Note: Descriptions are shown in the official language in which they were submitted.
CA 02221599 1997-11-19
WO 961331~i6 PCT/~b~rt~
BENZOPHENONE DERIYATIVES USEFUL AS PHOT~-INI~ATORS
This invention relates to novel benzophenone
derivatives, to their preparation, to their use as
5 photoinitiators in polymerisation processes, and to
polymeric products cured using such photoinitiators.
Photoinitiated curing processes may use
photoinitiators which generate photo-excited species,
10 wh;Lch react with the curing agents, commonly calledL
synergists, to produce radicals which are thought to be
the species responsible for the polymerisation reaction.
Commonly the curing agents are aromatic tertiary ~1-; neC.
Commercial amine curing agents are ethyl-4- (N, N -
15 dimethylamino) benzoate (EDB) and 2-n--butoxyethyl 4
( dimethylamino ) benzoate ( BEDB ) .
Photoinitiators which are available, and suitable for
use with amine curing agents, are thioxanthone initiators,
20 in particular isopropylthioxanthone (ITX); anthroquinone
ini.tiators; and benzophenone initiators, in particular 2-
methylbenz oylbenz oate ( 2 -MBB ) .
The invention concerns i~ /ements in benzophenone
25 ini.tiators. Whilst benzop~ ~rlone/amine curing systems have
been extremely important they are now less favoured. In
part this is because the benzophPnon~ components, for
example 2--MBB, tend to migrate relatively easily from the
cured polymers, and taint adjacent ma~erials. When the
30 polymer is, for example, a film, and the adjacent
materials are, for example, foodstuffs, this is very
undesirable .
Japanese published patent application 6263814 (Toyo
35 Inl:) proposes photoinitiators obtained by reacting
CA 02221~99 1997-11-19
Wo96133156 PCT/GB96/00911
dihydric polyol compounds with ortho-benzoylbenzoic acid.
These are said to be of use in curable coating
compositions said to have r~ c~ odour, and to be free
from deterioration from curability. However, in
experiments we have found the di(benzoyl benzoate)
compound of this type produced by reacting ortho-
benzoylbenzoic acid with poly(ethylene glycol)3~ to migrate
from a cured polymer at a relatively high rate.
We have discovered certain benzoylbenzoate
derivatives which appear to have a surprisingly low
propensity to migrate from a cured polymer.
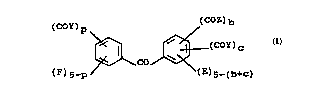
In accordance with a first aspect of the present
lS invention, there is provided a benzophenone c~o~,,d of
the general formula
~COY)p (C~z)b
~ ~ (C~Y)C
~ Co ~ (I)
(F)s_p / (E)s-(b+C)
wherein:
each Z indep~ tly represents an alkylene polyol moiety
or a polyalkylene polyol moiety, wherein hydroxyl groups
of the polyol moiety are optionally alkylated;
each Y independently L ~L esents an alkylene polyol moiety
or a polyalkylene polyol moiety, wherein hydroxyl ~L OU~
of the polyol moiety are optionally alkylated; or an
alkoxy group;
each E is independently selected ~rom hydrogen or halogen
atoms and alkyl, acyl, nitro, cyano, alkoxy, hydroxy,
amino, alkylamino, 5111 rh; ~yl ~ alkylslllph; nyl, sulphonyl,
=
CA 02221~99 1997-11-19
WO 96133156 ~ ~r~loo9ll
alkylsulphonyl, sulphonate, amido, alkylamido,
allcoxycarbonyl, halocarbonyl and haloalkyl groups;
ea~h F is independently selected from hyd.uyen or halogen
atoms and alkyl, acyl, nitro, cyano, alkoxy, hydroxy,
amino, alkylamino, sulphinyl, alkylsulphinyl, sulphonyl,
alkylsulphonyl, sulphonate, amido, alkylamido,
alkoxycarbonyl, halocarbonyl and haloalkyl ~l~U~;
b represents 1 to 5;
c represents O to 4; and
p represents O to 5.
Suitably, b represents 1 or 2, preferably 1.
Suitably, c represents O or 1, preferably 0.
Suitably, each E represents a hydrogen atom.
Suitably, p represents o or 1.
Suitably, each F represents a hydrogen atom.
Suitably, one or each group Z includes at least two
ether functionalities.
Suitably, the or each group Z independently
represents an alkylene glycol or polyalkylene glycol
moiety wherein hydroxyl groups of the moiety are
optionally alkylated.
Suitably, the or each group Z represents an ethylene
glycol or polyethylene glycol moiety, wherein hydroxyl
groups of the moiety are optionally alkylated.
Suitably, one or preferably each, group Z represents
a polyol moiety which is end-capped by an alkyl group.
CA 02221~99 1997-11-19
WO 96/33156 PCT/GB96/OOgll
Suitably, an alkyl group which end caps the group Z
is a Cl1 alkyl group. ~referably, it is a methyl group.
Suitably, one or preferably, each group Z represents
a polyol moiety wherein each l.ydhoxyl group is alkylated.
Suitably, one or preferably each group Z is of the
general formula -O-(CH2-CH2-O)e-alkyl where e has a mean
value of from 2 to 20, preferably 4 to 15, most preferably
6 to 13 and the alkyl group is suitably a Cl4 alkyl group,
preferably methyl.
Preferably, a said group COz is located in the 2- or
4-position, most preferably in the 4-position.
Suitably, one or each group Y includes at least two
ether functionalities.
Suitably, each Y in~pen~e~tly represents an alkylene
glycol or polyalkylene glycol moiety, wherein hydroxyl
groups of the moiety are optionally alkylated.
Suitably, Y represents an ethylene glycol or
polyethylene glycol moiety, wherein hydroxyl groups of the
moiety are optionally alkylated.
Suitably, one or preferably each, group Y represents
a polyol moiety which is end-capped by an alkyl group.
30Suitably, an alkyl group which end caps the group Y
is a Cl~ alkyl group. Preferably, it is a methyl group.
Suitably, one or preferably, each group Y represents
a polyol moiety wherein each hyd~oxyl group is alkylated.
,
CA 02221599 1997-11-19
WO 96133156 PCT/~ J*0~11
Preferably, one or preferably each group Y is of the
general formula -O-(CH2-CII2-O)c-alkyl where e has a mean
value of from 2 to 20, preferably 4 to 15, most preferably
6 1_o 13.
Preferably, a said group COY is located in the 2- or
4-position, most preferably in the 4-position.
In accordance wi~h a ~eco~ aspect of the present
invention there is provided a process for the preparation
of a compound of the general formula I, which comprises
the esterification or transesterification of a precursor
benzophenone compound to the compound of general formula
I, with an appropriate optionally alkylated polyol
compound having at least one hydroxyl group.
When p and c each represent 0, the preferred reaction
is an esterification, using the appropriate benzoyl
benzoic acid.
When p and/or c represents 1 and Y represents an
alkylene polyol or polyalkylene polyol moiety, optionally
end-capped by an alkyl group, the preferred reaction is a
transesterification, preferably from the appropriate
di~methoxycarbonyl) benzophenone compound.
When p and/or c represents 1 and Y represents an
alkoxy group the favou~ed reaction is an esterification,
using the appropriate precursor c~ u-.d having one or
more (as applicable) ester groups COY, where each Y
represents an alkoxy group, and one group -COOH.
Commercially available xylene derivatives may be
suitable reagents for the preparation of compounds of
general formula I.
CA 02221~99 1997-11-19
WO ~6/33156 PCT/~S'';~911
The esterification/transesterification may be carried
out under standard ~on~itions. The esterification
reaction may be carried out in an organic solvent, for
example toluene, at an elevated temperature, preferably
under reflux, with removal of water during the reaction
process. Suitably a catalyst is present. Suitable
catalysts are tin (II~ octanoate or an acid, suitably an
organic acid, for example a sulphonic acid. The
transesterification reaction may be carried out in an
organic solvent, for example toluene, in the substantial
absence of water, at an elevated temperature, preferably
under reflux. Suitably a catalyst is present, preferably
an alkyl titanate or an acid, suitably an organic acid,
for example a sulphonic acid.
In accordance with a further aspect of the present
invention there is provided a polymer curing composition,
which may be in kit form, comprising a compound of general
formula I as described above, together with a curing agent
with which the compound of general ~ormula I may react,
when irradiated, to generate a polymerisation radical.
In accordance with a further aspect of the present
invention there is provided a polymerisable composition
comprising a polymerisable material suitably present in an
amount from 80 to 97 wt. %, a curing agent suitably
present in an amount from 14 to 2 wt. ~, and a compound of
the general formula I, suitably present in an amount from
6 to 1 wt. %.
In accordance with a further aspect of the present
invention there is provided a polymeric composition
derived from said polymerisable composition by photo-
curing.
CA 02221599 1997-11-19
WO 96133156 ~ll~b5~l~D~ll
A suitable curing agent may, for example, be an
aromatic amine compound, for example ethyl-4-(N,N-
~di.methyl~;no~ benzoate (EDB) or 2-n-butoxyethyl 4-
(dimethyl~;n~) benzoate (BEDB). Preferably, howe~er, it
~5 is a novel amine compound of the type defined in our co-
filed patent application entitled "Novel Amine Curing
Asents", the conten~s of which are incorporated herein by
reference. Thus, such a compound is of the general
fcrmula
(Cox)n
(Q)6-(m+n) ~ (II)
(NRlR2)m
wherein:
each Rl independently represents an alkyl group;
each R2 independently represents an alkyl ~o~;
each group X independently represents a polyalkylene
polyol moiety wherein hydroxyl groups of the polyol moiety
are optionally alkylated;
n and m independently represent 1, 2 or 3; and
each Q is independently selected from hydrogen or halogen
atoms, and alkyl, acyl, nitro, cyano, alkoxy, hydroxy,
amino, alkylamino, sulphinyl, alkylsulphinyl, sulphonyl,
alkylsulphonyl, sulphonate, amido, alkylamido,
alkoxycarbonyl, halocarbonyl and haloalkyl groups.
Suitably, n and m i~ep~n~tly represent 1 or 2,
pre~erably 1.
Suitably, each Q represents a hydrogen atom.
Suitably, each Rt represents the same alkyl group.
-
CA 02221~99 1997-11-19
WO 96/33156 ~ 55.'~t~911
Suitably, each Rl represents a Cl1 alkyl group,
preferably methyl.
Suitably, each R2 represents the same alkyl group.
Suitably, each R2 represents a Cl~ alkyl group,
preferably methyl.
Suitably, one or each group X includes at least two
ether functionalities.
Suitably, each X independently represents a
polyalkylene glycol moiety, wherein hydroxyl groups of the
moiety are optionally alkylated.
Suitably, each X represents a polyethylene glycol
moiety, wherein hydroxyl groups of the moiety are
optionally alkylated.
20Suitably, one or preferably each, group X represents
a polyol moiety which is end-capped by an alkyl group.
Suitably, the alkyl group which end caps the group X
is a Cl~ alkyl group. Preferably, it is a methyl group.
Preferably, one or preferably each group X represents
a polyol moiety wherein each hydroxyl group is alkylated.
Preferably, one or each group X is of the general
formula -O-(CH2-CH2-O)z-alkyl where z has a mean value of
from 2 to 20, preferably 4 to lS, most preferably 6 to 13
and the alkyl group is suitably a Cl4 alkyl group,
preferably methyl.
-
CA 02221599 1997-11-19
WO 96/33156 PCT/CB~ 911
Preferably, at least one said group COX is located
para to a said dialkyl Ar; nP group. Where n = m = 1,
preferably the group COX is located para to the
r dialkylamine group.
Compounds of the general formula II may be prepared
by alkylation of a corresponding primary amine compound
(in which Rl = ~ = hydrogen). This may be carried out by
reductive alkylation, using the a~Lu~iate ~lk~n~. Such
a reaction can be carried out in a hyd oyenator, at
elevated temperature and pressure, and in the presence of
hydLuyen. The correspo~i n~ primary amine compound can
itself be prepared by similar hydrogenation, from the
correspon~;n~ nitro compound. The nitro compound may be
easily prepared by reaction of the appropriate
nitroberlzoic acid with ine a~p~u~.iate alkyl end-capped
alk:ylene glycol co~ound, ret~;~;ng a single hydroxyl
group. Alternatively, the appropriate nitrobenzoic acid
chloride may be employed, suitably with a base, for
example an amine base, suitably triethylamine.
Alternatively, the compounds of the general formula
II may be prepared by esterification of the appropriate
dialkylamine benzoyl chloride compound, with the
appropriate optionally alkylated polyol compound, having
at least one hydroxyl group. This reaction suitably takes
place in the preC~ce of a base, for example an amine
base, for example triethylamine. This reaction suitably
takes place at a t~r~ature in the range -20~C to 40~C,
preferably 0~C to ambient temperature. The benzoyl
chloride reactant may be prepared by the reaction of
thionyl chloride with the corresponding benzoic acid,
suitably at ambient temperature.
CA 0222l599 l997-ll-l9
WO 96/33156 PCT/C1~6J'~J91l
-- 10 --
A suitable polymerisable material is any material
whose polymerisation can be initiated by an amine radical.
Preferably the polymerisation is applied to acrylate
systems where the polymerisable material (monomer) may,
for example be 1,6-hexanediol diacrylate (HDDA), 2-
hydroxyethyl acrylate (HEA), hydroxy~o~yl acrylate (HPA)
and methyl methacrylate (MMA).
The polymerisable materials may be suitable for
surface/coating/film applications. They may be formulated
with other components, including inks, for printing
applications.
The invention will now be further described, by way
of example.
PreParation of ~hotoinitiators of the inventio~
ComPound 1: 4-BenzoYlbenzoYl ~oly(ethylene glYcol)~50
monomethvlether
2.1 g of poly(ethylene glycOl)350 mono methyl ether,
1 g of 4-benzoylbenzoic acid and 1 g of p-toluene
sulphonic acid were refluxed with 100 ml of toluene until
all water present had been driven over. Catalytic amounts
of tin (II) octanoate were added and the mixture refluxed
for 10 hours. After removal of the toluene, washing with
sodium carbonate (to neutralise any free acid present) and
drying under vacuum, a brown liquid was obtained (which
30 h~ extremely viscous on st~n~ ). The product
structure was confirmed by 100 MHz proton NMR, the results
being as follows: -
methyl ether 3.45 ppm singlet (3H)
ethylene protons 3.75 ppm singlet (-30H)
aromatic proton 7.60-8.50 ppm multiplet (9H)
-
CA 02221599 1997-11-19
WO 96/33156 PCTIGB96100911
ComPound 2: Di-r~olvfethYlene qlYCOl)3~ monomethvletherl
benzophenone-4.4-dicarboxvlate
The first step was to prepare 4,4 -dimethyl
benzophenone. To do this, 25 ~1 of p-toluoyl chloride was
~P~ slowly to a stirred mixture of 30 g of anhydLu~s
aluminium trichloride and llS ml of dry toluene. The
resulting solution was refluxed for 6 hours before the
product was isolated by addition of the reaction solution
to a solution of 200 ml water and lOo ml conc. HCl. The
resulting red solid was distilled (short path) at 141~C at
4 mm Hg. The distillate solidified on cooling to a white
solid.
~elting point: = 90-92~C~5 Elemental analysis: Cl5HI40
requires % C = 85.68, H = 6.71
found % C = 85.66, H = 6.44
100 MHz proton NMR: solvent = CD2C12; aromatic protons 7-8
ppm AAIBBl system (8H); methyl protons 2.4 ppm (6H).
The 4,4~-dimethyl benzophenone was then oxidised to
the corresponding dicarboxylic acid. A solution of
glacial acetic acid and the 4,4l-dimethyl benzophenone was
added to a solution of aq. acetic acid 80 % v/v and
chromium trioxide and stirred at ambient temperature for
24 hours. The addition of water facilitated a pale green
pre¢ipitate. Isolation and Hl NMR/elemental analysis
showed partial oxidation. The reaction was driven to
completion by refluxing, at 65~C, for 24 hours.
Ele~ental analy~is: C~5HI005
reguires % C = 66.67, H = 3.74
found % C = 66.71, H = 3.65
60 MHz Hl NMR aromatic protons exhibiting an AAIBB
splitting system 8-8.5 ppm.
% Yield = 67.43%
CA 02221~99 1997-ll-l9
WO 96/33156 ~ ~b~
-- 12 --
FTIR exhibited a large -OH stretch (persisted after
drying product in vacuum oven at 40~C for 48 hours).
The dicarboxylic acid compound was esterified to form
the dimethyl ester. The esterification was achieved by
refluxing in methanol and an acid for 24 hours. The crude
product was taken into THF and washed with water to remove
free acid. Recrys~A~ tion was from dry ethanol.
Characterisation was by Hl NMR which gave the AAIBBI
aromatic splitting pattern (8 . 20-8 . 70 ppm) and a singlet
assigned to the methyl protons (4. 20 ppm). Integration
was in the ratio 4 : 3. FTIR exhibited no -OH stretch
after drying.
Elemental analy~is:
requires % C = 68 .45, H - 4.73
found % C = 68.67, H = 4.54
Finally, the dimethyl ester was transesterified to
form the title compound. 2.5 g Dimethyl ester, 6 . 25 g
poly(ethylene glycol) 350 monomethylether and 500 ml of dry
toluene was refluxed in a Dean and Stark apparatus for 1
hour. 0.25 g of Tilcom [Trade Mark - Ti(OPr)2(0Bu) 2] was
injected and refluxing at 100~C was continued for 4 hours.
The methanol side product was removed using a
fractionating column and the reaction was driven to
completion by refluxing for a further 2.5 hours. Removal
of the catalyst was achieved by addition of 2 ml of water
to the vigorously stirred, cooled solution and filtration
of the resulting precipitate of titanium oxide. The
surplus water was removed by returning the toluene
solution back to the Dean and Stark apparatus for 1 hour.
Removal of the solvent produced a viscous, light brown
liquid (yield c. 2 g).
CA 02221599 1997-11-19
WO 96133156 PCT/~J,,,''~C911
13
Analysi.:
60 MHz Hl NMR;
methyl ether 3.45 ppm singlet (6H)
ethylene protons 3.75 ppm singlet (-58H)
S aromatic protons 7.95-8.50 ppm AAIBBI (8H)
HPLC; flow rate 1 ml/min, 254 nm, 20 micro litre sample
loop, a 50/50 acetonitrile water mobile phase. The
resulting trace showed two distinct ~L Ou~s of peaks,
attributed to the mono and di-substituted PEG esters.
Com~ound 3: 2-BenzovlbenzoYl polY(ethYlene qlYCOl) 3
monometh~lether
This compound was prepared by transesterification of
2-methyl benzoylbenzoate with poly(ethylene glycol) 350
monomethylether, in accordance with the method described
with reference to Compound 2. The product structure was
confirmed by 100 MHz proton NMR, the results being as
follows:
methyl ether 3.75 ppm singlet (3H)
ethylene protons 3.65 ppm singlet (~30H)
aromatic protons 7.20-7.80 ppm multiplet (9H)
comPari~on comPounds
Com~ound C1: Di-r2-(benzoylbenzovl ether)1 polY(ethYlene
~lvcol~ 3~
This compound was prepared by transesterification of
2-~ethylbenzoyl benzoate wi~h poly(ethylene glycol)3~, in
accordance with the method described with reference to
- Compound 2.
CA 02221~99 1997-ll-lg
WO961331~6 PCT/GB96/00911
- 14 -
Com~ound C2: 2-MethvlbenzoYl benzoate
This compound was the commercial product SPEEDCURE
MBB ob~;n~hle from Lambson Fine Chemicals Limited, of
Castleford, U.K.
ComPound C3: 4-Methvl benzovl benzoate
This was prepared by st~n~d esterification from 4-
benzoylbenzoic acid and methanol, in the presence ofconcentrated sulphuric acid. The product was confirmed by
NMR. mp 103-105~C.
Amine Curin~ A~ents
As described below, the standard compound N-
methyldiethanolamine (NMDA) was used in tests. However
also used was a novel compound, 4-N,N-dimethylaminobenzoyl
poly(ethylene glycol)~0monomethylether. This was prepared
as follows.
To a 25 ml round bottom flask equipped with a 50 ml
dropping funnel was added triethylamine (ex. Aldrich)
(1.22 g, 1.1 mol equiv.). Poly(ethylene glycol) 350
monomethyl ether (ex. Fluka) (3.38 g, 0.01 mol) was then
added. The mixture was dissolved in dry THF (20 ml). The
flask was then placed in an ice bath and cooled to 0~C.
4-Dimethyl~mino benzoyl chloride (2.00 g, 0.01 mol)
was also dissolved in dry THF (20 ml). This solution was
placed in the dropping funnel, and added dropwise to the
stirred cooled solution below. After the final addition
of the acid chloride the solution was allowed to stir for
17 hours at ambient t~mr~ature.
CA 02221599 1997-11-19
WO 96/33156
-- 15 --
The reaction was completed by removing the
triethylamine hydrochloride by filtration, and the
r~ ; n i ng solvent under r~ CD~ pressure. The residue
that was left was redissolved in chloroform (50 ml) and
washed with brine (15 ml). The organic layer was then
removed and dried with sodium sulphate, filtered and the
chloroform removed under re~t-ce~ pressure. This yielded
the novel amine compound as a yellow/brown semi-solid,
more liquid than solid.
Da~
Yi~ld: 3.21 g
N~OE~ IH (lOO MHZ, CDC13/TMS) ~ (ppm): 6.s-7.9 (m,Ar,4H); 3.6
(s,PEG,manyH[-28]; 3.3 (s,OMe,3H); 3.0 (~e2N-,6H).
IR (KBr) v cm~~: 2800-2960 (C-H); 1700 (C-O); 1620 (C=O);
770 (Ar)-
C24I~4l09N
Miarr-n~lysis: Requires C 59.13% H: 8.42% N: 2.87~
Found Co 57.81% H: 8.77% N: 2.35%
~omogeneity point: 50-60~C.
Extinction coe~ficient: 215600 @ 310.8 nm
Note on microanalysis
It has been noted that the microanalysis (CHN)
recults on all compounds con~in;ng polyethylene glycol
(PEG) are not as accurate as desired. This is not due to
impurities in the compounds, but to the fact that PEG
compounds contain average chain lengths. For example, a
PEG compound of average molPc~llAr weight 350 contains
chains that vary in length between 2 and 12 ethylene
glycol units. In order to calculate the percentage of
carbon and hydrogen present in the chain it was n~c~ ry
to determine the average number of units present. The
mel:hod of calculating this is shown below.
CA 0222l599 l997-ll-l9
WO 96t33156 PCTIGB96/00911
~ 16 --
PEG ch~;n = 350
Repeating unit (-OCH2CH20-) = 44. 053
Mono methyl ether (OMe) = 31. 034
350 - 31. 034 = 318 . 966
318 . 966 divided by 44 . 053 = 7 . 24
= ~7 repeating units
Therefore, one can assess the number of carbon,
hydrogen and oxygen atoms in the ~h~;n and it is also
possible to calculate the overall number of individual
carbon, hyd~en and oxygen atoms and hpncp the overall
percentage cQmrosition.
SummarY of Compounds
Compound 1 ~
~ C~-PEG350-OMe
Compound 2 l l
MeO--PEG350-O J ~ CO PEG3 5 O OMe
Compound 3 ~ ~G3so~~Me
CA 0222l599 l997-ll-l9
WO 96/33156 PCT/CI~9'J~,~91l
-- 17 --
Co,uE ~ -d Cl ~5PEG30
Co:mpound C2 O C02Me
Co~.npound C3 o
C~2~e
Compound A
NMe2~
O PEG350 OMe
CA 02221599 1997-11-19
PCT/CB~ 0~11
WO 96/33156
-- 18 --
Bffectiveness of Compounds as Curin~ A~ents
For the purpose of these tests, unless otherwise
stated, the st~ rd pre-polymer mixture was as follows:
1-6 hey~ne~iol Diacrylate (monomer) : 93 wt %
amine curing agent : 5 wt %
photoinitiator : 2 wt %.
It was found that all of the compounds ~-3 and
comr~rison compounds C1-C3 above, dissolved into the
prepolymer mixtures.
Curing was by a medium pressure W lamp. The results
presented below relate to the determination of whether the
polymer cures and if so, how quickly, and to the
propensity of the photoinitiators to migrate from a
polymer film after curing. The two methods employed to
address these aspects were Real Time Infra Red (RT-IR) and
High Pressure Liquid Chromography (HPLC).
Methods of Data Acouisition
RT-IR analvsis of the rate of cure of each of the ~re
~olYmer mixtures
The use of RT-IR allows the rate of cure of each of
the samples to be analysed. This method allows an infra-
red spectrum to be taken, then a frequency is chosen, one
at which the transmittance would change during
polymerisation. Commonly used is the acrylate stretch at
810 cm~~, the acrylate double bonds disappearing during
curing. Using the time drive facility, it allows infra-
red analysis while the sample is being irradiated with a
medium pressure W lamp, thus allowing the increase in
CA 02221599 1997-11-19
WO g6/331.56 PCT/GB96/00911
-- 19 --
transmittance to be monitored. This gives an indication
of the rate of polymerisation. A second spectrum was run.
This showed the final position of the acrylate stretch.
Two RT-IR's were run on each pre-polymer mixture.
The samples were prepared as follows. A drop of each pre-
polymer mixture was placed between two polyethylene sheets
in the centre of a polyether spacer of 25 ~m. This was to
ensure that all the films were of the same thickness.
This sandwiched sample was then placed between two NaCl
plates, which in turn were placed in a holder unit. This
entire unit was then placed into the infra-red machine and
the spectrum run.
~5 HPLC method used for analysin~ the ~ro~ensit~ of the
initiators to miqrate
The method for testing the propensity of the
initiators to migrate was the same throughout.
The migratable initiator content of each film was
analysed as follows. Initially a drop of the pre-polymer
mixture was placed on a piece of satinised paper. This
was then evenly spread over the surface using a "K" bar
which gave a film thi~nPcs of between 50-60 ~m.
For each of the inltlators samples of film were taken
after various passes o~ the Colordry unit (i.e., the W
lamp), as specified below in the tables.
The Colordry unit contains a medium pressure mercury
lamp. The samples are placed on a moving belt (in these
trials this was set at 24 metres/minute). It was
important to ensure two main factors during this stage of
the trial. Firstly, that all the samples taken, of
CA 02221~99 1997-11-19
WO 96/33156 PCT/~GI'
-- 20 --
satinised paper and film, were of the same size. It was
for this reason that a metal template was made that gave
samples of 21 x 28 mm. Secondly, that the curing of the
film was as unaffected by oxygen inhibition as possible.
This was ensured by placing the paper and uncured film in
a cell with a quartz window. This cell was then evacuated
with nitrogen and sealed. Only then was the sample passed
through the Colordry unit.
The 21 x 28 mm samples of each pre-polymer mixture
were then placed in individual 7 ml sample vials. To each
vial was ~e~ S ml of a de-gassed acetonitrile/water
50/50 mix, enough to immerse each sample. The vials were
then placed in a dark cupboard for 20 hours. After this
time the vials were removed and the sample extracted from
each vial. All that was left in each vial was the solvent
contAi n; ng the migratables that had leached out from the
film in the 20 hour period.
Method~ of Data ~nalvsis
RT-IR analvsis of the rate of cure of each of the ~re
Polvmer mixtures
2S For each sample three spectra were run, to allow an
average to be taken. Each spectrum had a decay curve
which represents the rate of polymerisation. The steeper
the gradient of the curve, the faster the rate. The angle
of the gradient was measured and this could then be used
to compare the rate of polymerisation achieved by
different curing agents.
. .
CA 02221599 1997-11-19
WO 96133156 ~ 96/
~ 21 --
HPIC method used for anal~sing the miqration of initiators
from films
These samples were then prepared for HPLC analysis by
filtering each one using Sartorus Millistart 0.45 ~m
disposable filters. This was to ensure that there were no
solid contaminants that would damage the HPLC column.
Chromatographs of the initiator components for each
of the films were run prior to the trials. This was to
ensure that firstly the elution time was known and,
secondly, to determine whether the respective initiator
components had any characteristic shaped peaks.
After filtration each sample was injected on to the
HPLC column. Each sample was run in acetonitrile/water
50/50 mixture. The data from each run was then used to
analyse the migratable content of each film.
On the chromatogram for the solution con~;n;ng
migrated initiator compound, the area under each of the
relevant peaks was noted.
It was assumed tha~ the contents of the solution that
had contained the film that had not passed the W lamp,
contained 100% migratable initiator co~o~.d, because the
pre-polymer mixture was not irradiated. The values for
the areas under the peaks from the solutions that had
cont~; n~ films that had passed the W lamp could then be
correlated respectively to the 100% migration value of the
uncured solution.
. . . ~
CA 02221599 1997-11-19
WO 96/33156 ~ l~b3CJ'1~91l
-- 22 --
Test set 1
In the first tests migration of the initiators from
the cured films were studied. The curing agent was N-
methyldiethanolamine (NMDA).
CA 02221599 1997~11~19
WO 96133156 ~23 ~ PCTI~ ~ 'r~,-~ll
V ~
CO O
~, O ~ O ~ ~
o ~1a~
N
q~ ~ O ~ ~ t~
o O
~ ~o
4~ U
o
~ ~1
U
~ ~ ~
r~ D. '3 ~ --I
r O
0 a U
o _I o a:~
O
r o
.,
.1
O ~ o ~ ~
O --I O u~ ~r
O
U
U
~~
~o ~
z o
~ o
CA 02221~99 1997-11-19
WO96133156 PCT/GB96/00911
- 24 -
It will be seen from Table l, that compound l
demonstrates a greatly r~ c~ % of migration, and that
even after 2 passes, the % migration is very low,
indicating a high rate of polymerisation. Compound 2 is
not so effective as compound l but nevertheless the %
migration after 8 passes is similar to that of the
commercial product, compound C2. Compound 2 unexpectedly
achieves significantly better properties after 8 p~c~s
than compound Cl, which is of the type disclosed in JP
6263814. A further significant aspect of the results is
that, whereas compound C3, the 4-methyl ester, has poorer
migration properties than compound C2, the corresponding
2-methyl ester, this pattern is not reproduced in the
analogous PEG ester, where it is the 4-ester, compound l,
which has the better migration properties than the 2-
ester, compound 2.
Test set 2
Further tests were carried out to explore further the
use of compounds of the present invention as
photoinitiators; and to assess their possible use with
end-capped PEG esters of dialkylamino benzoic acids, the
novel compounds of the co-filed application mentioned
above, those PEG esters themselves having very good self-
migration properties.
A) Migration studies as described above were carried out
to assess the degree of migration when the curing agent
was compound A described above (5 wt %), in comr~rison
with NMDA (5 wt %). The photoinitiator was compound 1 (2
wt %) and the monomer HDDA (93 wt %).
CA 02221599 1997-ll-lg
WO96133L56 PCTIGB96/00911
Compound A was found to be fully compatible with the
prepolymer formulation. The results are set out in Table
2 below.
J
CA 02221599 1997-11-19
WO 96133156 - 26 - PCT/GB96/00911
u
P
..
~n
~n
~n
C~
~r
o _I
0
q U
N ~ e
E~l 'I g
_I
1_
~ ~ '' ~
~r ~n ~ o
~ ~D
O o O
~ o o O
~r 'I ~1
30 3
o U
P~
~ o
CA 0222l599 l997-ll-l9
WO 96/33156 . ~ ,r '~00911
~ 27 ~
It is seen that in a well cured film the PEG
substituted aromatic curing compound A agent ~k~c little
difference to the % migration of the PEG-modified
photoinitiator compound 1. Therefore a formulation
combining a low migration initiator and the novel low
migration curing agent is possible.
B) The degree of migration from UV cured films of
compound 2 was ~cs~ss~, in co~p~rison with compound 1.
Due to the favourable results obt~; n~ when curing with
compound A, this was used as the curing agent in these
tests. The results are set out in Table 3 below.
Table 3
15% Mi~ration of compound 1 an~ 2
~o. of passes of 0 2 6 8
~V source
Compound 1 100 51.1 42.81 39.87
20Compound 2 100 30.1 32.53 28.76
It can be seen that compound 2 yields a lower degree
of migration than compound 1. It should be noted that
these results are not properly comp~able to the results
A) above, due to the replacement of the curing belt.
C) Studies of the ~ercentaqe cure of films using a ranqe
of curinq sYstems
Although the novel PEG-substituted photoinitiators
and curing agents have comparatively low migration from
cured films, it was also necessary to test their
efficiency in polymerising HDDA. This was assessed using
FTD~ as described above.
CA 02221~99 1997-11-19
WO 96/33156 PCT/~,;b5~ 'S~911
- 28 -
Curing Agent 5%, photoinitiator 2~, HDDA 93%
a) Ethyl-4-dimethylamino benzoate tEDB]/Compound 1/HDDA
b) Compound A/Compound 1/HDDA
c) Compound A/Compound 2/HDDA
Results
Percentaqe transmittance at 810 cml
No. of ~V O 2 ~ 6
passes
a 7.79 61.74 63.42 60.97
b 13.06 60.24 65.63 60.23
c 12.48 53.43 59.83 70.15
It will be seen that the novel PEG-substituted
initiators and curing agents provide good rates of
reaction and high degrees of polymerisation, in addition
to the good or excellent self-migration properties, shown
by the other tests.
The use of novel photoinitiators of the type of
compounds 1 to 3 can be expected to offer certain further
advantages. Firstly, the use of PEG can be expected to
have a plasticizing effect, useful to increase the
flexibility, and hence the durability, of films.
Secondly, the incorporation of PEG may be expected to
increase the compatibility of the polymers, with paper
surfaces. When polymer films cured using the novel
photoinitiators are used in conjunction with paper, the
extra adherence is desirable. Thirdly, PEG compounds are
likely to be highly soluble in water, such that the amine
curing agents could be used in conjunction with aqueous
CA 02221~99 1997-11-19
WO 96/33156 PCT/<.'rr)~
-- 29 --
curing formulations. These advantages are likely to ~e
furthered, when the novel PEG ~~ified amine curing agents
are also used.
The reader's attentio~ is directed to all papers and
documents which are filed concurrently with or previous to
this specification in connection with this application and
which are open to public inspection with this
specification, and ~he contents of all such papers and
documents are incorporated herein by reference.
All of the features disclosed in this specification
(including any accompanying claims, abstract and
drawings), and/or all of the steps o~ any method or
process so disclosed, may be combined in any combination,
except combinations where at least some of such features
and/or steps are mutually exclusive.
Each feature disclosed in this specification
(including any accompanying claims~ a~stract and
drawings), may be replaced by alternative features serving
the same, equivalent or similar purpose, unless expressly
stated otherwise. Thus, unless e~pressly stated
otherwise, each feature disclosed is one example only of
a ~3eneric series of equivalent or similar features.
The invention is not restricted to the details of the
foregoing embo~ir~nt(s). The invention extends to any
novel one, or any novel combination, of the features
disclosed in this specification (including any
accomr~nying claims, abstract and drawings), or to any
novel one, or any novel combination, of the steps of any
met:hod or process so disclosed.