Note: Descriptions are shown in the official language in which they were submitted.
CA 02221248 1997-11-14
W O96/36612 PCTAUS9~'~6
~ TRIS~ENZIM:IDAZOLES USEFUL AS TOPOISOMERASE I INIIIBITQRS
T3ackground of the Invention
DNA topoisomerases are nuclear enzymes that control and modify
the topological states of DNA by catalyzing the concerted breaking and rejoining of
DNA strands. See, for example, D'Arpa et al., Biochim. Biophys. Acta, 989, 163
(19~9). Topoisomerase II enzymes alter the topological state of DNA by means of
a double strand break in the DNA. ~mm~ n topoisomerase II represents an
effective ph~rm~c.ological target for the development of cancer chemotherapeutics.
(A. Y. Chen et al., Annu. Rev. Pharmacol. Toxicol., 34, 191 (1994)). Among the
clinical agents in use which are recognized as topoisomerase II inhibitors are
etoposide (VP-16), teniposide (VM-26), miLu~lLlune, m-AMSA, adriamycin
(doxorubicin), ellipticine and daunomycin.
In comparison to topoisomerase II inhibitors~ there are relatively few
known topoisomerase I inhibitors. Camptothecin represents the most extensively
studied m~mm~ n topoisomerase I inhibitor. See R C. Gallo et al., J. Natl.
Cancer Inst., 46, 789 (1971) and B. C. Giovanella et al., Cancer Res., 51, 3052
(1991). The broad spectrum of potent antineoplastic activity observed for
camptothecin has prompted further efforts to identify other agents which can
effectively poison m~mm~ n topoisomerase I.
It has recently been demonstrated that Hoechst 33342 (1), 2'-(4-
ethoxyphenyl)-5-(4-methyl-1-piperazinyl)-2,5'-bi-lH-benzimidazole, is an inhibitor
of topoisomerase I.
CA 02221248 1997-11-14
W O96/36612 pcTrus96lo6853
S E~3C ~
~ \>~N
1 OC2H,
This agent, which binds to the minor groove of DNA, traps the
reversible cleavable complex derived from DNA and topoisomerase I and produces
a limited number of highly specific single-strand DNA breaks. For example, see
A.Y. Chen et al., Cançer Res., 53, 1332 (1993) and A. Chen et al., PNAS, 90, 8131
(1993). A limit~tinn of Hoechst 33342 as an anticancer agent is the previously
reported observation that it is not effective against tumor cell lines which
overt;~less MDRl. While KB 3-1 cells are known to be quite sensitive to Hoechst
33342, with an ICso of a~plux,lllately 9 nM, this compound is applo~hllately 130-
fold less ~;yLOl~iC to KB V-l cells, which are known to overexpress MDRl.
Recently, several analogs of this bisbenzimidazole have been synthesized, to further
investigate the structure activity relationships associated with their potency as
topoisomerase I inhibitors and the related ~,y~oloxicity. For example, Q. Sun et al.,
Biorg. and Med. Chem. Lett., _, 2871 (1994) disclosed the preparation of bis-
benzimidazoles of formula (2):
C~H3
~-cN--(c~ N
~ OC H
CA 02221248 1997-11-14
W O 96/36612 PCTrUS96/06853
where n is 0, 1, 2, or 3. However, these compounds were found to be about one
order of magnitude less ~;ylo~ ic than Hoechst 33342. Therefore, a continuing
need exists for new compounds that can induce DNA cleavage in the presence of
m~mm~ n topoisomerase I.
Summary of the Invention
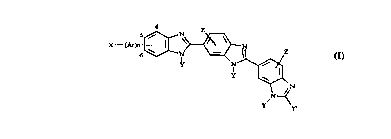
The present invention provides a compound of general formula (I):
X~ N
y' y~
wherein Ar is aryl or a nitrogen-, sulfur- or oxygen-cont~ining heteroaromatic
group; X is H, CN, CHO, OH, acetyl, CF3, O(Cl-C4)alkyl, NO2, NH2, halogen or
halo(CI-C,)alkyl; each Y is individually X (Cl-C4)alkyl or aralkyl; Y' is H or (Cl-
C4) alkyl; each Z is individually H, (Cl-C4)alkyl, halogen or halo(CI-C4)alkyl; and n
is 0 or 1; or a pharmaceutically acceptable salt thereof. Preferably, Ar is a (C6-
Cl2)aryl or a 5- to 12-membered heteroaryl group comprising 1-3 N, S or non-
peroxide O atoms in the ring, wherein each N is unsubstituted or is substituted with
(Cl-C.~)alkyl. As drawn, the Ar-group can occupy any position of the benzo moiety,
i.e., positions 4-7, preferably the 5 position, and X can occupy any available
position on Ar. The Ar-group may be optionally fused to the benzo moiety,
preferably at the 4, 5-, or 5, 6- positions. In one preferred embodiment, Ar is
phenyl, and X is H or is a 4-substituent. According to another preferred
embodiment, Ar is phenyl, and X is Cl or Br, preferably occupying thepara
CA 02221248 1997-11-14
W O96/36612 PCTrUS96/06853
position. As drawn, Z may occupy any position on the benzo moiety. Z is
preferably H, halogen, CH3 or CF3.
According to another embodiment, n is 0, and~X is preferably H,
CN, CHO or halogen, for example, F, Br, Cl or I, preferably Cl or Br, and
preferably occupies the S-position of the benzo moiety. Y is preferably H or CH3.
Y' is preferablv H or CH3.
Compounds of formula (I) are inhibitors of topoisomerase I, as
demonstrated bv their ability to promote DNA cleavage in the presence of
topoisomerase I. Furthermore, colllpuu~.ds of formula (I) also are cytotoxic to
m~mm~ n tumor cells, including camptothecin-sensitive and camptothecin-
resistant tumor cells and tumor cell lines exhibiting multi-drug resistance due to
expression of the P-glycoprotein.
Therefore, the present invention also provides a method for the
inhibition of m~mm~ n tumor cell growth, comprising contacting a susceptible
population of tumor cells with an effective growth-inhibiting arnount of a
compound of formula (I), preferably in combination with a ph~rm~seutically
acceptable carrier. The growth of the tumor cells can be inhibited in vitro, or vivo,
by ~-lmini.~tering the compound of formula (I) to a m~mm~l in need of such
treatment, such as a human cancer patient afflicted with a leukemia or solid tumon
The compounds of formula I can also be used to evaluate the activity of
topoisomerase I obtained from different sources, and are expected to exhibit at least
some of the other bioactivities observed for topoisomerase inhibitors, such as
antibacterial, antifungal, ~Liprotozoal, ~nthelmintic and/or antiviral activity. For
example, compound 14, shown on Figure 1, exhibits antifungal activity.
Brief Description of the Drawings
Figure 1 is a schematic depiction of the synthesis of compounds 10-16.
Figure 2 is a schematic depiction of the preparation of intermediates 4-8
used to prepare compounds of the invention.
CA 02221248 1997-11-14
W O96/36612 PCTrUS96/06853
~ Figure 3 is a schem~tic depiction of the preparation of intermediate 9.
Figure 4 is a schematic depiction of the synthesis of compounds JSKIV-68, -
37 and -47.
Figure 5 is a srhem~tic depiction of the plepala~ion of intermediate JSKIV-
44.
Figure 6 is a schematic depiction of the preparation of analogs modified on
the central benzimid~ole moiety.
Figure 7 is a schematic depiction of the plepal~Lion of analogs modified on
the terminal ben_imid~ole moiety.
Detailed Description of the Invention
The aryl groups (Ar) useful in the present compounds comprise
(C6=CI8) r.yl, p;~ .b!y (C6-Cl4) ~ry!, e.g., systems Gont~inin, aromatiG rings, whir-h
systems comprise a total of 6 to 12 carbon atoms. Thus, as used herein, the term"aryl" includes mono- or bis-(Cl-C4)aLkyl-substituted aryl, such as tolyl and xylyl;
ar(Cl-C4)alkyl, such as benzyl or phenethyl; and alkaralkyl. Preferably aryl is
phenyl, benzyl or naphthyl.
Heteroaromatic rings include aromatic rings c~ nt~ining up to 3 ring
heteroatoms such as N, S or non-peroxide O, and up to 12 ring atoms.
Representative aromatic rings include thiophene, benzothiophene,
n~phth~thiophene, trianthrene, furan, benzofuran, isobenzofuran, pyran, chromene,
xanthene, phenoxathiin, pyrrole, imid~ole, pyr~ole, pyridine, pyr~ine, tri~ole,
tetr~ole, pyr~ine, tri~7ine, pyrirnidine, pyrid~ine, indolizine, isoindole, indole,
ind~ole, purine, quinolizine, isoquinoline, quinoline, phth~l~7.ine, naphthyridine,
quinoxaline, quin~oline, cinnoline, pteridine, carb~ole, carboline, phenanthridine,
acridine, phen~nthroline, phen~ine, isothi~ole, phenothi~ine, ox~ole, isox~ole,
fur~an, phenox~ine and the like. Preferred heteroaromatic rings have a 5- or 6-
membered heteroaromatic ring which may or may not be fused to an aromatic ring
such as a benzo ring, e.g., the preferred 2-, 3- or 4-pyridyl substituents.
CA 02221248 1997-11-14
W O96/36612 PCTrUS9G/C~~~
The term "alkyl" includes straight-chain or branched alkyl, as well as
cycloalkyl and (cyloalkyl)aL~yl, e.g., methyl, ethyl, i-propyl, cyclopropyl or
cyclopropylmethyl.
- Ph~rm~ce~ltically acceptable salts include the acid addition salts of
basic ~H with organic or inorganic acids, e.g., hydrochloride, carbonate, sulfate,
acetate, phosphate, tartarate, citrate, malate, maleate, propionate, and the like.
The preparation of representative substituted trisb~n7imi(1~7.01es is
outlined in Figure 1. With the exception of phenylenediamine which was
commercially available, the a~plopliately substituted phenylene~ mines were
synthesized by catalytic hydrogenation of the respective o-nitroaniline deliv~ives.
These phenylenerli~minPs were then coupled with 5-formyl-2-(benzimidazo-5'-
yl)ben7imidazole, 2, by heating in nitrobenzene at 150~C to provide the various
trisb~n7imi(i~7nles, 10-16, in yields ranging from 43-96%, employing the generalmethodologies of M. P. Singh et al., Chem. Res. Toxicol., 5, 597 (1992) and Y.
Bathini et al., Synth Comm, ~, 955 (1990).
The requisite nitroanilines, as outlined in Figure 1, with the
exception of 3 which was commercially available, were synthesized from 4-bromo-
2-nitroaniline, 17. Compound 17 was prepared from o-nitroaniline in good yield,
94%, using 2,4,4,6-tetrabromo-2,5-cyclohexadienone as the bromination reagent.
G. J. Fox et al., Org. Svn, 55, 20 (1973). While allyltributyltin and
phenyltributyltin are commercially available, the pyridyltributyltin derivatives were
prepared from tributyltin chloride and 2-, 3-, and 4-bromopyridine, respectively.
See D. Peters et al., IIeterocyclic Chem., 27, 2165 (1990). These tril~uLyl~
deliv~L~ives were then coupled with 4-bromo-2-nitroaniline using PdCl2(PPh3)2 asthe catalyst in DMF as outlined in Figure 2 to provide compounds 4, 5, 6, 7, and 8,
respectively, in accord with the methodology of M. Iwao et al., Heterocycles, 36,
1483 (1993). This methodology can generally be applied to prepare 3-, 4-, 5- or 6-
aryl- and heteroaryl-substituted 2-nitroanilines from the corresponding
bromonitroanilines .
_
CA 02221248 1997-11-14
The preparation of 5-formyl-2-(benzimidazo-S'-yl)benzimidazole,
9, was accomplished as outlined in Figure 3. Reduction of 5-
benzimidazolecarboxylic acid to 5-hydroxymethylbenzimidazole was
accomplished using LiAlH4. Oxidation of the resulting crude benzylic alcohol
S with tetrapropylammonium perruthenate (TPAP) and N-methylmorpholine N-
oxide provided in two steps the desired 5-formylbenzimidazole in 32% an
overall yield. See, A. Cherif et al., J. Med. Chem.~ 35, 3208 (1992). Coupling of
5-formylbenzimidazole with 4-cyano-1,2-phenylene~i~mine provided 5-cyano-2-
(benzimidazol-S '-yl)benzimidazole, 19, which when treated with Ni-AI catalyst
10 in the presence of aqueous formic acid gave S-formyl-2-(benzimidazol-5 '-
yl)benzimidazole, 9, in 65% yield. (J. R. Pipier et al., J. Med. Chem., 31, 2164(1988)).
The compounds of the present invention can be formulated as
pharmaceutical compositions and ~lminictered to a m~mm~ n host, such as a
l S human cancer patient, in a variety of forms adapted to the chosen route of
~minictration, i.e., orally or parenterally, by intravenously, intramuscularly or
subcutaneous routes.
Thus, the present compounds may be orally ~(1minictered, for
example, in combination with a pharmaceutically acceptable vehicle such as an
20 inert diluent or an ~ccimil~ble edible carrier. They may be enclosed in hard or
soft shell gelatin capsules, may be compressed into tablets, or may be
incorporated directly with the food of the patient's diet. For oral therapeutic
~(lminictration, the active compound may be combined with one or more
excipients and used in the form of ingestible tablets, buccal tablets, troches,
25 capsules, elixirs, suspensions. syrups, wafers. and the like. Such compositions
and preparations should contain at least 0.1 % of active compound. The
percentage of the compositions and preparations may, of course, be varied and
may conveniently be between about 2 to about 60% of the weight of a given unit
dosage form. The amount of active compound in such therapeutically useful
30 compositions is such that an effective dosage level will be obtained.
AMENDED SttEET
lP~ P
CA 02221248 1997-11-14
W O 96/36612 PCTrUS~6/06~
The tablets, troches, pills, capsules, and the like may also contain the
following: A binder such as gum trag~nth, acacia, corn starch or gelatin;
excipients such as dicalcium phosphate; a tli.cintegrating agent such as corn starch,
potato starch, alginic acid and the like; a lubricant such as m~gnesium stearate; and
a sweet~ning agent such as sucrose, lactose or saccharin or a flavoring agent such as
peppermint, oil of wintergreen, or cherry flavoring may be added. When the unit
dosage form is a capsule, it may c~-nt~in~ in ~d~liti~n to materials of the above type,
a liquid carrier, such as a vegetable oil or a polyethylene glycol. Various other
materials may be present as coatings or to otherwise modify the physical form ofthe solid unit dosage form. For instance, tablets, pills, or capsules may be coated
with gelatin, wax, shellac or sugar and the like. A syrup or elixir may contain the
active compound, sucrose as a ~wet;l~llin~ agent, methyl and propylparabens as
presel vd~ives, a dye and flavoring such as cherry or orange flavor. Of course, any
material used in preparing any unit dosage form should be ~h~rm~ceutically
acceptable and substantially non-toxic in the amounts employed. In addition, theactive compound may be incorporated into sustained-release pl~aldLions and
devices.
The active compound may also be ~-imini~tered intravenously or
intraperitoneally by infusion or injection. Solutions of the active compound or its
salts can be prepared in water, optionally mixed with a nontoxic surfactant.
Dispersions can also be prepared in glycerol, liquid polyethylene glycols, triacetin,
and mixtures thereof and in oils. Under ordinary conditions of storage and use,
these plepald~ions contain a preselvd~ive to prevent the growth of microorg~ni.~m~.
The pharm~ce~ltical dosage forms suitable for injection or infusion
use can include sterile aqueous solutions or dispersions or sterile powders
comprising the active ingredient which are adapted for the extemporaneous
pl~pa~ ion of sterile injectable or infusable solutions or dispersions, optionally
encapsulated in liposomes. In all cases, the ultimate dosage form must be sterile,
fluid and stable under the conditions of m~nllf~ctllre and storage. The liquid carrier
CA 02221248 1997-11-14
W O 96/36612 PCT~US~6~
or vehicle can be a sol~-ent or liquid dispersion medium comprising, for example,
water, ethanol, a polyol (for example, glycerol, propylene glycol, and liquid
polyethylene ~lycols, and the like), vegetable oils, nontoxic glyceryl esters, and
suitable mixtures thereof. The proper fluidity can be maintained, for example, by
the formation of liposornes, by the m~int.-n~nce of the required particle size in the
case of dispersion or by the use of surfactants. The prevention of the action ofmicroorg~nicm~ can be brought about by various antibacterial and antifungal agents,
for example, parabens. chlorobutanol, phenol, sorbic acid, thimerosal, and the like.
In many cases, it will be preferable to include isotonic agents, for example, sugars,
buffers or sodium chloride. Prolonged absorption of the injectable compositions
can be brought about by the use in the compositions of agents delaying absorption,
for example, alllminl-m monostearate and gelatin.
Sterile injectable solutions are prepared by incorporating the active
compound in the required amount in the applupliate solvent with various of the
other ingredients enumerated above, as required, followed by filter sterilization. In
the case of sterile powders for the preparation of sterile injectable solutions, the
preferred methods of preparation are vacuum drying and the freezedrying
techniques, which yield a powder of the active ingredient plus any additional
desired ingredient present in the previously sterile-filtered solutions.
Useful dosages of the compounds of 1 can be ~let~rmined by
comparing their in vitro activity, and in vivo activity in animal models, to that of an
equivalent dosage of camptothecin (see, for example, B. C. Giovanella et al.,
Cancer Res., 51, 3052 (1991)) or Hoechst 33342 (see, A. Y. Chen et al., Cancer
Res, 53, 1332 (1993)) Methods for the extrapolation of effective anti-tumor
dosages in mice, and other ~nim~l~, to hllm~n~ are known to the art; for example,
see U.S. Pat. No. 4,938,949.
The present analogs can be used to treat cancers known to be
susceptible to topoisomerase I inhibitors, inclu~ling, but not limited to, Burkitt's
tumor, chronic lymphocytic leukemia, multiple myeloma, squamous cell and large
CA 02221248 1997-11-14
W O96/36612 PCTrUS~6/0
cell anaplastic carcinomas, adenocarcinoma of the lung, Ewing's sarcoma, non-
Hodgkins lymphoma, breast tumor, colon tumor, stomach tumor, oat-cell
bronchogenic carcinoma, squamous cell carcinoma of the cervix, ovarian tumors,
bladder tumors, testicular tumors, endometrial tumors, malignant melanoma and
acute lymphocytic le lkemi~, and prostatic carcinoma. The present compounds can
be ~rlmini~tered as single agents, or in combination with other antineoplastic drugs
commonly employed to treat these cancers.
The invention will be further described by reference to the following
detailed examples, wherein melting points were determined with a Thomas-Hoover
unimelt capillary melting point apparatus. Infrared spectral data (IR) were obtained
on a Perkin-Elmer 1600 Fourier transform spectrophotometer and are reported in
cm~l. Proton (IH NMR) and carbon (13C NMR) nuclear magnetic resonance were
recorded on a Varian Gemini-200 Fourier Transform spectrometer. NMR spectra
(200 MHz IH and 50 MHz 13C) were recorded in CDCl3 (unless otherwise noted)
with chemical shifts reported in ~ units downfield from tetramethylsilane (TMS).Coupling constants are reported in hertz. Mass spectra were obtained from Midwest
Center for Mass Spectrometry within the Department of Chemistry at the University
of Nebraska-Lincoln. Combustion analyses were performed by Atlantic Microlabs,
Inc., Norcross, GA, and were with in ~ 0.4%. T~ was freshly distilled from
sodium and benzophenone prior to use. Allyltributyltin and phenyltributyltin were
purchased from Aldrich Chemical Company.
Example 1. General Procedure for PdCI2(PPh~)2-catalyzed Coupling
Reaction of 4-Bromo-2-nitroaniline (13) with Tin Compounds.
(A) 4-Phenyl-2-nitroaniline (5). A solution of 4-bromo-2-nitroaniline 17
(1.0 g, 4.67 mmol), tributylphenyl tin (2.2 g, 6.07 mmol),
bis(triphenylphosphine)palladium (II) chloride (164 mg, 0.234 mmol), and
triphenylphosphine (613 mg, 2.34 mmol) in DMF (15 ml) was heated under N2 at
120~C overnight. After the solution was cooled to room temperature, the reaction
CA 0222l248 l997-ll-l4
W O96/36612 PCTrUS~CN~
mixture was directly chromatographed on silica gel eluting with 2-5%
EtOAc/Hexane to give 752 mg (75%) of 5 as a yellow solid: mp 169-171 ~C; IR
(CHCl3) 3517, 3398, 3022, 1635, 1525, 1250; IH NMR ~ 8.38 (lH, d, J= 2.2), 7.66
(lH, dd, J= 8.7, 2.2), 7.59-7.54 (2H, m), 7.49-7.34 (3H, m), 6.90 (lH, d, J= 8.8),
6.13 (NH, brs); '3CN~ 144.2, 139.3, 135.0, 130.9, 129.5, 127.8, 126.8, 124.4,
119.8, 112.8; Anal. Calcd for Cl2HIoN2O2 C, 67.28; H, 4.70; N, 13.08. Found: C,
67.38, H, 4.76; N, 13.01.
(B) 4-Allyl-2-nitroaniline (4). Prepared from 4-bromo-2-nitroaniline 17
(1.70 g, 7.84 mmol) and allyltributyltin (3.38 g, 10.2 mmol) as a yellow solid in
96% yield as described above for 5: mp 29-31 ~C; l:R (KBr) 3490, 3374, 1638,
1518, 1341, 1253; lHNMR~ 7.90 (lH, d, J = 2.0), 7.19 (lH, dd, J= 8.5, 2.0), 6.77(lH, d, J = 8,5), 6.05 (NH, brs), 6.00-5.80 (lH, m), 5.11 (lH, dd, J = 1.4, 1.4), 5.04
(lH, ddd, J = 6.6, 3.0, 1.5), 3.28 (lH, d, J = 6.6); 13C NMR ~ 143.81, 137.13,
129.34, 125.59, 119.49, 116.95, 39.18; HRMS (EI) calcd for C9HloN2O2 178.0742,
found 178.0746.
(C) 4-(2'-Pyridyl)-2-nitroaniline (6). Prepared from 4-bromo-2-
ni~roaniline 17 (597 mg, 2.75 mmol) and 2-tributylstannylpyridine (1.01 g, 2.75
mmol) as a yellow solid in 52% yield as described above for 5: mp 146-148 DC; IR(CHC13) 3516, 3397, 3020, 1634, 1524, 1341, 1250; lH NMR ~ 8.74 (lH, d, J=
2.2), 8.63 (lH, dd, J= 4.9, 1.5), 8.13 (lH, dd, J= 8.8, 2.1), 7.78-7.66 (2H, m), 7.20
(lH, ddd, J= 4.8, 4.7, 1.9), 6.92 (lH, d, J= 8.8), 6.37 (NH, brs); 13C NMR ~ 155.6,
150.1, 145.6, 137.4, 134.5, 129.1, 124.7, 122.4, 119.8, 119.7; Anal. Calcd for
CIlHgN3O2 C, 61.39; H, 4.21; N, 19.53. Found: C, 61.29; H, 4.23; N, 19.43.
(D) 4-(3'-Pyridyl)-2-nitroaniline (7). Prepared from 4-bromo-2-
nitroaniline 17 (1.42 g, 6.53 mmol) and 3-tributylstannylpyridine (3.60 g, 9.79
mmol) as a yellow solid in 32% yield as described above for 5: mp 177-179 ~C; IR(CHC13) 3515, 3399, 3052, 2983, 1638, 1524, 1341, 1259; lH NMR ~ 8.68 (lH, d, J
= 1.7), 8.42 (lH, dd, J= 4.8, 1.5), 8.22 (lH, d, J= 2.2), 7.74 (lH, ddd, J= 7.9, 2.4,
1.6), 7.50 (lH, dd, J= 8.7, 2.2), 7.23 (lH, ddd, J= 8.0, 4.8, 0.8), 6.92 (lH, d, J=
CA 02221248 1997-11-14
W O96/36612 PCTrUS96/06853
8.8), 6.56 (NH, brs); 13C N~ ~ 148.7, 147.8, 145.4, 135.0, 134.4, 133.8, 126.5,
124.4, 124.0, 120.4, Anal. Calcd for CllHgN3O2 C, 61.39; H, 4.21; N, 19.53.
Found: C, 61.28; H, 4.16; N, 19.40.
(E) 4-(4'-Pyridyl)-2-nitro~nilin~ (8). Prepared from 4-bromo-2-
nitroaniline 17 (165 mg, 0.76 mmol) and 4-tributylstannylpyridine (280 mg, 0.76
mmol) as a yellow solid in 25% yield as described above for 5: mp 230-232 DC; IR
(CHCl3) 3518, 3398, 3032, 1636, 1528, 1344; IH NMR (CD30D) ~ 8.55 (2H, d, J =
6.3), 8.52 (lH, d, J= 2.3), 7.84 (lH, dd, J= 8.9, 2.3), 7.71 (2H, d, J= 6.4), 7.13
(lH, d, J= 8.9); 13C NMR (CD30D) ~ 149.4, 133.4, 124.0, 120.7, 120.0; HRMS
(EI) calcd for CllHgN302 215.0695, found 215.0698.
E~ample 2. 5-Formyl-2-(benzimidazol-~'-yl)benzimidazole (9).
A mixture of 5-cyano-2-(benzimidazol-5'-yl)benzimidazole 19 (148 mg,
0.57 mmol), Ni-Al catalyst (500 mg), formic acid (7 ml) and water (3 ml) was
heated under refluxed under N2 for 4h. The hot reaction mixture was immediately
filtered through a plug of celite, and evaporated to give a yellow solid. The yellow
solid was then dissolved in hot water (5 ml), and the solution was neutralized to pH
9 by 2N NaOH. The solid precipitated was collected by suction filtration and further
purified by flash chromatography on silica gel (15% MeOH/EtOAc) to give 142 mg
(95%) of 9 as a white solid: mp > 275 ~; IR (KBr) 3106, 2835, 1685, 1618, 1432
1293; 'H NMR (CD30D) ~ 10.01 (lH, s), 8.39 (lH, s), 8.35 (lH, s), 8.13 (lH, s),
8.06 (lH, dd, J= 8.6, 1.6), 7.83 (lH, dd, J= 8.4, 1.4), 7.77 (lH, d, J= 8.5), 7.71
(lH, d, J= 8.3); ~MS (FAB) calcd for ClsHIlN40 263.0933, found 263.0932.
Example 3. General Procedures for Preparing ~-substituted
trisbenzimidazoles.
(A) 2-12'-(Ren7ir idazol-S"-yl)benzimidazol-S'-yl]benzimidazole (10).
A mixture of 5-formyl-2-(benzimidazol-5'-yl)benzimidazole 9 (121 mg, 0.46
mmol) and phenylene~ mine (60 mg, 0.55 mmol) in nitrobenzene (8 ml) was
CA 02221248 1997-11-14
W O 96/36612 PCTrUS~G/O~
heated at 150DC under N2overnight. The mixture was cooled to room temperature
arld chromatographed on silica gel (0-20% MeOHlEtOAc) to afford 155 mg (96%)
of 10 as a solid: mp > 275 DC; IR (KBr) 3400, 3157, 1630, 1542, 1438, 1294; IH
NMR (DMSO-d6 + 3 drops of CF3COOH) ~ 9.71 (lH, s), 8.75 (lH, s), 8.65 (lH, d,
J= 1.1), 8.48 (lH, dd, J= 8.7, 1.5), 8.21 (lH, dd, J= 8.6, 1.6), 8.14 (lH, d, J=8.8), 8.08 (lH, d, J= 8.7), 7.90 (2H, dd, J= 6.2, 3.1), 7.61 (2H, dd, J= 6.1, 3.1);
3C NMR (DMSO-d6 + 3 drops of CF3COOH) ~ 154.4, 149.8. 133.2, 132.0, 131.7,
1262, 125.5, 125.4, 123.9, 123.6, 116.3, 115.9, 114.23, 114.17, 114.13;HRMS
(FAB) calcd for C2lHI5N6351.1358, found 351.1367.
(B) 5-Cyano-2-[2'-(benzimidazol-5"-yl)ben7.imidazol-5'-
yl]l~en7.imidazole (11). Hydrogenation of 3 (70 mg, 0.43 mmol) was accomplished
at 40 psi H2at room temperature for 1 h using 10% Pd-C (30 mg) in EtOAc (10 ml).The reaction mixture was filtered and concenkated in vacuo to afford a solid. The
solution of this solid and 9 (87 mg, 0.33 mmol) in nitrobenzene (5 ml) was heated at
150 DC under N2 overnight. The mixture was cooled to room temperature, and
chromatographed directly on silica gel (0-10% MeOH/EtOAc) to give 107 mg
(86%) of 11 as a solid; mp > 280 DC; IR (KBr) 3416,3148, 2222, 1626, 1553,
1441, 1292; IH NMR (DMSO-d6 + 3 drops of CF3COOH) ~ 8.50 (lH, s), 8.46 (lH,
s), 8.40 (lH, s), 8.18-8.11 (3H, m), 7.81-7.75 (3H, m), 7.62 (lH, dd, J= 8.3, 1.5);
HRMS (FAB) calcd for C22HI3N7 376.1310, found 376.1309.
(C) ~-Propyl-2-[2'-(ben7.imidazol-5"-yl)b~n7.i~idazol-S'-
yl]benzimidazole (12). Prepared from 4-allyl-2-nitroaniline 4 (312 mg, 1.75
mmol) and 5-formyl-2-(ben7.imi~7t~1-5'-yl)ben7imi~ 1e 9 (121 mg, 0.46 mmol)
in 79% yield as described above for 11: solid; mp > 270 DC; IR (KBr) 3421, 3068,2957, 1434; IH NMR (DMSO-d6 ~ 3 drops of CF3COOH) ~ 9.66 (lH, s), 8.73 (lH,
s), 8.59 (lH, s), 8.48 (lH, dd, J= 8.7, 1.5), 8.13 (lH, dd, J = 8.7, 1.4), 8.11 (lH, d,
J = 8.7), 8.02 ( lH, d, J = 8.5), 7.79 ( lH, d, J = 8.4), 7.66 (lH, s), 7.45 (lH, dd, J =
8.5, 1.3), 2.80 ( 2H, t, J = 7.0), 1.70 (2H, m), 0.96 (3H, t, J = 7.2); 13C NMR
(DMSO-d6 + 3 drops of CF3COOH) ~ 153.84, 149.74, 141.64, 141.01, 139.37,
CA 0222l248 l997-ll-l4
W O96/36612 PCTAUS9G-'~~~
14
133.10, 132.26, 131.99, 130.34, 127.08, 126.26, 125.14, 141.64, 141.01, 139.37,
133.10, 132.26, 131.99, 130.34, 127.08, 126.26, 125.14, 122.91, 117.52, 116.32,
116.06, 115.76, 113.78, 112.99, 37.45, 24.73, 13.74;
(D) 5-Phenyl-2-[2'(b~n~imidazol-5"-yl)benzimidazol-5'-
yllbenzimidazole (13). Prepared from 4-phenyl-2-nitroaniline 5 (247 mg, 1.15
mmol) and 5-formyl-2-(benzimidazol-5'-yl)benzimidazole 9 (201 mg, 0.77 mmol)
in 89% yield as described for 11: solid; n~p 262-164 ~C dec; IR (KBr) 3402, 3104,
1627, 1552, 1442, 1290; lH NMR (DMSO-d6 + 3 drops of CF3COOH) 8 9.66 (lH,
s), 8.74 (lH, s), 8.65 (lH, s), 8.50 (lX dd, J= 8.8, 1.1), 8.21 (lH, dd, J= 8.7, 1.4),
8.12 (lH, d, J= 8.8), 8.06 (lH,s), 8.05 (lH, d, J= 8.4), 7.97 (lH, d, J= 8.7), 7.89
(lH, dd, J= 8.7, 1.5), 7.80 (2X d, J= 7.0), 7.61-7.47 (3H, m); HRMS (FAB) calcd
for C27HIgN6 427.1671, found 427.1666.
(E) 5-(2-Pyridyl)-2-[2'-(b~ ~ dazol-~ yl)benzimidazol-5'-
yllbenzimidazole (14). Prepared from 4-(2'-pyridyl)-2-nitroaniline, 6 (110 mg,
0.50 mmol), and 5-formyl-2-(b~n7:imi~701-5'-yl)benzimidazole 9 (51 mg, 0.25
mmol) in 84% yield as described above for 11: solid; mp > 275 DC; IR (KBr) 3411,3157, 1630, 1593, 1432; lH NMR (CD30D) 8 8.59 (lH, d, J= 4.8), 8.35 (lH, s),
8.31-8.25 (2H, m), 8.10 (lH, s), 8.04-7.94 (2H, m), 7.85-7.77 (3H, m), 7.72 (lH, d,
J= 8.6), 7.68 (lH, d, J= 8.7), 7.64 (lH, d, J= 8.7), 7.30 (lH, m); ~MS (FAB)
calcd for C~6HI8N7 428.1624, found 428.1611.
(F) 5-(3-Pyridyl)-2-[2'-(ber~imidazol-5"-yl)ben7:imidazol-5'-
yl]ben~imidazole (15). Prepared from 4-(3'-pyridyl)-2-nitroaniline 7 (183 mg,
0.85 rnmol) and 5-formyl-2-(benzimidazol-5'-yl)benzimidazole 9 in 46% yield as
described above for 11: solid; mp > 275 ~C; IR (KBr) 3400, 3070, 2836, 1438,
1289; lH NMR (CD30D) ~ 8.83 (lX d, J= 1.6), 8.49 (lH, dd, J= 4.9, 1.5), 8.38
(lX d, J=l.l), 8.31 (IH, d, J=l.l), 8.29 (lH, s), 8.11 (lH, ddd, J= 8.0, 2.3, 1.6),
8.05 (lH, dd, J= 8.5, 1.6), 8.00 (lH, dd, J= 8.5, 1.6), 7.81 (lH, d, J=l.l), 7.77-
7.68 (3H, m), 7.55-7.47 (2H, m); HRMS (FAB) calcd for C26HI8N7 428.1624, found
428.1612.
CA 02221248 1997-11-14
W O96/36612 PCTrUS9'~6
(G) 5-(4-Pyridyl)-2- [2 ' -(benzimidazol-5"-yl)benzimidazol-5 ' -
yl]benzimidazole (16). Prepared from 4-(4'-pyridyl)-2-nitroaniline 8 (35 mg, 0.16
mmol) and S-formyl-2-(b~n7imicl~7--1-5'-yl)benzimidazole 9 (50 mg, 0.19 mmol) in43% yield as described above for 11: solid; mp > 280 ~C; IR (KBr) 3411, 3118,
1600, 1552, 1439, 1290; IHN~(CD30D) ~ 8.51 (2H, d, J= 6.2), 8.33 (lH, d, J=
1.1), 8.27 (lH, s), 8.25 (lH, d,J= 1.1), 8.01 (lH, dd,J= 8.6, 1.7), 7.96 (lH, dd,J
= 8.9, 2.0), 7.87 (lH, d, J= 1.0), 7.74-7.56 (6H, m); HRMS (FAB) calcd for
C26HIgN7 428.1624, found 428.1625.
Example 4. 4-Bromo-2-nitroaniline (17).
A solution of 2-nitroaniline (5 g, 36.2 mmol) in CH2Cl2 (100 ml) was cooled
to -10~C, and treated by 90% 2,4,4,6-tetrabromo-2,5-cyclohexadienone (19.8 g,
43.5 mmol) in 5 portions. The mixture was stirred at -10~C-0~C for 1 hr. After
being warmed to room temperature, the reaction mixture was washed by 2N NaOH
(60 ml) and brine (50 ml), dried over Na2SO4 and evapo~ed. Flash
chromatography on silica gel (5% EtOAc/Hexane) gave 7.40 g (94%) of 17 as a
yellow solid: mp 109-110 (lit. mp 112-113 ~C); IH NMR ~ 8.27 (lH, d, J= 2.3),
7.43 (lH, dd, J= 8.9, 2.4), 6.73 (lH, d, J= 8.8), 6.09 (NH, brs).
Example 5. 5-Formylbenzimidazole (18).
A suspension of S-b~n7imitl~7~ 1ecarboxylic acid (1.57 g, 9.7 mmol) in dry
THF (50 ml) was cooled to -78~C under N2, and treated with LiAlH4 (736 mg, 19.4
mmol). After the addition, the mixture was allowed to warm slowly to room
temperature and then stirred at r.t. overnight. The mixture was quenched by MeOHand H2O cautiously, and passed through a short silica gel column eluting with 10%
MeOH/EtOAc. The eluate was conc~n~ ed to give 876 mg crude alcohol as a
solid. The crude alcohol (876 mg) was dissolved in a mixture of DMF (3 ml), THF
(10 ml) and CH2C12 (40 ml). 4-Methylmorpholine N-oxide (2.25 g, 19.2 mmol), 4A
molecular sieves (5 g), and TPAP (169 mg, 0.48 mmol) were subsequently added to
CA 02221248 1997-11-14
W O 96/36612 PCTrUS9''0C
16
the cmde alcohol solution. The mixture was stirred at room temperature overnight,
and filtered through a pad of silica gel eluting with 10% MeOH/EtOAc. The elute
was concentrated and further purified by flash chromatography on silica gel eluting
with 0-10% MeOH/EtOAc to give 452 mg (32%, 2 steps) of 17 as a white solid: mp
164-166 ~C; IR (KBr) 3087, 2818, 1690, 1292; IH N~ (CD30D) ~ 9.95 (lH, s),
8.34 (lH, s), 8.08 (lH, d, J= 1.5), 7.74 (lH, dd, J= 8.4, 1.5), 7.63 (lH, d, J= 8.4);
l3CNMR(CD30D)~ 194.2, 146.0, 143.0, 139.8, 133.6, 124.9, 120.7, 116.6;Anal.
Calcd for C8EI6N2O: C, 65.75; H, 4.14; N, 19.17. Found: C, 65.60; H, 4.17; N,
14.08.
F~ample 6. 5-Cyano-2-(benzimidazol-5'-yl)benzimidazole (19).
A mixture of 5-formylben7imid~7c-1e 18 (211 mg, 1.44 mmol) and 4-cyano-
1,2-phenylene~ mine (230 mg, 1.73 mmol) in nitrobenzene (10 ml) was heated at
150DC under N2 overnight. The mixture was cooled to room temperature and
directly chromatographed on silica gel eluting with 0-15% MeOH/EtOAc to give
244 mg (65%) of 18 as a solid: mp >270 ~C; IR (KBr) 3110, 2826, 2224, 1627,
1426, 1294; IH NMR (CD30D) ô 8.41 (lX s), 8.33 (lH, s), 8.07 (lH, dd, J= 8.6,
1.5), 7.98 (lX s), 7.78 (lH, d, J= 8.4), 7.73 (lH, d, J= 8.4), 7.56 (lH, dd, J= 8.4,
1.5); 13C NMR (DMSO-d6 + 3 drops of CF3COOH) ~ 153.4, 140.4, 138.3, 132.9,
131.6, 127.0, 125.8, 125.3, 120.8, 119.8, 116.0, 115.8, 113.9, 105.5; HRMS (FAB)calcd for Cl5HIoN5260.0936, found 260.0935.
Example 7.
(A) 5-Bromo-2-12'-(benzimidazol-5"-yl)benzimidazol-5'-yl]-
benzimidazole (JSK IV-37) A mixture of 5-formyl-2-(benzimid~ol-5'-
yl)benzimid~ole (118.8 mg, 0.45 mmol~ and 5-bromophenylene~ mine (169.6
mg, 0.90 mmol) in nitrobenzene (5 mL) was heated at 150~C under Nz overnight.
The mixture was cooled to room temperature and chromatographed using 0-10%
methanol/ethylacetate to afford 127.3 mg (66%) of brownish yellow solid:
CA 02221248 1997-11-14
W O96/36612 PCTAUS96/06853
mp>280~C; IR(KBr) 3101, 1626, 1547, 1440; IHNMR(DMSO-d6) ~ 7.34 (dd,
lH, J=7.0, 2.0), 7.57 (d, lH, J=9.0), 7.71-7.80 (m, 3H), 8.04-8.18 (m, 2H), 8.39 (s,
2H), 8.50 (s, lH); 13C NMR (DMSO-d6 + 3 drops CF3COOH) ~ 114.1 115.8, 116.2,
116.4, 11'7.0, 118.6, 123.5, 125.3, 126.2, 128.7, 128.9, 131.8, 132.0, 132.3, 133.1,
134.4, 138.3, 140.6, 151.1, 153.4.
(B) 5-Chloro-2-[2'-(benzimidazol-5"-yl)ben7.i idazol-5'-yll-
benzimidazole(JSK IV-68) A mixture of 5-formyl-2-(benzimidazol-5'-
yl)bPn~imicl~7ole (160 mg, 0.61 mmol) and 5-chlorophenylenerli~mine (174 mg,
1.22 mmol) in nitrobenzene (5 mL) was heated at 150~C under Nz overnight. The
mixture was cooled to room temperature and chromatographed using 0-10%
methanol/ethyl~cet~te to afford 167 mg (71%) of brownish yellow solid:
mp>280~C; IR(KBr) 3103, 2826, 1427, 1293; IHNMR(DMSO-d6) ~i 7.24 (dd,
lH, J=8.5, 2.0), 7.60-7.81 (m, 4H), 8.07-8.17 (m, 2H), 8.40 (s, 2H), 8.50 (s, lH);
13C NMR (DMSO-d6 +3 drops CF3COOH) ~ 114.3, 114.4, 115.3, 115.5, 115.6,
116.2, 118.5, 123.1, 125.4, 125.5, 125.6, 129.4, 132.4, 132.9, 133.0, 135.2, 138.9,
140.9, 151.8, 153.5.
(C) 5-~D-Chlorophenyl)- 2-12'-(benzimidazol-5"-yl)benzimidazol-5'-yll-
ben7.irnidazole (JSK IV-47) A mixture of 5-formyl-2-(benzimidazol-5'-
yl)benzimidazole (99 mg, 0.38 mmol) and 5-C~-chlorophenyl)-phenylene~i~mine
(154 mg, 0.71 mmol) in nitrobenzene (5 mL) was heated at 150~C under N2
overnight. The mixture was cooled to room temperature and chromatographed
using 0-10% methanol/ethylacetate to afford 85 mg (49%) of brownish yellow
solid: mp>280~C; IR (KBr) 3046, 2820, 1426, 1282; lH NMR (DMSO-d6 + 3 drops
CF3COOH) ~ 7.56 (d, 2H, J=8.5), 7.82 (d, 2H, J=8.5), 7.88-8.21 (m, 6H), 8.48 (d,lH, J=8.8), 8.63 (s, lH) 8.72 (s, lH), 9.69 (s, lH); '3C NMR (DMSO-d6 + 3 drops
CF3COOH)~ 111.8, 113.8, 114.7, 115.8, 116.1, 117.7, 123.0, 124.1, 125.2, 125.3,
129.2, 129.3, 131.9, 132.1, 133.0, 133.1, 137.2, 138,5, 139.3, 141.6, 150.8, 153.8.
-
CA 02221248 1997-11-14
W O96/36612 PCTrUS~6/OC
(D) 4-Bromophenylene~i~qmine (JSK ~V-35) To 2-nitro-4-br~ m~ aniline
(340 mg, 1.57 mmol) in absolute ethanol (20mL) was added SnCl2 (1.50g, 7.91
mmol) and refluxed overnight. The reaction mixture was then basified to pH 11
with 2N NaOH and extracted with other to give 275 mg (94%) of product. This
product was used without further purification for the synthesis of JSK IV-37.
(E) 4-Chlorophenylene~ mine (JSK IV-67) To 2-nitro-5-chloroaniline
(304 mg, 1.76 mrnol) in absolute ethanol (20 mL) was added SnCl2 (1.68g, 8.86
mmol) and refluxed overnight. The reaction mixture was then basified to pH 11
with 2N NaOH and extracted with ether to give 250 mg (quantitative yield) of
product. This product was used without further purification for the synthesis ofJSK IV-68.
(F) p-Chlorotributylphenyltin (JSK IV-42) 4-Bromochlorobenzene (3.2
g, 16.62 mmol) was dissolved in dry TH~ (20mL). After bringing the reaction
temperature down to -78~C with an acetone/dry ice bath, nBuLi(15.58 mL, 1.6M,
1.5 equiv.) was added slowly and stirred at -78DC for 30 min. Tributyltinchloride
(6.77 mL, 1.5 equiv.) was added and stirred overnight while bringing the reaction to
room temperature. Reaction mixture was quenched by stirring the reaction flask
open in air for 1 hour after which TE~ was rotav~ol~led off. Product was
obtained as an oil (7.35g, 97%) after passing the mixture through a quick silicagel
column eluting with 100% hexanes.
(G) 2-Nitro-5-~-chlorophenyl)aniline (JSK IV-44) To JSK IV-42 (2.02
g, 5.04 mmol) and 2-nitro-4-bromoaniline (730 mg, 3.36 mmol) in DMF (18 mL)
was added Pd(PPh3)2Clz(117.9 mg, 0.17 mmol) and PPh3 (440.2 mg, 1.70 mmol)
and heated at 120~C overnight. DMF was n~tavapol~ed off and the mixture was
separated on a silicagel column eluting with 5-10% ethylacetate/hexanes to give 270
mg (32%) of reddish solid.
CA 02221248 1997-11-14
W O96136612 PCTrUS96/06853
19
(I) 4-(p-Chlorophenyl)phenylene~ mine (JSK n~'-46) JSK IV-44 (190
mg, 0.77 mmol) was dissolved in ethylacetate (100 mL) and after adding 10% Pd-C
(40 mg) was reduced by hydrogenation (45 psi). Product (quantitative yield) was
used in JSK IV-47 without further purification.
S
Example 8. Bioas~,ays
A. Topoisomerase I-Mediated DNA Cleava~e Assays
DNA topoisomerase I was purified from calf thymus gland as reported
previously by B. D. Halligan et al., J. Biol. Chem., 260, 2475 (1985). Plasmid
YEpG was also purified by the aL~ali lysis method followed by phenol
deproteination and CsCl/ethidium isopycnic centrifugation as described by T.
Mariatis et al., Molecular Clonin~. A Laboratory Manual, Cold Spring Harbor Labs,
NY (1982) at pages 149- 185. The end-labeling of the plasmid was accomplished aspreviously described by L. F. Liu et al., J. Biol. Chem., 258, 15365 (1983). Thecleavage assays were performed as previously reported by A. Y. Chen et al., Cancer
Res., 53, 1332 (1993). Human topoisomerase was isolated as a recombinant fusion
protein using a T7 expression system.
~. Cytotoxicity assay
The ~;yLoLo~icity was determined using the as MTT-microtiter plate
tetrazolinium ~;yLutu~icity assay (MTA) following the procedures of F. Denizot et
al., J. Immunol. Methods. 89, 271 (1986); J. Carmichael et al., Cancer Res., 47, 936
(1987) and T. J. Mosmann et al., Immunol. Methods, 65, 55 (1983). The human
lymphoblast RPMI 8402 and its camptothecin-resistant variant cell line, CPT-K5
were provided by Dr. Toshiwo Andoh (Aichi Cancer Center Research Institute,
Nagoya, Japan). See, for example, T. Andoh et al., Adv. Pharmacol., 29B, 93
(1994). The cytotoxicity assay was performed using 96-well microtiter plates.
Cells were grown in suspension at 37 DC in 5% COz and m~int~ined by regular
CA 02221248 1997-11-14
W O96/36612 PCT~US96/06853
passage in RPMI medium supplemented with 10% heat inactivated fetal bovine
serum, L-glutamine (2 mM), penicillin (100 U/ml), and streptomycin (0.1 mg/ml).
For determination of IC50, cells were exposed continuously with varying
concentrations of drug concentrations and MTT assays were performed at the end of
the fourth day.
The drug sensitive human epiderrnoid carcinoma KB3-1 cell line (S.
Aliyama et al., Somatic Cell Mol. Genet., 11, 117 (1985)) and its vinblastine-
selected multidrug-resistant variant KBV-l cells (D. W. Shen et al., Science, 32,
643 (1986)) were provided by Dr. Michael Gott~m~nn (National Cancer Institute,
Bethesda, ML). These cells were grown as monolayer cultures at in 5% CO2 and
m~int~ined by regular passage in Dulbecco's minim~l essential medium
supplemented with 10% heat inactivated fetal bovine serum. KBV-l cells were
similarly m~int~inecl except they were gr~wn in the presence of 1 ~g/ml vinblastine.
C~. Results
As shown on Table 1, comparison of compounds 10-16 with Hoechst 33342
(1) as inhibitors of topoisomerase I demon~ ed that several of these
trisbenzimidazoles had similar potency.
_
CA 02221248 1997-11-14
W O 96136612 PCTrUS9ClOC~
Table 1.
Topoisomerase I-mediated DNA Cleavage and Cytotoxicity
of Bis- and Trisbenzimida_oles
Cytotoxicity IC50~ (~M)
Topo I- Cell Lines
mediated
Compound DNA cleavageb RPMI CPT-K5 KB3-1 KBV-l
Hoechst 33342 1 0.03 0.9 0.01 1.2
1.1 14 28 N.D. N.D.
11 1 > 25c > 25c N.D. N.D.
12 100 7.6 20 N.D. N.D.
13 2 0.09 0.58 0.58 0.35
14 3.3 0.16 5.8 0.05 0.09
2 0.035 2.5 0.02 0.02
16 2 0.035 2.5 0.02 0.01
19 1000 > 25c N.D. N.D. N.D.
JSKlV-37 1 1.40 1.40
JSKIV-47 10 0.09 0.20
JSKIV-68 1 1.04 0.65
a) IC50 has been calculated after 4 days of continuous drug exposure. N.D. = Notdetermined.
b) Topoisomerase I cleavage values are reported as REC, Relative Effective
Concentration, i.e. concentrations relative to Hoechst 33342, whose value is
albill~ily assumed as 1, that are able to produce the sarne cleavage on the plasmid
DNA in the presence of calf thymus topoisomerase I. Cleavage is c~lc~ ted from
the intensity of the strongest Hoechst specific band.
c) No indication of Cy~Otu~iCity were considered indicative of IC50 values
substantially greater than the highest doses assayed.
While 10 and 11 exhibited similar potency in their inhibition of
topoisomerase I as observed with Hoechst 33342, both of these compounds failed to
exhibit significant ~;y~OlO~iCity towards the human lymphoblast cell line, RPMI
8402. However, this may be due to the inability of the pure compound to penetrate
the target cells, which may be overcome by selection of a suitable carrier, such as
liposomes. The 5-phenyl substituted trisbP.n7.imi~ 0le, 13, was approximately one-
half as potent as Hoechst 33342 as a topoisomerase I inhibitor. In contrast to 10
and 11, however, it had significant ~;y~uto~icity towards the human lymphoblast cell
line, RPMI 8402 cells. As observed with Hoechst 33342, 13 was also effective
CA 02221248 1997-11-14
W O96136612 PCTrUS96/068S3
against camptothecin-resistant CPT-K5 cells. The relative resistance of Hoechst
33342 and 13, expressed as the ratio of the IC50 values of the resistant verses the
drug sensitive cell line, is approximately 30 fold as compared to the relative
resistance of camptothecin which is 2,500 fold, as reported by A. Y. Chen et al.,
S (:~ancer Res., 53, 1332 (1993). A similar effect was observed in another pair of cell
lines; 13 has an IC50 of 0.015 ,ug/ml in the human ovarian tumor cell line, A2780,
relative to an IC50 of 0.03 ,ug/ml in CPT-2000, a variant of A2780 selected for
camptothecin-resistance and known to contain a mutant camptothecin-resistant
topoisomerase I. The 5-n-propyl trisbenzimidazole d~liv~Live, 12, was much less
active than either 10, 11, or 13 as an inhibitor of topoisomerase I. Its weak activity
as a topoisomerase I inhibitor correlated with its weak ~;yLoLo~icity. The activity of
several of these compounds were also evaluated using recombinant human
topoisomerase I. Several of these analogs induced similar DNA cleavage in the
presence of human topoisomerase I as compared to that observed with
topoisomerase I isolated from calf thymus.
The ~;yLotox ic activity of Hoechst 33342 and 13 was also evaluated
against KB 3-1 and KB V-1 cells. The primary difference between these cell linesis in the degree to which human MDRl (P-glycopl~teil-) is expressed. Recent
studies have demonstrated that antineoplastic agents which are cationic at
physiological pH are more likely to serve as substrates for MDR1 and, therefore, are
likely to be less effective against cells that overexpress P-glycoploteill. In view of
the fact that Hoechst 33342 is extensively protonated at physiological pH, it is not
surprising that the IC50 differs by applo~ilnately two-orders of magnitude for KB 3-
1 as compared to KB V-1 cells, as reported by A.Y. Chen et al., Adv. Pharmacol
245, 29B (1994). In contrast to Hoechst 33342, there is little difference between the
IC50 values observed for 13 in these two cell lines. Thus, 13 appears not to be a
substrate for human MDR1. This data indicate that these trisbenzimidazole
derivatives may have significant chemotherapeutic advantages as compared to
CA 02221248 1997-11-14
W O96/36612 PCTrUS96/06853
Hoechst 33342 or pibenzimol (Hoechst 33258), 2'-(4-hydroxyphenyl)-5-(4-methyl-
1-piperazinyl)-2,5'-bi-lH-benzimidazole.
These data indicate that substitution of these trisbenzimidazole with a
S-Ar substituent can yield derivatives which are active as topoisomerase I inhibitors
- S and cylùtù~iC to tumor cells. Trisbenzimidazoles substituted at the 5- position with
either a 2-, 3-, or 4-pyridyl group, 14-16, were evaluated for their potency as
topoisomerase I inhibitors and for cytotoxicity as summarized in Table 1. These
ana1ogs, similar to 13, have activity as topoisomerase I inhibitors. The 3- and 4-
pyridyl analogs, 15 and 16, are somewhat more active than the 2-pyridyl derivative,
14, as topoisomerase I inhibitors as well as ~;y~Otu~iC agents. As was observed with
13, these pyridyl-substituted tribenzimidazoles had similar ~iy~uto?~icity to KB 3-1
cells as well as to KB V- l cells which ove~ ress MDR1. A principal advantage
of these heteroaryl substituted trisbenzimidazoles as compared to Hoechst 33342 is
their efficacy against cell lines which express MDR1.
All publications and patents are incorporated by reference herein, as
though individually incorporated by reference. The invention has been described
with reference to various specific and preferred embodiments and techniques.
However, it should be understood that many variations and modifications may be
made while r~m~ininJ~ within the spirit and scope of the invention.