Note: Descriptions are shown in the official language in which they were submitted.
CA 02218360 1997-11-10
WO 96135677 PCT/US9''~6~1
N~ tric Oxide Syntha~e Tr~h; ~itors~
Deri~red from Cyclic Amidine~
..
This application is a continuation-in-part of U.S. Serial
No. 08/438,321, filed May 10, 1995, the contents of which are
herein incorporated by reference.
Field of the Invention
The present invention relates to amidino derivative
compounds, pharmaceutical compositions containing these novel
compounds, and to their use in therapy, in particular their use
as nitric oxide synthase inhibitors.
R~ckarollnd of the Invention
It has been known since the early 1980's that the vascular
relaxation brought about by acetylcholine is dependent on the
presence of the endothelium and this activity was ascribed to a
labile humoral factor termed endothelium-derived relaxing factor
(EDRF). The activity of nitric oxide (NO) as a vasodilator has
been known for well over 100 years and NO is the active
component of amyl nitrite, glyceryl trinitrate and other
nitrovasodilators. The recent identification of EDRF as NO has
coincided with the discovery of a biochemical pathway by which
NO is synthesized from the amino acid L-arginine by the enzyme
NO synthase.
NO is the endogenous stimulator of the soluble guanylate
cyclase and is involved in a number of biological actions in
addition to endothelium-dependent relaxation including
cytotoxicity of phagocytic cells and cell-to-cell communicatio-.
in the central nervous system ~see Moncada et al, Biochemical
Pharmacolo~v, 38, 1709-1715 (1989) and Moncada et al,
ph~rmacolo~ical Reviews, 43, 109-142 (1991).
It is now thought that excess NO production may be involved in a
number of conditions, partic~llarly conditions which involve
CA 022l8360 l997-ll-l0
W096l3S677 PCT~S9G~'~ '
systemic hypotension such as toxic shock and therapy wi_h
certain cytokines.
The synthesis of NO from L-arginine can be inhibited by the
~-arginine analogue, L-N-monomethyl-arginine (L-NMMA) and the
therapeutic use of L-NMMA for the treatment of toxic shock and
other types of systemic hypotension has been proposed (WO
91/04024 and GB-A-2240041). The therapeutic use of certain
other NO synthase inhibitors apart from L-NMMA for the same
?urpo.se has also been proposed in WO 91/04024 and in EP-A-
0446699.
It has recently become apparent that there are at least
three types of NO synthase as follows:
(i) a constitutive, Ca++/calmodulin dependent enzyme,
loc~ted in the endothelium, that releases NO in response to
receptor or physical stimulation.
(ii) a constitutive, Ca++/calmodulin dependent enzyme,
located in the brain, that releases NO in response to receptor
or physical stimulation.
(iii) a Ca++ independent enzyme which is induced after
activation of vascular smooth muscle, macrophages, endothelial
cells, and a number of other cells by endotoxin and cytokines.
Once expressed this inducible NO synthase synthesizes NO for
long periods.
The NO released by the constitutive enzymes acts as a
transduction mechanism underlying several physiological
responses. The NO produced by the inducible enzyme is a
cytotoxic molecule for tumor cells and invading microorganisms.
It also appears that the adverse effects of excess NO
production, in particular pathological vasodilation and tissue
damage, may result largely from the effects of NO synthesized by
the inducible NO synthase.
There is also a growing body of evidence that NO may be
involved in the degeneration of cartilage which takes place in
certain conditions such as arthritis and it is also known that
NO synthesis is increased in rheumatoid arthritis. Accordingly,
-- 2 --
CA 02218360 1997-11-10
W O 96/3S677 PC~rrUS96/06831
urther conditions in which there is an advantage ~n inhiDiting
NO production from L-arginine inciude autoimmune and/or
inflammatory conditions affecting the joints, for example
ar~hri~is, inflammatory bowel disease, cardiovascular ischemia,
diabetes, hyperalgesia (allodynia), cerebral ischemia (both
focal ischemia, thrombotic stroke and global ischemia, secondary
to cardiac arrest), ar.d other CNS disorders mediated by N0,
including opiate tolerance in patients needing protracted opiate
analgesics, benzodiazepine tolerance in patients taking
benzodiazepines, and other addictive behaviors for example
nicotine and eating disorder.
Further conditions in which there is an advantage in
inhibiting NO production from L-arginine include systemic
hypotension associated with septic and/or toxic shock induced by
a wide variety of agents; therapy with cytokines such as TNF,
IL-1 and IL-2; and as an adjuvant to short term
;mml-nosuppression in transplant therapy. Further conditions in
whi.ch there is an advantage in inhibiting NO production from L-
arginine include autoimmune diseases and/or inflammatoryconditions such as those affecting the joints, for example
arthritis or ARDS or inflammatory bowel disease, or asthma,
cardiovascular ischemia, congestive heart failure, myocarditis,
artherosclerosis, migraine, reflux esophagitis, diarrhea,
irritable bowel syndrome, cystic fibrosis, emphysema, and
diabetes.
Some of the NO synthase inhibitors proposed for therapeutic
use so far, and in particular L-NMMA, are non-selective in that
~hey inhibit both the constitutive and the inducible NO
synthase. Use of such a non-selective NO synthase inhibitor
requires that great care be taken in order to avoid the
potentially serious consequences of over-inhibition of the
constitutive NO-synthase including hypertension and possible
thrombosis and tissue damage. In particular, in the case of the
therapeutic use of L-NMMA for the treatment of toxic shock t
has been reComm~n~e~ that the patient must be subject to
continuous blood pressure monitoring throughout the trea~ment.
Thus, while non-selective NO synthase inhibitors have
-- 3 --
CA 02218360 1997-11-10
therapeutic utility provided that appropriate precautions are
taken, N0 synthase inhibitors which are selective in the sense
that they inhibit the inducible N0 synthase to a considerably
greater e~tent than the constitutive is~forms of No synthase
would be of even greater therapeutic benefit and easier to use.
W094/12165, W094/14780, W093/13055, EP0446699A1 and U.S.
Patent No. 5,132,453 disclose compounds that inhibit nitric
oxide synthesis and preferentially inhibit the inducible iso~orm
o~ nitric o~ide synthase. The disclosures of which are hereby
incorporated by re~erence in their entirety as if written
herein.
WO-A 9511231 discloses cyclic amidino derivatives of the formula
R ~
2 /~ N ~NR4
R3
which are useful as nitric oxide synthase inhibitors.
US-A 2 049 582 describes the preparation of acid amidines.
US-A 3 121 093 is related to substituted iminopyrrolidines of the formula
R2 R~
Rl ~/~R
which are said to be useful as fungicides.
BRN 39493 describes the preparation of 7-imino-azepane-2-carboxylic acid.
BRN 6143647 describes the preparation of 7-(4-methoxy-phenyl)-<1,4>thiazepan-5-
yl-yiidineamine.
BRN 389517 describes the preparation of 6-imino-piperidine-2-carboxylic acid ethyl-
ester and its amide is disclosed in BRN 388043.
BRN 5981412 describes the preparation of the compound
)~ S~
;
CA 02218360 1997-11-10
4 2
HO e ~~=
BRN 778069 describes the preparation of 5-imino-thiomorpholine-3-carboxylic aid.BRN 880305 describes the preparation of (2-imino-1-methyl-hexahydropyridine-4-yl)-acetic
acid.
BRN 742055 describes the preparation of piperazine-2-ylidene amine.
BRN 881955 describes the preparation of amino-(2-imino-hexahydro-pyridimidine-4-yl)
acetic acid.
E3RN 777936 describes the preparation of 2-imino-hexahydro-pyrimidine-4-carboxylic acid.
BRN 507189 describes the preparation of 5-methyl-oxazolidine-2-ylideneamine.
BRN 507962 describes the preparation of 1-isopropyl-pyrazolidine-3-ylideneamine.BRN 5403979 describes the preparation of 4,5,6,7-tetrahydro-<1,3>thi~,epine-2-ylamine.
BRN 107123 describes the preparation of 4,5,6,7-tetrahydro-1H-<1,3>diazepine-2-ylamine.
BRN 141409 describes the preparation of 3-amino-2,5,6,7-tetrahydro-<1,4>thiazepine-5-
carboxylic acid.
BRN 5~08 describes the preparation of 5-amino-3,6-dihydro-2H-<1,4>thiazine-3-carboxylic
acid.
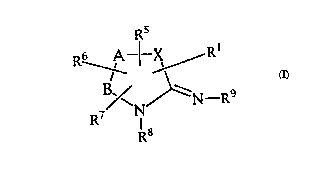
Summar~ of the Invention
In accordance with the present invention novel ~mi dino
derivatives are provided. These novel inhibitor compounds can
be represented by the following chemical formula (I):
Rs
RSB ~-----~Rl
~N~--N R
R7
. R8
(I)
0 S~
i
CA 022l8360 l997-ll-lO
W096/3S677 PCT~Sg~'OC~l
carboxyalkyl, CONR1OR11, S(O)R10, a (O) 2RlO, S02NRl~Rll,
PO(OR1O)(OR11), amidino, guanidino;
wherein all said substitutions may be optionally subs~i_uted with
one or more of the following: halogen, lower alkyl, am.ino,
alkylamino, dialkylamino, aminoalkyl, aminoacyl, carboxyl,
~ carboalkoxy, carboaryloxy, carboalkylaryloxy, hydroxy, lower alkoxy,
S(O)R10, S(O)2R10, amidino, guanidino;
X = NR2, O, S, SO, SO2, (CH2)p, CH=CH;
p = 0 to 6;
A = NR3, O, S, SO, SO2, (CH2)~, CH=C~;
q = 0 to 6;
B = NR4, O, S, SO, SO2, (CH2)~, CH=CH;
v = 0 to 6;
R2 = hydrogen, lower alkyl, aryl, heterocyclyl;
R3 = hydrogen, lower alkyl, aryl, heterocyclyl;
R4 = hydrogen, lower alkyl, aryl, heterocyclyl;
R5, R6, R7 are independently selected from hydrogen, lower
alkyl, lower alkenyl, lower alkynyl, heterocyclyl, hydroxy,
lower alkoxy, thiol, lower thioalkoxy, S(O)R9, S(O)2R9, halogen,
nitro, amino, alkylamlno, dialkylamino, Am~oAlkyl,
dialkylaminoalkyl, arylamino, aminoaryl, alkylaminoaryl,
acylamino, carboxyl, carboalkoxy, carboaryloxy,
carboarylalkyloxy, cyano, aminocarbonylalkoxy,
aminocarbonylamino, aminocarbonylaminoalkyl, haloalkyl,
SO2NRlORll~ wherein all said substitutions may be optionally
substituted with one or more of the following: lower alkyl,
amino, alkylamino, dialkylamino, aminoalkyl, aminoacyl,
carboxyl, carboalkoxy, carboaryloxy, carboalkylaryloxy, hydroxy,
lower alkoxy;
R5, R6, may optionally be taken together to form an alicyclic
hydrocarbon, heterocyclyl or aromatic hydrocarbon and said
optionally formed ring may be optionally substituted with one o-
more of the following:
lower alkyl, lower alkenyl, lower alkynyl which may be
optionally substituted with carboxyl, carboalkoxy, carboaryloxy,
carboxyalkylaryloxy and lower alkoxy;
CA 02218360 1997-11-10
6 .
R8 = hydrogen, hydr,oxy, O-Alkyl;
R9 = hydrogen, hydroxy, O-Alkyl;
R10 = hydro~en, lowe~ alkyl, alkylaryl, aryl
Rll = hydrogen, lower alkyl, alkylaryl, aryli
R1O and Rll, taken together, may be al~ylene, resulting in a N-
containing heterocycle;
with the proviso that when A is (CH2)q and B is (CH2)r, then only one of R1, Rs, R6, R7 can
be hydrogen;
with the proviso that when R1 is iower alkyl, lower alkenyl, lower alkynyl, alkyloxy, or
thioalkoxy, R' is not substituted by cycloalkyl, heterocyclyl, and aryl, unless one A, or B is
NR2, O, S, SO, SO2;
with the proviso that when A and B are(CH2)p or CH=CH, and R1 is lower alkyl, lower
aikenyl, lower alkynyl, alkyloxy, or thioalkoxy, R1 is not substituted by cycloalkyl,
heterocyclyl, or aryl and Rs and R5 are not H;
with the further proviso that when X=CH=CH, A=(CH2)q, B=(CH2)V and q+v=2, then none of
R1, Rs, R6 and R7 can be carboxy at the 6-position; and
with the further proviso that when X=NH, A=(CH2)ql B=(CH2)~ and q+v=4, then none of R1,
R5, R6 and R7 can be carboxy at the 7-position.
with the further proviso that when A or B is sulfur, R' cannot be aryl;
with the further proviso that when X=CH2, A=S and B=(CH2)V, and v= 1 or 2, one of R5, R6,
and R7 is carboxyl at position 6 if v=1, or position 7 if v=2, then at least one of R', R5, R6
and R' is not hydrogen;
with the further proviso that when A or B is N, at least one of R', R5, R6, and R7 is not H;
with the further proviso that when X is (CH2)p, A is (CH2)q, and p+q is 2, and B is N, R4 is not
alkyl;
o S~
CA 02218360 1997-11-10
6a
with the further proviso that when X is (CH2)p A is (CH2)ql p+q is 3, R1, R5, R6, and R7 are
each H, B is not SO2;
with the further proviso that when X is (CH2)p, A is (CH2)q, B is (CH2)v and p+q+v is 3, or one
of B is CH=CH and p+q is 1, then none of R', R5, R6, and R7 can be a lower alkyl, lower
alkenyl, lower alkynyl, cycloalkyl, or aryl at position 5, and no more than one of R1, R5, R5
and R7 can be alkoxy or cycloalkoxy at the ~position.
S~
,0
,. . CA 02218360 1997-11-10
.~. . ..
In another broad aspect, the present inventlon is directed
to inhibiting nitric oxide synthesis in a subject in need o~
such inhibition or treatment by administering a compound of
Formula (I) which preferentially inhibits the inducible iso~orm
S o~ nitric oxide synthase over the constitutive iso~orm of nitric
oxide synthase, in a nitric oxide synthesis inhibiting amount to
such subject.
The invention ~urther relates to a pharmaceutical
composition comprising a compound ~rom Formula (I)~including those
as described above in the 6th to 10th proviso.
Compounds and compositions de~ined above have use~ulness as
inhibitors of nitric oxide synthase. These compounds also
preferentially inhibit the inducible form.
Conditions in which there is an advantage in inhibiting NO
production ~rom L-arginine in disorders mediated by nitric.oxide
including amongst others, systemic hypotension associated with
septic and/or toxic shock induced by a wide variety of agents;
20 therapy with cytokines such as TNF, IL-l and IL-2; and as an
adjuvant to short term immunosuppression in transplant therapy.
Further conditions in which there is an advantage in inhibiting
NO production ~rom L-arginine include autoimmune diseases and/or
inflammatory conditions such as those affecting the joints, ~or
example arthritis or in~1ammatory bowel disease, cardiovascular
ischemia, diabetes, congestive heart ~ailure, myocarditis,
artherosclerosis, migraine, reflux esophagitis, diarrhea,
irritable bowel syndrome, cystic fibrosis, emphysema,
hyperalgesia (allodynia) cerebral ischemia (both focal ischemia,
thrombotic.stroke and global ischemia, secondary to cardiac.
arrest) and other CNS disorder mediated by NO, including opiate
tolerance in patients needing protracted opiate analgesics,
benzodiazepine tolerance in patients taking benzodiazepines, and
other addictive behaviors for example nicotine and eating
disorder.
,
The present invention includes compounds of ~ormula (I) in
~ the form o~ salts, in particular acid addition salts. suitable
s al ts include tho s e Eormed wi th b~ th ~rganic and inor~nic
CA 02218360 1997-11-10
WO 96/3S677 PCT/U~ Q~l
acids. Sucn acid addi.ion salts will normally be
pharmaceutically acceptable although salts of non-
pharmaceutically acceptable salts may be of utilit-~ in the
preparation and purification of the compound in question. Thus,
preferred salts include those formed from hydrochloric,
hydrobromic, sulfuric, citric, tartaric, phosphoric, lactic,
acetic, succinic, fumaric, maleic, methanesulfonic,
ethanesulfonic, p-toluenesulfonic, benzenesulfonic and the like.
(See, for example, S. M. Berge et al., Pharmaceutical Salts, J.
10 Pharm. Sci., 1977, 66, 1-19.) Salts of the compcunds of
formula (I) can be made by reacting the appropriate compound in
the form of the free base with the appropriate acid.
While it may be possible for the compounds of formula (I)
to be ~m; ni stered as the raw chemical, it is preferable to
present them as a pharmaceutical formulation. According to a
further aspect, the present invention provides a pharmaceutical
formulation comprising a compound of formula (I) or a
pharmaceutically acceptable salt or solvate thereof, together
with one or more pharmaceutically acceptable carriers thereof
and optionally one or more other therapeutic ingredients. The
carrier(s) must be "acceptable~ in the sense of being compatible
with the other ingredients of the formulation and not
deleterious to the recipient thereof.
The formulations include those suitable for oral,
inhalation, parenteral (including subcutaneous, intradermal,
intramuscular, intravenous and intraarticular), rectal and
topical (including dermal, buccal, sublingual and intraocular)
3 0 ~mi ni stration although the most suitable route may depend upon
for example the condition and disorder of the recipient. The
formulations may conveniently be presented in unit dosage form
and may be prepared by any of the methods well known in the ar~
of pharmacy. All methods include the step of bringing into
35 association a compound of formula (I) or a pharmaceutically
acceptable salt or solvate thereof ("active ingredient") with
the carrier which constitutes one or more accessory ingredients.
In general, the formulations are prepared by uniformly and
in~imately bringing into association the active ingredient with
-- 8 --
CA 02218360 1997-11-10
WO 96135677 PCT/US96/06831
' qui~ carr ers or finely divided solid carriers o~ both and
then, f necessary, shaping the product into the desired
formu ation.
~ormulations of the present invention suitable for oral
~nm; n stration may be presented as discrete units such as
capsuies, cachets or tablets each contA;n;ng a predetermined
amount of the active ingredient; as a powder or granules; as a
solution or a suspension in an aqueous liquid or a non-aqueous
liquid; or as an oil-in-water liquid 2mulsion or a water-in-oil
liquid emulsion. The active ingredient may also be presented as
a bolus, electuary or paste.
A tablet may be made by compression or moulding, optlonally
with one or more accessory ingredients. Compressed tablets may
be prepared by compressing in a suitable machine the active
ingredient in a free-flowing form such as a powder or granules,
optionally mixed with a binder, lubricant, inert diluent,
lubricating, surface active or dispersing agent. Moulded
tablets may be made by moulding in a suitable m~ch; ne a mixture
of the powdered compound moistened with an inert liquid diluent.
The tablets may optionally be coated or scored and may be
formulated so as to provide slow or controlled release of the
active ingredient therein.
Formulations for parenteral ~m; n; stration include aqueous
and non-aqueous sterile injection solutions which may contain
antioxidants, buffers, bacteriostats and solutes which render
~he formulation isotonic with the blood of the intended
recipient; and aqueous and non-aqueous sterile suspensions whic:-
may i-clude suspending agents and thickening agents. The
Formu_ations may be presen~ed in unit-dose or multi-dose
containers, for example sealed ampoules and vials, and may be
storea in a freeze-dried (lyophilized) condition requirir.g onl--
the addition of the sterile liquid carrier, for example, salir.ewater-for-injection, immediately prior to use. Extemporaneous
injec~ion solutions and suspensions may be prepared from ster- e
powders, granules and tablets of the kind previously described.
_ 9 _
CA 02218360 1997-ll-lO
WO 96t3S677 PCT/US9"."~'31
-ormulations for rectal admir stration may be Dresented as
a suppository with the usual carriers such as cocoa butte- or
polyethylene glycol.
~ormulations for topical administration in the mouth, for
exampie buccally or sublingually, include lozenges comprising
the active ingredient in a flavored basis such as sucrose and
acac a or tragacanth, and pastilles comprising the active
ingredient in a basis such as gelatin and glycerin or sucrose
and acacia.
Formulations for inhalation ~mi n; stration where the active
ingredient is inhaled into the lungs either as a mist or co-
~mi ni stered with an inert carrier agent.
Preferred unit dosage formulations are those containing an
effective dose, as hereinbelow recited, or an appropriate
fraction thereof, of the active ingredient.
It should be understood that in addition to the ingredients
particularly mentioned above, the formulations of this invention
may include other agents conventional in the art having regard
to the type of formulation in ~uestion, for example those
suitable for oral ~m;nistration may include flavoring agents.
The compounds of the invention may be administered orally
or via injection at a dose of from 0.001 to 2500 mg/kg per day.
The dose range for adult humans is generally from 0.005 mg to 10
g/day. Tablets or other forms of presentation provided in
discrete units may conveniently contain an amount of compound of
the i..vention which is effective at such dosage or as a multi2ie
of the same, for instance, units containing 5 mg to 500 mg,
usually around 10 mg to 200 mg.
The compounds of formula (I) are preferably administered
orally or by injection (intravenous or subcutaneous). The
precise amount of compound administered to a patient will be the
responsibility of the attendant physician. However, the dose
employed will depend on a number of factors, including the age
_ 10 -
CA 02218360 1997-11-10
WO 96/3S677 PCT/IJS9''0~ 1~31
and sex of the patient, the precise disorder being treated, and
its severity. Also, the route of ~m;nistration may vary
depending on the condition and its severity.
As utilized herein, the term "lower alkyl~, alone or in
combination, means an acyclic alkyl radical cont~in;ng from 1 to
about 10, preferably from 1 to about 8 carbon atoms and more
preferably 1 to about 6 carbon atoms. Examples of such radicals
include methyl, ethyl, n-propyl, isopropyl, n-butyl, iso~utyl,
sec-butyl, tert-butyl, pentyl, iso-amyl, hexyl, octyl and the
like.
The term "lower alkenyl" refers to an unsaturated acyclic
hydrocarbon radical in so much as it contains at least one
double bond. Such radicals cont~;n;ng from about 2 to about 10
carbon atoms, preferably from about 2 to about 8 carbon atoms
and more preferably 2 to about 6 carbon atoms. Examples of
suitable alkenyl radicals include propylenyl, buten-1-yl,
isobutenyl, pentenylen-l-yl, 2-2-methylbuten-1-yl, 3-
methylbuten-1-yl, hexen-1-yl, hepten-1-yl, and octen-1-yl, and
the like.
The term "lower alkynyl" refers to an unsaturated acyclic
hydrocarbon radical in so much as it contains one or more triple
bonds, such radicals contA;n;ng about 2 to about 10 carbon
atoms, preferably having from about 2 to about 8 carbon atoms
and more preferably having 2 to about 6 carbon atoms. Examples
of suitable alkynyl radicals include ethynyl, propynyl, butyn-1-
yl, butyn-2-yl, pentyn-1-yl, pentyn-2-yl, 3-methylbutyn-1-yl,
hexyn-1-yl, hexyn-2-yl, hexyn-3-yl, 3,3-dimethylbutyn-1-yl
radicals and the like.
- The term "alicyclic hydrocarbon" or "cycloalkyl" means a
aliphatic radical in a ring with 3 to about 10 carbon atoms, and
preferably from 3 to about 6 carbon atoms. Examples of suitable
alicyclic radicals include cyclopropyl, cyclobutyl, cyclopentyl,
cyclohexyl, cyclohexenyl and the like.
The term "aromatic hydrocarbon" means aromatic radical wi.h
4 to about 16 carbon atoms, preferably 6 to about 12 carbon
- 11 -
CA 02218360 1997-11-10
WO 96135677 PCI'lUS9f '0~
a~oms, more preferably 6 to about 10 carbon atoms. rxampies o
suitable aromatic hydrocarbon radicais include pher.yl, naphthyi,
and tre like.
The term ~aryl" as used herein means 5- and 6-membered
single-aromatic radicals which may include from zero to four
heteroatoms. Representative aryls include phenyl, thienyl,
furanyl, pyridinyl, (is)oxazoyl and the like.
The term DCM means dichloromethane.
The term DEAD means diethyl azodicarboxylate.
The term DIBAL-H means diisobutylall~m;nl~m hydride.
The term DMAP means dimethylaminopyridine.
The term DMSO means dimethylsulfoxide.
The term EDC means 1-(3-dimethylaminopropyl)-3-
ethylcarbodiimide hydrochloride.
The term ~heterocyclyl radical~ means a saturated or
unsaturated cyclic hydrocarbon radical including aromatic
systems with 4 to about 10 carbon atoms, preferably about 5 to
about 6; wherein 1 to about 4 carbon atoms are replaced by
nitrogen, oxygen, sulfur, or carbonyl. The "heterocyclic
radical n may be fused to an aromatic hydrocarbon radical.
Suitable examples include pyrrolyl, pyridinyl, pyrazolyl,
triazolyl, pyrimidinyl, pyridazinyl, oxazolyl, isoxazolyl,
thiazolyl, imidazolyl, indolyl, thienyl, furanyl, retrazolyl, 2-
pyrrolinyl, 3-pyrrolinyl, pyrrolindinyl, 1,3-dioxolanyl, 2-
imidazolinyl, imidazolidinyl, 2-pyrazolinyl, pyrazolidinyl,
isoxazolinyl, isothiazolyl, oxadiazolyl, triazolyl,
thiadiazolyl, 2H-pyranyl, 4H-pyranyl, piperidinyl, 1,4-dioxanyl,
morpholinyl, 1,4-dithianyl, thiomorpholinyl, pyrazinyl,
piperazinyl, triazinyl, 1,3,5-trithianyl, benzo(b)~hiophenyl,
benzimidazolyl, quinolinyl, and the like.
- 12 -
CA 02218360 1997-11-10
WO 9613S677 PCT/US96106831
~he term HOBT means N-hydroxybenzo~riazole.
The term "lower alkoxy", alone or in combination, means an
alkyl ether radical wherein the term alkyl is as defined above
and most preferably cont~;ning 1 to about 4 carbon atoms.
Examples of suitable alkyl ether radicals include methoxy,
ethoxy, n-propoxy, isopropoxy, n-butoxy, iso-butoxy, sec-butoxy,
tert-butoxy and the like.
L0 The term "lower thioalkoxy~, alone or in combination, means
an alkyl thioether radical wherein the term alkyl is as defined
above and most preferably contA;ning 1 to about 4 carbon atoms.
Examples of suitable alkyl thioether radicals include
thiomethoxy, thioethoxy, thio-n-propoxy, th o-i-propoxy, thio-n-
butoxy, thio-iso-butoxy, thio-sec-butoxy, thio-tert-butoxy and
the like.
The term alkoxycarbonyl as used herein means an alkoxy
group, as defined abo~e, having a carbonyl (C=O) group attached.
The term "halogen" means fluorine, chlorine, bromine or
iodine.
The term MCPBA means m-chloroperbenzoic acid.
The term NMM means N-methylmorpholine.
The term NMMO means 4-methylmorpholine N-oxide.
The term "prodrug" refers to a compound that is made more
active in vivo.
~ The term sulfinyl means SO.
The term sulfonyl means SO2.
The term TEA means triethylamine.
The term TMSN3 means azidotrimethylsilane.
- 13 -
CA 02218360 1997-11-10
WO 96/3S677 PCT/U~39G~
As used herein, reference to "treatment" of a patient is
intenaed to include prophylaxis.
All references, patents or applications, U.S. or foreign,
cited n the application are hereby incorporated by reference as
if wri~ten herein.
Compounds of the present invention can exist in geometric
or stereoisomeric forms. The present invention contemplates all
such compounds, including cis- and trans-geometric isomers, E-
and Z-geometric isomers, R- and S-enantiomers, diastereomers, d-
isomers, l-isomers, the racemic mixtures thereof and other
mixtures thereof, as falling within the scope of the invention.
Disclosed are eleven general synthetic processes useful in
the preparation of the compounds of the present invention.
- l4 _
CA 02218360 1997-11-10
Scheme 1:
~ R\J~ CO2Me (~
R~ A-¦- B 1 ( Z = halide) RX\~9
R7 ~ RS ~ RA ¦ B
RS '<1~~ R A-¦ B C02Me / z_
R6 A-¦ B C OH R ~ X\~~ R
a,b R6A ¦B R6A-¦ B
R7 R7
Rs ~ Rl g ~5 ~ ~ NH R5
R6--A ¦ B /~B R~ B/A~X
R7 / R7 R7 R6
RS~e f~h ~ 5
~gl Rl R /A\R6
i ~
RS NH .HCl NH .HCl
R6 X~ NH ~ NH RS
A/Bl Rl B/.A\X
a) Mg, THF; b) CuI, -30 ~C; c~ -30 ~C to O ~C or r. t.; d)
DMSO, oxalyl chloride, CH2Cl2, -70 ~C; e) Et3N, -70 ~C to 0 ~C;
~) NH20H, NaOAc, EtOH; g) PhSO2Cl, NaOH, H20, ace~one; h) Me30
BF4-; i) NH4Cl; j) ~2C03 ~r NaH, D~F; k) NaCN, DMSO, H20, heat
1) D~F, L- Rl (where L'-Rl is CH2=CHCO-Rl); m) iN LiOH, MeOH.
S~ '
O~
1 5
CA 02218360 1997-11-10
Scheme 2: ; ,
~ Rm/~n HQ~
R ~ ~ Rn 7~ RnY
R5 o Rm
R~ ~ + ~Rs
dr R ~d
5 OMe Rm OMe
R
e ¦ R le
R~5 ~ .HCl IRm NH .HCl
R6 X~\ NH R ~~~ ,NH R5
A/B~ y R B/A\X
(Y = CN, COOalkyl, NO2, 5O2alkyl,
SO2NH2, SO2NRL~Rll, heteroaryl)
Rm = H, alkyl, cycloalkyl, aryl, heterocycle
Rn = H, alkyl, aryl, heterocycle
Rm and Rn may be taken together to ~orm a rina
a) solvent (benzene); b) NH2OH, NaOAc, EtOH;_c) PhSO2Cl,
NaOH, ~2~, acetone; d) Me3otBFg-~ CH2C12; e) NH4Cl, MeOH
. ~i
,0
16
;
CA 02218360 1997-11-10
W09613S677 PCT/U~ 6~31
Scheme 3:
5~0R \~ J'OR R$~NH
- (R = alkyl or aryl) R6 RL
~H HCI ~ d
R~6 Rl
a) Base, RlCH2NO2 b) H2 / RaNi, 55'C c) Me3O+BF~~, CH~C12;
d) NH4Cl, MeOH
Scheme 4:
02N~Rl Rl b Rlj~'OMe
NO2 R7
~R6 Rl $R6 ~Rl
a) R5CoR6 base; b) Base, R7CH~CO~Me; c) H2/RaNi, 55~C; d)
Me3O BF~~, CH2Cl2; e) NH4Cl, MeOH.
CA 02218360 1997-11-10
W09613S677 PCT~S9'r~~~~l
S cheme 5:
O O
R7 ~ NH H R7 ~ b
R6~ ~ 6~ C02Me
'HN~ OCH~ Ph
~ ( Ph = phenyl )
O OMe
~ R7 ~ d
R6--~ ~ C02Me R~~ ~ CO2Me
R5 (CH2 ) p I R5 (CH2)
HN~ OCH2 Ph HN~ OCH2 Ph
O O
NH .HCl NH .HCl
R7_~NH ~ R _~NH
R6~ ~ C02Me R6~ ~ C02Me
R5 (CH2 ) I R5 (CH2 ) p I
HN~OCH2Ph NH2 . HCl
o
a) DBU, Z-a-phosphonoglycine trimethyl ester; b) H2/
[Rh~(COD)(R,R-DIPAMP)]~BF4- (antipod catalyst can be
used);
c) Me3O+ BF4-, CH2Cl2; d) NH4Cl,MeOH; e) H2, Pd/C.
CA 02218360 1997-11-10
Scheme 6:
O O O
NH a ~ NJ~ O~ b
--~ 1 Rs B~--6
a~ O ~ A/~ d
Me O ~ I e OMe
s~B~6 ~R
Ml e NH . HC 1 N~ . HC 1
Me ~/ ~ RI A/
Rs B~R6 s'B' 6
a) (t-butylOCO)2O, DMAP, THF; b) LiH~DS, ~MPA, THF, (lS)~
(10-camphorsulfonyl)oxaziridine or (1~)-(-)-(10-camphorsul~onyl)
oxaziridine; c) t-butyldimethylsilyl chloride, imidazole, DMF; d)
Mg(ClO4)2 (20%), CX3C~; e) Me3O BF~-, CH2C12; f) NH4Cl, MeOH; g)
(butyl)~N~F-, MeOH.
O
1 9
CA 02218360 1997-11-10
Scheme 7:
O - O ~ /--\
R ~ lR--~ 3 1 ~cOOEt
A lR - ~ f A g
CN 3 ~ i A
~, 1 R-- J 1 R ~C
R7 R7
oAo k A e
- ~= /~ON~ 1~ ~=NH
/--\ '
R ~=,'N--Z ~ lR ~=N-z 3
R R7 3) q
R ~,R7 1) r lR ~R7
MeO~N/~ 2) j ~ HN~N--
N-Z
a) Naff/TH~; b) BrCff2C~/THF; c) Ethylene glycol/p-toluenesul~onic
acid/toluene; d) LiAlH4/Et2O; e) Carbobenzoxy chloride/t-
S butanol/waCer/NaO~; f) p-Toluenesulfonyl chloride/CH2C12/pyridine: g)
2 0
CA 02218360 1997-11-10
W 0~6/35677 s~1~5-'~C~~l
:~CN/ace-oni~riie; h) .KOH/et:nylene glyco_; ~) MeI/~MF/NaHCO~i j) H~ ~d,'~eO:-;:~) B2H5,'THF; ~) HCli~cOH/H2O; n) NH2OH; ?) 3enzenesulfonyl chloride/
:-.2O/acetone NaOH; ~) T-ime~nyloxonium ~e~rafluorobora~e; r) NH4Cli~eOH;
Scheme 8:
~/~ R7 ~ R7
O N/~ O N~
N- Z BOC N- Z
/ _ o~ / d
N-Z O N
BOC ¦ N- Z
BOC
~SIi R7 i O~
o N~ MeO N--I
N- Z N- Z
Hl~ N-Z ~ N H
N- Z H H
CA 02218360 1997-11-10
W 096t35677 PCTrUS96/06831
a) (t-butylOCO)20, DMAP, THF; b) LiHMDs, HMPA, THF, (lS)-(+)- ~10-
hnr5ulfonyl)ox_ziridine or (lR)-(-)-(10-cAmrhorsulfonyl) oxaziridine;
c) t-butyldimethylsilyl chloride, imidazole, DMF; d) Mg~ClO4)2 ~20~),
CH3CN; e) Me30+ BF4-, CH2C12; f) NH4Cl, MeOH; g) ~butyl)4N+F~,MeOH; h) H2,
Pd~C.
- 22 - .
CA 02218360 1997-11-10
WO 9613S677 PCT/U~_ "0'~~l
Sche~ne 9:
J~C02Et xJ~JCN o ~ CN /--\O _N
A- B a }:. A B C02Et ' X1~JCO2Et O~OH
-~7 .~,7
e OX~NHZ f ~X~NHz For =CH2 ~X~
A- B OH A- B OTs A- B NZ
_.7 -~7 . 7
~ For E=cH2cH2
g
A X~X~ X\X~NHZ
A- B CN A- B COOH A- B COOMe
-~7 -~7 7
-- O~NH
~7 ~,7
For either E= CH2 o ~ m ~ n
or E= CH2CH2: X><~ XJ~
A- B E A- B E
7 ~?.7
NOH
A ~ ~ E
~7
- 23 -
CA 02218360 1997-11-10
W09613S677 PCT~5g~'nC~1
Scheme 9 (continued~
HO~ . , ~R ZN~-E ~
X ~ ~ /~B NZ + /A
A -B R7 R7
~7
~Me ~ q Z/N _ _ IMe
/B NZ ~R7 A
¦ r ¦ r
~H .HCl ~H .HCl N_:: NH .HCl N_ , NH HCl
B NB B NZ 7/A~ s B/A~X
a) NaH/THF; b) BrCH2CN/THF; c) Ethylene glycol/p-toluenesulfonic
acid/toluene; d) LiAlH4/Et2O; e) Carbobenzoxy chloride/t-
butanol/water/NaOH; f) p-Toluenesulfonyl
chloride/CH2C12/pyridine; g) KCN/acetonitrile; h) KOH/ethylene
glycol; i) MeI/DMF/NaHCO3; j) H2/Pd/MeOH; k) B2H6/THF; m)
HCl/AcOH/H2O; n) NH2OH; p) Benzenesulfonyl chloride/H2O/NaOH; q)
Trimethyloxonium tetrafluoroborate; r) NH4Cl/MeOH;s) H2/Pd/C.
- 24 -
CA 02218360 1997-11-10
W 096l3S677 PCTAUS96/06831
Scheme 1 ~:
O O O
~NH a ~NJ~O~ b
A' f E~Nz A/ f E~Nz
Ri
G ~ , A/~C~ d
e O Me Me
Me ~ ~ ~ Me Q~N
I e,NH . Hcl ,NH . HCl NH . HCl
~1 B ~NZ Rl B Rl
a) (t-bu~ylOCO)2O, DMAP, THF; b) LiHMDS, HMPA, THF, (lS)-(+)-
(10-camphorsulfonyl)oxaziridine or (lR)-(-)-(10-camphorsulfonyl)
oxaziridine: c) t-butyldimethylsilyl chloride, imidazole, DMF; d)
Mg(C104)2 (20%), CH3CN; e) Me~O~BF~~, CH2C12; ~) NH4Cl, ~eOH; ~)
(butyl)4N~F~, MeOH; h) H2, Pd/C.
- 25 -
CA 022l8360 l997-ll-lO
Scheme 11: '
Rs ~ R5
R~ ~\ ~4m /B~NH~
R = alkyl, cycloal~yl, aryl, heterocycle, b, c
C~I2CH (~JH2 )C~2~ or
Rm = H, alkyl, cycloalkyl, aryl, heterocycle d, e, f
or
Rn = H, alkyl, cycloalkyl, aryl, heterocycle
Rm and Rn may be taken together to form a ring
Z = leaving group X ~
n = 1-4 n
m = 1-4
Rs NH
A/B~4 N' R
a) catalytic hydrogenation; b) RCHO; c) reduction;
d) CH2=C(NHZ)CO2Me; e) reduction; ~) hydrolysis
Without further elaboration, it is believed that one s~illed in
the art can, using the preceding description, utilize the
present invention to its fullest extent. Therefore, the
following pre~erred specific embodiments are to be construed as
merely illustrative and not limitative of the remainder o~ the
disclosure in any way whatsoever.
All experiments were performed under either dry ni.trogen or
argon. A11 solvents and reagents were used without ~urther
puri~ication unless otherwise noted. The routine wor~-up o~ the
reactions involved the addition of the reaction mixture ta a
mixture o~ either neutral, or acidic, or basic aqueous
solutions and organic solvent. The a~ueous layer was extracted
26 ~ 9
CA 022l8360 l997-ll-l0
W096/3S677
-. times (x) with the indica~ed o-ganic solven~. The combined
organic extracts were washed n times (x) with ~he i.dicated
a~ueous solutions, dried over anhydrous Na2SO~, Ciltered,
concen~rated in vacuo, and purified as indicated. Separations
by column chromatography were achieved with conditions described
by S~ ill. (Still, W. C.; Kahn, M.; Mitra, A. Rapid
Chromatograhic Techrique for Preparative Separation with
Modera~e Resolution. ~. Org. Chem., 1978, 43, 2923-2925.) The
hydrocnloride salts were made from lN HCl, HCl in ethanol
(EtOH), 2 N in MeOH, or 6 N HCl in dioxane. Thin layer
chromatograms were run on 0.25 mm EM precoated plates of silica
gel 60 F254. High performance liquid chromatograms ~HPLC) were
obtained from C-8 or C-18 reverse phase columns which were
obtained from several vendors. Analytical samples were dried in
an Abderhalden apparatus at either 56 C or 78'C. lH NMR spectra
were obtA;ne~ from either General Electric QE-300 or Varian VXR
400 MHz spectrometer with tetramethylsilane as an internal
st~n~rd. 13C NMR were obtained from a Varian spectrometer at
125.8 MHz with tetramethylsilane as an internal st~n~Ard.
CA 022l8360 l997-ll-l0
Exam~le ~terme~iate )
2,2,6-trimethylcyclohexanone, oxime
N~OH
H3C ~ CH3
3 ~
A Sample o~ 2,2,6-trimethylcyclohexanone (Aldrich, 4 9 g, 39 0
mmol) was combined with hydroxylamine hydrochloride (NH2OH HCl,
3 6 g, 52.4 m~ol) and sodium acetate (NaOAc, 5 2 g, 62 9 mmol) in a
mixt~re o~ ethanol (EtOH, 35 mL) and water (25 mL). This mixture
was refluxed ~or 5 h under a nitrogen atmos-~here. After the
reaction was cooled to room temperature and stirred ~or an
additional 5 days, all solvent was removed under reduced pressure.
The residue was partitioned between ethyl acetate (EtOAc) and water
and the organic phase was washed with 1 x 75 mL o~ saturated NaCl
lS (brine), dried over Na2SO4, and stripped o~ all solvent under
reduced pressure. This provided 5 0 g (91~) of the title compound
as a white solid. This material showed a retention time of 9 6 min
(100% purity by peak area integration) on a Sh;m~U GC-14A gas
chromatograph (GC) with a 0 25 mm x 25 M methyl, 5% phenylsilicone
column using helium as the carrier gas and a temperature program
starting at 55 'C and increasing 10 /minute up to 200 C. The NMR
and IR spectra were also consistent with the assigned structure.
~lemental analysis: CgH17NO O.1 H2O (MW = lS7.04)
C H N
Calculated: 68.83 11.04 8.92
Found: 69 00 11 00 8.85
S~
' 28
r ' CA 022l8360 l997-ll-lO
~xample / Interme~iate)
Isomer-A: hexahydro-3,3,7-trimethyl-2H-azepin-2-one
Isomer-B: hexahydro-3,7,7-trimethyl-2H-azepin-2-one
c~3 c~3
_ H
\~-N ~ N
H3C 3 CH3
Isomer-A Isomer-B
A 4.9 g (34.3 mmol) sample ot the title material o~ Ex~mple 1 was
added to a dropping funnel containing 6 mL of 80~ H2so4~ ~~ter
using a stirring rod to obtain a turbid solution, thls mixture was
added dropwise (10 mi~) to 5 mL o~ 80~ H2S04 stirred magnetically
and maintained at 120 ~C with an external oil bath. Within S
minutes o~ the start o~ addition an exotherm was noted and the
temperature o~ the reaction rose to 160 ~C be~ore cooling again to
120 ~C. Ten minutes later the flask was removed ~rom the bath and
allowed to cool to room temperature. The product mixture was
diluted with water (20 mL) and brought to pH 6 with concentrated
NH40H. This solution was further diluted with 75 mL o~ water and
extracted with 3 x 75 mL o~ CH2Cl2. The combined organic phase was
washed with 1 x 50 mL of brine, dried (Na2S04), filtered, and
stripped of all solvent under reduced pressure. The oily residue
(2.9 g, 56~) is separated by HPLC on silica gel to yield the title
products.
" ~ D S~
~9
r CA 02218360 1997-11-10
.Exam~le ~ (Tnlerme~lat~)
- 3,4,5,6-tetrahydro-7-methoxy-2,6,6-trimethYl-2H-aZepine
CH3
~ CH3
~OMe
~ N
H3C
To a magnetically stirred slurry of trimethyloxonium
tetra~1uoroborate (Lancaster, 0.30 g, 2.0 mmol) and 3A molecular
sieves (2 g) in CH2C12 (lS mL) under argon (Ar) was added the
10 Isomer-A product of Example 2 (0.31 g, 1.5 mmol). This mixture was
stirred at room temperature for 3 days before it was diluted with
10 mL o~ CH2C12 and partitioned between 40 mL o~ saturated KHCO3
and 50 mL of EtOAc. The organic phase was separated, dried over
Na2SO4, ~iltered, and stripped o~ all solvent under reduced
pressure to provide the crude title product as a pale yellow oil.
This material was chromatographed on a short path Merck flash
silica column eluting with EtOAc/n-hexane (1:1). The title pale
yellow liquid product (308 mg, 93%) had a GC retention time o~ 15.5
min (lG0%) under the conditions of Example 1 and NMR and IR spectra
consistent with the indicated product.
Example 4 (Intermediate)
3,4,5,6-tetrahydro-7-methoxy-Z,2,6-trimethyl-2H-azepine
~ CH3
.~ /~OMe
~ N
CH3
The Isomer-B product of Example 2 is reacted with trimethyloxonium
tetrafluoroborate by the method of Example 3 to produce the title
material.
CA 022l8360 l997-ll-l0
W096/35677 PCT~S96/06831
Example 5
hexahydro-3,3,7-trimethyl-2H-azepin-2-imine, monohydrochloride
~ 5
CH3
- ~CH3
~= N H
~ H .HCl
H3C
The title product of Example 3 (0.30 g, 1.4 mmol) and 0.06 g (1.1
mmol) of ammonium chloride (NH4Cl) were refluxed in 13 mL of
methanol (MeOH) under a nitrogen atmosphere for 19 h. After
cooling the reaction to room temperature, it was filtered, stripped
of all solvent under reduced pressure, and partitioned between~l5
mL of water and 7 mL of CH2C12. The organic and aqueous phases
were separated and the a~ueous phase was washed with a 25 mL
portion of EtOAc before it was lyophilized to pro~ide 0.24 g (92~)
of the white solid title material.
Example 6
hexahydro-3,7,7-trimethyl-2H-azepin-2-imine, monohydrochloride
CH3
,)= N H
3 CH3
mhe product of Example 4 in MeOH is reacted with ammonium chloride
by the method of Example 5 to generate the title ma~erial.
- 31 -
CA 02218360 1997-11-10
Example 7 (Intermediate)
3,3,5,5-tetramethylcyclohexanone, oxime
N,OH
7C~
A sample o~ 3,3,5,5-tetramethylcyclohexanone (Aldrich, 6.2 g, 40.0
mmol) was converted to the title compound by the method of Example
1 using 5.6 g (80.0 mmol) o~ hydroxylamine hydrochloride and 6.7 g
(82.0 mmol) o~ NaOAc in a mixture o~ 60 mL o~ EtOH and 60 mL of
water. The procedure produced 7.5 g (100~) o~ the title material
as a white solid.
Example 8
hexahydro-4,4,6,6-tetramethyl-2H-azepin-2-one
~C~~
N
A sample o~ the product o~ Example 7 (7.5 g, 44.4 mmol) was
converted to the title compound by the method o~ Example 2 using 11
mL o~ 80% H2SO4. The procedure produced.5.6 g (75~) o~ the title
material as a pale yellow tacky solid.
~ S~
~ CA 022l8360 l997-ll-l0
~ ~xample 9 (InteI~me~ia~te)
3,4,5,6-tetrahydro-7-methoxy-3,3,5,5-tetramethYl-2H-azepine
~~ /> O M e
S N
The title product of Exampl~ 8 (8gS mg, S . 0 mmol) was reacted with
trimethyloxonium tetra~luoroborate (962 mg, 5.0 mmol) ~y the method
o~ Example 3 to yield 8I5 mg (100%) o~ the title material.
Example 10
hexahydro-4,4,6,6-tetramethyl-2H-azepin-2-imine, monohydrochloride
\l
~NH
N .HCl
H
The product o~ Example 9 (110 mg, 0.~ mmol) in 3.5 mL o~ MeOH was
reacted with ammonium chloride (32 mg, 0.6 mmol) by the method o~
Example 5 to yield 90 ms (67%) o~ the title material.
HRMS (EI) calcd ~or CloH20N2 m/e 168.163, 'ound m/e 168.162.
H NMR(CD3OD): ~ 3.21 (s, 2H), 2.62 (s, 2H), 1.54 (s, 2H), 1.1 (s,
6H), 1.01 (s, 6H).
25 Elemental analysis: CloH20N2 HCl 0.3 H20 - 0.25 NH4Cl (MW =
223.52)
C H N Cl
Calculated: 53.74 10.19 14.10 19.83
30 Found: 53.71 9.66 13.99 19.59
~ 9S~
33
CA 022l8360 l997-ll-lO
Example 11 (Intermediate)
Tetrahydro-4~-pyran-4-one, oxime
,OH
Tetrahydro-4H-pyran-4-one (5.0 g, 0.05 mole), hydroxylamine
hydrochloride (5.2 g, 0.075 mole) and sodium acetate (13.6 g,
0.1 mole) were re~luxed in ethanol (30 mL)/H20 (20 mL)
overnight. Contents were allowed to cool and concentrated in
vacuo to remove the ethanol. The aqueous solution le~t was
extracted with CH2C12 which was dried (MgS04) and concentrated
in vacuo leaving the title material as a white solid (5.4 g).
lH NMR (CDCl3): ~ 9.15 (br, lH); 3.85-3 70 (m, 4H); 2.72 - 2.60
(m, 2H); 2.40 - 2.3S (m, 2H).
Example 12 (Intermediate)
tetrahydro-1,4-oxazepin-5(2H)-one
N''~O
H
To the title material o~ Example 11 (5.4 g, 0.047 mole) in
2S acetone (30 mL) at 0 C was added lN sodium hydroxide. Benzene
sul~onyl chloride (6 mL, 0.047 mole) in acetone (lO mL) was
added dropwise with magnetic stirring. Contents were s~tirred 72
hours and concentrated in vacuo to remove the acetone. The
aqueous solution was extracted with CH2Cl2 (2 x 150 mL), dried
(MgSO4) and concentrated in vacuo leaving an am~er oil/solid
S~
34
CA 02218360 1997-11-10
WO 96/3S677 PCT/U' r ''~
(2.2 g). The residue was c~ystallized from hexanes ~o give the
.it:le material as a white solid (1.37 g).
lH NMR (CDCl3): ~ 6.90 (br, lH); 3.82 - 3.70 (m, 4H); 3.38 -
3.30 (m, 2H); 2.75 - 2.65 (m, 2H).
Example 13
tetrahydro-1,4-oxazepin-5(2H~-imine, tri~luoroacetate salt
~'
H~NH
~TFA
The title material of Example 12 (960 mg, 0.008 mole) and
trimethyloxonium tetrafluoroborate (1.5 g, 0.01 mole) were mixed
in CH2Cl2 (50 mL) and stirred 72 hours. Contents were
concentrated in vacuo and the residue was dissolved in methanol
(50 mL). Anhydrous 2~monia was bubbled through for 15 minutes.
Contents were stoppered and stirred overnight. After
concentrating in vacuo, the residue was partitioned between
CH~Cl2 and water. The aqueous layer was purified by C-18
reverse phase chromatography eluting with 100% H2O (0.05% TFA)
to give the title material as a white solid (730 mg).
lH NMR (D2O): ~ 3.78 - 3.72 (m, 2H); 3.68 - 3.63 (m, 2H); 3.49 -
3.44 (m, 2H); 2.85 - 2.80 (m, 2H).
- 35 -
CA 022l8360 i997-ll-l0
Ex~mple 14 ~Intermedia~e) ~.'
1-(5,6-dihydro-2H-pyran-4-yl)pyrrolidine
Tetrahydro-4H-pyran-4-one (5.0 g, 0.05 mole) and pyrrolidine
(4.6 mL, O 055 mole) were re~luxed in benzene (50 mL) with a
Dean Star~ trap to collect water ~or 2 hours. Contents were
concentrated in vacuo leaving a thick amber oil (7.6 g) which
was distilled o~ a kugelrohr apparatus at 40 C (0.1 mm) to give
the title material as a clear colorless oil (5.9 g).
1~ NMR (CDCl3): ~ 4.2~ - 4.20 (m, 2H); 4.20 - 4.13 (m, lH); 3.88
- 3.78 (m, 2~; 3.07 - 2.95 (L~ 4H); 2.35 - 2.22 (m, 2H~; i.90 -
1.80 (m, 4H).
Example 15 (Intermediate)
3-(2-Butenyl)tetrahydro-4H-pyran-4-one
o
,~
The title material o~. Example 14 (23 g, 0.15 mole) and crotyl
bromide.(15.4 mL, 0.15 mole) were mixed in benzene.(20~ mL) and
stirred 72 hours. Water (50 mL) was added and stirred 2 hours.
The benzene layer was removed and the aqueous layer was
extracted with EtOAc (150 mL). The organic extracts were
combined, dried (MgSO4) and concentrated in vacuo leaving an oil
36 ~ S~
CA 022l8360 l997-ll-l0
(20.8 g). The oil was chromatographed or .sllica gel eluti~g
with 5% EtOAc/hexanes to give the title material as a colorless
oil (12.3 g).
lH NMR (CDC13): ~ 5.52 - 5.25 (m, 2H); 4.20 - 4.07 (m, 2H); 3.82
- 3.70 (m, lH); 3.50 - 3.40 (m, lH); 2.68 - 2.40 (m, 4H); 2 .03 -
1.90 (m, lH); 1 65 (d, J = 6 Hz, 3H).
Example 16 ( Intermediate)
3-(2-Butenyl)tetrahydro-4H-pyran-4-one, oxime
HO--N
~\
To the title material o~ ~xample 15 (13.0 g, 0.0~4 mole) and
hydroxylamine hydrochloride (6.5 g, 0.093 mole) in methanol (100
mL) was added dropwise anhydrous pyridine (8.1 mL, 0.1 mole) in
methanol (50 mL). Contents were stirred overnight. Contents
were concentrated in vacuo and the residue was partitioned
between CH2C12 and water. The CH2Cl2 layer was dried (MgSO4)
and concen.trated in vacuo leaving the title material as an oil
(19.5 g)
lH NMR (CDCl3) as a mixture of syn and anti oximes: ~ [9.0, 8. 85
: (br, lH)]; [5.80 - 5.25, 5.20 - 4.85 (m, 2H)]; 4.20 - 2.90 (m,
5H); 2.80 - 2.00 ~m, 4Hl; [1.63 (d, J = 6 Hz), 1.20 - 0.90 (m)
(3H)~.
~. 30
~ ~~~
37
t~
~ CA 022l8360 l997-ll-l0 ~ -~-
?
;
Example 17 (Intermediate)
3-(2-Butenyl)tetrahydro-1,4-oxazepin-5(2H)-one
~ N O
~ H
To the title material of ~xample 16 (5.0 g, 0 03 mole) in
acetone (30 mL) at 0 C was added lN sodium hy~roxide (30 mL).
Benzene sul~onyl chloride (3.8 mL, 0.03 mole) i~ acetone (10 mL)
was added dropwise and after the reaction came, to room
temperature, it was stirred overnight The contents were
concentrated in vacuo to remove acetone and tke aqueous solution
left was extracted with CH2C12 (2 x 150 mL). The CH2Cl2
extracts were com~ined, dried (MgSO4), and concentrated in vac~o
leaving an oil. Hexane was added to the oil, the resulting
white solid was ~iltered and recrystallized from EtOAo~hexane to
give the title material as a white solid (812 mg) From the
mother liquor was isolated additional title material plus its
other regeoisomer, 6-(2-Butenyl)tetrahydro-1,4-oxazepin-5(2H)-
one, which was separated by chromatography.
lH NMR (CDCl3): ~ 5.75 (br, lH); 5.70 - 5.50 (m, lH); 5.40 -
5.23 (m, lH); 4.00 - 3.80 (m, 2H); 3.72 - 3.52 (m, 2H); 3.40 -
3.30 (m, lH); 2.95 - 2.80 (m, lH); 2.60 - 2.55 (m, lH); 2.30 -
25 2.15 (m, lH); 2.10 - 1.95 (m, lH); 1.70 (d, J = 6 Hz, 3H).
38
CA 02218360 1997-11-10
WO 9~13S677 PCr/U~ '06~1
Examp 1 e 18
3-(2-Butenyl)tetrahydro-1,4-oxazepin-5(2H)-imine,
trifluoroacetate salt
~~~
N NH
.TFA
To the title material of Example 17 (612 mg, 3.6 mmol) in CH2Cl2
(25 mL) was added trimethyloxonium tetrafluoroborate (540 mg,
3.6 mmol) and contents were stirred overnight. After
concentrating in vacuo, the residue was dissolved in me~hanol
(25 mL) and anhydrous ammonia was bubbled through the solution.
Contents were stoppered and stirred 72 hours. Contents were
concentrated in ~acuo and the residue was purified by C-18
lS reverse phase chromatography eluting with a CH3CN/H2O gradient
(0.05 ~ TFA) to give the title material as a white solid (404
mg).
Mass spectral analysis for CgH16N2O: M+H = 169.
lH NMR (CDCl3): ~ 9.7 (br, 2H); 8.9 (br, lH); 5.70 - 5.54 (m,
lH); 5.40 - 5.25 (m, lH); 4.03 - 3.92 (m, lH); 3.90 - 3.80 (m,
lH); 3.76 - 3.58 (m, 2H); 3.46 - 3.32 (m, lH); 3.04 - 2.76 (m,
2H); 2.42 - 2.18 (m, 2H); 1.67 (d, J = 6 Hz, 3H).
- 39 _
CA 02218360 1997-11-10
Example 19 (Intermediate)
l-Methyl-4-piperidin-4-one, oxime, monohydrochloride
,OH
¦ .HCl
To a slurry o~ l-methyl-4-pyridone (10 mL, 0.08 mole) and
hydroxylamine hydrochloride (6.1 g, 0.088 mole) in methanol (100
mL) was added anhydrous pyridine (7.8 mL, 0.097 mole) in
rnethallol (5G ~) dropwise. contents were stirred overnight and
the title material was ~iltered as a white solid (9.2 g). More
of the title material was recovered ~rom the methanol ~iltrate
(7.7 g).
15 lH NMR (D2oj: ~ 3.70 - 2.90 (m, SH)i 2.80 (s, 3H); 2.60 - 2. 45
(m, 2H); 2.40 - 2.10 (m, lH).
Example 20 (Intermediate)
hexahydro-l-methyl-5H-1,4-diazepin-S-one
\
To the title material o~ Example 19 (9.2 g, 0.056 mole) in
acetone (50 mL) at 0 'C was added dropwise lN sodium hydroxide.
A~ter stirring S minutes, ~enzene sul~onyl chloride (7.1 mL) in
acetone. ! S mL) was added dropwise. Contents were stirred 72
hours, coming to room temperature. Contents were concentrated
~ CA 022l8360 1997-ll-l0
~ ~ ~ t
in vacuo to remove the acetone, ~he a~ueous solucion was made
basic with lN sodium hydroxide and lyophilized leaving a solid.
The solid was triturated with CH2cl2 and ~iltered. The C~2Cl2
was concentrated in vacuo leaving the title material as a solid
(4.9 g).
lH NMR tcDcl3): ~ 6.85 (br, lH); 3.30 - 3.20 (m, 2H); 2.65 -
2.40 (m, 6H); 2.35 (s, 3H).
Example 21
hexahydro-1-methyl-5H-1,4-diazepin-5-imine, tri~1uoroacetate
salt
\
.TFA
The 5-Oxo-2,3,4,5,6,7-hexahydro-1,4-diazepine product o~ Example
20 was treated with Me30~BF4~ in CH2C12 and stirred overnight.
A~ter concentrating in vacuo, the residue was dissolved in
methanol and anhydrous ammonia was bubbled through the solution.
The contents were stirred overnight and concentrated in vacuo.
The residue was purified by C-18 reverse phase chromatography to
give the title product.
Mass spectral analysis for C6H13N3: M+H = 128.
1H NMR (DMSO-d6): ~ 9.80 - 9.40 ts, lH); 9.40 (s, lH); 9.10 (s,
lH); 8.60 (s, lH); 3.70 - 2.85 (m, 8H); 2.80 (s, 3H).
Example 22 (Intermediate)
p~
41
i
CA 022l8360 l997-ll-lO
W096l3S677 P~~ 'rO6831
~etranydro-3-(2-methoxyethyl)-4H-pyran-4-one
o
Me
~OJ
The title compound of Example 14 is reacted with bromoethyl
methyl ether by the method of Example 15 to generate the title
compcund.
- 42 -
CA 02218360 1997-11-10
Example 23 (Intermediate)
tetrahydro-3-(2-methoxyethyl)-4H-pyran-4-one, oxime
HO'N
~ ~ ~ Me
The title compound of Example 22 is reacted with hydroxyl~mine
by the method o~ Example 16 to generate the title compound.
Example 24 (Intermediate)
Isomer-A: tetrahydro-3-(2-methoxyethyl)-1,4-oxazepin-5(2H)-one
Isomer-B: tetrahydro-6-(2-methoxyethyl)-1,4-oxazepin-5(2H)-one
~ ~ N ~ HN ~
~ ~)'
O O
Isomer A. Isomer B
The title compound o~ Example 23 is reacted wlth benzenesulfonyl
chloride by the method o~ ~xample 17 to generate the title
! compounds. The isomers are separated by column chromatography.
p~ ~5~
43
~ CA 02218360 1997~ 10
.:
Example 25 (Intermediate)
2,3,6,7-tetrahYdro-3-(2-methoxyethyl)-s-methoxy-l~4-oxazepine
M e O~ N ~ O ~
The Isomer A o~ Example 24 is reacted with trimethyloxonium
tetrafluoroborate in methylene chloride by the method o~ Example
3 to generate the title compound.
Example 26 (Intermediate)
2,3,6,7-tetrahydro-6-(2-methoxyethyl)-5-methoxy-1,4-oxazepine
O Me
~~
o
The isomer B of Example 24 is reacted with trimethyloxonium
tetra~luoroborate in methylene chloride by the method o~ Example
3 to generate the title compound.
o~o
44
CA 022l8360 l997-ll-lO
Example 27 (Intermediate)
tetrahydxo-3-(2-methoxyethyl~-1,4-oxazepin-5(2H)-imine,
monohydrochloride
.HClH
HN~ N ~o
~ ~ .
The product o~ ~xample 25 is rcacted with ammonium chloride in
methanol by the method of Example 4 to generate the title
compound
Example 28
tetrahydro-6-(2-methoxyethyl)-1,4-oxazepin-5(2H)-imine,
15 monohydrochloride r
.HCI NH
HN ~/"--'
~O
The product of Example 26 is reacted with ammonium chloride in
methanol by the method of Example 4 to generate the title
compound.
~,9S~
i
CA 02218360 1997-11-lo
Wo 96l3S677 PCrtUS9G,~r~31
Example 29
4,4-dimethyl-S-pentylpyrrolidin-2-imine, monohydrochloride
. HCl
HN N
Example 29A ) Ethyl 3,3-dimethylacrylate (4.9 g, 38 mmol) was
mixed with nitrohexane (5.0 g, 38 mmol), lM
tetrabutylammoniumfluoride (in THF, 38 mL) and heated at 40 ~C for
24 hours. The reaction mixture was diluted with diethyl ether,
washed with brine, followed by water. Purif~ication by
chromatography on silica gel yielded the product, methyl 3,3-
dimethyl-4-nitrononanoate (6.6 g, 67%).
Example 29 B) The product of Example 29 A (5.6 g, 24 mmol) in
absolute MeOH was hydrogenated over RaNi at 55 C and 60 psi for
24h. The reaction product was purified by colum.n chromatography to
yield 4,4-dimethyl-5-pentylpyrrolidin-2-one (2.63 g, 60%).
Example 29 C) The product of Example 29 B (2.63 g, 14.3 mmol) was
treated with trimethyloxonium tetrafluoroborate (2.56 g, 17.4 mmol)
in DC~5 (20 mL) by the method of Example 3, to yield 3,4-dihydro-5-
methoxy-3,3-dimethyl-2-pentyl-2H-pyrrole (2.0 g, 7196).
Example 29) A solution of the title product of Example 29 C (2.0
g, 10 mmol) in MeOH (30 mL) was reacted with AmmoP;um chloride (529
mg, 9.9 mmol) by the method of Example 5 followed by chromatography
on reverse phase HPLC.
- 46 -
CA 02218360 1997-11-10
WO 9~513S677 P~ J' 3~ /06831
Example 30
5-pentyl-4,4-bis(trifluoromethyl)pyrrolidin-2-imine,
monohydrochloride
.,
HCl
H H
CF3
Example 30 A) Ethyl 4,4,4-trifluoro-3-(trifluromethyl)crotonate
(9.0 g, 38 mmol) was mixed with nitrohexane (5.0 g, 38 mmol),
potassium carbonate (5.3 g, 38 mmol) and Ali~uat 336 (20 drops).
The mixture was sonicated at room temperature. When the reaction,
monitored by G.C., was complete the mixture was acidified with HCl
(1 N) and the a~ueous phase extracted with ether. Purification by
chromatography on silica gel yielded the product, methyl 4-nitro-
3,3-bis(trifluoromethyl)nonanoate (3 g, 21~).
Example 30 B) The product of Example 30 A in absolute MeOH is
hydrogenated over RaNi at 55 C and 60 psi for 24h. The reaction
product is purified by column chromatography to yield 5-pentyl-4,4-
bis(trifluoromethyl)pyrrolidin-2-one.
Example 30 C) The product of Example 30 B is treated with
trimethyloxonium tetrafluoroborate in DCM (20 mL) by the method of
Example 3, to yield 3,4-dihydro-S-methoxy-2-pentyl-3,3-
bis(trifluoromethyl)-2H-pyrrole.
Example 30) A solution of the title product of Example 30 C in
MeOH (30 mL) is reacted with ammonium chloride by the method of
Example 5 followed by chromatography on reverse phase HPLC to
generate the title material.
- 47 -
CA 022l8360 l997-ll-l0
W0~6t3S677 PCT~59-'Q6~1
Example 31
e~hyl 2-imino-4-methyl-5-pentylpyrrolidine-3-carboxylate,
monohydrochloride
. .HCI H
HN~~,
O
Example 31 A) The diethyl ethyli~n~m~lonate (6.4 g, 33 mmol) is
mixed with nitrohexane (5 g, 38 mmol), potassium carbonate (2 g)
and Ali~uat 336 (10 drops). The mixture is sonicated at room
temperature. When the reaction, monitored by G.C., is complete the
mixture is acidified with HCl (1 N) and the a~ueous phase extracted
with ether. Purification by chromatography on silica gel yields
the product, diethyl 2-~1-methyl-2-nitroheptyl)propane-1,3-dioate.
Example 31 B) The product of Example 31 A in absolute EtOH is
hydrogenated over RaNi at 55 C and 60 psi for 24h. The reaction
product is purified by column chromatography to yield ethyl 4-
methyl-2-oxo-5-pentylpyrrolidine-3-carboxylate.
Example 31 C) The material 31 B is treated with trimethyloxonium
tetrafluoroborate in DCM by the method of Example 3, to yield ethyl
3,4-dihydro-5-methoxy-2-pentyl-2H-pyrrole-3-carboxylate.
Example 31) A solution of the title product of Example 31 C in
MeOH is reacted with ammonium chloride by the method of Example 5
followed by chromatography on reverse phase HPLC to generate title
material.
- 48 -
CA 02218360 1997-11-10
WO 96/3S677 PCrrUS96/06831
Example 3 2
2-imino-4-methYl-5-pentylpyrrolidine-3-carboxylic acid,
monohydrochloride
.HCI
.. H
HN N
HO~\
S O
Example 32 A) A solution of the title product of Example 31 B in
MeOH / 2N NaOH is stirred 6h followed by lyophilization. The
resulting solid is dissolved in water and EtOAc cont~;n;ng
benzylbromide added. The mixture is shaken in a separatory ~unnel.
The organic solution is separated, dried and evaporated. The
residue is purified by column chromatography to yield phenylmethyl
4-methyl-2-oxo-5-pentylpyrrolidine-3-carboxylate.
Example 32 B) The product of Example 32 A is treated with
lS trimethyloxonium tetrafluoroborate in DCM by the method of Example
3, to yield phenylmethyl 3,4-dihydro-5-methoxy-3-methyl-2-pentyl-
2H-pyrrole-4-carboxylate.
Example 32 C) A solution of the title product of Example 32 B in
MeOI~ is reacted with ammonium chloride by the method of Example 5
followed by chromatography on reverse phase HPLC to generate
phenylmethyl 2-imino-4-methyl-5-pentyl-3-carboxylate.
Example 32) A solution of product of Example 32 C in absolute MeOH
is hydrogenated over Pd~C. The reaction product is purified by
chromatography on reverse phase HPLC to generate title material.
_ 49 -
CA 02218360 1997-11-10
WO 9613S677 PCT/U59''~G~~l
Example 33
~-amino-4-hydroxy-5-imino-3-(trifluoromethyl)pyrroiidine-2-
butanoic acid, monohydrochloride
.HCl H .HCl
H N $~ O H
HO CF3 o
Example 33 A) The ethyl 4,4,4-trifluoromethyl crotonate (10 mmol)
and 2-(2-nitroethyl)-1,3-dioxolane (12 mmol) are reacted with,
potassium carbonate (5 mmol) and Aliquat 336 (3 drops), by the
method of Example 14. Purification by chromatography on silica gel
yields ethyl ~-nitro-~-(trifluoromethyl)-1,3-dioxolane-2-
pentanoate.
Example 33 B) The product of Example 33 A in MeOH is hydrogenated
over RaNi at 55 C and 60 psi for 6h. The reaction product is
purified by column chromatography to yield 5-[(1,3-dioxolan-2-
yl)methyl]-4-(trifluoromethyl)pyrrolidin-2-one as a mixture of
diasteromers.
Example 33 C) The product of Example 33 B is treated with di-t-
butyldicarbonate and DMAP in THF and refluxed for 2 h. The solvent
is removed and the product is purified by column chromatography to
yield 1,1-dimethylethyl 2-[(1,3-dioxolan-2-vl)methyl]-5-oxo-3-
(trifluoromethyl)pyrrolidine-1-carboxylate.
~xample 33 D) The product of Example 33 C with HMPA (1 equivalent)
in THF at -70 C is treated with Lithium hexamethyl disilazide (1.2
equivalents, lM in THF). The solution is allowed to warm to -40 'C
then cooled to -70 C, and a solution of camphor sulfonyl
oxaziridine in THF is added. The solution is stirred at -40 C for
2h then quenched onto saturated NH4Cl. The solution is then
extracted with EtOAc. The organics are combined. The solvent is
removed and the product is purified by column chromatography to
yield 1,1-dimethylethyl 2-[(1,3-dioxolan-2-yl)methyl]-4-hydroxy-5-
oxo-3-(trifluoromethyl)pyrrolidine-1-carboxylate.
- 50 -
CA 02218360 1997-11-10
WO g6/3S677 PCTrUS96/06831
Example 33 E) The product of Example 33 D is treared with NaH and
benzylbromide in THF. The product is purified by column
chromatography to yield l,l-dimethylethyl 2-[(l,3-dioxolan-2-
yl)methyl]-S-oxo-4-(phenylmethoxy)-3-(trifluoromethy')pyr olidine-
l-carboxylate.
Example 33 F) The product of Example 33 E in MeOH is treated
with HCl (lN) to yield 5-oxo-4-(phenylmethoxy)-3-
(trifluoromethyl)pyrrolidine-2-acetaldehyde which is used
directly in the next step.
Example 33 G) To a solution of produ-t of Example 33 F and z-a-
phosphonoglycine trimethyl ester in CH2Cl2 is added 3BU. The
solution is stirred for 2h The solvent is removed and the product
is purified by column chromatography to yield methyl 4- [5-oxo-4-
(phenylmethoxy)-3-(trifluoromethyl)pyrrolidin-2-yl]-2-
lS [[(phenylmethoxy)carbonyl]amino]-2-butenoate.
Example 33 H) The product of Example 33 G is hydrogenated with
[Rh (COD) (R, R-DIPAMP) ] + BF4- . The solvent is removed and the
product is purified by column chromatography to yield methyl 5-oxo-
a- [ [ (phenylmethoxy)carbonyl]amino]-4-(phenylmethoxy)-3-
(trifluoromethyl)pyrrolidine-2-butanoate.
Example 33 I) The product of Example 33 H is treated with
trimethyloxonium tetrafluoroborate in DCM by the method of Example
3, to yield methyl 3,4-dihydro-5-methoxy-a-
[[(phenylmethoxy)carbonyl]amino]-4-(phenylmethoxy)-3-
(trifluoromethyl)-2H-pyrrole-2-butanoate.
Example 33 J) A solution of the title product of Example 33 I in
MeOH is reacted with ammonium chloride by the method of Example 5
followed by chromatography on reverse phase HPLC to yield methyl 5-
imino-a-t~(phenylmethoxy)carbonyl]amino]-4-(phenylmethoxy)-3-
(trifluoromethyl)pyrrolidine-2-butanoate, monohydrochloride.
Example 33) The product of Example 33 J in absolute MeOH is
hydrogenated over Pd/C for 24h. The reaction product is purifiec
by chromatography on reverse phase HPLC to yield 33.
_ 51 -
CA 022l8360 l997-ll-lo
W096~S677 ~ PCT/U~ 6~1
Example 34
hexahydro-2-imino-4-methyl-7-(2-propenyl)-lH-azepin-3-ol
~ Me
~OH
~N NH
.HCl
Example 34 A) A THF solution of hexahydro-4-methyl-7-(2-
propenyl)-2H-azepin-2-one is treated with di-t-butyldicarbonate
and dimethylaminopyridine (DMAP, 1 e~) to generate the Boc
protecced lactam, 1,l-dimethylethyl hexahydro-4-methyl-2-oxo-7-
(2-propenyl)-lH-azepine-l-carboxylate.
Example 34B) To the product of Example 34 A above dissolved in
THF and cooled to a low temperature is added
hexamethylphosphoramide (HMPA, 1 eq) followed by lithium
hexamethyldisilylazide (LHMDS, 1.1 eq). To this is added 1.2
equivalents of either (lS)-(+)-(camphorsulfonyl)-oxaziridine or
(lR)-(-)-(camphorsulfonyl)-oxaziridine to generate a
chromatographically separable mixture of diastereomers Isomer-A
1,1-dimethylethyl hexahydro-3R-hydroxy-4-methyl-2-oxo-7-(2-
propenyl)-lH-azepine-1-carboxylate or Isomer-B 1,1-dimethylethyl
hexahydro-3S-hydroxy-4-methyl-2-oxo-7-(2-propenyl)-lH-azepine-1-
carboxylate.
Example 34 C) A product or product mixture from Example 34 B
above dissolved in DMF is treated with imidazole (2 eq) and t-
butyldimethylsilyl chloride yielding l,l-dimethylethyl 3-[(1,1-
dimethylethyl)dimethylsilyloxy]hexahydro-4-methyl-2-oxo-7-(2-
propenyl)-lH-azepine-1-carboxylate.
Example 34 D) To a product or product mixture from Example 34 C
above dissolved in acetonitrile and warmed to around 50 'C is
added magnesium perchlorate [Mg(ClO4)2, 0.2 eq] generating 3-
[(1,1-dimethylethyl)dimethylsilyloxy]hexahydro-4-methyl-2-oxo- -
(2-propenyl)-2H-azepin-2-one.
Example 34 E) The product or a product mixture from Example 34 3
above is treated with trimethyloxonium tetrafluoroborate in CH2Cl2
- 52 -
CA 022l8360 l997-ll-l0
W096l3S677 PCT/U~ ~~l
by the method of Example 3, to yield 6-[(1,1-
dimethylethyl)dimethylsilyloxy]-3,4,5,6-tetrahydro-7-methoxy-5-
methyl-2H-azepine.
Example 34 F) A solution of the title product or a product mixture
of Example 34 E in MeO~I is reacted with ~monium chloride by the
method of Example 5 to generate 3-[(1,1-
dimethylethyl)dimethylsilyloxy]hexahydro-4-methyl-7-(2-propenyl)-
2H-azepin-2-imine, monohydrochloride. This material is treated
with a source of fluoride ion and the crude product
chromatographed on reverse phase HPLC to yield the title material.
Example 35
6-butyl-3-hydroxy-4-methylpiperidin-2-imine, monohydrochloride
Me
~OH
~~ H ~NH
.HCl
Example 35 A) A THF solution of 6-butyl-4-methylpiperidin-2-one
is treated with di-t-butyldicarbonate and dimethylaminopyridine
(DMAP, 1 eq) to generate the Boc protected lactam, 1,1-
dimethylethyl 2-butyl-4-methyl-6-oxopiperidine-1-carboxylate.
Example 35 B) To the product of Example 35 A above dissolved in
THF and cooled to a low temperature is added
hexamethylphosphoramide (HMPA, 1 eq) followed by lithium
hexamethyldisilylazide (LHMDS, 1.1 eq). To this is added 1.2
equivalents of either (lS)-(+)-(camphorsulfonyl)-oxaziridine or
(lR)-(-)-(camphorsulfonyl)-oxaziridine to generate
chromatographically separable mixture of diastereomers Isomer-A
~ 1,1-dimethylethyl 6-butyl-3R-hydroxy-4-methyl-2-oxopiperidine-1-
carboxylate or Isomer-B 1,1-dimethylethyl 6-butyl-3S-hydroxy-4-
methyl-2-oxopiperidine-1-carboxylate.
Example 35 C) A product or product mixture from Example 35 B
above dissolved in DMF is treated with imidazole (2 e~) and t-
butyldimethylsilyl chloride yielding 1,1-dimethylethyl 6-butyl-
_ 53 -
CA 02218360 1997-11-10
WO 96/3S677 PCT/US9r'Qr~~l
3-lll,i-dimethylethyl)dimethylsilyloxy]-4-methyl-2-
oxopiperidine-1-carboxylate.
Example 35 D) To a product or producc mixture from Example 35 C
above dissolved in acetonitrile and warmed to around 50 C is
added magnesiu~ perchlorate tMg(ClO4)2, 0.2 eq] generating 6-
butyl-3-[(1,1-dimethylethyl)dimethylsilyloxy]-4-methylpiperidin-
2-one.
Exampie 35 E) The product or a product mixture ~rom.. Example 35 D
above is treated with trimethyloxonium tetrafluoroborate in CH2C12
by the method of Example 3, to yield 2-butyl-5-[(1,1-
dimethylethyl)dimethylsilyloxy]-6-ethoxy-2,3,4,5-tetrahydro-4-
methylpyridine.
Example 35 F) A solution of the title product or a product mixture
of Example 35 E in MeOH is reacted with ~mmonium chloride by the
method of Example 5 to generate 6-butyl-3-[(1,1-
dimethylethyl)dimethylsilyloxy]-4-methylpiperidin-2-imine. This
material is treated with a source o~ fluoride ion and the crude
produc~ chromatographed on reverse phase HPLC to yield the title
material.
Example 36
6-imino-2,4-dimethylpiperidine-3-meth~n~mine, dihydrochloride
CH3
.HCl
H2N ~
H3C ~ I ~ NH
H .HCl
6-amino-2,4-dimethylpyridine-3-carbonitrile (1.5 g) ana platinum
oxide (500 mg) in ethanol (30 mL) and conc HCl (1 mL) were
shaken on a Parr hydrogenation apparatus at 5S psi of hydrogen
at 55 'C for 48 hours. The contents were filtered and the
filtrate concentrated in vacuo leaving a waxy solid.
Trituration with ethanol gave the title material as a white
solid (191 mg).
_ 54 -
CA 02218360 1997-11-10
W096l3S677 PCT~S96/06831
~Iass spectral analysis ~or C8H17N3: M+H = 156
H NMR (D2O): ~ 3.63 - 3.40 (m, 2H); 3.20 - 3.07 (m, lH); 2.72 -
2.60 (m, lH); 2.40 - 2.25 (m, lH); 2.05 - 1.90 (m, 2H); 1.25 (d,
J = 6 Hz, 3H); l.00 (d, J = ~ Hz, 3H).
Example 37
4,6,6-trimethylpiperidine-2-imine, trifluoroacetate salt
H3 ~
H .CF3CO2H
Example 37 A) A solution of 2,2,4-trimethylcyclopentanone (5.5
g, 44 mmol) in 35 mL EtOAc/25 mL of water was refluxed with
hydroxylamine hydrochloride (4.6 g, 66 mmoles) and sodium
acetate trihydrate (10.8 g, 79 mmol) for 4 hrs under nitrogen.
After the solvent was l~,..oved by evaporation, the residue was
redissolved in 100 mL of EtOAc, washed with a saturated aqueous
sodium chloride solution, dried over m~nesium sulfate, and
stripped of all solvent to give 5.6 g of the white powder,
2,2,4-trimethylcyclopentanone oxime. FAB/MS: (MH+)=142.
Example 37 B) The product of Example 37 A was dissolved in 50
mL of acetone and 50 mL of 1 N sodium hydroxide at 0 ~C.
Benzenesulfonyl chloride (7.8 g, 44 mmol) was added over 5 min.
The reaction mixture was allowed to warm up and stirred for 18
hrs until complete as determined by shift in HPLC retention time
(Vydac C-18, linear gradient 5 % to 75 % acetonitrile/0.05 % TFA
in water/0.05 % TFA over 20 min). The solvent was removed by
evaporation and the residue redissolved in 100 mL EtOAc, washed
with saturated aqueous sodium chloride solution, dried over
magnesium sulfate and stripped of all solvent by evapora~ion.
The crude semisolid material was purified on Waters Deltapak C-
_ 55 -
CA 02218360 1997-11-10
WO 9613S677 PCT/u~ 6Q~l
18 using a linear gradient from 10 ~ to 15 ~ aceton trile(0.05 %
TFA) i~ water (0.05 ~ TFA) over 20 min. The lyophilized
product, 4,6,6-trimethylpiperidin-2-one, was a tan semisolid,
G.47 g. FAB/MS: (MH+)=142.
Example 37 C) To the product of Example 37 B (3.3 mmol) in 10
mL CH2Cl2 was added trime~hyloxonium tetrafluoroborate (0.6 g,
4.0 mmol). After stirring 18 hrs, the reactior. mixture was
diluted with an additional 10 mL of CH2Cl2, washed with a
saturated aqueous potassium carbonate solution, dried over
magnesium sulfate, and stripped of all solvent to generate
2,3,4,5-tetrahydro-6-methoxy-2,2,4-trimethylpyridine.
Example 35) The product of Example 37 C was dissolved in 25 mL
of methanol and refluxed with ammonium chloride for 3 hrs. The
solvent was removed by evaporation and the residue oil was
dissolved in 25 mL of EtOAc, washed with water, and stripped of
all solvent under reduced pressure to produce the crude product.
The material was purified on Waters Deltapak C-18 using a linear
gradient of 5% to 70% acetonitrile(0.05 % TFA) in water (0.05
TFA) over 30 min. and lyophilized to give 0.075 g white powder
title material. F~3/MS: (MH+)=141.
H NMR (CDCl3): ~ 10.4 (bs, lH); 9.7 (bs, lH); 7.5 (bs, lH); 2.6
(q, lH); 2.0 (g, 2H); 1.8 (d, 2H); 1.4 (s, 3H); 1.3 (s, 3H); 1.1
(d, 3H).
Example 38
4,4,6-trimethylpiperidin-2-imine, trifluoroacetate salt
H3C CH3
H3C ~ ~ NH
.CF3CO,H
Example 38 A) A solution of 2,4,4-trimethylcyclopentanone (5.5
g, 44 mmol) in 35 mL ethyl acetate/25 mL water was refluxed wi~;~
:~ydroxylamine hydrochloride (4.6 g, 66 mmol) and sodium acetate
_ 56 ~
CA 022l8360 l997-ll-l0
W096/3S677 PCT~S~''.'~~l
-rihydrate (10 8 g, 79 mmol) for 4 hrs under nitrogen. ~emoved
solvent by evaporation, redissolved in 100 mL ethyl acetate and
washed with saturated a~ueous sodium chloride solution, dried
over magnesium sulfate and then removed solvent to give 5.2 g of
2,4,4-trimethylcyclopentanone oxime as a white powder. FAB/MS:
(MH+)=142.
Example 38 B) The product of Example 38 A was dissolved in 50
mL acetone and 50 mL 1 N sodium hydroxide at 0 ~C.
Benzenesulfonyl chloride was then added (7.8 g, 44 mmol) over 5
min. The reaction mixture was allowed to warm up and stir for
18 hrs until complete, as determined by ~he shift in HPLC
retention time (Vydac C-18, linear gradient 5 % to 75 ~
acetonitrile/0.05 ~ TFA in water/0.05 % TFA over 20 min). The
solvent was removed by evaporation and the residue was
redissolved in 100 mL EtOAc, washed with saturated a~ueous
sodium chloride solution, dried over magnesium sulfate and
stripped of all solvent by evaporation. The semisolid product
was purified on a Wa~ers Deltapak C-18 using a linear gradient
from 10~ to 15% acetonitrile(O.05 % TFA) in water (0.05 % TFA)
over 20 min. The lyophilized product, 6,4,4-trimethylpiperidin-
2-one, was a tan semisolid, 0.75 g. FAB/MS: (MH+)=142.
Example 38 C) To the product of Example 38 B (5.3 mmol) in 15
mL CH2Cl2 was added trimethyloxonium tetrafluoro~orate (0.9 g,
6.0 mmol). After stirring 18 hrs, the reaction mixture was
diluted with an additional 15 mL of CH2Cl2, washed with
saturated aqueous potassium carbonate solution, dried over
magnesium sulfate, and stripped of all solvent to give 0.69 g
of 2,3,4,5-tetrahydro-6-methoxy-2,4,4-trimethylpyridine as an
oil.
Example 38) The product of Example 38 C was dissolved in 25 mL
of methanol and refluxed with ammonium chloride (0.25 g, 4.6
mmol) for 3 hrs. The solven. was removed by evaporation and t:e
residue oil was dissolved in 25 mL EtOAc, washed with water, a~.d
stripped of all solvent. The residue was purified on a Waters
Deltapak C-18 using a linear gradient of 5% to 70% acetonitrile
(0.05 % TFA) in water (0.05 ~ TFA) over 30 min. and lyophilizea
to give 0.66 g of the title material as a white powder. FAB/~S:
(MH+)=141.
_ 57 -
CA 022l8360 l997-ll-l0
WO 96/3S677 PCr/US96/06831
H NMR ~CDC13): ~ 10.4 (bs, lH); 9.5 (bs, lH~; 8.1 (bs, lH); 3.8
tm, lH); 2.3 (q, 2H); 1.75 (d, 2H); 1.3 (d, 3H); 1.1 (s, 3H);
1.0 (s, 3H).
Example 39
3-(2-butenyl)hexahydro-5-imine-1,4-oxazepin-6-ol,
trifluoroacetate salt
~H~NH
.TFA
Isomer A
Example 39 A) A sample of the 3-(2-Buten-l-yl)-5-oxo-
15 2,3,4,5,6,7-hexahydro-1,4-oxazepine product of Example 17 (6.6
g, 39 mmol), di-t-butyl dicarbonate (17.5 g, 80 mmol) and 4-
dimethylaminopyridine (200 mg) were re~luxed in anhydrous THF
(80 mL) overniçrht. The contents were allowed to cool, diluted
with EtOAc, and washed with 5% aqueous NaHCO3, dried over MgSO4,
20 and concentrated in vacuo leaving an oil (12.9 g). The oil was
purified by chromatography on silica gel eluting with 1096
EtOAc/hexanes to give 4-N-Boc-3-(2-buten-1-yl)-5-oxo-
2,3,4,5,6,7-hexahydro-1,4-oxazepine as a colorless oil (3.7 g).
Example 39 B) To the 4-N-Boc-3-(2-buten-1-yl)-5-oxo-
25 2,3,4,5,6,7-hexahydro-1,4-oxazepine product of Example 39 A (3.1
g, 12 mmol) in anhydrous THF (60 mL) at -78 ~C was added
dropwise lithium bis(trimethylsilyl)amide (lM in THF, 12 mL)
keeping the temperature below -70 ~C. The contents were allowed
to warm to -40 ~C and then cooled back to -78 ~C. A solution of
30 (lS)-(+)-(10-camphorsulfonyl)oxaziridine (3.0 g, 13 mmol) in THF
(30 mL) was added dropwise. The contents were warmed to -25 ~C
and stirred 3 hours before pouring into saturated NH4Cl and
extracting with EtOAc. The EtOAc layer was dried over MgSO4 and
concen~rated in vacuo to provide 4-N-Boc-3-(2-buten-1-yl)-6-
_ 58 -
CA 02218360 1997-11-10
WO g613S677 PCT/U~ 5
hydroxy-5-oxo-2,3,4,5,6,7-hexahydro-1,4-oxa~epine as a waxy
solid.
Example 39 C) The 4-N-Boc-3-(2-buten-1-yl)-6-hydroxy-5-oxo-
2,3,4,5,6,7-hexahydro-1,4-oxazepine product of Example 39 B (600
mg), t-butyldimethylsilyl chloride (2.0 g), imidazole (1.6 g)
and arhydrous THF (50 mL, were stirred overnight. The contents
were partitioned between EtOAc and water. The EtOAc layer was
dried over MgSO4 and concentrated in vacuo to generate an oil.
This oil was chromatographed on silica gel eluting with 25%
EtOAc/h~nes to give 4-N-Boc-3-(~-buten-1-yl)-6-(t-
butyldimethylsilyloxy)-5-oxo-2,3,4,5,6,7-hexahydro-1,4-oxazepine
as an oil (400 mg).
Example 39 D) The 4-N-Boc-3-(2-Buten-1-yl)-6-(t-
butyldimethylsilyloxy)-5-oxo-2,3,4,5,6,7-hexahydro-1,4-oxazepine
product of Example 39 C (400 mg, 1 mmol) and magnesium
perchlorate (45 mg) were heated at 50 ~C in CH3CN (25 mL) for 3
hours. The contents were allowed to cool and were partitioned
between EtOAc and water. The EtOAc layer was dried over MgSO4
and concentrated in vacuo leaving 3-(2-Buten-1-yl)-6-(t-
butyldimethyl-silyloxy)-5-oxo-2,3,4,5,6,7-hexahydro-1,4-
oxazepine as an oil (300 mg).
Example 39) The 3-(2-Buten-1-yl)-6-(t-butyldimethylsilyloxy)-5-
oxo-2,3,4,5,6,7-hexahydro-1,4-oxazepine product of Example 39 D
(300 mg, 1 mmol) and Me30+BF4~ (150 mg, 1 mmol) were stirred in
CH2Cl2 overnight. The contents were concentrated in vacuo, the
residue dissolved in methanol, and anhydrous ammonia bubbled
into the solution. The reaction was stoppered and stirred
overnight. The contents were concentrated in vacuo leaving a
yellow oil (366 mg). The oil was purified by C-18 reverse phase
chromatography eluting with a CH3CN/H2O to give the title
products of this Example 39 (isomer A , 16 mg) and Example 40
(isomer B, 11 mg) as oils.
Mass spectral analysis for CgH16N2O2: M+H = 185
1H NMR (D2O): ~ 5.60 - 5.42 (m, lH); 5.35 - 5.20 (m, lH); 4.75 -
4.60 (m, lH); 3.95 - 3.50 (m, 5H); 2.35 - 2.20 (m, 2H); 1.60 -
1.45 (m, 3H)
_ 59 -
CA 022l8360 l997-ll-lO
W096~S677 PCT~Ssr'0'~~l
Example 40
3-(2-butenyl)hexahydro-5-imine-1,4-oxazepin-6-ol,
trifluoroacetate salt
~_OH
~NH
.TFA
Isomer B
The crude product oil of Example 39 was purified by C-18 reverse
phase chromatography eluting with a CH3CN/H2O to give the title
products of Example 39 and title product of Example 40 (isomer
B, 11 mg).
Mass spectral analysis for CgH16N2O2: M+H = 185
1H NMR (D2O): ~ 5.65 - 5.45 (m, lH); 5.35 - 5.20 (m, lH); 4.90 -
4.75 (m, lH); 3.90 - 3.45 (m, 4H); 3.35 - 3.20 (m, lH); 2.25 -
2.05 (m, 2H); 1.60 - 1.45 (m, 3H).
Example 41
6-(2-butenyl)hexahydro-1,4-oxazepin-5-imine, trifluoroacetate
salt
o
~N~NH
.TFA
The title material was prepared according to the procedure of
Example 18, using the 6-(2-Butenyl)tetrahydro-1,4-oxazepin-
5(2H)-one isolated in Example 17.
_ 60 -
CA 02218360 1997-11-10
WO 96/3S677 PCrlUS96/06831
Mass spectral analysis for CgH16N2O: M+H = 159.
lH NMR (D2O): ~ 5.65 - 5.50 (m, lH); 5.40 - 5.20 (m, lH); 3.95 -
- 3.25 (m, 6H); 2.80 - 2.60 ~m, lH); 2.50 - 2.30 (m, 2H); 1.60 -
5 1. 50 (m, 3H) .
.
- 61 -
CA 02218360 1997-11-10
WO 9613S677 PCT/U' ~ 16
Example 42
3-butylhexahydro-1,4-oxazepin-5-imine, trifluoroacetate salt
--~N H
.TFA
The product of Example 18 (1.3 g, 4.6 mmole), 5% rhodium/carbon
(400 mg), ethanol (30 mL) and glacial acetic acid (30 mL) were
shaken on a Parr hydrogenator at 55 psi of hydrogen overnight.
The reaction contents were filtered and the filtrate was
concentrated in vacuo leaving an oil (l.1 g). The oil was
purified by C-18 reverse phase chromatography eluting with a
CH3CN/H2O to give the title product as an oil (701 mg, 54
yield).
Mass spectral analysis for CgH18N2O: M+H = 171.
H NMR (CDCl3): ~ 9.90 (s, lH); 9.50 (s, lH); 8.90 (s, lH); 4.00
- 3.40 (m, 6H); 3.00 - 2.70 (m, 2H); 1.80 - 1.20 (m, 6H); 1.00 -
0.80 (m, 3H).
Example 43
hexahydro-5-imino-1,4-oxazepine-3-eth~n~m;ne,
bis(trifluoroacetate) salt
'~'
H2N H N H
.2TFA
Example 43 A) To 2-nitroethanol (Aldrich, 50 mL, 0.7 mol) in
CH2C12 (50 mL) was added dropwise acetyl chloride (53.3 mL, 0.75
mol) n CH2Cl2 (50 mL). The contents were stirred overnight,
_ 62 -
CA 02218360 1997-11-10
WO 9613S677 - PCT/US96106831
washed with water, dried over MgSO4 and concentrated in vacuo
leaving 1-acetyl-2-nitroethanol as a light yellow oil (86 g).
Example 43 B) A sample of tetrahydropyran-4-one (Aldrich, 30 g,
0.3 mol) and morpholine (Aldrich~ 30.5 mL, 0.35 mol) were
refluxed in benzene (500 mL~ for 3 hr with a Dean Stark trap to
collect the water. The contents were allowed to cool and were
concentrated in vacuo. The residue was dissolved in
acetonitrile (250 mL) and added dropwise to a solution of the 1-
acetyl-2-nitroethanol product of Exam.ple 48 A (46.6 g, 0.35 mol)
in acetonitrile (250 mL) at -20 ~C. The reaction contents were
stirred overnight coming to room temperature and concentrated in
vacuo. The residue was partitioned between Et2O and water The
ether layer was dried over MgSO4 and concentrated in vacuo
leaving an oil. The oil was distilled on a Kugelrohr apparatus
at 100 ~C (0.1 mm) to give 2-nitroethyltrahydropyran-4-one as an
oil which p~rtially solidified (20.9 g).
Example 43 C) The 2-Nitroethyltetrahydropyran-4-one product of
Exa~ple 43 B, hydroxylamine-O-sulfonic acid, and formic acid
(98%) are refluxed for 0.5 hr. The contents are allowed to cool
and concentrated in vacuo. The residue is partitioned between
CH2Cl2 and water. The CH2Cl2 layer is dried over MgSO4 and
concentrated in vacuo. The residue is purified by C-18 reverse
phase chromatography to give 3-(2-nitroethyl)-5-oxo-2,3,4,5,6,7-
hexahydro-1,4-oxazepine.
Example 43 D) To the 3-(2-nitroethyl)-5-oxo-2,3,4,5,6,7-
hexahydro-1,4-oxazepine product of Example 43 C in CH2C12 (25
mL) is added Me30+BF4~ and the contents are stirred overnight.
After concentrating in vacuo, the residue is dissolved in
methanol (25 mL) and anhydrous ammonia is bubbled through the
solution. Contents are stoppered and stirred 72 hours.
Contents are concentrated in vacuo and the residue is purified
by C-18 reverse phase chromatography eluting with a CH3CN/H2O
gradient (0.05~ TFA) to give 3-(2-nitroethyl)-5-imino-
2,3,4,5,6,7-hexahydro-1,4-oxazepine.
Example 43) A sample of the 3-(2-Nitroethyl)-5-imino-
2,3,4,5,6,7-hexahydro-1,4-oxazepine product of Example 43 C and
palladium black in ethanol are shaken at 55 psi hydrogen on a
Parr hydrogenation apparatus overnight. The contents are
filtered and the filtrate is concentrated in vacuo. The residue
~63-
CA 022l8360 l997-ll-l0
W096/3S677 PCT~S96/06831
is purified by C-i8 reverse phase chromatography to give the
title compound.
Example 44
(~) 3a-methoxy-4a-methyl-5a-pentylpyrrolidin-2-imine,
monohydrochloride
MeO~ CH3
/~ _
HN N HCI
lo H
Example 44 was prepared from Example 45e, iodomethane, and
sodium hydride. The synthesis of Example 44 is completed in the
mAnn~r described in Example 45.
Example 45
(+) 2-imino-4a-methyl-Sa-pentyl-3a-pyrrolidinol,
monohydrochloride
H
HNlN ~
Example 45A) To a stirring solutlon of methyl crotonate
25 (3.28 g, 32.8 mmol) and nitromethane (1.08 g, 16.0 mmol) in
20 mL of CH3CN was added DBU (2.39 mL, 16.0 mL). After 72 h,
the reaction was concentrated under reduced pressure. The
residue was taken up in EtOAc. The EtOAc solution was washed
with 0.5 N HCl and brine, was dried over Na2SO4 anhydrous,
filtered, and concentrated under reduced pressure. The crude
product was purified by colum~n chromatography to give 3.05 g.
--64-
CA 02218360 1997-11-10
WO 96/3S677 PCI~/US96/06831
_xample 45B,C) Example 45A (34 g, 0.15 mol) was reducea
under catalytic hydrogenation conditions using Raney Ni ~n
MeOH. After heating the reaction mixture for 16 h at 55 C,
the solvent was removed under vacuum. The crude lactam was
separated by column chromatography into the cis (45B) and
tr~ns (45C) lactam.
Example 45D) A stirring solution of Example 45B (20 g, 0.12
mol), (BocO)20 (38.7 g, 0.18 mol), ~MAP (14.4 g, 0.12 mol) in
500 mL of THF was heated at reflux for 3 h. After
concen~rating reaction under vacuum, the residue was taken up
in EtOAc and washed with KHSO4 and brine. The organic layer
was dried over anhydrous Na2SO4, filtered, and stripped. The
crude product was purified by column chromatography to yield
31 g.
15 Example 45E) To a stirring solution of Exa~ple 45D (2.7 g,
9.9 mmol) and HMPA (1.8 g, 10.0 mmol) in 15 mL of THF cooled
to -70 'C was added lithium hexamethyldisilazide (1.7 g, 10.0
mmol). After 20 min, the reaction was warmed to -40 C and
cooled again to -70 C. To the stirring reaction was added
20 (R)-(-)-(camphorsulphonyl)oxaziridine (2.4 g, 10.4 mmol) in 7
mL of THF. After stirring at -70 C for 30 min, the reaction
was warmed to -30 C and stirred an additional 2.5 h. To the
reaction was added saturated NH4Cl solution followed by
EtOAc. The organic layer was washed with brine, dried over
anhydrous Na2SO4, filtered, and stripped. The crude product
was purified by column chromatography to yield 1.3 g of 3-
hydroxylactam.
Example 45F) To a solution of Example 45E (1.3 g) in CH2Cl2
was added TFA (6 mL). After 2 h, the reaction was
concntrated under vacuum to give 0.85 g of product.
Example 45G) To a stirring solution of Example 45F (0.85 g,
4.6 mmol) and imidazole (0.35 g, 4.6 mmol) in 15 mL was added
t-butyldimethylsilylchloride (0.70 g, 4.6 mmol). After 18 h,
the reaction mixture was concentrated under high vacuum. To~ 35 the residue was added EtOAc. The organic layer was washed
with KHCO3 solution, H2O, and brine, dried over anhydrous
Na2SO4, filtered, and stripped to yield 1.1 g of product.
Example 45H) A solution of Example 45G (1.1 g, 3.7 mmol) and
trimethyloxonium tetrafluoroborate (0.6 g, 4.7 mmol) in 30 mL
-65
CA 02218360 1997-11-10
WO 9613S677 PCI~/US96106831
was stirred for 72 h at ambient temperature. Afte~ remo~ing
solvent under vacuum, the residue was dissolved in EtOAc.
The organic layer was washed with KHCO3 solution and brine,
dried over anhydrous Na2SO4, filtered, and stripped to yield 1
g of product.
Exampie 45I) Example 45H (1 g) in MeOH w s treated with
NH4Cl (O.3 g) under 12 Kbar Ot pressure. The reaction was
concentrated under vacuum. The residue was taken up in
CH2Cl2, filtered, and stripped to give 0.8 g of product.
Example 45) To a solution of Example 45I (0.8 g) in 40 mL of
MeOH was added 10 mL of 1 N HCl. After 1.5 h, the reaction
mixture was concentrated under vacuum. The residue was
partitioned between 0.05 N HCl and CH2C12. The aqueous layer
stripped. The residue was purified by chromatography on a
reverse phase C-18 column to give two alcohols. The first
eluting was Example 46 and the second eluting was Example 45.
Elemental analysis: CloH2oN2o 1 HC1 ~0.2 H20 (MW=224.35)
C H N Cl
C~lculated: 53.80 9.61 12.49 15.80
Found: 53.80 9.47 12.14 15.46
Example 46
(+) 2-imino-4a-methyl-5a-pentyl-3~-pyrrolidinol,
monohydrochloride
H
~'
HN ~ ~ "'"~~~~'~'
.HCI H
The synthesis and isolation of Example 46 was described _~.
Example 45.
Elemental analysis: C10H2oN2o-l HCl 0.2 H20 (MW=224.35)
C H N Cl
Calculated: 53.80 9.61 12.49 15.80
~ound: 53.78 9.37 12.14 15.78
-66-
e
CA 022l8360 l997-ll-l0
W096l35677 PCT~S96/06831
Example 47
(') 2-imino-5a-pentyl-4~-(t~ifluoromethyl)-3a-pyrrolidinol,
monohydrochloride
H ~ ~CF3
HN
.HCl
Example 47A) A suspension of ethyl 4,4,4-trifluorocrotonate
(10.0 g, 59 mmol), 1-nitrohexane (7.86 g, 60 mmol), K2CO3 (4.1
g), and Aliquot 336 ( 6 drops) was sonicated for 5 h. To the
reaction was added Et20 (200 mL). The reaction mixture was
filtered, extracted with brine, dried over Na2SO4 (anhydrous),
filtered, and concen~rated under reduced pressure to give a
yellow liguid. The product was purified by column
chromatography to give 13.8 g (77~).
Example 47B,C) A solution o~ Example 47A (13.0 g) in MeOH
was reduced under catalytic hydrogenation conditions (60 psi,
C) using Raney nickel. The reaction was heated for 8 h
to effect cyclization after reduction of the nitro group.
After concentration of the reaction mixture under reduced
pressure, the residue was purified by column chromatography
to give 9.0 g of a light yellow liquid. A second column was
run to separate the cis (47B) and trans lactam (47C).
_xample 47D) Example 47C was treated in the manner desc~ibed
in Example 45D and following to prepare Example 47.
_lemental analysis: CloH17N2F30 ~ 1 HCl (MW=274.71)
C H N C1
Calculated: 43.72 6.60 10.20 12.91
-ound: 43.62 6.44 10.15 12.73
CA 02218360 1997-11-10
W096t3S677 PCT~S96/06831
Example 48
hexahyàro-5-imino-~-Phenyl-1,4-oxazepine-3-eth~n~mine,
bis(t-lfluoroacetate) salt
H2N~H~NH
TFA
~he title product is prepared according to the procedure of
Example 43, using ~-nitrostyrene instead of 1-acetyl-2-
nitroethanol to afford the title product.
Example 49
N-(3,4-dihydro-2H-pyrrol-5-yl)hexahydro-5-imino-1,4-oxazepine-3-
ethAn~mine, bis(trifluoroacetate) salt
O
C~H~H NH
.~TFA
_xample 43 is allowed to react with 2-methoxypyrroline to a~ford
_he t tle product.
Example 50
3-[[2-(hexahydro-5-imino-1,4-oxazepin-3-yl)ethyl]amino]alanine,
~ris(~_ifluoroacetate) salt
-68-
- =
CA 022l8360 l997-ll-lO
wOg6/35677 PCT~S96/06831
.3TFA ~ ~
HO2C~--H~--H NH
., NH2
rxample 50 A) Example 43 is allowed to react with ~-CBZ-
dehydroalanine methyl ester to afford the protected title
produc_.
Exampie 50) Removal of the CBZ protecting group from Example 50
A by hydrogenation followed by acid hydrolysis affords the title
produc~.
Example 51
3-~2-(hexahydro-5-imino-1,4-oxazepin-3-yl)-2-
pnenylethyl]amino]alanine, tris(trifluoroacetate) salt
.3TFA ~ ~
HO2C ~N~HN NH
NH2 ~3
Example 51 A) Example 48 is allowed to react ~ith N-CBZ-
dehydroalanine methyl ester to afford the protected title
product.
Example 51) Removal of the Cr3Z protecting group from example
51a by hydrogenation followed by acid hydrolysis affords the
itle product.
Example 52
2-(hexahydro-5-imino-1,4-oxazepin-3-yl)cyclohexAnAm;ne,
bis(t-ifluoroacetate) salt
_69
CA 02218360 1997-ll-lO
W096/35677 PCT~S96/06831
NH2 ~ ?~
~H NH
. 'TFA
~he ti.le product is prepared by the method of Example 43 using
,-nit_ocyclonexanol in place of 2-nitroethanol.
~70-
CA 022l8360 l997-ll-lO
W096/35677 PCT~S9C~'Q~l
Example 53
~-cyclopropylhexahydro-5-imino-1,4-oxazepine-3-eth~n~m;ne,
bis(tr-fluoroacetate) salt
..
~~~ ~
.2TFA
Example 53 A) 2-nitro-2-cyclopropylethanol is prepared f_om
cyclopropylcarboxaldehyde via the Henry reaction.
Example 53) The title product is prepared by the method of
Example 43 using the 2-nitro-2-cyclopropylethanol produc. of
Example 53 A in place of 2-nitroethanol.
Example 54
a-ethylhexahydro-5-imino-~-methyl-1,4-oxazepine-3-eth~n~m;ne,
bis(trifluoroacetate) salt
~~ ~'
.2TFA
?he ti~le product is preparea by the method of Example 43 usin
3-nit-o-4-hydroxypentane in piace of 2-nitroethanol.
~ ~xample 55
- 2-(hexanydro-5-imino-1,4-oxazepin-3-yl)cyclohex~n~;ne,
bis(t- fluoroacetate) salt
-7l-
CA 02218360 1997-11-lo
W096/35677 PCT~Ss'~
NH2 ~ ~
~N~NH
.2TFA
rxample 55 A) Tetrahydropyran-4-one is allowed to react with o-
nitrobenzyl ~romide under basic conditions to give 2-(o-
nitrobenzyl)tetrahydropyran-4-one.
Example 55 B) The 2-(o-nitrobenzyl)tetrahydropyran-4-one
product of Example 55 A is carried on as in Example 43c-d to
give 3-(o-nitrobenzyl)-5-imino-2,3,4,5,6,7-hexahydro-1,4-
oxazepine.
Example 55) The 3-(o-nitrobenzyl)-5-imino-2,3,4,5,6,7-
hexahydro-1,4-oxazepine product of Example 55 A is reduced under
hydrogen atmosphere utilizing platinum oxide catalyst to afford
the title product.
Example 56
hexahydro-5-imino-~-(2-thienyl)-1,4-oxazepine-3-eth~n~mine,
bis(trifluoroacetate) salt
H2N~--N NH
[~S .~TFA
Lhe ti.le material is prepared according to the procedure of
_xampie 48 using 1-nitro-2-(2-thiophenyl)ethene.
Example 57
a-aminonexahydro-5-imino-~-(2-thienyl)-l~4-oxazepine-3-pro2anc:
- 30 acid, ~is(t~ifluoroacetate) salt
-72-
CA 02218360 1997-11-10
W096/35677 PCT~S~6/0
O
H 02C ~ )
H2N~--HN~N H
~S .2TFA
.,
S Example 58
a- (aminomet:~yl)hexahydro-5-imino-1,4-oxazepine-3-methanoi,
bis(t-ifluoroacetate) salt
'~'
H2N~H NH
o O H .2TFA
Example 59
8-imino-3,7-diazaspiro~5.6]dodecan-9-ol, dihydrochloride
OH
~--H~NH
H N . 2HCl
,-(Spiro-4-piperidinyl-N-Z)caprolactam is treated as desc-ibed
n Example 34 to give the tille compound.
Example 60
3-(2-aminoethyl)hexahyaro-5-imino-1,4-oxazepin-6-oi,
bis(t-ifluoroacetate) salt
-73-
CA 022l8360 l997-ll-lO
W096/35677 PCT~S9'~~B~I
f ,_O H
, /~HN
., H2N
_xample 60 A) The produc~ of Example 43C is reacted as in
_xample 39 to afford 3-(2-nitroethyl)hexahydro-5-imino-1,4-
oxazepin-6-ol.
~xample 60) 3-(2-nitroethyl)hexahydro-5-imino-1,4-oxazepin-6-ol
,s reduced as in Example 43 to afford the title compound.
Example 61
hexahydro-5-imino-3-[2-[~2-pyrrolidinylidene)amino]ethyl]-1,4-
oxazepin-6-ol, bis(trifluoroacetate) salt
N H
~H H
The product of Example 60 is reacted as in Example 49 to afford
the title compound.
Example 62
-(2-amino-1-phenylethyl)hexahydro-5-imino-1,4-oxazepin-6-ol,
~is(t-ifluoroacetate) salt
-74-
CA 022l8360 l997-ll-lO
W096/35677 PCT~S96/06831
--N~N H
H
H2N
~3
Example 62 A) 3-(2-Nitro-1-phenylethyl)-5-oxo-2,3,4,5,6,7-
hexahydro-1,4-oxazepine is prepared as in Example 43, using ~-
nitrostyrene instead of 1-acetyl-2-nitroethanol.
Exampie 62 B) 3-(2-Nitro-1-phenylethyl)-5-oxo-2,3,4,5,6,7-
hexahydro-1,4-oxazepine is reacted as in Example 39 to afford 3-
(2-nitro-1-phenylethyl)-6-hydroxy-5-imino-2,3,4,5,6,7-hexahydro-
1,4-oxazepine.
Example 62) 3-(2-Nitro-1-phenylethyl)-6-hydroxy-5-imino-
2,3,4,5,6,7-hexahydro-1,4-oxazepine is reduced as in Example 43
to afford the title compound.
Example 63
(+) 2-imino-4a-(trifluoromethyl)-5~-pentylpyrrolidin-3a-ol
HO ~CF3
HN~
_xamp;e 63 's synthesized ar.d isolated _rom Example 47.
Example 64
(_) 2-imino-4~-(trifluorome~hyl)-5~-pentylpyr-olidin-3a-ol
_75-
CA 02218360 1997-11-10
W096/35677 PCT~S96/06831
HO~ ~CF3
HN~
., H HCI
_xample 54 is prepared from Example 47B in the manner
descriDed in Example 47.
Example 65
(+) 2-imino-4a-(trifluoromethyl)-5a-pentylpyrrolidin-3a
HO~ CF3
I \
HN~N~--
H HCI
Example 65 is prepared from Example 47B in the manner
described in Example 47.
Example 66
(+) 2-imino-4~-methyl-5a-pentylpyrrolidin-3a-ol
HO~ ~CH3
HN~--/--
H HCI
_xample 66 is prepared from Example 45C in the manner
described in Example 45.
Example 67
(=) 2-imino-4a-methyl-5~-pentylpyrro'idin-3~-ol
_76-
-
CA 02218360 1997-11-10
W096/3~677 PCT~S9C10
HO~ &H3
r~
HN~N~
H HCI
_xampie 67 is prepared from Example 45C in the manner
described in Example 45.
Example ~8
(+) 5a-(3-aminopropyl)2-imino-4a-methylpyrrolidin-3a-ol~
dihydrochloride
HO~ CH3
HN~--/NH2
H HCI
Example 69
(+) 5a-(3-aminobutyl)-2-imino-4a-methylpyrrolidin-3a-ol~
dihydrochloride
HO~_~CH3
HN N~/NH2
Example 70
(+) a-amino-4a-hydroxy-5-imino-3a-methylpyrrolidine-2a-
2utanol, dihydrochloride
-77-
CA 022l8360 l997-ll-lO
W096/35677 PCT~S96/06831
HO~ CH3
HN~N~NH2
H HCI OH
Example 71
(+) me~hyl a-amino-4a-hydroxy-5-imino-3a-methylpyrrolidin
2a-butanoate, dihydrochloride
HO~ &H3
HN~ ~,NH2
H HCI COOMe
Example 72
(+) 2-imino-4a-methyl-5a-pentylpyrrolidin-3a-amine,
dihydrochloride
H2~ CH3
HN~----
H HCI
_xample 72 -s prepared from Example 45E and Boc2~Hby
'~itsunobu reaction conditions. The synthesis of Example 72
is co.npleted in the manner desc-ibed in Example 45.
Example 73
(H 5-imino-4a-methyl-2a-pen=ylpyrrolidin-3a-Ol,
monohydroch_oride
7a-
CA 022l8360 l997-ll-lO
W096/35677 PCT~S9C/0~~~1
H3C~ 0H
HN~N~
H HCI
Example 74
(+) 5-imino-3a,4a-dimethylpyrrolidin-2a-prop~n~ine,
dihydroch oride
H3C~ ~CH3
HN~ NH2
H HCI
o
Example 75
(+) 2-imino-4a-methyl-5a-pentylpyrrolidine-3a-carboxylic
aci.d, monohydrochloride
HOOC~ CH3
HN~ N----
H HCI
Example 76
(+) 2-imino-4a-methyl-5a-pentylpyrrolidine-3a-methanol,
monohydrochloride
HOH2C~ CH3
HN~N ~
H HCI
-79-
CA 02218360 1997-11-10
W096t3S677 PCT~S96/06831
Example 77
(+) 5a-[3-(4,5-dihydro-lH-imidazoi-2-yi)propyl] -2-imino-~a-
methyipyrrolidin-3a-ol, dihydrochloride
S
HO~ ~CH3 HN~>
HN~N'\--J_ N
H HCI
Example 78
(+) 5a-[3-(lH-imidazol-2-yl)propyl]-2-imino-4a-
methylpyrrolidin-3a-ol, dihydrochloride
HN~
H HCI
Example 79
(+) sa- [ 3-amino-3-(lH-imidazol-2-yl)propyl]-2-imino-4a-
methylpyrrolidin-3a-ol, trihydrochloride
HO~ CH3 HN/~
~=N
HN~ N~
H HCI NH2
Example 80
(+) 2-imino-4a-methyl-Sa-[3-
,(pheryimet:yl)amino]propyl]pyrrolidin-3a-ol, dihydrocnio~ide
-80-
e
CA 022l8360 l997-ll-l0
W096/3S677 PCT~S9G/0~Q~l
HO~ ,CH3
HN~/- H--
H
Example 80 A) cis and trans-5-[(1,3-dioxol~n-2-yl)methy:]-4-
(methyl)pyr~olidin-2-one was prepared in the manner desc_ibed
in R. Ohrle n, W. Schwab, R. Ehrler, 'J. Jager, Synthesis
l9g6, 535-538) starting with 1,1-dimethoxy-3-nitropropane and
methyl crotonate.
Example 80 3,C) Example 80 A was reduced under catalytic
hydrogenation conditions using Raney Ni in MeOH. After
heating the reaction mixture for 16 h at 55 C, the solvent
was removed under vacuum. The crude lactam was separated by
column chromatography into the cis (80 s) and trans (80C)
lactam.
Example 80 D) A stirring solution of Example 80 B ,
(BocO)2O, DMAP in THF is heated at reflux for 3 h. After
concentrating reaction under vacuum, the residue is taken up
in EtOAc and washed with KHSO4 and brine. The organic layer
is dried over anhydrous Na2SO4, filtered, and stripped. The
crude product is purified by column chromatography.
Example 80 E) To a stirring solution of Example 80 D and
XMPA in THF cooled to -70 ~C is added lithium
hexamethyldisilazide. After 20 min, the reaction is warmed
to -40 C and cooled again to -70 C. To the stirring
reaction is added (R)-(-)-(camphorsulphonyl)oxaziridine in
THF. Afte- stirring at -~0 C for 30 min, the reaction s
warmed to -_0 C and stirred an additional 2.5 h. To the
react-on is added saturated NH4Cl soiution followed by E~OAc.
The organic layer is washed with brine, dried over anhyd-ous
~ 30 ~a~SO~, fi'_ered, and stripped. The crude product is purifiec
by column -:-romatography to yield 1.3 g of 3-hydroxylactam.
_xample 80 -) To a stirring solution of Example 80 E in CHC'-
is added H~~ and TFA. After stirring for 2 h, the reaction
mixture is ~oncentrate~ under reduced pressure. ~he res-due
is dissolved in EtOAc. The organic layer is washed with a
-81-
CA 02218360 1997-11-10
WO 96/3S677 PCT/US96/0~31
- i ni ml~ of satura~ed NaHCO3, driea ove~ MgSo4, Ciltered, a-.d
_oncen~rated under reduced pressure to recover crude aldehyde.
_xampie 80 G) To a sirring solution of Example 80 F in MeOH
-s added NaBH3CN. The reaction is main~ained at pH 4 by _he
aadition of HOAc. ~fter stirring for three days, the reac~ion
.ixture is concentrated under vacuum. ~o the residue is added
: N HCl and EtOAc. After separating the layers, the aqueous
phase is neutralized with NaHCO3 and extracted with EtO~c.
~fter concentrating the organic phase, the residue is treated
-.;ith 1 N HCl and lyophilized. The resulting solid is purified
by reverse phase column chromatography on a C-18 column.
_xample 80 H) The produ-t of Example 80 G is treated with
-rimethyloxonium tetrafluoroborate in CH2C12 as described in
_xample ~5.
_xample 80) A solution of the product of Example 80 H in MeOH
is reacted with ammonium chloride by the method of Example 5
Collowed by chromatography on reverse phase HPLC to generate
the title material.
Example 81
4a-methyl-5a-pentyl-3a-(methylthio)pyrrolidin-2-imine,
monohydrochloride
MeS~ CH3
HN~ N~
H HCI
Biological Data
~ he ac.ivity of the abo~Je listed compounas as NO sy.._hase
-nhibitors has been determinea in the following assays:
_itrllline .~.ssav ~or Nitric ~xide Svnthase
a~-
.
CA 02218360 l997-ll-l0
W096/35677 PCT~S96/0~Q~l
~'itr - oxide synthase (NOS~ ac~-v-ly was measurea _y mo--~or~
.he conversion o- [3H]-arginine tO [3H~-cicrulline ('3rea_ and
anyde~, Pro~ Natl. Acad. âci. -J.S.~., 87, 682-6~5, la90 and
~isko et al, ~ur. J Ph~m., 233, 119-125, 1993). ;iuman
-~duc ble NOS (hiNOS), human endothelial constitut~ve NOS
(hecNOS) and human neuronal consti-utive NOS (hncNOâ) were each
cloned from RNA extracted from human tissue. The cDNA for human
inducible NOS (hiNOS) was isolated from a ~cDNA library .,;ade
rom RNA extracted from a colon sample from a patient wi-h
uicerative colitis. The cDNA for human endotheliai cons.itut ve
NOS (hecNOS) was isolated from a ~cDNA library made from RNA
extracted from huma~ umbilical vein endothelial cells (H WEC)
and the cDNA for human neuronal constitutive NOS (hncNOS) was
isola~ed from a ~cDNA library made from RNA extracted from human
cerebellum obtained from a cadaver. The recombinant enzymes
were expressed in Sf9 insect cells using a baculovirus vector
(Rodi et al, in The ~iolo~v of Nitric Oxide. Dt. 4: F~nzvm
RiochPm;strv ~nd Immunoloav: Moncada, S., Feelisch, M., 3usse,
R., Higgs, E., Eds.; Portland Press Ltd.: London, 1995; pp 447-
450). Enzyme activity was isolated from soluble cell extractsand partially purified by DEAE-Sepharose chromatography. To
measure NOS activity, 10 ~L of enzyme was added to 40 ~L of 50
mM Tris (pH 7.6) in the presence or absence of test compounds
and the reaction initiated by the addition of 50 ~L of a
~eaction mixture cont~in;ng 50 mM Tris (pH 7.6), 2.0 mg/m~
bovine serum albumin, 2.0 mM DTT, 4.0 m~ CaC12, 20 ~M FAD, 100
~M te-rahydrobiopterin, 0.4-2.0 mM NADPH and 60 ~M L-arg nine
containing 0.9 ~Ci of L-[2,3-3H]-arginine. The final
concenrration of L-arginine in the assay was 30 ~M. For hecNOâ,
and hncNOS, calmodulin was included at a final concentrat-on o-
40-10~ nM. ~ollowing incuba~ion at 37~ C for 15 minutes, ~he
-eact_on was ~erminated by addition of 300 ~L of cold sto~
buffe_ containing 10 mM EGTA, 00 mM H~PES, pH 5.5 and 1 .-.M
c trul:ine. 3H]-Citrulline was separated by chromatography o..
~owex -0W X-~ cation exchange resin and radioactivity dete~mined
.~ith a liqu d scintillation counter. Results are repor-ea --.
mable ~ as ~:e ICso values o~ compounds for hiNOS, hecNOS anc
:ncNOâ. Co...pounds giving less than 50% inhibition a~ 10r UM
were repor~ed as having ICso ~alues of >100 ~M and compou.ds
_83-
CA 02218360 1997-11-10
W096/35677 PCT~S96/06831
givir.g grea-er than 50% inhiDi-ion a~ 100 UM were repor~e~ as
:-aving ICso values of <100 ,UM.
-84-
CA 02218360 1997-11-10
W096/3S677 PCT~S96/06831
~he foliowing Examp;es were assayea wit:- the -ollo:Jins ~esults.
Table I
, IC50 t~M]
~Ex~ple hiNOS hocNOS hncNOS
iO >100
13 cl00 <l00 <l00
18 <lO0 >l00 >l00
15 21 >l00 >l00 >l00
36 >l00
37 <l00 ~l00 <l00
38 >l00 <l00 <l00
39 <l00 >l00 <l00
25 40 <l00 >l00 <l00
41 >l00 >l00 >l00
42 <l00 >l00 <l00
44 >l00 >l00 >l00
<l00 >l00 <l00
~6 cl00 >l00 <l00
~7 <l00 >l00 <l00
:~iNoS rerers to human induci~le NOS
_85-
CA 02218360 1997-11-10
W096/35677 PCT~S96/06831
hecNOS refers to human endotheiial constitutive NOS
hncNOa refe-s to human neuronal constitutive NOS
~ rom the foregoing description, one skilled ~n the art can
easil~ ascertain the essential characteristics of this
-nven-ion, and without depar~ing from the spirit and scope
~hereof, can make various changes and modifications of the
inven~ion to adapt it to various usages ar.d conditions.
-86-