Note: Descriptions are shown in the official language in which they were submitted.
CA 02203791 1997-04-2~
W O 96/13S07 PCT/AU9S/00717
SYNTHETIC POLYUNSATURATED FA7'IYACID ANALOGUES
The present invention relates to new polyunsaturated fatty acids having
antimRl~rial activity and/or neutrophil stimulatory activity. In addition.
5 certain of the new polyunsaturated fatty acids depress cytokine activity.
Over half of the world s population is at risk from malaria. with about 500
million acute infections and approximately 1 million deaths recorded each
year. (Tropical Diseases Progress in International Research, 1987-1988.
10 Ninth Programme Report. UNDPlWorld BanklWHO, Geneva, 43-49;
Stevenson MM Preface In: Stevenson MM, Ed. Malaria: Host responses to
Infection. CRC Press, Inc). The use of antimRlRrial drugs is associated with
major problems because of increased resistance and toxic side-effects. Most
currently used antimalarials are unsuitable for use in children (most at risk
15 of potentiallv fatal cerebral malaria). pregnant women and the aged.
Neutrophil/macrophage stimulatory agents may have application in the
treatment of other infections including Candida sp. Trypanosoma,
Schistosomiasis, Tuberculosis, viruses eg herpes, Sindbis virus, Legionella,
20 Listeriosis, Pneumocystsis, Pseudomonas. They would also be useful as
adjunct therapy in immunocompromised individuals including those
undergoing cancer chemotherapy. transplant recipients and burns patients.
In addition, others, so called normal individuals may also be treated, eg the
aged, children under 2, alcoholics. who are known to have poor phagocytic
25 cell activity.
TnflRmmRtion may be caused by bacteria, viruses and!or other infective
agents, opportunistic infections (which may be consequent on an
immunodepressed state, for example resulting from cancer or therapy,
30 particularly cvtotoxic drug therapy or radiotherapy), autoimmunity or
otherwise. Septic shock is an illustration of a disease involving systemic
inflammation. Many of the clinical features of Gram-negative septic shock
- may be reproduced in RnimRl~ by the administration of LPS to Rnim~l~ can
prompt severe metabolic and physiological changes which can lead to death.
35 Associated with the iniection of LPS is the extensive production of pro-
inflammatorv cytokines such as tumour necrosis factor alpha (TNFa).
CA 02203791 1997-04-2~
Wo 96/13507 PCT/AUg5/00717
Chronic administration of TNF in mice, rats and/or humans causes anorexia,
weight loss and depletion of body lipid and protein within 7 to 10 days
(Cerami et al, 1985, Immunol. Lett. 11, 173: Fong et al, 1989 J. Exp. Med.
170, 1627. Moldawer et al, Am. J. Physiol. 254 G450-G456. 1988: Fong et al,
Am. J Physiol. 256, R659-R665 (1989): McCarthy et al, Am. J. Clin. Nature.
42. 1179-1182). TNF levels have been measured in patients with cancer and
chronic disease associated with cachexia.
TNFa has been implicated in the pathology of other diseases associated with
chronic inflammation apart form toxic shock and cancer-related cachexia.
TNF has been detected in synovial fluid in patients with both rheumatoid
and reactive arthritis and in the serum of patients with rheumatoid arthritis
(Saxne et al. 1988. Arthrit. Rheumat. 31. 1041). Raised levels of TNF have
been detected in renal transplant patients during acute rejection episodes
(Maurv and TeppG, 1987~ J. Exp. Med. 166. 113~). In ~nim~ , TNF has been
shown to be involved in he pathogenesis of graft-versus-host disease in skin
and gut following allogenic marrow transplantation.
Administration of a rabbit anti-murine TNF antibody was shown to prevent
the histological changes associated with graft-versus-host disease and to
reduce mortality (Piquet et Ql. 1987, ~. Exp. Med. 166, 12Z0). TNF has also
been shown to contribute significantly to the pathology of malaria (Clark et
a~. 1987. Am. J. Pathol. 129. 192-199). Further. elevated serum levels of TNF
have been reported in malaria patients (Scuderi et al, 1986. Lancet 2. 1364-
1365~.
Elevated pro-infl~mm:ltory cytokine levels have further been implicated in
causing the pathology and tissue destructlon in rheumatoid arthritis~
multiple sclerosis (MS) and Crohns disease. Experimentally~ anti-bodies
which neutralise the activity of cytokine producing cells (eg antibodies
against CD4+ T cells or antibodies against CD3) or of the cytokines
themselves (eg anti-TNF antibodies) have proved beneficial. High levels of
interferon ~ are known tc be associated with disease exacerbation in MS.
PUFA's have a ran,ge of useful biological activities (see for example
International Patent Application Nos. WO 93!00084 and WO 95/00607 and
CA 02203791 1997-04-2~
WO 96/13S07 PCT/AU9S/00717
the references cited therein). Unfortunately, due to their limited stability in
v~vo, PUFA's have not achieved widespread use as therapeutic agents. The
present inventors have developed a method for coupling amino acids to
PUFAs which, while retRining biological activity, have increased stability
5 and solubility. These new polyunsaturated fatty acid (PUFA) compounds
have direct antimalarial activity. In addition to their direct antimRlRrial
activity. certain of the novel PUFA activate human neutrophils causing
release of granule contents, and exhibit synergy with TNF in the production
of superoxide. Activation of human neutrophils by the PUFA results in
10 enhanced ability of these cells to kill malaria parasite (P. fa~5ciparum) within
red blood cells and also the bacteria Stczphylococcus aureus.
Further. the present inventors have also found that certain of the amino acid
coupled PUFA are anti-inflammatory in that they depress the production of
15 pro-inflammatory cytokines while failing to activate neutrophils.
Accordingly. the present invention consists in a polvunsaturated fattv acid
compound having antimalarial and/or neutrophil stimulatorv actlvitv. Ol
anti-inflammatory activity. the polyunsaturated fattv acid containlllg a lG-26
20 carbon chain. 3-6 double bands wherein the polvunsaturated fatt~ acid ls
covalently coupled at the carboxylic acid group to an aInino acl(l
In a preferred embodiment of the present inventioIl the iatt~ e~ olllalnS
18-22 carbons.
In a further preferred embodiment of the present invention the anliIlo acid is
glycine or aspartic acid.
In another preferred embodiment of the present invention the fatty acid is an
30 n-3 to n-6 compound.
In vet a further preferred embodiment of the present invention the
compound is y-linolenic acid-glycine~ a-linolenic acid-glycine. arachidonic
acid-glvcine. docosahexaenoic acid-glvcine. eicosapentaenoic glycine5 y
35 linolenic acid - aspartic acid, a-linolenic acid - aspartic acid, arachidonic
CA 02203791 1997-04-2~
WO 96/13507 PCT/AU9S/00717
acid - aspartic acid, eicosapentaenoic acid - aspartic acid and
docosahexaenoic acid - aspartic acid.
In order that the nature of the present invention may be more clearly
5 understood, a preferred form thereof will now be described with reference to
the following examples and figures in which:
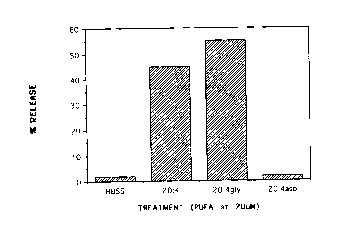
Figures 1 and 2 show the effects of PUFAs on release from azurophilic
granules;
10 Figure 3 shows release of neutrophil specific granule contents following
treatment with PVFAs; and
Figure 4 shows the effect of PUFA on neutrophil mediated killing of
S. aureus.
In these Figures the following abbreviations are used:
20:4 Arachidonic acid
20 :5 Eicosapentaenoic acid
22 :6 Docosahexaenoic acid
gly glycine
asp aspartic acid
Table 1 shows the direct anti-malarial activity of the amino-acid conjugated
PUFAs.
Table 2 shows the abilitv of amino acid conjugated PUFAs to suppress TNFa
production and interferon r production by PHA-stimulated peripheral blood
25 mononuclear cells.
Table 3 shows the abilitv of amino acid conjugated PUFAs to suppress PHA
stimulated proliferation (principally T cell proliferation ) of peripheral bloodmononuclear cells.
30 METHODS
Preparation of neutrophils
Heparinised blood from normal healthy individuals was layered onto Ficoll-
Hypaque medium of density 1.114 and centrifuged at 600~ for 30-40 min at
35 room temperature. The cells were washed three time in Hanks Balanced Salt
Solution (HBSS). Preparation were of 96-99% purity with respect to white
CA 02203791 1997-04-2~
WO 96/13507 PCT/AU95100717
blood cells and were >99% viable as judged by their ability to exclude
trypan blue. Red blood cell cont~minAtion was always less than 1 per
neutrophil with platelets being generally absent.
5 Prepara'don of Fatty Acid micelles and pretreatment of neutrophils
To overcome fatty acid insolubility in aqueous solution, mixed dipalmitoyl
phosphatidylcholine (DPC, 40011g):fatty acid (100~1g) micelles were prepared
in HBSS by sonication. Neutrophils were pretreated for 30 min at 37C. In
some experiments PUFA were solubilized in ethanol.
Measurement of neutrophil chemiluminescence
To 1oo~l of neutrophils (1 x 10~) in HBSS was added 1oo~ll of fatty acid
micelles or DPC aione and an additional 300,u1 of HBSS. This was followed
immediately by the addition of 500111 of lucigenin (0.25mg/ml in PBS) and
the resulting light output (mV) measured over time in a luminometer.
Experiments were performed in triplicate with cells from a separate
individual and values presented represent peak values of the responses.
20 Measurement of degranulation
Degranulation was determined by measuring vltamin B12 ~inding protein (as
described by Gottleib et al, 1965, Blood 25:875-883J and ~-glucuronidase
release (as described bv Kolodenev and Mumford. 1976. Clin. Chem. Act~
70 :247-257) .
Bactericidal assay
Neutrophil bactericidal activity against Staphyiococcus aureus was measured
according to the procedure described by Ferrante and Abell. 198G, Infect.
Immun. 51:607.
MononuGlear cell proliferation assays
Mononuclear cells were separated from peripneral blood of normal human
donors as described bv Ferrante and Thong (1978...). The mononuclear cells
were resuspended in RPMI-1640 cont~ining 20% human AB serum and
placed into 96 well microtrays (50~1 per well. cell density 4Xl06 cells/ml).
Fatty acid was then added in 50Ld and pre-incubated with the cells for 30
CA 02203791 1997-04-2~
WO 96/13507 PCT/AU95/00717
min at 37C in 5% CO2. Mitogen (PHA, ConA, PWM, Staph. Aureus) was
then added in 1oo~l and the cells incubated for 66 hours at 37C in 5% CO2
before the addition of tritiated thymidine (lllcilwe~ After a total of 72h in
culture, the cells were harvested and proliferation (thymidine incorporation)
and supernatants assayed for the presence of cytokines.
Cytokine assays
Cytokine levels in culture supernatants were determined by specific ELISA
using anti-cytokine antibodies. The following cytokine levels were
determined: TNFa, TNF,~, interferon-~, IL-1,~, IL-2.
Chemical syntheses
Arachidonic acid-glycine-OH
Arachidonic acid (0.50 g) was dissolved in DMF (2.0 mL), HOSu (0.38 g in
0.5 mL DMF) and H-Gly-OtBu.HCl (0.55 g in 1.5 mL DMF~ were added. The
mixture was cooled in ice bath. DCC (0.41 g in 0.5 mL DMF) was added. N-
MM was added and the mixture was stirred for 30 minutes in ice bath and
then stirred at room temperature for 20 hours. The reaction did not go to
completion and about 20-3-% arachidonic acid was not reacted. More DCC
(0.16 g), HOSu (0.19 g), H-Gly-OtBu.HCl (0.20 g) and N-MM (0.24 g) were
added and the mixture was stirred for 24 hours. DCU was filtered off and the
product was isolated by preparative HPLC and lyophilised to vield a pale
green oil (0.67 g, 98%). The oil of arachidonic-Glv-OtBu was redissolved in
neat trifluoroacetic acid (40 mL) in ice bath and stirred for 30 min and then
at room temperature for further 30 minutes. TFA was evaporated to yield
arachidonic-Gly-OH as a muddy green oil (0.53 g). It was purified by HPLC
and lyophilised to yield a light yellow gluey solid (0.23 g. 39%).
Purification
Preparative HPLC conditions:
buffer A: 0.1% TFA/H2O, buffer B: 0.1% TFA/10%H2O/90% CH3CN.
40 mLlmin, 214 nm. C18 semiPrepPak
Stepwise increments of %B: 10--20--30--40--50--60--70--80--90--100U/o
B.
CA 02203791 1997-04-2~
WO 96/13507 PCT/AU95/00717
Arachidonic acid eluted at 60% B, arachidonic-Gly-OH eluted at 75-
80% B, arachidonic-Gly-OtBu eluted at 80-85% B.
1. HPLC
buffer: 0.1% TFA / 10% H2O / 90% CH3CN
2 rnL/min, 214 nm, C18 NovaPak
isocratic
Retention times of components:
Arachidonic acid: Rt 4.14 min
Arachidonic-Gly-OH: Rt 2.78 min
Arachidonic-Gly-OtBu: Rt 5.23 min
2. 13C n.m.r.
Arachidonic-Gly-OH
_(DMSO-d6): 14.1. C20. 22.1. 25.4. 2~.4~ 26.8. 28.9. 31Ø 34.7. 10 x
CH2: 40.7. Ga; 127.7, 127~85. 127.93, 128.2, 128.3, 129.6, 130.1, 8 x CH;
171.5, C=O! G: 172.5, C1.
3. FAB-MS
mJz 362 (M + 1)
4. Amino acid analvsis
Gly present
25 Arachidonic-aspartic acid-OH
Arachidonic acid. HOSu and H-Asp(OtBu)-OtBu.HCl were dissolved together
in DMF (3 mL). The mixture was cooled in ice bath and DCC in DMF (0.7
mL) was added. N-MM was added and the mixture was stirred for 20 hours.
About 20% arachidonic acid remained. More HOSu (0.19 g), H-Asp(OtBu)-
30 OtBu.HCl (0.30 g). DCC (0.16 g) and N-MM (0.24 g) were added and the
mixture was stirred for further 20 hours. DCU was filtered off and the
product was isolated bv HPLC. The purified Ara-Asp(OtBu)-OtBu was
concentrated to an oil and TFA (25 mL) was added. After an hour stirring,
TFA was evaporated to yield a dark green oil. Arachidonic-Asp-OH was
35 purified by HPLC. The pure fractions of Ara-Asp-OH were combined.
concentrated and lyophilised (in tBu-OH) to yield brown oil (0.3~ g, 55%~.
CA 02203791 1997-04-2~
WO 96/13507 PCT/AU95tO0717
Purification
HPLC purification:
buffer A: 0.1% TFA,
buffer B: 0.1% TFA + 10% H2O + 90% CH3CN
40 mL/min, 214 nm, C18 SemiPrepPak
Stepwise increments of %B: 10%--20--30--40--50--60--70--80--85--
100% B.
Arachidonic acid eluted at 70% B.
Arachidonic-Asp(OtBu)-OtBu eluted at 80% B.
Arachidonic-Asp-OH eluted at 60% B.
Analysis
1. HPLC
buffer: 0.1% TFA ~ 10% H2O + 90% CH3CN
2 mL/min, 214 nm. C18 NovaPak
isocratic
Retention times:
Arachidonic acid: Rt 4.12 min
Arachidonic-Asp(OtBu)-OtBu: Rt 9.52 min
Arachidonic-Asp-OH: Rt 2.31 min
2. 13C n.m.r.
Arachidonic-Asp-OH
_(DMSO-d6): 14.1. CH3; 22.1, 25.4. Z6.4, 26.8~ 28.9, 31.0, 31.5, 34.8.
10 x CHz; 34.4, ??; 36.2. D~; 48.7, Da; 67.1. ??; 127.7, 127.88, 127.97. 128.18
128.23. 129.6, 130.1, 8 x CH; 171.6. D_: 172.1, C=O, Asp; 172.7, C=O~
30 Arachidonic.
Arachidonic acid
_(DMSO-d6): 14.1, CH3; 22.2, 24.6. 25.4, 26.3, 26.8, 26.9, 28.9, 31.1,
33.3, 10 x CH2; 127.7. 127.9, 128Ø 128.2, 128.3, 128.4, 129.3, 130.1, 8 x CH;
174.5, C=O.
CA 02203791 1997-04-2~
W O96/13S07 PCT/AU9S/00717
3. FAB-MS and CI-MS
m/z 420 (M + 1).
4. Amino acid analvsis
Asp present.
Eicosapentaenoic acid-glycine-OH
Eicosapentaenoic acid. H-Gly-OtBu.HCl and HOSu were dissolved to~ether
in DMF (4 mL). The mixture was cooled in ice bath and DCC (in 1 mL DMF)
was added. N-methylmorpholine was added and the mixture stirred in ice
bath for 20 minutes and then at room temperature for 20 hours. 36% of
eicosapentaenoic acid remained unreacted. More H-Gly-OtBu.HCl (0.22 g),
HOSu (0.15 g), DCC (0.16 g) and N-MM (0.27 g) were added and stirred for
further 20 hours. Some eicosapentaenoic acid remained (about 30% by
HPLC). The mixture was filtered and the crude product was purified hy
HPLC to yield Epe-Gly-OtBu as coloured oil (0.49 g, 71%). The oil was
redissolved in cold trifluoroacetic acid (30 mL) and stirred for an hour. TFA
was evaporated to leave a black oil. The crude Epe-Gly-OH was purified bv
HPLC to yield 0.13 g (22%) brown oil.
Purification
HPLC purification
buffer A: 0.1% TFA/HzO
buffer B: 0.1% TFA + 10% H20 + 90/cJ C H3CN
40 mL!min, 214 nm, C18 semiPreppak
Increments of %B: 10--20--30--40--50--55--60--65--6~--70%B.
Epe acid and Epe-Gly-OtBu eluted at 65-70% B. It was able to isolate
some pure fractions of Epe-Gly-OH. Fractions cont~inine the two compounds
were combined and repurified.
Under the same conditions as above. Epe-Gly-OH eluted at 60% B.
Analysis
1. Analvtical HPLC
Buffer: 0.1% TFA + 10% H2O + 90% CH3CN
CA 02203791 1997-04-25
WO 96/13507 PCT/AU95/00717
2 mIlmin, 214 nm, C18 Novapak
isocratic
Retention times of reaction components:
eicosapentaenoic acid: Rt 3.1 min
Epe-Gly-OtBu: Rt 3.9 min
Epe-Gly-OH: Rt 2.1 min
2. 13C n.m.r.
(DMSO-d6): 14.3, CH3: 20.2, 25.4, 26.4, 34.8, CH2; 40.7, Ga; 127.2,
127.9, 128.1, 128.2. 128.3, 129.7, 131.8, CH; 171.6, 172.5, C=O.
3. CI-MS
m~z 360 (M+1).
I~:icosapentaenoic acid-aspartic acid-OH
Eicosapentaenoic acid, H-Asp(OtBu)-OtBu.HCl and HOSu were dissolved
together in DMF (4 mL). The mixture was cooled in the ice bath and DCG (in
1 mL DMF) was added. N-Methvlmorpholine was added and the mlxture was
stirred in ice bath for 20 minutes and then at room temperature for 20 hours.
About 23% Epe acid by HPLG remained. More H-Asp(OtBu~-OLBu.HCl (0.28
g). HOSu (0.11 g), DCC (0.12 g) and N-MM (0.20 g) v~ere adde(l alld the
mixture stirred for further 20 hours. About 17~) Epe acid relllallle d I he
mixture was filtered and the crude Epe-Asp(OtB~l~-OtB~ s })l~rltle(l b~
HPLC and vielded 0.83 g (94%) brown oil. Cold trifhlolL)~ t~c .~ 0 mL)
was added to the brown oil and the mixture stirred for all llc~ul T~A was
evaporated to leave a dark brown oil which was redissolved ill CH3CN (10
mL) and was purified by HPLC. The pure Epe-Asp-OH weighed 0.50 g (72%).
Purification
Buffer A: 0.1% TFA/H2O
Buffer B: 0.1% TFA + 10% H2O + 90% GH3CN
40 mL/min, 214 nm. C18 semiprepPak
Increments of %B: 10%--20--30--40--50--52--55--57--60--65--68--70%
B.
CA 02203791 1997-04-2~
WO 96/13507 PCI/AU95/00717
Epe acid eluted at 65% B, Epe-Asp(OtBu)-OtBu eluted at 70% B, Epe-
Asp-OH eluted at 55% B.
Analysis
1. Analvtical HPLC
Buffer: 0.1% TFA + 10% H2O + 90% CH3CN
2 mL/min, 214 nm, C18 Novapak. isocratic
Retention times:
Epe acid: Rt 3.1 min
Epe-Asp(OtBu)-OtBu: Rt 6.7 min
Epe-Asp-OH: Rt 1.8 min
2. 13C n.m.r.
(DMSO-d6): 14.3. CH3: 20.2. 25.3. 25.4, 26.4. 31.5, 34.8, 8 x CH2:
36.3. D~; 48.7. Da: 127.2, 127.92, 127.97, 128.1, 128.2, 128.3~ 129.7, 131.8, 10x CH; 171.9, 172.1, 172.7, 3 x C=O.
3. CI-MS
m/z 418 (M+1).
Docosahexaenoic acid-glycine-OH
H-Gly-OtBu.HCl and HOSu were dissolved together in DMF (2 mL). The
mixture was cooled in ice bath and docosahexaenoic acid. DCG (in 0.4 mL
DMF), and N-methylmorpholine were added. The mixture stirred in ice bath
for 30 minutes and then at room temperature for 5 hours. 30%
docosahexaenoic acid (Dhe acid) remained. More DCC (0.11 g) was added
and the mixture stirred for further 20 hours. About 28% Dhe acid remained.
The mixture was filtered and the crude product was purified by HPLC. The
lyophilised Dhe-Gly-OtBu (light yellow oil) weighed 0.62 g (92%). Cold TFA
(30 mL) was added to the oil and the mixture stirred for an hour. TFA was
evaporated to leave a dark brown oil which was redissolved in CH3CN (10
mL) and was purified by HPLC. The purified Dhe-Gly-OH was lyophilised to
leave a dark brown oil (0.27 g. 46%).
CA 0220379l l997-04-2~
WO 96/13507 PCT/AU95/00717
PurificaJdon
HPLC conditions:
Buffer A: 0.1% TFA~H20
Buffer B: 0.1% TFA + 10% H2O + 90% CH3CN
40 mL/min, 214 nm, C18 semipreppak
manual increment of %B: 10%--20--30--40--50--55--60--65--70--73--
100%B.
Both Dhe acid and Dhe-Gly-OtBu eluted at 71-73%B. The acid
10 eluted slightly earlier than Dhe-Gly-OtBu.
Dhe-Gly-OH eluted at 60%B.
Analysis
1. Analvtical HPLC
Buffer: 0.1% TFA + 10% HzO + 90/u CH3CN
2 mL/min. 214 nm, C18 NovaPak
Retention hmes of reaction components
Dhe acid: Rt 3.6 min
Dhe-Gly-OtBu Rt 4.5 min
Dhe-Gly-OH: Rt 2.5 min
2. 13C n.m.r.
(DMSO-d6): 14.3. CH3; 20.2. 23.2, 25.3, 25.36, 25.42, 35.1. 8 x CH2;
40.8, Ga; 127.1, 127.90, 127.98, 128.06. 128.1, 128.27, 128.3, 129.1, 131.8, 6 xCH; 171.5, 172.0, 2 x C=O.
3. CI-MS
m/z 386 (M+1).
Docosahexaenoic acid-aspartic acid-OH
H-Asp(OtBu)-OtBu.HCl and HOSu were dissolved together in DMF (2 mL~.
The mixture was cooled in ice bath and docosahexaenoic acid, DCG (in 0.4
mL DMF), and N-methylmorpholine were added. The mixture stirred in ice
bath for 30 minutes and then at room temperature for 4 hours. 30%
docosahexaenoic acid ~Dhe acid) remained. More DCC ~0.11 g) was added
CA 0220379l l997-04-2~
WO 96/13507 PCT/AU95/00717
and the mixture stirred for further 20 hours. About 18% Dhe acid remained.
The m~xture was filtered and the crude product was purified by HPLC. The
~yophilised Dhe-Asp(OtBu)-OtBu (light yellow oil) weighed 0.73 g (86%).
Cold TFA (30 rnL) was added to the oil and the mixture stirred for an hour.
TFA was evaporated to leave a dark brown oil which was redissolved in
CH3CN (5 mL) and was purified by HPLC. The purified Dhe-Gly-OH was
lyophilised to leave a dark brown oil (0.33 g. 49%).
Purification
HPLC conditions:
Buffer A: 0.1% TFA/H20
Buffer B: 0.1% TFA + 10% H2O + 90% CH3CN
40 mL/min, 214 nm, C18 semipreppak
manual increment of %B: 10%--20--30--40--50--55--60--65--68--70--73-
-75%B.
Dhe acid eluted at 73% B. Dhe-Asp(OtBu)-OtBu eluted at 73-75%B.
Dhe-Asp-OH eluted at 58% B.
Analysis
1. Analvtical HPLC
Buffer: 0.1% TFA + 10% H2O + 90~" CH3C~
2 mLlmin. 214 nm. C18 NovaPak
Retention times of reaction components
Dhe acid: Rt 3.6 min
Dhe-Asp(OtBu)-OtBu: Rt 8.2 min
Dhe-Asp-OH: Rt 2.0 min
2. 13C n.m.r.
(DMSO-d6): 14.3. CH3: 20.2. 23.2, 25.3, 25.4, 25.4, 35Ø 8 x CH2:
36.4, DB; 48.7, Da; 127.1. 127.9~ 127.98, 128.0, 128.1. 128.22f 128.28~ 128.3,
129Ø 131.8. CH; 171.8. 171.8. 172.7. 3 x C=O~
3. CI-MS
m!z 444 (M+ 1) .
CA 0220379l l997-04-2~
WO 96/13507 PCT/AU95/00717
Linolenic acid-glycine-OH
Linolenic acid, HOSu and H-Gly-OtBu.HCl were dissolved together in DMF
(3 mL), the mixture cooled in ice bath and DCC (in 0.3 mL DMF) added. N-
MM was added and the mixture stirred for 20 hours, after which time some
unreacted linolenic acid remained. More DCC (0.10 g) was added and the
mixture stirred for further 20 hours. DCU was filtered off and the product
isolated by reversed phase HPLC. The purified product was concentrated to
an oil and TFA (30 mL) was added. After an hour stirring, the TFA was
evaporated to leave the product as a brown oil which was redissolved in
CH3CN (6 mL) and was purified by HPLC. The pure fractions obtained were
combined, concentrated and lyophilised (in t-butanol) to yield a brown oil
~0.24 g. 40%)
Purification
HPLC purification:
buffer A: 0.1% TFA / H2O
buffer B: 0.1% TFA + 10% H2O + 90 % CH3CN
40 mL/min, 214 nm, C18 small prep column
Lino-Gly-OH eluted at 65% B, linolenic acid eluted at 67% B,
linolenyl-Gly-OtBu eluted also at 67% B but slightly later.
Analysis and characterisation
1. Analvtical HPLC
Buffer A: 0.1% TFA, buffer B: 0.1% TFA/ 10% H2O! 90% CH3CN
Z mL/min, 214 nm, C18 Novapak
100% B isocratic, retention times of ingredients:
linolenic acid: Rt 3.96 min
linolenyl-Gly-OtBu: Rt 4.63 min
linolenyl-Gly-OH: Rt 2.59 min
2. 13C n.m.r.
(DMSO-d6): 14.2, CH3: 20.2, 25.26, 25.32, 26.8, 28.7, 28.8, 29.2,
35.2. CH2; 40.7, Ga: 127.1, 127.7. 128.1! 130.1, 131.7, CH; 171.6, 172.7, C=O.
CA 0220379l l997-04-2~
WO 96/13S07 PCT/AU95/00717
3. C.I.-M.S.
m/z 336 (M+1).
Linolenic acid-aspartic acid-OH
Linolenic acid, HOSu and H-Asp(OtBu)-OtBu.HCl were dissolved together in
DMF (3 mL), the mixture cooled in ice bath and DCC (in 0.3 mL DMF)
added. N-MM was added and the mixture stirred for 20 hours, after which
time some unreacted linolenic acid remained. More DCC (0.10 g) was added
and the mixture stirred for further 20 hours. DCU was filtered off and the
product isolated by reversed phase HPLC. The purified product was
concentrated to an oil (0.66 g) and TFA (30 mL) was added. After an hour
stirring, the TFA was evaporated to leave the product as a brown oil which
was redissolved in CH3CN (6 mL) and was purified by HPLC. The pure
fractions obtained were combined, concentrated and lyophilised (in t-
butanol) to yield a brown oil (0.38 g, 54%).
Purification
HPLC purification:
buffer A: 0.1% TFA / H2O
buffer B: 0.1% TFA + 10% H2O + 90% CH3CN
40 mL/min, 214 nm. C18 small prep column
Lino-Asp-OH eluted at 55% B. linolenic acid eluted at 65% BJ
linolenyl-Asp(OtBu)-OtBu eluted at 70% B.
Analvsis and characterisation
1. Analvtical HPLC
Buffer A: 0.1% TFA, buffer B: 0.1% TFA/ 10% H2O/ 90% CH3CN
2 mL/min, 214 nm, C18 Novapak
100% B isocratic. retention times of ingredients:
linolenic acid: Rt 4.14 min
linolenyl-Asp(OtBu)-OtBu: Rt 8.46 min
linolenyl-Asp-OH: Rt 2.04 min
CA 02203791 1997-04-2~
WO 96/13507 PC~r/A U95/00717
16
2. 13C n.m.r.
(DMSO-d6): 14.2, CH3; 20.2, 25.26, 25.34, 26.8, 28.69, Z8.72, 28.83,
29.2, 35.2, CH2; 36.3, DB; 48.7, Da; 127.1, 127.7, 128.1, 130.1, 131.7, CH;
171.8, 172.2, 172.7, C=O.
3. C.I.-M.S.
m/z 394 (M+1).
Gamma linolenic acid-glycine-OH
y-Linolenic acid, HOSu and H-Gly-OtBu.HCl were dissolved together in DMF
(3 mL), the mixture cooled in ice bath and DCC (in 0.3 mL DMF) added. N-
MM was added and the mixture stirred for 20 hours, after which time some
unreacted linolenic acid remained. More DCC (0.10 g) was added and the
mixture stirred for further 20 hours. DCU was filtered off and the product
isolated by reversed phase HPLC. The purified product was concentrated to
an oil (0.46 g) and TFA (30 mL) was added. After an hour stirring, the TFA
was evaporated to leave the product as a brown oil which was redissolved in
CH3CN (6 mL) and was purified by HPLC. The pure fractions obtained were
combined. concentrated and lyophilised (in t-butanol) to yield a brown oil
(0.35 g. 58%).
Purification
HPLC purification:
buffer A: 0.1% TFA / H2O
buffer B: 0.1 % TFA + 10% H2O + 90% CH3CN
40 mL/min, 214 nm, C18 small prep column
y-Lino-Gly-OH eluted at 66 % B. y-linoiellic acid eiuted at 66 % B, y-
linolenyl-Gly-OtBu eluted at 67 % B. Compounds eluted in the order listed.
Analysis and characterisation
1. Analvtical HPLC
Buffer A: 0.1% TFA. buffer B: 0.1% TFAI 10% H2O/ 90% CH3CN
2 mL/min, 214 nm, C18 Novapak
100% B isocratic. retention times of ingredients:
~-linolenic acid: Rt 4.07 min
CA 02203791 1997-04-2~
W 096/13507 PCT/AU95/00717
y-linolenyl-Gly-OtBu: Rt 4.85 min
~-linolenyl-Gly-OH: Rt 2.82 min
2. 13C n.m.r.
(DMSO-d6): 14.1, CH3; 22.2, 25.0, 25.4, 26.7, 26.8, 28.8, 28.9, 31.1,
35.1. CH2; 40.7, Ga; 127.7, 127.9, 128.1, 128.2, 129.9, 130.1, CH; 171.6, 172.6,C=O.
3. C.I.-M.S.
m/z 336 (M+ 1).
Gamma linolenic-aspartic acid-OH
Gamma linolenic acid. HOSu and H-Asp(OtBu)-OtBu.HCl were dissolved
together in DMF (3 mL), the mixture cooled in lce bath and DCG (in 0.3 mL
DMF) added. N-MM was added and the mixture stirred for 20 hours, after
which time some unreacted linolenic acid remained. More DCC (0.10 g) was
added and the mixture stirred for further 20 hours. DCU was filtered off and
the product isolated by reversed phase HPLC. The purified product was
concentrated to an oil (0.65 g) and TFA (30 mL) was added. After an hour
stirring. the TFA was evaporated to leave the product as a brown oil which
was redissolved in CH3CN (6 mL) and was purified by HPLC. The pure
fractions obtained were combined. concentrated and lyophilised (in t-
butanol~ to yield a brown oil (0.30 g. 42%).
Purification
HPLC purification:
buffer A: 0.1% TFA / H2O
buffer B 0.1% TFA + 10% H2O ~ 90% CH3CN
40 rnLImin. 214 nm. C18 small prep column
Gamma iinolenic-Asp-OH eluted at 50% B. linoienic acid eluted at
70O/o B. linolenvl-Asp!OtBu3-OtBu eluted at 75% B
-
CA 02203791 1997-04-2~ -
W 096/13507 PCT/AU95/00717
Analysis and characterisation
1. Analvtical HPLC
Buffer A: 0.1% TFA, buffer B: 0.1% TFA/ 10% H20/ 90% CH3CN
2 mLlmin, 214 nm, C18 Novapak
100% B isocratic. retention times of in~redients:
gamma linolenic acid: Rt 4.14 min
gamma linolenyl-Asp(OtBu)-OtBu: Rt 8.71 min
gamma linolenyl-Asp-OH: Rt 2.28 min
2. 13C n.m.r.
(DMSO-d6): 14.1. CH3; 22.2. 25.1, 25.4. 26.7. 26.8. 28.79 28.9. 31.08.
35.~. CH2: 3~.3. D~. 48 7. Oa; 127.8, 127.9. 128.1. 128.2, 130.0, 130.1. CH;
171.9, 172.2, 172.7. ~=O.5
3 C.I.-M.S.
m/z 394 (M+ 1).
It will be appreciated by persons skilled in the art that numerous variations
20 and/or modifications mav be made to the invention as shown in the specific
embodiments without departing from the spirit or scope of the invention as
broadlv described. The present embodiments are, therefore! to be
considered in all respects as illustrative and not restrictive.
CA 02203791 1997-04-25
WO 96/13507 PCT/AU95/00717
19
TABLE 1: Inhibition of chloroquine-resistant P. falciparum strain K by
amino acid conjugated PUFA.
COMPOUND % INHIBITION
Chloroquine 20. 1
Arachidonic acid-glycine-OH 84.2
Docosahexaenoic acid-glycine-OH 84.9
Linolenic acid-glycine-OH 81.5
All PUFA at 11~1m
CA 02203791 1997-04-25
WO 96/l3507 PCT/AU95/00717
TABLE 2: Effect of amino acid conjugated PUFAs on PHA-stimulated
TNFa and inte.~e~ production
COMPOUND TNFa IFN~
a-linolenic acid-glycine-OH 29.3 14.5
a-linolenic acid-aspartic acid-OH 0 o
v-linolenic acid-glycine-OH 21.5 0
y-linolenic acid-aspartic acid-OH 4.7 o
arachidonic acid-glycine-OH 26.6 35.9
arachidonic acid-aspartic acid-OH 38.3 68.4
eicosapentaenoic acid-glvcine-OH 11 68.2
eicosapentaenoic acid-aspartic acid-OH 17.1 66.1
docosahexaenoic acid-glvcine-OH 16.2 44
docosahexaenoic acid-aspartic acid-OH 17.4 8.3
All PUFA were at 20!1M
CA 02203791 1997-04-2~
WO 96/13507 PCT/AU9S/00717
TABLE 3: Effect of PUFA on cell proliferation induced by PHA
COMPOUND % INHIBITION OF
PROLIFERATION
linolenic acid-glycine-OH 15.6
linolenic acid-aspartic acid-OH 7.3
a linolenic acid-glycine-OH 29
a linolenic acid-aspartic acid-OH 15.4
arachidonic acid-glycine-OH
arachidonic acid-aspartic acid-OH 39.7
eicosapentaenoic acid-glycine-OH 5 .4
eicosapentaenoic acid-aspartic acid-OH 20.7
docosahexaenoic acid-glycine-OH 16.6
docosahexaenoic acid-aspartic acid-OH 21.1
All PUFA were at 20~M