Note: Descriptions are shown in the official language in which they were submitted.
CA 02212378 1997-08-06
W O96/25361 PCT~US96/O1910
COP]PER PRECIPITATION PROCESS
BACKGROUND OF THE INVENTION
Field of the Invention: The present invention relates to the separation of copper from
one or more metals. More particularly, the present invention relates to the precipitation of
copper as a s,ulfide from aqueous acidic solutions while retaining other metals in the liquid
phase for later precipitation as desired.
Description of Related Art: The need and desirability of precipitating a variety of
10 metals from solution is well documented. The metals may be in solution in waste water, for
in~t~nce, whiich must be discharged and dealt with in an environmentally responsible manner.
Typical prior art methods for the removal of such metals are described in U.K. Patent
Application GB 2216114A and in U.S. Patents 3,575,853; 3,617,559; 3,738,932; 4,186,088;
4,4659597; 4,543,189; 4,566,975; ~,606,829; 4,728,438; 4,572,882 and 5,039,428.
It is desi~able in cerlain circumstances to remove and separate copper from other
metals which are in solution. It is further desirable to carry out such processes by means of
readily available chemical compounds, at low process temperatures and pressures and at high
reaction rates; thereby minimizing the cost, process times and equipment size. Examples of
such copper removal processes are disclosed in U.S. Patents 3,218,161; 3,728,430 and
20 5,032,175, C~n~di~tl Patent 1,020,363 and Society of Mining Engineers of AIME, Paper No.
80-73, entitled "The Electroslurry Process for Copper Recovery from Smelter and Refinery
Wastes. "
Patent 3,218,161 (Kunda et al.) gives a method for precipitating values of metals which
form insoluble sulfides more readily than nickel in acid and neutral solutions having a pH
25 from 1 to 7. The method is aimed at the selective precipitation of metals from nickel and
cobalt at temperatures greater than 125 degrees Fahrenheit or 52 degrees Celsius (125~F or
52~C) by the use of sulfur dioxide and finely divided sulfur, the latter in excess of
stoichiometric requirements. The major metal to be precipitated is copper and the pH is
adjusted to be within pH 1 to 7 at a preferred temperature of 180~F (82~C). The method
30 requires an inert, or substanltially inert, atmosphere.
U.S. E'atent 3,218,16l1 claims a working range of pH 1 to pH 7 which is equivalent to
a theoretical acidity of approximately 5 grams per liter sulfuric acid to 0.05 milligrams per
liter sulfuric acid. The patent embraces solutions with acid strengths greater than the
CA 02212378 1997-08-06
W O 96/25361 PCTrUS96/019lO
equivalent of pH 1, e.g., sulfuric acid strengths greater than about 5 grams per liter, by
c!~iming prior adjustment of the pH. In other words, if faced with an industrial solution with
a pH of less than 1, the solution would need to be neutralized by an alkali to bring it within
the stated range.
Whereas waste waters are generally of low acidity, industrial process streams such as
refinery electrolyte and acid plant blowdown are high in acidity, being typically 150 to 200
grams per liter sulfuric acid. However, in these cases, it may not be economical or desirable
to partially neutralize the acidity of the solution to be purified so that it comes within a range
of relatively low acidity. For example, if a bleed stream of refinery electrolyte could be
10 purified without decreasing the acid content, it would be possible to return the purified
electrolyte to the refinery. By this means, the need to add fresh acid and dispose of large
quantities of neutralization product, whether as solids or liquids, would be avoided.
U.S. Patent 4,404,071 (Abe et al.) achieves precisely this outcome by the purification
of copper refinery bleed using hydrogen sulfide as the precipitating reagent, in which case
15 copper is precipitated but in combination with arsenic, antimony and bismuth. This precipitate
is commonly returned to the copper smelting stage where the impurity elements, especially
bismuth, are re-distributed into the products. If any of these impurities has reached a
maximum permissible level in the product copper, the new intake of these impurities in
concentrates and other new feeds will need to be controlled.
It would be advantageous in the art, therefore, to provide a method wherein the copper
in refinery bleed can be separated from other metals in the bleed.
Patent 3,728,430 (Clitheroe et al.) gives a method for the extraction of copper from
ores by leaching with sulfites or bisulfites and the precipitation of the extracted copper from
the leach solution with bisulfite, or sulfur dioxide, and elemental sulfur. A pH between 1 and
6 is preferred at a temperature between 80~ and 212~F (27~ to 100~C). The two operations
can occur in the same reactor. The method provides a way of beneficiating a copper ore and
the copper sulfide product is preferably floated away from the residual matter.
The hydrometallurgical extraction of copper from ore-bodies and concentrates, for
example, is well known. In particular, a solution of sulfuric acid is arranged to percolate
30 through metals-containing material to extract copper as copper sulfate from the copper
minerals present. Copper minerals may be present as oxides, sulfides, oxysalts and the like,
and may require the use of air, oxygen or bacteria to solubilize the copper. Typical equations
describing these extractions are:
CA 02212378 1997-08-06
W O96S25361 PCTAUS~6/01910
~ Cu2O + 2 H7SO4 + 0.5 O~ ~ 2 CuSO4 + 2 H70
~ Cu2S + 2.5 O~ + H[2SO4 ~ 2 CuSO4 + H20
~ CuCO3.Cu(OH)2 + 2.H~SO4 ~ 2.CuSO4 + CO~ + 3.H~O
- The copper sulfate solution produced from the reaction may be processed to produce a
5 valuable copper product in a number of ways. It can be chemically precipitated in various
forms or, more generally, it may be electrowon to produce metallic copper in the form of a
cathode. The electrowinning is usually performed after solvent extraction of the copper from
the leach solution followed by transfer into a strong acid electrolyte. The solvent extraction
step has the advantage of rejecting impurities in the copper sulfate solution and providing an
10 optimum ele,-trolyte for copper electrolysis.
Patent 3,728,430 elects to leach the oxidic, or mixed oxidic-sulfidic ore with sulfites
rather than sulfuric acid, then precipitate the copper as a sulfide. A feature of the patent is the
production of sulfuric acid by the copper precipitation process and the use of this excess acid
to meet, to a greater or less;er extent, the natural acid demand of basic components in the oxide
15 ore. However, the value of the copper sulfide precipitate is lower than cathode copper
obtained by electrowinning the copper sulfate and many copper oxide ore-bodies are exploited
by solvent ex~traction and electrowinning (SX-EW) to maximize the added value. It would be
advantageous in the art, therefore, to provide a method whereby high added value copper
could be produced and at thle same time provide acid for the natural acid demand of the ore.
An impediment to the universal application of SX-EW for ore-body extraction is the
poor leach behavior of the common copper mineral, chalcopyrite, whether by sulfite or acid
leach. It would be advantageous in the art, therefore, to provide a copper extraction process
that facilitates the leaching of chalcopyrite and other copper minerals to produce an added
value copper material.
Canada Patent 1,02(),363 (Hall et al.) provides a method for the separation of copper
from arsenic, antimony or bismuth in aqueous solutions and slurries. The method uses sulfur
and sulfur dioxide to precipitate copper sulfide from solutions that are adjusted to an acidity of
less than pH 4 at a preferred temperature between 60~ to 90~C. The method requires air to be
essenti~lly excluded.
Canada Patent 1,02(),363 claims a pH less than 4 to effect the separation of copper
from other more acid soluble metal sulfides. The description does not give a range for acidity
CA 02212378 1997-08-06
W O96/2~361 PCT~US96/0191
and, in effect, the method relates to slurries and solutions having sulfuric acid concentrations
of 15 grams per liter (patent examples 1 and 2), equivalent to a theoretical pH of 0.5.
In investigating the use of sulfur and sulfur dioxide as precipitation agents for copper
in sulfuric acid solutions, it is our experience that whereas low acidity (high pH) solutions may
5 have an acceptable precipitation rate, higher acid strength (low pH) solutions have a slow
reaction rate. It is also our experience that the inception of the copper precipitation reaction in
any acid strength solution is slow without a minimum level of solids in suspension. These
undesirable features are not indicated in the prior patent art described above.
In the operation of metallurgical processes it is known for copper-bearing solid and
10 liquid by-product streams, or in-process streams, to be produced. These streams often contain
valuable levels of copper and other metals which make beneficial extraction of the metals
worthwhile. It may also be necessary to selectively remove metals to benefit the overall
metallurgical flowsheet and improve the economics of operation.
In one such application, it is known for copper smelters and associated refining15 operations to generate flue dusts, acid plant liquid bleeds and refinery liquid bleeds. The dusts
can be processed by hydrometallurgical means to produce a liquid that contains copper and
other metals. In one instance, the liquid streams can be processed to extract copper and other
metals by precipitation as sulfides using hydrogen sulfide gas or liquids such as sodium
hydrosulfide. The cost of reagents based on hydrogen sulfide is high, however, and the
20 quality of the resulting precipitates can be poor.
It would be advantageous in the art, therefore, to provide improved sulfide
precipitation methods for copper over a wide range of acidity, which reduce the cost of
reagents while providing a readily filtered precipitate at an acceptable precipitation rate. The
ability to selectively extract copper would also be an advantage over the less selective nature
25 of extraction using hydrogen sulfide-based reagents.
SUMMARY OF THE INVENTION
In accordance with the present invention, copper is precipitated as a sulfide from
aqueous acidic solutions, such as those produced from the processing of ore concentrates or
30 non-ferrous smelter by-products, while retaining other metals in the liquid phase for optional
downstream processing. The copper is precipitated over a wide range of acidity with
relatively inexpensive reagents, at a desirable precipitation rate, and the copper precipitate is
CA 02212378 1997-08-06
WO 96/25361 PCT/US96/01910
readily filterable. This invention also provides more economical means of precipitating
copper by using less expensive and more readily available reagents.
As a broad statement of the present invention, copper sulfide is precipitated from an
aqueous acidic solution conltaining soluble copper values by contacting the aqueous acidic
5 solution with (i) sulfur, (ii) sulfur d;oxide, and (iii) an added copper sulfide. The sulfur is
usually supplied in the for m of elemental sulfur or chalcopyrite, the sulfur dioxide is usually
supplied as a soluble sulfite or bisulfite or as smelter gas, and the added copper sulfide as a
thickened recycled stream of copper sulfide (which was originally precipitated as a product of
the pr~cess). Sufficient copper sulfide is added to the process to ensure that the solids content
10 of the precipitation zone (the zone isl which the soluble copper is reacted with the precipitating
sulfur reagents) exceeds the maximum solids content that would result in the zone if the
precipitation reaction proceeded to completion (batch mode) or reached steady state
(continuous mode) under the particular conditions of the precipitation of interest.
In one embodiment of this invention, copper is precipitated as a sulfide from an15 aqueous solution of copper and other metals, the solution having a free acid range of about
0.05 to about 200 grams per liter (gpl). The solution is contacted with elemental sulfur and a
material selected from the group consisting of soluble sulfites or bisulfites, the contacting
conducted at a temperature from about 40~ to about 90~C. The precipitated copper sulfide is
removed from the precipitation zone, thickened by any conventional technology, optionally
20 filtered, and then a portion recycled to the precipitation zone.
A variable part of the thickened suspension is recycled to the precipitation stage to
promote the reaction, increase the operable range of acidity, increase the rate of reaction,
reduce the reaction time and enhance the degree of completion, all relative to a non-recycled
operation. The precipitation may occur in a multicompartmental reactor, such as four
25 compartments in series. Also, the method may use chalcopyrite as a source of sulfur or as a
material to be processed in its own right.
BRIEF DESCRIPTION OF THE DRAWINGS
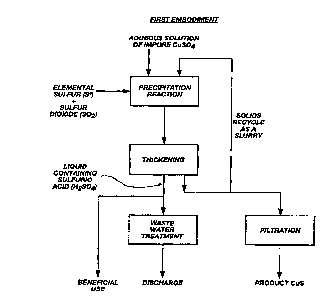
FIG. 1 is a flow diagram of a first embodiment of the invention in which an aqueous
30 solution of impure copper sulfate is processed using elemental sulfur and sulfur dioxide;
FIG. 2 is a flow diagram of a second embodiment of the invention in which a weak,
impure solul:ion of copper sulfate is processed into a strong, pure solution;
CA 02212378 1997-08-06
W O96/25361 PCT~US96/01910
FIG. 3is a flow diagram of a third embodiment of the invention in which previousembodiments of FIGS. 1 and 2 are used to extract copper from oxidic copper to produce
cathode copper;
FIG. 4is a flow diagram of a fourth embodiment of the invention, similar to the
5 previous embodiments of FIGS. 1 and 2, wherein elemental sulfur is substituted with sulfur
derived from chalcopyrite-bearing ore;
FIG.Sis a flow diagram of a flfth embodiment of the invention in which the
embodiment illustrated if FIG. 4is modified by routing a majority of the thickened solids and
liquids to an oxidation stage;
10FIG. 6is a flow diagram of a sixth embodiment of the invention in which the
embodiment of FIG.5 is modifed to include a leach of oxidic copper ore;
FIG. 7is a schematic diagram showing a seventh embodiment of the invention
illustrating operational parameters and conditions;
FIG.8is a schematic diagram of a vessel used to simulate a reactor of the present
15 invention;
FIG. 9is a drawing of a laboratory simulation of the present invention shown in FIG.
7;
FIG. lOis a correlation between rate and solids concentration in the reactors in Test 14
through 17 of the present invention;
20FIG. llis an illustration of separation of metals in each stage versus EMF for Test 14
of the present invention;
FIG. 12is an illustration of separation of metals in each stage versus EMF for Test 16
of the present invention;
FIG. 13is an illustration of copper concentration as a function of precipitation time of
the present invention for Test 18 through 20;
FIGS. 14(a) and (b) are summaries of the sedimentation test on the precipitate
produced in the process as set forth in Table 5 herein;
FIG. 15 is a s~mm~ry of cake weight versus thickness from the filtration tests on the
precipitate as set forth in Table 5 herein;
30FIG. 16is a summary of cake weight versus formation time of the precipitate as set
forth in Table 5 herein;
FIG. 17 is a s-lmm~ry of cake moisture versus dry time of the precipitate as set forth in
Table 5 herein;
CA 02212378 1997-08-06
W~ 96125361 PCT~JS96J~I911
FIG. 18 is a summ~ry of wash time versus cake weight versus wash volume of the
precipitate as set forth in Table 5 herein;
FIG. 19 is an illustration of residual copper concentration in solution as a function of
precipitation time of the present invention using chalcopyrite with various EMF potentials;
S FIGS. 20(a) and (b) illustrate settling data derived from thickening tests performed on
precipitation slurry using chalcopyrite;
FIG. 21 is a graph illustrating test results of the inventive process conducted at high
acidity levels; and
FIG. 22 is a schematic diagram of a generalized description of the invention.
DE'iCRTPTION OF THE INVENTION
FIG. 22 describes the process of this invention in a generic manner. Soluble copper
(e.g., copper sulfate, CuSC)4) iS contacted with elemental sulfur (S~) and sulfur dioxide (SO2)
in a reaction vessel to precipitate the soluble copper as copper sulfide. The contacting is
15 typically conducted with agitation (the stirrer) and at a temperature above ambient (e.g., in
excess of 25~C). The soluble copper is usually associated with other soluble metals in an
aqueous aciclic solution with a free acid content in excess of 0.05 gpl.
Copper sulfide will continue to precipitate until the soluble copper or the precipitating
reagents are depleted if the process is conducted in a batch mode, or until steady state is
20 achieved if the process is conducted in a continuous mode. In either event, one hallmark of
this invention is to increase the solids content, i.e., the copper sulfide, of the precipitation
zone (here the reaction ves,sel) by adding copper sulfide. This addition has the desirable effect
of increasing the precipitat;on rate.
The copper sulfide lthat is added to the reaction vessel is usually and preferably a
25 thic~ned recycled stream. As the copper sulfide is formed in the reaction vessel, it is
transferred to a thickening tank by any conventional means, e.g., gravity overflow, pumping,
etc. Here the copper sulficle content is thickened (in FIG. 22, from 30 gpl to 500 gpl). The
thickened ca,pper sulfide is then divided into two streams, a product stream for further
proceccing (e.g., smelting or converting) and a recycle stream for return to the reaction
30 vessel. Clear overflow is removed from the thickener as necessary to m~int~in a working
b~l~nce of liquids and solicls in the thickener.
CA 02212378 1997-08-06
W O96/25361 PCTrUS96/01910
While FIG. 22 depicts the thickener as a separate vessel, the action of thickening the
CuS can occur in the same vessel in which the CuS is precipitated. Vessels such as these will
have a precipitation zone and a thickening zone and will allow for the transfer of thickened
CuS from the latter to the former zone.
The use of sulfur and SO~ as the precipitating reagents produces a precipitate that has
desirable settling and filtering properties, and this is another hallmark of this invention.
Moreover, this combination of precipitating reagents affords a more selective precipitation of
copper than generally possible with the use of other precipitating a reagents, e.g., hydrogen
sulfide (H2S).
The conditions at which the process of this invention is operated will vary with the
reagents, equipment, starting materials and the like. As such, operating conditions such as
temperature, pressure, agitation, contact or residence time~ molar ratios, solids content, acid
strength, etc., are selected to optimize copper precipitation in terms of rate, selectivity and
completion.
Our attempts to duplicate the teachings of the above patents concerning separation of a
copper product, particularly at pH levels below 2 and at temperatures of 60~C have not been
able to reach complete copper precipitation in a reasonable length of time (less than 2 hours)
without employing the technique of increased solids content in the reactor. When we utilize
the practical measure of recycling of thickened suspension to build solids content in a
20 continuous operation, the reaction goes essentially to completion much more rapidly when
compared at similar conditions to those cited in the above patents.
Furthermore, the practicality of the process is enhanced in the present invention by the
application of smelter gas (SO~) containing appreciable quantities of free oxygen, whereas the
prior art claims the substantial absence of oxygen. Whereas the prior art uses the chemistry of
25 the method to separate copper from other metals by controlling the acidity of the system, the
present invention provides for the sequential separation of metals by the control of the
reduction potential, especially in those situations whereby copper, bismuth and antimony need
to be sequentially separated.
The chPmistry of the preferred embodiment of the present invention can be generally
30 described and schem~tically represented as follows:
CuSO4 + S + SO~ + 2 H~O ~ CuS + 2 H~SO4 (1)
CuFeS~ + 3 CuSO4 + 2 SO~ + 4 H~O ~ 2 Cu~S + 4 H~SO4 + FeSO4 (2)
= ~ =
CA 02212378 1997-08-06
WO 96J25361 PCT~US96~191
The first reaction (1) above represents one embodiment of the invention which utilizes
elemental sulfur (S) and S02 as reactants from suitable sources. The second reaction (2) above
represents an alternative source of sulfur-bearing reagent in the preferred embodiment. In the
description o,f the this invention, the term "copper sulfide" is used interchangeably for both the
5 cupric and cuprous forms of this sulfide. Notably, the cuprous form of this sulfide may be
formed as a ~secondary product during the precipitation reaction as described by equation (5).
The precipitation reaction or step in all of the embodiments of this invention can be practiced
using one or more, and preferably more, reaction vessels.
This firse embodiment of the invention, shown schematically in FIG. 1, is practiced by
10 employing the impure aqueous sulfate solution from the natural (e.g., mine run-off), or
engineered (e.g., heap leaching), leaching process of a copper-containing material.
Alternatively, a copper-conitaining aqueous sulfate stream from a production process (e.g.,
refinery bleed) may be employed. The copper-containing aqueous sulfate streams are
subjected to a precipitation process in a precipitation system. The solids suspension from the
15 precipitation process is thiclcened and a portion of the thickened suspension is recycled to the
precipitation stage as a slurry to initiate the reaction, increase the operable range of acidity,
increase the rate of reaction, reduce the reaction time, and enhance the degree of completion,
all r~lative to a non-recycled operation.
The product sulfide precipitate from the preferred embodiment may be separated from
20 the thickened suspension by filtration and processed to final copper-containing product by
other means, such as pyrometallurgical smelting methods. The residual filtrate solution
(cont~ining sulfuric acid, among others) from the reaction in this first case is either neutralized
and discardeci, or returned to the process for beneficial use (e.g., as described in FIG. 3).
Alternatively, the residual filtrate solution which may contain additional valuable metals, e.g.,
25 molybdenum, cadmium, zinc, antimony, bismuth, arsenic, etc., may be processed to recover
one or more of these metals.
In a second embodiment of this invention, shown schematically in FIG. 2, the product
sulfide precip,itate may be separated from the solution by filtration and re-oxidized to copper
sulfate in an additional reactor system:
CuS ~- 2 02 ~ Cu'~04 (3)
Cu2S + 2.5 02 + H:~S04 ~ 2 CuS04 + H20 ~4)
CA 02212378 1997-08-06
W O96/25361 PCTrUS96/01~10
By this means, the copper can be selectively removed from impure solutions and concentrated
in solution so that the resulting copper sulfate can be electrowon to copper cathode, or
processed to other products such as copper sulfate crystals.
In a third embodiment of this invention, shown schematically in FIG. 3, the sulfuric
5 acid solution remaining after removal of the copper sulfide precipitate may be utilized to
provide acid for reaction with the basic minerals of an oxidic copper ore-body (e.g., an ore-
body containing copper in a non-sulfidic form).
Oxidic copper ore-bodies may contain relatively high proportions of basic minerals that
consume acid from circulating leach solutions used to convert the oxidized form of copper to
10 soluble copper sulfate. Ore-bodies may consume inefficiently 10 to 20 kilograms (kg) of
100% sulfuric acid per ton of rock, and in extreme cases may consume as much as 100 kg of
100% sulfuric acid per ton of rock. This inefficiency has a large impact on the economics of
the extraction process because processing sites are often in remote areas thereby necessitating
the transport of acid to, or the generation of acid at, the site.
In this third embodiment, sulfur may be brought to site to produce acid according to
the preferred embodiment of the first reaction:
CuSO4 + S + SO~ + 2 H~O -~ CuS + 2 H7SO4 (1)
lt will be seen that for every weight unit of copper that reacts, 3.08 weight units of sulfuric
acid are produced. On return to the copper extraction process, 1.54 of those weight units of
20 sulfuric acid are consumed to produce a soluble weight unit of copper. Thus, 1.54 additional
weight units of sulfuric acid are available from each copper extraction cycle to meet the
consumption of acid by the basic components of the ore or concentrate.
At the limit, all of the copper will need to be precipitated as sulfide to supply the
maximum quantity of acid to meet the inefficient consumption of ore or concentrate. Hence,
25 for a common acid demand of 10 kg per ton of ore or concentrate, the extractable copper
content will need to be 6.5 kg per ton, or 0.65% copper in the material. This concentration of
copper is common in ore-bodies. Hence it is seen that there is a relationship between the
extractable copper present and the maximum acid demand of the rock. If the acid demand
e~c~ee-l~ the limit, then additional acid will need to be brought in or generated on site, if it is
30 econcmic~l to do so.
Under certain circumstances of this third embodiment, only a proportion of the
exLId~Ldble copper will need to be precipitated from solution to meet the acid demand of the
rock. The rem~ining solution can be solvent extracted and electrolyzed in the conventional
CA 022l2378 l997-08-06
W O 96/25361 PCTAUS96/01910
manner. Thus, the transport of acid from industrial centers to isolated areas and the high
capital costs of on-site acid plants are both avoided. If the preferred product is cupric sulfide
(CuS) the oxidation stage and the SX/EW stage are unnecessary. If the product sulfuric acid
from ~he process exceeds tllle demand made by the basic ore, then the excess acid can be
5 diverted to a waste water treatment operation.
If a second reactor system is provided in this third embodiment to the preferredreaction, the amount of suliur brought to site can be reduced by using the cupric sulfide to
react with aclditional copper sulfate and sulfur dioxide to produce cuprous sulfide:
CuSC)4 + CuS + S07 + 2 H20 ~ Cu~S + 2 H~S04 (5)
10 The same quantity of acid is still produced per weight unit of copper, but the sulfur quantity is
reduced by 25% when taking into account the sulfur in the sulfur dioxide.
In circumstances whereby it is not favorable to make a copper sulfide product, the
copper sulfides produced i~ the third embodiment may be oxidized according to the method
expounded in the second embodiment, and the copper sulfate so produced routed to the
15 SX/EW stage for production of cathode copper. As the SX/EW stage liberates an equivalent
quanLtity of sulfuric acid from the input copper sulfate for return to the ore le~ching step, an
additional equivalent quantity of acid is available from the precipitation stage to meet the
demands of the basic components of the ore-body.
In a t'ourth embodiment of the invention chalcopyritic-bearing ores or concentrates are
20 processed:
CuFeS2 + 3 CuS04 + 2 S07 + 4 H~0 ~ 2 Cu2S + 4 H~S04 + FeS04 (2)
In this fourth embodiment, a cuprous sulfide precipitate is produced that can be readily
oxidized to c:opper sulfate according to the reaction:
2 Cu2S + 5 0~ + 2 H~S04 ~ 4 CuS04 + 2 H~0 (6)
25 The cuprous sulfide precipitate may be removed from the solids suspension produced in
reaction (2) by filtration or flotation.
The sulfuric acid used in the oxidation reaction may be at least partially derived from
~ the precipitation reaction and as such will contain ferrous sulfate (FeS04). Depending on the
conditions of the oxidation reaction, some of the ferrous sulfate may be oxidized to ferric
30 sulfate (Fe2(S04)3). Since ferric ions can contaminate the SX/EW, the oxidation reaction is
preferably conducted to disfavor the production of ferric sulfate.
CA 02212378 1997-08-06
W O96125361 PCTrUS96/01910
The sulfuric acid required in the oxidation of the cuprous sulfide by reaction (6) may
be provided from the acid produced in reaction (2). According to the stoichiometry of the
reactions, half of the solution produced in reaction (2) is required to meet the acid demand of
reaction (6).
S The rern~ining half of the solution from reaction (2) is withdrawn as a bleed stream for
iron and any other solubilized species. As most copper ores and concentrates contain basic
minerals, such as limestone, dolomite and magnesite, the aqueous solution produced in
reaction (2) may have lost some of the indicated sulfuric acid through neutralization of these
minerals. The remainder may be treated with alkali to neutralize the excess acid and
10 precipitate iron.
Three fourths of the copper sulfate solution produced by reaction (6) is returned to
continue reaction (2) with new chalcopyrite. The other one fourth is removed and purified
from soluble iron. The resulting copper sulfate may be electrowon to copper cathode, or
processed to other products such as copper sulfate crystals. Sufficient sulfuric acid is
15 produced in the SX stage to meet the acid demands of the copper oxidation reaction.
In a fifth embodiment of the invention, shown schematically on FIG. 5, chalcopyritic-
bearing ores or concentrates are processed as in the fourth embodiment given above.
However, in this fifth embodiment, the whole solution and solids suspension produced by
reaction (2) is oxidized as follows:
20 2 Cu~S + 4 H2S04 + FeS04 + Sl~ 02 ~ 4 CuS04 + 1/2 Fe~(SO4)3 + 21/2 H~0 + ll/2 H2S04 (7)
As previously noted, the quantity of acid available for this reaction will be affected by the
basic materials present in the chalcopyritic-bearing ores or concentrates. The ferric sulfate
that may be produced by this reaction is a strong oxidant and will assist in the oxidation of
copper species in the concentrate or ore-body when three fourths of the copper sulfate solution
is returned to continue reaction (2) with new chalcopyrite, as in the fourth embodiment above.
The oxidation reaction product is separated into solid and liquid fractions using conventional
technology. The solid fraction may be subjected to flotation to separate gangue material from
a copper concentrate. This flotation operation is facilitated by the prece-ling precipitation and
oxidation reactions.
As in the fourth embodiment, one fourth of the above copper sulfate solution (i.e. the
liquid fraction from the thickening operation) is removed and purified from soluble iron by
solvent extraction. The resulting copper sulfate may be electrowon to copper cathode, or
processed to other products such as copper sulfate crystals.
12
CA 022l2378 l997-08-06
W O96/25361 PCT~US96JO19~0
In a sixth embodiment of the invention, shown schematically in FIG. 6, chalcopyritic-
bearing ores or concentrates are processed as-in the fourth and fifth embodiments given above.
In this sixth embodiment, the free acid produced by reaction (2) in the fourth case and
- reaction (7) in the sixth case, is utilized for the purpose of reacting with oxidic copper material
5 and acid-consuming basic components. These acid-consuming materials and components may
be part of the chalcopyritic-bearing material, or separate oxidic and basic material. If the
former, the rnaterial will be processed in the vessel. In the latter, the material will be
processed orl a heap.
On r~lany occasions, oxide ore-bodies are found associated with sulfides that contain
10 chalcopyrite Hence, the economics of copper extraction may be improved by the above-
described leaching of chalcopyrite-bearing ore, or concentrate, to provide acid for the basic
components of the ore or concentrate. It may be only necessary to leach that quantity of
chalcopyrite-bearing material needed to provide the required sulfuric acid, as the remaining
sulfide minerals containing the chalcopyrite can be concentrated and transported to
15 pyrometallurgical processing sites. Alternatively, copper concentrate may be extracted after
the chalcopyrite consuming stage of the sixth embodiment.
By the above invention, ores and concentrates that contain chalcopyrite may be
processed to cathode copper, at the mine site or in other locations, by simple
hydrometallurgical operations. The major reagent to be supplied is sulfur dioxide which can
20 be produced by the roasting of iron pyrite or the combustion of elemental sulfur. In a similar
manner, oxidic ores can be processed to produce saleable copper sulfide concentrates.
The process of this invention is preferably conducted such that a thermal balance is
m~int~inecl between the various operational steps. Each of the operational steps 13 conducted
at conditions and in equipmlent that maximize their individual performance. The m~int~n~nre
25 of the therm;al balance between the various operational steps is accomplished using known and
conventional techniques, i.e., heat exchangers.
The invention is further described by reference to FIGS. 7-21.
As shown schematically in FIG. 7, a precipitation vessel 20 containing four reaction
compartments 22 in series may be used. Alternatively, separate reactors may be used. Each
30 reaction compartment 22 includes a turbine 24 for mixing, baMes 26, reagent sparger 28,
suitable temperature and sample ports and level control system means. The aqueous solution
cont~ining copper is introduced into the precipitation vessel 20 and is subjected to the reagents
therein while being transferred from one compartment 22 to the next. The discharge from the
13
CA 02212378 1997-08-06
W O96/25361 PCT~US96/01910
precipitation vessel 20 has flocculent added thereto and flows to the continuous thickener 30
where part of the underflow 31 therefrom is recirculated to the precipitation process and part
(equivalent to the new copper fed to the process when in material balance) is fed to a filter 32
to produce a valuable filter cake. The overflow 33 from the thickener 30 is recirculated, in
5 whole or in part, to the copper leaching process, or is passed forward, in whole or in part, to
downstream systems 34 for removal of other metals. The liquid from these downstream
systems 34 may also be returned, in whole or in part, to the copper leaching process.
One aspect of the embodiment shown in FIG. 7 is the processing of secondary copper
materials such as flue dust, bag house dust, etc. These materials are leached in an acid media,
10 e.g., sulfuric acid, to produce an impure solution containing copper sulfate and soluble
species, e.g., bismuth, arsenic, antimony, cadmium, zinc, molybdenum, and iron, and a solid
residue depleted of these metals. The product from this leach operation is separated into solid
and liquid fractions. The solid fraction is routed for further processing, e.g., recovery of
precious metals. The liquid fraction is routed to a precipitation operation as shown in FIG. 1.
In a manner similar to that of the first embodiment, copper sulfide is selectively
precipitated from the impurities in the aqueous copper sulfate solution, more specifically
arsenic, antimony, molybdenum, bismuth, cadmium, zinc and iron. The solution resulting
from the separation of copper sulfide from the impurities is optionally in part recycled to
provide acid for the dust leach operation, or is bled to further processing (which may consist
20 of further removal of impurity elements for resale (e.g., As) or an environmentally acceptable
disposal, e.g., Bi or Sb).
Another feature of this embodiment is the introduction of copper-containing bleeds
(e.g., acid plant blowdown (APB), refinery electrolytes, etc.) into the precipitation operation
or stage to selectively precipitate the copper values. These bleeds may and usually do
25 introduce additional acidity into the process and this enhanced acidity promotes selective
precipitation of the copper values. Indeed, one hallmark of this invention is the ability to
precipitate copper at the high acidity while promoting the solubility of the impurities, e.g., Bi
and Sb.
The supply of sulfur dioxide in this embodiment may be provided, for example, from
30 an associated smelter off-gas stream and in part from various bleed streams, e.g., APB, that
contains soluble sulfur dioxide. The sulfur for the copper precipitation reaction may be
provided, like in the other embodiments of this invention, from elemental sulfur or
chalcopyrite-containing concentrates from an associated smelter.
14
CA 02212378 1997-08-06
WO 96/25361 PCTJIJS96~01911)
FIG. 23 shows an alternative embodiment of FIG. 7 whereby the leach and
precipitation operations are combined in a single reaction step. In this embodiment, copper-
containing solids, e.g., electrostatic precipitation dusts, are fed to a reaction vessel optionally
in conjunctic)n with copper-containing aqueous bleeds. In this reaction vessel, the copper-
5 containing solids are leached by sulfuric acid introduced as a separate reagent, or optionally
- recycled in an aqueous stre'am. The leachate so formed contains soluble copper, Bi, As, Sb,
Cd and Fe. The soluble copper so -formed is reacted in situ with sulfur dioxide and a source
of sulfur, surh as elemental sulfur or chalcopyrite to form a copper sulfide precipitate. The
aqueous suspension of coppler sulfide and leach residue is removed from the reaction vessel to
10 a thickening stage. In this stage the copper sulfide and leach residue are settled to form a
thickened suspension, part of which is recycled tO the reac~ion vessel. A portion of this
thickened suspension is removed, optionally filtered, and routed for further processing, e.g.,
through a copper smelter. The clear solution produced from the thickening stage is optionally
partially recycled to the reaction vessel to utilize sulfuric acid and a portion is removed for
15 further processing, e.g., removal of Bi, Sb, As, Cd and Fe, or to waste water tre~tment
A series of tests substantially embodying these conditions were run to simulate a
continuous plant arrangement as generally described in FIG. 7. As shown in FIG. 8, to
simlllate the precipitation v~ ssel 20 containing four reaction compartments 22, simulated
reactors were used comprising four 2-liter glass beakers 36 each having an effective operating
20 volume of 1.6 liters. Each simulated reactor was equipped with a Rushton turbine 38, baffles
40, reagent sparger 42, a temperature port 44 having a thermometer 46, an inlet 48 for feed
liquid, and an outlet 50. Four such glass beaker reactors 36 were used in series to simul~t~
the precipitation vessel 20 of FIG. 7.
The laboratory system is shown in its assembled arrangement in FIG. 9. Pumps 52
25 were used to provide a conl;inuous flow of reactants, copper sulfate feed liquor, sulfur slurry,
and sulfite solutions. Interstage transfer pumps 54 were also used to transfer slurry from one
glass beaker reactor 36 to the next, and other pumps (not shown) were used to add flocculent
to the final discharge and to recirculate the underflow from the continuous thickener. When
sulfur dioxide gas was used, rotameters (not shown) were provided to monitor the flow of gas.
- 30 To provide control of the acidity conditions in each reactor, a peristaltic pump was used to
add sodium hydroxide to each of the reactors. Not shown in FIG. 9 is the continuous
thickener.
CA 02212378 1997-08-06
W O96/25361 PCTrUS96/01910
In general, the present invention is directed to the following preferred conditions using
a reaction temperature of about 60~C and a solution pH corresponding to a free acid range of
about 0.05 to about 180 gpl (grams per liter) H~SO4, although 10-20 gpl may be preferred.
Approximately 10% excess sulfur may be added, although less may be required.
Test 4, as referenced in Table 1, shows that an excess amount of S07, when supplied
by NaHSO3, is not required for the recycle ratio and acidity conditions employed. However,
increased acidity, or reduced solids density, or shallow reactor vessel configuration may result
in the necessity of adding an excess of SO~, supplied in gaseous form. That is, increased
acidity will tend to decrease the solubility of SO~, reduced solids density will tend to decrease
10 the adsorption of S07, and shallow vessels will tend to decrease SO~ contact time and bubble
dispersion.
Furthermore, increased acidity will tend to oppose the production of copper sulfide
according to the chemistry of the invention thereby requiring greater concentrations of S02 to
compensate. To overcome this potential limitation, SO~ may be introduced into the reaction
15 vessels in a countercurrent manner to that of the copper-bearing solution, with the gas which
exits from the last stage being returned to the second to the last stage and so on sequentially to
the first stage. By this means, the efficiency of SO~ use can be maximized.
Solids precipitation substantially begins in the measured pH range of 2.7 to 3.0 for a
feed liquor containing approximately 20 gpl copper as sulfate, 1 gpl arsenic, 10 gpl iron, and
20 minor quantities of bismuth, cadmium, antimony and zinc. A solution containing the
constituents as described was fed into the glass beaker reactors 36 at a rate of about 40
milliliters per minute (40 ml/min.), equivalent to 48 grams per hour (48 gm/hr) of copper or,
in terms of equivalent copper sulfide, 72 gm/hr. Depending upon the flow conditions, other
reactants, including recycled underflow, could add 15 to 40 ml/min. reactive volume such that
25 the nominal retention time in the system could vary from 80 minutes to 116 minutes.
Elemental sulfur was provided having a particle size in the 10-20 micron range. An
elemental sulfur of the described particle size, designated "Super Fine", may be obtained from
a process licensed to Innochem Engineering Company of Canada, such sulfur being produced
by a process of injecting molten sulfur through a nozzle into a water bath. Alternately,
30 flowers of sulfur, a sulfur product from Geneva Steel, Provo, Utah, or sulfur made from
natural gas may be used. All types of elemental sulfur may require crushing and grinding
before use.
16
CA 02212378 1997-08-06
W O96125361 PC~US96J~1910
Sulfur dioxide sources may include sulfites and bisulfites, sulfur dioxide in a pure state
or a blend of sulfur dioxide, nitrogen and oxygen, such as from smelter gas. Additionally, the
sulfites and bisulfites may be derived from the sodium or calcium salts of sulfites and
bisulfites.
A first series of test~i, numbered 1 through 7, employed sodium bisulfite at variable
precipitate solids recycle raltios. The results of the tests using sodium bisulfate are
s--mm~rized in Table 1. Table 1 represents the various test conditions with sodium bisulfite as
the SO donor which were e mployed to evaluate the process in a continuous operating mode.
Different types of sulfur and varying dosages of the reactants were utilized while employing a
10 moderately high recycle ratio of precipitated solids to the reaction zone. This approach had
been found to be very critical in batch tests in making the reaction proceed at a practical rate
and with a high degree of completion. The continuous system confirmed the observations
made from tlle batch test results.
Notably, in Test 6 shown in Table 1, copper precipitation appeared to be lower due to
15 higher copper contents in thle reactors resulting from an extended down-time of the test
reactors. With SO~ not being added in the final reactor (see footnote 4 to Table 1), the system
was slow to come to equilibrium and the precipitation appeared low. If the test had been
continued at the same conditions beyond the test time of 2.35 hours, equilibrium would have
been reached and the results would be similar to those obtained in Test 7.
CA 02212378 1997-08-06
W O96/2~361 PCTAUS96/01910
C4 ~ ~ r~ ~ ~ ~ ~
E
.~
.~ E
t ~
t~ ' 1~ r
t~ -- _
.
u~ ,
-- -- ~ ~ 'D ~ 'D 'O
t~ ~ c4 -- ~ r~ ~ ~ ~ ~1
c4
a
o ~
a
tY x
E
t~ ~ t
Z - ~C
~ ~ E '~ ~ oo c~ r o~ c
E - E E
~ ~ ~ ~ .E_
E
L L L L r8~
C O O O o O ~ ~
o o Co, ... o
r~ ~ -c c~c ~ o~~ c~c ~~r r~- ~~ c~ co v~
r' C4 -- ~ ~ ~ 1' ~ --J' ~ ~ fi 3~
r ~ ~. _ c
e O g ~~ O. 8 O. ~ ~ _
18
CA 02212378 1997-08-06
WO 9612S361 PCTrU596/019~0
A second series of tests, numbered 8 through 13, employed pure sulfur dioxide gas
under conditions similar to those of the first series of tests. These test results are sl~mm~rized
in Tab~e 2. These tests demonstrate the effectiveness of gaseous S02 in the process, even
under conditions not conducive to efficient gas absorption, the high acidity of the solution and
- S the shallow lilquid depth in the reactors.
These tests were conducted to confirm the acceptability of gaseous S02 as a reductant
without attempts to optimize its utilization. Typically, 100% excess gas gave high
precipitation of copper when sufficient sulfur was present. From Test 10 on, the acid
produced during the reactior,l was neutralized to an extent by the addition of NaOH.
10 Precipitation lwas generally g~ood under these conditions.
It is important to note that the required quantity of sulfur can be provided in ~he
incoming copper solution and as long as it is present in sufficient excess in the final reactor,
the reaction will proceed to a high level of completion if S02 is present. On the other hand,
the addition of S02 in gaseous form has been demonstrated to be necessary to each and every
15 stage to reach maximum completion. That is, the retention of gaseous S02 between stages is
low, especially in higher acidity solutions, such that the reaction is suppressed.
19
CA 02212378 1997-08-06
W O 96/2S361 PCT~US96/01910
'' ~3 ~ G v~ ~ 50 ~ o ~,,
~1 L~
~ E ~ ~ ~,, ~
~ V' ~'-- ~ o
~e ~
g
.S o 'D ~ O ~ ~ ~
~ U~
~
E ~ ~ ~ ~ o o
L~
L~ O V~ o ~D
LU _ ~
' ~~
C ~ ~-- ~ O, ~ O o
L~ ~ 3 :- O
o2 2 2 2 2 2 e~ ~ ~ 2
3 o ~ ~ ~
E , ~ .
C~ L L ~ E L. ,, ~ ~ .o,
~ o
o ! w ~
~" o E _ E,- ~ E ~~
~_ C 0
C , o ~ _ . Z V~
- ~ ~ ~ ~ ~ ~ 8 ~ , ~ o
o x ~ ~ ~ ~ ~ _ _ ~ ~:
CA 02212378 1997-08-06
WO 96/2S361 PCT/~S96~019~0
The third series of tests confirmed the applicability of a simulated feed to a smelter
acid plant, such as may apply from a copper flash smelting process. It was shown that despite
the presence of a partial pressure of 5% oxygen in the simulated gas, there was little or no
interference with the reductiive power of the S02.
- 5 In adclition, the level of excess S02 was decreased to as low as 54%, and high
precipitation efficiencies were confirmed. As previously described, it is not considered that
this represents a lower limit to the S02 requirement since several factors are involved in its
utilization, not least of which is the shallowness of the test reactors.
This t;hird series of tests, numbered 14 through 17, utilized sulfur dioxide derived from
a simulated flash smelter ga's containing 35% S02, 60% N2 and 5% 02. In such a flash
smelting process, the smelting operation is continuous and the off-gas which passes to an acid
plant contains a high level of sulfur dioxide with a relatively low oxygen content. The gas
composition is also fairly constant due to the continuous nature of the process. Other
continuous processes like the Noranda, Mitsubishi, and Isasmelt Processes produce high
strength off-gases that may prove acceptable as a source of S02 reductant. The recycle ratios
for precipitated solids were varied from O to as high as 23, with the reaction times decreasing
proportionately. These results are summarized in Table 3. The similarity in tllese results and
those with pure sulfur dioxide (Table 2) indicates that this approach to copper precipitation
would be very practical in that a process gas stream containing sulfur dioxide gas, could be
employed jUSl as effectively as a purified (and more costly) reactant, such as liquefied S02 or
sulfites/bisulfites .
CA 022l2378 l997-08-06
W O96/25361 PCTrUS96/01910
,~ _ ~ ~D ~' =
~ ~ ~ ~~ 1-
t
.0 O. ~~ ~ O,
4 X ~ ~ ~~
O ,~
l V~
E ~ ~ ~ ~
.C
~ C~ ~ C~ X o
~ 2 ~ .c
o ~v; 3 3 ~ E
V~C V~ V7 , c o ~
~ E~,
~ C C~ t -- C
~ ~
u C~ ~ ~ ~ ~ e , ~ ~,
~~~ ~ ~ ~ ~ ~ XE~ ~ ~
~ Z
-- _ ~
22
CA 02212378 1997-08-06
WO 96125361 PCT/IJS96JI)191D
The final test in this third series, Test 17, was designed to verify the results when using
no recycled solids, which in previous studies had shown greatly reduced precipitation rates. It
was assumed that there could be some precipitation of copper sulfide, and at best, this might
be 25 ~ of the total quantity in each reactor, such that the last reactor would contain
S approximately 30 gpl sulfide solids if all the copper had precipitated. The reactors were
prepared prior to the beginning of the tests so that these possible equilibrium conditions would
exist initially by dosing with precipitated solids as if this degree of precipitation had occurred.
Caustic soda was then added during the run at the dose required to maintain constant acidity
assuming the precipitation would occur at a constant rate.
The results were initially unexpected, when compared to the original batch test work,
in that the fin;al eMuent from the system was almost as good as that with a higher seed
precipitated solids concentration. The initial precipitation rate was very low, as expected, but
in the second and third reaCt~DrS the rate accelerated greatly, actually exceeding the rates
observed in the other tests. Upon consideration, however, it appeared that the reaction rate
15 was due to the reduced acidity since caustic soda had been added earlier before any acid would
have been generated, thereby lowering the acid level to about 1 to 2 gpl by the time the
precipi~ation rate began to accelerate. Based on the earlier batch test, it is reasonable to
assume that, if seed solids had not been added nor had sodium hydroxide been added to
neutralize an assumed generation of acid, there would have been very little precipitaLtion
20 occurring and the final copper concentration in the liquor proceeding to the thickener would
have been close to that in the feed liquor.
A correlation between rate and solids concentration is best seen in FIG. 10 showing the
copper concentrations in each of the reactors for the four runs, as well as the calculated
in~t~nt~neous concentration a.s the feed liquor and recycled underflow entered the first reactor.
25 The samples ~were taken from each reactor and, therefore would represent the concentration
of copper leaving that reactor stage and entering the next reactor. These data are used to
calculate the rleaction rates shown in Table 4.
CA 02212378 1997-08-06
W O96/2S361 PCTrUS96/01910
,.,
~~_
c ~
~ I~ ~ ~<~
' U
O O OO O O O O O O O O O O
U ~ O ~ ~ ~ O OC X X
O
-- - .0
O~
O
011 ~ ~ o 00 00 0
O C~
24
CA 02212378 1997-08-06
WO 961~5361 PCTJUS96J01910
A good separation of copper from the other constituents of the solution is possible,
provided the electromotive force (EMF) does not drop too low. This is best illustrated in
FIGS. 5 and 6, representing the analytical results from Tests 14 and 16. In Test 14, where the
copper sulfide concentration was 490 gpl, the reaction was complete after only 40 minutes of
5 retention time, or as the slurry left the second stage reactor. It will be noted that bismuth
began to precipitate in the second reactor and was completely precipitated in the third reactor.
Antimony blegan to precipitate in the third reactor and it was almost completely removed from
the solution leaving the fourth reactor. The EMF had dropped below 200 millivolts in the
third stage and had dropped to 65 in the fourth. It had been indicated that the EMF remains
around 250-300 millivolts during most of the precipitation reaction and then drops down to
close to 200 millivolts as most of the copper is depleted. This provides a good control method
for a continulous circuit, modulating the rate of liquor or sulfur dioxide addition in order to
m~int~in the final EMF around 200 millivolts. It should be noted that these EMF measure-
ments were laken on solutions that had been filtered and cooled, in order to obtain values that
could be compared.
Test 16, which would represent a desirable operating level in terms of good separation
of copper from the other elements, illustrates these preferred conditions. The EMF in this
case was slightly lower than that noted in the previous test, although this may have been due to
a difference in the sulfur dioxide concentration in the different samples.
To determine if the system would be effective at higher acidities, Tests 18, 19 and 20,
as shown in 'Iable 4, were carried out on a batch basis, but with the simulated smelter gas as
the source of sulfur dioxide. Since the process solution could be present at a higher
temperature in a flue dust leach process, these tests were carried out at 80~C which would be
anticipated to increase the reaction rate. The amount of precipitate solids present in the
reactor corresponded to that which would exist with the recycle ratio of 23. In Tests 18 and
19, the acidity was allowed to increase due to the reaction, with the average acidity used in
- evaluating the tests. In Test 20, the acidity was controlled at a constant level averaging 37 gpl
by adding sodium hydroxide during the course of the test. These results are represented in
FIG. 13 which shows the copper concentration as a function of precipitation time. When the
acidity is held constant, as in Test 20, the precipitation rate is virtually constant, and the
reaction is complete in about 40 minutes. Precipitation rates calculated from these data are
CA 02212378 1997-08-06
W O96/25361 PCTrUS96/01910
also shown in Table 4. It should be noted from Figure 5 that the major impurities, iron and
arsenic, rem~ine~l in solution while all of the copper was precipitated. Of the minor
impurities, zinc and cadmium also remained in solution. Only bismuth and antimony appear
to be all or partially precipitated, and this begins after approximately 80% of the copper has
5 been precipitated.
The material produced in the inventive process was more easily thickened and filtered
than that, for example, resulting from the use of hydrogen sulfide precipitant. A settling test
and several filtration tests were carried out on the material produced during the continuous
precipitation study, with the test conditions and results shown in Tables 5 and 6, and FIGS. 8
10 through 12.
TABLE 5
Flocculent:PERCOL 351 from Allied Colloids
Concentratioll (gpl): 1.00
Volunle Added (ml): 2.00
Dosage (kilogran1s per n1etric toll): 0.0049
Vol (n11)Weight (g) Tare (g)
Un-:lec ~ntcd Slurry: 2000.00 2996.00 489.00
Decanted Slurry: 280.00 1114.60
Dry Solids: 407.50 0.00 .
Settling Vessel Size (n1illiliters per cpn~ lptpr):47.05
Ultin1ate Interface Height (n1illiliter per meter):886.00
Sp. Grav. Supernatant: 1.0940
Sp. Grav. Solids: 5.0500
Deptl~ Correction: AUTOMATED
Initial Final Ultimate
Miniml-m Desired Underflow Percent Solids:40.00 16.25 64.57 66.30
UF Concentration (kilogran1s per liter):0.05 0.02 0.12 0.13
Tm(min) Ht(ml)Tm(min) Ht(ml)
0.00 2000.09.00 310.0
0.50 1105.010.00 302.0
1.00 630.012.00 295.0
2.00 470.015.00 290.0
3.00 430.020.00 285.0
4.00 395.00.00 0.0
5.00 370.00.00 0.0
6.00 350.00.00 0.0
7.00 335.00.00 0.0
8.00 322.00.00 0.0
26
CA 02212378 1997-08-06
WO 96/25361 PC'rJUS96JO~911
Underflow Underflow Test Results Scale-Up
Weight Copper UnitArea, Unit Area,
Percent (Kilograms Square Meter Square Meter Per
Solids Per Liter) Per Metric Metric Ton Per Day
Ton Per Day (Depth-Corrected)
64.57 0. L194 0.1581 0.0395
64.00 O. L173 0.1248 0.0319
63.00 0. :L 136 0.0915 0.0235
62.00 0. L 101 0.0716 0.0192
61.00 0. :l067 0.0652 0.0170
60.00 0.1034 0.0551 0.0155
59.00 0. L002 0.0473 0.0146
S8.00 0.()971 0.0453 0.0143
57.00 0.()941 0.0437 0.0142
~6.00 0.()911 0.0394 0.0142
55.00 0.()883 0.0355 0.0142
54.00 0.()855 0.0320 0.0141
53.00 0.()828 0.0288 0.0138
52.00 0.()802 0.0259 0.0135
51.00 0.()776 0.0232 0.0131
50.00 0.()751 0.0207 0.0123
d,9.00 0.()727 0.0186 0.0117
TABLE 6
Material: COPPER SULPIDE PRI,CIPITATE
Filter Cloth: PCIPR 929
Feed Tot Solids (~Iveight percent): 66.000 Liquid Sp. Gr: 1.0900
Feed Diss Solids (weight percen~): 0.000 Slurry Sp. Gr: 2.2600
Calcd Feed Susp Slds (weight pcrcent): 66.000 Calcd Solid Sp. Gr.: 5.0555
Filter Aren (squnre meter): 0.09 Barometric pressure (- - Hg): 65.354
Slurry pH: 2.0 Filtrate Susp Solids ( ' = per liter): N/A
Slurry Temp (Decree C): 50.0 Air Leal;age (squnre meter per minule): N/A
Slurry Feed Technique:TOP Air Flow Meter: GAS METER
Test No. 1 2 3 4
Form Vncuum (c - Hg) 38.100 38.100 38.100 38.100
Wnsh Vacuum (~ - Hg) 38.100 38.100 38.100 38.100
Dly Vacuum (c - - Hg) 38.100 38.100 38.100 38.100
Form rm1e (seconds) 2.000 3.000 4.000 2.000
Wash rlme (seconds) 8.000 10.000 12.000 8.000
Dly rzme (seconds) 60.000 45.000 30.000 45.000
Air Reading rdef (cubic me~ers) 0.000 0.000 0.000 0.000
Air Reading Afl (cobic meters) 0.000 0.000 0.000 0.000
Filter Vol (incl ~. b)' -- ' ) 160.000169.000 168.000 220.000
Wnsh Volume (milliliter) 100.000100.000 100.000 100.000
Cnlce Thickness (I ~--- - ) 13.000 15.000 17.000 22.000
Cnke Tnre Weight (gram) 7.170 7.100 7.220 7.180
27
CA 02212378 1997-08-06
W O96/25361 PCTrUS96/01910
C~ke Tot~l Wet Weigh~ (grum)226.480 268.200312.690 432.160
Cllke P~ l Wet Weight (gr~m)0.0000.000 0.000 0.000
C~ke Pnrlid Dry Weight (gr~m)191.810 226.580260.780 382.260
Net Dly C~ke Wt (gr~m)18$.640219.480 253.560375.080
C~kc Moisture (weight percent)15.809 15.940 16.993 11.742
Air Flow (cubic meters per minute0.000 0.000 0.000 0.000
per squ~re meter)
Bk C~lc ~:eed S.S. (weight percent) 64.852 65.261 66.798 67.487
C-ke Londing (Kilogr~ms Per Meters0.199 0.236 0.273 0.403
Squnred)
Moist F~ctor ( - ~ .G meter5.0393.1751.844 1.864
per kilogram)
Moist F~clor 2 (grAph 4) 0.0000.000 0.000 0.000
W~sh 1:~ . ' 2.8842.403 1.9262.004
WVw (l;ilogr~ms per square meler)23.8328.34 32.75 48.44
(lilers per squllre meler)
Grnph Symbol STAR STAR STAR PLUS
TIIICKENER CYCLONE
UNDER~LOW UNDI~RI~LOW
It should be noted that the quantity of recycled precipitate solids appears to be less
important in m~int~ining a high reaction rate if the measured pH is maintained close to 2.
However, high recycle ratios that may be used to maintain a productive reaction rate
S incidentally will also produce a substantially coarser particle that will yield lower cake
moisture and give higher filtration rates on the product slurry. If the system is to be used
following a pH reduction stage (e.g., a bismuth precipitation stage), then it may be
advantageous to add caustic soda or other suitable alkaline material to maintain the pH
constant since this will maximize the precipitation rate. However, if this reaction were to be
10 employed directly after a metals solubilization stage, it may be preferable to allow the acidity
to increase during the precipitation reaction so that all, or a portion, of the resulting effluent
could be recycled back to the leaching process as the lixiviant. This is especially true in the
case of leaching ores and concentrates where the acid generated can be usefully employed in
meeting the natural acid demand of the solids being leached.
As an alternative to the use of elemental sulfur, chalcopyrite (CuFeS2) may be used. A
test was run under the conditions previously described using chalcopyrite as a substitute. The
chalcopyrite used in the test was obtained from sources located at Bingham Canyon, Utah.
Test 21, shown in FIG. 19, illustrates the test results. A sample of 250 grams of chalcopyrite
was rod-milled for about 30 minutes to reduce the average particle size, and this was
20 employed in the test. The reaction rate curve as shown in Test 21 was somewhat different
from the results using sulfur, since there appeared to be a lag time before the reaction
CA 02212378 1997-08-06
W O96/25361 PCT~US96101910
commenced, although subsequently the rate was relatively high. The test was also conducted
at 80~C.
A thick~ning test W2LS run on the slurry produced, the results being shown in FIGS.
14(a) and (b). Notably, the upper line in FIG. 20(b) represents the settling rate values
calculated from the settling test data illustrated in FIG. 20(a), while the lower line represents a
scale-up application to a full-sized thickener of the values calculated in the upper line. The
underflow from this test was filtered at a rate/ of about 2.53 kilograms per hour per square
meter (60 pounds per hour per square foot) yielding a filter cake having a solids concentration
of 74.6%.
An alternative approach to grinding chalcopyrite for use in the precipitation process
described above is to process an amount of chalcopyrite through a cyclone of a diameter
suitable to remove only the fine particles in the slurry--for example, the particles sized at
equal to or less than 15 microns. This material may be thickened and added to the reactors.
To s~lmm~rize, the test work indicated that a retention time of about 80 minutes in a
continuous circuit with a recycled solids concentration in the reactors of 300-400 gpl copper
sulfide solids would yield almost 100% recovery of the copper and at the same time provide a
good separation from the innpurities, arsenic, iron, and other minor elements. The tests show
that all the sulfur and sulfur dioxide sources tested are equally applicable. Sulfur dioxide at
the concentration present in the smelter gas reacted as rapidly as did pure sulfur dioxide gas.
An excess dose of 50-100% sulfur dioxide was required due in part to the shallowness of the
test reactors and, in part, to varying acidity of the solutions. Approximately 10% excess
elemental sulfur is sufficient to ensure reaction completion.
The solution acidity was found to be a major factor in controlling the rate of reaction.
During precipitation, approximately 3 gpl H~SO4 was produced for every gram per liter of
copper that was precipitated using sulfur dioxide. The reaction rate in a solution at 60~C and
an initial pH of about 2 will decrease from approximately 10 kilograms copper per cubic
meter per hour to about half that rate as the reaction progresses. To compensate for this,
caustic soda or other suitable alkali can be added to maintain acidity at a constant level to yield
a rela~ively c:onstant precipitation rate. It should be noted that the use of NaHSO3 to provide
the necess~ry SO~ for the reaction, also results in a simultaneous neutralization of the acidic
29
-
CA 022l2378 l997-08-06
W O96/2S361 PCTrUS96/01910
solution to help m~int~in acidity at a constant level. Sodium bisulfite can be obtained
commercially or by reaction of SO~ with caustic soda.
A soluble neutralization product, such as is obtained by use of caustic soda or sbdium
bisulfite, is preferred to prevent the dilution of the copper sulfide by insoluble products of
5 neutralization. Under these conditions, the effect of recycled solids concentration on the
reaction rate is less pronounced, and the reaction will go to completion at concentrations as
low as 20-40 gpl copper sulfide. A high concentration, in the range of 500 gpl copper sulfide,
effectively doubles the reaction rate. Higher concentrations also produce a precipitate with a
subst~nti~lly coarser particle size.
To simulate the use of the process on liquor at a much higher acidity, tests were
conducted at 80~C and with a recycled solids concentration of 500 gpl. The average acidity
varied from approximately 125 gpl to 40 gpl of H~SO4 acid resulting in the reaction rate
increasing from 5 to 30 kilograms copper per cubic meter per hour. This concentration of
solids requires a recycle ratio of about 20 times the amount of solids being precipitated on
each pass.
To determine the efficacy of the present invention at high acidity levels such as those
likely to be encountered in electrolyte, for example in copper tankhouse circuits, Tests 22 and
23 were conducted. In these tests the acidity of a solution containing a final 20 grams per liter
copper as copper sulfate was adjusted to 180 grams per liter sulfuric acid prior to reaction at
80~C.
In both tests, an excess of elemental sulfur was added just before commencement of the
batch reaction. Sodium bisulfte was added as a 30% solution as a sulfur dioxide donor, with
sulfuric acid being added to compensate for the dilution and neutralizing effects of this
addition. By this means, the sulfuric acid level throughout the two tests was m~int~inPd
around the 180 grams per liter level. Test 22 had no solids apart from the elemental sulfur
added to the solution whereas Test 23 had 500 grams per liter of copper sulfide precipitate
added to the solution.
Results from the two tests are shown on FIG. 21. In Test 22 with no added coppersulfide, there was only a small decrease in the copper concentration of the solution over a
period of 4 hours. In Test 23 there was almost a complete precipitation of copper in 2 hours
demonstrating the benefits of the present invention.
CA 02212378 1997-08-06
WO 96125361 PCT/U596/01910
The precipitate resulling from the process was easily thickened and filtered, with a
thickener unit area in the range of 0.015 square meter per metric ton per day (0.15 square feet
per ton per day) and filtration rates varying from 5.06 to 12.64 kilograms per hour per square
meter ~120 to 300 pounds per hour per square feet). Filter cake moistures ranged from 25%
5 to as low as 12% when a sinnulated cyclone underflow was dewatered.
These high acidity tests show that not only do iron, cadmium, arsenic and zinc
remained in solution while copper sulfide is precipitated, but that Bi and Sb actually increased
in soluble concentration (as a result of being leached from the seed material used in these
experiments). This illustrates that increasing the concentration of the acidity of the copper
10 precipitating ~solution facilitates the selective precipitation of copper (which is particularly
useful when operating with materials that contain many other metals, e.g., APB).In a mlethod as descriibed, the preferred temperature of the precipitation reaction is
between 60~ ~md 80~C whereas the temperature of the leach solutions may be in the range 5~
to 80~C depending on source. The heat required for the precipitation reaction can be provided
15 by recuperation of the heat c:ontent in the outgoing stream with the incoming stream, or the
sensible heat of the SO2-bearing gas stream or both.