Note: Descriptions are shown in the official language in which they were submitted.
CA 02218816 1997-11-10
W096/37497 PCTICA96/00318
LOW MOLECULAR WEIGHT BICYCLIC-UREA TYPE
TFRO~RIN INHIBITORS
FIELD OF THE lNv~N-lloN
This invention relates to compounds use~ul ~or the treatment
of thrombotic disorders, and more particularly to novel
heterocyclic inhibitors of the enzyme thrombin.
10R~CK~:ROUND
Inordinate thrombus formation on blood vessel walls
precipitates acute cardiovascular disease states that are the
chie~ cause o~ death in economically developed societies.
Plasma proteins such as fibrinogen, proteases and cellular
receptors participating in hemostasis have emerged as
important factors that play a role in acute and chronic
coronary disease as well as cerebral artery disease by
contributing to the formation of thrombus or blood clots that
effectively diminish normal blood ~low and supply. Vascular
aberrations stemming from primary pathologic states such as
hypertension, rupture of atherosclerotic plaques or denuded
endothelium, activate biochemical cascades that serve to
respond and repair the injury site. Thrombin is a key
regulatory enzyme in the coagulation cascade; it serves a
pluralistic role as both a positive and negative feedback
regulator. However, in pathologic conditions the former is
amplified through catalytic activation of cofactors required
for thrombin generation as well as activation of factor XIII
necessary ~or fibrin cross-linking and stabilization.
In addition to its direct effect on hemostasis, thrombin
exerts direct effects on diverse cell types that support and
CA 022188l6 lss7-ll-lo
W096/37497 PCT/CA96/00318
amplify pathogenesis o~ arterial thrombus disease. The enzyme
is the strongest activator of platelets causing them to
aggregate and release substances (e.g. ADP TXA2 NE) that
further propagate the thrombotic cycle. Platelets in a fibrin
mesh comprise the principal framework o~ a white thrombus.
Thrombin also exerts direct effects on endothelial cells
causing release of vasoconstrictor substances and
translocation of adhesion molecules that become sites for
attachment of immune cells. In addition, the enzyme causes
o mitogenesis of smooth muscle cells and proliferation of
fibroblasts. From this analysis, it is apparent that
inhibition of thrombin activity constitutes a viable
therapeutic approach towards the attenuation of proliferative
events associated with thrombosis.
The principal endogenous neutralizing factor for thrombin
activity in m~mm~l S is antithrombin III (ATIII), a circulating
plasma macroglobulin having low affinity for the enzyme.
Heparin exerts clinical efficacy in venous thrombosis by
enhancing ATIII/thrombin binding through catalysis. However,
heparin also catalyzes inhibition of other proteases in the
coagulation cascade and its efficacy in platelet-dependent
thrombosis is largely reduced or abrogated due to
inaccessibility of thrombus-bound enzyme. Adverse side effects
such as thrombocytopenia, osteoporosis and triglyceridemia
have been observed following prolonged treatment with heparin.
Hirudin, derived from the glandular secretions of the leech
hirido medicinal is is one of the high molecular weight natural
anticoagulant protein inhibitors of thrombin activity
(Markwardt F. Cardiovascular Drug Reviews, 10, 211, 1992). It
is a biopharmaceutical that has demonstrated ef~icacy in
CA 02218816 1997-11-10
wos6/374s7 PCTICA96/00318
experimental and clinical thrombosis. A potential drawbac~ to
the use of Hirudin as a therapeutic agent is likely
antigenicity and lack of an effective method of
neutralization, especially in view of its extremely tight
binding characteristics toward thrombin. The exceedingly high
affinity for thrombin is unique and is attributed to a
simultaneous interaction with the catalytic site as well as a
distal ~anion binding exosite" on the enzyme.
Thrombin activity can also be abrogated by Hirudin-like
molecules such as hirulog (Maraganore, J.M. et al.,
Biochemistry, 29, 7095, l990) or hirutonin peptides (DiMaio,
J. et al., J. Med. Chem., 35, 3331, 1992).
Thrombin activity can also be inhibited by low molecular
weight compounds that compete with fibrinogen for thrombin's
catalytic site, thereby inhibiting proteolysis of that protein
or other protein substrates such as the thrombin receptor. A
common strategy for designing enzyme inhibitory compounds
relies on mimicking the specificity inherent in the primary
and secondary structure of the enzyme's natural substrate.
Thus, Blomback et al. first designed a thrombin inhibitor that
was modeled upon the partial sequence of the fibrinogen Aa
chain comprising its proteolytically susceptible region
(Blomback, et al., J. Clin. Lab. Invest., 24, 59, 1969). This
region of fibrinogen minimally includes the residues
commencing with phenylalanine:
Ala-Asp-Ser-Gly-Glu-Gly-Asp-Phe-Leu-Ala-Glu-Gly
-Gly-Gly-Val-Arg-Gly-Pro-Arg
~ scissile bond
~=
CA 02218816 1997-11-lo
W096l37497 PCT/CA96/00318
~ystematic replacement o~ amino acids within this region has
led to optimization o~ the tripeptidyl inhibitory sequence
exempli~ied by the peptide (D)-Phe-Pro-Arg which corresponds
to interactions within the P3-Pz-Pl local binding sites on
thrombin (Bajusz S. et al. in Peptides: Chemistry Structure
and Biology: Proceedings of the Fourth American Peptide
Symposium, Walter R., Meienhofer J. Eds. Ann Arbor Science
Publishers Inc., Ann Arbor MI, 1975, pp. 603).
o Bajusz et al. have also reported related compounds such as
(D)Phe-Pro-Arg-(CO)H (GYKI-14166) and (D)MePhe-Pro-Arg-(CO)H
(GYKI-14766) (Peptides-Synthesis, Structure and Function:
Proceedings of the Seventh American Peptide Symposium, Rich,
D.H. & Gross, E. eds., Pierce Chemical Company , 1981, pp.
417). These tripeptidyl aldehydes are effective thrombin
inhibitors both in vi tro and in vivo. In the case of both
GYKI-14166 and GYKI-14766, the aldehyde group is presumed to
contribute strongly to inhibitory activity in view of its
chemical reactivity toward thrombin's catalytic Serl95 residue,
generating a hemiacetal intermediate.
Related work in the area o~ thrombin inhibitory activity has
exploited the basic recognition binding moti~ engendered by
the tripeptide (D)Phe-Pro-Arg while incorporating various
functional or reactive groups in the locus corresponding to
the putative scissile bond (i.e. Pl-P1').
In U.S. Patent 4,318,904, Shaw reports chloromethyl-ketones
(PPACK) that are reactive towards Ser195 and His57. These two
residues comprise part of thrombin's catalytic triad (Bode, W.
et al., EMBO Journal 8, 3467, 1989).
=
CA 022l88l6 lss7-ll-lo
W096/37497 PCTICA96/00318
u~ner examples o~ thrombin inhibitors bearing the (D)Phe-Pro-
Arg general motif are those incorporating COOH-terminal
boroarginine variants such as boronic acids or boronates
(Kettner, C. et al., J. Biol. Chem., 268, 4734, 1993).
Still other congeners of this motif are those bearing
phosphonates (Wang, C-L J., Tetrahedron Letters, 33, 7667,
1992) and a-Keto esters (Iwanowicz, E.J. et al.,Bioorganic and
Medicinal Chemistry Letters, 12, 1607, 1992).
lo
Neises, B. et al. have described a trichloromethyl ketone
thrombin inhibitor (MDL-73756) and Attenburger, J.M. et al.
have revealed a related difluoro alkyl amide ketone
(Tetrahedron Letters, 32, 7255, 1991).
Maraganore et al. (European 0,333,356; WO 91/02750; U.S.
5,196,404) disclose a series of thrombin inhibitors that
incorporate the D-Phe-Pro- moiety and hypothesize that this
preferred structure fits well within the groove adjacent to
zo the active site of thrombin. Variations on these inhibitors
are essentially linear or cyclic peptides built upon the D-
Phe-Pro moiety.
Another series of patents and patent applications have
described attempts to develop effective inhibitors against
thrombosis by using alpha-ketoamides and peptide aldehyde
analogs (EP 0333356;WO 93/15756; WO 93/22344; WO 94/08941; WO
94/17817).
Still others have focused their attention on peptides, peptide
derivatives, peptidic alcohols, or cyclic peptides as anti-
thrombotic agents (WO 93/22344, EP 0276014; EP 0341607; EP
CA 02218816 1997-ll-10
W096/37497 PCT/CA96/00318
0291982). Others have examined amidine sulfonic acid moieties
to achieve this same end (U.S. 4,781,866), while yet others
have examined para or meta substituted phenlyalanine
derivatives (WO 92/08709; WO 92/6549).
A series of Mitsubishi patents and patent applications have
disclosed apparently effective arg; n; n~m; de compounds for use
as antithrombotic agents. The chemical structures described
in these documents represent variations of side groups on the
0 argininamide compound (U.S. 4,173,630; U.S. 4,097,591; CA
1,131,621; U.S. 4,096,255; U.S. 4,046,876; U.S. 4,097,472; CA
2,114,153).
Canadian patent applications 2,076,311 and 2,055,850 disclose
cyclic imino derivatives that exhibit inhibitory effects on
cellular aggregation.
Many of the examples cited above are convergent by maintaining
at least a linear acyclic tripeptidyl motif consisting of an
arginyl unit whose basic side chain is required for
interaction with a carboxylate group located at the base of
the Pl specificity cleft in thrombin. Two adjacent hydrophobic
groups provide additional binding through favourable Van der
Waals interactions within a contiguous hydrophobic cleft on
the enzyme surface designated the P3-P2 site.
Accordingly, it is an object of the present invention is to
provide thrombin inhibitors that display inhibitory activity
towards the target enzyme, thrombin.
-
CA 02218816 1997-11-10
WO 96/37497 PCT/CA96/00318
S~MMARY OF T~IE lNv~;NlloN
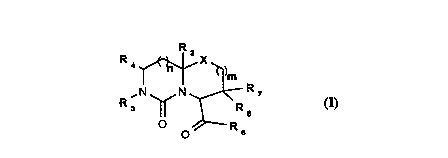
The present invention provides ~or novel compounds that
display thrombin inhibitory activity as represented by ~ormula
(I):
R ~ )m
R3 ~N~R7
~ o R6
whereln:
X is selected from CH-R5, O, S, SO, SO2 and NRg wherein R5 is
0 hydrogen, C16 alkyl optionally interrupted with 1 or 2
heteroatoms; C6l6 aryl, C37 cycloalkyl or heterocyclic ring
or a hydrophobic group;
R2 is selected ~rom H, NH2 and Cl6 alkyl optionally substituted
wlth C6 aryl, a 6 member heterocycle or a C37 cycloalkyl
r1ng;
R3 and R4 are independently selected ~rom H; NR6R7; C6l6 aryl or
C37 cycloalkyl optionally substituted with Cl 6 alkyl; Cl l6
alkyl optionally interrupted by one or more heteroatom or
carbonyl group and optionally substituted with OH, SH, NR6R7
or a C6l6 aryl, heterocycle or C37 cycloalkyl group
optionally substituted with halogen, hydroxyl, Cl 6 alkyl; an
amino acid side chain; and a hydrophobic group;
R6 is a polar amino acid residue, arginyl moiety or an analog
or derivative thereo~ optionally substituted with an amino
acid, a peptide or a heterocycle;
R7 and R8 are independently hydrogen or Cl 6 alkyl;
m is an integer between 0 and 2; and
n is an integer between 0 and 2.
CA 02218816 1997-11-10
W096/37497 PCT/CA96/00318
According to another aspect o~ the invention, there is
provided pharmaceutical compositions comprising compounds o~
the formula (I) in combination with pharmaceutically
acceptable carriers, diluents or adjuvants.
In yet another aspect, there is provided a method for the
treatment or prophylaxis of thrombotic disorders in a m~mm~l,
comprising administering to said mammal an effective amount of
a compound according to formula (I).
DETZ~TT~T~n DESCRIPTION OF THE INVENTION
The present invention relates to compounds which inhibit the
enzyme, thrombin. These molecules are characterized by a
heterobicyclic moiety as illustrated in formula (I):
R 4 ~ X ~I)m
R 3 ~ N ~ R 7
~ o R6
wherein X, Rl to R8, m and n are as previously defined.
The term "hydrophobic group" (HG) as used hereinafter, refers
to any group which lacks affinity for, or displaces water.
Hydrophobic groups include but are not limited to Cl20 alkyl,
C220 alkenyl (e.g. vinyl, allyl) or C220alkynyl (e.g.
propargyl) optionally interrupted by a carbonyl group, (e.g.
forming an acyl group); C6l6 aryl, C3 7 cycloalkyl, C620
aralkyl, C620 cycloalkyl substituted Cl20 alkyl, wherein the
aliphatic portion is optionally interrupted by a carbonyl
group (e.g. forming an acyl group) and the ring portion is
optionally substituted with Cl6 alkyl such as methyl ethyl or
CA 02218816 1997-ll-10
W096/37497 PCT/CA96/00318
t-butyl; or a hydrophobic amino acid side chain. Preferred
hydrophobic groups include cyclohexyl, benzyl, benzoyl,
phenylmethyl, phenethyl and para-t-butyl-phenylmethyl.
The term ~arginyl moiety~ represents an arginine amino acid
v residue Ol- an analogue or derivative thereof For example, an
analogue or derivative of the natural residue may incorporate
a longer or shorter methylene chain ~rom the alpha carbon
(i.e. ethylene or butylene chain); replacement of the
guanidino group with a hydrogen bond donating or accepting
group (i.e. amino, amidino or methoxy); replacement o~ the
methylene chain with a constrained group (i.e. an aryl,
cycloalkyl or heterocyclic ring); elimination of the terminal
carboxyl (i.e. des-carboxy) or hydroxyl (i.e. an aldehyde); or
a combination thereof.
The term "alkyl" represents a straight or branched, saturated
or unsaturated chain having a specified total number of carbon
atoms.
The term "aromatic" or "aryl" represents an unsaturated
carbocyclic ring(s) of 6 to 16 carbon atoms which is
optionally mono- or di-substituted with OH, SH, amino (i.e.
NR6R7) halogen or Cl6 alkyl. Aromatic rings include benzene,
napththalene, phenanthrene and anthracene. Preferred aromatic
rings are benzene and naphthalene.
The term "cycloalkyl" represents a carbocyclic ring of 3 to 7
carbon atoms which is optionally mono- or di-substituted with
OH, SH, amino (i.e. NR6R7) halogen or Cl6 alkyl. Cycloalkyl
groups are generally saturated but may be partially
=
CA 02218816 1997-11-10
W096/37497 PCT/CA96/00318
unsaturated and include cyclo-propyl, butyl, pentyl, hexyl and
heptyl. A preferred cycloalkyl group is cyclohexyl.
The term "aralkyl" represents a substituent comprising an aryl
moiety attached via an alkyl chain (e.g. benzyl, phenethyl)
wherein the sum total of carbon atoms for the aryl moiety and
the alkyl chain is as specified. The aryl or chain portion of
the group is optionally mono- or di-substituted with OH, SH,
amino ~i.e. NR6R7) halogen or Cl6 alkyl
The term "heteroatom" as used herein represents oxygen,
nitrogen or sulfur (O, N or S) as well as sulfoxyl or sulfonyl
(SO or SO2) unless otherwise indicated. It is understood that
alkyl chains interrupted by one or more heteroatoms means that
a carbon atom of the chain is replaced with a heteroatom
having the appropriate valency. Preferably, an alkyl chain is
interrupted by 0 to 4 heteroatoms and that two adjacent carbon
atoms are not both replaced.
The term "heterocycle" represents a saturated or unsaturated
mono- or polycyclic (i.e. bicyclic) ring incorporating l or
more (i.e. 1-4) heteroatoms selected from N, O and S. It is
understood that a heterocycle is optionally mono- or di-
substituted with OH, SH, amino (i.e. NR6R7), halogen, CF3, oxo
or C16 alkyl. Examples of suitable monocyclic heterocycles
include but are not limited to pyridine, piperidine, pyrazine,
piperazine, pyrimidine, imidazole, thiazole, oxazole, furan,
pyran and thiophene. Examples of suitable bicyclic
heterocycles include but are not limited to indole, quinoline,
isoquinoline, purine, and carbazole.
CA 02218816 1997-ll-lo
W096/37497 PCT/CA96100318
The term ~hydrophobic amino acid~ represents an amino acid
residue that bears an alkyl or aryl group attached to the a-
carbon atom. Thus glycine, which has no such group attached to
the a-carbon atom is not a hydrophobic amino acid. The alkyl
or aryl group can be substituted, provided that the
~ substituent or substituents do not detract from the overall
hydrophobic character of the amino acid. Examples of
hydrophobic amino acids include natural amino acid residues
such as alanine; isoleucine; leucine; phenylalanine; and non-
lo naturally occurring amino acids such as those described in
"The Peptides", vol. 5, 1983, Academic Press, Chapter 6 by
D .C. Roberts and F. Vellaccio. Suitable non-naturally
occurring amino acids include cyclohexylalanine and 1-
aminocyclohexane-carboxylic.
By "amino acid side chain" is meant the substituent attached
to the carbon which is a to the amino group. For example, the
side chain of the amino acid alanine is a methyl group and
while benzyl is the side chain for phenylalanine.
Preferably, X is CH-R5, S or O wherein Rs is preferably H or
Cl4 alkyl and most preferably H.
More preferably, X is S.
Preferably R2 is H, methyl or ethyl. Most preferably, R2 is H.
Preferably, one of R3 or R4 is a carboxyl group or a
hydrophobic group such as a saturated or unsaturated
carbocycle of 5 or 6 members optionally fused to another
carbocyclic group while the other is H, C116 alkyl optionally
substituted by NR6R7 or carboxy. The carboxy group or
hydrophobic group may be linked via a spacer such as a Cll6
11
CA 02218816 1997-11-10
W096/37497 PCT/CA96/00318
alkyl chain optionally interrupted with l or more (i.e. l-4)
heteroatoms, carbonyl or sulfonyl (SO2) groups. More
preferably, one of R3 and R4 is an optionally substituted
aromatic ring such as phenyl, cyclohexyl, indole, thienyl,
quinoline, tetrahydroisoquinoline, naphthyl or benzodioxolane
linked via Cll6 alkyl optionally interrupted with a heteroatom
or a carbonyl while the other is H, carboxymethyl or
carboxyethyl. Optional aromatic ring substituents include OH,
carboxy, Cl4 alkyl and halogen. In another more preferred
embodiment, one of R3 and R4 is optionally substituted phenyl
or cyclohexyl linked via a Cl4 alkyl optionally interrupted
with carbonyl while the other is H, carboxymethyl or
carboxyethyl. In a most preferred embodiment, R3 is benzyl,
phenylethyl, phenylpropyl or cyclohexyl-methyl and R4 is H.
Preferably R7 and R8 are independently hydrogen, methyl or
ethyl. More preferably R7 and R8 are independently hydrogen or
methyl. Most preferably R7 and R8 are both hydrogen.
Preferably, m is 0 or l. More preferably, m is 0.
Preferably, n is O or l. More preferably, n is l.
In a preferred embodiment, R6 is represented by one of formula
VIa to VId:
CA 022l88l6 l997-ll-l0
WO 96/37497 PCT/CA96/00318
R"N ~ (J)n R,lN
Vla K~J )0-7 Vlb ~ )0-8
G K~
U~ ~(J)n Vld ~(J)nT
wherein:
R1l is hydrogen or C16 alkyl;
K is a bond or -NH-;
G is Cl 4 alkoxy; cyano; -NH2; -CH2-NH2; -C(NH)-NH2; -NH-C(NH)-
NH2; -CH2-NH-C (NH) -NH2; a C6 cycloalkyl or aryl substituted
with cyano, -NH2, -CH2-NH2, -c (NH) -NH2, -NH-C (NH) -NH2 or -CH2-
NH-C (NH) -NH2; or a 5 or 6 member, saturated or unsaturated
heterocycle optionally substituted with cyano, -NH2, -CH2-
o NH2, -C(NH)-NH2, -NH-C (NH)-NH2 or -CH2-NH-C(NH)-NH2;
U is cyano, -NH2, -C(NH)-NH2 or -NH-C(NH)-NH2;
P is a bond, -C(O)- or a bivalent group:
~ CH' OH ~ ~ ~¦~ ~
J is C16 alkylene optionally substituted with OH, NH2 and C16
alkyl and optionally interrupted by a heteroatom selected
from 0, S and N;
n is O or 1; and
T iS H, OH, amino, a peptide chain, C116 alkyl, C116 alkoxy, C6
20 aralkyl, or heterocycle optionally substituted.
- Pre~erably R11 is H or methyl and most pre~erably H.
Pre~erably K is a bond.
CA 022l88l6 lss7-ll-lo
W096/37497 PCT/CA96/00318
Preferably G is -NH-C (NH) -NH2 attached via a methylene chain of
3-7 carbons or phenyl substituted with -C (NH) -NH2 attached via
a methylene chain of 0 to 3 carbons. More preferably G is
-NH-C (NH) -NH2 attached via a methylene chain of 3 atoms.
Preferably P is -C(O)-.
Preferably J is selected from: -CH2-S-CH2-CH2-; -CH2-O-CH2-CH2-;
-CH2-NH-CH2-CH2-; and a bond when n is 0. More preferably, J
is a bond while n is 0.
o In particular embodiments of the invention, R6 is selected from
the following amino acid derivatives prepared according to the
procedures described in Bioorg. Med. Chem., 1995, 3:1145 :
"N~JI~ NJi~ "NJ~T
(~NH (~ (~
NJ\NMe N\~H
NH2 )=N //S--N
H2N o nl
T "N~J~ , ~T
( ~?NH (~NH (~NH
N~ NH2 N/~r N~O
S--N ,~o )=N
H2N H2N
CA 02218816 1997-ll-lO
WO 96/37497 PCT/CA96/00318
O O O
N~JI~ HN~JI~T
N~\NH ~ NH
H2N
H2N
N~,JI~ ~N~JI~ H~J~
N~N (~ NH2 H3~ NH2
NH2 CH3
~.1T ~N X~"
n2 NH2 n2 NH
HN~NH2
NH2
T~O T~O
N~ N~NH)n~
HN~NH H2N NH HNe~
NH2 NH2
CA 02218816 1997-ll-lO
WO 96/37497 PCT/CA96/00318
,_N~
1~NH~H/N~
HN NH
H2N NH
N\J~ T.N~J¦~
~1 ~NH2 H2N~
H2N
~N ~Jl~ N~JI~
4~ HNq~N~3
Jl~ NH2
H2N H
.~T - T~N~JI~
9~NH2 9~NH2 ~NH
HN
,,N~JI~ T . ~T
H ~ ~N~NH2
HN
CA 02218816 1997-11-10
W096/37497 PCT/CA96100318
T H~J~ H~Jl~
~0 H,!N~ ~H
H2N NH NH
O O
,.N~JI~ N~JI~T "N~J~T
~N~NH
NH2
O O
.~' ~J~T N~JI~T
HN~
H2N ~NH
O O
T ~ N ~I~
~NH2 NH ~NH2
NH NH
0 wherein n=l-6, nl=l-2, n2=0-7 and T is as previously defined.
In a preferred embodiment, T is a peptide of l to 4 amino acid
residues in length and preferably fibrinogen's A or B chain or
fragment or derivative thereof. In another preferred
17
CA 02218816 1997-ll-lo
W096t37497 PCTICA96/00318
embodiment, T is a heterocycle selected from the group
consisting of:
X6 ~ X5 ~ ~R'
''I~ ~ '<X,3 ~X,~
wherein
X5, X10, Xl1 and X12 are each independently selected from the
group consisting of N, or C-X7 where X7 iS hydrogen, C14 alkyl,
or C616 aryl;
X6 and X13 are each independently selected from the group
o consisting of C, O, N, S, N-X7, or CH-X7;
R' is hydrogen, C116 alkyl optionally carboxyl substituted,
carboxyl, -C0l6 alkyl-CO2-Cll6 alkyl, C620 aralkyl, C37
cycloalkyl, aryl or an aromatic heterocycle.
Preferably T is selected from the group consisting of:
CA 02218816 1997-11-lo
W096/37497 PCT/CA96/00318
~ f ~ ~ ~N~ ~N, , ,~
;~1 N~ 3 N ~ N R
~N~ R ~ R' R'~
- _ _~ \ _ _<N ? ~ s~
wherein R' is as defined above.
More preferably T is selected from the group consisting of:
- ~ ~N~ , ~ R' j~
wherein R~ is as de~ined above.
More preferably T is selected from the group consisting of:
' ' 1/ ~ ~ ~ ' ;~ ' ~ N ~ ~ I J
lo wherein R' is as defined above.
CA 02218816 1997-11-10
W096/37497 PCT/CA96/00318
Most pre~erably T is
S \~ or R~ ~
wherein R' is H or Cl4 alkyl such as methyl, ethyl, propyl or
butyl and most preferably wherein R' is hydrogen,. In another
embodiment, T is a l,2 thiazole optionally substituted with R~
and/or is attached to J at the 2, 3, 4 or 5 position of the
ring.
A more preferred embodiment of the present invention is
illustrated by compounds having Formulae II, III, IV, and v
wherein R3, R~, R6, R7 and R8 are as defined in each of the
above embodiments.
R ~o
II
R41 I S
, N ~ N ~ R,
o R6
III
R,
IV
CA 02218816 1997-11-10
WO 96/37497 PCT/CA96/00318
R4~X~
, N ~ N ~8
~
V
It will be appreciated by those skilled in the art that the
compounds of formulae (I) to (V), depending of the
substituents, may contain one or more chiral centers and thus
exist in the form of many different isomers, optical isomers
(i.e. enantiomers) and mixtures thereof including racemic
mixtures. All such isomers, enantiomers and mixtures thereof
including racemic mixtures are included within the scope of
o the invention.
Preferred compounds of the invention include:
6 (3S)-6-benzyl-5-oxo- ~ s
hexahydro-imidazo[5,1- \-~ ~ . N~,N~NH !' N~
b]thiazole-3-carboxylic o .
acid [1-(benzothiaozle-2-
HN
carbonyl)-4-guanidino-
H2N ~NH
butyl]-amide
7 (S)-6-benzyl-5-oxo- ~1 ~ O
hexahydro-thiazolo(3,2- ,~~,N~N~NH N~ -
c)pyrlmldlne-3-carboxylic 0 J
acid (4-guanidino-1-
(benzothiazole-2- ~
carbonyl)-butyl)-amide H2N NH
CA 02218816 1997-11-lo
W096/37497 PCT/CA96/00318
6-(para-tBu-phenylmethyl)-
5-oxo-hexahydro- ~ , ~ r-~ >
imidazo[5,1-b]thiazole-3-~-~' ,N~,N~NH~
carboxylic acid [1-
(benzothiaozle-2- lN
carbonyl)-4-guanidino- H2N NH
butyl]-amide
9 6-(3-phenyl-prop-2-enyl)- -"~ ! s
5-oxo-hexahydro- \~ N ~, N ~ ) N
imidazo[5,1-b]thiazole-3- ~ ' <s
carboxylic acid [1-
H l~i
(benzothiaozle-2-
H
carbonyl)-4-guanidino-
butyl]-amide
lQ 6-(cyclohexylmethyl)-5- ~ s
oxo-hexahydro-imidazo[5,1- ~ N~ .N~NH ~ ~1 N~
b]thiazole-3-carboxylic~ O j ~ ~ i
acid [1-(benzothiaozle-2-
carbonyl)-4-guanidino- H~
butyl]-amide ~2N N~
11 6-(2-trifluoromethyl ~ ~-~ ~ 5
quinolin-7-yl)-5-oxo- F3C N J ~ ~ NH ~ N
hexahydro-thiazolo(3,2- ~ s
c)pyrimidine-3-carboxylic HN'
acid (4-guanidino-1- HIN NH
(thiazole-2-carbonyl)-
butyl)-amide
CA 022l88l6 lss7-ll-lo
W096/37497 PCTICA96/00318
12a (3S,9S)-6-phenylpropyl-5- ~ ~~~ s
oxo-hexahydro- ~3 ~ NH ~0 N
thiazolo(3,2-c)pyrimidine- O ~/ 3
3-carboxylic acid (4-
guanidino-1-(thiazole-2- HN
carbonyl)-butyl)-amide H2N~NH
12b (3S,9S)-6-phenylpropyl-5- ~ j~~J.s>
oxo-hexahydro- ~~ ) N~ ,N--~_NH N-
thiazolo(3,2-c)pyrimidine- O O ~ <
3-carboxylic acid (4-
guanidino-1-(thiazole-2- HN
carbonyl)-~utyl)-amide .!;
12c (3S,9R)-6-phenylpropyl-5- ~, ~ s
oxo-hexahydro- ~ l ,N~ ,N_ ~ _NH N
thiazolo(3,2-c)pyrimidine- o O ~/ <~
3-carboxylic acid (4-
guanidino-1-(thiazole-2- HN
carbonyl)-butyl)-amide H2N~NH
12d (3S, 9R) -6-phenylpropyl-5- ~ s
oxo--hexahydro- ~3 ~i ~ NH i N
thiazolo(3,2-c)pyrimidine- o
3-carboxylic acid (4-
guanidino-l-(thiazole-2- HN
carbonyl)-butyl)-amide H2N~NH
13a (3S,9S)-6-benzyl-5-oxo- ~ ~ H
hexahydro-thiazolo(3,2- ~ J ~N~r~N--~NH 1~, N
c)pyrimidine-3-carboxylico ~ ~ -~/ 3
acid (4-guanidino-1-
(thiazole-2-carbonyl)- HN
butyl)-amide (~ast moving H2N~NH
isomer on HPLC)
CA 022188l6 lss7-ll-lo
W096/37497 PCT/CA96/00318
13b (3S,9S)-6-benzyl-5-oxo- ~ H
hexahydro-thiazolo(3,2- ~ ~,1\ N~ N ~ NH il N _
c)pyrimidine-3-carboxylic o O ''~~ </ 1¦
acid (4-guanidino-1-
(thiazole-2-carbonyl)- HN
butyl)-amide (slow moving H2N iNH
isomer on HPLC )
13c (3S,9R)-6-benzyl-5-oxo- ~ H s~
hexahydro-thiazolo(3,2- ~ N~ N~NH N'
c)pyrimidine-3-carboxylic 0 '~
acid (4-guanidino-1-
(thiazole-2-carbonyl)- HN
butyl)-amide (fast moving H2N 'NH
isomer on HPLC)
1~ (3S,9R)-6-benzyl-5-oxo- H
hexahydro-thiazolo(3,2- -~ .. N, N~ O
c)pyrimidine-3-carboxylic '' ~NH~ ~,' N
acid (4-guanidino-1-
(thiazole-2-carbonyl)- HN
butyl)-amide (slow moving H2N~NH
isomer on HPLC )
Compounds of the present invention are further characterized
by their ability to inhibit the catalytic activity of
thrombin, which can be demonstrated in the assay as follows.
Compounds of the present invention may be prepared for assay
by dissolving them in buffer to give solutions ranging in
concentrations from 0 to lOO~M. In an assay to determine the
inhibitory dissociation constant, Ki, for a given compound, a
chromogenic or fluorogenic substrate of thrombin would be
o added to a solution containing a test compound and thrombin;
the resulting catalytic activity of the enzyme would be
24
CA 02218816 1997-ll-lo
W096/37497 PCT/CA96/00318
spectrophotometrically determined. This type of assays lS
well known to those skilled in the art.
Accordingly, compounds o~ the invention may be used in the
treatment and/or prophylaxis o~ thrombotic disorders mediated
by the activity of thrombin. Such thrombotic disorders
include venous thrombosis, pulmonary embolism, arterial
thrombosis, myocardial in~arction and cerebral in~arction.
Methods of treatment or prophylaxis according to the invention
o comprise administering to a mammal, more particularly human,
an effective amount of compounds of the present invention. By
"effective" is meant an amount of the compound sufficient to
alleviate or reduce the severity o~ the disorder as measured
by parameters established for the particular indication i.e.
blood flow (patency), clot size or density.
The compounds of the present invention may be used as anti-
coagulants in vitro or ex vivo as in the case o~ contact
activation with foreign thrombogenic surfaces such as is ~ound
in tubing used in extracorporeal shunts. The compounds of the
invention may also be used to coat the sur~ace of such
conduits. To this end, the compounds of the invention are
obtained as lyophilized powders, redissolved in isotonic
saline and added in an amount sufficient to maintain blood in
an anticoagulated state.
The therapeutic agents of the present invention may be
administered alone or in combination with pharmaceutically
acceptable carriers, diluents or adjuvants. The proportion o~
0 each carrier, diluent or adjuvant is determined by the
solubility and chemical nature of the compound, the route of
administration, and standard pharmaceutical practice. For
CA 02218816 1997-11-10
W096/37497 PCT/CA96/00318
example, the compounds may be in~ected parenterally; this
being intramuscularly, intravenously, or subcutaneously. For
parenteral administration, the compound may be used in the
form of sterile solutions containing other solutes, for
example, sufficient saline or glucose to make the solution
isotonic. The compounds may be administered orally in the
form of tablets, capsules, or granules containing suitable
excipients such as starch, lactose, white sugar and the like.
The compounds may also be administered sublingually in the
o form of troches or lozenges in which each active ingredient is
mixed with sugar or corn syrups, flavouring agents and dyes,
and then dehydrated sufficiently to make the mixture suitable
for pressing into solid form. The compounds may be
administered orally in the form of solutions which may contain
colouring and/or flavouring agents.
Physicians will determine the dosage of the present
therapeutic agents which will be most suitable. Dosages may
vary with the mode of administration and the particular
compound chosen. In addition, the dosage may vary with the
particular patient under treatment. For parenteral
administration, typical dosage is about O.l to 500 mg/kg body
weight per day, and preferably about 0.5 to lO mg/kg body
weight per day.
When the composition is administered orally, a larger quantity
of the active agent will typically be required to produce the
same effect as caused with a smaller quantity given
parenterally.
CA 02218816 1997-11-10
WO 96/37497 PCT/CA96/00318
For preparation of the compounds of this invention, various
methods can be employed depending upon the particular starting
materials and/or intermediates involved.
Successful preparation of these compounds is possible by way
o~ several synthetic routes one of which is outlined below.
27
CA 02218816 1997-ll-lO
WO 96t37497 PCTICA96/00318
S CHEME
OH o
~J STEP 1 ~~ H
HN R4 N R4
R3 R3
X XI
H R7 Ra O
X ~ ~ o R 2 ol STEP 2
X I I ~
R ~ ~ STEP 3 ~aR 7
O O ORzo
XIV
R 6H ~ XIII
STEP4 !
R4~ ~)m
o
xv
Wherein;
Pg is a nitrogen protecting group;
R20 is a Cl6 alkyl; and X, n, m, R3, R4, R6, R7, and R8 are as
de~ined above.
The process depicted in scheme 1 can be briefly describe as
follows:
28
CA 022l88l6 lss7-ll-lo
Wog6/374s7 PCT/CA96/00318
ST~p 1
The amino function of the alkylaminoalcohol of formula (X) is
protected with an appropriate amino protecting group. A
variety of protecting groups known for reactive functional
groups and suitable protection and deprotection protocols may
be found in T. Greene, Protective Groups In Or~anic Synthesis,
(John Wiley & sons, 1981). The appropriate protecting group
to use in a particular synthetic scheme will depend on many
lo factors, including the presence of other reactive functional
groups and the reaction conditions desired for removal. The
protected aminoalkylalcohol is then sub]ected to oxidation,
using an appropriate oxidizing agent, such as a catalytic
amount of tetrapropylammonium perruthenate (TPAP) along with
N-methylmorpholine oxide (NMO)in an inert solvent such as
dichloromethane tCH2C12) to yield to a protected amino alkyl
aldehyde of formula (XI).
STEP 2
The protected amino alkyl aldehyde of formula (XI) is coupled
with an amino acid alkyl ester of formula (XII) with an
appropriate base such as potassium carbonate in an inert
solvent such as dichloromethane to yield to a cyclic
intermediate of formula (XIII).
STEP 3
The amino protecting group of the cyclic intermediate of
formula (XIII) is removed under appropriate condition and the
resulting compound is then contacted with a reagent
appropriate for internal ring closure such as phosgene,
triphosgene or carbonyldiimidazole in an inert solvent such as
tetrahydrofuran to yield to a bicyclic intermediate of formula
(XIV).
29
CA 02218816 1997-11-10
W096/37497 PCTtCA96/00318
ST~P 4
The ester function (-C(O)O-R20) of the bicyclic intermediate of
formula (XIV) is subjected to hydrolysis using an appropriate
reagent such as LiOH to yield to the free carboxylic acid. The
resulting compound is then coupled to R6H with a peptide
coupling agent such as BOP in an appropriate solvent such as
dimethylformamide to yield to a coupled bicyclic compound of
formula (XV). Suitable conditions for peptide bond formation
are well known in the art of peptide chemistry. For example
see Principles of Peptide Synthesis, Bodanszky M., Springer-
Verlag, Berlin, Heidelberg, New York, Tokyo 1984; and The
Peptides. Analysis. Synthesis. Biology. Vol. ~.edited by Gross
E., and Meienhofer J., Academic Press , New York, San
Francisco, London, 1979.
In a particular embodiment wherein X is S, the following
scheme 2 may be followed:
CA 02218816 1997-11-10
W096/37497 PCT/CA96/00318
SCHEME 2
L-cysteine ethyl ester
Ph~N~VOH 1) (BOC)20- CH7C12 ~ Ph~N~O K2C03, M9504
H 2) TPAP, NMO or PCC BOC H
Ph N~ S 1) HCI 4.0 M / dioxane Ph ~,~5
BOC HN ~ 2) L, iul~osgt:ne or N,N-carbonyl ~ ~
COzEt O CO2Et
1) LiOH
2) thiazole or ben~utl lid~OIe Ph H
keto arginine, BOP ~r N~S
4) HPLC purification ~N~ ~
Ar = thiazole, be~l,uUIid~ult:
HN rn-; 2
,1~ CF3C02H
HN ~ NH2
The compounds of this invention may be puri~ied during their
synthesis and/or after their preparation by standard
techniques well known to the skilled artisan. One pre~erred
puri~ication technique is silica gel chromatography. In
particular, the flash chromatographic technique may be used.
However, other chromatographic methods, including HPLC, may be
used ~or purification of the compounds. Crystallization may
also be used to purify the products, as may washing procedures
with appropriate organic solvents.
Where the compound o~ ~ormula (I) is desired as a single
isomer, it may be obtained either by resolution o~ the final
CA 02218816 1997-ll-lo
W096/37497 PCTICA96/00318
product or by stereospecific synthesis from isomerically pure
starting materlal or any convenient intermediate.
Resolution of the final product, or an intermediate or
starting material therefor, may be effected by any suitable
method known in the art: see for example, "Stereochemistry of
Carbon Compounds", by E.L. Eliel (McGraw Hill, 1962), and
"Tables of Resolving Agents", by S.H. Wilen. Resolution of the
final compound can also be achieved using chiral HPLC
techniques.
To further assist in understanding the present invention, the
following non-limiting examples of such thrombin inhibitory
compounds are provided. The following examples, of course,
should not be construed as specifically limiting the present
invention, variations presently known or later developed,
which would be within the purview of one skilled in the art
and considered to fall within the scope of the present
invention as described herein. The preferred compounds as of
the present invention can be synthesized using conventional
preparative steps and recovery methods known to those skilled
in the art of organic and bio-organic synthesis, while
providing a new a unique combination for the overall synthesis
of each compound. Preferred synthetic routes for
intermediates involved in the synthesis as well as the
resulting anti-thrombotic compounds of the present invention
follow.
CA 022l88l6 l997-ll-l0
WO 96/37497 PCT/CA96/00318
EXAMPLE 1
O O
Boc~ ~ ~OCH3 1. Zn/Cu couple; ultrasound Boc~ ~ ~OCH3
CH3 2. Io-cH3c6H4)3pl2pdcl~ l H3
4-iodobenzonitrile
NC~
A solution of tert-butyloxycarbonyl-iodo-alanine-N,O-
dimethylamide (2.68 g, 7.5 mmol) (J. Org. Chem. 1992, 57,
3397-3404) in dry benzene (30 mL), and dry N,N-
dimethylacetamide (2.0 mL) was added to a dry nitrogen-purged
round bottom flask charged with zinc-copper couple (0.90 g).
The resulting mixture was sonicated under nitrogen until no
starting material remained (as judged by TLC). Bis(tri-o-
tolylphosphine)palladium dichloride (0.35 g, 0.40 mmol) was
added followed by 4-iodobenzonitrile (1.72 g, 7.5 mmol). The
resulting mixture was stirred under a nitrogen atmosphere with
heating, allowed to cool, ethyl acetate (100 mL) was added,
and the mixture filtered into a separatory funnel. Sequential
washing with aqueous HCl (50 mL; 0.1N), distilled H20 (3 x 50
mL), drying over Na2SO4, ~iltration, and concentration under
reduced pressure yielded the crude product. Flash
chromatography over silica gel (light petroleum-ethyl acetate
gradient) a~orded the puri~ied compound.
CA 02218816 1997-11-10
WO ~6/37497 PCT/CA96/00318
O O
~N~ ~OCH3 1. Zn/Cu couple; ultrasound _ ~OCH3
2. Io-CH3C6H4)3Pl2Pdc12 ~ ~ CH3
3-iodobellzooitrile~~
CN
A solution o~ tert-butyloxycarbonyl-iodo-alanine-N,O-
dimethylamide (2.68 g, 7.5 mmol) (J. Org. Chem. 1992, 57,
3397-3404) in dry benzene (30 mL), and dry N,N-
dimethylacetamide (2.0 mL) was added to a dry nitrogen-purged
round bottom flask charged with zinc-copper couple (0.90 g).
The resulting mixture was sonicated under nitrogen until no
starting material remained (as judged by TLC). Bis(tri-o-
tolylphosphine)palladium dichloride (0.35 g, 0.40 mmol) was
added followed by 3-iodobenzonitrile (1.72 g, 7.5 mmol). The
resulting mixture was stirred under a nitrogen atmosphere with
heating, allowed to cool, ethyl acetate (100 mL) was added,
and the mixture filtered into a separatory ~unnel. Sequential
washing with aqueous HCl (50 mL; 0.1N), distilled H20 (3 x 50
mL), drying over Na2SO4, filtration, and concentration under
reduced pressure yielded the crude product. Flash
chromatography over silica gel (light petroleum-ethyl acetate
gradient) afforded the purified compound.
34
CA 02218816 1997-ll-10
WO 96/37497 PCT/CA96/00318
O O
N~ 3 1. Zn/Cu couple; ~ .sol.lld Boc~ ~ ~OCH3
CH3 2. lo-CH3C6H~)3Pl~PdCIz ~ ~' CH3
2-iodobenzonitrile Ir ~r
~CN
solution of tert-butyloxycarbonyl-iodo-alanine-N,O-
dimethylamide (2.68 g, 7.5 mmol) (J. Org. Chem. 1992, 57,
3397-3404) in dry benzene (30 mL), and dry N,N-
dimethylacetamide (2.0 mL) was added to a dry nitrogen-purged
round bottom flask charged with zinc-copper couple (0.90 g).
The resulting mixture was sonicated under nitrogen until no
starting material remained (as judged by TLC). Bis(tri-o-
tolylphosphine)palladium dichloride (0.35 g, 0.40 mmol) was
added followed by 2-iodobenzonitrile (1.72 g, 7.5 mmol). The
resulting mixture was stirred under a nitrogen atmosphere with
heating, allowed to cool, ethyl acetate (100 mL) was added,
and the mixture filtered into a separatory funnel. Sequential
washing with aqueous HCl (50 mL; 0.1N), distilled H20 (3 x 50
mL), drying over Na2SO4, filtration, and concentration under
reduced pressure yielded the crude product. Flash
chromatography over silica gel (light petroleum-ethyl acetate
gradient) afforded the purified compound.
N'~l~ ~OCH3 1. NH20H, DlEA/EtOH N~ ~OCH3
NC~¢~ CH3 2 11~, I'd/C/EtOE~:llOAc CH,
NH
CA 022l88l6 lss7-ll-lo
W096l37~97 PCTtCA96/00318
To a solution of tert-butyloxycarbonyl-para-cyano-
phenylalanine-N,O-dimethylamide ~1.33 g, 4.0 mmol) in dry
ethanol (20 mL) was added hydroxylamine hydrochloride (0.416
g, 6.0 mmol), and diisopropylethylamine (1.02 mL, 6.0 mmol).
The mixture was refluxed and then cooled. The precipitate was
filtered, washed with cold ethanol, diisopropylether, dried
with MgSO4, concentrated under reduced pressure, and used
directly in the next step. The semi-solid was suspended in a
mixture of acetic acid (20 mL), and dry ethanol (40 mL) with
0 warming. Subsequently, Pd/C catalyst (0.30 g, 10~ Pd) was
added, and hydrogen was bubbled through the mixture with
warming. The hydrogenation was continued until no starting
material could be detected as judged by TLC. The catalyst was
removed by filtration, the solution was concentrated under
reduced pressure (50 mL), HCl (50 mL, 1 N) was added, and the
mixture was concentrated once again to 50 mL. The solution
was chilled overnight yielding the title compound.
O O
~N~ ~OCH3 1. NH2OH,DIE~EtOH Boc~ ~ ~OCH3
CH3 2. H2 I d/C/EtOH:HOAc S~ CH3
NC
H2N NH
To a solution of tert-butyloxycarbonyl-meta-cyano-
phenylalanine-N,O-dimethylamide (1.33 g, 4.0 mmol) in dry
ethanol (20 mL) was added hydroxylamine hydrochloride (0.416
g, 6.0 mmol), and diisopropylethylamine (1.02 mL, 6.0 mmol).
The mixture was refluxed and then cooled. The precipitate was
36
CA 02218816 1997-ll-lo
W096/37497 PCTICA96/00318
~iltered, washed with cold ethanol, diisopropylether, drle~
with MgSO4, concentrated under reduced pressure, and used
directly in the next step. The semi-solid was suspended in a
mixture of acetic acid (20 mL), and dry ethanol (40 mL) with
warming. Subsequently, Pd/C catalyst (0.30 g, 10~ Pd) was
added, and hydrogen was bubbled through the mixture with
warming. The hydrogenation was continued until no starting
material could be detected as judged by TLC. The catalyst was
removed by filtration, the solution was concentrated under
o reduced pressure (50 mL), HCl (50 mL, 1 N) was added, and the
mixture was concentrated once again to 50 mL. The solution
was chilled overnight yielding the title compound.
o o
H ,1~ ~OCH3 I NHzOH~ DlEA/EtOH Boc~ ~ ~OCH3
¢~CN 2. H2, Pd/C/EtOH:HOAc NH2
To a solution of tert-butyloxycarbonyl-ortho-cyano-
phenylalanine-N,O-dimethylamide (1.33 g, 4.0 mmol) in dry
ethanol (20 mL) was added hydroxylamine hydrochloride (0.~16
g, 6.0 mmol), and diisopropylethylamine (1.02 mL, 6.0 mmol).
The mixture was re~luxed and then cooled. The precipitate was
filtered, washed with cold ethanol, diisopropylether, dried
with MgSO4, concentrated under reduced pressure, and used
directly in the next step. The semi-solid was suspended in a
mixture of acetic acid (20 mL), and dry ethanol (40 mL) with
warming. Subsequently, Pd/C catalyst (0.30 g, 10~ Pd) was
added, and hydrogen was bubbled through the mixture with
37
CA 022l88l6 lss7-ll-lo
W096/37497 PCT/CA96/00318
warming. The hydrogenation was continued until no starting
material could be detected as judged by TLC. The catalyst was
removed by filtration, the solution was concentrated under
reduced pressure (50 mL), HCl (50 mL, 1 N) was added, and the
mixture was concentrated once again to 50 mL. The solution
was chilled overnight yielding the title compound.
CH ~ Lith iu m th iA~ole/THF 130c~ ~J~N,~
H2N ~J~ H2N~/
NH NH
To a solution of thiazole (1.28 g, 15.0 mmol) in anhydrous THF
(30 mL) was added n-BuLi (1.6 M/hexane, 8.9 mL, 13.9 mmol)
dropwise at -78~ C, and the solution stirred. tert-
Butyloxycarbonyl-para-amidino-phenylalanine-N,O-dimethylamide
(1.15 g, 3.3 mmol) in THF (15 mL) was then added dropwise, and
the resulting mixture stirred. The reaction was quenched with
saturated aqueous ammonium chloride. The mixture was diluted
with ethyl acetate (150 mL), and the organic layer washed with
saturated aqueous ammonium chloride ~2 x 50 mL), brine (50
mL), dried with MgSO4, filtered, and concentrated under reduced
pressure. The crude material was purified on silica gel
(ethyl acetate/hexane), and concentrated under reduced
pressure.
CA 022l88l6 lss7-ll-lo
W096/37497 PCT/CA96/00318
Bcc ~ IN Llthiumthiazole~HF Boc~
~ , CH, ~ N
HN NH2 HN~NH2
To a solution of thiazole (1.28 g, 15.0 mmol) in anhydrous THF
(30 mL) was added n-BuLi (1.6 M/hexane, 8.9 mL, 13.9 mmol)
dropwise at -78~ C, and the solution stirred. tert-
Butyloxycarbonyl-meta-amidino-phenylalanine-N,O-dimethylamide
(1.15 g, 3.3 mmol) in THF (15 mL) was then added dropwise and
the resulting mixture stirred. The reaction was quenched with
saturated a~ueous ammonium chloride. The mixture was diluted
with ethyl acetate (150 mL), and the organic layer washed with
0 saturated aqueous ammonium chloride (2 x 50 mL), brine (50
mL), dried with MgSO4, filtered, and concentrated under reduced
pressure. The crude material was purified on silica gel
(ethyl acetate/hexane), and concentrated under reduced
pressure.
O O
~OCH3 Lithium thiazole/THF ~ Boc~ ~_~
¢~NH3 ¢~NH~
To a solution of thiazole (1.28 g, 15.0 mmol) in anhydrous THF
=
CA 02218816 19s7-ll-lo
W096/37497 PCT/CA96/00318
(30 mL) was added n-BuLi (1.6 M/hexane, 8 9 mL, 13.9 mmol
dropwise at -78~ C, and the solution stirred. tert-
Butyloxycarbonyl-ortho-amidino-phenylalanine-N,O-dimethylamide
(1.15 g, 3.3 mmol) in THF (15 mL) was then added dropwise, and
the resulting mixture stirred. The reaction was quenched with
saturated aqueous ammonium chloride. The mixture was diluted
with ethyl acetate (150 mL) and the organic layer washed with
saturated aqueous ammonium chloride (2 x 50 mL), brine (50
mL), dried with MgSO4, filtered, and concentrated under reduced
pressure. The crude material was purified on silica gel
(ethyl acetate/hexane), and concentrated under reduced
pressure.
Boc ~OCH3 1. H2, RaNi/EtOH:NH3 H~y~ H3
~ CH3 ~e OCH3
tert-Butyloxycarbonyl-para-cyano-phenylalanine-N,O-
dimethylamide (1.33 g, 4.0 mmol) was dissolved in ethanol
saturated with ammonia (30 mL), and sponge Raney Ni (100 mg)
added. The solution was shaken under H2 at room temperature
(40 psi). The solution was ~iltered through celite, and
concentrated under reduced pressure to yield a clear residue.
The residue was dissolved in ethyl acetate (250 mL), and
washed with 1 N NaOH (2 x 50 mL), and brine (2 x 50 mL). The
solution was dried with MgSO4, ~iltered, and concentrated under
reduced pressure.
CA 02218816 1997-11-lo
W096137497 PCT/CA96/00~18
O O
Boc~ ~ ~OCH3 1. H2,RaN~EtOH:NH3 H~ I ~CH3
~- CH3 ~ OCH3
H2N
tert-Butyloxycarbonyl-meta-cyano-phenylalanine-N,O-
dimethylamide (1.33 g, 4.0 mmol) was dissolved in ethanol
saturated with ammonia (30 mL), and sponge Raney Ni (100 mg)
added. The solution was shaken under H2 at room temperature
(40 psi). The solution was ~iltered through celite, and
concentrated under reduced pressure to yield a clear residue.
The residue was dissolved in ethyl acetate (250 mL), and
washed with 1 N NaOH (2 x 50 mL), and brine (2 x 50 mL). The
solution was dried with MgSO4, filtered, and concentrated under
reduced pressure.
O o
Boc~N~J~ ~OCH3 1. H2 RaNi/EtOH:NH3 H J~ IN~cH3
~-- CH ~ OCH3
tert-Butyloxycarbonyl-ortho-cyano-phenylalanine-N,O-
dimethylamide (1.33 g, 4.0 mmol) was dissolved in ethanol
saturated with ammonia (30 mL), and sponge Raney Ni (100 mg)
added. The solution was shaken under H2 at room temperature
(40 psi). The solution was filtered through celite, and
concentrated under reduced pressure to yield a clear residue.
CA 02218816 1997-11-lo
W096/37497 PCT/CA96/00318
The residue was dissolved in ethyl acetate (250 mL), and
washed with 1 N NaOH (2 x 50 mL), and brine (2 x 50 mL). The
solution was dried with MgSO4, ~iltered, and concentrated under
reduced pressure.
¦ H3CSJ~NHZ Boc ~ IN
OCH3 HgCI2/THF ~Ei OCH3
H2N~J~ ~
NZ
tert-Butyloxycarbonyl-para-aminomethyl-phenylalanine-N,O-
dimethylamide (1.00 g, 3.1 mmol) was dissolved in dry THF (10
mL) under nitrogen with stirring. The solution was cooled,
N,N'-bis-(benzyloxycarbonyl)-S-methyl-isothiourea (1.14 g, 3.2
mmol), and HgCl2 (0.95 g, 3.5 mmol) added. The solution was
concentrated under reduced pressure, the re~ining residue was
suspended in ethyl acetate (200 mL), and filtered through
celite. The filtrate was concentrated under reduced pressure.
Flash chromatography over silica gel (hexane/ethyl acetate
gradient) afforded the purified compound.
CA 022l88l6 lss7-ll-lo
W096/37497 PCTlCA96/00318
~N~J~ ~Ch, H~CSJ~NHZ Eioc~ ~N~CH,
ç~ OCH3 HgCI2/THF 13~ OCH3
NH2 HN
ZHN~NZ
tert-Butyloxycarbonyl-meta-aminomethyl-phenylalanine-N,0-
dimethylamide (1.00 g, 3.1 mmol) was dissolved in dry THF (10
mL) under nitrogen with stirring. The solution was cooled,
N,N'-bis-(benzyloxycarbonyl)-S-methyl-isothiourea (1.14 g, 3.2
mmol), and HgCl2 (0.95 g, 3.5 mmol) added. The solution was
concentrated under reduced pressure, the r~m~;n'ng residue was
suspended in ethyl acetate (200 mL), and filtered through
celite. The filtrate was concentrated under reduced pressure.
o Flash chromatography over silica gel (hexane/ethyl acetate
gradient) afforded the purified compound.
H3CSJ~NHZ ~ l
OCH3 HgCI2/THF ~= OCH3
I.~NH2 ~NH~NHZ
ZN
tert-Butyloxycarbonyl-ortho-aminomethyl-phenylalanine-N,O-
dimethylamide (1.00 g, 3.1 mmol) was dissolved in dry THF (10
43
CA 022188l6 lss7-ll-lo
W096/37497 PCT/CA96/00318
mL) under nitrogen with stirring. The solution was cooled,
N,N'-bis-(benzyloxycarbonyl)-S-methyl-isothiourea (1.14 g, 3.2
mmol), and HgCl2 (0.95 g, 3.5 mmol) added. The solution was
concentrated under reduced pressure, the re~A;n;ng residue was
suspended in ethyl acetate (200 mL), and ~iltered through
celite. The filtrate was concentrated under reduced pressure.
Flash chromatography over silica gel (hexane/ethyl acetate
gradient) afforded the purified compound.
~ 1. I,ilhi~mthiazol~/TllF ~~
HN~NHZ NZ
NZ
lo To a solution o~ thiazole (1.28 g, 15.0 mmol) in anhydrous THF
(30 mL) was added n-BuLi (1.6 M/hexane, 8.9 mL, 13.9 mmol)
dropwise at -78~ C, and the solution stirred. The protected
amino acid (1.36 g, 3.3 mmol) in THF (15 mL) was then added
dropwise, and the resulting mixture stirred. The reaction was
quenched with saturated aqueous ammonium chloride. The
mixture was diluted with ethyl acetate (150 mL), and the
organic layer washed with saturated aqueous ammonium chloride
(2 x 50 mL), brine (50 mL), dried with MgSO4, ~iltered, and
concentrated under reduced pressure. The crude material was
purified on silica gel (ethyl acetate/hexane), and
concentrated under reduced pressure.
44
CA 022188l6 lss7-ll-lo
W096/37497 PCT/CA96/00318
o o
~CH, 1. Lithiumthi~/olerrElF
NH HN
ZN~NHZ ZHN~NZ
To a solution of thiazole (1.28 g, 15.0 mmol) in anhydrous THF
(30 mL) was added n-BuLi (1.6 M/hexane, 8.9 mL, 13.9 mmol)
dropwise at -78~ C, and the solution stirred. The protected
amino acid (1.36 g, 3.3 mmol) in THF (15 mL) was then added
dropwise, and the resulting mixture stirred. The reacti~n was
quenched with saturated aqueous ammonium chloride. The
mixture was diluted with ethyl acetate (150 mL), and the
organic layer washed with saturated aqueous ammonium chloride
(2 x 50 mL), brine (50 mL), dried with MgSO~, filtered, and
concentrated under reduced pressure. The crude material was
purified on silica gel (ethyl acetate/hexane), and
concentrated under reduced pressure.
o o
Boc ~ Nl 1. Lithium thiazole/THF ~N~
OCH ~N~l~NHZ
ZN ZN
CA 022l88l6 lss7-ll-lo
W096t37497 PCT/CA96/00318
To a solution o~ thiazole (1.28 g, 15.0 mmol) in anhydrous THF
(30 mL) was added n-BuLi (1.6 M/hexane, 8.9 mL, 13.9 mmol)
dropwise at -78~ C, and the solution stirred. The protected
amino acid (1.36 g, 3.3 mmol) in THF (15 mL) was then added
dropwise, and the resulting mixture stirred. The reaction was
quenched with saturated aqueous ammonium chloride. The
mixture was diluted with ethyl acetate (150 mL), and the
organic layer washed with saturated aqueous ammonium chloride
(2 x 50 mL), brine (50 mL), dried with magnesium sul~ate,
filtered, and concentrated under reduced pressure. The crude
material was purified on silica gel (ethyl acetate/hexane),
and concentrated under reduced pressure.
o o
~N~D~ ~OCH3 1. Zn/Cu couple; ull. s~ ~ ~NJ~ ~OCH3
2. Io-CH3C6H4)3PI2PdCI2 ~ ~ CH3
2-amino-5-~ b -T
H2N~N
A solution of tert-butyloxycarbonyl-iodo-alanine-N,O-
dimethylamide (2.68 g, 7.5 mmol) (J. Org. Chem. 1992, 57,
3397-3404) in dry benzene (30 mL), and dry N,N-
dimethylacetamide (2.0 mL) was added to a dry nitrogen-purged
round bottom flask charged with zinc-copper couple (0.90 g).
The resulting mixture was sonicated under nitrogen until no
starting material remained (as judged by TLC).. Bis(tri-o-
tolylphosphine)palladium dichloride (0.35 g, 0.40 mmol) was
added followed by 2-iodobenzonitrile (1.72 g, 7.5 mmol). The
resulting mixture was stirred under a nitrogen atmosphere with
heating, allowed to cool, ethyl acetate (100 mL) was added,
and the mixture filtered into a separatory funnel. Sequentlal
46
CA 022188l6 lss7-ll-lo
W096/37497 PCTICA96/00318
washing with aqueous HCl (50 mL; O.lN), distilled H2O (3 X 50
mL), drying over Na2SO4, filtration, and concentration under
reduced pressure yielded the crude product. Flash
chromatography over silica gel (light petroleum/ethyl acetate
gradient) afforded the purified compound.
o o
NJI~ ~OCH3 1. Lithium thiazole/THF Boc~ ~N
H2N ~ H2NJ~
To a solution of thiazole (1.28 g, 15 . O mmol) in anhydrous THF
(30 mL) was added n-BuLi (1.6 M/hexane, 8.9 mL, 13.9 mmol)
dropwise at -78~ C, and the solution stirred. The amino acid-
lo N,O-dimethylamide (1.07 g, 3.3 mmol) in anhydrous THF (15 mL)
was then added dropwise and the resulting mixture stirred.
The reaction was quenched with saturated aqueous ammonium
chloride. The mixture was diluted with ethyl acetate (150
mL), and the organic layer washed with saturated aqueous
ammonium chloride (2 x 50 mL), brine (50 mL), dried with MgSO4,
filtered, and concentrated under reduced pressure. The crude
material was purified on silica gel (ethyl acetate/hexane),
and concentrated under reduced pressure.
B ~N~l~ ~OCH3 1. ZnlCu couple; ulll s~ ~ NH~J~ ~OCH3
CH3 2. Io-cH3c6H4)3pl2pdcl2 ~ CH3
2-cyano-~
NC~N~
47
CA 02218816 1997-11-lo
W096/37497 PCT/CA96/00318
A solution o~ tert-butyloxycarbonyl-iodo-alanine-N,O-
dimethylamide (2.68 g, 7.5 mmol) (J. Org. Chem. 1992, 57,
3397-3404) in dry benzene (30 mL), and dry N,N-
dimethylacetamide (2.0 mL) was added to a dry nitrogen-purged
round bottom flask charged with zinc-copper couple (0.90 g).
The resulting mixture was sonicated under nitrogen until no
starting material remained (as judged by TLC). Bis(tri-o-
tolylphosphine)palladium dichloride (0.35 g, 0.40 mmol) was
added followed by 2-iodobenzonitrile (1.72 g, 7.5 mmol). The
resulting mixture was stirred under a nitrogen atmosphere with
heating, allowed to cool, ethyl acetate (100 mL) was added,
and the mixture filtered into a separatory ~unnel. Sequential
washing with aqueous HCl (50 mL; 0.1N), distilled H2O (3 x 50
mL), drying over Na2SO4, filtration, and concentration under
reduced pressure yielded the crude product. Flash
chromatography over silica gel (light petroleum/ethyl acetate
gradient) afforded the purified compound.
o o
~N~JI~ ~OCH3 1. I~H20H, DlEAJEtOH Boc~ ~ ~OCH3
NC~-- CH, 2.El~,l'd/C/E:tOll:llOAc b~ CH,
NH
To a solution of tert-butyloxycarbonyl-(4-cyano)3-
pyridylalanine-N,O-dimethylamide (1.34 g, 4.0 mmol) in dry
ethanol (20 mL) was added N,O-hydroxlyamine hydrochloride
(0.416 g, 6.0 mmol), and diisopropylethylamine (1.02 mL, 6.0
mmol). The mixture was refluxed and then cooled. The
precipitate was ~iltered, washed with cold ethanol,
48
CA 022l88l6 lss7-ll-lo
W096/37497 PCT/CA96/00318
diisopropylether, dried with MgSO4, concentrated under reduced
pressure, and used directly in the next step. The semi-solid
was suspended in a mixture of acetic acid (20 mL), and dry
ethanol (40 mL) with warming. Subsequently, Pd/C catalyst
(0.30 g, 10~ Pd) was added, and hydrogen was bubbled through
the mixture with warming. The hydrogenation was continued
until no starting material could be detected as judged by TLC.
The catalyst was removed by filtration, and the solution was
concentrated under reduced pressure (50 mL), HCl (50 mL, 1 N)
was added, and the mixture was concentrated once again to 50
mL. The solution was chilled overnight yielding the title
compound.
o o
~N~ IN~OcH3 1. Lithium thiazole/THF Boc~ ~r4
~¢~ H2N ~,~ N
NH NH
To a solution of thiazole (1.28 g, 15.0 mmol) in anhydrous THF
(30 mL) was added n-BuLi (1.6 M/hexane, 8.9 mL, 13.9 mmol)
dropwise at -78~ C, and the solution stirred. The amino acid-
N,O-dimethylamide (1.16 g, 3.3 mmol) in anhydrous THF (15 mL)
was then added dropwise, and the resulting mixture stirred.
The reaction was quenched with saturated aqueous ammonium
chloride. The mixture was diluted with ethyl acetate (150
mL), and the organic layer washed with saturated aqueous
- ammonium chloride (2 x 50 mL), brine (50 mL), dried with MgSO4,
filtered, and concentrated under reduced pressure. The crude
material was purified on silica gel ethyl acetate/hexane), and
49
CA 022188l6 lss7-ll-lo
W096/37497 PCTICA96/00318
concentrated under reduced pressure.
HN~J~N~ 3 1. H2, PtO2/ACOH N~ ~CH3
N~ OCH3 HN~= 1CH3
tert-Butyloxycarbonyl-3-(4-pyridyl)alanine-N,O-dimethylamide
(4.50 g, 14.4 mmol) was dissolved in acetic acid (100 mL), and
PtO2 (100 mg) added. The solution was shaken under H2 until
gas uptake ceased. The solution was filtered through celite,
and concentrated under reduced pressure yielding tert-
butyloxycarbonyl-3-(4-piperidyl)alanine-N,O-dimethylamide.
0 The residue was dissolved in ethyl acetate (250 mh), washed
with 1 N NaOH (2 x 50 mL), brine (2 x 50 mL), dried with MgSO4,
filtered, and concentrated under reduced pressure to yield the
title compound.
o o
Boc~ ~ ~CH3 1. H2, PtO~/AcOH HN~JI~ ~CH3
OCH3 Q~ OCH3
tert-Butyloxycarbonyl-3-(3-pyridyl)alanine-N,O-dimethylamide
(4.50 g, 14.4 mmol) was dissolved in acetic acid (100 mL), and
PtO2 (100 mg) added. The solution was shaken under H2 until
gas uptake ceased. The solution was filtered through celite,
CA 02218816 1997-11-lo
W096/37497 PCT/CA96/00318
and concentrated under reduced pressure yielding tert-
butyloxycarbonyl-3-(3-piperidyl)alanine-N,O-dimethylamide.
The resiclue was dissolved in ethyl acetate (250 mL), washed
with 1 N NaOH (2 x 50 mL), brine (2 x 50 mL), dried with MgSO4,
~iltered, and concentrated under reduced pressure to yield the
title compound.
o o
Boc , ~CH3 1. H2, PtO2/AcOH Boc~ J~ ~CH3
OCH3 Cl~-- OCH3
N NH
tert-Butyloxycarbonyl-3-(2-pyridyl)alanine-N,O-dimethylamide
(4.50 g, 14.4 mmol) was dissolved in acetic acid (100 mL), and
PtO2 (100 mg) added. The solution was shaken under H2 until
gas uptake ceased. The solution was ~iltered through celite,
and concentrated under reduced pressure yielding tert-
butyloxycarbonyl-3-(2-piperidyl)alanine-N,O-dimethylamide.
The residue was dissolved in ethyl acetate (250 mL), washed
with 1 N NaOH (2 x 50 mL), brine (2 x 50 mL), dried with MgSO4,
filtered, and concentrated under reduced pressure to yield the
title conpound.
CA 022l88l6 lss7-ll-lo
W096/37497 PCT/CA96/00318
H3CSJ~NHZ ~N~J~N~ H3
OCH3HgCI2rrHF ~ 1CH3
HN~I ZHN~N
NZ
tert-Butyloxycarbonyl-3-(4-piperidyl)alanine-N,O-dimethylamide
(1.00 g, 3.2 mmol) was dissolved in dry THF (10 mL) under
nitrogen with stirring. The solution was cooled, N,N'-bis-
(benzyloxycarbonyl)-S-methyl-isothiourea (1.14 g, 3.2 mmol),
and HgCl2 (0.95 g, 3.5 mmol) added. The solution was
concentrated under reduced pressure, the remaining residue was
suspended in ethyl acetate (200 mL), and filtered through
celite. The filtrate was concentrated under reduced pressure.
lo Flash chromatography over silica gel (hexane/ethyl acetate
gradient) afforded the title compound.
~JI~N~ 3 H3CSJ~NHZ ~N~J~N~ 3
OCH3 HgCI2rrHF ~ 1CH3
ZN~NHZ
tert-Butyloxycarbonyl-3-(3-piperidyl)alanine-N,O-dimethylamide
(1.00 g, 3.2 mmol) was dissolved in dry THF (10 mL) under
nitrogen with stirring. The solution was cooled, N,N'-bis-
CA 022l88l6 lss7-ll-lo
W096/37497 PCT/CA96/00318
(benzyloxycarbonyl)-S-methyl-isothiourea (1.14 g, 3.2 mmol),
and HgCl2 (0.95 g, 3.5 mmol) added. The solution was
concentrated under reduced pressure, the r~m~n~ng residue was
suspended in ethyl acetate (200 mL), and filtered through
celite. The ~iltrate was concentrated under reduced pressure.
Flash chromatography over silica gel (hexane/ethyl acetate
gradient) afforded the title compound.
N~ 3 H3CSJ~NHZ N~J~N~ 3
Cl' OCH3 HgCI2/THF ~-- OCH3
NH ~N~NHZ
lo tert-Butyloxycarbonyl-3-(2-piperidyl)alanine-N,0-dimethylamide
(l.oo g, 3.2 mmol) was dissolved in dry THF (10 mL) under
nitrogen with stirring. The solution was cooled, N,N'-bis-
(benzyloxycarbonyl)-S-methyl-isothiourea (1.14 g, 3.2 mmol),
and HgCl2 (0.95 g, 3.5 mmol) added. The solution was
concentrated under reduced pressure, the rem~;n'ng residue was
suspended in ethyl acetate (200 mL), and filtered through
celite. The filtrate was concentrated under reduced pressure.
Flash chromatography over silica gel (hexane/ethyl acetate
gradient) af~orded the title compound.
,
CA 02218816 1997-11-10
WO 96/37497 PCT/CA96/00318
~y 1. Lithium thiazole/THF N~
1--~ OCH3 ~ N
ZHN~N~J ZHN~N~J
NZ NZ
To a solution of thiazole in anhydrous THF (1.23 g, 14.4 mmol)
was added n-BuLi (1.6 M/hexane, 8.4 mL, 13.4 mmol) dropwise at
-78~ C and the solution stirred. The guanidylated 4-
piperidylalanine derivative (2.00 g, 3.2 mmol) in anhydrous
THF (15 mL) was added dropwise, and the resulting mixture
stirred. The reaction was quenched with saturated aqueous
ammonium chloride. The mixture was diluted with ethyl acetate
(150 mL), and the organic layer washed with saturated aqueous
ammonium chloride (2 x 50 mL), brine (50 mL), dried with MgSO4,
filtered, and concentrated under reduced pressure.
~y 1. Lith;um thi:l~ole/TB F Boc~ ~N
~ ~=
~N ~N
ZN~NHZ ZN~NHZ
To a solution of thiazole in anhydrous THF (1.23 g, 14.4 mmol)
was added n-BuLi (1.6 M/hexane, 8.4 mL, 13.4 mmol) dropwise at
-78~ C with stirring. The mixture was stirred at -78~ C for 1
h. The guanidylated 3-piperidylalanine derivative (2.00 g,
54
CA 022l88l6 lss7-ll-lo
wos6/374s7 PCT/CA96/00318
3.2 mmol) in THF (15 mL) was added dropwise, and the resulting
mixture stirred. The reaction was quenched with saturated
aqueous ammonium chloride. The mixture was diluted with ethyl
acetate (150 mL), and the organic layer washed with saturated
aqueous ammonium chloride (2 x 50 mL), brine (50 mL), dried
with MgSO4, ~iltered, and concentrated under reduced pressure.
o o
1. Lithium thiazole/TH~ Boc~ ~4
C~' OCH3 Cl' N
N~NHZ N~NHZ
ZN ZN
To a solution of thiazole in anhydrous THF (1.23 g, 14.4 mmol)
lo was added n-BuLi (1.6 M/hexane, 8.4 mL, 13.4 mmol) dropwise at
-78~ C with stirring. The mixture was stirred at -78~ C for 1
h. The guanidylated 2-piperidylalanine derivative (2.00 g,
3.2 mmol) in THF (15 mL) was added dropwise, and the resulting
mixture stirred. The reaction was quenched with saturated
aqueous ammonium chloride. The mixture was diluted with ethyl
acetate (150 mL), and the organic layer washed with saturated
aqueous ammonium chloride (2 x 50 mL), brine (50 mL), dried
with MgSO4, ~iltered, and concentrated under reduced pressure.
CA 022188l6 lss7-ll-lo
W096/37497 PCT/CA96/00318
o o
HN~JI~IN~CH3 1. H2,PtO2/AcOH~N~JI~Nl~cH3
o2NJ3'"= OCH3 H2NJ~ OCH3
tert-Butyloxycarbonyl-para-nitro-phenylalanine-N,O-
dimethylamide (13.88 g, 39.3 mmol) was dissolved in acetic
acid (100 mL), and PtO2 (100 mg) added. The solution was
shaken under H2 until gas uptake ceased. The solution was
filtered through celite, concentrated under reduced pressure,
taken up in H2O (150 mL), and lyophilized. The semi-solid was
dissolved in ethyl acetate (350 mL), washed with 1 N NaOH (3 x
50 mL), and brine (3 x 50 mL). The solution was dried with
MgSO4, filtered, and concentrated under reduced pressure
yielding the title compound.
Boc~ ~ ~CH3 1. H2, PtO2/AcOH ~ l
-- OCH~ ~ OCH,
02N H2
tert-Butyloxycarbonyl-meta-nitro-phenylalanine-N,O-
dimethylamide (13.88 g, 39.3 mmol) was dissolved in acetic
acid (100 mL), and PtO2 (100 mg) added. The solution was
shaken under H2 until gas uptake ceased. The solution was
~iltered through celite, concentrated under reduced pressure,
taken up in H2O (150 mL), and lyophilized. The semi-solid was
56
CA 02218816 1997-11-10
WO 96/37497 PCT/CA96/00318
dissolved in ethyl acetate (350 mL), washed with 1 N NaOH (3 x
50 mL), and brine (3 x 50 mL). The solution was dried with
MgSO~, ~iltered, and concentrated under reduced pressure
yielding the title compound.
~CH3 1. H~, PtO2/AcOH ~N~Il~N~ 3
~NO2 ~ H2 OCH3
tert-Butyloxycarbonyl-ortho-nitro-phenylalanine-N,O-
dimethylamide (13.88 g, 39.3 mmol) was dissolved in acetic
acid (100 mL), and PtO2 (100 mg) added. The solution was
o shaken under H2 until gas uptake ceased. The solution was
~iltered through celite, concentrated under reduced pressure,
taken up in H2O (150 mL), and lyophilized. The semi-solid was
dissolved in ethyl acetate (350 mL), washed with 1 N NaOH (3 x
50 mL), and brine (3 x 50 mL). The solution was dried with
MgSO4, filtered, and concentrated under reduced pressure
yielding the title compound.
o . o
HN V~ IN~CH3 l.Z-CI, NaHCO,l/THF:H20 Boc~ ~N
H2N~ OCH3 2. Lithium thia~olerrHF NHZ~
1. tert-Butyloxycarbonyl-3-( cis/ trans-4-
aminocyclohexyl)alanine-N,O-dimethylamide (1.00 g, 3.0 mmol)
20 was dissolved in saturated aqueous sodium bicarbonate, and THF
S7
CA 02218816 1997-ll-lo
W096/37497 PCT/CA96/00318
[60 mL, (1:1)] with stirring. The solution was cooled and a
solution o~ benzyl chloro~ormate (0.43 mL, 3.0 mmol) in THF
(10 mL) was added dropwise. Excess solid sodium bicarbonate
was added, the THF was removed under reduced pressure, and the
remaining aqueous phase was poured into ethyl acetate (250
mL), and mixed thoroughly. The aqueous phase was discarded
and the rem~in~ng solution was washed with saturated aqueous
sodium bicarbonate (2 x 50 mL), 4 N aqueous sodium bisulfate
(2 x 50 mL), and brine (2 x 50 mL). The solution was dried
o with MgSO4, filtered, and concentrated under reduced pressure.
The semi-solid was chromatographed on silica gel (ethyl
acetate/ hexane).
2. To a solution of thia~ole (1.16 g, 13.7 mmol) in anhydrous
THF was added n-BuLi (1.6 M/hexane, 8.0 mL, 12.8 mmol)
dropwise at -78~ C and the solution stirred. The above
protected amino acid amide (1.41 g, 3.0 mmol) in THF (15 mL)
was added dropwise, and the resulting mixture stirred. The
reaction was quenched with saturated aqueous ammonium
chloride. The mixture was diluted with ethyl acetate (150
mL), and the organic layer washed with saturated aqueous
ammonium chloride (2 x 50 mL), brine (50 mL), dried with MgSO4,
filtered, and concentrated under reduced pressure. The crude
material was purified on silica gel (ethyl acetate/hexane),
and concentrated under reduced pressure.
58
CA 02218816 1997-ll-10
WO 96/37497 PCT/CA96/00318
Boc ~ I I. ~cl, N-HCo~ o N~,_
Q~ OCH~ Z.Lithiumthia~ole/lliF Q~ N
H2 ZHN
1 . tert - Butyloxycarbonyl- 3 - ( ci s/ trans - 3 -
aminocyclohexyl)alanine-N,O-dimethylamide (1.00 g, 3.0 mmol)
was dissolved in saturated aqueous sodium bicarbonate, and THF
[60 mL, (1:1)] with stirring. The solution was cooled and a
solution of benzyl chloroformate (0.43 mL, 3.0 mmol) in THF
(10 mL) was added dropwise. Excess solid sodium bicarbonate
was added, the THF was removed under reduced pressure, and the
remaining aqueous phase was poured into ethyl acetate (250
lo mL), and mixed thoroughly. The aqueous phase was discarded
and the r~m~ining solution was washed with saturated aqueous
sodium bicarbonate (2 x 50 mL), 4 N aqueous sodium bisul~ate
(2 x 50 mL), and brine (2 x 50 mL). The solution was dried
with MgSO~, filtered, and concentrated under reduced pressure.
The semi-solid was chromatographed on silica gel (ethyl
acetate/ hexane).
2. To a solution of thiazole (1.16 g, 13.7 mmol) in anhydrous
THF was added n-BuLi (1.6 M/hexane, 8.0 mL, 12.8 mmol)
dropwise at -78~ C and the solution stirred. The above
protected amino acid amide (1.41 g, 3.0 mmol) in THF (15 mL)
was added dropwise, and the resulting mixture stirred. The
reaction was quenched with saturated aqueous ammonium
chloride. The mixture was diluted with ethyl acetate (150
mL), and the organic layer washed with saturated aqueous
59
CA 022l88l6 lgg7-ll-lo
W096/37497 PCT/CA96/00318
ammonium chloride (2 x 50 mL), brine (50 mL), dried with MgSO4,
filtered, and concentrated under reduced pressure. The crude
material was puri~ied on silica gel (ethyl acetate/hexane),
and concentrated under reduced pressure.
I. Z-Cl, NallCO3TNF~ O 130c~ ~N
OCH3 2. Lithium thiazole/THF
~NH2 ~NHZ
1. tert-Butyloxycarbonyl-3-( cis/ trans-2-
aminocyclohexyl)alanine-N,O-dimethylamide (1.00 g, 3.0 mmol)
was dissolved in saturated aqueous sodium bicarbonate, and THF
0 [60 mL, (1:1)] with stirring. The solution was cooled and a
solution of benzyl chloroformate (0.43 mL, 3.0 mmol) in THF
(10 mL) was added dropwise. Excess solid sodium bicarbonate
was added, the THF was removed under reduced pressure, and the
remaining aqueous phase was poured into ethyl acetate (250
mL), and mixed thoroughly. The aqueous phase was discarded
and the r~m~; n; ng solution was washed with saturated aqueous
sodium bicarbonate (2 x 50 mL), 4 N aqueous sodium bisulfate
(2 x 50 mL), and brine (2 x 50 mL). The solution was dried
with MgSO~, filtered, and concentrated under reduced pressure.
The semi-solid was chromatographed on silica gel (ethyl
acetate/ hexane).
2. To a solution of thiazole (1.16 g, 13.7 mmol) in anhydrous
THF was added n-BuLi (1.6 M/hexane, 8.0 mL, 12.8 mmol)
dropwise at -78~ C and the solution stirred. The above
protected amino acid amide (1.41 g, 3.0 mmol) in THF (15 mL)
CA 022l88l6 lss7-ll-lo
W096/37497 PCTICA96/00318
was added dropwise, and the resulting mixture stirred. The
reaction was quenched with saturated aqueous ammonium
chloride. The mixture was diluted with ethyl acetate (150
mL), and the organic layer washed with saturated aqueous
ammonium chloride (2 x 50 mL), brine (50 mL), dried with MgSO4,
~iltered, and concentrated under reduced pressure. The crude
material was purified on silica gel (ethyl acetate/hexane),
and concentrated under reduced pressure.
1. H3CSJ~NHZ ~N ~>
OCH3 HgCk/THF ZN ~ N
J 2. Lithium thiazole/THF J~ ~,L J
H2N~ ZHN H
1. tert-Butyloxycarbonyl-3-( cis/trans-4-
aminocyclohexyl)alanine-N,O-dimethylamide (2.0 g, 6.1 mmol)
was dissolved in dry THF (20 mL) under nitrogen with stirring.
The solution was cooled to 0~ C, N,N'-bis-(benzyloxycarbonyl)-
S-methyl-isothiourea (2.18 g, 6.1 mmol), and HgCl2 (1.81 g, 6.7
mmol) adcled. The solution was concentrated under reduced
pressure, the rem~;n;ng residue was suspended in ethyl acetate
(300 mL), and filtered through celite. The filtrate was
concentrated under reduced pressure. Flash chromatography
over silica gel (hexane/ethyl acetate gradient) afforded the
purified product.
.
2. To a solution of thiazole (2.32 g, 27.3 mmol) in anhydrous
THF was added n-BuLi (1.6 M/hexane, 15.9 mL, 25.4 mmol)
dropwise at -78~ C and the solution stirred. The above
-
CA 022l88l6 lss7-ll-lo
W096/37497 PCT/CA96/00318
guanidylated amino acid (3.88 g, 6.1 mmol) in THF (15 mL) was
added dropwise, and the resulting mixture stirred. The
reaction was quenched with saturated aqueous ammonium
chloride. The mixture was diluted with ethyl acetate (150
mL), and the organic layer washed with saturated aqueous
ammonium chloride (2 x 50 mL), brine (50 mL), dried with MgSO4,
filtered, and concentrated under reduced pressure. The crude
material was purified on silica gel (ethyl acetate/hexane),
and concentrated under reduced pressure.
BOC .~N 1. H3CSJ~NHZ ~N~ ,,S
1CH3 HgCI2rrHF _ N_~
2. Lithium thiazole/THF Q~
H2 ~NZ
ZHN
1. tert-Butyloxycarbonyl-3-( cis/ trans-3-
aminocyclohexyl)alanine-N,O-dimethylamide (2.0 g, 6.1 mmol)
was dissolved in dry THF (20 mL) under nitrogen with stirring.
The solution was cooled to 0~ C, N,N'-bis-(benzyloxycarbonyl)-
S-methyl-isothiourea (2.18 g, 6.1 mmol), and HgCl2 (1.81 g, 6.7
mmol) added. The solution was concentrated under reduced
pressure, the rem~;n;ng residue was suspended in ethyl acetate
(300 mL), and filtered through celite. The filtrate was
concentrated under reduced pressure. Flash chromatography
over silica gel (hexane/ethyl acetate gradient) afforded the
purified product.
CA 022l88l6 lss7-ll-lo
W096/37497 PCT/CA96/00318
2. To a solution of thiazole (2.32 g, 27.3 mmol) in anhydrous
THF was added n-BuLi (1.6 M/hexane, 15.9 mL, 25.4 mmol)
dropwise at -78~ C and the solution stirred. The above
guanidylated amino acid (3.88 g, 6.1 mmol) in THF (15 mL) was
added dropwise, and the resulting mixture stirred. The
reaction was quenched with saturated aqueous ammonium
chloride. The mixture was diluted with ethyl acetate (150
mL), and the organic layer washed with saturated aqueous
lo ammonium chloride (2 x 50 mL), brine (50 mL), dried with MgSO~,
filtered, and concentrated under reduced pressure. The crude
material was purified on silica gel (ethyl acetate/hexane),
and concentrated under reduced pressure.
1. H~C5 NHZ ~N"~ ' N~C~
OCH3 HgCI2/THF ~
U ~N~2 2. Lithium thiazole/THF [~ NH
ZN~NHZ
1. tert-Butyloxycarbonyl-3-( cis/ trans-2-
aminocyclohexyl)alanine-N,O-dimethylamide (2.0 g, 6.1 mmol)
was dissolved in dry THF (20 mL) under nitrogen with stirring.
The solution was cooled to 0~ C, N,N'-bis-(benzyloxycarbonyl)-
S-methyl-isothiourea (2.18 g, 6.1 mmol), and HgCl2 (1.81 g, 6.7
mmol) added. The solution was concentrated under reduced
pressure, the remaining residue was suspended in ethyl acetate
(300 mL), and ~iltered through celite. The ~iltrate was
63
CA 02218816 1997-11-lo
W096/37497 PCT/CA96/00318
concentrated under reduced pressure. Flash chromatography
over silica gel (hexane/ethyl acetate gradient) afforded the
puri~ied product.
2. To a solution of thiazole (2.32 g, 27.3 mmol) in anhydrous
THF was added n-BuLi (1.6 M/hexane, 15.9 mL, 25.4 mmol)
dropwise at -78 C and the solution stirred. The above
guanidylated amino acid (3.88 g, 6.1 mmol) in THF (15 mL) was
added dropwise, and the resulting mixture stirred. The
reaction was quenched with saturated aqueous ammonium
chloride. The mixture was diluted with ethyl acetate (150
mL), and the organic layer washed with saturated aqueous
ammonium chloride (2 x 50 mL), brine (50 mL), dried with MgSO4,
filtered, and concentrated under reduced pressure. The crude
material was purified on silica gel (ethyl acetate/hexane),
and concentrated under reduced pressure.
EXAMPLE 2 SYNTHESIS OF COMPOUND # 6
;l
~ S
h~ N ~> ~
/~ NH
(6) ~'
NH
~!= NH
NH2 ~ 2HBr
ST~P 1 ~
OH OH
(BOC) 2~
CH2C12 ~ R . T -
BnNH BnNBOC
64
CA 022l88l6 lss7-ll-lo
W096/37497 PCT/CA96/00318
(1) (2)
One equivalent of di-tert-butyl dicarbonate (5.56 g; 25.0
mmols) was added to a solution of N-benzylethanolamine (1)
(3.92 g; 26.0 mmols) in CH2c12 (75 ml).The solution was stirred
at room temperature overnight. Evaporation of the solvent
gave the N-Boc protected amine (2) (6.61 g; 100~).
STEP 2
OH O
TPAP, NMO
r ~ H
i MS4A,CH2C12
BnNBOC BnNBOC
(2) (3)
Tetrapropylammonium perruthenate (TPAP)(67 mg; 4.23 mmoles)
was added to a well stirred mixture of the alcohol (2) (608
mg; 4.23 mmols) and powdered 4A molecular sieves (1.5 g) in
dichloromethane (10 ml). After being stirred for 20 minutes,
the mixture was ~iltered through celite~. Evaporation of the
solvent gave a black oil which was then purified on silica gel
(ethyl acetate(EtOAc) 30~; Hexanes 70~) to give the aldehyde
(3) (410 mg; 68~) as a colorless oil.
STEP 3
Bn ~ N ''--~ S
~ Cysteine ethyl ester - HC1 BOCHN ~
BnNBOC K2CO3/Mgs04~cH2cl2 2Et
~) (4)
-
A mixture of the aldehyde (3) (387 mg; 1.82 mmols), cysteine
ethyl ester hydrochloride (370 mg; 1.99 mmols), potassium
CA 02218816 1997-11-10
W 096/374g7 PCT/CA96/00318
carbonate (1.2 g) and magnesium sulphate (1.2 g) in
dichloromethane (10 ml) was stirred at room temperature for 18
hours. The resulting mixture was then trans~erred into an
aqueous saturated solution of saturated NaHCO3 (30 ml) and
extracted with dichloromethane (3 x 30 ml). The combined
organic layers were dried (MgSO4) and the solvent evaporated to
give an oil which was purified on silica gel (EtOAc 20~,
Hexanes 80~) to give the thioamine (4) 439 mg; 70~) as a
mixture of diastereoisomers.
STF.P 4
Bn-N ~_-S H!
~ . ~ S
HN __ ~ 1) 4.0 M HCl in dioxane BnN
~ ~ N
CO Et 2) triphosgene/base/THF o
2 C02Et
(4) ~)
4.0 M HCl in dioxane (15 ml; 60 mmols) is added to a mixture
of the thioamine (4) (367 mg; 1.07 mmols) and ethylmethyl
sulfide (1 ml). The resulting solution is stirred 3 hours.
Evaporation of the solvent gives the crude deprotected amine
as a mixture of isomers. The crude deprotected amine is
solubilized in THF (15 ml) and diisopropylethylamine (0.7 ml).
Triphosgene (400 mg; 1.34 mmols) is added to the solution and
the resulting mixture is stirred at room temperature
overnight. The mixture is then transferred into aqueous
saturated NaHCO3 (30 ml) and extracted with dichloromethane (3
x 30 ml). The combined organic layers are dried (MgSO4) and
evaporation of the solvents gives an oil that is-purified on
silica gel to give the compound (5).
66
CA 02218816 1997-11-10
WO g6/37497 PCTICA96/00318
STE:I~ 5
1) ~iOH H20 Bn-N/--~ S
s THF / H2 0
0~ ~ ~ ~,1 ",~ 5
(5) j--NH NH
3 ) BBr3 / CH2C12 NH2 2HBr
The isolated urea (5a) is hydrolyzed with one equivalent of
LiOH-H20 in a 1:1 mixture of THF and H20. The mixture is
stirred at room temperature for 1 hour and the resulting
solution is poured into 10~ citric acid and extracted with
dichloromethane to yield the crude carboxylic acid. The crude
carboxylic acid is coupled with benzothiazole keto arginine in
DMF using BOP as the coupling reagent in the presence of
diisopropylethylamine. Extraction with EtOAc gives a solid
that is purified on silica gel to give the protected amide.
The CBZ protecting group is removed with BBr3 in
dichloromethane at room temperature finally gives the bicyclic
benzothiazole keto arginine inhibitors (6).
EXAMPLE 3 Synthesis of compound 1
67
CA 022188l6 lss7-ll-lo
W096/37497 PCT/CA96/00318
~-~ s~
N ~N ~ ~
O ,~5 /J
(7) NH
,,~ NH
H N (2H3r)
The urea of formula (7) is produced according to the same
method as for the bicyclic benzothiazole keto arginine
inhibitors (6) with the difference that the N-
benzylethanolamine (1) is substituted by N-Benzyl-3-amino-
propanol.
EXAMPLE 4 Synthesis of compounds 13a and 13b
OH H~O
" (BOC)20 CH2CI2 J
r 2) NMO, TPAP,
Ph~NH M.S. 4A, CH2CI2 Ph~NBOC
A solution of the amine (5.32 g, 0.032 mol) in CH2Cl2 (100 ml)
was treated with (BOC)2O (7.10 g, 0.038 mol). The solution
was stirred at room temperature for 24 hours to afford after
evaporation of the solvent the crude protected amine (8.25 g,
97~) which was used in the next step without further
purification. To the amine (5.18 g, 0.0195 mol) in CH2Cl2 (100
ml) was added powdered 4A M.S. (12.6 g) and treated with NMO
(4 . 21 g, 0.0359 mol) and TPAP (341 mg, 0.972 mmol). The
mixture was stirred at room temperature for 30 minutes, then
. 68
CA 022l88l6 lss7-ll-lo
W096/37497 PCT/CA96/00318
filtered on a path of celite. Puri~ication on silica gel (20
EtOAc/80~ hex) afforded the aldehyde (2.76 g, 54~).
H ~ o L-cysteine-OEtHCl BnBOCN ~S
/ K2CO3/Mgso4, HN
CH2CI2
BnNBOC ~
O OEt
A mixture of the aldehyde (2.76 g, 0.0105 mols), L-cysteine
ethyl ester ~ HCl (2.91 g, 15.7 mmols), potassium carbonate (9
g) and magnesium sulphate (9 g) in CH2Cl2 (75 ml) were stirred
at room temperature ~or 17 hours. The mixture was poured into
NaHCO3(s) (200 ml) and extracted with CH2Cl2 (3x200 ml). The
combined organic phases were dried (MgSO4). Puri~ication of
the oil on silica gel (20~ EtOAc/80~ hex) af~orded the
thio~m; n~l (3.54 g, 86~).
H : H
BnBOCN~ ' S
> 1) 4.0MHCI/dioxane ~ + ~ S
HN ~ 2) Triphosgene, Na2CO3BnN ~f N BnN ~ N
A B
42% 34%
The protected amine (1.87 g, 0.0047 mol) dissolved in EtSMe (4
ml) was treated with 4.0 M HCl in dioxane (50 mL) and stirred
for 75 minutes. The mixture was poured into a saturated
solution of NaHCO3 (200 ml) and an additional 20 g of NaHCO3
was added. The aqueous mixture was extracted with EtOAc
(3x200 ml) and the combined organic phases were washed
(brine), dried (Na2SO4). Evaporation of the volatiles le~t the
crude deprotected amine (1.35 g, 97~). The crude amine (1.23
g, 4.17 mmols) dissolved in THF (15 ml) and treated
successively with a solution of Na2CO3 (885 mg) in water (15
69
CA 022188l6 lss7-ll-lo
W096/37497 PCT/CA96/00318
ml) ~ollowed by a solution o~ triphosgene (408 mg, 1.37
mmols). The mixture was stirred at room temperature for 19
hours, then poured into water (60 ml) and extracted with CH2Cl2
(4x60 ml). The combined organic phases were washed with HCl
5~ (200 ml), NaHCO3(s) (200 ml), and dried (MgSO4).
Purification on silica gel afforded the urea A (568 mg, 42~)
and the urea B (470 mg, 34~). The stereochemistry at the
angular position is arbitrarily assigned.
~ S LiOH- H20 ~~ S
BnN ~ N ~ THF/H,O BnN ~ N
~ O OEt ~ O OH
A solution of the ester (517 mg, 1.61 mmols) in THF (10 ml)
was treated with LiOH-H2o (74 mg, 1.8 mmols) in water (10 ml).
The solution was stirred at room temperature for 60 minutes
then poured into 5~ HCl (50 ml) and extracted with CH2Cl2 (4x70
ml) and dried (MgSO4). Evaporation of the solvent left the
crude carboxylic acid (418 mg, 89~).
H H
1~ s LiOH- H20 ~ S
BnN ~f N THF/H20 BnN ~N ~
~ O OEt ~ O OH
A solution of the ester (432 mg, 1.35 mmols) in THF (10 ml)
was treated with LioH-H2O (161.6 mg, 3.85 mmols) in H20 (10
ml). The mixture was stirred at room temperature overnight.
The mixture was poured into HC1 5~ (50 ml) and extracted with
CH2Cl2 (4x70 ml) and dried (MgSO4). Evaporation of the solvent
CA 022188l6 lss7-ll-lo
W096/37497 PCT/CA96/00318
le~t a residue that was puri~ied on silica gel (1~ AcOH, 99
AcOEt) to afford the pure carboxylic acid (125 mg, 32~).
~ S I ) Cl H,N ~< ~: BOP: DIEA ~ S ~,_ S
l~nN ~ N ~> I~ BnN ~ N > 13nN ~ N ~>
2) BCI, ~ O~NII~N~ 11 ~ ~
3) HrLC purification
NH ~ NH ~
)~ NH CFlCO2H ~ NH CF,CO2H
NH2 NH2
# 441-143-1 # 441-143-2
A 7.6% (fast monnl ) B 4.7 ~/O (slow monng)
To a solution of the acid (310 mg, 1.06 mmol) in DMF (3 ml)
was added successively DIEA (1 ml), the arginine (277 mg,
0.601 mmol) and a solution of BOP (355 mg, 0.841 mmol) in DMF
(2 ml). The solution was stirred at room temperature for 20
hours, then poured into water (130 ml), extracted with EtOAc
(3x30 ml). The combined organic phases were washed with
citric acid 10~ (90 ml), NaHCO3(s) (90 ml) then brine and
dried (Na2SO4). Purification of the foam afforded the amide
(190 mg, 63~). The amide was dissolved in CH2Cl2 (10 ml) and
treated at -78~C with BCl3 lM (2.8 ml). The solution was
stirred at room temperature for 2 hours and quenched with dry
methanol (3.0 ml) at -78~C. The solution was stirred at room
temperature for one hour, then volatiles were evaporated.
Purification by HPLC afforded the pure compound A (59.8 mg,
fast mov.ing component, one pure isomer) and B (37.4 mg, slow
moving component, one pure isomer). *Stereochemistry
arbitrarily assigned.
In a like manner, the compounds 13c and 3d were prepared.
EXAMPLE S Synthesis of compounds 12a and 12b
71
CA 022188l6 lss7-ll-lo
W096l37497 PCTICA96100318
O ~ H 1 ) H 2 ~ PtO 2, ettlanol [~ N ~~~ OH
HzN--~OH ~ [~3 2) (BOC) 2~ DCM BOC
To a solution of the amine (5.0 g, 66.0 mmols) in absolute
ethanol (25 ml) was added distilled benzaldehyde (9.0 mL g, 68
mmols). The solution was stirred at room temperature for 10
minutes and PtO2. (80 mg) was added. The mixture was
hydrogenated at 60 psi for 8 hrs at room temperature. The
mixture was filtered on celite and volatiles removed. A
portion of the crude oil (5.92 g, 30 6 mmols) was dissolved in
10 DCM and treated with (BOC) 2~ (6.8 g). The solution was
stirred for 15 hrs at room temperature. Volatiles were
removed i~ vacuo and the oil purified on silica gel (EtOAc30%,
hexanes 70%) to afford the protected amine (6.40 g; 71%).
N--~--OH PCC, AcONa ~ N ~O
BOC MS 4 A, DCM . EtOC
To a solution of the alcohol (6.21 g, 21.1 mmols) in DCM (200
ml) was added successively sodium acetate (2.6 g)and powdered
M . S 4 A (7.0 g). The mixture was cooled to 0 ~C then PCC (6.9
g; 32 mmols) was added. The mixture was stirred at O ~C for 45
minutes then at room temperature for 1 hour. The mixture was
filtered through florisil and washed several times with DCM.
Evaporation of the solvent left an oil that was purified on
silica gel (EtOAc20~, hexanes 80%) to afford the aldehyde
(2.85 g; 46%).
H
¦~~ N ~~ L-cyste~e-OEt ~ ~ N ~~ S >
~ BOC tC2C03, M~S04 . OCM ~J 30C CO2Et
CA 022l88l6 lss7-ll-lo
W096/37497 PCTICA96/00318
A mixture of the aldehyde (2.71 g, 9.30 mmols), L-cysteine
ethyl ester ~ HCl (2.91 g, 15.7 mmols), potassium carbonate (9
g) and magnesium sulphate (9 g) in CH2Cl2 (75 ml) were stirred
at room temperature for 17 hours. The mixture was poured into
NaHCO3(s) (200 ml) and extracted with CH2Cl2 (3x200 ml). The
combined organic phases were dried (MgSO4). Purification of
the oil Oll silica gel (20~ EtOAc/80~ hex) afforded the
thio~ml n~l (3.62 g, 92~).
BOC S 1) 40MHCI/dioxane ~ H ~ H
HN 2) T.; Lu ~ ,N~,COI ~N~ ~N~ J
27 % 1 7 %
The protected amine (3.2g) dissolved in EtSMe (4 ml) was
treated with 4.0 M HCl in dioxane (50 mL) and stirred for 75
minutes. The mixture was poured into a saturated solution of
NaHCO3 (200 ml) and an additional 20 g of NaHCO3 was added.
The aqueous mixture was extracted with EtOAc (3x200 ml) and
the combined organic phases were washed (brine), dried
(Na2SO4). Evaporation of the volatilès left the crude
deprotected amine (1.35 g, 97~). The crude amine (1.23 g,
4.17 mmols) dissolved in THF (15 ml) and treated successively
with a solution of Na2CO3 (885 mg) in water (15 ml) followed by
a solution of triphosgene (408 mg, 1.37 mmols). The mixture
was stirred at room temperature for 19 hours, then poured into
water (60 ml) and extracted with CH2Cl2 (4x60 ml). The
combined organic phases were washed with HCl 5~ (200 ml),
NaHCO3(s) (200 ml), and dried (MgSO4). Purification on silica
gel (EtOAC 50~, hexanes 50~) afforded the urea A (745 mg, 27~)
CA 022188l6 lss7-ll-lo
W096/37497 PCT/CA96/00318
and the urea B (457 mg, 17~). The stereochemistry at the
angular position is arbitrarily assigned.
LiOH H 2~
A solution of the ester (698 mg) in THF (10 ml) was treated
with LioH-H2O (74 mg, 1.8 mmols) in water (10 ml). The
solution was stirred at room temperature for 60 minutes then
poured into 5~ HCl (50 ml) and extracted with CH2Cl2 (4x70 ml)
lo and dried (MgSO4). Evaporation of the solvent left the crude
carboxylic acid (614 mg, 96~).
I THF/H2O ~ N ~ N
A solution of the ester (418 mg) in THF (10 ml) was treated
with LiOH-H2O (161.6 mg, 3.85 mmols) in H2O (10 ml). The
mixture was stirred at room temperature overnight. The
mixture was poured into HCl 5~ (50 ml) and extracted with
CH2Cl2 (4x70 ml) and dried (MgSO4). Evaporation of the solvent
2n left a residue that was purified on silica gel (1~ AcOH, 99
AcOEt) to afford the pure carboxylic acid (221 mg, 57
74
CA 02218816 1997-11-10
WO 96t37497 PCTICA96/00318
~ ~ I) cl n,N~ DOI': DIEA \~ +h~ N ~ N ~
2) BCI3 ~ ~ N~ O O N~
3) HPLCpurific~tion
NH
NH CF,CO,I~ ~--NH CF~CO2H
# 551-36-1 # 551-36-2
A ~ 5% (f~n mo~ing) B l o % (dow movinE )
To a solution of the acid (208 mg, 0.647 mmol) in DMF (3 ml)
was added successively DIEA (1 ml), the arginine (277 mg,
0.601 mmol) and a solution of BOP (355 mg, 0.841 mmol) in DMF
(2 ml). The solution was stirred at room temperature for 20
hours, then poured into water (130 ml), extracted with EtOAc
(3x30 ml). The combined organic phases were washed with
citric acid 10~ (90 ml), NaHCO3(s) (90 ml) then brine and
dried (Na2SO4). Purification of the foam afforded the amide
(190 mg, 63%). The amide was dissolved in CH2Cl2 (10 ml) and
treated at -78~C with BCl3 lM (2.8 ml). The solution was
stirred at room temperature for 2 hours and quenched with dry
methanol (3.0 ml) at -78~C. The solution was stirred at room
temperature for one hour, then volatiles were evaporated.
Purification by HPLC afforded the pure compound A (62 mg, fast
moving component, one pure isomer) and B (44.4 mg, slow moving
component, one pure isomer). *Stereochemistry arbitrarily
assigned.
In a like manner, compounds 12c and 12d were prepared.
EXAMPLE 6 Thrombin Affinity
The affinity of inhibitors for thrombin was measured according
to the procedures described in ~DiMaio et al, J. Bio. Chem.,
1990, 265:21698) Inhibition of amidolytic activity of human
CA 022l88l6 lss7-ll-lo
W096/37497 PCT/CA96/00318
thrombin was measured fluorometrically using Tos-Gly-Pro-Arg-AMC
as a fluorogenic substrate in 50 mM Tris-HCl buffer (pH 7.52 at
37~C) containing 0.1 M NaCl and 0.1~ poly(ethylene glycol) 8000
at room temperature, and (Szewczuk et al., Biochemistry, 1992
31:9132).
The hydrolysis of the substrate by thrombin was monitored on a
Varian-Cary 200 oTM spectrophotometer in the fluorescence mode
(~eX = 383 nm, ~em = 455 nm) or on a Hitachi F2000TM
lo fluorescence spectrophotometer (~eX = 383 nm, ~em = 455 nm), and
the fluorescent intensity was calibrated using AMC. The
reaction reached a steady-state within 3 minutes after mixing
thrombin with the substrate and an inhibitor. The steady-state
velocity was then measured for a few minutes. The compounds of
this invention were also pre-incubated with thrombin for 20
minutes at room temperature before adding the substrate. The
steady-state was achieved within 3 min and measured for a few
min. The kinetic data (the steady-state velocity at various
concentrations of the substrate and the inhibitors) of the
competitive inhibition was analyzed using the methods described
by Segel (1975). A non-linear regression program, RNLIN in the
IMSL library (IMSL, 1987), LMDER in MINPACK library (More et
al., 1980) or MicrosoftTM ExcellTM , was used to estimate the
kinetic parameters (Km Vm~ and Ki).
76
CA 02218816 1997-11-10
WO 96137497 PCT/CA96100318
Table 1
Compound Ki ( nM )
6 80
13a 90
13b soo
13c 500
13d looO
12c 460
12d 100
12a 4 0
12b 320