Note: Descriptions are shown in the official language in which they were submitted.
CA 02226668 1998-O1-13
WO 97/43358 PCT/US97/07990
-1-
O1 AROMATIC ESTERS OF POLYALKYLPHENOXYALKANOLS
02 AND FUEL COMPOSITIONS CONTATNING THE SAME
03
04 BACKGROUND OF THE INVENTION
05
06 Field of the Invention
07
Og This invention relates to aromatic esters of
Og polyalkylphenoxyalkanols and derivatives thereof. In a
further aspect, this invention relates to the use of these
11 compounds in fuel compositions to prevent and control engine
12 deposits.
13
14 Description of the Related Art
16 It is well known that automobile engines tend to form
17 deposits on the surface of engine components, such as
ig carburetor ports, throttle bodies, fuel injectors, intake
19 ports and intake valves, due to the oxidation and
polymerization of hydrocarbon fuel. These deposits, even
21 when present in relatively minor amounts, often cause
22 noticeable driveability problems, such as stalling and poor
23 acceleration. Moreover, engine deposits can significantly
24 increase an automobile's fuel consumption and production of
exhaust pollutants. Therefore, the development of effective
26 fuel detergents or "deposit control" additives to prevent or
27 control such deposits is of considerable importance and
2g numerous such materials are known a.n the art.
29
For example, aliphatic hydrocarbon-substituted phenols are
31 known to reduce engine deposits when used in fuel
. 32 compositions. U.S. Patent No. 3,849,085, issued
33 November 19, 1974 to Itreuz et al., discloses a motor fuel
34 composition comprising a mixture of hydrocarbons in the
CA 02226668 1998-O1-13
WO 97/43358 PCT/fJS97/07990
-2-
O1 gasoline boiling range containing about 0.01 to 0.25 volume
02 percent of a high molecular weight aliphatic
03 hydrocarbon-substituted phenol in which the aliphatic
04 hydrocarbon radical has an average molecular weight in the
05 range of about 500 to 3,500. This patent teaches that
06 gasoline compositions containing minor amounts of an
aliphatic hydrocarbon-substituted phenol not only prevent or
Og inhibit the formation of intake valve and port deposits in a
Og gasoline engine, but also enhance the performance of the
ZO fuel composition in engines designed to operate at higher
11 operating temperatures with a minimum of decomposition and
12 deposit formation in the manifold of the engine.
13
14 Similarly, U.S. Patent No. 4,134,846, issued January 16,
15 lg~g to Machleder et al., discloses a fuel additive
16 composition comprising a mixture of (1) the reaction product
1~ of an aliphatic hydrocarbon-substituted phenol,
lg epichlorohydrin and a primary or secondary mono- or
lg polyamine, and (2) a polyalkylene phenol. This patent
20 teaches that such compositions show excellent carburetor,
21 induction system and combustion chamber detergency and, in
22 addition, provide effective rust inhibition when used in
23 hydrocarbon fuels at low concentrations.
24
25 wino phenols are also known to function as
26 detergentsjdispersants, antioxidants and anti-corrosion
2~ agents when used in fuel compositions. U.S. Patent
2g No. 4,320,021, issued March 16, 1982 to R. M. Lange, for
29 example, discloses amino phenols having at least one
30 substantially saturated hydrocarbon-based substituent of at
31 least 30 carbon atoms. The amino phenols of this patent are
32 taught to impart useful and desirable properties to
33 oil-based lubricants and normally liquid fuels.
34
CA 02226668 1998-O1-13
W~ 97/43358 PCT/US97I07990
-3-
OZ Similarly, U.S. Patent No. 3,149,933, issued September 22,
02 1964 to K. Ley et al., discloses hydrocarbon-substituted
03 amino phenols as stabilizers for liquid fuels.
04
05 U.S. Patent No. 4,386,939, issued June 7, 1983 to
06 R- M. Lange, discloses nitrogen-containing compositions
O7 prepared by reacting an amino phenol with at least one 3- or
Og 4-membered ring heterocyclic compound in which the hetero
Og atom is a single oxygen, sulfur or nitrogen atom, such as
ethylene oxide. The nitrogen-containing compositions of
11 this patent are taught to be useful as additives for
12 lubricants and fuels.
13
14 Nitro phenols have also been employed as fuel additives.
For example, U.S. Patent No. 4,347,148, issued August 31,
16 1982 to K. E. Davis, discloses vitro phenols containing at
17 least one aliphatic substituent having at least about 40
lg carbon atoms. The vitro phenols of this patent are taught
lg to be useful as detergents, dispersants, antioxidants and
demulsifiers for lubricating oil and fuel compositions.
21
22 Similarly, U.S. Patent No. 3,434,814, issued March 25, 1969
23 to M. Dubeck et.al., discloses a liquid hydrocarbon fuel
24 composition containing a major quantity of a liquid
hydrocarbon of the gasoline boiling range and a minor amount
26 sufficient to reduce exhaust emissions and engine deposits
27 of an aromatic vitro compound having an alkyl, aryl,
2g aralkyl, alkanoyloxy, alkoxy, hydroxy or halogen
29 substituent.
31. More recently, certain poly(oxyalkylene) esters have been
32 shown to reduce engine deposits when used in fuel
33 compositions. U.S. Patent No. 5,211,721, issued May 18,
34 1893 to R. L. Sung et al., for example, discloses an oil
CA 02226668 1998-O1-13
WO 97/43358 PC'.~/US97/07990
-4-
01 soluble polyether additive comprising the.reaction product
02 of a polyether polyol with an acid represented by the
03 formula RCOOH in which R is a hydrocarbyl radical having
04 6 to 27 carbon atoms. The poly(oxyalkylene) ester compounds '
05 of this patent are taught to be useful for inhibiting
06 carbonaceous deposit formation, motor fuel hazing, and as '
p7 ORI inhibitors when employed as soluble additives in motor
pg fuel compositions.
09
Poly(oxyalkylene) esters of amino- and nitrobenzoic acids
il are also known in the art. For example, U.S. Patent
12 No. 2,714,607, issued August 2, 1955 to M. Matter, discloses
13 polyethoxy esters of aminobenzoic acids, nitrobenzoic acids
14 and other isocyclic acids. These polyethoxy esters are
taught to have excellent pharmacological properties and to
16 be useful as anesthetics, spasmolytics, analeptics and
17 bacteriostatics.
18
ig Similarly, U.S. Patent No. 5,090,914, issued February 25,
igg2 to D. T. Reardan et al., discloses poly(oxyalkylene)
21 aromatic compounds having an amino or hydrazinocarbonyl
22 substituent on the aromatic moiety and an ester, amide,
23 carbamate, urea. or ether linking group between the aromatic
24 moiety and the poly(oxyalkylene) moiety. These compounds
are taught to be useful for modifying macromolecular species
26 such as proteins and enzymes.
27
2g U.S. Patent No. 4,328,322, issued September 22, 1980 to
29 R~ C. Baron, discloses amino- and nitrobenzoate esters of
3p oligomeric polyols, such as polyethylene) glycol. These
31 materials are used in the production of synthetic polymers
32 bY reaction with a polyisocyanate.
33
34
CA 02226668 1998-O1-13
WO 97/43358 PCT/US97/07990
-5-
OZ U.S. Patent No. 4,859,210, issued August 22, 1989 to Franz
02 et al., discloses fuel compositions containing (1) one or
03 more polybutyl or polyisobutyl alcohols wherein the
04 polybutyl or polyisobutyl group has a number average
05 molecular weight of 324 to 3,000, or (2) a poly(alkoxylate)
' 06 of the polybutyl or polyisobutyl alcohol, or (3) a
07 carboxylate ester of the polybutyl or polyisobutyl alcohol.
Og This patent further teaches that when the fuel composition
09 contains an ester of a polybutyl or polyisobutyl alcohol,
the ester-forming acid group may be derived from saturated
11 or unsaturated, aliphatic or aromatic, acyclic or cyclic
12 mono- or polycarboxylic acids.
13
14 U.S. Patent Nos. 3,285,855, and 3,330,859 issued
November 15, 1966 and July 11, 1967 respectively, to Dexter
16 et al., disclose alkyl esters of dialkyl hydroxybenzoic and
17 hydroxyphenylalkanoic acids wherein the ester moiety
lg contains from 6 to 30 carbon atoms. These patents teach
lg that such esters are useful for stabilizing polypropylene
and other organic material normally subject to oxidative
21 deterioration. Similar alkyl esters containing hindered
22 dialkyl hydroxyphenyl groups are disclosed in U.S. Patent
23 No. 5,196,565, which issued March 23, 1993 to Ross.
24
U.S. Patent No. 5,196,142, issued March 23, 1993 to Mollet
26 et al., discloses alkyl esters of hydroxyphenyl carboxylic
27 acids wherein the ester moiety may contain up to 23 carbon
2g atoms. This patent teaches that such compounds are useful
2g as antioxidants for stabilizing emulsion-polymerized
polymers.
31
32 MY prior U.S. Patent No. 5,407,452, issued April 18, 1995,
33 and corresponding International Application Publication No.
34 W~ 95/04118, published February 9, 1995, disclose certain
CA 02226668 1998-O1-13
WO 97/4338 PCf/iJS97l07990
-6-
O1 poly(oxyalkylene) vitro and aminoaromatic esters having from
02 5 to 100 oxyalkylene units and teach the use of such
03 compounds as fuel additives for the prevention and control
04 of engine deposits. '
05
06 Similarly, my prior U.S. Patent No. 5,427,591, issued
07 June 27, 1995, and corresponding International Application
Og Publication No. WO 94/14926, published July 7, 1994,
09 disclose certain poly(oxyalkylene) hydroxyaromatic esters
which are useful as fuel additives to control engine
11 deposits.
12
13 In addition, my prior U.S. Patent No. 5,380,345, issued
14 January 10, 1995, and corresponding International
Application Publication No. WO 95j15366, published June 8,
16 1995, disclose certain polyalkyl vitro and aminoaromatic
17 esters useful as deposit control additives for fuels.
ig Moreover, my prior International Application Publication No.
ig WO 95/11955, published May 4, 1995, discloses certain
2o polyalkyl hydroxyaromatic esters which are also useful as
21 deposit control fuel additives.
22
23 SUMMARY OF THE INVENTION
24
I have now discovered certain aromatic esters of
26 polyalkylphenoxyalkanols which provide excellent control of
27 engine deposits, especially intake valve deposits, when
2g employed as fuel additives in fuel compositions.
29
3o The compounds of the present invention include those having
31 the following formula and fuel soluble salts thereof:
32
33
34
CA 02226668 1998-O1-13
WO 97/43358 PCT/L1S97/07990
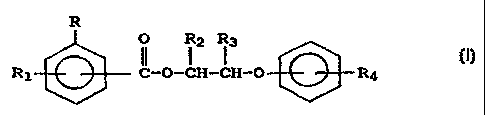
01 R
O R2 Rg
02 II I I .
03 R1 C-O-CH-CH-O R4 (I)
04
05 wherein R is hydroxy, vitro or -(CH2)X-NR5R6, wherein R5 and
' 06 R6 are independently hydrogen or lower alkyl having 1 to 6
O~ carbon atoms and x is 0 or 1;
08
p9 Rl is hydrogen, hydroxy, vitro or -NR7Rg, wherein R7 and Rg
are independently hydrogen or lower alkyl having 1 to 6
11 carbon atoms;
12
13 R2 and R3 are independently hydrogen or lower alkyl having 1
14 to 6 carbon atoms; and
16 R4 is a polyalkyl group having an average molecular weight
1~ in the range of about 450 to 5,000.
18
i9 The present invention further provides a fuel composition
comprising a major amount of hydrocarbons boiling in the
21 gasoline or diesel range and a deposit-controlling effective
22 amount of a compound of the present invention.
23 . .
24 The present invention additionally provides a fuel
concentrate comprising an inert stable oleophilic organic
26 solvent boiling in the range of from about 150°F. to 400°F.
27 and from about 10 to 70 weight percent of a compound of the
28 present invention.
29
Among other factors, the present invention is based on the
31 surprising discovery that certain aromatic esters of
32 polyalkylphenoxyalkanols provide excellent control of engine
33 deposits, especially on intake valves, when employed as
34 additives in fuel compositions.
CA 02226668 1998-O1-13
WO 97/43358 PCT/US97J07990
_g_
Ol DETP~ILED DESCRIPTION OF THE INVENTION
02
03 Based on performance (e. g. deposit control), handling
04 properties and performance/cost effectiveness, the preferred
05 compounds of the invention are those wherein R is vitro,
06 amino, N-alkylamino, or -CH2NH2 (aminomethyl). More
07 preferably, R is a vitro, amino or --CH2NH2 group. Most
O8 preferably, R is an amino or -CH NH rou
2 2 g po especially
Og amino. Preferabl R is h dro en h dro
Y. 1 Y g . Y xY. vitro or amino.
More referabl R is h dro en or h dro
p Y. 1 Y 9 y xy. Most
il preferably, R1 is hydrogen. Preferably, R4 is a polyalkyl
12 group having an average molecular weight in the range of
13 about 500 to 3 000 more
preferably about 700 to 3,000, and
14 most preferably about 900 to 2,500. Preferably, the compound
has a combination of preferred substituents.
16
17 Preferably, one of R2 and R3 is hydrogen or lower alkyl of 1
i$ to 4 carbon atoms, and the other is hydrogen. More
ig referabl
p y, one of R2 and R3 is hydrogen, methyl or ethyl,
and the other is h dro en. Most
Y g preferably, R2 is hydrogen,
21 methyl or ethyl, and Rg is hydrogen.
22
23 yen R and or R ' -is an N-alk lamino
/ 1 Y group, the alkyl group
24 of the N-alk lamino moiet
Y y preferably contains 1 to 4 carbon
atoms. More preferably, the N-alkylamino is N-methylamino
26 or N-ethylamino.
27
2$ Similarly, when R and/or R1 is an N,N-dialkylamino group,
29 each alkyl group of the N,N-dialkylamino moiety-preferably
contains 1 to 4 carbon atoms. More preferably, each alkyl
31 group is either methyl or ethyl. For example, particularly
32 referred N N-dialk lamino
p , y groups are N,N-dimethylamino,
33 N-ethyl-N-methylamino and N,N-diethylamino groups.
34
CA 02226668 1998-O1-13
WO 97/43358 PCT/CIS97/07990
-9-
01 A further preferred group of compounds are those wherein R
02 is amino, vitro, or -CH2NH2 and Rl is hydrogen or hydroxy.
03 ~1 particularly preferred group of compounds are those
' 04 wherein R is amino, Rl, R2 and R3 are hydrogen, and R4 is a
05 polyalkyl group derived from polyisobutene.
06
0~ It is preferred that the R substituent is located at the
Og mete or, more preferably, the pare position of the benzoic
Og acid moiety, i.e., pare or mete relative to the carbonyloxy
group. When R1 is a substituent other than hydrogen, it is
11 particularly preferred that this R1 group be in a mete or
12 pare position relative to the carbonyloxy group and in an
13 ortho position relative to the R substituent. Further, in
14 general, when Rl is other than hydrogen, it is preferred
that one of R or Rl is located pares to the carbonyloxy group
16 and the other is located mete to the carbonyloxy group.
1~ Similarly, it is preferred that the R4 substituent on the
ig other phenyl ring is located pare or mete, more preferably
lg pare, relative to the ether linking group.
21 The compounds of the present invention will generally have a
22 sufficient molecular weight so as to be non-volatile at
23 normal engine intake valve operating temperatures
24 (about 200°-250°C). Typically, the molecular weight of the
compounds of this invention will range from about 700 to
26 about 3,500, preferably from about 700 to about 2,500.
27
2g Fuel-soluble salts of the compounds of formula I can be
2g readily prepared for those compounds containing_an amino or
substituted amino group and such salts are contemplated to
31 be useful for preventing or controlling engine deposits.
32 Suitable salts include, for example, those obtained by
33 protonating the amino moiety with a strong organic acid,
34
CA 02226668 1998-O1-13
WO 97/43358 PCT/US97/07990
-10-
O1 such as an alkyl- or arylsulfonic acid. Preferred salts are
02 derived from toluenesulfonic acid and methanesulfonic acid.
03
04 When the R or Rl substituent is a hydroxy group, suitable "
05 salts can be obtained by deprotonation of the hydroxy group
06 with a base. Such salts include salts of alkali metals, '
p7 alkaline earth metals, ammonium and substituted ammonium
Og salts. Preferred salts of hydroxy-substituted compounds
Og include alkali metal, alkaline earth metal and substituted
ammonium salts.
11
12 pefinitions
13
14 As used herein, the following terms have the following
meanings unless expressly stated to the contrary.
is
17 The term "amino" refers to the group: -NH2.
18
19 The term "N-alkylamino" refers to the group: -NHRa wherein
~Ra is an alkyl group. The term "N,N-dialkylamino" refers to
21 the group: -~TRbR~, wherein Rb and R~ are alkyl groups.
22
23 The term "alkyl°!-refers to both straight- and branched-chain
24 alkyl groups.
26 The term "lower alkyl" refers to alkyl groups having 1 to
27 about 6 carbon atoms and includes primary, secondary and
28 tertiary alkyl groups. Typical lower alkyl groups include,
29 for example, methyl, ethyl, n-propyl, isopropyl, n-butyl,
sec-butyl, t-butyl, n-pentyl, n-hexyl and the like.
31
32 The term "polyalkyl" refers to an alkyl group which is
33 generally derived from polyolefins which are polymers or
34 copolymers of mono-olefins, particularly 1-mono-olefins,
CA 02226668 1998-O1-13
WO 97!43358 PCT/US97/07990
-11-
O1 such as ethylene, propylene, butylene, and the like.
02 Preferably, the mono-olefin employed will have 2 to about
03 24 carbon atoms, and more preferably, about 3 to 12 carbon
04 atoms. More preferred mono-olefins include propylene,
05 butylene, particularly isobutylene, 1-octene and 1-decene.
06 polYolefins prepared from such mono-olefins include
O~ polypropylene, polybutene, especially polyisobutene, and the
Og -polyalphaolefins produced from 1-octene and 1-decene.
09
The term "fuel" or °'hydrocarbon fuel" refers to normally
11 liquid hydrocarbons having boiling points in the range of
12 gasoline and diesel fuels.
13
14 General Synthetic Procedures
16 The polyalkylphenoxyalkyl aromatic esters of this invention
1~ may be prepared by the following general methods and
lg procedures. It should be appreciated that where typical or
lg preferred process conditions (e. g., reaction temperatures,
times, mole ratios of reactants, solvents, pressures, etc.)
21 are given, other process conditions may also be used unless
22 otherwise stated. Optimum reaction conditions may vary with
23 the particular reactants or solvents used, but such
24 conditions can be determined by one skilled in the art by
routine optimization procedures.
26
2~ Those skilled in the art will also recognize that it may be
2g necessary to block or protect certain functional groups
2g while conducting the following synthetic procedures. In
such cases, the protecting group will serve to protect the
31 functional group from undesired reactions or to block its
32 undesired reaction with other functional groups or with the
33 reagents used to carry out the desired chemical
34 transformations. The proper choice of a protecting group
CA 02226668 1998-O1-13
WO 97/43358 PC~YITS97/07990
-12-
O1 for a particular functional group will be readily apparent
02 to one skilled in the art. Various protecting groups and
03 their introduction and removal are described, for example,
04 in T. W. Greene and P. G. M. Wuts, Protective Groups in
05 Organic Synthesis, Second Edition, Wiley, New York, 1991,
06 and references cited therein.
07
Og In the present synthetic procedures, a hydroxyl group will
pg preferably be protected, when necessary, as the benzyl or
tert-butyldimethylsilyl ether. Introduction and removal of
11 these protecting groups is well described in the art. Amino
12 groups may also require protection and this may be
13 accomplished by employing a standard amino protecting group,
14 such as a benzyloxycarbonyl or a trifluoroacetyl group.
Additionally, as will be discussed in further detail
16 hereinbelow, the aromatic esters of this invention having an
17 amino group on the aromatic moiety will generally be
ig prepared from the corresponding vitro derivative.
ig accordingly, in many of the following procedures, a vitro
group will serve as a protecting group for the amino moiety.
21
22 Moreover, the compounds of this invention having a -CH2NH2
23 group on the aromatic moiety will generally be prepared from
24 the corresponding cyano derivative, -CN. Thus, in many of
the following procedures, a cyano group will serve as a
26 protecting group for the -CH2NH2 moiety.
27
2g Synthesis
29
The polyalkylphenoxyalkyl aromatic esters of the present
31 invention may be prepared by a process which initially
32 involves hydroxyalkylation of a poiyalkylphenol of the
33 formula:
34
CA 02226668 1998-O1-13
WO 97/4338 PCT/LTS97/07990
-13-
Ol
02
03 Ho R4 (=I)
04
05
06 wherein R4 is as defined herein, with an alkylene carbonate
07 of the formula:
O8
0
09
O (III)
Z1
12
13 R2 R3
14
wherein R2 and R3 are as defined herein, in the presence of
16 a catalytic amount of an alkali metal hydride or hydroxide,
17 or alkali metal salt, to provide a polyalkylphenoxyalkanol
lg of the formula:
19
2 0 R2 R3
21 HO-CH-CH-O R4 (IV)
22
23 . .
24 wherein R2, R3 and R4 are as defined herein.
26 The polyalkylphenols of formula II are well known materials
27 and are typically prepared by the alkylation of phenol with
28 the desired polyolefin or chlorinated polyolefin. A further
29 discussion of polyalkylphenols can be found, for example, in
U.S. Patent No. 4,744,921 and U.S. Patent No. 5,300,701.
31
32 Accordingly, the polyalkylphenols of formula II may be
33 prepared from the corresponding olefins by conventional
34 procedures. For example, the poiyalkylphenols of formula II
CA 02226668 1998-O1-13
W~ 97/43358 PC~YUS97/07990
-24-
01 above may be prepared by reacting the appropriate olefin or
02 olefin mixture with phenol in the presence of an alkylating
03 catalyst at a temperature of from about 25°C. to 150°C., and
04 preferably 30°C. to 100°C. either neat or in an essentially
05 inert solvent at atmospheric pressure. A preferred
06 alkylating catalyst is boron trifluoride. Molar ratios of
07 reactants may be used. Alternatively, molar excesses of
pg phenol can be employed, i.e., 2 to 3 equivalents of phenol
Og for each equivalent of olefin with unreacted phenol
recycled. The latter process maximizes monoalkylphenol.
11 Examples of inert solvents include heptane, benzene,
12 toluene, chlorobenzene and 250 thinner which is a mixture of
13 aromatics, paraffins and naphthenes.
14
The polyalkyl substituent on the polyalkylphenols employed
16 in the invention is generally derived from polyolefins which
17 are polymers or copolymers of mono-olefins, particularly
ig 1-mono-olefins, such as ethylene, propylene, butylene, and
19 the like. Preferably, the mono-olefin employed will have 2
to about 24 carbon atoms, and more preferably, about 3 to 12
21 carbon atoms. More preferred mono-olefins include
22 propylene, butylene, particularly isobutylene, 1-octane and
23 1-decene. Polyolefins prepared from such mono-olefins
24 include polypropylene, polybutene, especially polyisobutene,
and the polyalphaolefins produced from 1-octane and
26 1-decene.
27
2g The preferred polyisobutenes used to prepare the presently
29 employed polyalkylphenols are polyisobutenes which comprise
at least about 20% of the more reactive methylvinylidene
31 isomer, preferably at least 50% and more preferably at least
32 70%. Suitable polyisobutenes include those prepared using .
33 BF3 catalysts. The preparation of such polyisobutenes in
34 which the methylvinylidene isomer comprises a high
CA 02226668 1998-O1-13
WO 97/43358 PCT/US97/07990
-15-
01 percentage of the total composition is described in U.S.
02 Patent Nos. 4,152,499 and 4,605,808. Such polyisobutenes,
03 known as "reactive" polyisobutenes, yield high molecular
04 weight alcohols in which the hydroxyl group is at or near
05 the end of the hydrocarbon chain. Examples of suitable
06 PolYisobutenes having a high alkylvinylidene content include
07 Ultravis 30, a polyisobutene having a number average
pg molecular weight of about 1300 and a methylvinylidene
pg content of about 74%, and Ultravis 10, a polyisobutene
having a number average molecular weight of about 950 and a
11 methylvinylidene content of about 76%, both available from
12 British Petroleum.
13
14 The alkylene carbonates of formula III are known compounds
35 which are available commercially or can be readily prepared
16 using conventional procedures. Suitable alkylene carbonates
17 include ethylene carbonate, propylene carbonate, 1,2-
ig butylene carbonate, 2,3-butylene carbonate, and the like. A
19 preferred alkylene carbonate is ethylene carbonate.
21 The catalyst employed in the reaction of the polyaklyphenol
22 and alkylene carbonate may be any of the well known
23 hydroxyalkylation catalysts. Typical hydroxyalkylation
24 catalysts include alkali metal hydrides, such as lithium
hydride, sodium hydride and potassium hydride, alkali metal
26 hydroxides, such as sodium hydroxide and potassium
27 hydroxide, and alkali metal salts, for example, alkali metal
2g halides, such as sodium chloride and potassium chloride, and
2g alkali metal carbonates, such as sodium carbonate and
potassium carbonate. The amount of catalyst employed will
31 generally range from about 0.01 to 1.0 equivalent,
32 preferably from about 0.05 to 0.3 equivalent.
33
34
CA 02226668 1998-O1-13
WO 97/43358 PCTlUS97/07990
-16-
O1 The polyalkylphenol and alkylene carbonate are generally
02 reacted in essentially equivalent amounts in the presence of
03 the hydroxyalkylation catalyst at a temperature in the range
04 of about 100°C. to 210°C., and preferably from about
I50°C.
05 to about 170°C. The reaction may take place in the presence
06 or absence of an inert solvent. -
07
Og The time of reaction will vary depending on the particular
Og alkylphenol and alkylene carbonate reactants, the catalyst
used and the reaction temperature. Generally, the reaction
11 time will range from about two hours to about five hours.
12 The progress of the reaction is typically monitored by the
13 evolution of carbon dioxide. At the completion of the
14 reaction, the polyalkylphenoxyalkanol product is isolated
using conventional techniques.
16
17 The hydroxyalkylation reaction of phenols with alkylene
ig carbonates is well known in the art and is described, for
ig example, iri U.S. Patent NOS. 2,987,555; 2,967,892; 3,283,030
~ and 4,341,905.
21
22 Alternatively, the polyalkylphenoxyalkanol product of
23 formula IV may be prepared by reacting the polyalkylphenol
24 of formula II with an alkylene oxide of the formula:
26
27
28 R2-CH CH-R3 (V)
29
wherein RZ and R3 are as defined herein, in the'presence of
31 a hydroxyalkylation~catalyst as described above.
32
33 Suitable alkylene oxides of formula V include ethylene
34 oxide, propylene oxide, 1,2-butylene oxide, 2,3-butylene ,
CA 02226668 1998-O1-13
WO 97/43358 PCT/fTS97/07990
-17-
O1 oxide, and the like. A preferred alkylene oxide is ethylene
02 oxide.
03
04 In a manner similar to the reaction with alkylene carbonate,
05 the polyalkylphenol and alkylene oxide are reacted in
06 essentially equivalent or equimolar amounts in the presence
07 of 0.01 to 1.0 equivalent of a hydroxyalkylation catalyst,
pg such as sodium or potassium hydride, at a temperature in the
Og range of about 30°C. to about 150°C., for about 2 to about
l0 24 hours. The reaction may be conducted in the presence or
11 absence of a substantially anhydrous inert solvent.
12 Suitable solvents include toluene, xylene, and the like.
13 Generally, the reaction conducted at a pressure sufficient
Z4 to contain the reactants and any solvent present, typically
15 at atmospheric or higher pressure. Upon completion of the
16 reaction, the polyalkylphenoxyalkanol is isolated by
~ conventional procedures.
18
lg The polyalkylphenoxyalkanol of formula IV is subsequently
20 reacted with a substituted benzoic acid of formula VI to
21 provide the aromatic ester compounds of formula I. This
22 reaction can be represented as follows:
23 . _
24 ~ R
2 5 ~' ~ 2 'R3
26 Rl C-OH + HO-CH-CH-O R4 -->
27
28
29 (VI) (IV)
31 R
32
33 Rl C-O-CH-CH-O' R4 (I)
34
CA 02226668 1998-O1-13
WO 97/43358 PCT/ITS97/07990
-18-
O1 wherein R, R1, R2, R3 and R4 are as defined herein, and
02 wherein any hydroxy or amino substituent on the substituted
03 benzoic acid of formula VI is preferably protected with a
04 suitable protecting group, for example, a benzyl or nitro
05 group, respectively. Moreover, a -CH2NH2 substituent on the
06 aromatic ring will preferably be protected by the use of a
07 cyano group, CN.
08
09 This reaction is typically conducted by contacting a
polyalkylphenoxyalkanol of formula IV with about 0.25 to
il about 1.5 molar equivalents of the corresponding substituted
12 and protected benzoic acid of formula VI in the presence of
13 an acidic catalyst at a temperature in the range of about
14 70°C. to about 160°C. for about 0.5 to about 48 hours.
Suitable acid catalysts for this reaction include p-toluene
16 sulfonic acid, methanesulfonic acid and the like.
17 Optionally, the reaction can be conducted in the presence of
18 an inert solvent, such as benzene, toluene and the like.
19 The water generated by this reaction is preferably removed
during the course of the reaction, for example, by
21 azeotropic distillation.
22
23 The substituted benzoic acids of formula VI are generally
24 known compounds and can be prepared from known compounds
using conventional procedures or obvious modifications
26 thereof. Representative acids suitable for use as starting
27 materials include, for example, 2-aminobenzoic acid
28 (anthranilic acid), 3-aminobenzoic acid, 4-aminobenzoic
29 acid, 3-amino-4-hydroxybenzoic acid,
4-amino-3-hydroxybenzoic acid, 2-nitrobenzoic acid,
31 3-nitrobenzoic acid, 4-nitrobenzoic acid,
32 3-hydroxy-4-nitrobenzoic acid, 4-hydroxy-3-nitrobenzoic
33 acid. When the R substituent is ~H2-NR5R6, suitable
34
CA 02226668 1998-O1-13
WO 97/43358 PCT/US97/07990
-19-
O1 starting materials include 4-cyanobenzoic acid and
02 3-cyanobenzoic acid.
03
04 preferred substituted benzoic acids include 3-nitrobenzaic
05 acid, 4-nitrobenzoic acid, 3-hydroxy-4-nitrobenzoic acid,
06 4-hydroxy-3-nitrobenzoic acid, 3-cyanobenzoic acid and
07 4-cyanobenzoic acid.
08
Og The compounds formula I or their suitably protected analogs
also can be prepared by reacting the polyalkylphenoxyalkanol
11 of formula IV with an acid halide of the substituted benzoic
12 acid of formula VI such as an acid chloride or acid bromide.
13 This can be represented by the following reaction equation:
14
R
16 ~~ i 2 ~R3
17 R~ C-X + HO-CH-CH-O R4 ->
18
19 (VII) (IV)
21 R
22
23 R1 . C-O-CH-CH-O R4 (I)
24
26 wherein X is halide, typically chloride or bromide, and R,
27 Rl, R2, R3 and Rq are as defined herein above, and wherein
28 any hydroxy or amino substituents on the acid halide of
29 formula VII are preferably protected with a suitable
protection group, for example, benzyl or nitro,
31 respectively. Also, when R is --CH2NR5R6, a suitable
32 starting material is a cyanobenzoyl halide.
33
34
CA 02226668 1998-O1-13
WO 97/43358 ~CT/LTS97/U7990
-20-
01 Typically, this reaction is conducted by contacting the
02 polyalkylphenoxyaikanol of formula IV with about 0.9 to
03 about 1.5 molar equivalents of the acid halide of
04 formula VII in an inert solvent, such as, for example,
05 toluene, dichloromethane, diethyl ether, and the like, at a
06 temperature in the range of about ~5°C. to about 150°C. The
reaction is generally complete in about 0.5 to about
08 48 hours. Preferably, the reaction is conducted in the
O9 presence of a sufficient amount of an amine capable of
neutralizing the acid generated during the reaction, such
11 as, for example, triethylamine, di(isopropyl)ethylamine,
12 pyridine or 4-dimethylaminopyridine.
13
14 When the benzoic acids of formula VI or acid halides of
formula VII contain a hydroxyl group, protection of the
16 aromatic hydroxyl groups may be accomplished using
1~ well-known procedures. The choice of a suitable protecting
18 group for a particular hydroxybenzoic carboxylic acid will
19 be apparent to those skilled in the art. Various protecting
groups, and their introduction and removal, are described,
21 for example, in T. W. Greene and P. G. M. Wuts, Protective
22 Groups in Organic Synthesis, Second Edition, Wiley, New
23 York, 1991, and.references cited therein.
24
After completion of the esterification, deprotection of the
2s aromatic hydroxyl group can also be accomplished using
2~ conventional procedures. Appropriate conditions for this
28 deprotection step will depend upon the protecting groups)
2g utilized in the synthesis and will be readily apparent to
those skilled in the art. For example, benzyl protecting
31 groups may be removed by hydrogenolysis under 1 to about 4
32 atmospheres of hydrogen in the presence of a catalyst, such -
33 as palladium on carbon. Typically, this deprotection
34 reaction is conducted in an inert solvent, preferably a
CA 02226668 1998-O1-13
WO 97143358 PCTIUS97l07990
-21-
O1 mixture of ethyl acetate and acetic acid, at a temperature
02 of from about O°C. to about 40°C. for about 1 to about
03 24 hours.
04
05 When the benzoic acids of formula VI or acyl halides of
06 formula VII have a free amino group (-NH2) on the phenyl
07 moiety, it is generally desirable to first prepare the
Og corresponding vitro compound (i.e., where R and/or R1 is a
Og vitro group) using the above-described synthetic procedures,
including preparation of the acyl halides, and then reduce
11 the vitro group to an amino group using conventional
12 procedures. Aromatic vitro groups may be reduced to amino
13 groups using a number of procedures that are well known in
14 the art. For example, aromatic vitro groups may be reduced
under catalytic hydrogenation conditions; or by using a
16 reducing metal, such as zinc, tin, iron and the like, in the
17 presence of an acid, such as dilute hydrochloric acid.
lg Generally, reduction of the vitro group by catalytic
lg hydrogenation is preferred. Typically, this reaction is
~ conducted using about 1 to 4 atmospheres of hydrogen and a
21 platinum or palladium catalyst, such as palladium on carbon.
22 The reaction is typically carried out at a temperature of
23 about OQC. to about 100QC. for about 1 to 24 hours in an
24 inert solvent, such as ethanol, ethyl acetate and the like.
Hydrogenation of aromatic vitro groups is discussed in
26 further detail in, for example, P. N. Rylander, Catalytic
27 Hydrogenation in Organic Synthesis, pp. 113-137, Academic
2g Press (1979); and Organic Synthesis, Collective Vol. I,
29 Second Edition, pp. 240-241, John Wiley & Sons,_Inc. (1941);
and references cited therein.
31
_ 32 Likewise, when the benzoic acids of formula VI or acyl
33 halides of formula VII contain a -CH2NH2 group on the phenyl
34 moiety, it is generally desirable to first prepare the
CA 02226668 1998-O1-13
WO 97/43358 PCT/US97/07990
-22-
O1 corresponding cyano compounds (i.e., where R and/or R1 is a
02 ~N groupj, and then reduce the cyano group to a ~H2NH2
03 group using conventional procedures. Aromatic cyano groups
04 may be reduced to --CH2NH2 groups using procedures well
O5 known in the art. For example, aromatic c ano
06 Y groups may be
reduced under catalytic hydrogenation conditions similar to
O? those described above for reduction of aromatic vitro groups
08 to amino groups. Thus, this reaction is typically conducted
09 using about 1 to 4 atmospheres of h dro en and a
l0 y g platinum or
palladium catalyst, such as palladium on carbon. Another
11 suitable catalyst is a Lindlar catalyst, which is palladium
12 on calcium carbonate. The hydrogenation may be carried out
13 at temperatures of about 0°C. to about 100°C. for about 1 to
14
24 hours in an inert solvent such as ethanol, ethyl acetate,
and the like. Hydrogenation of aromatic cyano groups is
16
further discussed i.n the references cited above for
1?
reduction of aromatic vitro groups.
18
19
The acyl halides of formula VII can be prepared by
contacting the corresponding benzoic acid compound of
21
formula VI with an inorganic acid halide, such as thionyl
22
chloride, phosphorous trichloride, phosphorous tribromide,
23
or phosphorous pentachloride; or with oxalyl chloride.
24
Typically, this reaction will be conducted using about 1 to
5 molar equivalents of the inorganic acid halide or oxalyl
26
chloride, either neat or in an inert solvent, such as
27
diethyl ether, at a temperature in the range of about 20°C.
28
to about 80°C, for about 1 to about 48 hours. A catalyst,
29
such as N,N-dimethylformamide, may also be used in this
31 reaction. Again it is preferred to first protect any
32 hydroxy or amino substituents before converting the benzoic
33 acid to the acyl halide.
34
CA 02226668 1998-O1-13
WO 97/43358 PCT/US97/07990
-23-
O1 Fuel Comt~ositions
02
03 The compounds of the present invention are useful as
04 additives in hydrocarbon fuels to prevent and control engine
05 deposits, particularly intake valve deposits. The proper
06 concentration of additive necessary to achieve the desired
O~ deposit control varies depending upon the type of fuel
Og employed, the type of engine, and the presence of other fuel
pg additives.
11 In general, the concentration of the compounds of this
12 invention in hydrocarbon fuel will range from about 50 to
13 about 2500 parts per million (ppm) by weight, preferably
14 from 75 to 1,000 ppm. When other deposit control additives
are present, a lesser amount of the present additive may be
16 used.
17
lg The compounds of the present invention may be formulated as
lg a concentrate using an inert stable oleophilic (i.e.,
dissolves in gasoline) organic solvent boiling in the range
21 of about 150°F. to 400°F. (about 65°C. to
205°C.).
22 Preferably, an aliphatic or an aromatic hydrocarbon solvent
23 is used, such as_benzene, toluene, xylene or higher-boiling
24 aromatics or aromatic thinners. Aliphatic alcohols
containing about 3 to 8 carbon atoms, such as isopropanol,
26 isobutylcarbinol, n-butanol and the like, in combination
2~ with hydrocarbon solvents are also suitable for use with the
28 present additives. In the concentrate, the amount of the
2g additive will generally range from about l0 to about
70 weight percent, preferably 10 to 50 weight percent, more
31 preferably from 20 to 40 weight percent.
32 In gasoline fuels, other fuel additives may be employed with
33 the additives of the present invention, including, for
34 example, oxygenates, such as t-butyl methyl ether, antiknock
CA 02226668 1998-O1-13
WO 97/43358 PCT/US97/07990
-24-
01 agents, such as methylcyclopentadienyl manganese
02 tricarbonyl, and other dispersants/detergents, such as
03 hydrocarbyl amines, hydrocarbyl poly(oxyalkylene) amines,
04 hydrocarbyl poly(oxyalkylene) aminocarbamates, or
05 succinimides. Additionally, antioxidants, metal
06 deactivators and demulsifiers may be present.
07
Og In diesel fuels, other well-known additives can be employed,
09 such as pour point depressants, flow improvers, cetane
improvers, and the like.
11
12 A fuel-soluble, nonvolatile carrier fluid or oil may also be
13 used with the aromatic esters of this invention. The
14 carrier fluid is a chemically inert hydrocarbon-soluble
liquid vehicle which substantially increases the nonvolatile
16 residue (NVR), or solvent-free liquid fraction of the fuel
17 additive composition while not overwhelmingly contributing
ig to octane requirement increase. The carrier fluid may be a
lg natural or synthetic oil, such as mineral oil, refined
petroleum oils, synthetic polyalkanes and alkenes, including
21 hydrogenated and unhydrogenated polyalphaolefins, and
22 synthetic polyoxyalkylene-derived oils, such as those
23 described, for example, in U.S. Patent No. 4,191,537 to
24 Lewis, and polyesters, such as those described, for example,
in U.S. Patent Nos. 3,756,793 to Robinson and 5,004,478 to
26 Vogel et al., and in European Patent Application
27 Nos. 356,726, published March 7, 1990, and 382,159,
2g published August 16, 1990.
29
These carrier fluids are believed to act as a carrier for
31 the fuel additives of the present invention and to assist in
32 removing and retarding deposits. The carrier fluid may also
33 exhibit synergistic deposit control properties when used a.n
34
CA 02226668 1998-O1-13
WO 97/43358 PCT/LTS97/07990
-25-
O1 combination with a polyalltyl aromatic ester of this
02 invention.
03
04 The carrier fluids are typically employed in amounts ranging
05 from about 100 to about 5000 ppm by weight of the
06 hydrocarbon fuel, preferably from 400 to 3000 ppm of the
07 fuel. Preferably, the ratio of carrier fluid to deposit
Og control additive will range from about 0.5:1 to about,10:1,
Og more preferably from 1:1 to 4:1, most preferably about 2:1.
to
il When employed in a fuel concentrate, carrier fluids will
12 generally be present in amounts ranging from about 20 to
13 about 60 weight percent, preferably from 30 to 50 weight
14 Percent.
16 PREPARATTONS AND EXAMPLES
17
ig A further understanding of the invention can be had in the
ig following nonlimiting Examples. Wherein unless expressly
stated to the contrary, all temperatures and temperature
21 ranges refer to the Centigrade system and the term "ambient"
22 or "room temperature" refers to about 20°C.-25°C. The term
23 "percent" or "~"--refers to weight percent and the term
24 "mole'° or "moles" refers to gram moles. The term
"equivalent" refers to a quantity of reagent equal in moles,
26 to the moles of the preceding or succeeding reactant recited
27 in that example in terms of finite moles or finite weight or
2g volume. Where given, proton-magnetic resonance spectrum
29 (p.m.r. or n.m.r.) were determined at 300 mHz, signals are
assigned as singlets (s), broad singlets (bs), doublets (dj,
31 double doublets (dd), triplets (t), double triplets (dt),
32 quartets (q), and multiplets (m), and cps refers to cycles
33 per second.
34
CA 02226668 1998-O1-13
WO 97!43358 PCT/US97/07990
-26-
O1 Examt~le 1
02
03 Pret~aration of Polvisobutyl Phenol
04
05 To a flask equipped with a magnetic stirrer, reflux
06 condenser, thermometer, addition funnel and nitrogen inlet
07 was added 203.2 grams of phenol. The phenol was warmed to
Og 40°C. and the heat source was removed. Then, 73.5
p9 milliliters of boron trifluoride etherate was added
dropwise. 1040 grams of Ultravis 10 Polyisobutene
11 (molecular weight 950, 76% methylvinylidene, available from
12 British Petroleum) was dissolved in 1,863 milliliters of
13 hexane. The polyisobutene was added to the reaction at a
14 rate to maintain the temperature between 22°C-27°C. The
reaction mixture was stirred for 16 hours at room
16 temperature. Then, 400 milliliters_of concentrated ammonium
17 hydroxide was added, followed by 2,000 milliliters of
18 hexane. The reaction mixture was washed with water (3 X
19 2,000 milliliters), dried over magnesium sulfate, filtered
and the solvents removed under vacuum to yield 1,056.5 grams
21 of a crude reaction product. The crude reaction product was
22 determined to contain 80% of the desired product by proton
23 NMR and chromatography on silica gel eluting with hexane,
24 followed by hexane: ethylacetate: ethanol (93:5:2).
26
27
28
29
31
32
33
34 _
CA 02226668 1998-O1-13
WO 97/43358 PCT/US97/07990
-27-
01 Example 2
02
03 preparation of
04
05 OOH
06
07
08
09
1o P IB (molecular weight ~ 950)
il
12
1.1 grams of a 35 weight percent dispersion of potassium
13
hydride in mineral oil and 4- polyisobutyl phenol (99.7
14
grams, prepared as in Example 1) were added to a flask
equipped with a magnetic stirrer, reflux condensor, nitrogen
16
inlet and thermometer. The reaction was heated at 130°C for
17
one hour and then cooled to 100°C. Ethylene carbonate (8.6
18 grams) was added and the mixture was heated at 160°C for 16
19
hours. The reaction was cooled to room temperature and one
milliliter of isopropanol was added. The reaction was
21
diluted with one liter of hexane, washed three times with
22
water and once with brine. The organic layer was dried over
23
.anhydrous magnesium sulfate, filtered and the solvents
24
removed in vacuo to yield 98.0 grams of the desired product
as a yellow oil.
26
27
28
29
31
32
33
34
CA 02226668 1998-O1-13
WO 97/43358 PCT/I7S97/07990
-28-
O1 Example 3
02
03 Preparation of
04 '
05 OH
06 O "
07
08
09
la P!B (molecular weight ~ 950)
11
12
15.1 grams of a 35 weight percent dispersion of potassium
13
hydride in mineral oil and 4- polyisobutyl phenol (1378.5
14
grams, prepared as in Example 1) were added to a flask
16 e~lpped with a mechanical stirrer, reflux condenser,
nitrogen inlet and thermometer. The reaction was heated at
17
13o°C for one hour and then cooled to 100°C. Propylene
18
carbonate (115.7 milliliters) was added and the mixture was
19
heated at 160°C for 16 hours. The reaction was cooled to
room temperature and ten milliliters of isopropanol were
21
added. The reaction was diluted with ten liters of hexane,
22
washed three times with water and once with brine. The
23
organic layer was dried over anhydrous magnesium sulfate,
24
filtered and the solvents removed in vacuo to yield 1301.7
grams of the desired product as a yellow oil.
26
27
28
29
31
32
33
34
CA 02226668 1998-O1-13
WO 97/43358 PCT/US97/07990
-29-
O1 Example 4
02
03 Preparation of
04
05 ~~2
06
0 7 ~~o
08
09
11
12 P IB (molecular weigi~ ~ 950)
13
14 To a flask equipped with a magnetic stirrer, thermometer,
Dean-Stark trap, reflux condensor and nitrogen inlet was
16 added 15.0 grams of the alcohol from Example 2, 2.6 grams of
17 4-nitrobenzoic acid and 0.24 grams of p-toluenesulfonic
18 acid. The mixture was stirred at 130°C for sixteen hours,
19 cooled to room temperature and diluted with 200 mL of
hexane. The organic phase was washed twice with saturated
21 aqueous sodium bicarbonate followed by once with saturated
22 aqueous sodium chloride. The organic layer was then dried
23 over anhydrous magnesium sulfate, filtered and the solvents
24 removed in vacuo to yield 15.0 grams of the desired product
as a brown oil. The oil was chromatographed on silica gel,
26 eluting with hexane/ethyl acetate (9:1) to afford 14.0 grams
27 of the desired ester as a yellow oil. ~'H NMR (CDClg) d 8.3
28 (AB quartet, 4H), 7.25 (d, 2H), 6.85 (d, 2H), 4.7 (t, 2H),
29 4.3 (t, 2H), 0.7-1.6 (m, 137H). _
31
32
33
34
CA 02226668 1998-O1-13
WO 97/43358 PCT/US97/U7990
-30-
O1 Examt~le 5
02
03 Preparation of
04 '
05
06 ~ ~ "
07 O O
08
Q9 / O
~
11
12 PIB (molecularweigf~ ~ 950)
13
14 To a flask equipped with a magnetic stirrer, thermometer,
Dean-Stark trap, reflux condensor and nitrogen inlet was
16 added 15.0 grams of the alcohol from Example 3, 2.7 grams of
17 4-nitrobenzoic acid and 0.23 grams of p-toluenesulfonic
18 acid. The mixture was stirred at 130°C for sixteen hours,
ig cooled to room temperature and diluted with 200 mL of
hexane. The organic phase was washed twice with saturated
21 aqueous sodium bicarbonate followed by once with saturated
22 aqueous sodium chloride. The organic layer was then dried
23 over anhydrous magnesium sulfate, filtered and the solvents
24 removed in vacuo to yield 16.0 grams of the desired product
as a brown oil. The oil was chromatographed on silica gel,
26 eluting with hexane/ethyl acetate (8:2) to afford 15.2 grams
27 of the desired ester as a brown oil. IH NMR (CDC13) d 8.2
28 (AB quartet, 4H), 7.25 (d, 2H), 6.85 (d, 2H), 5.55 (hx, 1H),
2g 4.1 (t, 2H), 0.6-1.8 (m, 140H).
31
32
33
34
CA 02226668 1998-O1-13
WO 97/43358 PCT/(JS97/07990
-31-
O1 Example 6
02
03 Preparation of
04
05 NH2
06 ( \
07 ~/~~ /
08
09 /
\
11
12 P1B (molecular weight ~ 950}
13
14 A solution of 9.4 grams of the product from Example 4 in 100
milliliters of ethyl acetate containing 1.0 gram of lob
16 palladium on charcoal was hydrogenolyzed at 35-40 psi for 16
17 hours on a Parr low-pressure hydrogenator. Catalyst
18 filtration and removal of the solvent in vacuo yield 7.7
19 grams of the desired product as a yellow oil. 1H NMR
(CDClg) d 7.85 (d, 2H), 7.3 (d, 2H), 6.85 (d, 2H), 6.6 (d,
21 2H), 4.6 (t, 2H), 4.25 (t, 2H), 4.05 (bs, 2H), 0.7-1.6 (m,
22 137H) .
23 . .
24
26
27
28
29
31
32
33
34
CA 02226668 1998-O1-13
WO 97/4338 PCT/CTS97/07990
-32-
O1 Example 7
02
03 Preparation of
04 NH2
05
06 O o ~
O7
0
09
11 P!B (molecular weight ~ 950}
12
13 A solution of 15.2 grams of the product from Example 5 in
14 200 milliliters of ethyl acetate containing 1.0 gram of 10~
palladium on charcoal was hydrogenolyzed at 35-40 psi for 16
16 hours on a Parr low-pressure hydrogenator. Catalyst
1~ filtration and removal of the solvent in vacuo yield 15.0
18 grams of the desired product as a brown oil. 1H NMR
19 (CDC13/DZO} d 7.85 (d, 2H), 7.25 (d, 2H}, 6.85 (d, 2Hj, 6.6
(d~ 2H}, 5.4 (hx, 1H), 3.8-4.2 (m, 4H), 0.6-1.8 (m, l4oH).
21
22
23 .. Example 8
24
Sinctle-Cylinder Enctine Test
26
2~ The test compounds were blended in gasoline and their
28 deposit reducing capacity determined in an ASTM/CFR
2g single-cylinder engine test.
31 A Waukesha CFR single-cylinder engine was used. Each run
32 was carried out for 15 hours, at the end of which time the
33 intake valve was removed, washed with hexane and weighed.
34 The previously determined weight of the clean valve was -
CA 02226668 1998-O1-13
WO 97/43358 PCT/US97/07990
-33-
O1 subtracted from the weight of the value at the end of the
02 run. The differences between the two weights is the weight
03 of the deposit. A lesser amount of deposit indicates a
04 superior additive. The operating conditions of the test
05 were as follows: water jacket temperature 200°F; vacuum of
06 12 in Hg, air-fuel ratio of 12, ignition spark timing of
07 400 BTC; engine speed is 1800 rpm; the crankcase oil is a
08 commercial 30W oil.
09
The amount of carbonaceous deposit in milligrams on the
11 intake valves is reported for each of the test compounds in
12 Table I.
13
TABLE I
14
Intake Valve Deposit Weight
16 (in milligrams)
17 Sampler Run 1 Run 2 Average
18 Base Fuel 354.9 333.5 344.2
1g Example 4 169.0 178.0 173.5
Example 6 13.4 12.2 12.8
21
22 lAt 150 parts per million actives (ppma).
23
24 The base fuel employed in the above single-cylinder engine
tests was a regular octane unleaded gasoline containing no
26 fuel detergent. The test compounds were admixed with the
27 base fuel to give a concentration of 150 ppma (parts per
28 million actives).
29 _
The data in Table I illustrates the significant reduction in
31 intake valve deposits provided by the aromatic esters of
32 polYalkylphenoxyalkanols of the present invention (Examples
33 4 and 6) compared to the base fuel.
34