlu GlyArgArg ArgGlyTyr ValGlu
705 710 715
720
Thr Leu Phe Gly Arg Arg Arg Tyr Val Pro Asp Leu Asn Ala Arg Val
725 730 735
Lys Ser Val Arg Glu Ala Ala Glu Arg Met Ala Phe Asn Met Pro Val
740 745 750
Gln Gly Thr Ala Ala Asp Leu Met Lys Leu Ala Met Val Arg Leu Phe
755 760 765
Pro Arg Leu Gln Glu Leu Gly Ala Arg Met Leu Leu Gln Val His Asp
770 775 780
Glu Leu Val Leu Glu Ala Pro Lys Asp Arg Ala Glu Arg Val Ala Ala
785 790 795
800
-247-
DEMANDES OU BREVETS VOLUMlNEUX
LA PRESENTS PART1E DE CETTE DEMANDS OU CE BREVET
COMPREND PLUS D'UN TOME_
CEC1 EST LE TOME ~ DE
NOTE: Pour les tomes additionels, veuiilez contacter le Bureau canadien des
brevets
3 ~ ~.'7
JUMBO APPL1CATIONS/PATENTS
THiS SECTION OF THE APPLICAT10N/PATENT CONTAINS MORE
'THAN ONE VOLUME ~ ,
THIS 1S VOLUME y~_ OF
' NOTE: For additional volumes-please contact the Canadian Patent Office .