Note: Descriptions are shown in the official language in which they were submitted.
CA 02215930 2006-09-12
1
A PROCESS FOR THE PREPARATION OF ~,NTHRl-CYCL.ZNF AIfTIBIO-
TZrS
The present invention relates to a process for the
preparation of anthracycline antibiotics.
=IELD OF THE INVENTION
Epirubicin (epidoxorub=cin) and epidaunomycin are
antibiotics belonging to the anthracycline class having
antitumoral activity.
These compounds differ from the antitumorals
doxorubicin and daunorubicin in the configuration of the
hydroxy group at the C-4' position of the glycosidic
moiety of the molecule, which configuration is
respectively axial in doxo- and daunorubicin and
equatorial in epirubicin and epidaunorubicin.
Doxorubicin has beez used for a long time in the
antineoplastic treatment, for a review see Arcamone, ed.
"Doxorubicin", Acad. Press, New York 1981. A serious
side-effect of doxorubicin is the onset of often
irreversible myocardiopathies.
Epirubicin was found to have advantageous
pharmacological properties compared with its analogue,
showing an equivalent antitumoral activity but less
side-effects (R. B. Weiss et al., Cancer Chemother.
Pharmacol. 18, 185-97 (1986)).
The starting synthesis of epirubicin involved the
condensation between daunorubicin aglycone of formula
(B)
CA 02215930 2006-09-12
2
HO O 0 0
p (B)
0 O% O
and the 1-chloro-derivative of acosamine protected as
trifluoroacetamide (Arcamone, F. et al., J. Med. Chem.,
18, 7, 703-707, (1975)), subsequent deprotection and
working up of the side chain with a method well known in
literature, already used for the transformation of
daunorubicin into doxorubicin (E. M. Acton, J. Med.
Chem., 17, 65 (1974); DE 1917874).
Trifluoroacetylacosaminyl chloride N,0 derivative
was obtained by synthetic transformation of various
natural sugars with a number of. rather complex,
expensive processes.
Italian Patent 1,163,001 and, subsequently, G.
Bonadonna in "Advances in Anthracycline Chemotherapy
Epirubicin", Masson Ed., Milan, Italy, 1984, disclose a
synthesis process carried out on the whole N-
trifluoroacetyldaunorubicin glycoside. This process
comprises the oxidation of the hydroxy group at C-4' to
keto group, then its stereoselective reduction to
hydroxy group by means of sodium borohydride. The
oxidation reaction has to be performed at exceeding1y
low temperatures (-70'C). The keto derivative is
delicate and unstable. Moreover, the reduction with
sodium borohydride must be carried out at a i.c~
temperature to minimize the competitive reduction of the
aglycone carbonyl group. Although not stated, the
maximum isomerization yields which can deduced from sai:
CA 02215930 1997-09-19
WO 96/29335 PCT/EP96/01174
3
Patent are of about 48%.
Another method for the epimerization of C-4' is
described by B. Barbieri et al. Cancer Research, 47,
4001 (1987). This method aims at obtaining 4'-halogen-
daunorubicin, then effects the epimerization from the
equatorial configuration to the axial one, i.e. in a
direction opposite to that desired in the present
invention. The epimerization reaction is carried out by
nucleophilic substitution of the triflate group
(equatorial) with a tetrabutylammonium halide.
DISCLOSURE OF THE INVENTION
Now a novel method has been found for the
isomerization of the 4' hydroxy group of the daunosamine
residue on the whole anthracycline antibiotic molecule,
in reaction conditions which can be regulated in more
easily, particularly with respect to the reaction
temperature, and with easier purification procedures.
According to this method, the epimerization of the
4' hydroxy group from the axial configuration to the
equatorial one of the daunosamine residue, which is
suitably protected at the amino group, is obtained by
introducing a strong leaving group and subsequently
substituting it with a carboxylate group, with inversion
of the configuration of the 4' carbon atom, subsequently
hydrolysing the carboxylic ester, restoring the hydroxy
group, removing the amino-protective group.
During the study of the epimerization reaction, the
yields in the desired epimer were found to be not very
high, due to a side product formed in competition with
the substitution reaction of the triflate leaving group.
Surprisingly, the protection of the hydroxy groups
CA 02215930 1997-09-19
WO 96/29335 PCT/EP96/01174
4
of the aglycone moiety, particularly that at the 9
position, made it possible to obtain the desired epimer
without competitive formation of the side product.
For the purposes of the process according to the
invention, it is essential that the hydroxy group at the
9 position of the aglycone moiety is protected. The
protection of the hydroxy groups at the 6 and 11
positions can improve the yields.
Therefore, it is an object of the present invention
process for the preparation of antibiotics of the
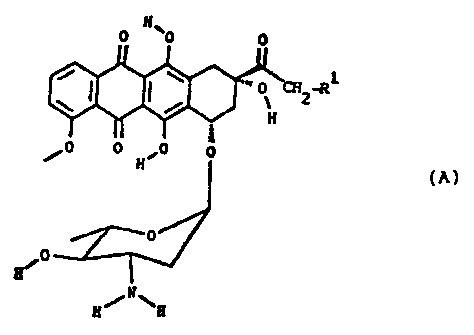
anthracycline class of formula (A)
0 0 0
o ~2~
H
0-le O 0 ~O 0
H
(A)
0
JO
H/N\H
wherein R1 is hydrogen, OH, or OCOR2, in which R2 is a
C1-C4-alkyl group,
which comprises the epimerization of the 4' hydroxy
group by means of nucleophilic substitution of the 4'
triflate group with a carboxylate group, upon protection
of the hydroxy groups of the aglycone moiety of the
molecule.
DETAILED DISCLOSURE OF THE INVENTION
According to a first embodiment of the invention,
the preparation process comprises:
CA 02215930 1997-09-19
WO 96/29335 PCT/EP96/01174
a) reaction of N-protected daunorubicin of formula (I)
O ~'O O
~
1 2
N. 5
0 O H 00 0
(I)
O
a/~ R
3
or a derivative thereof,
wherein RI is hydrogen, halogen, suitably protected
hydroxy; R3 is an amino-protective group;
with triflic acid or a reactive derivative thereof, to
give the compound of formula (II)
O H% 0 O
O . 2-~t
H
R ( I I)
.01 O O - O
0
F %% o x
S,' g~ R
__ *0 3
F
wherein R1 and R3 are as defined above;
b) protection of the hydroxy group at the 9 position,
and optionally those at 6 and 11, to give the
intermediate of formula (III)
CA 02215930 1997-09-19
WO 96/29335 PCT/EP96/01174
6
0 Or 0
1
OT
Je0 0 Or 0
(III) ,
0
F 0
F y0 H~
~F! ti 3
wherein T is a protective group, R1 and R3 are as
defined above;
c) treatment of the compound obtained in step b) with a
salt of a secondary or tertiary amine with a carboxylic
acid of formula RCOOH, wherein R is an aliphatic
residue, optionally substituted or interrupted by
heteroatoms, or an optionally substituted aromatic
residue, to give the ester of formula (IV)
0 kp O
0 -aZ1
1 20 T
/0 A '.0
(IV)
0
R
0 /N-
'
R3
wherein R, R1, R3 and T are as defined above;
d) deprotection of the hydroxy group at the 9 position,
to give the intermediate of formula (V)
CA 02215930 1997-09-19
WO 96/29335 PCT/EP96/01174
7
0 0 0
0 CH 2-R
/p p H~0 0
(V)
R p O
~ ~x
0 H R3
wherein R, R1 and R3 are as defined above;
e) hydrolysis of the ester to give N-protected
epidaunorubicin of formula (VI)
0 H% 0 0
v 0 2~
IH
0 0 H .00 0
(VI)
0
e0
H R
3
wherein R1 and R3 are as defined above;
f) removal of the amino-protective group and, if
desired,
g) transformation into epirubicin or an ester thereof of
= formula (A)
CA 02215930 1997-09-19
WO 96/29335 PCT/EP96/01174
8
0 H 0 O
0 CH2-R
H
0 0 0 =
H
(A)
0
,O
H
RI0, N\H
wherein R1 is OH or OCOR2, in which R2 is as defined
above.
The starting compound is preferably daunorubicin
trifluoroacetamide, or a derivative thereof, known in
the art and anyhow preparable according to known methods
starting from daunorubicin and trifluoroacetic anhydride
(e.g. J. Med. Chem., 18, 7, 703-707, (1975)).
Other amino-protective groups can be used as well,
e.g. carbamate esters such as fluorenylmethoxy carbamate
(Fmoc) and the like.
As the leaving group, the triflate
(trifluoromethylsulfonic) ion is used herein, which is
commercially available and can be used in the form of
the corresponding anhydride. The reaction is carried out
in a suitable solvent which does not affect the
reactants and the final product, such as poorly polar
aprotic solvents, for example dichloromethane,
dicloroethane, chloroform, dioxane, tetrahydrofuran,
benzene.
The reaction is carried out at a low temperature
(about 0 C) for a time sufficient to complete it.
CA 02215930 2006-09-12
9
The resulting intermediate compound of formula (II)
can be used directly in the subsequent step, without
recovering it. After that, in the same reaction vessel
the suitable reagent is added, which provides the
protective group for the hydroxy groups at the 9
position and, if desired, at 6 and 11, of the aglycone
moiet'y. This intermediate (III) can also be directly
used in the subsequent step. Suitable protective groups
in this reaction are known to those skilled in the art.
The trimethylsilyl group is preferred due to its low
commercial cost, but other silyl derivating groups are
equally valid.
In the subsequent step the carboxylate is added in
the form of a salt with a secondary or tertiary amine.
The reaction was found to proceed unsatisfactorily with
ammonium quaternary salts.
Examples of carboxylates are formate, acetate,
isobutyrate, trimethylsilylacetate, p-nitrobenzoate,
haloacetates, such as trifluoroacetate. Any substituent
groups of the aliphatic or aromatic residue or any
heteroatoms in the aliphatic chain of the RCOOH acid
should not inte=fere with the nucleophilic substitution
reaction. A preferred amine is triethylamine. The
reaction is carried out at a temperature ranging from 0'
~0 50'C, preferably at room temperature, for a time
sufficient to complete it. At the end, the product of
formula (IV) is recovered according to conventional
procedures, then it is suitably treated with a
conventional procedure to deprotect the hydroxy group at
9, thereby obtaining the compound of formula (V).
The hydrolysis of the ester is carried out in
CA 02215930 1997-09-19
WO 96/29335 PCT/EP96/01174
alkaline conditions as it is well known to those skilled
in the art, for example dissolving compound (V) in a
polar solvent, such as methanol, and treating it with an
alkali or alkaline-earth metal hydroxide, such as sodium
5 hydroxide. N-Protected epidaunorubicin (VI) is thereby
obtained and after hydrolysis of the protective group on
the nitrogen, the epidaunorubicin (A; R1=H) is obtained
which, if desired, is transformed into epirubicin or an
ester thereof (A; R1=0H or OCOR2) with processes known
10 in the art (e.g. A. Suarato et al. Carbohydrate Res.,
98, cl-c3 (1981)).
According to another embodiment of the present
invention, epirubicin is prepared starting from
doxorubicin of formula (C),
0 O O
0 CH2OH
t
H
/0 0 0 0
(C)
0
H'0 H'Ie N
said process comprising:
a) reaction of doxorubicin with a suitable protective
agent for the hydroxy groups at 9 and 14, and subsequent
protection of the 3' amine, to give the compound of
formula (VII)
CA 02215930 1997-09-19
WO 96/29335 PCT/EP96/01174
11
0 0 0
"0
0
/0 0 Hlo0 0 M2
(VII)
0
~ R
3
wherein M1 is a C1-C4-alkoxy group and M2 is hydrogen or
a C1-C4-alkyl group;
b) reaction of the intermediate (VII) with triflic acid
or a reactive derivative thereof, to give the compound
of formula (VIII)
0 0 O
coo
- x
14,
/0 0 H'0 0 z ( VI I I)
0
j
F ~%
Fs0!0 H/NR 3
E
wherein M1, M2 and R3 are as defined above;
c) protection of the hydroxy groups at 6, 11, to give
the intermediate of formula (IX)
CA 02215930 2006-09-12
12
0 OT O
0, _0
.01 0 0 OT 0 "2 ( I X)
0
S
F~,S-O N\R
F b 3
wherein T is a protective group, M1, M2 and R3 are as
defined above;
d) treatment of the compound obtained in step c) with a
salt with a secondary or tertiary amine with a
carboxylic acid of formula RCOOH, wherein R is an
aliphatic residue optionally substituted or interrupted
by heteroatoms; an optionally substituted aromatic
residue, to give the ester of formula (X)
O O
o
0 0 0 0 Ml N2
(X)
0
R 0
0 g~ R3
wherein R, R3, M1 and M2 are as defined above;
e) hydrolysis of the ester and subsequent removal of the
protective group of the hfdroxyl groups at 9 and 14 and
of the amino group.
The starting compound is prepared conventionally. A
CA 02215930 2006-09-12
13
preferred example is the reaction of doxorubicin with
triethyl orthoformate (M1=C2H50, M2=H) (see Arcamone,
ibid. p. 21). The subsequent steps are carried out as
described for the first embodiment of the invention. In
particular, step d) is effected at a temperature ranging
from 0' to 50'C, preferably at room temperature. The
protecting groups are removed according to well known
methods.
The following examples further illustrate the
invention.
Example 1
A mixture of a compound of formula (I)(5 g) in
dichloromethane (500 ml) and pyridine (2.5 ml), cooled at 0 C,
is slowly added with a solution of trifluoromethanesulfonic
anhydride (2.5 ml) in dichloromethane (125 ml). The mixture is
left to react for about 1 hour to form the triflate (compound
of formula (II) ) .
N,0-bis-trimethyls.ilylacetamide (10 ml) is added,
heating at room temperature and stirring for 4 hours.
The tris silylate (compound of formula (III), T = trimethylsilyl is
added with a 0:1 molar solution of triethylamine
isobutyrate in diczloromethane (500 ml) and stirring at
room temperature for a further 15 hours. The reaction
mixture is washed with 500 ml of 0.25 N hydrochloric
acid, then with a 2% solution of sodium bicarbonate and
finally with 500 ml x 2 of water. The organic phase is
dried over sodium sulfate, filtered and evaporated to
dryness.
5 g of compound of formula (IVc)(i.e. formula (IV)
wherein R= (CH3) 2CH, T = Si (CH3) 3r R3 = CF3CO) are obtained
1H-NMR (CDC13): ppm: 13.92 (s, 1H, phenol OH); 13.40 (s,
CA 02215930 2006-09-12
14
1H, phenol OH) ; 8. 05 (d, 1H, H-1) ; 7. 80 (t, 1H, H-2) ; 7. 40 (d,
1H, H-3) 6.55 (d, 1H, CONH); 5.37 (d, 1H, H-1'); 5.05 (m, 1H,
H-7); 4.65 (t, 1H, H-4'); 4.55-4.40 (m, 2H, H-3' and H-5');
4.08 (s, 3H, OCH3); 3.25 (q, 2H, 10-CHZ); 2.65-2.55 (m, 1H,
CHCOO); 2.45-2.20 (m, 2H, 8-CH2); 2.37 (s, 3H, COCH3); 2.10-
1.70 (m, 2H, 2' -CHZ) ; 1.30-1.10 (m, 9H, 5' -CH3 and (CH3) Z) ;
0. 15 (s, 9H, Si (CH3) 3)
Example 2
Following the procedure described in Example 1, using
triethylamine acetate, 5 g of compound of formula (IVa)(i.e.
formula (IV) wherein R=CH3, T = Si (CH3) 3r R3 = CF3CO) are obtained.
Example 3
Following the procedure described in Example 1, using
diethylamine formate, 5.1 g of compound of formula (IVd)(i.e.
formula (IV) wherein R=H, T = Si(CH3)3, R3 = CF3CO) are obtained.
Example 4
Following the procedure described in Example 1, using
triethylamine p-nitrobenzoate, 4.9 g of compound of formula (IVe)
(i.e. formula (IV) wherein R=p-02NC6H4r T = Si(CH3)3, R3 = CF3CO)
are obtained.
Example 5
Following procedure described in Example 1, using
triethylamine trimethylsilyl acetate, 5.1 g of compound of
formula (IVb) (i.e. formula (IV) wherein R=(CH3)3 SiCH2, T
Si (CH3) 3r R3 = CF3CO) are obtained.
Example 6
A solution of compound of formula (IVc) (5 g) in
dichloromethane (1000 ml) is added with a 48% aqueous solution
of potassium fluoride (20 ml) and 1 g of triethylamine
acetate. The mixture is stirred at room temperature for 2
days, then the phases are separated. The organic phase, after
drying over sodium sulfate, is evaporated to dryness.
The residue is purified through a column (silica gel:
eluent dichloromethane-acetone 95:5) to yield 4.5 g of
compound of formula (Vc) (i.e. formula (V) wherein R
(CH3) 2CH) .
CA 02215930 2006-09-12
1H-NMR (CDC13) : ppm: 14.00 (s, 1H, phenol OH); 13.28 (s, 1H,
phenol OH); 8.03 (d, 1H, H-1) ; 7.79 (t, 1H, H-2); 7.40 (d, 1H,
H-3) 6. 68 (d, 1H, CONH) ; 5.53 (d, 1H, H-1' ); 5.30 (m, 1H, H-7) ;
4.65 (t, 1H, H-4'); 4.40-4.10(m, 2H, H-3' and H-5'); 4.10 (s,
3H, OCH3); 3.35-2.85(m, 2H, 10-CH2); 2.65-2.55 (m, 1H, CHCOO);
2.45 (s, 3H, COCH3); 2.45-1.70 (m, 4H, 8-CH2and 2'-CH2); 1.27
(d,3H, 5'-CH3) ; 1.15 (t, 6H, (CH3) 3) .
Example 7
Starting from a compound of formula (IVa) (5 g), with the
same procedure as in Example 6, 4.2 g of compound of formula
(Va) (i.e. formula (V) wherein R = CH3r R3=CF3C0) are obtained.
Example 8
Starting from a compound of formula (IVd) (5.1 g), with
the same procedure as in Example 6, 4.4 g of compound of
formula (Vd) (i.e. formula (V) wherein R = H, R3=CF3C0) are
obtained.
Example 9
Starting from a compound of formula (IVe) (4.9 g), with
the same procedure as in Example 6, 4.3 g of compound of
formula (Ve) (i.e. formula (V) wherein R = p-02NC6H4r R3=CF3CO)
are obtained.
Example 10
Starting from a compound of formula (IVb) (5.1 g), with
the same procedure as in Example 6, 3.9 g of compound of
formula (Va) (i.e. formula (V) wherein R = CH3r R3=CF3CO) are
obtained.
Example 11
A solution of compound of formula (Vc) (4.5 g) in
methanol (270 ml) is treated with 0.3m1 of a 10% solution of
sodium hydroxide for 2 hours. The mixture is neutralized with
0.1 ml of acetic acid and evaporated to dryness. The residue
is dissolved in 20 ml of dichloromethane and 0.5 ml of water.
The solution is left to crystallize for 10 hours, to
obtain 3 g of compound of formula (VI) (Rl = H, R3=CF3C0) .
'H-NMR (CDC13) : ppm: 14.00 (s, 1H, phenol OH); 13.25 (s, 1H,
phenol OH).; 8.05 (d, 1H, H-1) ; 7.80 (t, 1H, H-2) ; 7.40 (d, 1H,
CA 02215930 2006-09-12
16
H-3) 6.55 (d, 1H, CONH); 5.55 (d, 1H, H-1'); 5.25
(m, 1H, H-7); 4.10 (s, 3H, OCH3); 4.10-3.85 (m, 3H, H-3', H-4'
and H-5' ) ; 3. 35-2. 85 (dd, 2H, 10-CH2) ; 3. 30 (s, 1H, 9-OH) ; 2. 40
(s, 3H, COCH3) ; 2. 40-1. 75 (m, 4H, 8-CH2 and 2' -CH2) ; 1.40 (d,
3H, 5' -CH3) .
Example 12
Starting from a compound of formula (Va) (4.2 g), with
the same procedure as described in Example 11, 3.5 g of
compound of formula (VI) are obtained (Rl = H, R3=CF3CO) .
Example 13
Starting from a compound of formula (Vd) (4.4 g), with
the same procedure as described in Example 11, 3.9 g of
compound of formula (VI) are obtained (R1 = H, R3=CF3C0).
Example 14
Starting from a compound of formula (Ve) (4.3 g), with
the same procedure as described in Example 11, 2.9 g of
compound of formula (VI) are obtained (R1 = H, R3=CF3C0) .
Example 15
A mixture of doxorubicin hydrochloride (1.6 g)
in dimethylformamide (32 ml) is added with triethyl
CA 02215930 2006-09-12
17
orthoformate (8 ml) and trifluoroacetic acid (0.8 ml).
The resulting solution is stirred at room temperature
for 3 hours, then diluted with dichloromethane (60 ml)
and added with N-methyl-morpholine (2.5 ml). After
cooling at 0'C, a solution of trifluoroacetic anhydride
(0.8 ml) in dichloromethane (6 ml) is added. The mixture
is reacted for 3 hours at 0'C, then 3 g of sodium
bicarbonate and 30 ml of methanol are added. After about
20 minutes, the reaction mixture is washed with 50 ml of
water, 50 ml of 0.25 N hydrochloric acid and 50 ml of
water. The organic phase is dried over sodium s=slfate
and evaporated to dryness.
The residue (compound of formula (VII), M1 = C2H50, M2 = H) is
dissolved in 100 ml of dichloromethane and 5 ml of
pyridine, cooled at 0'C and added drop by drop with a
solution of trifluoromethanesulfonic anhydride (0.5 ml)
in dichioromethane (20 ml). The mixture is reacted at
0'C t:or about 1 hour to obtain the triflate (compound
of formula (VIII) M1=CZH50, Ml = H, R3=CF3CO) .
2 ml of bis-trimethylsilylacetamide are added,
warming at room temperature for 4 hours to obtain the
bis silylate (compound IX, M1 = C2H50, M1 = H, T =
Si(CH3)31 R3=CF3CO), then a 1M solution of triethylamine
formate in dichloromethane (100 ml) is added. The
mixture is reacted for 15 hours to obtain compound of formula (X)
( R=si , M1=C2H50, M.)=H ) .
10 ml of a 48% solution of potassium fluoride and
20 ml of methanol are added, stirring for 2 days. The
organic phase is washed with 100 ml of 0.5 N
hydrochloric acid, then with 100 ml of a 3% solution of
sodium bicarbonate and finally with 100 ml of water. The
CA 02215930 2006-09-12
18
organic phase is dried over sodium sulfate and
evaporated to dryness. The residue is treated with 250
ml of a 0.1 M NaOH aqueous solution at 5'C for 3 hours,
then the product is extracted with 4 x 250 ml of CHC13,
the combined organic phases are dried over Na2SO4 and
evaporated to dryness.
The residue is dissolved in 100 ml of methanol and
adjusted to pH 2 with hydrochloric acid.
The hydrolysis of the orthoester is complete in
about 30 minutes.
The mixture is evaporated to dryness under vacuum
at room temperature and the residue is ground with
diisopropyl ether, to obtain 0.5 g of crude epirubicin
hydrochloride.
The purification is carried out according to
Italian Patent n. 1,237,202, to yield pure epirubicin
hydrochloride.
1H-NMR (DMSO): ppm: 14..00 (s, 1H, phenol OH); 13.25 (s,
1H, phenol OH); 8.20-8.00 (broad, s, 3H, NH3+); 7.90 (m,
'2H, H-1 and H-3); 7.65 (m, 1H, H-2) 5.80 (d, 1H, 4' OH);
5.55 (s, 1H, 9-OH); 5.30 (broad, s, 1H, H-1' ); 4.95 (m,
2H, H-7 and 14-OH); 4.60 (m, 2H, 14-CH2); 4.00 (broad s,
4H, OCH3 and H-5' ); 3.15 (m, 2H, H-3' and H-4' ); 2.95
(q, 2H, 1OH-CH2); 2.30-1.70 (m, 4H, 8-CH2 and 2'-CH2);
1.25 (d, 3H, 5'-CH3).
Example 16
A mixture of a compound of formula (I) (R'=H) (5 g) in
dichloromethane (500 ml) and pyridine (2.5 ml), cooled
at 0'C, is slowly added with a solution of trifluorome-
thanesulfonic anhydride (2.5 ml) in dichloromethane (125
ml). The mixture is reacted for about 1 hour to obtain
CA 02215930 2006-09-12
19
the triflate (compound of formula (II), R1 = H), then pyridine (6
ml) and triethylsilyl trifluoromethenesulfonate (15m1) are
added, warming at room temperature and stirring for 4
hours.
The tris silylate (compound of formula (III); R1 = H, T
(C2H5)3Si) is added with a 0.1M solution of
triethylamine formate in dichloromethane (500 ml),
stirring at room temperature for a further 15 hours.
The reaction mixture is washed with 500 ml of 0.25
N hydrochloric acid, then with a 2% solution of sodli!m
bicarbonate and finally with 500 ml x 2 of water. Thz
organic phase is dried over sodium sulfate, filtered and
evaporated to dryness.
5.2 g of compound of formula (IVf) (i.e. formula (IV) wherein
Rl = H, R = H, R = Si (CH2CH3) 3, R3=CF3C0, R3 = CF3CO) are obtained.
1H-NMR (CDC13): ppm: 13.96 (s, 1H, phenol OH); 13.39 (s,
1H, phenol OH); 8.14 (s, IH, HCO2); 8.02 (d, iH,-H-i);
7.77 (t, 1H, H-2); 7.36 (d, 1H, H-3); 6.39 (d, jJ(,
CONH); 5.41 (d, 1H, H-1'); 5.09 (m, 1H, H-7); 4.74
1H, H-4'); 4.51-4.31 (m, 2H, H-3' and H-5'); 4.07 (s,
3H, OCH3); 3.43-3:00 (q, 2H, 10-CH2); 2.31-1.64 (m, 2H,
8-CH2 and 2'-CH2-); 2.29 (s, 3H, COCH3) 1.29 (d, 3H, 5'-=
CH8); 0.90 (t, 9H, Si(CH2-Qi3)3; 0.62 (q, 6H,
Si(-CH2CH3)3'
Example 17
Starting from a compound of formula (IVf) (5.2 g), with the
same procedure as in example 6, 4.2 g of compound of formula (Vd)
are obtained.
Example 18
145 mg of n-trifluoroacetyladriamycin 14-valerate (0.2 mM)
(AD32), obtained as described in US Pat. 4,033,566, are dissolved
in 10 ml of anhydrous CH2C12;
CA 02215930 1997-09-19
WO 96/29335 PCT/EP96/01174
the mixture is cooled at 5 C and 48 pl of pyridine and
49 pl of triflic anhydride (0.3 mM) are added. After 2
hour cooling, when the reaction is completed, 49 pl (0.2
mM) of bis-trimethylsilylacetamide are added, heating at
5 room temperature and stirring for 4 hours. The silylated
product is added with 450 pl (3.3 mM) of triethylamine
and 124 pl of formic acid (3.3 mM). The mixture is
stirred at room temperature for a further 15 hours, then
2 ml of methanol and 2 ml of a 48o potassium fluoride
i0 aaueous solution are added. After two days, the phases
are separated and the organic one is extracted with 10
ml of water. The organic phase is evaporated and the
residue is crystallized from pure dichloromethane. The
crystalline epiadriamycine 14-valerate-3-trifluoro-
15 acetamide has m.p. 230 C; NMR (DMSO) and MS in agreement
with the structure.
The recovered product has Rf slightly lower than
the starting compound AD32.
The hydrolysis of the product with a 0.05 M sodium
20 hydroxide aqueous solution for 10 min. at 5 C shows a
partial formation of epirubicin, which is separated by
chromatography and turns out to be the same as that
already described.
Example 19
1 g of daunorubicin hydrochloride in 40 ml of
anhydrous dichloromethane is reacted at room temperature
with 0.4 ml of N-methylmorpholine and 0.6 g of FMOC
chloride. After one hour, 3 ml of methanol are added
leaving at room temperature for 12 hours. The reaction
mixture is washed, extracted with 20 ml of 0.2M HC1 then
with 20 ml of water, then it is dried over sodium
CA 02215930 1997-09-19
WO 96/29335 PCT/EP96/01174
21
sulfate and evaporated crystallizing the residue from
diisopropyl ether. 0.5 g of a first crop are obtained,
having melting point 174-175 C.
150 mg are reacted as in Example 18, but the final
hydrolysis is carried out with 1 ml of dimethylformamide
and 0.15 ml of diethylamine, instead of 0.05M sodium
hydroxide, for about 1 hour at -10 C. The reaction
mixture is precipitated in 20 ml of ethyl ether and the
obtained gum is filtered and suspended in water. 0.1 M
Hydrochloric acid is added q.s. to dissolve the residue
and to adjust pH to 3.7. The solution is purified on
RP18, analogously to what described in Italian Pat.
1,237,202 obtaining pure epidaunorubicin which is the
same as that already described in literature.