Note: Descriptions are shown in the official language in which they were submitted.
CA 02210532 2005-10-07
METHOD AND PACKAGE DESIGN FOR CRYOPRESERVATION
AND STORAGE OF CULTURED TISSUE EQUIVALENTS
BACKGROUND OF THE INVENTION
1. Field of the Invention:
This invention relates to the cryopreservation of both harvested tissue
and cultured tissue equivalents made using in vitro technology. This
invention also relates to a cryopreservation package design for both harvested
tissue and cultured tissue equivalents that is both a cost effective and easy
to
handle package design that allows for maximal viability of the tissue or
tissue
equivalent to be cryopreserved. By use of the cryopreservation technology,
either cryopreserved harvested tissue or cryopreserved cultured tissue may be
stored for indefinite perioas of time prior to use. The cultured tissue is an
in
vitro model of the equivalent human tissue, which, when retrieved from
storage, can be used for transplantation or implantation, in vivo, or for
screening compounds in vitro.
2. Brief Description of the Background of the Invention:
In: vitro technology has developed tissue equivalents for the purposes of in
vitro testing or in vivo grafting for wound repair. Methods of producing such
tissue equivalents are disclosed in US Patent Nos. 4,485,096, 4,604,346,
4,835,102 and 5,374,515 and US Patent Nos. 5,536,656 and 5,827,641.
The shelf life of living tissues is limited and, subsequently, their window
of use is short, resulting in much waste. There is a need to preserve such
tissues for extended periods of time, as for shipping and storage, until their
use. Both the development of a cryopreservation method and a package for
cryopreservation and storage would extend the window of use indefinitely,
ease shipping and allow for the maintenance of an inventory. To enable an
-1-
CA 02210532 1997-07-15
WO 96/24018 PCT/US96/01217
inventory of tissue at burn care centers and hospitals is also desirable.
Other
advantages are that samples can be retained from different stages of the
manufacturing cycle for quality control archives and larger production
batches can be made as they can be maintained in a frozen state.
Currently, the storage time of cellular biological materials is extended by
cooling to "cryogenic" temperatures. The transition from the liquid into the
solid state by lowering the temperature of the system can take place either as
crystallization (ice), involving an orderly arrangement of water molecules, or
as vitrification or amorphization (glass formation), in the absence of such an
orderly arrangement of crystalline phase. The challenge for a cryobiologist is
to bring cells to cryogenic temperatures and then return them to physiological
conditions without injuring them.
There are two basic approaches to cryopreservation of cells and tissues:
freeze-thaw and vitrification. In freeze-thaw techniques, the extracellular
solution is frozen (i.e., in crystalline form), but steps are taken to
minimize
the intracellular ice formation. In vitrification procedures, there is an
attempt to prevent ice formation throughout the entire sample. The former
approach is problematic in that if ice crystals are formed inside the cells,
they
are detrimental to cell viability upon thawing. However, cells could survive
a freeze-thaw cycle if they are cooled at controlled rates in the presence of
non-toxic levels of cryoprotectants. The latter approach of vitrification
seeks
to avoid potentially damaging affects of intra- and extracellular ice by
depressing ice formation using very high concentrations of solutes and/or
polymers. However, the cell damage may occur to long exposure to toxic
levels of these additives required for vitrification.
Cryoprotectants protect living cells from the stresses involved in the
freezing process. One way cryoprotectants protect cells is by diluting the
salt
-2-
CA 02210532 1997-07-15
WO 96/24018 PCT/US96101217
that becomes increasingly concentrated in the unfrozen solution as water is
transformed to ice. The amount of ice is dictated by the temperature and
initial composition of the solution; whereas the amount of unfrozen fraction
is a function of temperature only. Cryoprotectants have several other
functions. An important one is that they usually reduce the intraceIlular ice
formation temperatures. Another function is that they stabilize membranes
and proteins.
All solutions will supercool below their freezing point until they find a
random nucleation site for crystal formation. When cryopreserving by a
freeze-thaw method, ice formation in the extracellular medium should be
deliberately initiated by seeding at low degrees of supercooling. If ice
formation is not induced by seeding, ice will form spontaneously when the
solution is cooled sufficiently far below its equilibrium freezing point.
Because this process is random in nature, ice formation will occur at random,
unpredictable temperatures; consequently, survival rates will be highly
variable between repeated trials with the same freezing protocol.
Furthermore, the extremely rapid crystallization which results when ice
forms in a highly supercooled solution can cause damage to cells and tissues.
Moreover, it has been shown that if extracellular ice formation is initiated
at
high degrees of supercooling, the probability of intracellular ice formation
is
drastically increased. This phenomenon results from the delayed onset of
freeze-induced cell dehydration, which results in increased retention of
intracellular water, and thus higher likelihood of ice formation in the cell.
Once the extracellular ice is seeded and the sample is surrounded by the ice
phase, it is necessary to cool the sample to a cryopreserved state. The
cooling
step is one of the most critical steps in a freeze-thaw protocol. Due to the
formation of ice, i.e., pure water, the partially frozen extracellular
solution is
-3-
CA 02210532 1997-07-15
WO 96/24018 PCT/US96/01217
more concentrated than the intracellular compartment. As a consequence,
the cell will dehydrate by losing water in an attempt to restore
thermodynamic equilibrium. As the system cools, more extracellular ice is
generated and the concentration of solutes rises and forces the cells to
dehydrate further. There are three characteristics of the cells that control
their
rate of dehydration. One is the cell membrane water permeability; the lower
the water permeability, the longer it takes for the cells to dehydrate.
Another
is the temperature dependence of the cell membrane water permeability; all
cells decrease their water permeability with decreasing temperatures. The
final is cell size; larger cells take longer to dehydrate than smaller cells.
Given
that each cell type may have drastically different characteristics, the
optimal
cryopreservation conditions can vary by orders of magnitude for different cell
types.
Although the exact mechanisms of cell damage during cryopreservation
has not yet been completely elucidated, characteristic survival signatures
generated by measuring cell survival as a function of cooling rate appear to
be
qualitatively similar for all cell types and displays an inverted U-shaped
curve. Cell survival is low ai: very slow and very fast cooling rates, and
there
is an intermediate cooling rate yielding optimal survival. Even though the
optimal cooling rate and the width of the curve can vary drastically for
different cell types, the qualitative behavior appears to be universal. Faster
cooling rates do not allow cells enough time to dehydrate and cells form ice
internally. Cell injury at fast cooling rates is attributed to intracellular
ice
formation. At slow rates of cooling, cell injury is thought to be due to the
effects of exposure to highly concentrated intra- and extracellular salt and
cryoprotectant solutions or to the mechanical interactions between cells and
the extracellular ice.
-4-
CA 02210532 1997-07-15
WO 96/24018 PCTIUS96101217
It is necessary to dehydrate the cells as much as possible before they cross
the intracellular ice nucleation curve. It is at this point that practically
all
water remaining in the cell will nucleate and form ice. It is impractical to
determine the exact temperature where this will happen but it is
approximately -40 C to -50 C when the cells are slowly frozen in the presence
of 1M to 2M concentrations of cryoprotectants. It is important to note that
the
amount of water that turns to ice inside a cell at this point may be innocuous
when frozen, but if not thawed fast enough, it will expand and kill the cell
upon thawing. (The Biophysics of Organ Cryopreservation, Pg. 117-140, edited
by
David E. Pegg and Armand M. Karow, Jr. NATO ASI Series A: Life Sciences
Vol. 147 1987 Plenum Press, New York 233 Spring St., New York, NY 10013).
Before the development of a commercially viable skin equivalent, cadaver
skin was used for the purposes of grafting. Cryopreservation protocols were
developed so that burn centers and hospitals could maintain skin banks. A
number of different protocols were developed utilizing different
cryoprotectants, freeze rates, packaging formats and storage conditions. Most
researchers agreed upon a fast thaw protocol. The success or failure of the
protocol was measured either by graft take to a wound bed or by cell viability
assay.
In U.S. 3,842,831 to Beisang is disclosed a method for the cryopreservation
of cadaver skin patches. The method involves the attachment of the cadaver
skin to a loosely woven scrim or backing and, together, the skin patches and
the scrim are rolled prior to freezing. No cryoprotectant is employed, though
the inventors suggest the use of either glycerin or DMSO. The freezing
protocol employs a fast uncontrolled (fixed temperature) freeze rate protocol
to a cryogenic temperature of -70 C.
-5-
CA 02210532 1997-07-15
WO 96/24018 PCT/US96/01217
May SR and FA DeClement (Skin Banking Methodology, 17, 33-45 (1980))
performed an evaluation of packaging geometry and cooling and warming
rates using dermatome cadaver skin. The results suggested that cadaver skin
be flat, rather than rolled, and that a slower controlled rate of freezing be
5 employed.
U.S. 5,040,677 to Tubo discloses a gas-tight sealable container for individual
grafts of epithelial cell sheets. The container requires that the epithelial
cell
sheet be attached to an adhesive substrate sheet or backing by use of clips.
U.S. 5,145,770 to Tubo discloses a cryopreservation method for keratinocyte
sheets that employs a cryoprotectant of a non-cell penetrating agent, such as
dextran, and a cell penetrating reagent, such as glycerol, with a cooling rate
of
about -1 C/minute. Similarly, EP 0 364 306 to Chao et al, discloses a method
for cryopreserving a sheet of living, cultured epithelial cells but utilizing
both
DMSO and glycerol as a cryoprotectant and a freezing protocol of preferably
-1 C/minute.
U.S. 5,298,417 to Cancedda et al, discloses a cryopreservation protocol
developed for single layer constructs such as epithelial sheets prepared as
described in US 4,016,036, 4,304,866 and 4,456,687. Epidermal sheets were
incubated with a cryoprotectant of either 8-15% glycerol or DMSO and were
cryopreserved by employing a controlled rate protocol where the cooling rate
is slower at the start than at the end of the protocol and is characterized by
an
increase in temperature before the culmination of the freezing procedure.
A method for the cryoprotection of dermal fibroblasts in a collagen gel was
investigated by Teasdale et al, Burns, 19 (5) 406-410 (1993). Teasdale
determined that optimum cell viability could be obtained by freezing at
-0.5 C/minute with DMSO as a cryoprotectant.
-6-
CA 02210532 1997-07-15
WO 96/24018 PCT1II896/01217
Nanchahal et al., "Cultured composite skin grafts: Biological skin
equivalents permitting massive expansion," The Lancet, 2 (8565), 191-193 (July
22, 1989), discusses a technique for storage of composite cultured tissue
grafts
utilizing a cryoprotectant of 15% glycerol arid 10% FCS in Medium 199. The
grafts and the cryoprotectant, were incubated at 37 C for two hours and were
then frozen at -1 C per minute to -70"C and then stored in liquid nitrogen.
After fast thawing of the grafts, their viability was determined by culturing
for
two weeks and by grafting to hairless mice. A final evaluation was made by
grafting to three patients undergoing tattoo excision.
Johnstone et al. "Cryopreservation of Rabbit and Cat Corneas at -18 to
-24 C;" Cornea, 11(3): 211-220 (1992), is directed to a simple procedure for
cryopreservation of rabbit and cat corneas which utilizes a domestic freezer
rather than liquid nitrogen or very low temperature freezers. Perfusion of
cryopreservative is obtained by placing corneas in successive solutions of 50%
fetal calf serum and McCarey-Kaufman medium with increasing glycerol and
glucose content.
Using prior art methods, it is not possible to cryopreserve cultured tissue
equivalents, in part because they are relatively thick and of heterogeneous
cell
layers. One of the functions of these tissues in vivo are to provide a
permeability barrier. Tissue functions have to be considered in the
development of a cryopreservation protocol.
The present inventors have discovered a method for cryopreservation
and, in particular, a package design, that is applicable to a number of
cultured
tissue equivalents and to mammalian skin, one that is a surprisingly effective
and commercially practical package for the cryopreservation of cultured tissue
equivalents.
-7-
CA 02210532 1997-07-15
WO 96/24018 PCT/US96/01217
SUMMARY OF THE INVENTION
The present invention provides a method for the successful
cryopreservation of cultured tissue equivalents at. very low temperatures
which avoids the formation of harmful intracellular ice crystals, minimizes
the effective concentration of potentially harmful chemicals, and permits the
rapid introduction and removal of cryoprotectants at feasible temperatures
using programmable freezing equipment.
The present invention also provides a new package design developed for
the cryopreservation, storage and distribution of cultured tissue equivalents.
The new design offers many advantages over existing package designs. At
present, there are no commercially available cryopreserved cultured tissue
equivalents, therefore there are no off-the-shelf package designs available.
The design of the new package enables better temperature tracking of the
package interior with the exterior temperature of the freezing chamber
because of more efficient heat transfer. An improved heat transfer rate allows
for a more controlled process; thus, uniform cooling and warming rates of the
cultured tissue equivalent, thereby reducing variability in cell viability.
The inventors have discovered a method for cryopreserving cultured
tissue equivalents made from in vitro techniques so that the tissues maintain
their viability and utility as equivalents of human tissues. The invention
includes the use of agitation to enhance the penetration of an effective
amount of cryoprotectant. The present method provides for the
cryopreservation of both harvested tissue and cultured tissue equivalents in a
manner which protects structural integrity and cellular viability.
The method of this invention involves the following steps:
1) The harvested tissue or cultured tissue equivalent is immersed in a
cryoprotectant solution and the cryoprotectant solution and the immersed
-8-
CA 02210532 1997-07-15
WO 96/24018 PCT/[TS96/01217
tissue are agitated to achieve effective penetration of the cryoprotectant
solution into the tissue (perfusion of the tissue); and,
2) After the perfusion of the cryoprotectant solution into the tissue,
cooling to solid-liquid phase equilibrium temperature range for the
cryoprotectant; seeding extracellular ice and cooling to a cryopreserved state
by
freezing the tissue at a slow freezing rate to a temperature at or below at
least
about -70 C, more preferably at or below -120 C, even more preferably at
-140 C and most preferably, at -196 C.
Once frozen, the cryopreserved tissue can be stored for indefinite time
periods between temperatures of about -120 to about -196 C, the temperature
of liquid nitrogen.
Thawing the cryopreserved tissue is accomplished by warming the frozen
tissue at a high rate, which is done in about 1 to 2 minutes. The frozen
tissue
may be thawed by direct application of warmed culture media or by
physiologic buffered solution or by another rapid heating method. The
package design allows for thawing of the tissue by immersion of the package
in a waterbath as the package design ensures both a fast warming rate and
controlled thermal uniformity while maintaining sterility during he critical
thawing procedure.
Prior to use as an equivalent for human tissue, for grafting or in vitro
testing, the thawed cultured tissue equivalent is rinsed to remove the
cryoprotectant solution. The cryoprotectant solution may be removed by
rinsing with, for example, an isotonic buffer solution at physiological pH.
The cultured tissue equivalents can then be stored temporarily in such a
buffer solution or recultured in an appropriate cell medium before use.
BRIEF DESCRIPTION OF THE FIGURES
Figures 1A and 1B show views of the lid.
-9-
CA 02210532 2005-10-07
Figure 2 shows a top view of the seal.
Figures 3A and 3B show views of the seal mounted to the lid.
Figures 4A and 4B show views of the carrier.
Figures 5A and 5B show views of the tray.
Figures 6A and 6B show side views of the assembly, exploded and
completed.
Figure 7 shows a close view of the assembly.
Figure 8 shows a top view of the assembly, completed.
Figure 9 shows a graph of the viability of cryopreserved cultured tissues
compared that of non-cryopreserved cultured tissues. The viability of
cryopreserved cultured tissues as measured by MTT is shown. LSE was
additionally measured by LDH assay. All cultured tissues are compared to
age-matched non-cryopreserved controls and are expressed as a percent of
control.
DETAILED DESCRIPTION OF THE INVENTION
Tissue engineering is an emerging area which utilizes cultured tissue cells
to construct tissue equivalents which can be used to examine the response to
injury by chemical agents or pharmaceutical compounds. The cultured tissue
may also be used to form graftable human tissue.
Tissue equivalents have been described extensively in many patents,
including U.S. Patent Nos. 4,485,096; 4,485,097; 4,539,716; 4,546,500;
4,604,346;
4,837,379; and 5,374,515. One successful application of the tissue
equivalent is called the "Living Skin Equivalent," which has a
morphology similar to actual human skin. The Living Skin Equivalent
(LSE) is composed of two layers: the upper portion is made of
differentiated and stratified human epidermal keratinocytes that cover a
lower layer of human dermal fibroblasts in a collagen matrix.
-10-
CA 02210532 2005-10-07
Parenteau, et al., "Epidermis Generated In Vitro: Practical Considerations and
Applications," J. of Cellular Biochemistry, 45:245-251 (1991); Parenteau, et
al.,
"The organotypic culture of human skin keratinocytes and fibroblasts to
achieve form and function," Cytotechnology, 9:163-171 (1992); and Bell et al.,
"The Living Skin Equivalent: Its Manufacture, Its Organotypic Properties and
Its Responses to Irritants," Toxic. in Vitro, 5:591-596 (1991). LSE for
grafting is
under investigation in clinical trials for indications relating to partial and
full
thickness skin wounds: excision surgery, burns, venous stasis ulcers, diabetic
ulcers, decubitus ulcers, and chronic inflammatory ulcers. The LSE is a full-
thickness, bilayered, in vitro engineered skin tissue
An in vitro organ equivalent of the cornea of the eye has been
developed as described in U.S. Patent No. 5,374,515. The cornea
tissue equivalent has three distinct cell layers, the external layer, a
stratified squamous epithelium; the middle layer of collagen fibers and
stromal cells; and an inner layer, simple squamous epithelium, also called the
corneal endothelium. An in vitro cornea equivalent can be used for in vitro
toxicity assays to serve as accurate and inexpensive non-animal predictive
models of in vivo ocular and dermal irritation potential for many types of
products and raw materials.
The goal of cryopreservation is to preserve the structural integrity and
viability of biological materials for an indefinite period of time so that
these
materials can be available and used as needed. Complex tissues of finite life
span will require cryopreservation to expand product availability and utility.
The history of cryopreservation of biological material, however, has shown
that the optimization of a cryopreservation protocol for a particular cell
does
not necessarily give good results when used with another cell type or with
other cells in a tissue. The development of more specialized methods due to
-11-
CA 02210532 1997-07-15
WO 96/24018 PCT/US96/01217
the differences in cell density, water content and level of structural
organization of the full-thickness tissue equivalents was required. The
cryopreservation protocols of this invention are surprisingly applicable to
the
single layer epidermal and dermal layers alone, bilayer tissue equivalents,
trilayered cornea equivalents and harvested mammalian skin. The
development of a package design makes cryopreservation of these tissues
practical for production scale processes.
As used herein, the term "cultured tissue equivalents" means tissue
equivalents of mammalian tissues, wherein the tissue equivalents are made
by in vitro techniques and are meant to include monolayer skin equivalents,
either a dermal equivalent or an epidermal sheet; bilayered skin equivalents,
particularly LSE; and trilayered cornea equivalents and skin equivalents. The
morphology of the cultured tissue equivalents are similar to the in vivo
mammalian organ, typically the human organ. For illustration, the
morphology of the LSE bears many similarities to human skin. Metabolically
and mitotically active human dermal fibroblasts (HDF) are found throughout
the dermal layer of the construct, and have been shown to secrete collagen
and other matrix components into the lattice. The epidermis consists of a
basal layer shown to divide with a mitotic rate similar to that of human skin.
The suprabasal epidermis shows the same strata as skin in vivo, with well
defined spinous and granular layers containing keratohyalin and lamellar
granules covered by a stratum corneum. Immunohistochemistry
demonstrates the presence of extracellular matrix components routinely
found at the dermo-epidermal junction in normal human skin, such as
laminin, Type IV collagen and kalanin (GB3).
Cultured tissue equivalents obtained by organotypic culture methods, such
as the Living Skin Equivalent (LSE), utilize a carrier as a framework on which
-12-
CA 02210532 2005-10-07
to form the equivalents. Cultured tissue equivalents are fabricated on a
carrier which allows for the manipulation of the construct during
manufacturing, shipping and end use without directly contacting the
construct. Methods for producing cultured tissue equivalents on a carrier are
disclosed in US Patent No. 5,374,515 and US Patent Nos. 5,536,656 and
5,827,641.
A carrier includes a flat permeable membrane which is attached to one
end of an essentially tubular support. The other end of the tubular support
includes an otutwardly extending flange which in turns carries a rim that can
hang upon the upper end of a well in a tissue culture dish and in the package
design of the present invention. The membrane-support assembly is sized to
be used with a culture dish having at least one well of a specific size so as
to
provide the proper dearance about the support and engage the top end of the
well. The membrane-support assembly may be made in different sizes so as
to be used with different sizes of both culture dishes and cryopreservation
and
storage packages. Examples of these carriers is a TRANSWELL (Costar) as
described in US Patent No. 5,466,602. Modifications to the package design may
be made by the skilled artisan to accommodate similar carriers that employ
different means of support and engagement in the culture vessels. Similar
carriers are described in US Patent Nos. 5,358,871 and 5,366,893 or those
described in US Patent No. 4,871,674.
The preferred package design of this invention incorporates the carrier
framework and defines an environment around the tissue equivalent that is
able to be controlled to predetermined specifications. The present invention
uses materials that are preferably of medical grade quality and are able to
withstand a wide range of temperatures. These materials are molded to form
a tray and a lid that are able to be sealed together by a sealing means. The
new
-13-
CA 02210532 1997-07-15
WO 96/24018 PCT/US96/01217
package is designed to incorporate a carrier support on which tissue
equivalents are formed.
The new package design comprises a tray, a lid and a tight seal
therebetween. The tray and the lid are designed to fit the existing carrier
containing the attached tissue equivalent. The carrier with the attached
tissue
equivalent are placed into the tray and cryoprotectant solution is added. The
lid is then placed onto the tray and the two parts are sealed together to form
a
tight seal between the interior and exterior environments of the package.
The package design allows for an equal distribution of cryoprotectant
below the carrier and above the surface of the tissue equivalent while
maintaining contact of cryoprotectant to the tissue equivalent and both the
tray and the lid. The design of the package is based on the concept that
controlled cooling and freezing rates can be maintained with an equal
volume of cryoprotectant, arranged with a similar geometry above and below
the cultured tissue to be cryopreserved. This arrangement provides a
uniform distance, measured from both top and bottom surfaces of the
cultured tissue equivalent to the exterior wall of the package (provided that
the package also has uniform thickness). This uniformity should provide
uniform heat exchange from the tissue equivalent to the freezing chamber
upon cooling; and from the external environment to the tissue equivalent,
upon thawing.
The tray comprises a bottom surface that is contiguous with a sidewall and
a flange. The flange provides a planar surface for sealing of the tray to the
lid.
The sidewall of the tray has a singular continuous or a plurality of
discontinuous supports inwardly extending from and integral with the
sidewall. The upper edge of the carrier containing the cultured tissue
equivalent, when positioned in the tray, is susperided on the supports
-14-
CA 02210532 1997-07-15
WO 96/24018 PCT/US96101217
provided by the sidewall of the tray. A space is defined between the
suspended carrier and the bottom surface of the tray measuring about 1.0 mm
in thickness with an area about the size of the tissue equivalent.
The lid comprises a planar bottom surface contiguous with a sidewall and
a flange. The diameter of the lid flange is slightly smaller that the diameter
of
the tray flange. The lid flange, when placed on the tray containing a carrier,
rests on the carrier so that the top surfaces of both the tray flange and the
lid
flange are in contact oriented on or about the same plane. A second space is
defined by the lid and the surface of the cultured tissue equivalent also
measuring about 1.0 mm in thickness with an area about the size of the tissue
equivalent.
The design allows for a total of about 1 mm between the tray and bottom
of the carrier containing the tissue equivalent disposed thereon and about 1
mm between the lid and tissue equivalent with the cryoprotectant above and
below the tissue equivalent in contact with both the lid and tray. When the
lid is placed in the tray containing medium, the lid displaces any air in the
package to the peripheral space containing the tubular support of the carrier.
The lid is sealed to the tray along the exposed plane created by the top
surfaces of the tray and lid flanges. Sealing may be accomplished by heat,
adhesive or other means known in the art. The seal between the lid and the
tray prevents leakage of cryoprotectant and maintains the sterility of the
interior of the unit. Preferably an annular sheet of heat sealable lid stock
is
heat sealed to both the tray and lid flanges thereon. The lid stock preferably
has a tab for peelably removing, and therefor unsealing, the lid from the tray
to access the tissue equivalent. In another embodiment, a lid with a flange of
about the same diameter as the tray flange is placed on a tray with a carrier
disposed within so that the bottom surface of the lid flange intimately
-15-
CA 02210532 1997-07-15
WO 96/24018 PCT/US96/01217
contacts the top surface of the tray flange. The flanges are then sealed
together
by a sealable means.
Figures 1A and 1B show the lid 10; lid flange 11; lid sidewall 12; and
bottom surface 13. Lid flange 11, lid sidewall 12 and bottom surface 13 form a
contiguous surface.
Figure 2 shows the annular seal 20 and pull 21.
Figures 3A and 3B show the seal 20; lid 10; lid flange 11. Seal 20 is
mounted to the lid flange 11 of the lid 10 prior to use to facilitate
alignment of
the seal during the sealing process.
Figures 4A and 4B show the carrier 30; carrier rim 31; permeable
membrane 32; and tubular support 33. The permeable membrane 32 contains
a cultures tissue equivalent disposed thereon bound by the tubular support
33. The carrier 30 allows transfer of the tissue equivalent without contact or
removal of the tissue equivalent throughout the cryopreservation process.
Figures 5A and 5B show tray 40; tray bottom surface 41; tray sidewall 42;
tray flange 43; and carrier supports 44. The tray bottom surface 41, tray
sidewall 42, tray flange 43, and carrier supports 44 form a contiguous
surface.
Carrier supports 44 contact and support the carrier within the tray.
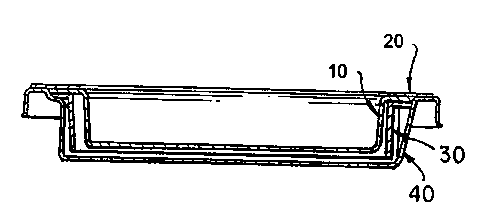
Figures 6A and 6B show seal 20; lid 10; carrier 30; and tray 40. Carrier 30
containing a tissue equivalent theron is disposed in tray 40 and is suspended
within the tray. Lid 10 with seal 20 is placed over carrier 30 and is
suspended
over the carrier.
Figure 7 shows seal 20; lid flange 11; lid 10; carrier 30; carrier rim 31;
carrier
supports 44; and tray 40. Carrier 30 containing a tissue equivalent theron is
disposed in tray 40 and is suspended within the tray by contact of the bottom
surface of carrier rim 31 to the top surfaces of the carrier supports 44. Lid
10
with seal 20 is placed over carrier 30 and is suspended over the carrier by
-16-
CA 02210532 1997-07-15
WO 96/24018 PCTlUS96101217
contact of the bottom surface of lid flange 11 to the top surface of carrier
rim
31. The top surfaces of both lid flange 11 and tray flange 43 form a planar
surface for seal 20.
Figure 8 shows a top view of the assembly.
Materials that trays and lids may be manufactured from are rigid or
semiflexible thermoplastic materials that, when heated, are molded by a
vacuum or injection molded such as polytetrafluoroethylene (PTFE) or
polyethylene terephthalate glycol (PETG). The package trays and lids are
preferably sterilized prior use by methods that are known in the art of
sterilization. The materials used to fabricate the tray and lid will determine
the method of sterilization that can be used. To sterilize PTFE or PETG,
Gamma radiation at 2.5 to 3.0 megarads is preferable. Electrical and chemical
methods of sterilization known by those skilled in the art, may alternatively
be employed.
Cultured tissue equivalents comprise a dermal equivalent of a hydrated
collagen lattice contracted by an agent, preferably fibroblasts. In one
embodiment, a stratified layer of epidermal cells is cultured on the surface
of
the dermal equivalent. In another embodiment, the hydrated contracted
collagen lattice, contracted by keratocytes, is disposed on a layer of corneal
endothelial cells with a stratified layer of corneal epithelial cells cultured
on
the surface of the lattice. Alternatively, epidermal cells alone may be
cultured
and induced to stratify to form an epidermal sheet. The cultured tissue
equivalent is formed either on a porous membrane of a carrier or on a
collagen gel that is adhered to a porous membrane of the bottom surface of a
carrier. In addition, other cultured tissue equivalents may be cryopreserved
according to the methods of this invention, including, but not limited to, any
-17-
CA 02210532 1997-07-15
WO 96/24018 PCT/US96/01217
cultured epidermal sheet, any cultured dermal equivalent, any cultured
cornea equivalent, or harvested mammalian skin.
The present invention will now be described using tissue equivalents and
the preferred package design as an illustration. It will be understood by
those
of skill in the art that modifications can be made to the described method and
still be within the scope of this invention.
The method of perfusing a tissue equivalent with a cryoprotectant
solution is to submerge the tissue equivalent and its attached carrier in a
volume of cryoprotectant solution sufficient to submerge the sample and to
have equal volume of cryoprotectant solution above and below the tissue
equivalent. To a 100 mm petri disk containing the tissue equivalent and
carrier is added 25 mL of 2M Glycerol in DMEM for a period of time sufficient
to completely perfuse the sample, preferably between one and two hours, but
most preferably for about one hour. Extended periods of time in
cryoprotectant solution result in reduced cell viability in the tissue, while
too
short of a time does not ensure complete permeation of cryoprotectant into
the tissue. Monolayer constructs will typically require less time for
perfusion
as they have fewer cell layers and reduced barrier function. During this hour,
penetration of cryoprotectant solution is enhanced by agitating the sample
and cryoprotectant solution, typically by shaking the petri dish on an orbital
platform shaker (Bellco orbital shaker) at 70 rpm in a 10% C02 gassed
chamber. The 10% C02 environment, the same as the culture environment
in which the tissue equivalent was fabricated, prevents the media from
degassing, thus maintaining the pH of the base media component of the
cryoprotectant. Agitation allows for a faster and a more complete perfusion of
cryoprotectant into the tissue equivalent and better reproducibility of
results
between frozen tissue equivalents. One is able to substitute an orbital shaker
-18-
CA 02210532 1997-07-15
WO 96/24018 PCT/US96/01217
with an apparatus that performs an agitative motion in other spatial planes.
Additionally, other methods of mechanically enhanced perfusion include,
but are not limited to, rocking the construct with cryoprotectant in a vessel
on
a platform or centrifugation of the construct *ith cryoprotectant and
perfusing cryoprotectant around the construct using a pump.
The carrier and attached tissue equivalent is placed into the tray and a total
of about 16.0 ml of cryoprotectant solution is added. The package design
allows for equal distribution of cryoprotectant: About 8.0 ml below the
carrier
and about 8.0 ml on the surface of the tissue equivalent. The lid is then
placed onto the tray and the two parts are heat sealed, preferably to the lid
stock.
Packaged tissue equivalents are then placed into a programmable freezer
(Cryomed or Planer) set at a starting temperature of about room temperature,
preferably at 20.0 C. The freezer chamber containing packaged tissue
equivalents is cooled at -10.0 C/minute to the solid-liquid phase equilibrium
temperature range for the cryoprotectant which is typically, for 2.0 M
glycerol
in DMEM, between about -5.3 C and -6.0 C. The solid-liquid phase
equilibrium temperature is the temperature necessary for seeding ice in the
cryoprotectant. The chamber temperature is held for a time for the purpose of
equilibrating the internal temperature of the packaged tissue equivalents to
the chamber temperature. The cooling rate used to obtain the seeding
temperature is not critical. A hold time of about 40 minutes is sufficient to
ensure thermal equilibration but the time will vary depending on chamber
size, air circulation within the chamber, and number of packages to be frozen.
While at least 30 minutes is typically needed, up to one hour can be taken to
ensure equilibration. After the hold time, extracellular ice formation by
seeding is initiated.
-19-
CA 02210532 2005-10-07
Ice seeding is defined as a method of initiating ice formation in the
extracellular cryoprotectant. One preferred method of ice seeding is by
contacting the side of the tray containing the tissue with a chilled probe.
such
as a liquid nitrogen chilled (-196 C) steel rod. The contact site of the rod
to
the package must be below the level of the cryoprotectant freeze media in the
package. Another preferred method is to directly release an expanding gasses
such as freon or C02 to the outside of the package. Ice formation can be
initiated by a chamber spike where the temperature of the chamber is lowered
and raised witlun a range sufficient to form an ice crystal. Other methods of
ice seeding known in the art may be substituted.
After all tissue equivalents are seeded with ice crystals, the units are held
an additional one hour to allow for thermodynamic equilibration and
propagation of the ice seed crystal throughout the cryoprotectant. Cooling is
then resumed at a rate of preferably between about -0.02 to about -0.3 C per
minute more
15. preferably between about -0.05 to about -0.1 C/minute; and most preferably
at
about -0.07 C/minute to a final temperature preferably at least or below
-70.0 C; more preferably at -120 C; even more preferably at -140 C; or most
preferably at -196 C. As the final freezing temperature approaches the glass
transition temperature of water, -120 C, the less likely there will be
detrimental temperature fluctuations during transfers to final storage
locations.
Cryopreserved tissue equivalents are transferred from the programmable
freezer to storage in a vapor phase liquid nitrogen storage tank (Dewar) at a
temperature between about -120 to -150 C, or in liquid nitrogen at -196 C
until use.
To thaw, cryopreserved cultured tissue equivalents are removed from
vapor phase liquid nitrogen storage and transferred to dry ice to warm to
-20-
CA 02210532 1997-07-15
WO 96/24018 PCT/1JS96/01217
about -75 C. Once equilibrated at -75 C, cultured tissue equivalents are then
transferred to an environment, preferably a waterbath, preferably set at 37 C;
or more preferably set at 4 C; or most preferably set at room temperature.
Once it is visible that all the cryoprotectant medium is turned to liquid
phase,
the cultured tissue equivalent package units are preferably transferred to a
biological safety cabinet, or other aseptic area and sanitized with ethanol.
The
lid of the dishes are removed by peeling or cutting the lid stock from the
bottom of the unit. The carriers containing cultured tissue equivalents are
then each transferred to petri dishes while pouring off excess cryoprotectant.
To rinse the cultured tissue equivalent of residual cryoprotectant, 25 mL of
rinse solution, preferably DMEM at room temperature, is then added to the
petri dish containing the cultured tissue equivalent for about 30 minutes.
Other sufficient rinse agents, preferably physiological strength solutions,
such
as cell culture media or phosphate buffered saline, may be determined by the
skilled artisan. The rinse solution is exchanged a second time for an
additional 30 minutes. Rinse times may vary depending on the complexity of
the tissue.
The cultured tissue equivalent unit may be then transferred back to its
original culture dish and recultured in culture maintenance medium at
37 C/10%CO2. Alternatively, the equivalent may be applied to a patient or
tested for a response to contact with a substance as described in US Pat.
4,835,102.
The following examples are provided to better elucidate the practice of the
present invention and should not be interpreted in any way to limit the scope
of the present invention. Those skilled in the art will recognize that various
modifications can be made to the methods described herein while not
departing from the spirit and scope of the present invention.
-21-
CA 02210532 2005-10-07
EXAMPLES
Examples 1 to 3
Examples 1 to 3 show the cryopreservation and thawing techniques of this
invention with the preferred package design.
Example 1: Cryopreservation of Living Skin Equivalent (LSE)
With the Package Design
Living Skin Equivalent (LSE) constructs were prepared in accordance to
U.S. Patent No. 5,536,656. LSEs and attached 75 mm carrier inserts
(TRANSWELL , Costar, Cambridge), at 9 to 10 days post air lift, were placed
in 100 mm petri dishes (Costar). LSE constructs were perfused with
cryoprotectant by submerging the constructs and the transwell with 25 mL of
cryoprotective media, 2M Glycerol in DMEM, in the 100 mm petri dish for
one hour. During perfusion, the constructs were agitated for one hour on an
orbital shaker (Belico) at 70 rpm in a 10% C02 gassed chamber. Agitation
allows for more complete perfusion and better reproducibility of the
cryopreservation method. After LSE were perfused, the petri dishes
containing LSE, carrier inserts and extracellular freezing media (2M Glycerol
and DMEM). were placed in a cryopreservation package and heat sealed.
The chamber of a programmable freezer (Planar) was outfitted with the
invention described herein. The freezer was set at a starting temperature of
20.0 C. The tubing lines were purged with freon for a single one second
interval to remove any air in the lines. Packaged LSE units were placed
securely into racks accommodating eight LSE units each. The racks, guided by
locating pins, were placed adjacent to the spray rails. The chamber door of
the
freezer was dosed to seal the chamber from the external environment.
-22-
CA 02210532 1997-07-15
WO 96/24018 PCT[US96101217
LSE units were cooled at -10 C/minute to -6 C and the chamber
temperature was held at -6 C for 40 minutes to equilibrate the cryoprotectant
and perfused constructs to the chamber temperature. After the 40 minute
hold, extracellular ice was initiated by directly discharging freon for one
second at close proximity to the side of the package. The freon, as it
evaporated from the surface of the package, caused the contact area of the
freon to drop in temperature enough to initiate extracellular ice formation.
After all LSE units were seeded with ice crystals, the units are allowed to
equilibrate for one hour at -6 C. The chamber temperature was then cooled at
-0.07 C/minute to a final temperature of -20 C. The chamber was then cooled
at -0.5 C/min to a final temperature of -70 C. Once the LSE units were
cryopreserved, they were transferred to a vapor phase storage tank (Dewar) at
a temperature of -120 to -150 C.
Example 2: Thawing Cryopreserved LSE
Cryopreserved LSE were removed from vapor phase liguid nitrogen
storage and transferred to dry ice to warm to about -75 C. Once equilibrated
at
-75 C, cultured tissue equivalent are then transferred to a waterbath
preferably set at 37 C; or more preferably set at 4 C; or most.preferably set
at
room temperature. Once it is visible that all the cryoprotecant medium is
turned to liquid phase, the. cultured tissue equivalent package units are
prefeably transferred to a biological safety cabinet, or other aseptic area
and
sanitized with ethanol. The lid of the dishes are removed by peeling or
cutting the lid stock from the bottom of the unit. The carriers containing
cultured tissue equivalents are then each transferred to petri dishes while
pouring off excess cryoprotectant. To rinse the cultured tissue equivalent of
residual cryoprotectant, 25 mL of (room temp) rinse solution, preferably
DMEM is then added to the petri dish containing the cultured tissue
-23-
CA 02210532 1997-07-15
WO 96/24018 PCT/US96/01217
equivalent for about 30 minutes. Other sufficient physiological rinse
solutions, such as other cell culture media or phosphate buffered saline, may
be determined by the skilled artisan. The rinse solution is exchanged a second
time for an additional 30 minutes.
Thawed cryopreserved and control samples were processed by histological
methods and evaluated by light microscopy for morpohological organization
and viability and showed near 100% viablility and were almost
indistinguishable from control.
Example 3: Evaluation of the Cryopreservation Package Design
for Warming Rate and Thermal Uniformity
A cryopreservation package of the present invention was evaluated for
uniform heat transfer. To a carrier membrane, without a cultured tissue
equivalent, were mounted five thermocouples, one in the center and four
equally spaced 1.0 mm from the peripheral wall of the carrier. Thermocouple
leads exited the package from the side between lid and tray flanges to an
temperature recorder (Azonix Scanner Plus). The package was filled with 17
mL of cryoprotectant and the package was heat sealed. The package was
equilibrated to -70 C and then transferred to a water bath set at 20 C. The
temperature recorder recorded the temperature at each thermocouple every 1
to 2 seconds during warming. The package warmed at a rate of 700 C/min.
initially and slowed down as it approached the melting point of the
cryoprotectant solution. The thermal uniformity was between 2 C and 6 C of
the average thermocouple temperature during the fastest warming rates and
approached 8 C as it reached the melting point of the solution.
Examples 4 to 12 show the cryopreservation method of this invention on
different tissues.
Example 4: Cryopreservation of Epidermal Sheets
-24-
CA 02210532 1997-07-15
WO 96/24018 PCT/US96/01217
Epidermal sheets were procured from mature LSE at 12 days post air-lift.
Removal of the epidermal sheet was accomplished by peeling the dermal
substrate layer from the epidermal layer with forceps and discarding the
dermal layer. Each sheet was cut in to three equivalerit pieces. One piece
from each sheet was fixed as a control. The remaining two pieces form each
sheet were placed on top of a 75mm polycarbonate transwell membrane
(Costar) in 100 mm culture dishes (Costar). Each piece was perfused with 25
mL of DMEM and 2 Molar Glycerol for one hour. The dishes containing
constructs were placed on an orbital shaker (Bellco) at 70 rpm for one hour in
a 10% C02 gassed chamber. After the epidermal sheet was perfused, the petri
dish containing epidermal sheet, transwell and extracellular freezing media
(2M Glycerol and DMEM) were placed in a bag and vacuum sealed
(Audiovox) programmed for a 5 second vacuum and a 3.9 second seal time.
Packaged epidermal sheets were placed into the programmable freezer
(Planer) at a starting temperature of 20.0 C. Epidermal sheets were cooled at
-10.0 C/minute to -6.0 C. The temperature was allowed to hold for 20
minutes at -6.0 C to equilibrate to chamber temperature. After the 20 minutes
hold, extracellular ice was initiated by contact of the outside of the bag,
below
the level of the freeze media, with a liquid nitrogen chilled probe. After all
epidermal sheets had been seeded with ice crystals, the temperature was held
an additional 5 minutes at -6.0 C. The chamber was then cooled at
-1.0 C/minute to -8.0 C. The temperature was then allowed to hold for 30
minutes at -8.0 C to allow for uniform distribution of ice throughout the
sample. The chamber was then cooled at -0.1 C/minute to a final
temperature of -70.0 C.
Cryopreserved epidermal sheets were removed from the freezer and the
bags were cut from the petri dishes and the lids removed. To thaw, 40 mL of
-25-
CA 02210532 1997-07-15
WO 96/24018 PCT/US96/01217
warmed (37 C) DMEM was aseptically poured into each petri dish. After 45
seconds, all liquid was removed from the dishes and an additional 40 mL
aliquot was added to each dish for two minutes After all ice was thawed,
epidermal sheets were rinsed with 25 mL of DMEM for 30 minutes. The
media was exchanged a second time for an additional 30 minutes. The
epidermal sheets were then incubated in culture maintenance medium at
37'C/10%C02 for 24 hours prior to analysis. The incubation time was
allowed for the lag typically seen for frozen cells to reestablish steady
state
conditions. Again, the incubation time was allowed for the lag typically seen
for frozen cells to reestablish steady state conditions.
Of the two remaining pieces from each epidermal sheet, one from each
was processed for histology and one piece from each sheet was assayed
following the MTT assay protocol. Comparing photomicrographs of non-
cryopreserved epidermal sheet and cryopreserved epidermal sheet of Example
4 showed that the three basic features of the epidermis: the basal layer of
the
epidermis, the suprabasal layers of the epidermis and the stratum corneum of
the epidermis were all preserved by this method. All layers were intact and
the overall morphology of the cryopreserved epidermal sheet was identical to
that of the non-cryopreserved epidermal sheet.
Example 5: Cryopreservation of Dermal Equivalent
Dermal equivalents used in this study were the non-epidermalized
component of the LSE. Dermal equivalents were frozen at 8 days post cast.
The dermal equivalents were perfused with cryoprotectant by submerging
the dermal equivalents attached to 75mm transwell (Costar) placed in 100mm
petri dishes (Costar) with 25 mL of DMEM and 2 Molar Glycerol for one hour.
The petri dishes were placed on an orbital shaker (Bellco) at 70 rpm for one
hour in a 10% C02 gassed chamber. After dermal equivalents were perfused,
-26-
CA 02210532 1997-07-15
WO 96/24018 PCT/US96/01217
the petri dishes, dermal equivalents, transwells and extracellular freezing
media were placed into the programmable freezer (Planer) at a starting
temperature of 20.0 C. The chamber was then cooled at -10.0 C/minute to
-6.0 C. The temperature was held for 20 minutes to equilibrate to chamber
temperature. After the 20 minutes hold, extracellular ice was initiated by
contact of the outside of the dishes, below the level of the freeze media,
with a
liquid nitrogen chilled probe. After all dermal equivalents had been seeded
with ice crystals, the temperature was held an additional 5 minutes at -6.0 C.
The chamber was then cooled at -1.0 C/minute to -8.0 C. The chamber
temperature was held again for 30 minutes at -8.0 C to allow for uniform
distribution of ice throughout the sample. Dermal equivalents units were
then cooled at -0.1 C/minute to a final temperature of -70.0 C.
Cryopreserved dermal equivalents were then removed from the freezer
and the lid of the dish was removed. To thaw, 40 mL of warmed (37 C)
DMEM was aseptically poured into each petri, dish. After 45 seconds, all
liquid
was removed from the dishes and an additional 40 mL aliquot was added to
the dishes for two minutes After all ice was thawed, dermal equivalents were
rinsed with 25 mL of DMEM for 30 minutes The media was exchanged a
second time for an additional 30 minutes The dermal equivalents, still
attached to the transwells, were then transferred back to the culture dishes
and incubated in culture maintenance medium at 37 C / 10%C02 for 24 hours
prior to analysis. The incubation time was allowed for the lag typically seen
for frozen cells to reestablish steady state conditions. Samples were assayed
using the MTT assay protocol.
Example 6: Cryopreservation of Cornea Equivalents
Cornea equivalents, attached to 24mm culture transwells (Costar), 9 days
post moist air lift, were placed into six well cluster dishes (Costar). The
-27-
CA 02210532 1997-07-15
WO 96/24018 PCT/US96/01217
method of perfusing cornea equivalents with cryoprotectant was to submerge
each cornea equivalent and transwell with 4 mL extracellular freezing media
(2M glycerol in DMEM) in the six well cluster dishes for one hour. The six
well duster dishes containing the cornea constructs were shaken for one hour
on an orbital shaker (Bellco) at 70 rpm in a 10% C02 gassed chamber. After
cornea equivalents were perfused, the petri dishes containing cornea
equivalents, transwells and extracellular freezing media were placed into the
programmable freezer (Planer) at a starting temperature of 20.0 C. Cornea
equivalents were cooled at -10.0 C/minute to -6.0 C. The temperature was
held for 20 minutes to equilibrate to chamber temperature. After the 20
minutes hold, extracellular ice was initiated by contact of the outside of
each
well in the cluster plate, below the level of the freeze media, with a liquid
nitrogen chilled probe. After all cornea equivalents had been seeded with ice
crystals the temperature was held an additional 5 minutes at -6.0 C prior to
cooling at -1.0 C/minute to -8.0 C. The temperature was held again for 30
minutes at -8.0 C to allow for uniform distribution of ice throughout the
sample. Cornea equivalent units were cooled at -0.1 C/minute to a final
temperature of -70.0 C.
Cryopreserved cornea equivalents were removed from the freezer. The
lid of the dish was removed. To thaw, 6 mL of warmed (37 C) DMEM was
aseptically poured into each well of the cluster plate. After 45 seconds, all
liquid was removed from the dish and an additional 6 mL aliquot was added
to the dish for two minutes Using sterile forceps the cornea equivalents and
attached transwells was transferred to a new cluster dish. To rinse, 4 mL of
DMEM was added to each well for 30 minutes. The media was exchanged a
second time for an additional 30 minutes The cornea units were then
transferred back to the/same culture dish and incubated in cornea
-28-
.-
CA 02210532 1997-07-15
WO 96/24018 PCTlUS96/01217
maintenance medium at 37 C / 10%C02 for 24 hours prior to analysis. The
incubation time was allowed for the lag typically seen for frozen cells to
reestablish steady state conditions. Samples were assayed using the MTT
assay protocol.
Example 7: Cryopreservation of Harvested Murine Skin
Wild type mice, strain B6CB6YF1, were euthanized by Nembutal overdose.
The skin was harvested aseptically. Excess blood vessels, fat and connective
tissue were removed from the dermis. Murine skin was trimmed to a
rectangular 1 cm x 2 cm piece. Murine skin pieces were then placed on a 75
mm transwell (Costar) in 100 mm petri dishes (Costar). Murine skin was
perfused with cryoprotectant by submerging in 25 mL of 2M Glycerol in
DMEM in the 100 mm petri dishes for one hour. During perfusion, the petri
dishes each containing the murine skin and the transwell were shaken for
one hour on an orbital shaker (Bellco) at 70 rpm in a 10% C02 gassed
chamber. Control, non-cryopreserved mouse skin was kept in nutrient
media at 4 C for the duration of the cryopreservation and thawing process (2
days) before skin grafting.
After murine skin is perfused, the petri dishes containing murine skin,
transwells and extracellular freezing media are placed into the Planer
programmable freezer at a starting temperature of 20.0 C. Murine skin was
cooled at -10.0 C/minute to -6.0 C and were allowed to hold at -6.0 C for 20
minutes to equilibrate to chamber temperature. After the 20 minute hold,
extracellular ice was initiated by contact of the outside of the dish with a
liquid nitrogen chilled probe. The contact site must be below the level of the
freeze media. After all murine skin had been seeded with ice crystals, the
temperature was held an additional 5 minutes at -6.0 C prior to cooling at
-1.0 C/minute to -8.0 C. The temperature was held again for 30 minutes at
-29-
CA 02210532 1997-07-15
WO 96/24018 PCT/US96/01217
-8.0 C to allow for uniform distribution of ice throughout the sample. The
units were then cooled at -0.1 C/minute to a final temperature of -70.0 C.
Cryopreserved murine skin was removed from the freezer and the lid of
the dish was removed. To thaw, 40 mL of warmed (37 C) DMEM was
aseptically poured into the petri dishes. After 45 seconds, all liquid was
removed from the dishes and an additional 40 mL aliquot was added to the
dishes for two minutes After all ice was thawed, murine skin was rinsed in
the dishes with 25 mL DMEM for 30 minutes The media was exchanged a
second time for an additional 30 minutes.
Thawed cryopreserved and control samples were processed for histology
or were grafted to mice. The photomicrographs of non-cryopreserved mouse
skin compared with cryopreserved mouse skin showed that the four basic
features of mouse skin, the dermis with fibroblasts, the basal layer of the
epidermis, the suprabasal layers of the epidermis and the stratum corneum of
the epidermis were preserved by this method. All layers were intact and the
overall morphology of cryopreserved mouse skin was identical to that of
non-cryopreserved mouse skin.
Example 8: Cryopreservation of ATS SKIN2TM
SKIN2TM, model ZK 1300 (Advanced Tissue Sciences, La Jolla, CA), was
removed from the packaging according to shipping inserts upon arrival and
were placed in culture dishes (Costar). Transwell inserts were placed above
the SKIN2TM to keep the skin construct submerged. SKIN2TM was perfused
with cryoprotectant by submerging SKIN2TM in 2M Glycerol in DMEM in the
culture dishes for one hour. During perfusion, the culture dishes containing
the construct and the transwell were shaken for one hour on an orbital
shaker (Bellco) at 70 rpm in a 10% C02 gassed chamber.
-30-
CA 02210532 1997-07-15
WO 96/24018 PCT/US96/01217
After SKIN2TM is perfused, the units were placed into the programmable
freezer (Planar) at a starting temperature of 20.0 C. SKIN2TM units were
cooled at -10.0 C/minute to -6.0 C and were allowed to hold at -6.0 C for 20
minutes to equilibrate to the chamber temperature. After the 20 minute hold,
extracellular ice was initiated by contact of the outside of the dishes below
the
level of the freeze media with a liquid nitrogen chilled probe. After all
SKIN2TM units were seeded with ice crystals, the temperature was held for an
additional 5 minutes at -6.0 C. The chamber temperature was then cooled at
-1.0 C/minute to -8.0 C. SKIN2TM units were allowed to hold for 30 minutes
at -8.0 C to allow for uniform distribution of ice throughout the sample.
SKIN2TM units were then cooled at -0.1 C/minute to a final temperature of
-70.0 C.
Cryopreserved SKIN2TM were removed from the freezer, the lids of the
dishes was removed and warmed (37 C) DMEM was aseptically poured into
each culture dish. After 45 seconds, all liquid was removed from the dishes
and another addition of DMEM was added to each dish for two minutes.
Once thawed, the SKIN2TM unit and attached transwell were transferred to a
new culture dishes, using sterile forceps. To rinse the SKIN2TM of
cryoprotectant, DMEM was added to each culture dish containing the
SKIN2TM for 30 minutes. The media was exchanged a second time for an
additional 30 minutes. The SKIN2TM unit was then transferred back to the
same type culture dish and was incubated in culture maintenance medium at
37 C/10%CO2 for 24 hours prior to analysis. Again, the incubation time was
allowed for the lag typically seen for frozen cells to reestablish steady
state
conditions.
Cryopreserved and control constructs were assayed following the MTT
assay protocol for SKIN2TM model ZK1300, enclosed with the product, and
-31-
__
CA 02210532 1997-07-15
WO 96/24018 PCTiUS96/01217
also by histological evaluation. The photomicrographs of non-cryopreserved
SKIN2TM compred with cryopreserved SKIN2TM showed that the four basic
features of SKIN2TM, the collagen lattice with fibroblasts, the basal layer of
the
epidermis, the suprabasal layers of the epidermis and the stratum corneum
of the epidermis were all preserved by this method. All layers were intact and
the overall morphology of cryopreserved SKIN2TM was similar to that of non-
cryopreserved SKIN2TM.
Example 9: Metabolic Mitochondrial Activity Assay (MTT)
Frozen and non-frozen (control) LSE, epidermal sheet, dermal equivalents
and corneal equivalents were tested using the MTT assay. [SKIN2TM
constructs were assayed according to "MTT Assay Protocol for use with Model
ZK1300", enclosed with the shipping inserts.] Cell viability of the constructs
were measured using the MTT assay, a colorimetric MTT conversion assay
developed to measure cellular growth and viability. This assay is described in
detail in Gay et al., "The Living Skin Equivalent as a Model In Vitro for
Ranking the Toxic Potential of Dermal Irritants," Toxic. in Vitro, 6:303-315
(1992). The metabolic reduction of the soluble tetrazolium salt to a blue
formazan precipitate is dependent on the presence of viable cells with intact
mitochondrial function. This assay is used to quantitate cytotoxicity in a
variety of cell types, including cultured human keratinocytes.
To the culture dishes containing LSE, epidermal sheet and dermal
equivalent, 40 mL of assay medium and to the wells containing corneal
equivalents, 1.5 mL of assay medium containing 0.33 mg /mL MTT (Sigma
Chemical Co., St. Louis, MO) was added. The tissue equivalents were
incubated in the MTT assay medium for 3-4 hours. At the end of the
conversion period, the tissue equivalent was biopsied using an 8-mm
diameter skin biopsy punch. The punch biopsies were then extracted for 2-3
-32-
CA 02210532 2005-10-07
hours at room temperature in 0.3 mL isopropanol, acidified with 0.04 N HCI.
At the end of the extraction period, 0.2 mL of each extract was transferred to
a
well of a 96-well plate. The absorbencies were read on a plate reader
(Dynatech) at 570 run with the isopropanol extraction medium as a blank.
MTT values obtained from thawed cryopreserved samples were compared to
corresponding control samples and expressed in terms of percent of control
(Figure 9).
Example 10: Lactose Dehydrogenase Assay (LDH)
LDH is an enzyme typically found in a viable cell. Damage to the cell
causes a release of the enzyme and a corresponding decrease in the enzyme
activity detected by this assay.
Thawed cryopreserved and control samples of LSE were punched with an
8 mm diameter skin biopsy punch. Three samples were taken from each LSE
unit. Punch samples were placed in a 15 mm tubes with 1 mL of 0.1 M
triethanolamine buffer (on ice) and homogenized with an electric tissue
homogenizer for one minute. The samples were then centrifuged at 1000g at
4.0 C. The supernatant was then assayed. LDH cocktail reagent was prepared
by mixing together the following: 3.00 mL phosphate buffer (0.1 mol/l;pH
7.0), 0.1 mL pyruvate, Na salt (2.5 mg/mL), and 0.05 mL NADH, Na salt (10
mg/mL). l00 L of sample supernatant was added to 900 .L of the reagents
and allowed to react for two minutes. The change in absorbance over the two
minutes was recorded. The average of three samples per cryopreserved LSE
unit were compared to the non-frozen controls. Sample values were
compared to corresponding control values and expressed- in terms of percent
of control (Figure 9).
Example 11: Bioequivalence of Cryopreserved LSE
To Non-Cryopreserved LSE
-33-
CA 02210532 1997-07-15
WO 96/24018 PCT/US96/01217
To demonstrate the bioequivalence of cryopreserved and non-
cryopreserved LSE, a grafting study to athymic mice was performed.
LSE units, cryopreserved according to the method outlined in Example 1,
were thawed one day before grafting and recovered in maintenance media for
twenty-four hours.
Four experiments were performed in which a total of 66 athymic mice of
the strain B6CB6YF1/J-nu (Jackson Harbor Labs) were grafted with either
cryopreserved LSE (n= 43) or non-cryopreserved (control) LSE (n=23).
Animals were anesthetized with Nembutal. A 2 x 2 cm full thickness skin
section was excised from the dorsum of each mouse, sparing the panicculus
carnosus. LSE grafts, either control or cryopreserved, were placed on the
wound and trimmed to fit. All grafts were dressed with one layer of
petrolatum impregnated gauze (vendor) and covered by two adhesive
bandages (vendor). The dressings were removed at 7 days post-graft.
At 14 days post-graft, all animals were euthanized and photographed. The
graft site was then excised for histological analysis and evaluation. The
gross
photographs and micrographs showed no difference in graft integration
between control LSE or thawed, cryopreserved LSE. No significant difference
was seen in rates of wound contracture between control or cryopreserved
grafts.
Example 12: Syngenaic Skin Grafting of Cryopreserved and
Control Mouse Skin
To demonstrate the bioequivalence of cryopreserved and non-
cryopreserved mouse skin, a syngenaic skin grafting study was performed.
Wild type mice, strain B6CB6YF1, were euthanized by Nembutal overdose.
The skin was harvested aseptically. Excess blood vessels, fat and connective
tissue were removed from the dermis in preparation for skin grafting. Mouse
-34-
CA 02210532 1997-07-15
WO 96124018 PCT/1JS96/01217
skin was cryopreserved and thawed following the method in Example 7,
Control, non-cryopreserved mouse skin was kept in a nutrient media at 4 C
for the duration of the cryopreservation and thawing process, a total of two
days, before skin grafting.
Six mice of the same strain received skin grafts, two received control, non-
cryopreserved mouse skin and four received thawed, cryopreserved mouse
skin. The mice were anesthetized with Nembutal. A 2 x 2 cm full thickness
skin section was excised from the. dorsum.of each mouse, sparing the
panicculus carnosus. Mouse skin grafts, either control or cryopreserved, were
placed on the wounds and trimmed to fit. All grafts were dressed with one
layer of petrolatum impregnated gauze, covered by two adhesive bandages.
These dressings were removed at 7 days post-graft.
At thirty days post-graft, all animals were euthanized and photographed.
The graft site was then excised for histological analysis and evaluation. The
gross photographs and micrographs showed that there was no difference in
graft integration between control mouse skin or thawed, cryopreserved
mouse skin. No significant difference was seen in rates of wound
contracture between control or cryopreserved grafts.
Although the foregoing invention has been described in some detail by
way of illustration and example for purposes of clarity of understanding, it
will be obvious to one skilled in the art that certain changes and
modifications may be practiced within the scope of the appended claims.
-35-