Note: Descriptions are shown in the official language in which they were submitted.
CA 022161~1 1997-09-23
W 096130353 PCTrUS96/03844
REVERSIBLE PROTEASE INHIBITORS
FIELD OF THE INVENTION
The invention relates to novel reversible protease inhibitors. The inhibitors are selective for cysteine
proteases.
BACKGROUND OF THE INVENTION
Cysteine or thiol proteases contain a cysteine residue at the active site responsible for proteolysis.
Since cysteine proteases have been illlplicd~:d in a number of dis~ s, including arthritis, muscuiar
dystrophy, i"nd"""dLion, tumor invasion, glomerulonephritis, malaria, and other parasite-borne
infections, methods for selectively and irreversibly inactivating them provide opportunities for new drug
candidates. See, fore)~d",~lc, Esser, R.E. etal., Arthritis & Rheumatism (1994) 37, 236; Meijers,
M.H.M. et a/., Agents Actions (1993), 39 (Special Conference Issue), C219; Machleidt, W. et al,
Fibrinolysis (1992), 6 Suppl. 4, 125; Sloane, B.F. ef al., Biomed. Biochim. Acta (1991), 50, 549; Duffy,
M.J., Clin. Exp. Mei-lsl~si~ (1992), 10, 145; Rosenthal, P.J., Wollish, W.S., Palmer, J.T., Rasnick, D.,
J. Clin. Investigations (1991), 88, 1467; Baricos, W.H. et al, Arch. Biochem. Biophys. (1991), 288, 468;
Thornberry, N.A. et al., Nature (1992), 356, 768.
Low molecular weight inhibitors of cysteine proteases have been described by Rich, P, utei"ase
Inhibitors (Chapter4, "Inhibitors of Cysteine P,uL~i,,ases''), Elsevier Science Publishers (1986). Such
inhibitors include peptide aldehydes, which form hemithioacetals with the cysteine of the p~ul~:ase
active site. See, for instance, Cheng, H., Keitz, P., and Jones, J.B., J. Org. Chem. (1994), 59, 7671.
The disadvantage of aldehydes is their ~ys~ and chemical il ' ~- -5
Aldehydes have been l,dnsrc,r",ed into a,r~-unsaturated esters and sulfones by means of the
Wadsworth-Emmons l lo" ,er mo~iification of the Wittig reaction, shown below (Wadsworth, W.S. and
CA 022161~1 1997-09-23
W 096/30353 PCT~US96/03844
Ll~lllons, W.D. (J. Am. Chem. Soc. (1961), 83, 1733).
O O
~ + EtO - P~_ " EWG b~e ~ EWG
R H EtO
where R = alkyl, aryl, etc.
EWG = COOEt, SO2Me, etc.
a,B-unsaturated esters (Hanzlik et al., J. Med. Chem., 27(6):711-712 (1984), Thompson et al., J. Med.
Chem. 29:104-111 (1986), Liu eta/., J. Med. Chem., 35(6):1067 (1992)) and a,B-unsaturated sulfones
(Thompson et al., supra, Liu et al., supra) were tested as inhibitors of two cysteine proteases, papain
and dipeptidyl amino-peptidase I (also called cathepsin C). However, the inhibition of papain by these
a,B-unsaturated compounds showed poor inhibition, evidenced by second order rate constants from
less than 1 M~1sec~' to less than 70 M~'sec~' for the a,B-unsaturated esters, and from less than 20 M-
'sec~' to less than 60 M~'sec~' for the sulfone.
In addition, this chemistry has not been demonstrated with derivatives of a-amino acids other than
those corresponding to glycine, or in the case of the ester, phenylalanine Thus the chirality of these
compounds is non-existent for the glycine derivatives and unclear for the phenylalanine derivatives.
This is significant since inhibition of an enzyme generally requires a chiral compound
Alpha-amino sulphonic acids were suggested as potential inhibitory compounds, and several were
made, although their inhibitory effects were not reported (Mcllwain et al, J. Chem. Soc. 75 (1941)).
In addition, the Mannich condensation of sulfinic acid, aldehyde, and ethyl carbamate, to form
urethanes has been reported (Engberts et al., Recueil 84:942 (1965).
Additional methods for selectively and irreversibly inhibiting cysteine proteases have relied upon
alkylation by peptide a-fluoromethyl ketones (Rasnick, D., Anal. Biochem. (1985), 149, 416),
di~onl~Lllyl-ketones (Kirschke, H., Shaw, E. Biochem. Biphys. Res. Commun. (1981), 101, 454),
acyloxymethyl ketones (Krantz, A. etal., Biochemistry, (1991), 30, 4678; Krantz, A. etal., U.S. Patent
5,055,451, issued October 8, 1991), and ketosulfonium salts (Walker, B., Shaw, E., Fed. Proc. Fed.
Am. Soc. Exp. Biol., (1985), 44, 1433).
Other families of cysteine pr.,tease inhibitors include epoxysuccinyl peptides, including E-64 and its
analogs (Hanada, K. etal., Agric. Biol. Chem (1978), 4Z, 523; Sumiya, S. etal., Chem. Pharm. Bull.
((1992), 40, 299 Gour-Salin, B.J. etal., J. Med. Chem., (1993), 36, 720), a-dicarbonyl compounds,
CA 022161~1 1997-09-23
W 096/303~3 PCTAUS96103844
3 .
reviewed by Mehdi, S., Bioorganic Chemistry, (1993), 21, 249, and N-peptidyl-O-acyl hyclluxdllldL~:s
(Bromme, D., Neumann, U., Kirschke, H., Demuth, H-U., Biochim. Biophys. Acta, (1993), 1202, 271.
An additional summary of methods for reversibly and irreversibly inhibiting cysteine proL~:ases has
recently been con,, 'ed; see Shaw, E., Advances in Enzymology and Related Areas of Al o'_c
Biology (1990), 63, 271.
.
SUMMARY OF THE INVENTION
An aspect of this invention is a protease inhibitor comprising a targeting group linked through a two
carbon atom chain to an electron withdrawing group, wherein the dissociation constant for inhibition of
the protease with said inhibitor (K,) is no greater than about 100 ,uM.
An additional aspect of this invention is a protease inhibitor comprising a targeting group linked either
directly or through a linker selected from the group consisting of an intermediate carbon atom or a two
carbon atom chain to a sulfone group group, wherein the dissociation constant for inhibition of the
protease with said inhibitor (K,) is no greater than about 100 ,uM.
A further aspect of this invention is a compound, preferably a protease inhibitor, of Formula l:
Rl / Z ~ B \A~ zl S(O~R~
in which: I
nisOto13;
A-B represents a linkage selected from -C(O)NR3-, -CH2NR3-, -C(O)CH2- and -NR3C(O)-, wherein R3
is hydrogen or as defined below;
X represents a bond, methylene or the linkage -CH2CH(R4)-, wherein R4 is hydrogen, alkyl or arylalkyl;
Y is -CH(Rs)- or -NRs-, wherein Rs is hydrogen or as defined below;
Z is -(CH2)2-, -C(R5)(R7)- or-N(R7)-, wherein R3 is hydrogen or methyl and R7 is as defined below;
Z' is -(CH2)2-, -C(R5)(R8)- or-N(Ra)-, wherein R6 is hydrogen or methyl and R8 is as defined below;
R1 is hydrogen, alkyloxycarbonylalkanoyl, alkyloxycarbonyl, alkanoyl (optionally sllhstitllt~d with a
radical selected from carboxy, alkyloxycarbonyl and heterocycloalkylalkanoylamino),
cycloalkylcarbonyl, heterocycloalkylcarbonyl (opLion-"y substituted with a radical selected from
hydroxy, alkyl, alkanoyl, alkyloxycarbonyl, arylalkyloxycarbonyl and heterocycloalkylcarbonyl),
arylalkyloxycarbonyl, ca,L,ar"oyl, alkylca,ba",uyl, dialkylcd,bal"oyl, arylca,L,a",uyl, arylalkylca~l,a",oyl,
arylalkanoyl, aroyl, alkylsulfonyl, dialkylc,,, ' ,osulfonyl, arylsulfonyl or heteroarylsulfonyl;
R7 and R8 are independently hydrogen, alkyl (opLion-"y substituted with a radical selected from
hydroxy, amino, alkylamino, dialkylamino, uriedo, mercapto, alkylthio, carboxy, ca,Lan,oyl,
CA 022161~1 1997-09-23
W 096/30353 PCTrUS96/03844
alkylcarbamoyl, dialkylcal ban,oyl, alkylsulfonyl and guanidino, or a protected derivative thereof),
cycloalkyl, cycloalkylalkyl, heteroaryl, heteroarylalkyl, a group selected from aryl and arylalkyl (which
group is optionally substituted at its aryl ring with one to three radicals selected from hydroxy, amino,
guanidino, halo, optionally halo-substituted alkyl, alkyloxy and aryl, or a protected derivative thereof) or
together with an adjacent R3 or Rs forms a divalent radical selectPd from (C34)methylene and
1 ,2-phenylenedimethylene (which radical is optionally substituted with hydroxy, or a protected
derivative thereof, or oxo); and
R2 is hydrogen, alkyl (optionally substituted with one or more radicals selected from amino, guanidino,
halo, hydroxy, alkyloxy, nitro, alkylsulfonyl and arylsulfonyl, or a protected derivative thereof),
cycloalkyl, cycloalkylalkyl or a group selected from aryl and arylalkyl (which group is optionally
substituted at its aryl ring with one to two radicals selected from amino, halo, hydroxy, optionally
halo-substituted alkyl, alkyloxy, nitro, alkylsulfonyl and arylsulfonyl, or a protected derivative thereof);
and the pha""aceutically acceptable salts, individual isomers and mixtures of isomers thereof,
preferably wherein the di~so~ia~ion constant for inhibition of the protease with said inhibitor (K,) is no
greaterthan about 100 ,uM.
An additional aspect of this invention is a compound, preferably a protease inhibitor, of Formula ll:
Ri / \z~ ~ B/ \A~ B~z! ~ R9
in which:
the groups are as defined above and
R9 is cyano, -C(O)OR'~, -P(O)(OR'~)2, -S(O)(NR'0)R10, C(O)R", -S(O)R", -C(o)NR'2R'3,
-S(O)2NR'2R'3, -C(o)NHR~4 or-S(O)2NHR'4, wherein each R'~ is independently hydrogen, alkyl
(optionally substitlltPd with one or more radicals selected from amino, halo, hydroxy, alkyloxy, nitro,
alkylsulfonyl and arylsulfonyl, or a protected derivative thereof), cycloalkyl, cycloalkylalkyl or a group
selected from aryl and arylalkyl (which group is optionally substituted at its aryl ring with one to two
radicals selected from amino, halo, hydroxy, op~ionally halo-substitlltPd alkyl, alkyloxy, nitro,
alkylsulfonyl and arylsulfonyl, or a prulecl,:d derivative thereof), Rll is hydrogen, alkyl, perfluoroalkyl,
cycloalkyl, cycloalkyialkyl, per~uoroaryl, perfluoroarylakyl or a group selected from aryl and arylalkyl
(which group is oplionally substituted at its aryl ring with one to two radicals selected from amino, halo,
hydroxy, oplion-'ly halo-substituted alkyl, alkyloxy, nitro, alkylsulfonyl and arylsulfonyl, or a p,ule.;l~d
derivative thereof), R12 and R13 are independently hydrogen, alkyl, cycloalkyl, cycloalkylalkyl, aryl or
aralkyl, and R14 is -C(O)OR10, in which R10 is as defined above, or a group selected from
CA 022161~1 1997-09-23
W 096/30353 PCTrUS96103844
5 _
Formulae (a) and (b):
\ ~A~B~ ~A~ ~ZlORI~ ~'b~ ~ 3n~
(a) . (b)
wherein each n, A, B, Y, Z, R' and R'~ are as defined above, and the pha~ ~"aceutically acceptable
salts; individual isomers and mixtures of isomers thereof, preferably wherein the di~socidlion consLd"L
for inhibition of the protease with said inhibitor (Kj) is no greater than about 100 ,uM.
A further aspect of this invention is a compound, preferably a protease inhibitor, of Formula lll:
Rl/ Z ~B \A~ Zl~RIs
~ 16
III
in which:
the groups are as defined above and
R's is hydrogen, methyl, fluoro or a group selected from Formulae (a) and (b) as defined above, and
R16 is a group selected from phenyl or (Css)heteroaryl (which group is optionally sl~hstitllt~d with at
least one radical selected from alkylcarbamoyl, dialkylcarbamoyl, alkyloxycarbonyl, alkylsulfinamoyl,
dialkylsulfinamoyl, alkylsulfonyl, carboxy, nitro, sulfinamoyl, sulfo, calba",oyl, phosphono,
alkyloxyphosphinyl, dialkyloxyphosphinyl, alkanoyl, cyano, alkylsulfinyl, sulfamoyl, alkylsulfamoyl,
dialkylsulfamoyl, alkyloxysulfonyl, aryl, heteroaryl, hydroxy, alkyloxy, optionally halo-substituted alkyl,
arylalkyl, halo, -~N(R'7)3, wherein each R'7 is independently alkyl, aryl or arylalkyl, or -N(R'3)2, wherein
each R1s is independently hydrogen, alkyl, aryl or arylalkyl); and the pharmaceutically acceptable salts;
individual isomers and mixtures of isomers thereof, prer~rdbly wherein the dissociation consldllt for
inhibition of the protease with said inhibitor (Kj) is no greater than about 100 ,uM.
An additional aspect of this invention is a phar".~ceutic~l composition cor"~.ri~i"g a ther~peut;c-"y
effective amount of a cysteine protease inhibitor of the invention, or of an individual isomer, a mixture
of isomers, or the pharrn~ceutic~lly aCCept~h'e salt or salts thereof, in co" Ibil ,dlion with one or more
pharmaceutically acceplable excipients.
A further aspect of this invention is a method for treating a condition capable of amelioration by
inhibition of a cysteine protease in an animal in need thereof, which method comprises administering
to such animal a therapeutically effective amount of a cysteine p, u~ase inhibitor of the invention, or of
an individual isomer, mixture of isomer, or the pharmaceutically accepldble salt or salts thereof.
CA 022161~1 1997-09-23
W 096/303S3 PCTrUS96/03844
Another aspect of this invention is a method for detecting a cysteine protease in a sample, which
method comprises:
(a) assaying said sample for protease activity using a protease substrate:
(b) assaying for protease activity in the presence of a known concenl,dlion of cysteine
protease inhibitor on the invention; and
(c) c~lculating the difference between a) and b) to determine the prult:ase activity due to
cysteine pruhase.
An aspect of this invention are the processes for preparing the cysteine protease inhibitors of
this invention.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 depicts Scheme 1, the synthesis of Formula I compounds when X is a bond. The synthetic
steps are as follows: a) HC02H, H20; b) HBr/acetic acid; c) 4-methylmorpholine, isobutyl
chloroformate, Mu-ROH; and d) chro",dlug,dphic purification. The groups are as defined herein.
Figure 2 depicts Scheme 2, the synthesis of Formula I compounds when X is a methylene group. The
synthetic steps are as follows: a) 4-methylmorpholine, isobutyl chlororur",dlt:, followed by NaBH4
reduction in water/THF; b) CH3SO2CI, triethylamine, CH2CI2; c) R1SH, NaH, CH30H, THF, heat;
d) 4-chloroperbenzoic acid, CH2CI2; e) HCI/dioxane or p-CH3C6H4SO3H/ether; and fl Mu-ROH, 4-
methylmorpholine, isobutyl chloroformate.
Figure 3 depicts Scheme 3, the synthesis of Formula I compounds when X is a methylene group. The
synthetic steps are as follows: a) (CH3)3CH2CH2SH, NaH, MeOH, THF, heat; b) 4-chloroperbenzoic
acid; c) (n-C4Hg)4N+F-, THF, followed by BrCH2CI, heat; d) HCI/dioxane or 4-CH3C6H4SO3H/ether, and;
e) 4-methylmorpholine, isobutyl chloroformate, Mu-PheOH.
Figure 4 depicts Scheme 4, the synthesis of Formula 11 compounds. The synthetic steps are as
follows: a) Cl-H2N+(CH3)0CH3, dicyclohexylcarboiimide, Et3N/CH2CI2; b) LiAlH4/THF; c) NaH/THF;
d) Hcl/dioxane/CH2CI2; e) 4-methylmorpholine, isobutyl chloruru""dl~/THF; and fl H2, 5% Pd/C.
Figure 5 depicts Scheme 5, the synthesis of Formula I compounds when X is an ethylene. The
synthetic steps are as follows: a)(CH20)n, HCI, dioxane, for instance where Ar = 2-naphthyl; b)
(EtO)3P; c~ CH3C03H, CH2C12; d) NaH, THF; e) p-CH3C6H4SO3H, Et20; fl 4-methylmorpholine, isobutyl
chlo,uru~ dLe; and g) H2, Pd/C.
Figure 6 depicts the synthesis of compounds of Formula 11 in which R9 is -COOH.
CA 022161~1 1997-09-23
W 096/30353 PCTrUS96/03844
7 .
Figure 7 depicts the synthesis of compounds of Formuia ll in which R9 is -P(O)(R'~)2. The synthetic
scheme is as follows: a) NaH/THF; b) anhydrous p-CH3C6H4SO3H/ether; c) 4-methylmorpholine,
isobutyl chlolurur,lldle/THF, and; d) H2, Pd/C.
Figure 8 depicts the synthesis of compounds of Formula ll in which R9 is -C(O)NHR'4. The synthetic
scheme is as follows: a) NaOH/EtOH, followed by Hcl/H20; b) benzylamine, dicyclohexylcarbodiimide,
CH2CI2; c) NaH/THF, diethyl benzylamidomethylenephosphonate; d) HCI/dioxane; e)
4-methylmorpholine, isobutyl chlolururllldle~ THF; f) H2, Pd/C, and as an alternative preparation from
carboxylates as s~lllhesi~ed via Scheme 6, above; and 9) aniline, dicyclohexylcarbodiimide, CH2CI2.
Figure 9 depicts the general synthesis of compounds of Formula ll.
Figure 1û depicts the synthesis of compounds of Formula lll. The synthetic steps are as follows: a)
CH3CN or other suitable solvent, reflux; b) H20, NaOH, followed by extraction into organic medium;
c) phosphorane, THF (Wittig reaction); d) p-CH3C6H4SO3H, ether; e) Mu-PheOH, 4-methylmorpholine,
isobutyl chlorururllldLe~ THF; and fl H2, Pd/C.
DEFINITIONS
Unless otherwise stated, the following terms used in the speciricdlion and claims are defined for the
purposes of this applicdLion and have the meanings given below:
"Alkyl", as in alkyl, alkyloxy, alkylthio, alkylsulfonyl, alkylcalL,anluyl, dialkylcall,amuyl, h~ ualylalkyl~
arylalkyl, and the like, mear.s a straight or branched, saturated or unsaturated hydrocarbon radical
having from 1 to 10 carbon atoms or the number of carbon atoms indicated (e.g., methyl, ethyl, propyl,
isopropyl, butyl, sec-butyl, isobutyl, tert-butyl, vinyl, allyl, 1-propenyl, isopropenyl, 1-butenyl, 2-butenyl,
3-butenyl, 2-methylallyl, ethynyl, 1-propynyl, 2-propynyl, etc.).
"Alkyloxyphosphinyl" and "dialkyloxyphosphinyl" mean the radicals -P(O)(OH)OR and -P(O)(OR)2,
respectively, wherein R is alkyl as defined above.
aAlkanoyl", as in alkanoyl, alkanoyloxy, heterocycloalkylalkanoylamino, and the like, means the radical
-C(O)R, wherein R is alkyl as defined above, having overall from 1 to 11 carbon atoms or the number
of carbon atoms illdi~idL~:d (e.g., (C,4)alkanoyl includes the radicals formyl, acetyl, propionyl,
isoplupionyl, butyryl, isobutyryl, crotonoyl, isocrotonyl, etc.).
aAryl" means an dlullldlic monocyclic or polycyclic hydrocarbon radical conldi,l lg 6 to 14 carbon
CA 022161~1 1997-09-23
W 096/30353 PCTrUS96/03844
atoms or the number of carbon atoms indicated and any carbocylic ketone or thioketone derivative
thereof, wherein the carbon atom with the free valence is a member of an a~ ur, IdLic ring, (e.g., aryl
includes phenyl, naphthyl, anthracenyl, phenanlllr~r,yl, 1,2,3,4-tetrahydro-5-naphthyl,
1-oxo-1,2-dihydro-5-naphthyl, 1-thioxo-1,2-dihydro-5-naphthyl, etc.).
"Aroyl" means the radical -C(O)Ar, wherein Ar is aryl as defined above, having overall from 7 to 15
carbon atoms or the number of carbon atoms indicated (e.g., (C7 ")aroyl includes benzoyl, naphthoyl,
etc.).
"Cycloalkyl", as in cycloalkyl and cycloalkylalkyl, means a saturated or unsaturated, monocyclic or
polycyclic hydrocarbon radical containing 3 to 20 carbon atoms or the number of carbon atoms
indicated, wherein the carbon atom with the free valence is a member of a non-aromatic ring, and any
carbocyclic ketone and thioketone derivative thereof (e.g., the term cycloalkyl is meant to include
cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclohexenyl, bicyclo[2.2.2]octyl, 1,2,3,4-tetrahydro-
1-naphthyl, oxocyclohexyl, dioxocyclohexyl, thiocyclohexyl, 9-fluorenyl, etc.).
"Halo" means fluoro, chloro, bromo or iodo.
"Heterocycloalkyl", as in heterocycloalkyl, heterocycloalkylalkancylamino, heterocycloalkylcarbonyl,
heterocycloalkylcarbonyl, and the like, means cycloalkyl as defined above wherein 1 to 5 of the
indicated carbon atoms is replaced by a heteroatom chosen from N, O, S, P or As, wherein the atom
with the free valence is a member of a non-aromatic ring, and any heterocyclic ketone, thioketone,
sulfone or sulfoxide derivative thereof, (e.g., the term heterocycloalkyl is meant to include piperidyl,
pyrrolidinyl, pyrrolinyl, imid~7olidirlyl, indolinyl, quinuclidinyl, morpholinyl, piperazinyl,
N-methylpiperazinyl, piperadinyl, 4,4-dioxo4-thiapiperidinyl, 1,2,3,4-tetrahydro-3-isoquinolyl,
2,4-diaza-3-oxo-7-thia~-bicyclo[3.3.0]octyl, etc.). Thus, hetero(C6)cycloalkyl includes the radicals
morpholinyl, piperazinyl, piperidinyl and the like.
"Heteroaryl" means an arol"dlic monocyclic or polycyclic hydrocarbon radical containing overall from
5 to 14 atoms or the number of atoms indicated, wherein 1 to 5 of the indicated carbon atoms are
replaced by a h~ :leroalcl", chosen from N, O, S, P or As, wherein the atom with the free valence is a
member of an ~IUllldlic ring, and any heterocyclic ketone and thioketone derivative thereof (e.g., the
term heteroaryl is meant to include thienyl, furyl, pyrrolyl, pyrimidinyl, isoxazolyl, oxaxolyl, indolyl,
benzo[b]thienyl, isobenzofuranyl, purinyl, isoquinolyl, pterdinyl, pyrimidinyl, ill,;dd~ulyl, pyridyl,
pyrazolyl, pyrazinyl, 4-oxo-1,2-dihydro-1-naphthyl, 4-thioxo-1,2-dihydro-1-naphthyl, etc.). Thus,
hetero(C6)aryl includes the radicals pyridyl, pyrimidinyl, and the like.
CA 022161~1 1997-09-23
W 096130353 PCTrUS96/03844
"1,2-Phenylenedimethylene" means a divalent radical of the formula -CH2C6H4CH2-. For example, the
group R'Y-Z-A- in which Y is -N(R5), Z is -CH(R7)-, A is carbonyl and R7 together with Rs forms
1,2-diphenylenedimethylene" means a group of following formula:
and substituted derlvatives and individual stereoisomers and mixture of stereoisomers thereof.
Substituted derivabves of the 1,2-phenylenedimethylene divalent radical may contain a hydroxy group
on any carbon within the ring system or an oxo group on either of the unsaturated ring carbon atoms.
"Phosphono" means the radical -P(O)(OH)2.
"Methylene" as in U(C34)methylene''and "(C3 7)methylene" mean a straight, saturated divalent radical
having the number of carbon atoms indicated; "(C3 4)methylene" includes trimethylene (-(CH2)3-) and
tetramethylene (-(CH2)4-). For example, a preferred embodiment herein utilizes a proline residue as
an A-B-Z group, wherein A-B represents CH2-NR3 and R3 together with either R7 or R3 form a C3
methylene. Thus, the group R'-Y-Z-A- in which Y is -(NRs)-, Z is -CH(R7)-, A is carbonyl and R7
together with Rs forms trimethylene means a group of following formula:
<~
N ~
and the individual stereoisomers and mixtures of stereoisomers thereof. Substituted derivatives of the
trimethylene and l~ dl I ,ethylene divalent radicals may contain a hydroxy group, or a protected
derivative thereof, or an oxo group on any of the ring carbon atoms. Suitable hydroxy protective
groups are defined below.
"Oxa(C3 7)methylene" and "aza(C3 7)methylene" mean methylene as defined above wherein one of the
indicated carbon atoms is replaced by an oxygen or nitrogen atom, respectively For example,
"oxa(Cs)methylene" includes 3-oxapenldr"~:ll,ylene (-CH2CH20CH2CH2-) and 2-oxdpenld",~:ll,ylene
(-CH20CH2CH2CH2-). Thus, -C(O)NR2'R22 means the radical 4-morpholinylcarbonyl when R2l and R22
together form 3-oxapenld",ell ,ylene and the radical 1-piperazinylca, L an.,yl when R2' and R22 together
form 3-azape, lldl, l~ll ,ylene.
CA 022161~1 1997-09-23
W 096/30353 PCTrUS96/03844
UAdjacent'', as use in the phrase "R7 together with an adjacent R3", means that the atoms to which the
R7 and R3 groups are respectively attached are in turn attached to one another.
"Animal" includes humans, non-human ",a"""als (e.g., dogs, cats, rabbits, cattle, horses, sheep,
goats, swine, deer, etc.) and non-",a",l"al . (e.g., birds, etc.).
"Disease" specific~"y includes any unhealthy condition of an animal or part thereof and includes an
unhealthy condition which may be caused by, or incident to, medical or veL~ i"ary therapy applied to
that animal, i.e.~ the "side effects" of such therapy.
"Electron withdrawing group" (EWG) means a functional group that in its broadest sense is a group
able to exert a polarizing force on the bond between itself and the carbon to which it is attached, such
that electrons are polarized in favor of the electron withdrawing group. While not being limited to any
particular theory, it is believed that the polarizing property enables the electron withdrawing group to
pdlii~ Jdl~ in hydrophobic or hydrogen bonding i"le~d~lions with an active site of the cysteine
protease, resulting in inhibition of the enzyme. In general, a moiety is suitable as an electron
withdrawing group if when present in the a-position of a phosphonium ylide of the general structure
Ph3P=C(R)EWG it exerts sufficient polarization to stablize the ylide against undergoing decomposition
reactions with oxygen, water, hydrohalic acids, alcohols, and the like. Preferred electron withdrawing
groups are those which would similarly ' 'I ' e ylides of the general formula (RO)2P(O)C(R)EWG.
Suitable electron withdrawing groups include cyano, -S(0)2R2, -C(O)OR'~, -P(O)(OR1~)2,
-S(O)(NR10)R10, C(O)R'1, -S(o)R1',-C(o)NR12R'3, -S(o)2NR'2R'3,-C(o)NHR'4 -S(O) NHR14 phenyl
and (Cs~)heteroaryl, wherein each R2, R10, R1', R'2, R13 and R14 are as defined in their broadest
definitions set forth in the Summary of the Invention. When the electron withdrawing group is phenyl
or (Cs~)heteroaryl the ring may be substituted with one or more meta directing groups
(e.g., alkyloxycarbonyl, alkylsulfinamoyl, dialkylsulfinamoyl, alkylsulfonyl, carboxy, nitro, sulfinamoyl,
sulfo, phosphono, alkyloxyphosphinyl, dialk5rloxy~hospl,i"yl, alkanoyl, cyano, alkylsulfinyl, sulfamoyl,
alkylsulfamoyl, dialkylsulfamoyl, alkyloxysulfonyl, rlisuh~stitllt~d amino, tris~hstitllt~d a"""on c, and the
like), ortho and para directing groups (e.g., hydroxy, alkyloxy~ optionally halo-s~hstit~t~d alkyl~ aryl,
arylalkyl~ ha!o, and the like) and electron withdrawing moieties (e.g., alkylca,ban,uyl, dialkylcarbd",oyl~
alkyloxycarbonyl~ alkylsulfinamoyl, dialkylsulfinamoyl, alkylsulfonyl, carboxy, nitro, sulfinamoyl, sulfo,
ca,L,a",oyl, phospho"o, alkyloxyphosphinyl, dialkyloxyphosphinyl, alkanoyl, cyano, alkylsulfinyl,
sulfamoyl, alkylsulfamoyl, dialkylsulfamoyl, alkyloxysulfonyl, aryl, heteroaryl, and the like).
"Leaving group" has the meaning conventionally ~so~ d with it in synthetic organic chemistry, i.e.,
an atom or group ~ p'c ~~~'!e under alkylating condiLions, and includes halo and alkane- or
arenesulfonyloxy, sucha mesyloxy, ethanesulfonyloxy. benzenesulfonyloxy and tosyloxy, and
CA 022161~1 1997-09-23
W O96/303Ci3 PCTrUS96/03844
11
alkaesuifonylamino alkal-ecdll,orlylamino aminosulfonylamino aminocarbonylamino and the like.
Isomerism is the phenomenon wherein compounds have identical molecular formulae but differ in the
nature or sequence of bonding of their atoms or in the arrangement of theri atoms in space. Isomers
that differ in the arrangement of their atoms in space are termed "sl~rl,iso~"e(~". Stereoisomers that
are not mirror images of one another are termed "diastereomers" and stereoisomers that are
nonsupe, i" ,posab,le mirror images are termed "enantiomers" or sometimes "optical isomers". A
carbon atom bonded to four nonidentical substituents is termed a "chiral center".
A compound with one chiral center has two enantiomeric forms of opposite chirality is termed a
"rdcei";c mixture". A compound that has more than one chiral center has 2n-1 enantiomeric pairs
where n is the number of chiral centers. Compounds with more than one chiral center may exist as
ether an individual diasteromer or as a mixture of diastereomers termed a "diastereomeric mixture".
Compounds of Formulae 1 11 and 111 can exist as individual steroisomers or mixtures of stereoisomers.
For example compounds of Formulae 1 11 and 111 contain a chiral center at the carbon to which the
substituent Ra is attached. Furthermore compounds of Formulae 1 11 and 111 in which Z is -C(R6)(R7)
contain a chiral center at the carbon to which the R7 substituent is attached. Thus for ~Xdl " !e
compounds of Formulae 1 11 and 111 in which n is O and Z is -C(R6)(R7) will have two chiral centers and
can exist as four individual stereoisomers or any mixture thereof.
Individual stereoisomer may be cl-alduLeri~ed by the absolute configuration of their chiral centers.
Absolute configuration refers to the dl, dnge" ,ent in space of the sl Ih5titl ~ents attached to the chiral
center. The siubstitl ~ents attached to the chiral center under consideration are ranked in accordance
with the Sequence Ru/e of Cahn Ingold and Prelog and then the ~hsol~.lf~ desc, i,ulOr R is assigned if
the three highest ranked s~bstit~ents are arranged in space (with the fourth lowest ranked sut)stitllent
directed away from the observer) from high to low priority in a clockwise sequence and the -'scl-lt~
desc, il-Lor S is assiy"ed for a counler- Iockwise a" dngel "eul. When an individual Ste,~:GiSol "er
collld;ll 19 one chiral center is described the aLsn' ~t~ des.,i~,lor R or S is cited in parenthesis followed
by a hyphen and the chemical name of the compound. For the purposes of this invention when an
individual sl~r~o;~ul~ler or mixture of ~ oi~ o" ,er:, co~,i ~ ,i~ ,g two or more chiral centers is desu, iLed
the ab s c '-lt~ desc, i~,lur R or S is cited i" " "edidlely after the apprupridle locant Acyl radicals derived
from naturally occurring amino acids are referred to as their amino acid radicals preceded by the
descri~,lor L (e.g. L-phenylalanine). The nonnatural end, llio" ,ers of amino acid acyl radicals are
preceded by the descriptor D. P~:r~rdbly the amino acid side chains are the (S) or L-form due to the
stereospeciri~ i~y of enzymes although the D-forms may be used in some cases. When no aLs clu
desc, i~.lur is cited for a chiral center the desc;, i~lion is meant to include both configurations and
CA 022l6l~l l997-09-23
W 096/303S3 PCT~US96/03844
12
mixtures thereof, racemic or otherwise. Thus, for e,~d~ "ple, a compound of the following formula:
R~ y ~RI9
is named:
N2-(4-morpholinylcarbonyl)-N1-[3-phenyl-1 S-(2-phenylsulfonylethyl)propyl]-L-phenylalaninamide, when
R1 is 4-morpholinylcarbonyl, R8 is 2-phenylethyl and lies on the same side of the reference plane as
the R7 substituent, R7 is benzyl and R19 is phenylsulfonyl;
N9-4-morpholinylcarbonyl-N~-[3-phenyl-1-(2-phenylsulfonylethyl)propyl]-L-phenylalaninamide, when R1
is 4-morpholinylcarbonyl, R8 is 2-phenylethyl and lies on either or both sides of the reference plane, R7
is benzyl and R19 is phenylsulfonyl;
/\~2-4-morpholinylcarbonyl-N-[3-phenyl-1 S-(2-phenylsulfonylethyl)propyl]-,~-(2-naphthyl)-L-alaninamide,
when R1 is 4-morpholinylcarbonyl, Ra is 2-phenylethyl lies on the same side of the reference plane as
the R7 substituent, R7 is 2-naphthylmethyl and R19 is phenylsulfonyl and
ethyl 4S-(N-4-morpholinylcarbonyl-L-phenylalanylamino)-6-phenylhexanoate, when R1 is
4-morpholinylcarbonyl, R8 is 2-phenylethyl and lies on the same side of the reference plane as the R7
substituent, R7 is benzyl and R19 is ethoxycarbonyl.
In a preferred embodiment, the compositions of the invention are pure diasteromers. Alternatively, the
compositions contain mixtures of diasteromers. Preferred embodiments have greater than about 70%
of a single disasteromer, with at least about 90% being particularly preferred.
"Protective group" has the meaning conventially associated with it in synthetic organic chemistry, i.e.,
a group which blocks a reactive site in a compound. See for example Greene et al., Protective Groups
in Organic Synthesis, 2nd Ed., John Wiley & Sons, 1991, hereby inco"uo,dl~d by reference.
Examples of hydroxy protective groups include heterocycloalkyl-carbonyl such as
4-morpholinylcarbonyl and the like, aroyl such as benzoyl and arylalkyl such as benzyl and the like.
Examples of amino protective groups include aryloxycarbonyl such as benzyloxycarbonyl and the like,
aroyl such as benzoyl and the like and oxycarbonyl such as ethoxycarbonyl and
9-fluorenylmethoxycarbonyl and the like. Examples of guanidino protective groups include sulfonyl
such as 2,3,5-trimethyl-4-methoxyphenyl-sulfonyl and the like. Examples of suitable carboxy
protective groups that form ester ", . t - 5 are alkoxylcarbonyl of overall 4 to 8 carbon atoms,
particularly tert-butoxycarbonyl (BOC) or benzyloxycarbonyl (CBZ, Z), espec~ "y
cycloalkyla" ,' ,oca, boriyl or oxacycloalkylaminocarbonyl of overal 4 to 8 atoms in the ring, particularly
4-morpholinecarbonyl (Mu) and the like.
CA 022l6l~l l997-09-23
W O 96/30353 PCTrUS96/03844
13
"Protected" in reference to a compound of a group means a derivative of a compound or group in
which a reactive site or sites are blocked with protective groups.
"Optional" or "optionally" means that the subsequently described event or circumstance may or may
not occur, and that the description includes instances wherein the event or circu" ,~ldnce occurs and
instances in which it does not. For example, "optionally further substituted with one or more functional
groups" means that the substituents may or may not be present in order for the compound described
to fall within the invention, and the invention includes those compounds wherein one or more
functional groups are present and those compounds in which no functional groups are present.
By "cysteine protease-associated disorders" herein is meant pathological conditions associated with
cysteine proteases. In some disorders, the condition is associated wtih increased levels of cysteine
proteases; for t:xdmple, arthritis, muscular distrophy, i"na"""dLion, tumor invasion, and
glomerulonephritis are all associ~t~d with increased levels of cysteine proteases. In other disorders or
diseases, the condition is associated with the appearance of an extracellular cysteine protease activity
that is not present in normal tissue. In other embodiments, a cysteine protease is associated with the
ability of a pathogen, such as a virus, to infect or replicate in the host organism.
Specific examples of cysteine protease associated disorders include, but are not limited to, arthritis,
muscular distrophy, inna" ,r"dLion, tumor invasion, glomerulonephritits, malaria, Alzheimer's disease,
cancer metastasis, trauma, i"rlar"r"aLion, gingivitis, leishmaniasis, filariasis, and other bacterial and
parasite-borne infections. In particular, disorders associated with interleukin 1~ converting enzyme
(ICE) are included.
"Pl,ar",aceutically acceptable" means that which is useful in preparing a pharmaceutical composition
that is generally safe, non-toxic and neither biologic~lly nor otherwise undesirable and includes that
which is acceptable for veterinary use as well as human pharmaceutical use.
"Pharmaceutic~lly acce~i 'le salts" means salts which are pharrn~ceutic.~lly acceptable, as defined
above, and which possess the desired pha""a c!cg ' activity. Such salts include acid addition salts
formed with inorganic acids such as hy.l,u~ -ric acid, hydrobromic acid, sulfuric acid, nitric acid,
phosphoric acid, and the like; or with organic acids such as acetic acid, propionic acid, hexanoic acid,
hepLdnGic acid, cyclopentaneprupionic acid, glycolic acid, pyruvic acid, lactic acid, malonic acid,
succinic acid, malic acid, maleic acid, fumaric acid, tartatic acid, citric acid, benzoic acid,
o-(4-hydroxybenzoyl)benzoic acid, ci"na",: acid, madelic acid, methanesulfonic acid, ethanesulfonic
acid, 1,2-~Ll,dnedi:,ulfonic acid, 2-hydroxyeLl,anesulfonic acid, benzenesulfonic acid,
p-chlorobenzenesulfonic acid, 2-naphLl,~'~nesulfonic acid, p-toluenesulfonic acid, ca",pho,:julfonic
CA 022161~1 1997-09-23
W 096/30353 PCTIUS96/03844
14
acid, 4-methylbicyclo[2.2.2]oct-2-ene-1-carboxylic acid, glucoheptonic aicd,
4,4'-methylenebis(3-hydroxy-2-ene-1-carboxylic acid), 3-phenyl~rupionlc acid, trimethylacetic acid,
tertiary butylacetic acid, lauryl sulfuric acid, gluconic acid, glutamic acid, hydroxynaphthoic acid,
salicylic acid, stearic acid, muconic acid and the like.
Fl,ar",aceutically acceptable salts also include base addition salts which may be formed when acidic
protons present are capable of reacting with inorganic or organic bases. Acceptabale inorganic bases
include sodium hydroxide, sodium carbonate, potassium hydroxide, aluminum hydroxide and calcium
hydoxide. ~:cept~hle organic bases include ethanolamine, diethanolamine, triethanolamine,
tromethamine, N-methylglucamine and the like.
"Therapeutically effective amount" means that amount which, when administered to an animal for
treating a disease includes:
(1 ) preventing the disease from occurring in an animal which may be predisposed to the disease but
does not yet experience or display syl"~Lu",s of the disease,
(2) inhibiting the disease, i.e., arresting its development, or
(3) ameliorate the disease, i.e., causing regression of the disease.
DETAILED DESCRIPTION OF THE INVENTION
The present invention relates to novel cysteine protease inhibitors. Without being bound by theory, it
is believed that the inhibitors bind to cysteine proteases based on the following scheme.
Enz
Gln-H----O R-
R7 ~ S ~ Enz
B ~ O----H - His enymeinhibito.-
.nz
It is believed that the enzyme is thus reversibly inhibited by means of i"I~:rd~;Lions between the R, Y
and Z moieties of the inhibitor and the surface of the binding sites of the enzyme, and by means of
hydrogen bonding i"Lt:r~,Lions between the sulfone and active site amino acid side chains.
This mechanism of reversible inhibition permits specificity of the enzyme inhibitors for cysteine
p,uL~ases. Generally, the inhibitors of the present invention inhibit cysteine pruL~:ases and do not
inhibit serine, aspartyl, and zinc proteases. However, in some embodiments, the proL~ase inhibitors of
the present invention may have activity against other types of ~luL~ases, such as serine, asparLyl or
CA 022l6l~l l997-09-23
W O 96/30353 PCT~US96/03844
other met,-"~pr~ ases, but to a lesser extent.
In addition, the electron withdrawing properties of the sulfone group of Formula I polarize the electrons
between the sulfone group and the carbon to which it is attached, thus permitting hydrogen bonding
between itself and active site residues of a cysteine protease, to allow tight binding between the
inhibitor and the cysteine p~utease~ as is generally described below. It is to be understood that there is
presumably additional electron withdrawing or electron polari,dLion occurring between the sulfur atom
and the oxygen atoms, which allows the oxygen atoms to pa~ iic;l-aL~: in hydrogen bonding with active
site residues of the p, ~l~ase and thus contributing even further to the inhibition of the enzyme.
The present invention generally provides new peptide-based and peptidomimetic cysteine protease
inhibitors for use as reversible cysteine protease inhibitors. By "cysteine protease inhibitor" herein is
meant an inhibitor which inhibits cysteine proteases. In a preferred embodiment, the cysteine
protease inhibitors are specific to cysteine proteases; that is, they do not inhibit other types of protease
such as serine, aspartyl, or other metalloproteases. However, in alternative embodiments, the
cysteine p,uhase inhibitors of the invention may inhibit other types of proteases as well.
By "reversible" herein is meant that the inhibitor binds non-covalently to the enzyme, and is to be
distinguished from irreversible inhibition. See Walsh, Enzymatic Reaction Mechanisms, Freeman &
Co., N.Y., 1979. "Reversible" in this context is a term understood by those skilled in the art. In
addition, the reversible cysteine protease inhibitors are competitive inhibitors, that is, they compete
with substrate in binding reversibly to the enzyme, with the binding of inhibitor and substrate being
mutually exclusive. In addition, the stoichiometry of inhibition is 1:1; that is, a single inhibitor molecule
is sufficient to inhibit a single enzyme molecule.
The cysteine protease inhibitors herein are designed to bind reversibly to cysteine proteases. This
binding is acco",, ' ~hed by using peptide-based or peptidol" lleLic structures as targeting groups that
mimic naturally occurring suL,~ L~s and/or inhibitors. "Peptido",;-,lelic", for the purposes of this
invention, means amino acid or peptide-like in structure but wherein one or more of the peptide
linkages (i.e., -C(O)NR-) is substituted by an isosleric form, i.e. -CH2NR-, -C(O)CH2- or -NRC(O)-
and/or wherein non-naturally occurring amino acid substituents are present.
"Targeting group", for the purposes of this app' - "~ n, means a peptide or peptidol ";",eLic residue of
the cysteine pruL~ase inhibitor that allows the binding of the inhibitor to a cysteine pr~,lease. In a
preferred ~" ,bod;",enL, the targeting group of a cysteine protease inhibitor comprises at least two
amino acid side chains or side chain analogs, linked via a peptide bond or isostere. The targeting
group may co, . ~priae up to about 15 amino acids or analogs, although inhibitors are generally from
CA 022l6l~l l997-09-23
W O 96/303~3 PCTAUS96/03844
16
about 1 to 7 amino acids or analogs, since smaller inhibitors are usually desired in therapeutic
applications. Thus, in Formulae 1, ll and lll, n is preferably from O to 13, with from O to 5 being
preferred, and from O to 3 being particularly prer~r,~d.
As depicted in Formulae 1, ll and lll, the targeting group can be represented by a naturally or
non-naturally occurring peptide residue of the following formula:
R7
1A F~/
wherein the R3 and R~ components represent naturally or non-naturally occurring amino acid analogs
or substituents as is more fully described below. The targeting group of the inhibitor may also contain
additional functional groups, as depicted by R' and described herein.
While not being limited to any particular theory, it is believed that the amino acid substituents of the
targeting group interact with the surface binding sites of the protease to promote binding. It is also
believed that the amino acid sl Ihstitl l~nt proximal to the electron withdrawing group (e.g., R3 of the
above formula) will occupy the S1 position of the substrate binding site and therefore is designated the
P, residue of the inhibitor. Similarly, the next adjacent amino acid substituent (e.g., R7 of the above
formula) will occupy the S2 position of the substrate binding site and is designated the P2 residue of the
inhibitor If present, additional amino acid substituents will occupy the S3, S4, etc. posilions of the
substrate binding site and be designated as the P3, P4, etc. residues of the inhibitor. An additional
targeting group may be attached to the electron withdrawing group and, if present, its amino acid
substituents will occupy the S,', S2', etc. positions of the substrate binding sites and are designated the
P3', P4', etc. residues of the inhibitor, respectively.
In general, targeting groups for specific enzymes are determined by rules governing substrate
specificity in cysteine proteases (e.g., see "P,ut~i"ase Inhibitors", in Research Monographs in Cell and
tissue Physiology (1986), ed. Barret eta/., Vol 12, Chapter4: Inhibitors of Cysteine P,~ i"ases,
Daniel Rich, Elsevier, New York; and Thornberry et al., supra., hereby expressly incorl-ordled by
reference). For exd",,~le, interleukin-1 converting enzyme (ICE) accepts an aspartic acid s~ ~hstitllent
(i.e., 2-carboxyethyl) at the P, position and an alanine (methyl), valine (isopropyl) or histidine
(4-i", ~' - Iylmethyl) 5~h5tjtl ~ent at the P2 position. Papain accepts a arginine, Iysine, N-
benzyloxycarbonyllysine (i.e. 4-benzyloxycarbonylaminobutyl), homophenylalanine (i.e. 2-phenylethyl),
Guanidino-phenylalnine (i.e., 4-guanidi"oben~yl) or norluecine (i.e., butyl) s' 'hstitl ~t~nts at the P,
position and phenylalnine, tyrosine, ~-)2-naphthyl)alanine (i.e., 2-naphthyl), leucine, norleucine,
CA 022l6l~l l997-09-23
W 096/30353 PCTAUS96/03844
17
isoleucine or alanine suhstitllents at the P2 position. Cathepsin B accepts a arginine, Iysine, N-
benzyloxycarbonyllysine, guanidino-phenylalanine, homophenylalanine or norleucine s~hstitl~ents at
the P, position and phenylalanine, tyrosine, 3,5-diiodotyrosine (i.e., 3,5-diiodo-4-hydroxybenzyl), ~-(2-
naphthyl)alanine, arginine, guanidino-phenylalanine or citrulline (i.e., 3-ureidopropyl) sybstituents at
the P2 position. Cathepsin L and cruzain accept arginine, Iysine, homophenylalanine, guanindino-
phenylalanine, citrulline or norleucine substituents at the P1 position and phenylalanine, tyrosine or
~-(2-naphthyl)alanine substituents at the P2 position. Cathepsin S accepts a arginine, iysine,
homophenylalanine, guanidino-phenylalanine, citrulline or norleucine sllhstitllents at the P, position
and phenylalanine, tyrosine, ,B-(2-naphthyl)anine, valine, leucine, norleucine, isoleucine or alanine
substituents at the P2 position. DPP-1 accepts phenylalanine or tyrosine substituents at the P,
position and no subsutituent or alanine at the P2 position. Calpain accepts phenylalanine, tyrosine,
methionine, ,B-methylsulfonylmethylalanine (i.e., 2-methylsulfonylethyl) or valine substituent at the P,
position and valine, leucine, norleucine or isoleucine substituents at the P2 position.
Thus, R7 and R3 are independently hydrogen, alkyl (optionally substituted with a radical selected from
hydroxy, amino, alkylamino, dialkylamino, uriedo, mercapto, alkylthio, carboxy, carbamoyl,
alkylcarbamoyl, dialkylca,bal"oyl, alkylsulfonyl and guanidino, or a protected derivative thereofl,
cycloalkyl, cycloalkylalkyl, heteroaryl, heteroarylalkyl, a group seiect~d from aryl and arylalkyl (which
group is optionally substituted at its aryl ring with one to three radicals selected from hydroxy, amino,
guanidino, halo, optionally halo-substituted alkyl, alkyloxy and aryl, or a protected derivative thereofl or
together with an adjacent R3 or Rs forms a divalent radical selected from (C3~,)methylene and
1 ,2-phenylenedimethylene (which radical is optionally substituted with hydroxy, or a protected
derivative thereof, or oxo).
Accordingly, preferred R7 and R3 groups are the naturally occuring amino acid side chains and
homologous derivatives. These include, but are not limited to, alanine (methyl), arginine (3-
guanidinopropyl), aspa, dyil ,e (carbamoylmethyl), citurlline (3-ureidopropyl), aspartic acid
(carboxymethyl), cysteine (mercaptomethyl), glutamic acid (2-carboxyethyl), glutamine (2-
ca,l.d",oylethyl), glycine (hydrogen), histidine (4-imidazolylmethyl), homophenylalanine (2-
phenylethyl), hol"oserine (2-hydroxylethyl), isoleucine ((1-methylpropyl), leucine (isobutyl), Iysine (4-
aminobutyl), r"~Ll.ioni"e (2-methylthioethyl), ~-(1-naphthyl)alanine (1-napthylmethyl), ~-(2-
naphthyl)alanine (2-napthylmethyl), norleucine (butyl), norvaline (propyl), ornithine (3-a",;.,op,upyl),
phenylalanine (benzyl), proline (as described herein), sarcosine (methylaminomethyl), serine
(hydroxymethyl), threonine (1-hydroxyethyl), tryptophan (3-indolymethyl), tyrosine (4-hydroxybenzyl),
and valine (isopropyl).
While the broadest definition of this invention is set forth in the Summary of the Invention, certain
CA 022161~1 1997-09-23
W 096/303S3 PCTrUS96/03844
18
compounds of the invention are preferred. For example, generally preferred compounds of Formulae
1, ll and lll are those in which n is 0 to 5; A-B represents a linkage selected from -C(O)NR3-, wherein
R3 is hydrogen or as defined below; Y is -N(R5)-, wherein Rs is hydrogen or as defined below; Z is
-(CH2)2- or -C(R6)(R7)-; Z' is -CH(R8)-; R1 is hydrogen, alkyloxycarbonylalkanoyl of overall 3 to 10
carbon atoms, (C, g)alkoxycarbonyl, (C2 10)alkanoyl (optionally sl Ihstitl lt~d with a radical selected from
carboxy, (C,9)alkyloxycarbonyl and hetero(C4 8)cycloalkyl(C2 ,0)alkanoylamino), (C4 9)cycloalkyl-
carbonyl, hetero(C4 8)cycloalkylcarbonyl (optionally substituted with a radical selected from hydroxy,
(C, s)alkyl, (C, s)alkanoyl, (C1 s)alkyloxycarbonyl, (C6 10)aryl(C, 5)alkyloxycarbonyl and
hetero(C48)cycloalkylcarbonyl), (C6,0)aryl(C,s)alkyloxycarbonyl, calL,a~,,uyl, (C~5)alkylCdllJdllluyl~
di(C, s)alkylcarbamoyl, (Cs ,O)arylcarbamoyl, (C6 ,0)aryl(C, s)alkylcarbamoyl, (C6 ,0)aryl(C, s)alkanoyl,
(C7 ")aroyl, (C, s)alkylsulfonyl, di(C, s)alkylaminosulfonyl, ~C6 ,0)arylsulfonyl or hetero(Cs 8)arylsulfonyl;
and R7 and Rs are independently (C, s)alkyl (optionally substituted with a radical selected from
hydroxy, amino, alkylamino, dialkylamino, uriedo, mercapto, alkylthio, carboxy, carbamoyl,
alkylcarbanloyl, dialkylca, ba",uyl, alkylsulfonyl and guanidino, or a protected derivative thereofl,
(C3 7)cycloalkyl, (C3 7)cycloalkyl(C, s)alkyl, pyridyl, thienyl, furyl, imidazolyl, indolyl, pyridyl(C,4)alkyl,
thienyl(C, 6)alkyl, furyl(C, 6)alkyl, i",idd~olyl(C, 6)alkyl, indolyl(C, 6)alkyl, a group selected from or a
group selected from phenyl, naphthyl, phenyl(C, 6)alkyl, naphthyl(C, 6)alkyl, (which group is optionally
substituted at its aryl ring with one to three radicals selected from hydroxy, amino, chloro, bromo, iodo,
fluoro, methyl, trifluoromethyl, methoxy and phenyl, or a protected derivative thereofl or together with
an adjacent R3 or R4 forms a divalent radical selected from (C34)methylene and
1,2-phenylenedimethylene (which radical is optionally substituted with hydroxy, or a protected
derivative thereof, or oxo).
More preferred compounds of Formulae 1, ll and lll are those in which n is 0 to 2; A-B represents a
linkage selected from -C(o)NR3-, wherein R3 is hydrogen or as defined below; Y is -N(Rs)-, wherein Rs
is hydrogen or as defined below; Z is -(CH2)2- or -C(R5)(R7)- (with the proviso that when n is 0, Z is not
-(CH2)2-); Z' is -CH(Rs)-; R' is hydrogen, (C4 8)alkoxycarbonyl, (C2.6)alkanoyl (oplionally 5llhstihlt~d
with a radical selected from carboxy, (C, s)alkyloxycarbonyl and hetero(C4 8)cycloalkyl(C4 6)
alkanoylamino), -C(O)NR2'R22 wherein R21 and R22 together form aza(C2-6)methylene,
oxa(C2 6)methylene or (C3 7)methylene, (C4 8)cycloalkylcarbonyl, benzyloxycarbonyl, acetyl, benzoyl or
dimethylaminosulfonyl; and R8 and R7 are independently (Cs 6)cycloalkyl, (Cs 6)cycloalkylmethyl,
3-pyridyl, 2-thienyl, 2-furyl, 4-illl;dd~olyl, 3-indolyl, 3-pyridylmethyl, 2-thienylmethyl,
2-furylmethyl,4-i",: ' Iylmethyl, 3-indolylmethyl, (C, s)alkyl (oplional'y substituted with a radical
selected from ,,,erca,~,lu, carboxy, amino, methylthio, methylsulfonyl, ca,bar"uyl, dimethylcd,L,ar"uyl,
guanidino and hydroxy, or a plute~l~d derivative thereofl, a group select~d from phenyl, 1-naphthyl,
2-naphthyl, benzyl, 1-naphthylmethyl, 2-naphthylmethyl and 2-phenylethyl (which group is optionally
Sl Ih5titl It,~d at its aryl ring with one radical s~lec~d from hydroxy, amino, chloro, bromo and fluoro, or a
CA 022161~1 1997-09-23
W O 96/30353 PCTrUS96/03844
19
protected form thereofl or together with an adjacent R3 or Rs forms a divalent radical selected from
(C34)methylene and 1,2-phenylenedimethylene (which radical is optionally substituted with hydroxy, or
a protected derivative thereof, or oxo).
Particularly preferred compounds of Formulae 1, ll and lll are those in which n is O to 1; A-B represents
a linkage selected from -C(o)NR3-; Y is -N(Rs)-, wherein Rs is hydrogen or as defined below; Z is
-C(R5)(R7)-; Z' is -CH(R8)-; R' is hydrogen, fert-butoxycarbonyl, benzyloxycarbonyl, acetyl,
3-carboxypropionyl, 3-methoxycarbonyll,rupiol-yl, biotinylaminohexanoyl, phenylacetyl, benzoyl,
dimethylaminosulfonyl, benzylsulfonyl, 1-piperazinylcarbonyl, 4-methylpiperazin- 1-ylcarbonyl or
4-morpholinylcarbonyl, R7 is 3-pyridylmethyl, 2-thienylmethyl, 2-furylmethyl, 4-imidazolylmethyl,
3-indolylmethyl, (C,.s)alkyl (optionally substitllt~d with a radical selected from mercapto, carboxy,
amino, methylthio, methylsulfonyl, cal L,ar"uyl, dimethylcarbamoyl, guanidino and hydroxy, or a
protected derivative thereofl, a group select~d from benzyl, 1-naphthylmethyl, 2-naphthylmethyl and
2-phenylethyl (which group is optionally substituted at its aryl ring with one radical selected from
hydroxy, amino, chloro, bromo and fluoro, or a protected form thereofl or together with an adjacent R3
or Rs forms a divalent radical selected from (C3,,)methylene and 1,2-phenylenedimethylene (which
radical is optionally substituted with hydroxy, or a protected derivative thereof, or oxo); and R8 is butyl,
2-phenylethyl, 2-methylsulfonylethyl, 2-tert-butoxycarbonylethyl, 2-tert-butoxycarbonylmethyl,
4-tert-butoxycarbonylaminobutyl, 4-benzoylaminobutyl or benzyloxymethyl.
More particularly preferred compounds of Formulae 1, ll and lll are those in which n is 0; A-B
represents a linkage selected from -C(O)NH-; Y is -NH-; Z is -CH(R7)-; Z1 is -CH(R8)-; R' is hydrogen,
tert-butxoycarbonyl, benzyloxycarbonyl, biotinylaminohexanoyl, benzoyl, piperizin-1-ylcarbonyl,
4-methylpiperazin-1-ylcarbonyl or 4-morpholinylcarbonyl; R7 is (C1 s)alkyl, optionally suhstitllt~d benzyl,
1-naphthylmethyl, 2-naphthylmethyl, 3-pyridinylmethyl or 2-methylsulfonylethyl; and R8 is butyl,
2-phenylethyl or 2-methylsulfonylethyl.
Most preferred compounds of Formula 1, ll and lll are those in which n is 0; A-B ,~p,~se,lL~ a linkage
selected from -C(O)NH-; Y is -NH-; Z is -CH(R7)-; Z1 is -CH(Rs)-; R' is 1-piperizinylcarbonyl, 4-methyl-
1 -piperazinylcarbonyl or 4-morpholinylcarbonyl; R7 is oplionally sl Ihstitllted benzyl, 1-naphthylmethyl
or 2-naphthylmethyl; and R8 is 2-phenylethyl.
Generally preferred compounds of Formula I are those in which R2 is independently (C, s)alkyl
(optionally substituted with one or two radicals sele,l~d from amino, chloro, bromo, fluoro, hydroxy
and methoxy, or a protected derivative thereofl, perhalo(C1 s)alkyl, (C3 7)cycloalkyl,
(C3 7)cycloalkyl(C1 s)alkyl or a group selectpd from phenyl, pentafluorophenyl, naphthyl and
phenyl(C, 6)alkyl (which group is optionally suhstitllt~d at its aryl ring with one to two radicals selected
CA 022161~1 1997-09-23
096/30353 PCTrUS96/03844
from amino, chloro, bromo, fluoro, hydroxy, methoxy and optionally halo-substituted methyl, or a
protected derivative thereofl and R4 is hydrogen, (C1 s)alkyl or (C6 10)aryl(C1 s)alkyl. More preferred
compounds of Formula I are those in which in which R2 is (C1 s)alkyl (optionally substituted with one or
two radicals selected from amino, chloro, bromo, fluoro and hydroxy, or a protected derivative thereof),
perfluoro(C1 s)alkyl, (Cs6)cycloalkyl, (Cs6)cycloalkylmethyl or a group selected from phenyl, naphthyl
and benzyl (which group is optionally substituted with one radical selected from amino hydroxy, chloro,
bromo or fluoro, or a protected derivative thereofl and R4 is hydrogen or methyl. Par~icularly pr~r~r,~d
compounds of Formula I are those in which R2 is methyl, trifluoromethyl, optionally substituted phenyl,
2-naphthyl or 2-phenylethyl. Most preferred compounds of Formula I in which R2 is phenyl, 2-naphthyl
or 2-phenylethyl, particularly phenyl or 2-naphthyl, and R4 is hydrogen.
Generally preferred compounds of Formula ll in which R9 is -C(O)OR10, -P(O)(OR1~)2, -S(O)(NR10)R10,
-C(O)NHC(O)R10 or-S(0)2NHC(O)R'~ are those in which each R'~ is independently (C1 s)alkyl
(optionally sl ~hstitl ~ted with one or two radicals selected from amino, chloro, bromo, fluoro, hydroxy
and methoxy or a protected derivative thereofl, (C3 7)cycloalkyl, (C3 7)cycloalkyl(C, s)alkyl, or a group
selected from phenyl or phenyl(C1.6)alkyl (which group is optionally s~bstitllt~d at its phenyl ring with
one to two radicals selected from amino, chloro, bromo, fluoro, hydroxy, methoxy and optionally
halo-substituted methyl, or a protected derivative thereofl. More preferred compounds of Formula ll in
which R9 is -C(O)OR'~, -P(O)(OR'~)2, -S(O)(NR10)R'0, -C(O)NHC(O)R'~ or -S(0)2NHC(O)R'~ are those
in which in which R'~ is ethyl, (Cs 6)cycloalkyl, (Cs 6)cycloalkylmethyl or a group selected from phenyl
and benzyl (which group is optionally substituted at its phenyl ring with one radical selected from
amino hydroxy, chloro, bromo or fluoro, or a protected derivative thereofl.
Generally preferred compounds of Formula ll in which R9 is C(O)R'1 or -S(O)R" are those in which R"
is (C~ s)alkYI~ (C3 7)cycloalkyl, (C3 7)cycloalkyl(C, s)alkyl or a group selected from phenyl and
phenyl(C, 6)alkyl (which group is optionally substituted at its aryl ring with one to two radicals selected
from amino, chloro, bromo, fluoro, hydroxy, methyl, trifluoromethyl and methoxy). More prer~:r,t:d
compounds of Formula ll in which R" is C(O)R" or-S(O)R1' are those in which in which R" is ethyl,
cyclo(C5 6)alkyl, cyclo(Cs 6)alkylmethyl or a group selected from phenyl and benzyl (which group is
optionally substituted at its phenyl ring with one radical select~-d from amino hydroxy, chloro, bromo or
fluoro, or a protected derivative thereofl.
Generally preferred compounds of Formula ll in which R9 is -C(o)NR'2R'3 or -S(0)2NR'2R'3 are those
in which R12 and R'3 are independently (C1 s)alkyl, (C3 7)cycloalkyl, (C3 7)cycloalkyl (C1 s)alkyl or a group
selected from phenyl and phenyl(C1 6)alkyl (which group is optional!y 5,,h5tit,,t~d at its phenyl ring with
one to two radicals selected from amino, chloro, bromo, fluoro, hydroxy, methoxy and optionally
halo-sllhstitllt.od methyl). More preferred compounds of Formula ll in which R9 is -C(o)NR'ZR'3 or
CA 022161~1 1997-09-23
W 096/303S3 PCTrUS96/03844
21
-S(o)2NR'2R'3 are those in which R12 and R'3 are independently ethyl, (Cs 6)cycloalkyl,
(Cs s)cycloalkylmethyl or a group selected from phenyl and benzyl (which group is optionally
substituted at its phenyl ring with one radical selected from amino hydroxy, chloro, bromo or fluoro, or
a protected derivative thereof).
Preferred compounds of Formula ll in which R9 is -C(o)NHR'4 or -S(0)2NHR'4 wherein R'4 is a group
selected from Formulae (a) and (b) are those in which each n, A, B, Y, Z, R' and R10 are as defined
above with respect to preferred compounds of Formulae 1, ll and lll.
Generally preferred compounds of Formula lll are those in which R's is a group selected from 2-furyl,
2-thienyl, 2-pyrrolyl, 2-phospholyl, 2-arsoyl, 3-pyridyl or 3-phosphorinyl (which group is optionally
substituted with at least one radical selected from (C, s)alkylcarbamoyl, di(C, 5)alkylcarba",oyl,
(C, s)alkyloxycarbonyl, (C, s)alkylsulfinamoyl, di(C, s)alkylsulfinamoyl, (C, s)alkylsulfonyl, carboxy, nitro,
sulfinamoyl, sulfo, carbamoyl, phosphono, (C, s)alkyloxyphosphinyl, di(C, s)alkyloxyphosphinyl,
(C, s)alkanoyl, cyano, (C, s)alkylsulfinyl, sulfamoyl, (C, s)alkylsulfamoyl, di(C, s)alkylsulfamoyl,
(C, s)alkyloxysulfonyl, (C~ s)l phenyl, naphthyl, pyridyl, thienyl, fury', imidazolyl, indolyl, hydroxy,
(C, s)alkyloxy, optionally halo-substituted (C~ s)alkyl, benzyl, halo, -~N(R'7)3, wherein each R'7 is
independently (C, s)alkyl, phenyl or benzyl, or -N(R'8)2, wherein each R1s is independently hydrogen,
(C, s)alkyl, phenyl or benzyl). More preferred compounds of Formula lll are those in which R1s is a
group selected from 2-furyl, 2-thienyl, 2-pyrrolyl, 2-phosholyl, 2-arsolyl, 3-pyridyl or 3-phosphorinyl
(which group is optionally substituted with at least one radical selected from methylcarbamoyl,
dimethylcarbamoyl, methyloxycarbonyl, methylsulfinamoyl, dimethylsulfinamoyl, methylsulfonyl,
carboxy, nitro, sulfinamoyl, sulfo, ca,L,an,uyl, phosphono, methyloxyphosphinyl,dimethyloxyphosphinyl, formyl, cyano, methylsulfinyl, sulfamoyl, methylsulfamoyl, dimethylsulfamoyl,
methoxysulfonyl, methylsulfonimidoyl, phenyl, naphthyl, pyridyl, thienyl, furyl, imidazolyl, indolyl,
hydroxy, methoxy, methyl, trifluromethyl, benzyl, halo, -~N(R'7)3, wherein each R'7 is independently
methyl, phenyl or benzyl, or-N(R's)2, wherein each R18 is independently hydrogen, methyl, phenyl or
benzyl). Generally pr~t~r,~d compounds of Formula lll in which R15 is a group selected from
Formulae (a) and (b) are those in which each n, A, B, Y, Z, R' and R'~ are as defined above with
respect to preferred compounds of Formulae 1, ll and lll.
In general, preferred cysteine protease inhibitors of the invention are those in which the ~hsolute
configuration of each chiral center present is the (S)-configuration. However, preferred compounds of
Formula I in which n is O are those in which the ahs - l- ltp configuration of chiral center to which the R7
substituent is attached is in the (R)-configuration. For example, preferred compounds of Formula I
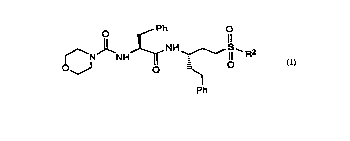
include: N2-(4-morpholinylcarbor"~l)-A/ -(3-phenyl-1 R-phenylsulfonylpropyl)-L-phenylaldni~ ,al " ~c
(compound 1), N2-(4-morpholinylcarbonyl)-Al-(3-phenyl-1s-phenylsulfonylpropyl)-L-phenyalaninamide
CA 022161~1 1997-09-23
W 096/303~3 PCT~US96/03844
22
(compound 2) N2-(4-morpholinylcarbonyl)-N'-(3-phenyl-1 -phenylsulfonylpropyl)-L-phenylalan;. ,a", :' ?
(compound 3) N2-(4-morpholinylcarbonyl)-N'-(3-phenyl-1 -benzylsulfonylpropyl)-L-leuci"ar"' ~e
(compound 4) N2-(4-morpholinylcarbonyl)-N'-(3-phenyl-1 -trifluoromethylsulfonylpropyl)-
L-phenylala" Idl ~ I;de (compound 5) N2-(4-morpholinylcarbonyl)-N'-(3-phenyl-1 -benzylsulfonylpropyl)-
L-phenylalaninamide (compound 6) N2-(4-morpholinylcarbonyl)-N'-(3-phenyl-1-phenylsulfonylpropyl)-
L-leucinamide (compound 7) N2-(4-morpholinylcarbonyl)-N'-(3-phenyl-1-fluolul"~ll,ylsulfonylpropyl)-
L-phenylalani"a",;de (compound 8) N2-(4-morpholinylcarbonyl)-N'-(3-phenyl-
1 S-phenylsulfonylmethylpropyl)-L-phenylalaniamide (compound 9); N2-(4-morpholinylcarbonyl)-
N'~3-phenyl-1S-[2-(2-phenylethylsulfonyl)ethyl] propyl}-L-phenylalanil,a"l,de (compound 10);
N2-(4-morpholinylcarbonyl)-N'-{3-phenyl-1 S-[2-(2-naphthylsulfonyl)ethyl]propyl}-,B-(2-naphthyl)-
L-alaninamide (compound 11); N2-phenylacetyl-N'-[3-phenyl-1S-(2-phenylsulfonylethyl)propyl]-
L-phenylalani"al ~ I;de (compound 12) N2-(N-benzyloxycarbonyl-~-alanyl)-N'-[3-phenyl-
1 S-(2-phenylsulfonylethyl)propyl]-L-phenylalaninamide (compound 13) 3-{2-phenyl-1 S-[3-phenyl-
1 S-(2-phenylsulfonylethyl)propylca, ba" ,oyl]ethylca, l.ar"oyl}propionic acid (compound 14);
3-{2-naphthyl-1 S-[3-phenyl-1 S-(2-phenylsulfonylethyl)propylcarL)al ~ luyl] ethylcarl,an ,oyl}prupionic acid
(compound 15); N2-(4-morpholinylcarbonyl)-N'-{3-phenyl-1S-[2-(2-naphthylsulfonyl)ethyl]propyl}-
L-tyrosi"an,' 'e (compound 16); methyl 3-{2-phenyl-1S-[3-phenyl-
1 S-(2-phenylsulfonylethyl)propylcarba" ,c yl]ethylcarL,an n~yl}propionate (compound 17);
N2-(4-morpholinylcarbonyl)-N'-[3-phenyl-1 S-(2-phenylsulfonylethyl)propyl]-L-phenylalani"al ": J '
(compound 18); N2-(~-alanyl)-N'-[3-phenyl-1S-(2-phenylsulfonylethyl) propyl]-L-phenylalani"a", ~e
(compound 19); and 5-phenylsulfonyl-3S-{N-[N-(N-acetyl-L-tyrosyl)-L-valyl]-L-alanylamino}valeric acid
(compound 20). Preferred compounds of Formula ll include: ethyl 4S-(N-benzylsulfonyl-
~-(2-naphthyl)-L-alanylamino)-6-phenylh~xdlloaLe (compound 21); ethyl 4S-(N-benzylcarL,a"loyl-
~-(2-naphthyl)-L-alanylamino)-6-phenylhexanoate (compound 22); ethyl
4S-[N-(4-morpholinylcarbonyl)-~-2-(naphthyl-L-alanylamino]-6-phenylhexanoate (compound 23); ethyl
4S-(N-benzylcarban,oyl-L-phenylalanylamino)-6-phenylhexanoate (compound 24); ethyl
4S-[N-(4-morpholinylcarbonyl)-L-phenylalanylamino]-6-phenylhexanoate (compound 25);
N2-(4-morpholinylcarbonyl)-N'-[3-phenyl-1 S-(2-phenylcarba" ,uylethyl)propyl]-L-phenylalaninamide
(compound 26); and N2-(4-morpholinylcarbonyl)-N'-[3-phenyl-15-(2-benzylcarbd",-)ylethyl)propyl]-
L-phenylalar ~a",' 'e (compound 27). Preferred compounds of Formula lll include:N2-(4-rl lol ~Jh t' ,ylcarbonyl)-N'-{3-phenyl-1 S-[2-(4-methoxyphenyl)ethyl]propyl}-L-phenylalaninamide
(compound 28); and /\~-(4-morpholinylcarbonyl)-Nt~{3-phenyl-1 S-[2-(4-aminophenyl)ethyl]propyl}-
L-phenylalani"d" ' 'e (compound 29).
As will be appr~. idLed by those in the art Formula I includes structures rep,~senLed by preferred
CA 022161~1 1997-09-23
W 096/303~3 PCTfUS96/03844
23
Species IV as deF ' -' below.
R7 1~
- \ Q 1 A~ ~ M RZ
IV
wherein M is zero, one or two carbon atoms, A-B are as defined above, R1, RZ, R7 and R8 are as
defined above, and Q is NH or CH2. Preferred embodiments utilize A-B linkages which contain
nitrogen at the B position. In this embodiment, the number of carbon atoms between the carbon to
which the R~ group is attached and the sulfur atom of the sulfone group determines whether the
compound is an a-aminosulfone, a ~-aminosulfone, or a y-aminosulfone. As is ~iiscussed below in the
Examples, compounds may be named as aminosulfones using the names of the amino acids or using
the chemical names.
Thus, for example, Species V is an a-aminosulfone:
Rl~ 1 B~ ~RZ
Species Vl is a ~-aminosulfone:
R7
\ Q ~ A~ ~ ~ RZ
R8 O
Vl
Species Vll is a y-aminosulfone:
R7 RZ
VII
Formula 11 includes structures of Species Vlll, referred to as y-amino groups, particularly when R9 is an
electron withdrawing group:
R7
Q ~ A~ ~ R9
VIII
CA 022161~1 1997-09-23
096/30353 PCTrUS96/03844
24
In a preferred er"bodi",ent, the di~socidlion constant for inhibition of a protease with an inhibitor of the
invention, generally referred to by those in the art as K" is at most 100 ,uM. By the term "binding
consldr"" or "di:,socidLion constant" or ~ dl "" ~atical equivalents herein is meant the equilibrium
dissociation constant for the reversible association of inhibitor with enzyme. The d i:,so~,ialion
constants are defined and determined as below.
The determination of dissociation constants is known in the art. For example, for reversible inhibition
,~a~Lions such as those of the present invention, the reaction scheme is as follows:
Equation 3
E +l ~ E
The enzyme and the inhibitor combine to give an enzyme-inhibitor complex, E-l. This step is assumed
to be rapid and reversible, with no chemical changes taking place, the enzyme and the inhibitor are
held together by non-covalent forces In this reaction, k, is the second order rate constant for the
formation of the E-l reversible complex. k2 is the first order rate constant for the disassociation of the
reversible E-l complex. In this reaction, K, = k2/k,.
The measurement of the equilibrium constant K, proceeds according to techniques well known in the
art, as described in the examples. r or example, assays generally use synthetic chromogenic or
fluorogenic sub~L,dL~s.
The respective K, values may be estimated using the Dixon plot as described by Irwin Segel in
Enzyme Kinetics: Behavior and analysis of rapid equilibrium and steady-state enzyme systems, 1975,
Wiley-l"L~:r:,cience Pllbliç~tion, John Wiley ~ Sons, New York, or for competitive binding inhibitors
from the following c~lcl li:~tion:
Equation 4
1-(vj/vo) = [1]/([1] + K,(1+([S]/KM)))
wherein
vO is the rate of substrate hydrolysis in the absence of inhibitor, and v; is the rate in the presence of
competitive inhibitor.
It is to be understood that dissociation constants are a particularly useful way of quantifying the
CA 022161~1 1997-09-23
W 096/30353 PCTAUS96/03844
effficiency of an enzyme with a particular substrate or inhibitor, and are frequently used in the art as
such. lf an inhibitor exhibits a very low K" it is an effficient inhibitor. Accordingly, the cysteine protease
inhibitors of the present invention have di~socidLion constants, K" of at most about 100,uM. Preferred
embodiments have inhibitors that exhibit ~ so- ;alion constants of at most about 10 ~M, with the most
preferred e,l,bocl;",enL:, having ~issoriation consLd,lL:, of at most about 1 ,uM.
CHEMISTRY
The synthesis of the inhibitors of the invention proceeds as follows. Compounds of Formula I in which
X represents a bond can be prepared by the process depicted in Scheme 1 of Figure 1.
Treatment of tert-butylcalL,an,dL~ or benzyl carbamate with an appropriate aldehyde, such as
isobutyraldehyde or hydrocinnamaldehyde, along with the sodium salt of a suitable sulfinic acid, such
as benzenesulfinic acid (Aldrich Chemical Co.), in the presence of aqueous formic acid affords the
corresponding N-protected aminomethyl sulfone. Benzyloxycarbonyl protected aminomethyl sulfones
are deprotected with hydrogen bromide in acetic acid. Coupling with a suitable N-protected amino
acid or peptide or a peptidomimetic derivative thereof affords a compound of Formula I in which X
represents a bond. Alternatively, an appropriate N-terminal protected amino acid or peptide of
peptidomimetic derivative thereof, such as N-(4-morpholinylcarbonyl)phenylalaninamide, is reacted
with an appropriate aldehyde along with the sodium salt of a suitable sulfinic acid, in the presence of
aqueous formic acid to aflord a compound of Formula I in which X repr~senL:, a bond.
Compounds of Formula 1 in which X represents a methylene bond can be prepared by the processes
depicted in Schemes 2 and 3, Figures 2 and 3, respectively.
Treatment of a suitable N-protected amino acid or peptidon,i",eLic derivative thereof with sodium
borohydride affords the corresponding ~-aminoethanol. Treatment of the alcohol with methanesulfonyl
chloride in the presence of triethylamine affords the corresponding mesylate. Nucleophilic
displacementwith the anion of a thiol, such as thiophenol, accol.lillg to the method of Spall~n:,L~i", A.,
Carpion, P., Miyake, F., and Hopkings, P.B., J. Org. Chem (1987) 52, 3759, affords the cor,~sponding
~-aminosulfide. The sulfide is oxidized by means of 4-chloruperL,enzoic acid to give the corresponding
N-protected ,B-aminoethyl sulfone. In a special instance, the mesylate is treated with thiolate ion such
as that derived from 2-(trimethylsilyl)ethanethiol, the synthesis of which is described by Anderson,
M.B., Ranasinghe, M.B., Palmer, J.T., Fuchs, P.L., J. Org. Chem. (1988) 53, 3125, to give the
corresponding ~-aminoethyl 2-trimethylsilylethyl sulfide. The 2-trimethylsilylethyl sulfide is reduced to
the corresponding ,B-aminoethyl 2-trimethylsilylethyl sulfone, which is then s~ to
fluoride-mediated cleavage, extruding trimethylsilyl fluoride and ethene as gaseous by-products, and
CA 022161~1 1997-09-23
W 096/30353 PCTrUS96/03844
26
an intermediate sulfinate, which sulfinate is alkylated in situ with an appropriate halogen-col,ldi"i.,g
species such as bru",ochlorun,etlla(le to give the corresponding N-protected ~-aminoethyl h '-n,~:ll,yl
sulfone. The N-protected ,B-aminoethyl sulfones are deprotected and then coupled with a suitable
N-protected amino acid or peptide or a peptidon ,;. "t:lic derivative thereof to afford a compound of
Formula I in which X is methylene.
Compounds of Formula ll and Formula I in which X represents ethylene can be prepared by the
processes depicted in Equations 5, 6 and 7.
Equation 5
O O o
Bo~ N ~ OH H ~ ~ ~ \ Boc--N ~ H
R8 a) R8 I b) Rs
wherein a) is a) Cl-H2N+(Me)OMe, dicyclohexylcarbodiimide, triethylamine; and b) lithium aluminum
hydride.
Equation 6
R'O~¦¦
Boc--N ,Jl~ R'O~ ~ Boc--N EWG
R"
R8 R'~ I R5
R~ ~ EWG
Equation 7
B N EWG H,~d B H EW
R8 , R--8
An app,upridl~ N-tert-butoxycarbonyl amino acid or peptidomimetic derivative thereof is converted to
the corresponding aminomethyl aldehyde (e.g., see method of Fehrentz, J-A. and Castro, B.
(Synthesis, (1983), 676; Equation 5). The aldehyde is converted to the corresponding vinylogous
compound via aWittig reaction or a Wadsworth-E~I "" ,ons-Horner "~odiricdLion of the Wittig reaction
(e.g., see Wadsworth et al., J. Amer. Chem. Soc. 83: 1733 (1991); Equation 6). The vinylogous
compound is reduced by catalytic h~,d,ugendlior! (e.g., see Equation 7) and then deprc,L~cLion and
coupling with a suitable N-protected amino acid or peptide or a peptidomimetic derivative thereof gives
CA 022161~1 1997-09-23
W 096/30353 PCT~US96/03844
Z7
the col,~spond;~lg compound of Formula I or ll. Alternatively, the vinylogous compound is deprotected
and coupled with the N-protected amino acid or peptide or a peptidonl lletic derivative thereof to give
the corl~uonding vinyloguous condensation product, which is then reduced to give the corresponding
compound of Formula I or ll.
Compounds of Formula ll can be prepared by the processes depicted in Scheme 4, Figure 4.
Preferably, the conversion of N-tert-butoxycarbonyl amino acid or peptidomimetic derivative thereof to
the corresponding aminomethyl aldehyde is carried out with N,O-dimethylhydroxylamine hydrochloride
in the presence of triethylamine and dicyclohexylcarbodiimide in dicloromethane. Alternatively, the
conversion is carried out by treating the amino acid or peptidomimetic derivative with triethylamine and
the coupling agent benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate (BOP)
and then reducing with lithium aluminum hydride to give the conesponding aldehyde (e.g., see methos
of Fehrentz, J-A. and Castro, B.; Synthesis, (1983), 676-678). The conversion of the aldehyde to the
corresponding vinylogous ester can be carried out with the sodium anion of triethyl phosphonoacel~
Deprotection of the vinylogous ester can be carried out with hydrogen chloride in dioxane. The
hydrogenation is typically carried out in the presence of palladium.
Compounds of Formula I in which X is ethylene are conveniently prepared by the process depiGt~d in
Scheme 5, Figure 5.
Treatment of a suitable N-tert-butoxycarbonyl-~-aminoaldehyde, prepared as described in Equation 5,
with the sodium anion of an apprupl i~l~ sulfonylmethanephosphonate (SMP) (e.g, diethyl
phenylsulfonyllllelhanephosphonate, diethyl 2-naphthylsulfonylmethane-phosphonate, diethyl
methylsulfonylmethanephosphonate, etc.) gives the corl,:sponding vinylogous sulfone. The sulfone is
deprotected with anhydrous p-toulenesulfonic acid in ether and then coupled with N-protected amino
acid or peptide or a peptidol 1, 1 letic derivative thereof to give the corresponding vinyloguous
condensdliol, product, which is then reduced to give the corresponding compound of Formula 1.
Suitable arylsulfonylllletllanephsophonates can be pl~par~d by treating arylthiols with
pararurlll-ldehyde in the pl~sence of hydrogen chloride and reacting with triethyl phosphite to give the
corresponding diethyl phsophonomt:Lllyl aryl sulfide and then oxidizing the sulfide. Alternatively,
suitable sulfides can be obtained collllllen ~"y (e.g., diethylphosphonolllelhyl methyl sufide obtained
from Aldrich Chemical Co., diethylphosphononl~:Lllyl phenyl sulfide, etc.) and oxidized to their
corresponding sulfones.
Compounds of Formula ll in which R9 is -COOH can be pl~pal~d by the process depicted in
Scheme 6, Figure 6.
CA 022161~1 1997-09-23
W 096/30353 PCT~US96/03844
28
Generally, saponiricdLion of a compound of Formula 11 in which R9 is -COOR'~ results in the
coo,~spondil ,9 carboxylate, which upon L,~dLI"er,L with acid gives the corresponding carboxylic acid.
Compounds of Formula ll in which R9 is -P(O)(R'~)2 can be prepared as depicted in Scheme 7 (Figure
7) by proceeding as in Scheme 6, but substituting for the SMP the sodium anion of an appropriate
methylenedi,.llosphonate (e.g., tetraethyl methylenediphosphonate, etc.).
Compounds of Formula 11 in which R9 is -C(O)NHR14 can be prepared as depicted in Scheme 8
(Figure 8) by proceeding as in Scheme 5, but substituting for the SMP an app,upridLe diethyl
amidomethylenephsophonate (e.g, diethyl benzylamidomethylenephosphonate, etc.).
Suitable amidomethylenephosphonates can be prepared by reacting the saponification product of
triethyl phosphonoacel,,~ with an appruplidL~ amine. Altenatively, compounds of Formula 11 in which
R9 is -C(O)NHR'4 can be prepared by reacting a compound of Formula I in which R9 is -COOH with an
al,prupridlt: amine. For example, the reaction can be carried out in the presence of
dicyclohexylcarbodiimide in dichloromethane or by any other peptide coupling reaction sequences
known to those of skill in the art.
In general, compounds of Formula 11 can be prepared by the process depicted in Scheme 9 (Figure 9)
and substituting the starting materials represented by Structures l-VII.
Structure I
O O
~ / Rl
Synthesis of ketones is performed by means of the Wadsworth-Emmons reaction between Boc-~-
amino aldehydes and the apprupridL~ phosphonate, followed by catalytic reduction with hydrogen in
the presence of palladium. Generally, the aldehyde portion is synthesized as outlined above. The
pho:",hondL~ :, if not cor ,. . .er~ ~ Ily available, is synthesized by l- ~dL- . .~. .L of the enolate anion of methyl
or 5~~hstit~~t~d methyl ketones, such as acetone or acetophenone, with diethyl chlo,uphosphonate.
The enolate anion is generated, for ~xdr"~,,le, by L, ~dL. "er,L of a tetrahydrofuran solution of
di;sop,upylamine with butyllithium, followed by addition of the ketone to the lithium diisopropylamide
(LDA) solution (H.O. House, Modern Synthetic Rea~,Lions, 2nd Ed. (W. Benjamin, Inc., Menlo Park,
CA, Chapter 9). Following rul " IdLion of the enolate, diethyl cl-loruphospl-ond~ is added. The
Wadsworth ~"""ons reagent forms as a consequence of coupling of the enolate with diethyl
chlorophosphate.
CA 022161~1 1997-09-23
W 096/303~3 PCTrUS96/03844
29
For the synthesis of cysteine protease inhibitors with nitriles as the EWG, structure ll is used:
Structure ll
o
C ~O~ I
' C - N
C2~0 ~/
Synthesis of nitriles is performed by means of the Wadsworth-Emmons reaction between Boc-a-
amino aldehydes and the appropriate phosphonate, followed by hydrogenation in the presence of a
suitable catalyst. Generally, the aldehyde portion is synthesized as outlined above. The phosphonate
is commercially available.
For the synthesis of cysteine protease inhibitors with sulfoxides as the EWG, structure lll is used:
Structure lll
O O
C_~
C.~O \/ Rl
Synthesis of sulfoxides is performed by means of the Wadsworth-Emmons reaction between Boc-a-
amino aldehydes and the appropriate phosphonate, followed by hydrogenation in the presence of a
suitable catalyst. Generally, the aldehyde portion is synthesized as outlined above. The phosphonate
is synthesized by treatment of the anion of methyl sulfoxides with diethyl chlorophosphate. The anion
is generated by addition of BuLi to diisopropylamine, followed by addition of the methyl sulfoxide.
For the synthesis of cysteine protease inhibitors with sulr~na" ,:~es as the EWG, structure IV is used:
Structure IV
o
C,H50 ~¦¦
C~H50 ~'~' NHRIJ
)
Synthesis of sulr~ al, :'es is performed by means of the Wadsworth-EmmGns reaction between Boc-
a-amino aldehydes and the appr~,pridL~ phosphonate, followed by hydrogenation in the presence of a
suitable catalyst. Generally, the aldehyde portion is synthesized as outlined above. The phosphonate
is synthesized, for instances, by a method such as the rc~ ,, i"y. a) diethylphosphoryl
methanesulfonates, as prepared by the method of Carretero and Ghosez (Tetrahedron Lett., 28:1104-
1108 (1987)), are converted to sulfonyl chlorides by L,~:dL",enlwith phosphorus penldchloride (M.
Quaedvlieg, in "Methoden der O(yanische Chemic (Houben-Weyl)", ed. E. Muller, Thieme Verlag,
Stuttgart, 4th Ed., 1955, Vol. IX, Chapter 14); or b) treatment of the sulfonyl chloride with an amine,
CA 022161~1 1997-09-23
W 096/30353 PCT~US96/03844
such as ammonia, a primary amine (including an amino acid derivative), or a secondary amine, that
results in the rur" ,dlion of the sulfonamide (Quaedvlieg, supra, Chapter 19). The sulfonamide-
phosphonate is then reacted with Boc-a-aminoaldehydes to form the target compounds as per the
Wadsworth-Emmons reaction.
For the synthesis of cysteine protease inhibitors with sulfinamides as the EWG, structure V is used:
Structure V
O O
C,H~O~II 11
~,o \/ NHE~-
Synthesis of sulfinamides is performed by means of the Wadsworth-Emmons reaction between Boc-a-
amino aldehydes and the appropriate phosphonate, followed by hydrogenation in the presence of a
suitable catalyst. Generally, the aldehyde portion is synthesized as outlined above. The phosphonate
may be synthesized using one of the following methods. Treatment of methyl dialkyl phosphonates
such as the commercially available methyl diethyl phosphonate (Aldrich), with thionyl chloride in the
presence of aluminum chloride gives the dialkylphosphoryl methanesulfinyl chloride (Vennstra et a/.,
Synthesis (1975) 519. See also Anderson, "Comprehensive Organic Chemistry (Pelya",on Press)",
Vol. 3, Chapter 11.18, (1979). Alternatively, treatment of the dialkyl phosphoryl sulfinyl chloride with
amines (Stirling, Internat. J. Sulfur Chem. (B) 6:277 (1971)), yields the dialkyl phosphoryl sulril,d",;de.
For the synthesis of cysteine protease inhibitors with sulfoximines as the EWG, structure Vl is used:
Structure Vl
O O
C,~O\ ¦ ¦ --Rl~
C~O
Rl~
Synthesis of sulfoximines is performed by means of the Wadsworth-EI,,,,,ons reaction between Boc-a-
amino aldehydes and the apprupridL~ phosphonate, followed by hydrogenaiion in the presence of a
suitable catalyst. Generally, the aldehyde portion is synthesized as outlined above. The phos~,hondL~
may be synthesized in several ways. For example, N-alkyl or N-aryl phenyl methyl sulfoximines are
made by the Illt:lhods described by Johnson, in "Comprehensive Organic Chemistry (~erydllloll
Press), supra, Chapter 11.11. Alternatively, the lithium anion of compounds such as N-alkyl phenyl
methyl sulfoximine is prepared by the treatment of the neutral compound with buthyl lithium in THF
(Cram et al., J. Amer. Chem. Soc. 92:7369 (1970)). Reaction of this lithium anion with dialkyl
chlorophosphdLt s such as the co"""e,l -"y available diethyl cl,lor~.phosphdLe (Aldrich) results in the
CA 022l6l~l l997-09-23
W 096/30353 PCTrUS96/03844
31
Wadsworth-Emmons reagent necessary for synthesis of the sulfoximine compounds.
For the synthesis of cysteine protease inhibitors with sulfonates as the EWG, structure Vli is used:
Structure Vll
o
C~H~jO~ ¦ ¦
C2H,O / SO3-M+
Synthesis of sulfonates is performed by means of the Wadsworth-Emmons reaction between Boc-a-
amino aldehydes and the appropriate phosphonate, for instance diethylphosphoryl methanesulfonate,
followed by hydrogenation in the presence of a suitable catalyst, such as Raney nickel. The
phosphonate may be synthesized as follows. The anion of methyl dialkyl phosphonates such as the
commercially available methyl diethyl phosphonate (Aldrich) is generated by treatment of said
phosphonate with a strong base such as LDA. The resulting anion is sulfonated with sulfur
trioxide/trimethylamine complex (Carreto et a/., Tetrahedron Lett., 28:1104-1108 (1987)) to form
diethylphosphoryl methanesulfonate, which is capable of reacting in the Wadsworth-Emmons
procedure with aldehydes to form a,l3-unsaturated sulfonates.
Compounds of Formula ll can be pr~pa,t:d by the process depicted in Scheme 10 (Figure 10).
The chloride compounds containing R8 and Rg groups are generally made using commercially
available reagents and products using techniques well known in the art. The reaction generally
produces a mixture of cis and trans configurations, favoring the trans isomer. Upon reduction to the
cysteine protease inhibitors of this embodiment, the cis-trans isomerism disappears by definition as a
single compound is formed.
In one embodiment, the cysteine protease inhibitors of the invention are further purified if necessary
after synthesis, for example to remove unreacted Illdtt:r;~ls. For example, the cysteine pruL~ase
i"hibiLùr~ of the present invention may be cry~' " 3d, or passed through silica clllullldLuyldphy
columns using solvent mixtures to elute the pure inhibitors.
In summary, the prucesses for preparing compounds of the invention are as follows:
(A) for the p~e:pa~ ~Lion of Formula IV:
o RX
R Z ~ B~ ~A~ --z JI N 1 20
H
IV
CA 022161~1 1997-09-23
W 096/30353 PCTrUS96/03844
in which n is O to 12; R20 is cyano, -S(0)2R2, -CH2S(0)2R2, -CH2CH(R4)S(0)2R2, -(CH2)2C(O)OR1~,
-(CH2)2P(O)(OR'~)2, -(CH2)2S(O)(NRl~)Rl~, -CH2)2C(O)Rll, -(CH2)2S(O)Rll, -(CH2)2C(O)NRl2Rl3,
-(CH2)2S(o)2NR12R13, -(CH2)2C(o)NHR14, -(CH2)2S(o)2NHR14 or -CH2CHR15R16 and each A, B, X, Y, Z,
R', R8 R', R8, R2, R'~, R", R'2, R'3, R'4, R'5 and R16 are as defined in the Summary of the Invention
with respect to compounds of Formulae 1, ll and lll, and the pha",~ceutic~lly acc-~pl~hle salts,
individual isomers and mixtures of isomers thereof,
reacting an amine of Formula V:
R~
H I N
with a compound of the Formula Vl:
~y~ ~ A~ ~ Z\A~ \Z ~ OH
Vl
in which each n, A, B, X, Y, Z, R', R8 and R20 are as defined above,
(B) for the preparation of a compound of Formula IV in which R20 is -S(0)2R2, and the
pharrn~cel~ti~:~lly acce~Ldble salts, individual isomers and mixtures of isomers thereof,
reacting a compound of Formula Vll:
Z ~A~ ~Z ~ NH.
Vll
with an aldehyde of the formula R8CHO and a sodium sulfinate of the formula R2S(O)ONa, in which
each n, A, B, X, Y, Z, R' and R3 are as defined above;
(C) for the p,~pd,dLion of a compound of Formula IV in which R20 is -S(0)2R2, and the
phdl,l'aceutically acce,t-''o salts, individual isomers and mixtures of isomers thereof,
(1) reacting a compound of the formula NH2P, wherein P is a protective group, with an
aldehyde of the formula R8CHO and a sodium sulfinate of the formula R2S(O)ONa and then
depluLe-,Lil,g to give a compound of Formula Vlll:
H~N lS(O) 2R2
CA 022161~1 1997-09-23
W 096/30353 PCTrUS96/03844
33
in which each R2 and R3 are as defined in the Summary of the Invention with respect to Formula l; and
(2) reacting the compound of Formula Vlll with a compound of Formula Vl in which each
n, A, B, X, Y, Z and R' are as defined above;
(D) for the preparation of a compound of Formula IV in which R20 is -CH2S(0)2R2 and the
pharmaceutic~lly accepL~L,le salts, individual isomers and mixtures of isomers thereof,
(1) reacting a compound of Formula IX:
R~
PHN ,
with a thiolate anion of the formula R2S-, in which L is a leaving group R2 and R3 are as defined above,
to give a compound of Formula X:
~l~,SR2
PHN
(2) oxidizing the compound of Formula X to give a compound of Formula Xl:
R~
P~N ~l' ~s(~) ~R-
and
(3) reacting the compound of Formula Xl with a compound of Formula Vl in which each n,
A, B, X, Y, Z and R' are as defined above;
(E) for the prt:pardlion of a compound of Formula IV in which R20 is cyano, -(CH2)2S(0)2R2,
-(CH2)2C(O)OR'~,-(CH2)2P(O)(OR'~)2,-(CH2)2S(O)(NR'~)R'~,-(CH2)2C(O)R",-(CH2)2S(O)R",
-(CH2)2C(O)NR'2R'3, -(CH2)2S(0)2NR'2R'3, -(CH2)2C(O)NHR'4 or -(CH2)2S(0)2NHR'4, and the
pharmaceutically acceptable salts, individual isomers and mixtures of isomers thereof,
(1) reacting an aldehyde of Formula Xll:
R~
PHN CHO
with a compound select~d from Formulae Xlll and XIV:
O R-l
R210~¦¦ R2o R-l\ll R~~
XIII XIV
in which each R3 and R23 are as defined above, and then deprotecting to give a
CA 022161~1 1997-09-23
W 096/30353 PCTrUS96103844
34
compound of Formula XV: R8
o
XV
(2) reacting the compound of Formula XV with a compound of Formula Vl in which each
n, A, B, X, Y, Z and R' are as defined above, and
(3) reduang;
(F) for the pl~pa,dlion of a compound of Formula IV in which R20 is -CH2CHR'sR'6 and the
pharmaceutically accepldble salts, individual isomers and mixtures of i~omers thereof,
(1 ) reacbng an aldehyde of Formula Xll with compound of Formula XVI:
Rl~
= P(Ph)3
R16
XVI
in which each R8, R's and R16 are as defined above, and then deprotecting to give a compound of
Formula XVII:
R8 Rl S
N~/~R,6
XVII
(2) reacting the compound of Formula XVII with a compound of Formula Vl in which each
n, A, B, X, Y, Z and R' are as defined above, and
(3) reducing;
(G) optiona"y further converting a non-salt form of a compound of Formula IV into a
phal".~ceutic?"y accep' '' salt;
(H) oplionally further converting a salt form of a compund of Formula IV into non-salt form; and
(H) opLionally further separdli"g a compound of Formula IV into individual slereoi~ullle,~.
In one embodiment, the cysteine protease in~ b ' -:, of the present invention are labelled. By a
"labelled cysteine prul~ase inhibitor" herein is meant a cysteine pl uL~ase inhibitor that has at least one
element, isotope or chemical compound attached to enable the detection of the cysteine pl ult:ase
inhibitor or the cysteine pl ul~ase inhibitor bound to a cysteine ~lutease. In general, labels fall into
three classes: a) isotopic labels, which may be ,dLlioa~;Li~e or heavy iso~upes; b) immune labels, which
CA 022161~1 1997-09-23
W 096/303~3 PCTfUS96/03844
may be anLiLJodies or antigens; and c) colored or fluor~scenL dyes. The labels may be incorporated
into the cysteine protease inhibitor at any position. For example, a label may be attached as the "R"'
group in Formula 1, or a Iddioisolope incor~ordLed into any position. Examples of useful labels include
14C, 3H, biotin, and fluorescent labels as are well known in the art.
PHARMACOLOGY AND UTILITY
Once produced, the cysteine protease inhibitors of the present invention may be easily screened for
their inhibitory effect. The inhibitor is first tested against the cysteine protease for which the targeting
group of the inhibitor was chosen, as outlined above. Alternatively, many cysteine proteases and their
corresponding chromogenic substrates are commercially available. Thus, a variety of cysteine
proteases are routinely assayed with synthetic chromogenic substrates in the presence and absence
of the cysteine protease inhibitor, to confirm the inhibitory action of the compound, using techniques
well known in the art. The effective inhibitors are then 5llhject~d to kinetic analysis to c~lclllatf~ the K,
values, and the dissociation constants determined.
If a compound inhibits at least one cysteine prc,t~:ase, it is a cysteine protease inhibitor for the
purposes of the invention. Preferred embodiments have inhibitors that exhibit the correct kinetic
parameters against at least the targeted cysteine protease.
In some cases, the cysteine protease is not commercially available in a purified form. The cysteine
protease inhibitors of the present invention may also be assayed for efficacy using biological assays.
For example, the inhibitors may be added to cells or tissues that contain cysteine proteases, and the
biological effects measured.
In one embodiment, the cysteine protease inhibitors of the present invention are synthesized or
modified such that the in vivo and in vitro proteolytic deyldddLion of the inhibitors is reduced or
prevented. Generally, this is done through the incorporation of synthetic amino acids, derivatives, or
substituents into the cysteine protease inhibitor. Preferably, only one non-naturally occurring amino
acid or amino acid side chain is incorporated into the cysteine protease inhibitor, such that the
Ldlyelillg of the inhibitor to the enzyme is not significantly affected. However, some embodilllerlL~ that
use longer cysteine protease il IhiL,itor:, containing a number of Ldry~Lil lg residues may tolerate more
than one synthetic derivative. In addition, non-naturally occurring amino acid substituents may be
designed to mimic the binding of the naturally occurring side chain to the enzyme, such that more than
one synthetic substituent is tolerated. Alternatively, peptide isosteres are used to reduce or prevent
inhibitor deyl dddLion.
CA 022161~1 1997-09-23
W 096/30353 PCTrUS96/03844
36
In this embodiment, the resistance of the modified cysteine p~ ut~ase inhibitors may be tested against a
variety of known commercially available proteases in vitro to determine their proteolytic stability.
Promising candidates may then be routinely screened in animal models, for example using labelled
inhibitors, to determine the in vivo stability and efficacy.
Specific cysteine pruL~ases that may be inhibited by the inhibitors of the present invention are those of
the family of cysteine p~uL~:ases that bear a thiol group at the active site. These pluL~ ases are found in
bacteria, viruses, eukaryotic r~ uOryal1iSmS~ plants, and animals. Cysteine plul~ases may be
generally ~ s~ified as belonging to one of four or more distinct supelrdlll;!ics. Examples of cysteine
proteases that may be inhibited by the novel cysteine protease inhibitors of the present invention
include, but are not limited to, the plant cysteine proteases such as papain, ficin, aleurain, oryzain and
auli"idai"; mammalian cysteine proteases such as cathepsins B, H, J, L, N, S, T, O, and C, (cathepsin
C is also known as dipeptidyl peptidase 1), interleukin converting enzyme (ICE), calcium-activated
neutral proteases, calpain I and ll; bleomycin hydrolase, viral cysteine proteases such as picoll,ian 2A
and 3C, aphthovirus endopeptidase, cardiovirus endopeptidase, comovirus endopeptidase, potyvirus
endopeptidases I and ll, adenovirus endopeptidase, the two endopeptidases from chestnut blight
virus, togavirus cysteine endopeptidase, as well as cysteine proteases of the polio and rhinoviruses;
and cysteine proteases known to be essential for parasite lifecycles, such as the proteases from
species of Plasmodia, Entamoeba, Onchocera, Trypansoma, Leishmania, Haemonchus,
Dictyostelium, Therileria, and Schisfosoma, such as those associated with malaria (P. falciparium),
trypanosomes (T. cruzi, the enzyme is also known as cruzain or cruzipain), murine P. vinckei, and the
C. elegans cysteine protease. For an extensive listing of cysteine prul~ases that may be inhibited by
the cysteine pr~L~ase i, Ihibilur~ of the present invention, see Rawlings et al., Biochem. J. 290:205-218
(1993), hereby expressly incorporated by reference.
Accordingly, inhibitors of cysteine proteases are useful in a wide variety of a~JplicdLions. For example,
the i, IhiL,ilc,r~ of the present invention are used to quantify the amount of cysteine p, ul~ :ase present in
a sample, and thus are used in assays and diagnostic kits for the qud"liricdlion of cysteine p,ul~ases
in blood, Iymph, saliva, or other tissue samples, in addition to bacterial, fungal, plant, yeast, viral or
Illdllllllalidll cell cultures. Thus in a preferred embodiment, the sample is assayed using a sldnda,-l
protease substrate. A known concentration of cysteine protease inhibitor is added, and allowed to
bind to a particular cysteine protease present. The protease assay is then rerun, and the loss of
activity is correlated to cysteine protease activity using techniques well known to those skilled in the
art.
The cysteine protease inhibitors are also useful to remove or inhibit cor,La,l,;"dLi"g cysteine pruL~ases
in a sample. For e,~dn, ' , the cysteine p, olt:ase inhibitors of the present invention are added to
sa", 1- s where proteolytic dey~ dddLion by conLd" ,;, IdLil ,9 cysteine pruL~ases is undesirable.
CA 022161~1 1997-09-23
W O 96/30353 PCTrUS96/03844
37
Alternatively, the cysteine protease inhibitors of the present invention may be bound to a
chromatographic support, using techniques well known in the art, to form an affinity clllullldaluyld~Jlly
column. A sample containing an undesi,dl;lc cysteine protease is run through the column to remove
the protease.
In a preferred embodiment, the cysteine protease inhibitors are useful for inhibiting cysteine proteases
i""~licated in a number of diseases. In particular, cathepsins B, L, and S, cruzain, calpains I and ll,
and interleukin 1 C~ converting enzyme are inhibited. These enzymes are examples of Iysosomal
cysteine proteases illl~JlicdL~d in a wide spectrum of ~lise~ses chard-.L~rized by tissue dey,dddLion.
Such diseases include, but are not limited to, arthritis, muscular dystrophy, inflan " "dLion, tumor
invasion, glomerulonephritis, parasite-borne infections, Alzheimer's disease, periodor,Ldl disease, and
cancer metdstdsis For example, mammalian Iysosomal thiol proteases play an i" ,,,~o, Ldnt role in
intracellular dey,ddatio,l of proteins and in the processing of some peptide hormones. Enzymes
similar to cathepsins B and L are released from tumors and may be involved in turnor ",etd~Lasis.
Cathepsin L is present in diseased human synovial fluid and transformed tissues. Similarly, the
release of cathepsin B and other Iy:,osol"al p,uL~:ases from polymorphonuclear granulocytes and
macrophages is observed in trauma and i"rla",n,dLion.
The cysteine protease inhibitors also flnd ~l~plicc1~ion in a multitude of other diseases, including, but not
limited to, gingivitis, malaria, leiahr"aniasis, filariasis, and other bacterial and parasite-borne i"r~ :..Lions.
The compounds also offer applicaLion in viral diaeases, based on the approach of inhibiting pruL~ases
necessary for viral replication. For example, many picornoviruses including poliovirus, foot and mouth
disease virus, and rhinovirus encode for cysteine pruL~ases that are essential for cleavage of viral
polyproteins.
Additionally, these compounds offer applicaLion in disorders involving interleukin-1 r~ converting
enzyme (ICE), a cysteine pruLt:ase responsible for processing interleukin 113; for ~:Xdlll, le, in the
treatment of illrldlllllldLion and immune based disorders of the lung, airways, central nervous system
and surrounding ",e",brdnes, eyes, ears, joints, bones, connective tissues, cardiovascular system
including the perica,di-lm, gasL,~i"L~aLi"al and urogenital systems, the skin and the mucosal
l"el"L"dnes. These conditions include infectious ~ -.cs where active infection exists at any body
site, such as meningitis and - ~ . IgiLi:,; co" r ~ "-ns of i"rt:cLions including septic shock,
diaser";"dl~d intravascular coagulation, and/or adult ,. r:.dLury distress syndrome; acute or chronic
illnd"""aLion due to antigen, antibody and/or cor", '~."ent deposiLion; i"nd"""aLury condiLions
including arthritis, ~,I,alanyiLis, colitis, encep~ ' endocarditis, glomerulonepl1riLi~, hepatitis,
myocd,.liLi:,, panc;,~dLili:,, perica,dili~, reperfusion injury and vascu' . Immune-based ~I;;eases
include but are not limited to condilions involving T-cells and/or macrophages such as acute and
CA 022161~1 1997-09-23
W 096/30353 PCTAJS96/03844
38
delayed hypersensitivity, graft rejection, and graft-versus-host disease; auto-immune d;~ases
including Type I diab~l~s mellitus and multiple sclerusia Bone and cal lilage reabso, ~Lion as well as
seases resulting in excessive deposition of extracellular matrix such as interstitial pulmonary fibrosis,
cirrhosis, systemic sclerosis, and keloid rul ,l ,dlion may also be treated with the inhibitors of the present
invention. The inhibitors may also be useful in the L,~:dl",ent of certain tumors that produce IL 1 as an
autocrine growth factor and in preventing the cachexia associated with certain tumors. Apoptosis and
cell death are also associdL~d with ICE and ICE-like activities and may be treated with the inhibitors of
the present invention.
Furthermore, the cysteine protease inhibitors of the present invention find use in drug pot~nlidlion
applications. For example, therapeutic agents such as dnliL,iotics or antitumor drugs can be
inactivated through proteolysis by endogeneous cysteine proteases, thus rendering the administered
drug less effective or inactive. For example, it has been shown that bleomycin, an antitumor drug, can
be hydrolyzed by bleomycin hydrolase, a cysteine protease (see Sebti et al., Cancer Res. January
1991, pages 227-232). Accordingly, the cysteine protease inhibitors of the invention may be
administered to a patient in conjunction with a therapeutic agent in order to potentiate or increase the
activity of the drug. This co-administration may be by simultaneous administration, such as a mixture
of the cysteine protease inhibitor and the drug, or by separate simultaneous or sequential
administration .
In addition, cysteine protease inhibitors have been shown to inhibit the growth of bacteria, particularly
human pathogenic bacteria (see Bjorck et al., Nature 337:385 (1989)). Accordingly, the cysteine
protease inhibitors of the present invention may be used as anliba-,L~rial agents to retard or inhibit the
growth of certain bacteria.
The cysteine protease inhibitors of the invention also find use as agents to reduce the damage of
bacterial cysteine proteases to host organisms. For example, staphylococcus produces a very active
extracellular cysteine protease which degrades insoluble elastin, possibly contributing to the
connective tissue destruction seen in bacterial infections such as septicer,-id, septic arthritis and otitis.
See Potempa et al., J. Biol. Chem 263(6):2664-2667 (1988). Accorcli"yly, the cysteine protease
i"hibiLura of the invention may be used to treat bacterial infections to prevent tissue damage.
ADMINISTRATION AND PHARMACEUTICAL COMPOSITION
In general, cysteine protease inhibitors of this invention will be ad" ,;. ,ialert:d in therapeutically effective
amounts via any of the usual and acc~ptz~e modes known in the art, either singly or in co.-,' ldLio,
with another cysteine protease inhibitor of the invention or with another therapeutic agent. A
CA 022161~1 1997-09-23
W 096130353 PCTrUS96/03844
39
therapeutically effective amount may vary widely depending on the severity of the disease, the age
and relative health of the subject, the potency of the compound used and other factors.
Ther~reutic~lly effective amounts of the cysteine protease inhibitors of this invention may range from
10 micrograms per kilogram body weight (,ug/kg) per day to 10 ~ dlll per kilogram body weight
(mg/kg), typically 100 ,ug/kg/day to 1 mg/kg/day. Thus, a ther~reutic~lly effective amount for a 80 kg
human may range from 1 mg/day to 1000 mg/day, typically 10 mg/day to 100 mg/day.
One of ordinary skill in the art of treating such ~iseases will be able, without undue experimentation
and in reliance upon personal knowledge and the ~Jicclos~ ~re of this application, to ascertain a
therapeutically effective amount of a cysteine prut~ase inhibitor of this invention for a given disease.
In general, the cysteine protease inhibitors of this invention will be administered as pharmaceutical
compositions by one of the following routes: oral, systemic (e.g., transdermal, i"L,dnasal,
intrapulmonary, or by surpositiory) or parenteral (e.g., intramuscular, intravenous, intrapulmonary or
5~hcut~neous). Col"posiLions can take the form of tablets, pills, c~rs~les, sen ~ 's, powders,
sustained release formulations, solutions, suspensions, elixiers, aerosols or any other appropriate
cor"posiiL~,n and are co" "., i:,ed of, in general, a cysteine protease inhibitor of the invention in
COI "bindLion with at least one pharmaceutically acceptable P~ , ?nt. Acceptable excipients are
non-toxic, aid adl";"i~L,dLion, and don not adversely affect the therapeutic benefit of the cysteine
protease inhibitor of this invention. Such ~, , .srll may be any solid, liquid, semisolid or, in the case of
an aerosol co" "~osiLion, gaseous excipient that is generally available to one of skill in the art.
Solid pharrn~ceutic~l excipients include starch, ~ ?" ~loce, talc, glucolse, lactose, sucrose, gelatin, malt,
rice, flour, chalk, silica gel, magnesium stearate, sodium sterate, glycerol mono~L~a,dL~, sodium
chloride, dried skim milk and the like. Liguid and semisolid excipients may be select~d from water,
ethanol, glycerol, propylene glycol and various oils, includingthose of petroleum, animal, vegetable or
synthetic origin (e.g., peanut oil, soybean oil, mineral oil, sesame oil, etc.). Preferred liquid carriers,
particularly for injectable solutions, include water, saline, aqueous dextrose and glycols.
Co,,,,u,t:ssed gases may be used to disperse the cysteine prul~ase inhibitor of this invention in aerosol
form. Inert gases suitable for this purpose are nitrogen, carbon dioxide, n~trous oxide, etc. Other
suitable pharmaceutical carriers and their formulations are described in A.R. Alfonso Reminton's
Pharmaceutical Sciences 1985, 17th ed. Easton, Pa.: Mack Pll~l '~ ,9 Company, hereby ~x,ur~ssly
i"corl,o~dl~d by ,~rt,~ nce.
The amount of a cysteine prult:ase inhibitor of this invention in the co" "~osiLiun may vary widely
depending upon the type of formulation, size of a unit dosage, kind of ex ,: ? )L~ and other factors
CA 022161~1 1997-09-23
W O 96/30353 PCTrUS96/03844
known to those of skill in the art of pha, r~ ,aceutic~l sciences. In general the final co, ~~ osiLion will
comprise from 0.1%w to 10%w of the cysteine protease inhibitor preferably 1%w to 10%w with the
remainder being the e,~ .ie, IL or e: nl:,.
Preferably the pharmaceutical composition is ad, ., fi ,L~red in a single unit dosage form for continuous
teatment or in a single unit dosage form ad libitum when relief of sy---~ --s is specifically required.
Representative pha",.~ceutic~l formulations co"Ldi"i"g a cysteine protease inhibitor of the invention
are described in Example 20 infra.
The following examples serve to more fully describe the manner of using the above-described
invention as well as to set forth the best modes contemplated for carrying out various aspects of the
invention. It is understood that these examples in no way serve to limit the true scope of this invention
but rather are presented for illustrative purposes. All references cited herein are expressly
incorporated by reference.
EXAMPLES
The following abbreviation conventions have been used to simplify the examples.
Mu = morpholine urea
Xaa, = amino acid at P1 position relative to active site of the enzyme
Xaa2 = amino acid at P2 position relative to active site of the enzyme
y-CO2Et= y-amino ethyl ester
y-SO2Ph= y-aminosulfone with phenyl terminus
y-CO2H = y-aminocarboxylate
y-PEt = y-aminophosphonate
y-AM = y-aminoamide
y-Ar(sub)= y-ar., lodlullldlic compound (s~hstitllt~d as appropriate)
~-SO2Ph= ~-aminosulfone with phenyl sl Ihstitl ~ent
~-SO2Ph= a-aminosulfone with phenyl s~ Ihstitl lent
Hph = homophenylalanine
PSMP = diethyl phenylsulfonylmethylenephosphonate
Np2 = 2-naphthyldldn le
SO22Np = sulfone with 2-maphthyl terminus
Phac = phenylacetyl
B-Ala = 13-alanine
MeOSuc = methoxysuccinyl
CA 022l6l~l l997-09-23
W 096/30353 PCTrUS96/03844
41
For instance, Mu-Phe-Hph-~-SO2Ph where Xaa2 = Phe (phenylalanine) and Xaa, = Hph(homophenylalanine,), L,dn~rur",ed to the ~-amino phenyl sulfone accordi"g to the procedure
described in the Examples.
Example 1
Synthesis of Cysteine Protease Inhibitor
Containing a y-aminoester as the EWG
Un!ess otherwise indicated, all reactions were performed under an inert atmosphere of argon or
nitrogen at room temperature. THF was distilled from sodium benzophenone ketyl. All other solvents
and commercially available reagents were used without further purification.
Synthesis of Ethyl (S)-4-(4-morpholinecarbonyl-phenylalanyl)-amino-6-phenylhexanoate, abbreviated
Mu-Phe-Hph-y-CO2Et, was as follows. Unless otherwise noted, all reagents were obtained from
Aldrich, Inc. 0.393 9 of a 60% mineral oil dispersion (9.82 mmol) of sodium hydride was added to a
solution of triethyl phosphonoacet~t~ (2.20 9, 9.82 mmol) in THF (50 mL) at -10~C. The mixture was
stirred for 15 minutes, whereupon a solution of Boc-homophenylalaninal (Boc-HphH) (2.35 9, 9.82
mmol, prepared by conversion of Boc-homophenylalanine (Synthetech) to its
N,O-dimethylhyd,u~d,,,;de, using the Fehrentz method. followed by lithium aluminum hydride
reduction) in THF (20 mL) was added. The mixture was stirred for 45 minutes. 1 M HCI (30 mL) was
added. The product was extracted with ethyl acetate (50 mL), washed with saturated aqueous
NaHCO3 (30 mL) dried over MgSO4, filtered, and evaporated to dryness. The dried material was
di~solved in CH2CI2 (10 mL), and a 4.0 M solution of HCI in dioxane (20 mL) was added. The mixture
was stirred for 30 minutes. The solvents were removed under reduced pressure and the residue, ethyl
(S)-4-amino-6-phenyl-2-hexenoate hydrochloride, was pumped dry.
4-Morpholinecarbonylphenylalanine (Mu-PheOH, 2.74 9, 9.82 mmol, prt:pared according to the
method described in Esser, R. et.al., Arthritis ~ Rheumatism (1994), 37, 236) was dissolved in THF
(50 mL) at-10~C. 4-methylmorpholine (1.08 mL, 9.82 mmol) was added, followed by isobutyl
chlorurur,,,dLe (1.27 mL, 9.82 mmolj. The mixed anhydride was stirred for 10 minutes, whereupon a
solution of ethyl (S)-4-amino-6-phenyl-2-hexenoate hydrochloride from the previous step in DMF (10
mL) was added, followed by 4-methylmorpholine (1.08 mL, 9.82 mmol). The mixture was stirred for 1
hour. 1 M HCI (50 mL) was added. The product was extracted with ethyl acetate (100 mL), washed
with saturated aqueous NaHCO3 (50 mL), dried over MgSO4 and decolorizing charcoal (DARCO),
filtered, and evdpo,dL~d to dryness, giving 3.80 9 of intermediate (80% yield from Boc-homo-
phenylalar,il ,al).
CA 022161~1 1997-09-23
W 096/303S3 PCTrUS96/03844
42
To a solution of this intermediate (1.45 9, 3.09 mmol) in ethanol (25 mL) was added 5% palladium on
active carbon (0.5 9). The mixture was reduced on a Parr hydrogenator for 36 hours. The solution
was filtered and the solvent was removed under reduced pressure, giving 1.19 9 (82%) of the product.
Thin-layer chromatography (TLC) was performed on each sample. Vicu~ ion was accol"~,l.;,hed by
means of UV light at 254 nm, followed by ninhydrin, bromocresol green, or p-a~ 'ehyde stain. The
retention factor (Rfl of the Mu-Phe-Hph-y-CO2Et was 0.35 (5% MeOH/CH2CI2).
NMR spectra were recorded on a Varian Gemini 300 MHz instrument. All 'H NMR data of this and
subsequent examples are reported as delta values in parts per million relative to internal
tetramethylsilane, peak assignments in boldface. The following abbreviations are used: s, singlet; d,
doublet; t, triplet; q, quartet; br, broad. An asterisk (~) implies that a signal is obscured or buried under
another resonance.
Example 2
Synthesis of a Cysteine Protease Inhibitor containing a y-aminosulfone as the EWG
Synthesis of (S)-3-tert-butoxycarbonylamino-5-phenyl-1-phenylsulfonylpentane (Boc-Hph-y-SO2Ph).
To a solution of PSMP (8.87 9, 30.34 mmol) in THF (150 mL) at 0~C was added sodium hydride (1.21
g of a 60% mineral oil dispersion). The mixture was stirred for 20 minutes, whereupon a solution of
Boc-homophenylalaninal, synthesized by the method of Fehrentz and Castro, above, (7.99 9, 30.34
mmol) in THF (20 mL) was added. The solution was stirred for 30 minutes at 0~C. 1M HCI (100 mL)
was added. The product was extracted into ethyl acetate (100 mL), washed with saturated aqueous
sodium bicarbonate (100 mL), brine (50 mL), dried over MgSO4, filtered, and the solvent was removed
under reduced pressure. The residue was dissolved in ethanol (100 mL) and transferred to a Parr
bottle charged with 5% palladium on active chaluoal (0.92 9). The mixture was reduced on a Parr
apparatus for 24 hours. The solution was filtered through Celite and the solvent was removed under
reduced pressure. TLC of the product indicated a single product in quantitative yield, R, = 0.29 (30%
ethyl acetate/hexane) that stained white with pa,~anicaldehyde spray.
Example 3
Synthesis of a Cysteine Protease Inhibitor CO~ l9 a y-aminosulfone as the EWG.
Synthesis of (S)-3-amino-5-phenyl-1-phenylsulfonyl-pentane hydrucl7'~ ide (HCl.Hph-y-SO2Ph). To a
solution of Boc-Hph-y-SO2Ph (12.24 9, 30.34 mmol) in dichloromethane (20 mL) was added hydrogen
chloride in dioxane (50 mL of a 4.0M solution). The mixture was stirred for 90 minutes. The solvent
was removed under reduced pressure, and the residue was dissolved in CH2CI2 (50 mL). The solution
CA 022161~1 1997-09-23
W 096/30353 PCTrCfS96/03844
43
was carefully added to ether (500 mL) with stirring. The solid was filtered, washed with ether (50 mL)
and dried in vacuo.
Example 4
Synthesis of a Cysteine Protease Inhibitor corlLdil ,il ,9 a y-aminosulfone as the EWG.
-
Synthesis of (S)-3-(4-morpholinecarbonylphenylalanyl)-amino-5-phenyl-1-phenylsulfonylpentane (Mu-
Phe-Hph-y-SO2Ph). To a solution of Mu-PheOH (2.94 9, 10.56 mmol) in THF (75 mL) at -10~C were
added 4-methylmorpholine (1.16 mL, 10.56 mmol) and isobutyl chlolu~oll"al~ (1.37 mL, 10.56 mmol).
The mixture was stirred for 5 minutes. (s)-(E)-3-amino-5-phenyl-1-phenylsulfonyl-1-pentene p-
toluenesulfonate, synthesized by Wadsworth-Emmons condensation between Boc-
homophenylalaninal and p-toluenesulfonic acid depru~cLion (5.00 9, 10.56 mmol) was added, followed
by 4-methylmorpholine (1.16 mL, 10.56 mmol). The mixture was stirred for 45 minutes. The solution
was diluted with ethyl acetate (100 mL), washed with 1M HCI (2x50 mL), saturated aqueous sodium
bicarbonate (50 mL), brine (50 mL), dried over MgSO4, filtered, and the solvent was removed under
reduced pressure. The residue was crystallized from CH2CI2/ether to give 4.27 9 (72%) of
intermediate. 1.17 9 of this material (2.08 mmol) was dissolved in ethanol (25 mL). The solution was
transferred to a Parr bottle charged with 5% palladium on active charcoal (0.30 9). The mixture was
hydrogenated at room temperature overnight on a Parr shaker. Ethyl acetate was added to the
suspension of product, which had crystallized from the reaction mixture. 1~he solution was filtered and
concentrated in vacuo, and then was recrystallized from CH2CI2/hexane. M.p. = 176-178~C. TLC:
(50% ethyl acetate/CH2CI2) Rf= 0.24.
Example 5
Synthesis of a Cysteine Protease Inhibitor containing a y-aminosulfone as the EWG.
Synthesis of (S)-3-(4-morpholinecarbonyltyrosyl)-amino-5-phenyl-1-phenylsulfonylpentane (Mu-Tyr-
Hph-y-SO2Ph). To a solution of 4-morpholinecarbonyltyrosine (Mu-TyrOH, synthesized according to
the method d~s-;,ibed in Esser, R. et.al., Arthritis ~ Rheumatism (1994), 37, 236, 0.50 9, 1.70 mmol) in
THF (10 mL) at-10~C were added 4-methylmorpholine (0.187 mL, 1.70 mmol) and isobutyl
clllolururllldLt: (0.220 mL, 1.70 mmol). After 5 minutes, HCl.Hph-y-SO2Ph (0.577 9, 1.70 mmol,
desc,iL,ed in Example 3) was added, followed by 4-methyl",or,~,h-' ,e (0.187 mL, 1.70 mmol). The
mixture was stirred for 45 minutes. Ethyl acetate (50 mL) was added. The solution was washed with
1M HCI, saturated ~queous sodium ~icarbondLe, and brine (30 mL each), dried over MgSO4, filtered,
and the solvent was removed under reduced pressure. The residue was pr~c;~ d from
CH2CI2/ether to give û.58 9 (59%) of Mu-Tyr-Hph-y-SO2Ph. M.p. 104-107~C. TLC: (1 û%
MeOH/CH2CI2)Rf=0.59.
CA 022161~1 1997-09-23
W O 96/30353 PCTrUS96/03844
44
Example 6
Synthesis of a Cysteine Protease Inhibitor containing a y-aminosulfone as the EWG.
Synthesis of (S)-3-(4-morphlinecarbonyl-2-naphthyl-alanyl)amino-5-phenyl-1-(2-
naphthylsulfonyl)pentane (Mu-Np2-Hph-y-SO22Np). 2-naphthalenethiol (9.64 9, 60.16 mmol) was
dissolved in toluene (75 mL). Paraformaldehyde (3.97 9, 132 mmol) and HCI/dioxane (33 mL of a
4.0M solution) were added. The mixture was stirred for several days at room temperature. The
solvent was removed under reduced pressure, and the residue was suspended in hexane (200 mL),
dried over MgSO4. filtered, and e\/dlJordL~d to dryness. This material, crude chloromethyl 2-naphthyl
sulfide, was comblned with triethyl phosphite (10.93 9, 65 mmol) and was heated at reflux for 4 hours.
The mixture was cooled to room temperature, diluted with ether (200 mL), washed with 1 M HCI,
saturated aqueous sodium bicarbonate, and brine (150 mL each), dried over MgSO4, filtered, and
concentrated in vacuo to give 17.35 g (93% crude yield) of diethyl 2-naphthylthiomethylene
phosphonate. This material was dissolved in CH2CI2 (300 mL) and cooled to 0~C. Peracetic acid (23.5
mL of a 32% dilute acetic acid solution (Aldrich Chemical Co.) was carefully added. The mixture was
stirred overnight while warming to room temperature. The solution was washed with freshly prepared,
saturated aqueous sodium bisulfite solution (100 mL), then with several portions of saturated aqueous
sodium bicarbonate, until the aqueous phase became basic. The organic phase was dried over
MgSO4, filtered, and the solvent was removed under reduced pressure. Cl1lullldlugld,~hy on 60-200
mesh silica gel (0-10% ethyl acetate!CH2CI2) afforded 6.5 9 (34%) of the pure Wadsworth-EI"",ons
reagent, diethyl 2-naphthylsulfonyl-methylene phosphonate along with an approximately equal mass of
impure material. TLC: (20% ethyl acetate/CH2CI2) R, = 0.37.
To a solution of diethyl 2-naphthylsulfonylmethylene phosphonate (3.91 9, 11.42 mmol) in THF (60
mL) at 0~C was added sodium hydride (0.457 g of a 60% mineral oil di:,pe,:,ion. The mixture was
stirred for 15 minutes, whereupon a solution of Boc-homophenylald"i"al (3.00 9, 11.42 mmol) in THF
(5 mL) was added. The mixture was stirred for 30 minutes. 1 M HCI (100 mL) was added. The
product was extracted with ethyl acetate (100 mL), washed with saturated aqueous sodium
bicarbonaLe (75 mL), brine (50 mL), dried over Mg~04, filtered, and the solvent was removed under
reduced pressure. The residue was dissolved in dichloro",~lllane (10 mL), to which was added
HCI/dioxane (25 mL of a 4.0M solution). The mixture was stirred at room temperature for 1 hour,
poured into ether (300 mL), and filtered. The solids were washed with ether (2x50 mL) and dried in
vacuo to give 3.30 g (74% from Boc-homophenyldld"..,al) of (S)-(E)-3-amino-5-phenyl-1-(2-
naphthylsulfonyl)-1 -pentene.
To a solution of Boc-2-naphthylalanine (2.68 g, ~.51 mmol, (Synthetech, Oregon) in THF (50 mL) at-
10~C were added 4-methylmorpholine (0.936 mL, 8.51 mmol) and isobutyl chlorurur,-,d~e (1.103 mL,
CA 022161~1 1997-09-23
W 096/303~3 PCTrUS96/03844
8.51 mmol). The mixture was stirred for 5 minutes, whereupon (S)-(E)-3-amino-5-phenyl-1-(2-
naphthylsulfonyl)-1-pentene (3.30 9, 8.51 mmol) was added, followed by 4-methylmorpholine (0.936
mL, 8.51 mmol). The mixture was stirred for 45 minutes, diluted with ethyl acetate (100 mL), washed
with 1 M HCI (50 mL), saturated aqueous sodium bicarbonate (50 mL), and brine (50 mL), dried over
MgSO4, filtered, and the solvent was removed under reduced pressure. The intermediate, (S)-(E)-3-
(tert-butoxycarbonyl-2-naphthylalanyl)amino-5-phenyl-1-(2-naphthylsulfonyl)-1-pentene, was
crystallized from a suitable mixture CH2CI2/ether/hexane in 69% yield. The resulting material (3.83 9,
5.90 mmol) was dissolved in CH2CI2 (5 mL) and was treated with HCI/dioxane (15 mL of a 4.0M
solution. The mixture was stirred at room temperature for 1 hour. The solution was poured, with
stirring, into ether (500 mL) and filtered. The solids were washed with ether (2x50 mL) and dried in
vacuo to give the intermediate, (S)-(E)-3-(2-naphthylalanyl)-amino-5-phenyl-1-(2-naphthylsulfonyl)-1-
pentene, 3.419, 99% yield.
2.00 9 of this material (3.42 mmol) was dissolved in THF (15 mL), and cooled to 0~C. 4-
methylmorpholine-carbonyl chloride (0.400 mL, 3.42 mmol) and triethylamine (0.953 mmol) were
added. The mixture was stirred for 1 hour at 0~C, and then at room temperature for 2 hours. Ethyl
acetate (50 mL) was added. The solution was washed with 1 M HCI (30 mL), saturated aqueous
sodium bicarbonate (30 mL), brine (30 mL), dried over MgSO4, filtered, and evaporated to dryness,
giving 1.58 9 (69%) of intermediate, (S)-(E)-3-(4-morpholinecarbonyl-2-naphthylalanyl)amino-5-
phenyl-1-(2-naphthylsulfonyl)-1-pentene. TLC: (50% ethyl acetate/hexane) R, = 0.37.
0.73 9 (1.10 mmol) of this material was dissolved in ethanol (20 mL) and L,dn~r~r,~:d to a Parr bottle
charged with 5% palladium on carbon (0.30 9). The mixture was reduced on a Parr hydrogenator for
36 hours. The solution was filtered and the solvent was removed under reduced pressure. The
productwas purified by column chrol"d~ug,dphy on 60-200 mesh silica gel (50% ethyl acetate/CH2CI2
as eluent) to give 0.14 9 (19%) of pure product, Mu-Np2-Hph-y-SO22Np, along with impure material.
TLC. (50% ethyl acetate/CH2CI2) R~ = 0.34.
Example 7
Synthesis of a Cysteine Protease Inhibitor cor' ,ing a y-aminosulfone as the EWG.
Synthesis of 3-acetyltyrosylvalylalanylamino4-hydroxy-carbonyl-1-phenylsulfonylbutane (Ac-Tyr-Val-
Ala-Asp-y-SO2Ph). Sodium hydride (0.489 9 of a 60% mineral oil dispersion, 12.23 mmol) was added
to a solution of diethyl phenylsulfonylmethylene phosphonate (3.58 9, 12.23 mmol) in 50 mL of THF at
0~C. The mixture was stirred for 15 minutes. A solution of Boc-AspH(13-Ot-Bu), (3.04 gm, 11.12
mmol, prepared by converting Boc-Asp(,3-O-t-Bu) to its N,O-dimethyld,~.,.d", ~e and reducing with
lithium aluminum hydride), in THF (10 mL) was added. The mixture was stirred for 1 hour, whereupon
CA 022161~1 1997-09-23
W 096/30353 PCT~US96/03844
46
1 M HCI (30 mL) was added. The product was extracted with ethyi acetate (100 mL), washed with
saturated aqueous NaHCO3 (30 mL), brine (30 mL), dried over MgSO4. filtered, and e\~dpoldled to
dryness, giving the intermediate, . Chromatography on silica gel (20-30% ethyl acetate/hexane,
gradient elution) afforded 2.07 9 45%) of the intermediate, (S)-(E)-3-tert-butoxycarbonylamino4-tert-
butoxycarbonyl-1-phenylsulfonyl-1-butene. This material was dissolved in ether (2 mL) and was
treated with a solution of anhydrous p-toluenesulfonic acid (1.0 9, 5.87 mmol) in ether (2 mL). The
mixture was stirred at room temperature overnight, then diluted with ether (25 ml). The white
precipitate, was filtered, washed with ether, and dried in vacuo to give 0.80 9 (95%) of the next
intermediate, (S)-(E)-3-amino4-tert-butoxycarbonyl-1-phenylsulfonyl-1-butene-p-toluenesulfonate.
This material was coupled, using mixed anhydride chemistry, to Ac-Tyr-Val-AlaOH, itself prepared by
standard peptide chemistry, giving the next intermediate, (S)-(E)-3-acetyltyrosylvalylalanylamino-4-
tert-butoxycarbonyl-1 -phenylsulfonyl-1 -butene.
This material was treated with trifluoroacetic acid to remove the t-butyl ester of the aspartic acid side
chain, giving (E)-3-acetyltyrosylvalylalanylamino4-hydroxycarbonyl-1-phenylsulfonyl-1-butene. 0.28 9
(0.444 mmol) of this material was dissolved in ethanol (10 mL). The solution was transferred to a Parr
bottle, charged with 5% palladium on carbon (0.1 9). The solution was reduced on a Parr
hydrogenator overnight. The solution was filtered and the solvent was removed under reduced
pressure. The residue, when dissolved in methanol (5 mL) and diluted with 40x 1:1 CH2CI2/ether,
formed a gelatinous precipitate, which was --I'-cted on a Buchner funnel to give 0.18 9 (64%) yield.
The isomer ratio of S to R with respect to the Asp residue was estimated as approximately 3:1 based
on the integration of the doublets asso~,idl~d with the aromatic region of the NMR as pertains to the
Tyr residue.
Example 8
Synthesis of a Cysteine Protease Inhibitor with a y-aminocarboxylate as the EWG.
Synthesis of (S)4-(4-morpholinecarbonylphenylalanyl)amino-6-phenylhexanoic acid, Mu-Phe-Hph-y-
CO2H. To a solution of Mu-Phe-Hph-y-CO2Et, p,~pa,~d accordi~,g to the procedure described in
Example 1 (0.59, 1.06 mmole) was added aqueous NaOH (1 mL of a 2M solution). After 4 hr the
reaction was cor, ~ I_t~. 1 M HCI (4mL) was added along with water (10 mL). The product was
exL,d.,Led with CH2C12 (2 x 10 mL), THF (15dried over MgSO4 the solvent was removed under reduced
pressure, and the residue, Mu-Phe-Hph-y-CO2H, was pumped to a solid Yield = 0.309 (60%).
Example 9
Synthesis of a Cysteine Protease Inhibitor with a y-aminophosphondL~: as the EWG.
CA 022161~1 1997-09-23
W 096/303S3 PCTrUS96103844
47
Synthesis of diethyl (S)-4-(4-morpholinecarbonyl-phenylalanyl)amino-6-phenylhexanepl1osphondle
(Mu-Phe-Hph-y-SO2Ph) was as follows. To a solution of tetraethyl methylenediphosphonate (2.00 9,
6.94 mmol) in THF (30 mL) was added sodium hydride (0.278 9 of a 60% mineral oil dispersion, 6.94
mmol). The mixture effervesced rapidly and then clarified. After 5 minutes, a solution of Boc-HphH
(1.83 9, 6.94 mmol) in THF (5 mL) was added. The mixture was stirred for 1 hour. 1 M HCI (20 mL)
was added. The product was extracted into ethyl acetate (50 mL), washed with saturated aqueous
NaHCO3 (20 mL), brine (10 mL), dried over MgS04, filtered, and evaporated to dryness, giving 2.46 9
(89%) of inte""e.liate, diethyl (S)-(E)-4-tert- butoxycarbonylamino-6-phenyl-2-hexenephosphonate.
To a solution of this material in CH2CI2 (3 mL) was added 10 mL of a 4.0 M solution of HCI in dioxane.
The mixture was stirred at room temperature for 1.5 hours. The solvents were removed under
reduced pressure and the residue was dissolved in methanol (10 mL). The solution was poured into
ether (400 mL). The precipitate was collectf~d on a Buchner funnel, washed with ether (2x20 mL), and
was pumped dry to give 1.25 9 (60%) of intermediate, diethyl (S)-(E)-4-amino-6-
phenyl-2-hexenephosphonate hydrochloride. To a solution of Mu-PheOH (1.04 q, 3.74 mmol) in THF
(15 mL) at -10~C was added 4-methylmorpholine (0.412 mL, 3.74 mmol), followed by isobutyl
chloroformate (0.486 mL, 3.74 mmol). The mixed anhydride was stirred for 5 minutes, whereupon a
solution of (S)-(E)-4-amino-6-phenyl-2-hexene-phosphonate hydrochloride (1.25 9, 3.74 mmol) in DMF
(5 mL) was added, followed by 4-methylmorpholine (0.412 mL, 3.74 mmol). The mixture was stirred
for 1 hour. Ethyl acetate (50 mL) was added. The solution was washed with 1 M HCI (25 mL),
saturated aqueous NaHCO3 (25 mL), and brine (10 mL), dried over MgSO4, filtered, and evaporated to
dryness. The product, upon treatmentwith CH2CI2/ether/hexane (315 mL in a 15:200:100 ratio)
formed an oil that solidified on drying in vacuo to give 1.44 9 (69%) of diethyl (S)-
(E)-4-(4-morpholinecarbonyl-phenylalanyl)-amino-6-phenyl-2-hexenephosphonate. 0.85 9 of this
material was dissGlved in ethanol (10 mL) and was transferred to a Parr bottle charged with 5%
palladium on active charcoal. The solution was reduced on a Parr hydrogenator for 36 hours. The
solution was then filtered through Celite, and the solvent was removed under reduced pressure to give
0.66 9 (76%) of the final product as an oil. TLC: (5% MeOH/CH2CI2) R, = 0.27.
Example 10
Synthesis of a Cysteine Protease Inhibitor with a y-aminoamide as the EWG.
Synthesis of benzyl (S)-3-(4-~"o,,uholi,,ecarbonylphenyl-alanyl)-amino-6-phenylhexdnalll: 'e (Mu-Phe-
Hph-y-AMBzl). The Wadsworth C" " "ons reagent diethyl benzylamido-carbonylmethylenephosphonate
was synthesized in two steps, first by saponirica~ion of triethyl phos~,honoac~ to diethyl
phosphonoacelic acid, which then was .li~solved in ethyl acetate to a 0.2 M conce~ll,dlion, treated
with an equivalent of benzylamine, 0.1 equivalents of 4-dimethylamino-pyridine, and one equivalent of
dicyclohexyl-cd"~ _ ' " ~'~ To a solution of this Wadsworth-Emmons reagent (2.59 9, 9.08 mmol) in
CA 022161~1 1997-09-23
W 096t30353 PCTrCfS96/0384
48
THF (40 mL) at 0~C was added sodium hydride (0.363 9 of a 60% mineral oil disper:,ion, 9.08 mmol).
The mixture was stirred at room temperature for 15 minutes, whereupon a solution of Boc-
homophenylalaninal (2.39 9, 9.08 mmol) in THF (10 mL) was added. The mixture was stirred for 1
hour. 1 M HCI (30 mL) was added. The product was extracted into ethyl acetate (100 mL), washed
with saturated aqueous sodium bicarbonate (50 mL), brine (30 mL), dried over MgSO4, filtered,
concentrated, and crystallized from ether/hexane to give 1.81 9 (51 %) of benzyl (S)-(E)-3-
tertbutoxycarbonylamino-6-phenyl-2-hexenamide. This material was di~solved in CH2CI2 (5 mL). To
the solution was added HCI/dioxane (10 mL of a 4.0M solution). The mixture was stirred for 3 hours at
room temperature. The solvents were removed under reduced pressure. The residue was dissolved
in methanol (5 mL) and poured into ether (300 mL), whereupon the intermediate, benzyl (S)-(E)-3-
amino-6-phenyl-2-hexenamide hydrochloride, separated out as an oil, in 82% yield (1.25 9). To a
solution of Mu-PheOH (1.05 9, 3.78 mmol) in THF (15 mL) at -10~C were added 4-methylmorpholine
(0.416 mL, 3.78 mmol) and isobutyl chlo~urulll,dl~ (0.490 mL, 3.78 mmol). The mixture was stirred for
10 minutes, whereupon a solution of benzyl (S)-(E)-3-amino-6-phenyl-2-hexenamide hydrochloride
(1.25 9, 3.78 mmol) in THF (3 mL) was added, followed by 4-methylmorpholine (0.416 mL, 3.78
mmol). The mixture was stirred for 45 minutes. Ethyl acetate (40 mL) was added. The solution was
washed with 1M HCI (10 mL), saturated aqueous sodium bicarbonate (10 mL~, brine (5 mL), dried over
MgS04, filtered, and evaporated to dryness. The intermediate, benzyl (S)-(E)-3-(4-
morpholinecarbonyl-phenylalanyl)amino-6-phenyl-2-hexenamide, was precipit~'f~d from CH2CI2/ether
in 56% yield. 0.48 9 (0.865 mmol) of this material was dissolved in ethanol (10 mL) and L,dnsr~r,~d to
a Parr bottle charged with 5% palladium on active carbon. The mixture was reduced on a Parr
hydrogenator for 4 hours. The solution was filtered through Celite, and the solvent was removed
under reduced pressure. The final product (Mu-Phe-Hph-y-AMBzl) was cr~,- ' " ed from
ethanol/hexane, giving 0.25 9 (52%). TLC: (50% ethyl acetate/hexane) Rf= 0.45.
Example 11
Synthesis of a Cysteine Protease Inhibito~ with a y-aminoamide as the EWG.
Synthesis of phenyl (S)-3-(4-morpholinecarbonylphenyl-alanyl)-amino-6-phenylhexanamide (Mu-Phe-
Hph-y-AMPh). To a solution of Mu-Phe-Hph-y-CO2H, (0.309, as p, u pa,~d accor.li"g to Example 8), in
THF (5 mL) at-10~C was added triethylamine (90,uL, 1 eq.) followed by addition of isobutyl
chloroformate (0.083 mL, 1 eq.). After 5 min, aniline (0.058 mL) was added. The cooling bath was
removed and the reaction stirred at room temp for 2 hr. CH2CI2 (30 mL) was added. The solution was
washed with 1M HCI and saturated aqueous sodium L,ca,bondl~: (10 mL each), dried over MgSO4,
filtered, and the solvent was removed under reduced pressure. The residue was triturated with Et2O,
filtered, and dried in vacuo to give 0.29 9 (85%) of the product, Mu-Phe-Hph-y-AMPh. TLC (10%
MeOH/CH2CI2) R, = 0.70, strongly absorbs UV (254 nm), 12.
CA 022161~1 1997-09-23
W 096/30353 PCT~US96/03844
a~g
Example 12
Synthesis of a Cysteine Protease Inhibitor with a y-aromatic as the EWG.
Synthesis of (S)4-aminophenyl-3-(4-morpholine-carbonylphenylalanyl)amino-5-phenylpentane
hydrochloride, (Mu-Phe-hPhe-y-C6H4NH2.HCI).
Triphenylphosphine (38.17 9, 0.146 mole) and 4-nitrobenzyl chloride (259, 0.146 mole) were
dissolved in CH3CN (100 mL) and heated at reflux for 2 hours, and then allowed to cool to room
temperature. The reaction mixture was diluted with Et2O (300 mL), the white solid was filtered,
washed with Et2O (200 mL), and dried in vacuo, giving 53.3 9 (84%) of 4-
nitrobenzyltriphenylphosphonium chloride as a single spot on TLC: (Rf=0.71, 4:1:1 butanol:acetic
acid:water).1H-NMR (d6-DMSO): 5.40-5.50 (2H, d, CH2P, J=20Hz); 7.20-7.40 (2H, dd, aromatic);
7.40-7.80 (12H, m, aromatic~; 7.90-8.00 (3H, m, aromatic); 8.10-8.20 (2H, d, aromatic).
To a stirred suspension of 4-nitrobenzyltriphenylphosphonium chloride (10.02 9, 23.1 mmol) in
CH2CI2 (100 mL) was added 4-methylmorpholine (2.54 mL, 23.1 mmol) was added. When all the
solid had dissolved, Boc-HphH (4.04 9, 15.4 mmol) was added. After 24 hrs., the reaction
mixture was diluted with CH2CI2 (200 mL), and filtered. The filtrate was washed with 1 M HCI
(200 mL) saturated aqueous sodium bica,bond~e (200 mL); dried over MgSO4, filtered and
concentrated under reduced pressure, giving 4.00 9 of crude intermediate, a portion of which
was purified by chromatography (gradient elution: 10-30% ethyl acetate/hexane) to permit NMR
analysis of the intermediate, (S)-t-butoxycarbonyl-3-amino-1-(4-nitrophenyl)-5-phenyl-1-pentene.
TLC: (30% EtOAc /hexane) Rf 0.49. To a solution of this material (2.76 9, 7.2 mmol) in Et2O
(25 mL), was added a solution of anhydrous e-toluenesulfonic acid (2.76 9, 16.0 mmol) in Et2O
(10 mL). The reaction was left to stir 16 hrs, filtered; washed solid with Et2O (25 mL), and dried
in vacuo, giving 2 9 (61%) of (S)-3-amino-1-(4-rliL,upl1el,yl)-5-phenyl-1-pentene as a single spot
on TLC: (Rf=0.49, 10% MeOH/CH2CI2).
To a solution of Mu-PheOH (1.29 9, 4.63 mmol) in THF (20 mL) were added 4-methyl~"o"~h-1 ,e
(0.51 mL, 4.63 mmol) and isobutyl chlorùru""dle (0.61 mL, 4.63 mmol). After 3 minutes, a
solution of (S) 3-amino-1-(4-nitrophenyl)-5-phenyl-1-pentene hydrochloride, prepared by
HCI/dioxane-mediated dep,uL~-,Lion of (S) 3-tert-butoxycarbonylamino-1-(4-r,iL,uphenyl)-5-
phenyl-1-pentene precursor, ~,.34 9, 4.20 mmol) in CH2CI2 (20 mL), followed by 4-
methylmorl.hGl",e (0.51 mL, 4.63 mmol). The mixture was stirred overnight while warming to
room L~"")e,dLure. The solution mixture was diluted with CH2CI2 (100 mL), washed with 1M HCI
(200 mL), saturated aqueous sodium bicdllJondL~: (200 mL); dried over MgSO4, filtered, and
CA 022161~1 1997-09-23
W 096/30353 PCTrUS96/03844
concentrated under reduced pressure to give a yellow oil. This material was cry~ d from
CH2CI2/ether (Z:100, 20 mL) to give 1.00 9 (40%) of (S)-3-(4-
morpholinecarbonylphenylalanyl)amino-1-(4-nitrophenyl)-5-phenyl-1-pentene as an
approximately 4:1 E/Z mixture. 0.27 9 (0.49 mmol) of this material was dissclvcd in ethanol (50
mL), transferred to a Parr bottle charged with 5% palladium on carbon (0.10 9) and reduced on a
Parr hydrogenator for 8 hours/ The mixture was filtered and the solvent was removed under
reduced pressure. The residue was di~solved in in 4:1 ether/CH2CI2 (100 mL), to which
HCI/dioxane (0.136 mL of a 4.0 M solution) was added. The product, Mu-Phe-Hph-y-C6H4NH2.HCI, was filtered and dried in vacuo. Yield = 0.15 9 (54%). TLC: (10%
",t:ll,anol/CH2CI2) R,= 0.31.
Example 13
Synthesis of a Cysteine Protease Inhibitor with a ~-aminosulfone as the EWG.
Synthesis of (S)-2-(4-morpholinecarbonylphenyl-alanyl)amino-4-phenyl-1-phenylsulfonylbutane
(Mu-Phe-Hph-,~-SO2Ph). Preparation of Boc-homophenylalaninol (Boc-Hph-,~-OH) and (S)-2-
tert-butoxycarbonylamino-1-methanesulfonyloxy-1-phenylbutane (Boc-Hph-~-OMs or Boc-
homophenylalaninol mesylate) followed a similar scheme to that reported by Spaltenstein,
Carpino, Miyake, and Hopkins, above. To a solution of Boc-homophenylalanine (10.29 9, 36.84
mmol) in THF (100 mL) at -10~C were added 4-methylmorpholine (4.05 mL, 36.84 mmol) and
isobutyl chlo,ur~nlldle (4.78 mL, 36.84 mmol). The solution was stirred for 10 minutes, and then
was filtered. The filtrate was carefully added to a stirred solution of sodium borohydride (2.77 9,
73.67 mmol) in water (100 mL) at 0~C. The mixture was stirred for 30 minutes. Saturated
aqueous sodium bicarbonate (200 mL) was added. The product was extracted with CH2CI2
(2x100 mL), dried over MgSO4, filtered, and the solvent was removed under reduced pressure to
give 9.78 9 (100%) Boc-homophenyl-alaninol. TLC: (30% ethyl acetate/hexane) R, = 0.15. 5.83
9 (21.97 mmol) of this material was di~solved in CH2CI2 (150 mL), cooled to 0~C, and treated with
methanesulfonyl chloride (4.15 mL, 53.71 mmol), and triethylamine (9.24 mL, 66.3 mmol). The
mixture was stirred for 30 minutes. Water (100 mL) was added; the mixture was stirred
vigorously. The organic phase was sepa, dl~d, dried over MgSO4, filtered, and the solvent was
removed under reduced pressure, giving 7.31 9 (97%) yield. TLC: (30% ethyl acetate/hexane)
R, = 0.21. A similar procedure was employed to prepare the cor,~:sponding benzenesulfonate
ester of Boc-Hph-~-OH.
To a solution of thiophenol (0.653 mL, 6.36 mmol) in THF (5 mL) was added sodium hydride
(0.254 9, 6.36 mmol as a 60% mineral oil di~pe,:,ion. The mixture was stirred for 10 minutes. A
solution of Boc-homophenylalaninol benzenesulfonate (2.58 9, 6.36 mmol) in THF (5 mL) was
CA 022l6l~l l997-09-23
W 096/30353 PCTrUS96/03844
51
added. The solution was stirred at room temperature for 10 minutes. Methanol (2 mL) was then
added, and the mixture was heated at reflux for 1 hour. The solution was cooled, diluted with 1 M
NaOH (25 mL), extracted with CH2C12 (100 mL), dried over MgS04, filtered, and the solvent was
~ removed under reduced pressure. The residue was .I;asolved in CH2CI2 (35 mL) and cooled to
0~C. To the solution was added 4-chloroperbenzoic acid (3.71 9, 13.99 mmol, esLillldlt:d peracid
content 65% by weight). The mixture was stirred for 1 hour, whereupon 10% NaOH (35 mL) and
saturated aqueous NaHSO3 (35 mL) were added. The mixture was extracted with CH2CI2 (3x50
mL portions), dried over MgSO4, filtered, and the solvent was removed under reduced pressure
to give a waxy solid, (S)-2-tert-butoxycarbonylamino~-phenyl-1-phenylsulfonylbutane. TLC:
(30% ethyl acetate/hexane) Rf = 0.32. 1.25 9 of this material was dissolved in CH2CI2 (5 mL) and
treated with HCI/dioxane (5 mL of a 4.0M solution). The mixture was stirred for 2 hours at room
temperature. The solution was poured into ether (200 mL), forming an oily residue. The
supernatant was discarded. The residue was again dissolved in CH2CI2 (10 mL), and poured
into ether (200 mL). The intermediate, (S)-2-amino4-phenyl-1-phenylsulfonylbutane
hydrochloride pl~ d out. The solid was filtered and dried in vacuo to give 0.40 9 of
material (38% yield from Boc-homophenylalaninol benzenesulfonate.
To a solution of Mu-PheOH (0.342 9, 1.23 mmol) in THF (10 mL) at -10~C were added 4-
methylmorpholine (0.135 mL, 1.23 mmol) and isobutyl chlorurunlldL~ (0.159 rnL, 1.23 mmol).
The mixture was stirred for 10 minutes, whereupon (S)-2-amino-4-phenyl-1-phenylsulfonylbutane
hydrochloride (0.40 9, 1.23 mmol) was added, followed by 4-methylmorpholine (0.135 mL, 1.23
mmol). The mixture was stirred for 45 minutes. 1 M HCI (15 mL) was added. The product was
extracted with ethyl acetate (30 mL), washed with saturated aqueous sodium b: -I,onal~ (15
mL), brine (15 mL), dried over MgSO4, filtered, and the solvent was removed under reduced
pressure. The final product, Mu-Phe-Hph-~-SO2Ph, weighed 0.68 9 (100% yield).
Example 14
Synthesis of a Cysteine Protease Inhibitor with a ,B-aminosulfone as the EWG.
Synthesis of (S)-2-tert-butoxycarbonylamino-4-phenyl-1-(1'-trimethylsilylethyl)-sulfonylbutane
(Boc-Hph-,B-SO2CH2CH2TMS). To a solution of 2-trimethylsilylethanethiol (0.86 9, 6.41 mmol),
synthesis desclibed by Anderson, Ranasinghe, Palmer, and Fuchs, above) in THF (10 mL) was
added sodium hydride (0.256 9, 6.41 mmol as a 60% mineral oil diapelaion). The mixture was
stirred for 10 minutes. Boc-homophenylalaninol mesylate (2.00 9, 5.82 mmol, synthesis
described in Example 13, above) was added. The solution was stirred for 2 hours. Ethyl acetate
(50 mL) was added. The solution was washed with 30 mL each of 1M HCI, saturated aqueous
sodium bicarbonate, and brine, dried over MgSO4, filtered, and the solvent was removed under
CA 022161~1 1997-09-23
W 096/30353 PCT~US96/03844
52
reduced pressure, giving the intermediate (S)-2-tert-butoxycarbonylamino-4-phenylbutyl
trimethylsilylethyl sulfide. TLC: (5% ethyl acetate/hexane) Rf = 0.22. This material was
dissolved in CH2CI2 (50 mL), cooled to -10~C, and treated with 4-chloroperbenzoic acid (3.24 9,
12.22 mmol, e~ "d(~:d 65% peracid content). The mixture was stirred overnight. The
suspension was filtered, and saturated aqueous NaHSO3 (40 mL) and saturated aqueous
sodium bicarbonate (50 mL) were carefully added to the filtrate. The organic phase was
separdLed, dried over MgSO4, filtered, and the solvent was removed under reduced pressure to
give the product, Boc-Hph-~-SO2CH2CH2TMS in quantitative mass recovery from the mesylate.
TLC: (30% ethyl acetate/hexane) R~ = 0.49.
Example 15
Synthesis of a Cysteine Protease Inhibitor with a ~-aminosulfone as the EWG
Synthesis of (S)-2-(4-morpholinecarbonylphenylalanyl)-amino-1-chloromethylsulfonyl-4-
phenylbutane (Mu-Phe-Hph-,B-SO2CH2CI. To a solution of Boc-Hph-~-SO2CHzCH2TMS (0.90 9,
2.18 mmol as described in Example 14) in THF (2 mL) were added tetrabutylammonium fluoride
(8.7 mL of a 1.0M THF solution) and several molecular sieves. The mixture was stirred overnight
at room temperature. Bromochloromethane (5 mL) was added. The mixture was heated at
reflux for 1 hour, cooled, and the volatile components were removed under reduced pressure.
The residue was dissolved in ethyl acetate (75 mL), washed with 1 M HCI (50 mL), dried over
MgS04, filtered, and the solvent was removed under reduced pressure. The residue, crude (S)-
2-tert-butoxycarbonylamino-1-chloromethylsulfonyl4-phenylbutane, was dissolved in ether (3
mL). A solution of anhydrous 4-toluenesulfonic acid (0.80 9, 4.70 mmol) in ether (3 mL) was
added. The mixture was stirred at room temperature overnight. Ether (100 mL) was added.
The solid inter"~edidl~, (S)-2-amino-1-chloromethyl-sulfonyl4-phenylbutane 4-toluenesulfonate
(TsOH.Hph-~-SO2CH2CI), was filtered, the solids were washed with ether (2x20 mL), and dried
in vacuo to give 0.193 9 of material (24% from Boc-Hph-,~-SO2CH2CH2TMS).
To a solution of Mu-PheOH (0.109 9, 0.392 mmol) in THF (3 mL) at -10~C were added 4-
methylmorpholine (43 ~L, 0.392 mmol) and isobutyl chloroformate (51 ~cL, C.392 mmol). The
mixture was stirred for 10 minutes, whereupon TsOH.Hph-~-SO2CH2CI (0.17 9, 0.392 mmol) was
added, followed by 4-methyll"or~,h-' ,e (43,uL, 0.392 mmol). The mixture was stirred for 45
minutes. Ethyl acetate (20 mL) was added. The solution was washed with 1 M HCI, saturated
aqueous sodium bicarbonate, and brine (2 mL each) dried over MgSO4, filtered, and the solvent
was removed under reduced pressure, to give the final product, Mu-Phe-Hph-,3-SO2CH2CI (90
mg, 48% yield.
CA 022161~1 1997-09-23
W 096/30353 PCT~US96/03844
53
Example 16
Synthesis of a Cysteine Protease Inhibitor with an a-aminosulfone as the EWG.
Synthesis of 1-(tert-butoxycarbonyl)amino-2-methyl-1-phenylsulfonylpropane (Boc-Val-a-
SO2Ph). To a stirred suspensionof t-butylca~ b dl I IdL~ (2.34 9, 20 mmol) and sodium
benzenesulfinate (3.28 9, 20 mmol) in water (20 mL) was added a solution of isobutyraldehyde
(2.00 mL, 22 mmol) in formic acid (5 mL). The mixture was stirred at room temperature
overnight. The p,~:cil,ildle was filtered, washed with water (2x50 mL) and crystallized from
i:,oprupanol/water to give 4.72 9 (75%) of the product.
Example 17
Synthesis of a Cysteine Protease Inhibitor with an a-aminosulfone as the EWG.
Synthesis of 1-benzyloxycarbonylamino-3-phenyl-1-phenylsulfonylpropane (Z-Hph-a-SO2Ph).
To a suspension of sodium benzenesulfinate (10 9, 60.9 mmol) and benzyl carbamate (9.21 9,
60.9 mmol) in water (40 mL) was added hydrocinnamaldehyde (8.8 mL, 67 mmol) in formic acid
(10 mL). The mixture was heated at 70~C for 1 hour, then permitted to cool to room temperature
overnight. The product crystallized out; it was filtered and recr~,~l " ed from hot isoprupanol,
giving 23 9 (100%) yield. TLC: (30% ethyl acetate/hexane) Rf = 0.37.
Example 18
Synthesis of a Cysteine Protease Inhibitor with an a-aminosulfone as the EWG.
Synthesis of (R)-1-(4-morpholinecarbonylphenylalanyl)amino-3-phenyl-1-phenylsulfonylpropane
and (S)-1-(4-morpholinecarbonylphenylalanyl)amino-3-phenyl-1-phenylsulfonylpropane (Mu-
Phe-Hph-a-SO2Ph, epimers separdLt:d). Method A: Z-Hph-a-SO2Ph (1.0 9, 2.44 mmol) was
treated with 30% hydrogen bromide in acetic acid (5 mL). After 30 minutes, the mixture was
diluted with ether (300 mL), filtered, washed with ether (2x30 mL), and dried in vacuo to give
0.74 9 (86%) 1-amino-3-phenyl-1-phenylsulfon~l,u,upane hydrobromide (HBr.Hph-a-SO2Ph). To
a solution of Mu-PheOH (0.64 9, 2.3 mmol) in THF (15 mL) were added 4-methylmorpholine
(0.302 mL, 2.3 mmol) and isobutyl chlo,ururllldle (0.312 mL, 2.3 mmol). The mixture was stirred
- for 10 minutes. HBr.Hph-a-SO2Ph (0.74 9, 2.1 mmol) was added, followed by 4-
methylmorpholine (0.302 mL, 2.3 mmol). After 45 minutes, the mixture was diluted with ethyl
acetate (30 mL), washed with 15 mL each of 1M HCI, saturated aqueous sodium ~ c- L,ondle,
and brine, dried over MgSO4, filtered, and the solvent was removed under reduced pressure to
give 0.75 9 (65%) of the product, Mu-Phe-Hph-a-SO2Ph.
CA 022161~1 1997-09-23
W 096/303~3 PCTrUS96/03844
54
Method B: To a solution of phenylalanine amide hydrochloride (10 9, 50 mmol) in DMF (50 mL)
and CH2CI2 (50 mL) were added triethylamine (13.9 mL, 100 mmol) and 4-lllo~,.,h-' ,ecarbonyl
chloride (5.9 mL, 50 mmol). The mixture was stirred overnight. The solvents were removed
under reduced pressure. The residue was dissolved in ethyl acetate (50 mL), and filtered. Ether
was added to the hltrate until the solution became turbid. 7.2 9 (80% yield) of the i"l~:r" ,edidl, :,
4-morpholinecarbonylphenylalanine amide (Mu-Phe-NH2) crystallized from the solution after 3
days To a solution of Mu-PheNH2 (2.24 9, 8.1 mmol) in formic acid (5 mL) was added, with
stirring, hydroc;"na",aldehyde (1.17 mL, 8.9 mmol). The mixture was stirred for 5 hours,
whereupon sodium benzenesulfinate (1.33 9, 8.1 mmol) was added. The mixture was quickly
heated to reflux over a five minute period, and was allowed to cool to room temperature. The
solution was then permitted to stir for three days. An equal volume of water was added. The
product was extracted with CH2CI2 (3x100 mL), dried over MgSO4, filtered, and the solvent was
removed under reduced pressure. The yield of product, diastereomeric (R)- and (S)-1-(4-
",or~holi"e-carbonylphenylalanyl)amino-3-phenylsulfonyl-1-phenyl-propane, was 3.9 9 (90%).
TLC (50% ethyl acetate/CH2CI2) R, = 0.27,0.34.
The diastereomers were separated by flash chromatography on 230-400 mesh silica gel (20-
50% ethyl acetate/CH2CI2, gradient elution).
Example 19
Inhibition of Cysteine Proteases with the
Inhibitors of the Invention
Conditions for cathepsin B: 50 mM phosphate, pH 6.0, 2.5 mM EDTA, 2.5 mM DTT. substrate-
[Z-Arg-Arg-AMC] = 50 mM (Km = 190 mM). The assay at 25~ was started by the addition of cat B
(final concentration approx 10 nM) and the increase in fluorescence at 450 nm with excitation at
380 nm was followed over 2 min. The depr~:ssion in the rate of substrate hydrolysis following
addition of varying conce"L,dlions of inhibitors was noted. The assay was linear throughout the
range observed. Duplicate runs were measured.
CondiLions for cathepsin L: 50 mM acetate, pH 5.5, 2.5 mM EDTA, 2.5 mM DTT. substrate:
[Z-Phe-Arg-AMC] = 5 mM (Km = 2 mM). The assay at 25~was started by the addition of cat L
(final concer,l,dlion approx 1 nM) and the increase in fluo,t:scence at 450 nm with ~x~ on at
380 nm was followed over 2 min. The dep,~:sslon in the rate of substrate hydrolysis following
addition of varying concer,L, dLions of inhibitors was noted. The assay was linear throughout the
range observed. Duplicate runs were measured.
CA 022161~1 1997-09-23
W O 96/30353 PCT~US96/03844
Conditions for cathepsin S: 50 mM phosphate, pH 6.5, 2.5 mM EDTA, 2.5 mM DTT. substrate:
[Z-Val-Val-Arg-AMC] = 10 mM (Km = 18 mM). The assay at 25~ was started by the addition of
cat S (final concer,L,dLion approx 30 pM) and the increase in fluorescence at 450 nm with
excitation at 380 nm was followed over 2 min. The depression in the rate of substrate hydrolysis
following addition of varying concentrations of inhibitors was noted. The assay was linear
throughout the range observed. Duplicate runs were measured.
Conditions for cruzain were the same as for cathepsin L with the exception that the Km for the
substrate was 1 mM.
The respective K, values were e~Li",dL~:d by using the Dixon plot as described by Irwin Segel in
Enzyme Kinetics: Behavior and analysis of rapid equilibrium and steady-state enzyme systems,
1975, Wiley-lnt~:r:,~,ience Publication, John Wiley & Sons, New York.
The results are shown in Table 2.
Tab e 2
Inhibitorcathepsin B cathepsin L cathepsin S cruzain
(K, ~M)
Mu-Phe-(DL)HphaSO2Ph 3,000 13 5 15
Mu-Phe-HphaSO2Ph - - 2.6
Mu-Phe-(DL)HphaSO2Ph, - - 3.7
Mu-Leu-HphaSO2Ph 16 1.3 1.6 0.27
Mu-Leu-HphaSO2Bzl 54 0.60 4.2 0.76
Mu-Phe-HphaSO2CH2F 170 1.5 2.8 2.0
Mu-Phe-HphaSO2Bzl 77 1.2 5.2 0.92
Mu-Phe-HphaSO2CR3 50 0.18 2.2 0.23
Mu-Phe-Hph~SO2Ph1,100 29 0.94 5.7
Mu-Phe-HphySO2Ph 48 10 0.16 4.3
Phac-Phe-HphySO2Ph~10 0.41 12.5 1.8
Mu-Np2-HphySO22Np20 0.26 0.53 0.10
Mu-Phe-HphySO2EtPh190 0.17 0.082 5.9
Suc-Phe-HphySO2Ph~1000 1.0 0.07 1.5
MeOSuc-Phe-HphySO2Ph 81 3.2 1.2 4.7
Suc-Np2-HphySO2Ph~1000 2.3 0.50 0.78
Suc-Np2-HphySO?2Np~1000 0.11 0.24 0.23
CA 022l6l~l l997-09-23
WO 96/30353 PCTrUS96/03844
56
Z-~-Ala-Phe-HphySO2Ph 520.79 3.0 0.54
~-AIa-Phe-HphySO2Ph ~>50 14 11 14
Mu-Tyr-HphySO2Ph - 2.3 9.520
Mu-Phe-HphyCO2Et 1.6 0.48 0.190.91
Mu-Phe-HphyCONHPh 2.6 2.0 1.30.13
Mu-Phe-HphyCONHBzl >>50 19 307.7
Mu-phe-Hphypo(o2Et)2 17 3.0 1.415
Mu-Phe-Hph-yPh-OMe 4.8 0.94 0.370.89
Mu-Phe-Hph-yPh-NH2 >>50 2.9 9.13.0
EXAMPLE 20
The following are representative pham~ceutic~l formulation containing a cysteine protease
inhibitor of this invention.
ORAL FORMULATION
A representative solution for oral administration contains:
Cysteine protease inhibitor 100 to 1000 mg
Citric Acid Monohydrate 105 mg
Sodium Hydroxide 18 mg
Flavoring
Water q.s. to 100 mL
INTRAVENOUS FORMULATION
A rep~ser,ldli~e solution for intravenous adminstration con- ~ I~.
Cysteine protease inhibitor 10 to 100 mg
Dextrose Monohydrate q.s. to make isotonic
Citric Acid Monohydrate 1.05 mg
Sodium Hydroxide 0.18 mg
Saline for Injection q.s. to 1.0 mL
TABLET FORMULATION
CA 02216151 1997-09-23
PCTrUS96/03844
W 096/30353
57
A rep,~:senLd~i~/e tablet form may contain:
Cysteine p,~ ase inhibitor 1%
Microcrystalline Cellulose 73%
Stearic Acid 25%
Colloidal Silica 1%